

Abstract
Expression of the 17β-estradiol (E2) synthesis enzyme aromatase is highly upregulated in astrocytes following brain injury. However, the precise role of astrocyte-derived E2 in the injured brain remains unclear. In the current study, we generated a glial fibrillary acidic protein (GFAP) promoter-driven aromatase knock-out (GFAP-ARO-KO) mouse model to deplete astrocyte-derived E2 in the brain and determine its roles after global cerebral ischemia (GCI) in male and female mice. GFAP-ARO-KO mice were viable and fertile, with normal gross brain structure, normal morphology, intensity and distribution of astrocytes, normal aromatase expression in neurons, and normal cognitive function basally. In contrast, after GCI, GFAP-ARO-KO mice: (1) lacked the normal elevation of astrocyte aromatase and hippocampal E2 levels; (2) had significantly attenuated reactive astrogliosis; and (3) displayed enhanced neuronal damage, microglia activation, and cognitive deficits. RNA-sequencing (RNA-seq) analysis revealed that the ischemic GFAP-ARO-KO mouse hippocampus failed to upregulate the “A2” panel of reactive astrocyte genes. In addition, the JAK-STAT3 pathway, which is critical for the induction of reactive astrogliosis, was significantly downregulated in the GFAP-ARO-KO hippocampus following GCI. Finally, exogenous E2 administration fully rescued the compromised JAK-STAT3 pathway and reactive astrogliosis, and reversed the enhanced neuronal damage and microglial activation in the GFAP-ARO-KO mice after GCI, suggesting that the defects in the KO mice are because of a loss of E2 rather than an increase in precursor androgens. In conclusion, the current study provides novel genetic evidence for a beneficial role of astrocyte-derived E2 in reactive astrogliosis, microglial activation, and neuroprotection following an ischemic injury to the brain.
SIGNIFICANCE STATEMENT Following cerebral ischemia, reactive astrocytes express the enzyme aromatase and produce 17β-estradiol (E2), although the precise role of astrocyte-derived E2 is poorly understood. In this study, we generated a glial fibrillary acidic protein (GFAP) promoter-driven aromatase knock-out (GFAP-ARO-KO) mouse to deplete astrocyte-derived E2 and elucidate its roles after global cerebral ischemia (GCI). The GFAP-ARO-KO mice exhibited significantly attenuated reactive astrogliosis, as well as enhanced microglial activation, neuronal damage, and cognitive dysfunction after GCI. Transcriptome analysis further revealed that astrocyte-derived E2 was critical for the induction of the JAK-STAT3 signaling pathway, as well as the A2 reactive astrocyte phenotype after ischemia. Collectively, these findings indicate that astrocyte-derived E2 has a key role in the regulation of reactive astrogliosis, microglial activation, and neuroprotection after cerebral ischemia.
Keywords: 17β-estradiol, aromatase, astrocyte activation, estrogen, global cerebral ischemia, hippocampus
Introduction
The steroid hormone, 17β-estradiol (E2) is well known to be produced in the ovaries of females, where it is released into the bloodstream to exert actions throughout the body (Simpkins et al., 2005; Brann et al., 2007). In addition to this gonadal source of E2, there is growing evidence that E2 is also produced locally in both the male and female brain through the conversion of androgen precursors to E2 by the biosynthetic enzyme, aromatase (Hojo et al., 2004; Roselli et al., 2009; Fester et al., 2011). Although the expression of aromatase is almost exclusively detected in neurons under physiological conditions, it can be highly upregulated in reactive astrocytes in the injured or ischemic brain (Garcia-Segura et al., 1999; Zhang et al., 2014). Recent work from our group showed that neuron-derived E2 functions as a neuromodulator in the forebrain to control synaptic plasticity and cognitive function (Lu et al., 2019). However, the precise role of astrocyte-derived E2 in the brain has not been fully addressed.
The production of E2 and aromatase expression in astrocytes was first detected in primary cultures from the developing zebra finch telencephalon (Schlinger et al., 1994) and also in astrocytes cultured from the cerebral cortex of neonatal rats (Zwain et al., 1997). Later, an in vivo study using a neurotoxic lesion model and a penetrating brain injury model reported a significant induction of aromatase expression in reactive astrocytes surrounding the injured lesion of both the adult mouse and rat brain (Garcia-Segura et al., 1999). Robust upregulation of glial aromatase expression following different forms of brain injuries has been extensively reported in songbird (Peterson et al., 2001; Duncan et al., 2013) as well as in mammals (Garcia-Segura et al., 2003; Zhang et al., 2014). In the adult rat brain, aromatase was shown to be highly expressed in glial fibrillary acidic protein (GFAP)-positive astrocytes in the hippocampus and brain E2 levels were found to be significantly increased at 2–3 d after global cerebral ischemia (GCI) reperfusion (Zhang et al., 2014). Knock-down of aromatase in rat hippocampus blocked the increase of E2 levels, and resulted in increased neuronal damage and enhanced microglia activation after GCI (Zhang et al., 2014), suggesting that brain-derived E2 has a neuroprotective role and anti-inflammatory actions against ischemic brain injury. However, the knock-down experiments reduced both neuronal and astrocyte aromatase expression, making it difficult to differentiate between the contributions of neuron-derived E2 versus astrocyte-derived E2 in the ischemic brain.
In the current study, we generated an astrocyte-specific aromatase knock-out (ARO-KO) mouse line, in which the deletion of aromatase occurs under the control of the GFAP promoter. Herein, we show that the induction of aromatase expression in astrocytes and the elevation of hippocampal E2 levels following GCI in the GFAP-ARO-KO mice were blocked, while neuronal aromatase expression was maintained. Basally, GFAP-ARO-KO mice exhibited no cognitive deficits, and the morphology and physical functions of astrocytes were not significantly changed in GFAP-ARO-KO mice as compared with FLOX mice. In contrast, examination of GFAP-ARO-KO after GCI revealed significantly increased neuronal damage in the hippocampal CA1 region, as well as enhanced cognitive decline, as compared with FLOX-GCI controls. In addition, an obvious astrocyte activation deficit was detected in the GFAP-ARO-KO mice following GCI, along with significant inhibition of the “A2” panel of reactive astrocyte genes and the leukemia inhibitory factor (LIF)/STAT3 pathway. Furthermore, the hemostatic microglial signature genes were suppressed, while inflammatory microglial activation was enhanced in the GFAP-ARO-KO mice after GCI, as compared with the FLOX-GCI control. Importantly, the cellular and molecular phenotypes could be rescued by reinstating forebrain E2 levels via exogenous E2 administration, indicating that the defects in the GFAP-ARO-KO mice are because of loss of E2 rather than an increase in androgen precursors. Altogether, our results demonstrate a neuroprotective role of astrocyte-derived E2 against GCI, and suggest a novel mechanism for astrocyte activation in response to ischemic brain injury through the LIF/STAT3 signaling pathway.
Materials and Methods
Generation of astrocyte-specific ARO-KO mouse model
To conditionally inactivate the estrogen synthetic enzyme, aromatase, in astrocytes, we generated Cyp19a1 Cre/LoxP conditional KO mice under the control of the GFAP promoter. Specifically, the GFAP-Cre (purchased from The Jackson Laboratory) mice were crossed with Cyp19a1 flox/flox mice, which contain LoxP sites flanking exons 10 and 11 of the Cyp19a1 gene. The Cyp19a1 flox/flox (FLOX) mice were created by Genoway. Inc (Lyon, France) as previously described (Lu et al., 2019), and were maintained in the C57BL/6 background. To avoid the potential recombination in the ova of GFAP Cre+/− mice, the female Cyp19a1 flox/flox mice were crossed with male Cyp19a1 flox/flox; GFAP-Cre+/− to generate needed mice for experiments. The genotyping was conducted by genomic DNA PCR using the following primers: FLOX forward: 5′-GCCATATTCCTGCAACAGTTTATTTGAGG-3′ and FLOX reverse: 5′-GTAAAACCATTCCTGGAAAATTCATAACAACC-3′; Cre forward: 5′-GCGGTCTGGCAGTAAAAACTATC-3′ and Cre reverse: 5′-GTGAAACAG CATTGCTGTCACTT-3′; Cre excised FLOX forward: 5′-GCATGATCTCATCGACTCTGTAGCAGC-3′ and Cre excised FLOX reverse: 5′-ATGAGCAAGTGTGCCTATGCCAAGC-3′. All experiments with mice were performed using approved IACUC protocols and are within the guidelines of the Laboratory Animal Services and Institutional Animal Care and Use Committee of Augusta University and University of Texas Health San Antonio.
Ovariectomy and GCI
Surgeries were performed on anesthetized mice (1–4% isoflurane) given preanalgesic (16 h before) and postanalgesic (0–16 h after surgery; carprofen). Bilateral ovariectomy was conducted to three- to four-month-old female mice at 7–10 d before GCI or sham surgery. Three- to four-month-old intact male and ovariectomized (ovx) female mice were subject to a two-vessel occlusion GCI model as previously described (Sareddy et al., 2015). Briefly, two common carotid arteries (CCAs) were isolated and occluded with artery clips for 20 min under anesthesia. Afterward, the artery clips were removed to allow reperfusion, and reperfusion was confirmed by visual inspection of blood flow in CCAs. The core body temperature of mice before, during and after ischemia was maintained at 37°C by a homoeothermic blanket. Five minutes after reperfusion, the incisions were closed, and mice were kept in a warm chamber to maintain the body temperature at ∼37°C. Mice which showed dilated pupils during surgery, and lost the righting reflex for at least 1 min after incision closure were included for additional experiments.
Astrocyte purification and flow cytometry
Brain hemispheres were collected from sham or GCI mice at indicated time points postsurgery. The acutely isolated brain tissue was dissociated into a single-cell suspension using an adult brain dissociation kit (MiltenyiBiotec, 130-107-677) as previously described (Wang et al., 2017). The dissociated cells were then processed with MACS sorting using ACSC2 MicroBeads (MiltenyiBiotec, 130-093-388) to generate a purified astrocyte population. Purified ACSC2+ cells were then stained with anti-ACSC2-APC (MiltenyiBiotec), and subjected to flow cytometry analysis on a FACS-Canto flow cytometer (BD Bioscience).
Glutamate uptake assay
Glutamate uptake assay was conducted on purified astrocytes obtained from three-month-old FLOX and KO mouse brain following a modified protocol described previously (Duan et al., 1999). Briefly, acutely isolated astrocytes (50 000 cells/ml) were seeded on ploy-D-lysine (Sigma) coated 24-well plates in DMEM (Corning) with 10% fetal bovine serum (HyClone) and 1% penicillin streptomycin (Invitrogen), followed by incubating at 37% with 5% CO2 for 24 h. At 24 h in culture, adult astrocytes were washed twice with HBSS (Sigma) at room temperature for 5 min. Afterward, 300 µl of HBSS containing 100 μm glutamate was added into each well, and the media were harvested at 5-, 10-, and 30-min intervals. At each time point, medium from a well without astrocytes was collected and served as a control. Protein lysates of astrocytes in each well were collected afterward, and subjected to protein concentration analysis with a microplate BCA protein assay kit (Thermo Fisher Scientific). Glutamate uptake was assessed by measuring the glutamate concentration in the medium using a Glutamate Assay kit (Abcam) following the manufacturer's instruction. A total of 50 µl of medium samples was used for measurements by the Glutamate Assay kit (with a measurement range of 2–10 nmol). Glutamate uptake rate for FLOX and KO astrocytes was calculated by normalization to the astrocyte lysate protein concentration (measured as described above). Duplicated wells and samples media from two independent experiments were used for measurement and statistical analysis.
Tissue lysates preparation and Western blotting
Mice were killed under deep anesthesia at different time points, as detailed in the experiments. Mice were perfused with ice-cold 0.9% saline, and the cortex and hippocampus were isolated. Total proteins were extracted from the dissected mouse brain tissue using RIPA buffer (50 mm TrisHCl at pH 7.4, 150 mm NaCl, 1% Triton X-100, 1% sodium deoxycholate, 0.1%SDS, and 1 mm EDTA) supplemented with protease and phosphatase inhibitors (Thermo Fisher Scientific). The protein concentrations of samples were determined by a microplate BCA protein assay kit (Thermo Fisher Scientific). Equal amounts of protein (20 or 40 µg) were subjected to an SDS-PAGE and transferred onto a nitrocellulose membrane for Western blot analysis as previously described (Wang and Yu, 2013). Antibodies used for Western blot analyses include: aromatase (rabbit, 1:1000, ThermoFisher, #PA1-16 532), Iba1 (goat, 1:1000, Abcam, #ab5076), GFAP (mouse, 1:2000, Millipore, #MAB360), GFAP (goat, 1:2000, Abcam, #ab53554), S100A10 (mouse, 1:800, Thermo Fisher Scientific, #MA5-24789), GAPDH (mouse, 1:2000, Santa Cruz Biotechnology, #sc-32233), β-actin (mouse, 1:4000, Sigma, #A5441), Cleaved Caspase-3 (Asp175; rabbit, 1:1000, Cell Signaling, #9661), Caspase-3 (rabbit, 1:1000, Cell Signaling, #9662), phospho-Stat3 (Tyr705; rabbit, 1:800, Cell Signaling, #9131), STAT3 (mouse, 1:1000, Cell Signaling, #9139), and anti-glutamate transporter 1 (GLT-1; rabbit, 1:1000, Abcam, #ab106289). Blots were visualized using a LI-COR Odyssey imager, and the ImageJ analysis software (version 1.49; NIH) was used to determine the intensity of each band. Band densities for the indicated proteins were normalized to the corresponding loading controls.
Immunohistochemistry (IHC)
Mouse brain sections were prepared as previously described (Wang et al., 2017). Briefly, after transcardially perfused with ice-cold saline, the brain was harvested and postfixed in 4% paraformaldehyde at 4°C for 16 h and cryoprotected with 30% sucrose. Coronal sections of 30-µm thickness were collected between −0.9 and −3.0 mm from bregma. Cresyl violet staining was performed as previously described (Wang et al., 2017). For immunofluorescence (IF) analysis, brain sections were blocked for 1 h with 10% normal donkey serum at room temperature, followed by overnight incubation at 4°C with primary antibodies. The primary antibodies used in this study include: aromatase (rabbit, 1:1000, Thermo Fisher Scientific, #PA1-16532), GFAP (goat, 1:2000, Abcam, #ab53554), Iba1 (goat, 1:400, Abcam, #ab5076), NeuN (rabbit, 1:1000, Millipore, #ABN78), GFAP (mouse, 1:800, Millipore, #MAB360), Cleaved Caspase-3 (Asp175; rabbit, 1:400, Cell Signaling, #9661), phospho-Stat3 (Tyr705; rabbit, 1:800, Cell Signaling, #9131), S100A10 (mouse, 1:400; Thermo Fisher Scientific, #MA5-24789), anti-GLT-1 (rabbit, 1:400, Abcam, #ab106289), and anti-EAAT1 (GLAST, rabbit, 1:400, Abcam, #ab416). Sections were subsequently washed and incubated with appropriate Alexa Fluor-tagged secondary antibodies (ThermoFisher) for 1 h at room temperature. After another three washes, the sections were mounted in Vectashield mounting medium with DAPI (Vector Laboratories). Images of each marker were acquired using a Zeiss 510 confocal microscope at 10×, 40×, or 63× Plan-apochromat objective. Exposure time was kept constant for each marker in all sections for analysis. For DAB staining, sections were incubated with 0.3% H2O2 for 30 min before blocking with 10% normal horse serum and then incubated with anti-S100β (mouse, 1:400, Sigma, #S2523) at 4°C overnight. After washing, sections were incubated with biotin-conjugated anti-mouse antibody and ABC reagent (VECTASTAIN Elite ABC kit, #PK-6100), and further developed using ImmPACT DAB Peroxidase Substrate (VECTOR, #SK-4105). The intensity of S100β staining and the number of S100β+ cells in the hippocampal CA1 were quantified with ImageJ software.
ELISA
E2 levels were measured in 100 μl freshly prepared hippocampal tissue lysates using a high-sensitivity ELISA kit (Enzo Life Sciences, #ADI-900-174), as previously described (Lu et al., 2019). The brain LIF and interleukin-6 (IL-6) levels were measured in 50-μl hippocampal tissue lysates using Quantikine ELISA kits (R&D Systems, #MLF00 and #M600B) following the manufacturer's instruction. The values of E2, LIF, and IL-6 levels were calculated by their concentrations in the lysate normalized to the protein concentration of each sample.
Barnes maze task
All the behavioral tests were conducted on three- to four-month-old male or ovx-females at two weeks after ovariectomy surgery. For the sham and GCI animal groups, the Barnes maze task was started at 7 d after GCI surgery following a protocol as previously described (Lu et al., 2019). Briefly, this behavior task was divided into two stages: a 180-s training trial for 3 d and a 90-s probe trial on day 4. On each day of the training trial stage (days 1–3), the escape latency of mice from the center of the platform to enter the hidden chamber was recorded by an overhead video camera controlled by ANY-maze video tracking software (Stoelting Co). For the probe test (day 4), the time the mouse spent in the target quadrant where the hidden box had been was recorded for 90 s.
Open field test
The open field test was conducted the next day following the Barnes maze task and was assessed on animals at 12 d after the GCI/sham surgery as previously described (Lu et al., 2019). A black open box with 56 × 56 cm was used, and the mice were traced by an overhead video camera controlled by the ANY-maze video tracking software. At the beginning of the test, a mouse was placed at one corner of the box, making it face the wall. During a 5-min period, the travel distance, average speed, as well as time spent in a delineated center zone (26 × 26 cm) were recorded.
Novel object recognition test
The novel object recognition test was conducted the next day following the open field test and was assessed on animals at 13 and 14 d after the GCI/sham surgery as previously described (Lu et al., 2019). On the first day (stage 1), the mice were placed in the recognition box containing two identical objects at an equal distance for 5-min exploration. On the second day (stage 2), the mice were returned to the same recognition box in the presence of one familiar object and one novel object to test the long-term recognition memory. Time spent exploring each object was recorded using ANY-maze video tracking software, and the discrimination index (Ennaceur and Delacour, 1988), calculated by the ratio of (time devoted to novel object – time devoted to the familiar project)/(time devoted to novel object + time devoted to the familiar project), was analyzed afterward.
Fear conditioning test
Fear conditioning was conducted for the basal level studies and was conducted as previously described (Lu et al., 2019) using a fear-conditioning chamber (17 × 17.5 × 33 cm3). Briefly, the task was divided into two sessions: conditioning (day 1) and the fear memory test (day 2). On day 1, mice were placed in the conditioning chamber for a 2-min acclimation, followed by three 30-s tones co-terminated with a 2-s shock at 0.5 mA for every 2 min. Mice were returned to their home cages immediately after conditioning. At day 2, a contextual fear memory was evaluated by putting the mice into the same conditioning chamber for 5 min without shock. Freezing time was recorded during this period.
RNA-sequencing (RNA-seq) and quantitative (q)RT-PCR
Total RNA was isolated from the hippocampal tissue of ovx-female ARO-FLOX and GFAP-ARO-KO mice subjected to GCI and reperfusion for 24 h. Bilateral ovariectomy of female mice was conducted one week before GCI surgery. Total RNA was isolated using an RNeasy mini kit (QIAGEN). Samples were run on an Illumina HiSeq 3000 in duplicate. Illumina TruSeq RNA sample preparation and sequencing were conducted using University of Texas Health San Antonio genomics core facility protocol. RNA-seq data has been deposited in the GEO database under a GEO accession number (GSE154383). DEseq was used to identify differentially expressed genes (DEGs) and the significant genes with fold change >2, and adjusted p < 0.05 were used for subsequent analysis. The DEGs were further interpreted for biological pathways using Ingenuity Pathway Analysis (IPA) software and gene enrichment analysis were performed using Gene Ontology (GO). Gene set enrichment analysis (GSEA) was used to analyze the association of GFAP-ARO-KO with specific signaling pathways. Quantitative real-time PCR was conducted to verify the RNA-seq results using SybrGreen on an Illumina Real-Time PCR system using the following primers:
Steap4 forward: 5′-CAGAGTCAAATGCGGAATACCT-3′,
Steap4 reverse: 5′-GGCTTGCATCTAGTGTTCCTG-3′;
Serpina3n forward: 5′-ATTTGTCCCAATGTCTGCGAA-3′,
Serpina3n reverse: 5′-TGGCTATCTTGGCTATAAAGGGG-3′;
Timp1 forward: 5′-GCAACTCGGACCTGGTCATAA-3′,
Timp1 reverse: 5′-CGGCCCGTGATGAGAAACT-3′;
Cxcl10 forward: 5′-CCAAGTGCTGCCGTCATTTTC-3′,
Cxcl10 reverse: 5′-GGCTCGCAGGGATGATTTCAA-3′;
Cd44 forward: 5′-TCGATTTGAATGTAACCTGCCG-3′,
Cd44 reverse: 5′-CAGTCCGGGAGATACTGTAGC-3′;
Osmr forward: 5′-GCATCCCGAAGCGAAGTCTT-3′,
Osmr reverse: 5′-GGGCTGGGACAGTCCATTCTA-3′;
Aspg forward: 5′-CTGACCAGAGGATCATCTACACG-3′,
Aspg reverse:5′-CTCTATGGTTTGGGCAATCTGAA-3′;
Vimentin forward: 5′-CGTCCACACGCACCTACAG-3′,
Vimentin reverse: 5′-GGGGGATGAGGAATAGAGGCT-3′;
Gfap forward: 5′-CGGAGACGCATCACCTCTG-3′,
Gfap reverse:5′-AGGGAGTGGAGGAGTCATTCG-3′;
Clcf1 forward: 5′-GACTCGTGGGGGATGTTAGC-3′,
Clcf1 reverse:5′-CTAAGCTGCGGAGTTGATGCT-3′;
Tgm1 forward: 5′-TCTGGGCTCGTTGTTGTGG-3′,
Tgm1 reverse: 5′-AACCAGCATTCCCTCTCGGA-3′;
Ptx3 forward:5′-CCTGCGATCCTGCTTTGTG-3′,
Ptx3 reverse: 5′-GGTGGGATGAAGTCCATTGTC-3′;
S100a10 forward: 5′-TGGAAACCATGATGCTTACGTT-3′,
S100a10 reverse: 5′-GAAGCCCACTTTGCCATCTC-3′;
Emp1 forward: 5′-TTGGTGCTACTGGCTGGTCT-3′,
Emp1 reverse: 5′-CATTGCCGTAGGACAGGGAG-3′;
Slc10a6 forward: 5′-GGAGGGCCATGCGAATCTAAA-3′,
Slc10a6 reverse: 5′-TGTCAGAGGCATAAGTCCAAAC-3′;
Tm4sf1 forward: 5′-ACATCGGATACTCTCTGGTGTG-3′,
Tm4sf1 reverse: 5′-GCATACTTTGTCTCCCCATTAGG-3′;
Cd14 forward: 5′-CTCTGTCCTTAAAGCGGCTTAC-3′,
Cd14 reverse:5′-GTTGCGGAGGTTCAAGATGTT-3′;
S1pr3 forward: 5′-ACTCTCCGGGAACATTACGAT-3′,
S1pr3 reverse: 5′-CCAAGACGATGAAGCTACAGG-3′;
Ptgs2 forward: 5′-TGAGCAACTATTCCAAACCAGC-3′,
Ptgs2 reverse: 5′-GCACGTAGTCTTCGATCACTATC-3′;
Serping1 forward: 5′-TAGAGCCTTCTCAGATCCCGA-3′,
Serping1 reverse: 5′-ACTCGTTGGCTACTTTACCCA-3′;
Fbln5 forward: 5′-GCTTGTCGTGGGGACATGAT-3′,
Fbln5 reverse: 5′-TGGGGTAGTTGGAAGCTGGTA-3′;
Fkbp5 forward: 5′-TGAGGGCACCAGTAACAATGG-3′,
Fkbp5 reverse: 5′-CAACATCCCTTTGTAGTGGACAT-3′;
H2-t23 forward: 5′-ACAGTCCCGACCCAGAGTAG-3′,
H2-t23 reverse: 5′-CCACGTAGCCGACAATGATGA-3′;
P2ry12 forward: 5′-CATTGACCGCTACCTGAAGACC-3′,
P2ry12 reverse: 5′-GCCTCCTGTTGGTGAGAATCATG-3′;
Tmem119 forward: 5′-ACTACCCATCCTCGTTCCCTGA-3′,
Tmem119 reverse: 5′-TAGCAGCCAGAATGTCAGCCTG-3′;
Gpr34 forward: 5′-GTGCCAAATGTCACTAGCTGTCC-3′,
Gpr34 reverse: 5′-TCCAACCAGTCCCACGAGGAAT-3′;
Olfml3 forward: 5′-TGGATGCCTGTGAATGGCTCTG-3′,
Olfml3 reverse: 5′-ACCACCAAACCGCTTCAGGATC-3′;
Csf1r forward: 5′-TGGATGCCTGTGAATGGCTCTG-3′,
Csf1r reverse: 5′-GTGGGTGTCATTCCAAACCTGC-3′;
Hexb forward: 5′-GCTGTTGGTGAGAGACTCTGGA-3′,
Hexb reverse: 5′-GAGGTTGTGCAGCTATTCCACG-3′;
Mertk forward: 5′-ATCATCCTCGGCTGCTTCTGTG-3′,
Mertk reverse: 5′-ACGACCAGTTGGGAATCCTCCT-3′;
Rhob forward: 5′-TCTGGCTGGTCACGCTGAGAAT-3′,
Rhob reverse: 5′-GGCTCTTTAGAAAAGGATGCTCAG-3′;
Cx3cr1 forward: 5′-GAGCATCACTGACATCTACCTCC-3′,
Cx3cr1 reverse: 5′-AGAAGGCAGTCGTGAGCTTGCA-3′;
Tgfbr1 forward: 5′-TGCTCCAAACCACAGAGTAGGC-3′,
Tgfbr1 reverse: 5′-CCCAGAACACTAAGCCCATTGC-3′;
C1qa forward: 5′-GTGGCTGAAGATGTCTGCCGAG-3′,
C1qa reverse: 5′-TTAAAACCTCGGATACCAGTCCG-3′;
C1qb forward: 5′-CAACCAGGCACTCCAGGGATAA-3′,
C1qb reverse: 5′-CCAACTTTGCCTGGAGTCCCAG-3′;
Spp1 forward: 5′-TCACCATTCGGATGAGTCTG-3′,
Spp1 reverse: 5′-ACTTGTGGCTCTGATGTTCC-3′;
Sall1 forward: 5′-GCTTGCACTATCTGTGGAAGAGC-3′,
Sall1 reverse: 5′-CTGGGAACTTGACAGGATTGCC-3′.
Exogenous E2 administration and aromatase inhibition
Three- to four-month-old ovx-female mice were used for an exogenous E2 “rescue” experiment. E2 was dissolved in 20% β-cyclodextrin at a concentration of 0.1 mg/ml. E2 or placebo (20% β-cyclodextrin, Sigma-Aldrich, C0926) infused Alzet minipumps (model 10014D, 14-d release; Durect Corporation) were implanted subcutaneously in the upper midback region at the time of ovariectomy. The dose of E2 used here yields serum levels of 10–15 pg/ml, which represents the physiological low diestrus 1 level of E2 (Zhang et al., 2008). GCI or sham surgery were conducted at 7 d after the minipump implantation. For the aromatase inhibition study, the aromatase inhibitor letrozole (Sigma-Aldrich, #L6545) was dissolved in 20% β-cyclodextrin at a concentration of 5 mg/ml and stored in an aliquot of 1 ml/tube at −20°C. Letrozole (10 mg/kg) or vehicle (20% β-cyclodextrin, 2 µl/g) treatments were given to three-month-old male wild-type (WT) mice daily through intraperitoneal injection from 1 d before the GCI or sham surgery until the day of killing. The dose of letrozole was selected based on a previous study demonstrating it effectively inhibited E2 synthesis in the mouse brain (Ishihara et al., 2017).
Statistical analyses
GraphPad Prism 6 software was used to analyze all the data. Data are presented as mean ± SEM. Multiple t tests were applied to the expression analysis of the RNA-seq data and compared the fold change difference between FLOX-GCI and KO-GCI. The two variable factors involved are group factors (e.g., FLOX vs KO; vehicle vs letrozole inhibitor; and vehicle vs estrogen infusion) and treatment factors (e.g., sham vs GCI). Unpaired t test was used for two-group and single-factor analysis. One-way ANOVA tests were conducted when there was a single variable factor in the analysis for three or more groups. Two-way ANOVA was used when interactions of both variable factors were analyzed. When the ANOVA test was found to be significant, the post hoc Dunnett's test, Sidak's multiple comparison, test and Tukey's test were conducted to make pairwise comparisons to determine the significance between the two groups. A value of p < 0.05 was considered statistically significant.
Results
Characterization and validation of aromatase deletion in astrocytes of the GFAP-ARO-KO mouse
A transgenic mouse line that expresses a floxed allele of cyp19a1 was established previously (Lu et al., 2019). Conditional ARO-KO mice were generated using Cre/loxP recombination system to study the role of astrocyte-derived estrogen. To conditionally inactivate the estrogen synthetic enzyme, aromatase, in astrocytes, we crossed Cyp19a1flox/flox; GFAP-Cre−/− females with Cyp19a1flox/flox; GFAP-Cre+/− males that express Cre under the control of the GFAP promoter (Fig. 1A). Mice genotypes for Cre and floxed alleles were confirmed with PCR on genomic DNA of tail biopsies and cerebral cortex (Fig. 1B,C). The Cyp19a1 flox/flox (FLOX) mice served as a control in our studies. Both the FLOX and GFAP-ARO-KO (KO) mice are viable and fertile, without obvious gross immunologic, reproductive or neurologic abnormality or phenotype. We next examined the gross morphology and structure of the adult FLOX and KO brains in both three- to four-month-old male and ovx-female mice. Histologic analyses of cresyl violet staining revealed that the adult KO mice exhibited normal gross anatomy of the cerebral cortex and hippocampus (Fig. 1D) in terms of the thickness of and cellularity of the cortex and CA1 neurons, as compared with the FLOX controls (p = 0.3771, unpaired t test; Fig. 1D,F). IHC for GFAP demonstrated that the morphology, intensity and distribution of astrocytes did not have any significant difference between the adult FLOX and KO brains under physiological conditions (p = 0.3870, unpaired t test; Fig. 1E,G). For all the characterization and validation tests (Figs. 1, 2), we compared FLOX versus KO in male and ovx-female animals. We statistically analyzed data from male and ovx-female mice, both of which demonstrate similar phenotypes.
Figure 1.
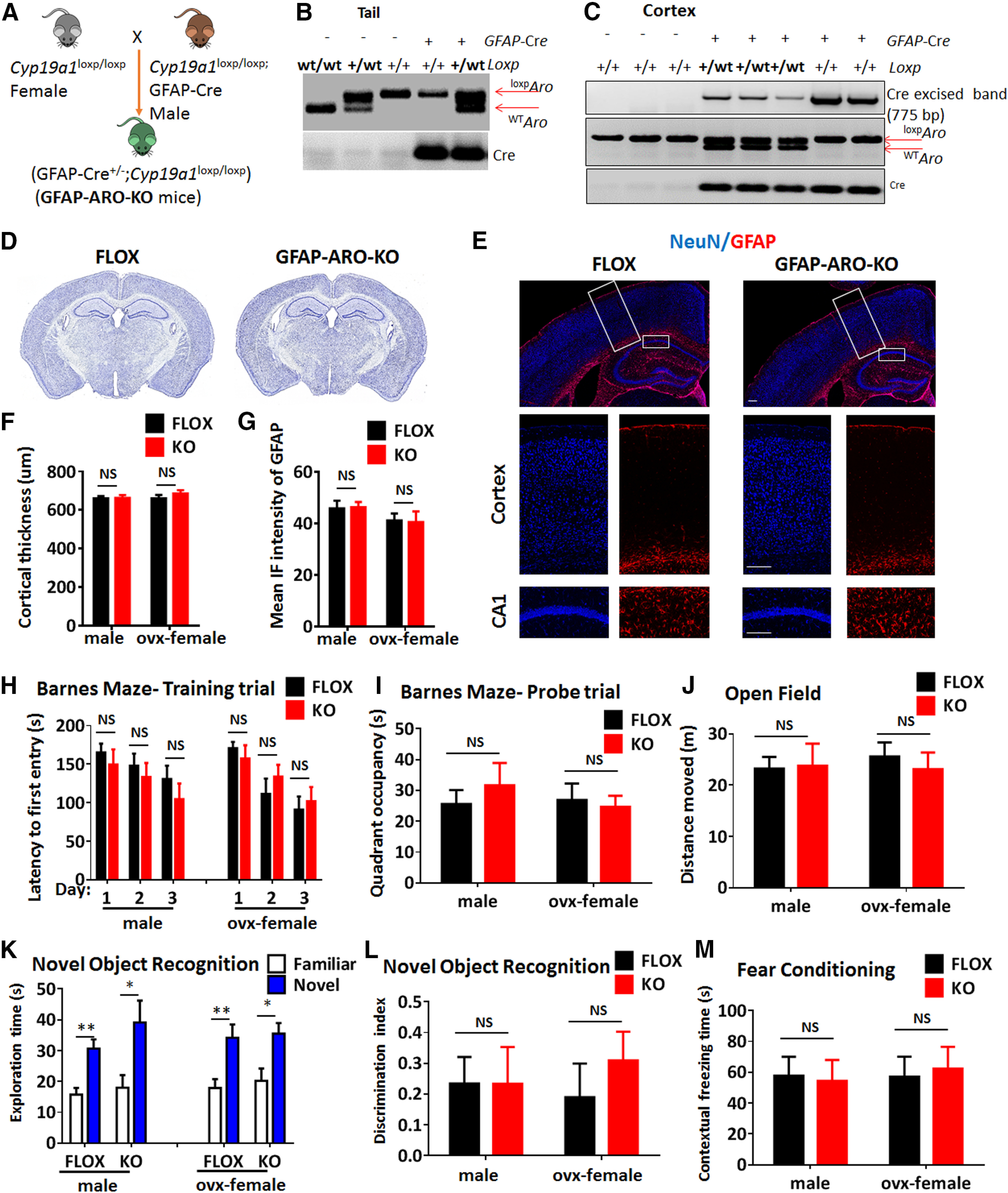
Generation and characterization of adult FLOX and GFAP-ARO-KO mice. A, Schematic representation of breeding strategy. B, Tail DNA PCR confirmation of bitransgenic GFAP-ARO-KO mice expressing GFAP-Cre and loxpCYP19a1. C, Confirmation of excision of floxed region in cerebral cortex of FLOX control and GFAP-ARO-KO mice using genomic PCR. D–G, Gross brain structure was examined by cresyl violet staining (D) and IHC of NeuN and GFAP (E) on coronal sections from both FLOX and GFAP-ARO-KO (KO) mice. Quantification of cortical thickness (F) and GFAP fluorescence intensity (G) demonstrate no significant difference between FLOX and KO mice in both male and ovx-female groups. Data are mean ± SEM; n = 6; unpaired t test. Scale bars: 100 μm. H–M, Behavioral tests. Quantification on latency to the first entry in the training trial (H) and quadrant occupancy (I) in Barnes maze tests on male and ovx-female animals. (J) Quantification of total distance moved in the open field test. K, L, Exploration time on the familiar or novel object was recorded in the novel object test (K), and discrimination index was analyzed (L). M, Contextual freezing time was recorded in the fear conditioning test. Data are mean ± SEM; n = 8–10; *p < 0.05, **p < 0.01 versus familiar with novel object exploration time, unpaired t test. NS, not significant.
Figure 2.
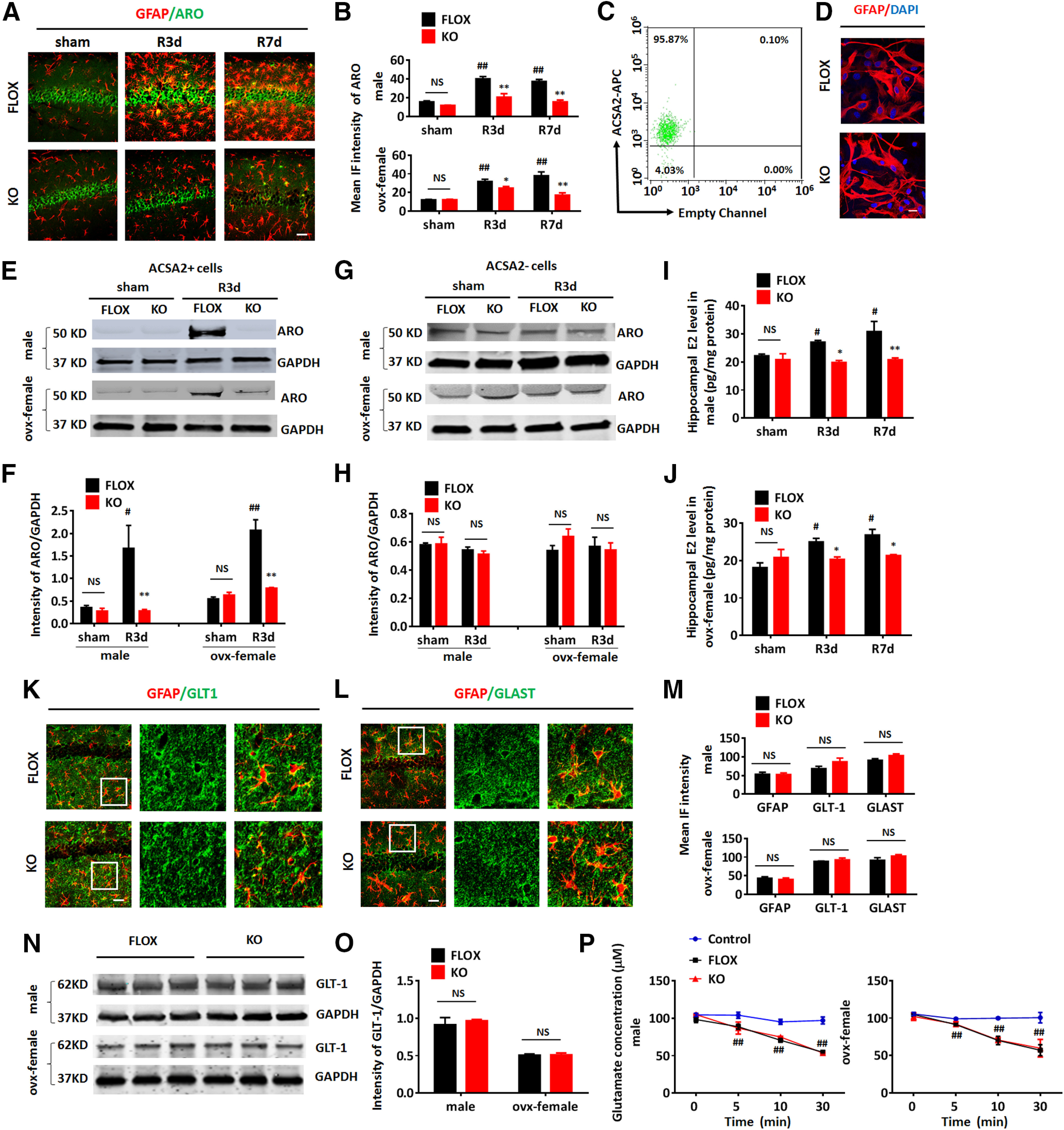
Confirmation of aromatase deletion in astrocytes of GFAP-ARO-KO mice. A, Representative image of GFAP and aromatase (ARO) co-immunostaining in the hippocampal CA1 region of sham and reperfusion 3 and 7 d after GCI (R3d and R7d) of FLOX and GFAP-ARO-KO (KO) in male and ovx-female mice. Scale bar: 20 μm. B, Quantification of mean fluorescence intensity (IF) of aromatase in male and ovx-female hippocampal CA1. C, Flow cytometry analysis for ACSA2-APC in ACSA2+ cells isolated by magnetic cell sorting from the hemispheres of adult FLOX male mice. D, Immunocytochemistry demonstrates that purified adult astrocytes express GFAP at 24 h in vitro. Scale bar: 10 μm. E–H, Western blot analysis of aromatase on protein lysates of ACSA2+ astrocytes (E) and ACSA– cells (G) isolated from the hemispheres of male and ovx-female FLOX and KO mice in sham and R3d, and densitometry analysis (F, H) by ImageJ. Intensities of the aromatase band were normalized to the intensity of GAPDH from two independent experiments. Samples from three mice were pooled for one collection for each experiment. I, J, Hippocampal E2 levels were measured by ELISA assay on hippocampal tissue from sham, R3d, and R7d FLOX and KO mice in male (I) and ovx-female (J). K–L, Representative images of GFAP and GLT-1 (K) or GLAST (L) co-immunostaining in the hippocampal CA1 region of FLOX and KO mice under basal conditions. Scale bars: 20 μm. M, IF quantification of GFAP, GLT-1, and GLAST from IHC in male and ovx-female. N, O, Western blot analysis of FLOX and KO hippocampal tissue lysates from male and ovx-female and quantification of GLT-1 expression. P, Glutamate uptake analysis. Glutamate concentrations were measured after the glutamate containing media (100 μm) were incubated with FLOX/KO astrocytes isolated from male and ovx-female hemisphere for 0–30 min. Data are mean ± SEM; n = 4; #p < 0.05, ##p < 0.01 versus sham; *p < 0.05, **p < 0.01 versus KO with FLOX, two-way ANOVA followed by post hoc tests. NS, not significant.
We next performed cognitive behavioral tests on three to four-month-old males and ovx-females to examine whether aromatase deletion in astrocytes affected the learning and memory capacity of the KO mice. Performance of the Barnes maze test results revealed that KO mice did not have a significant spatial memory and learning difference as compared with their FLOX controls, as evidenced by no significant differences detected in escape latency to find the hidden chamber on 3 d of the training trial (n = 8–10, F(5,90) = 0.6062, p = 0.6953, two-way ANOVA; Fig. 1H), and no significant difference in quadrant occupancy on probe trial (n = 8–10, F(1,24) = 0.2714, p = 0.6072, two-way ANOVA; Fig. 1I). Performance of the open field test also did not detect any difference in total travel distance between FLOX and KO males and ovx-females (n = 8–10, F(1,29) = 0.0888, p = 0.7678, two-way ANOVA followed by Sidak's test; Fig. 1J). Furthermore, performance of the novel object recognition test revealed that both the FLOX and KO mice spent more time exploring the novel object (n = 8–10, familiar vs novel F(1,55) = 37.19, p < 0.0001, two-way ANOVA followed by Sidak's test; Fig. 1K), however there was no difference in the exploring time (n = 8–10, F(3,55) = 0.7914, p = 0.5039, two-way ANOVA; Fig. 1K) or discrimination index (n = 8–10, F(1,29) = 0.3569, p = 0.5549, two-way ANOVA; Fig. 1L) between FLOX and KO in both males and ovx-females. Finally, performance of the fear conditioning test revealed that male and ovx-female KO mice did not have a significant difference in the contextual freezing rate as compared with FLOX controls (n = 8–10, F(1,26) = 0.003854, p = 0.9510, two-way ANOVA; Fig. 1M). Altogether, these behavioral data demonstrate that the male and ovx-female KO mice have comparable learning and memory capacity as their FLOX controls, indicating that aromatase deletion under the GFAP promotor does not have a significant effect on the cognitive functions of the mouse brain at the basal level.
We have previously demonstrated that the expression of aromatase in astrocytes was undetectable basally in the non-injured brain; however, it was highly induced in astrocytes at 3 d after GCI reperfusion (Zhang et al., 2014). To confirm the deletion of aromatase in astrocytes, we first measured the expression of aromatase in astrocytes by co-immunostaining of aromatase with GFAP in sham, 3 d after GCI reperfusion (R3d), and 7 d after GCI reperfusion (R7d) mouse brains. Aromatase expression in neurons was also assessed. As expected, the expression of aromatase in astrocytes was not detected in the sham groups (Fig. 2A); however, at R3d and R7d, the expression of aromatase was strongly induced in the hippocampal CA1 astrocytes of the FLOX mice, but not in the astrocytes of the KO group (Fig. 2A). Note that despite the failure of aromatase induction in KO astrocytes after GCI, expression of aromatase was well preserved in hippocampal CA1 layer neurons in both sham and GCI KO mice. Quantification of the IF intensity of aromatase staining in male and ovx-female demonstrated a significant increase of aromatase expression in the FLOX-R3d and FLOX-R7d hippocampus compared with FLOX-sham (n = 3–4, p < 0.01, two-way ANOVA followed by Dunnett's test; Fig. 2B). However, the IF intensity of aromatase in the hippocampus of KO-GCI mice was significantly lower than in the FLOX-GCI mice at R3d and R7d (n = 3–4, male, F(2,16) = 6.610; ovx-female, F(2,16) = 8.356, p < 0.01, two-way ANOVA followed by Sidak's test; Fig. 2B). To confirm these histologic findings, purified astrocytes labeled with anti-ACSA-2 microbeads from adult mouse brains were acutely isolated with magnetic-activated cell sorting. The beads-bonded (ACSA2+) and beads-depleted (ACSA2–) cells were collected separately. The purity of beads-bonded cells was verified by flow cytometry (Fig. 2C), which demonstrated that 95.87% of the microbeads labeled cells were ACSA2+. Immunocytochemistry confirmed that over 80% beads-bonded cells from both FLOX and KO brains express GFAP when seeded on poly-D-lysine coated dishes for 24 h (n = 6, FLOX: 87.28 ± 2.33 vs KO: 82.93 ± 2.22; p = 0.2095, unpaired t test; Fig. 2D). Western blot analysis on ACSA2+ cell lysates demonstrated that the expression of aromatase was highly upregulated in FLOX-R3d as compared with FLOX-sham, but there was no significant increase observed in KO-R3d ACSA2+ cells as compared with KO-sham (n = 3 of male and n = 3 of ovx-female mice in two independent experiments, FLOX-sham vs FLOX-GCI, p < 0.01; KO-sham vs KO-GCI, p > 0.05, two-way ANOVA followed by Sidak's test; Fig. 2E,F). Furthermore, the aromatase protein expression level in KO-GCI ACSA2+ cells was significantly less than that in the FLOX-GCI samples (n = 3 of male and n = 3 of ovx-female mice in two independent experiments, male, F(1,4) = 6.445; female, F(1,4) = 31.08 p < 0.05, two-way ANOVA followed by Sidak's test; Fig. 2E,F). In the ACSA2– cell lysates, no significant expression change of aromatase was detected between FLOX and KO in both male and ovx-female (n = 3 of male and n = 3 of ovx-female mice in two independent experiments, male, F(1,4) = 0.3746, p = 0.5736; ovx-female, F(1,4) = 1.413, p = 0.3003, two-way ANOVA; Fig. 2G,H), indicating aromatase is specifically deleted in astrocytes. We also measured the local E2 level of male and ovx-female hippocampal tissue in sham, R3d, and R7d groups by ELISA, and found that the E2 level showed a significant increase in the FLOX-R3d and FLOX-R7d hippocampus, as compared with FLOX-sham (n = 4 of male and n = 3 of ovx-female, R3d, p < 0.05; R7d, p < 0.05, two-way ANOVA followed by Dunnett's test; Fig. 2I,J). Interestingly, the E2 level in the KO-GCI hippocampus did not show any increase as compared with the KO-sham (n = 4 of male and n = 3 of ovx-female, p > 0.05, two-way ANOVA followed by Dunnett's test; Fig. 2I,J), and was significantly lower than that in FLOX-GCI hippocampus (n = 4 of male and n = 3 of ovx-female, male, F(2,16) = 2.643, R3d, p < 0.05, R7d, p < 0.01; ovx-female F(2,12) = 6.306 R3d, p < 0.05; R7d, p < 0.05; two-way ANOVA followed by Sidak's test; Fig. 2I,J). Moreover, no difference was detected in the sham controls between FLOX and KO (n = 4 of male and n = 3 of ovx-female, male and female p > 0.05, two-way ANOVA followed by Sidak's test; Fig. 2I,J), indicating aromatase deletion in GFAP+ cells does not affect the hippocampal E2 level under physiological (non-ischemic) conditions.
To further confirm that aromatase deletion by the GFAP-cre does not affect basal physiological properties of astrocytes in the GFAP-ARO-KO mouse brain, we examined the expression of the astrocyte protein GFAP and the glutamate transporters GLT-1, and GLAST in FLOX and KO hippocampus in the male and ovx-female mice. No significant difference in the expression of GFAP (male, p = 0.8988; female, p = 0.6285, unpaired t test), GLT-1 (male, p = 0.1758; female, p = 0.4615, unpaired t test) or GLAST (male, p = 0.0914; female, p = 0.2334, unpaired t test) was detected between FLOX and KO in male and ovx-female mice as measured by IF intensity (Fig. 2K–M). Western blot analysis confirmed that the expression level of GLT-1 in hippocampal tissue do not show any difference between FLOX and KO in male or ovx-female (male, p = 0.6197; female, p = 0.8743 unpaired t test; Fig. 2N,O). Next, we performed a glutamate uptake assay on astrocytes isolated from adult male and female FLOX and KO mouse brain after the astrocytes were seeded in culture for 24 h. Glutamate uptake capacity was measured by the glutamate concentration in the glutamate-containing media (started with 100 μm) after incubation with the astrocyte cultures from 0 to 30 min. We observed a significant decrease of glutamate concentration in the media after incubation with FLOX and KO astrocytes compared with control media at 5, 10, and 30 min (male, F(6,12) = 14.75; female, F(6,12) = 8.774, p < 0.01, two-way ANOVA followed by Tukey's test; Fig. 2P). However, there was no significant difference in the glutamate concentration between FLOX and KO at any time point examined (p > 0.05, two-way ANOVA followed by Tukey's test; Fig. 2P). Moreover, there was no significant difference in glutamate uptake capacity between FLOX and KO astrocyte after normalizing the reduction of glutamate concentration rate to the protein concentration of the FLOX and KO astrocytes lysates (n = 3 mice/group, male, p = 0.0998; ovx-female, p = 0.723, unpaired t test). Altogether, these data support that aromatase gene KO mediated by the GFAP-cre specifically deletes aromatase expression in astrocytes. Because the expression of aromatase in astrocytes is very low to undetectable under physiological conditions (Garcia-Segura et al., 1999), it is not surprising that the morphologic and functional properties of KO astrocytes were not significantly changed at the basal level as compared with FLOX astrocytes.
Ischemia-induced reactive astrogliosis is significantly decreased in the male GFAP-ARO-KO hippocampus and after aromatase inhibition
We next examined the effect of loss of astrocyte-derived E2 on reactive astrogliosis in the hippocampus after GCI in male mice. IHC for the astrocyte cell marker, GFAP, revealed the characteristic increased GFAP intensity and hypertrophy of astrocytes in the FLOX-R3d and R7d hippocampus (n = 6, p < 0.01; two-way ANOVA followed by Dunnett's test; Fig. 3A,B). In contrast, increases in GFAP intensity and GFAP+ astrocyte hypertrophy were significantly attenuated in the KO hippocampus at R3d and R7d (n = 6, F(2,31) = 10.26, p < 0.01, two-way ANOVA followed by Sidak's test; Fig. 3A,B), indicating that KO mice exhibit reduced reactive astrogliosis in the hippocampus after GCI. Note that in sham animals, there was no significant difference in GFAP immunostaining intensity between the FLOX and KO groups (n = 6, p > 0.05, two-way ANOVA followed by Sisak's test; Fig. 3B). To evaluate whether the decreased GFAP intensity is a result of reduced astrocyte cell number, we compared the GFAP+ cell number in FLOX and KO mice in the hippocampal region. The results revealed that the hippocampal GFAP+ cell number was not significantly different between FLOX and KO mice in either sham, R3d, or R7d groups (n = 6, F(2,56) = 0.2482, p = 0.7811, two-way ANOVA; Fig. 3C). To further confirm the decreased GFAP expression observed in KO-GCI mice by immunostaining, we performed Western blot analysis (Fig. 3D,E), which also demonstrated a significant decrease of GFAP levels in the KO R3d and R7d hippocampus, as compared with the FLOX (n = 4–6, F(2,16) = 7.203, p < 0.01, two-way ANOVA followed by Sidak's test; Fig. 3F). Note that in the sham group, the expression level of GFAP was not significantly different between FLOX and KO groups (n = 4–6, p > 0.05, two-way ANOVA followed by Sidak's test; Fig. 3F). To further confirm reduced astrocyte activation in KO mice after GCI, we analyzed astrocyte morphology by 3-D reconstruction of confocal z-stack images and cell volume measurement using Imaris software (Tang et al., 2020). As shown in Figure 3G, most astrocytes in FLOX hippocampus demonstrate a hypertrophic morphology with enlarged cell body and branches after ischemia at R3d and R7d. In contrast, astrocytes in the KO hippocampus appeared to show a much reduced hypertrophy after ischemia. Quantification of these results using cell volume measurements found that the percentage of hypertrophic astrocytes (defined by the Imaris software based on a cell volume pixel >750,000 units; Fig. 3H, yellow, orange, and red cells) was significantly decreased in the KO hippocampus as compared with FLOX at R3d and R7d (n = 6, F(2,30) = 37.17, p < 0.01, two-way ANOVA followed by Sidak's test; Fig. 3H,I). We also conducted IHC of S100β, another astrocyte marker, to further confirm changes in the astrocyte number and morphology in FLOX and KO hippocampus. Similar to the staining assay result for GFAP, the intensity of S100β staining was significantly enhanced in the FLOX hippocampus at R3d and R7d, as compared with sham (n = 6, p < 0.01, two-way ANOVA followed by Dunnett's test; Fig. 3J,K); however, the intensity of S100β was significantly decreased in the hippocampus of KO-R3d mice as compared with the FLOX-R3d mice (n = 6, F(2,34) = 16.03, p < 0.01, two-way ANOVA followed by Sidak's test; Fig. 3J,K). Quantification of S100β+ cell numbers in the hippocampal region did not detect any significant difference between FLOX and KO in either sham, R3d, or R7d (n = 6, F(2,42) = 0.2107, p = 0.8109, two-way ANOVA; Fig. 3L).
Figure 3.
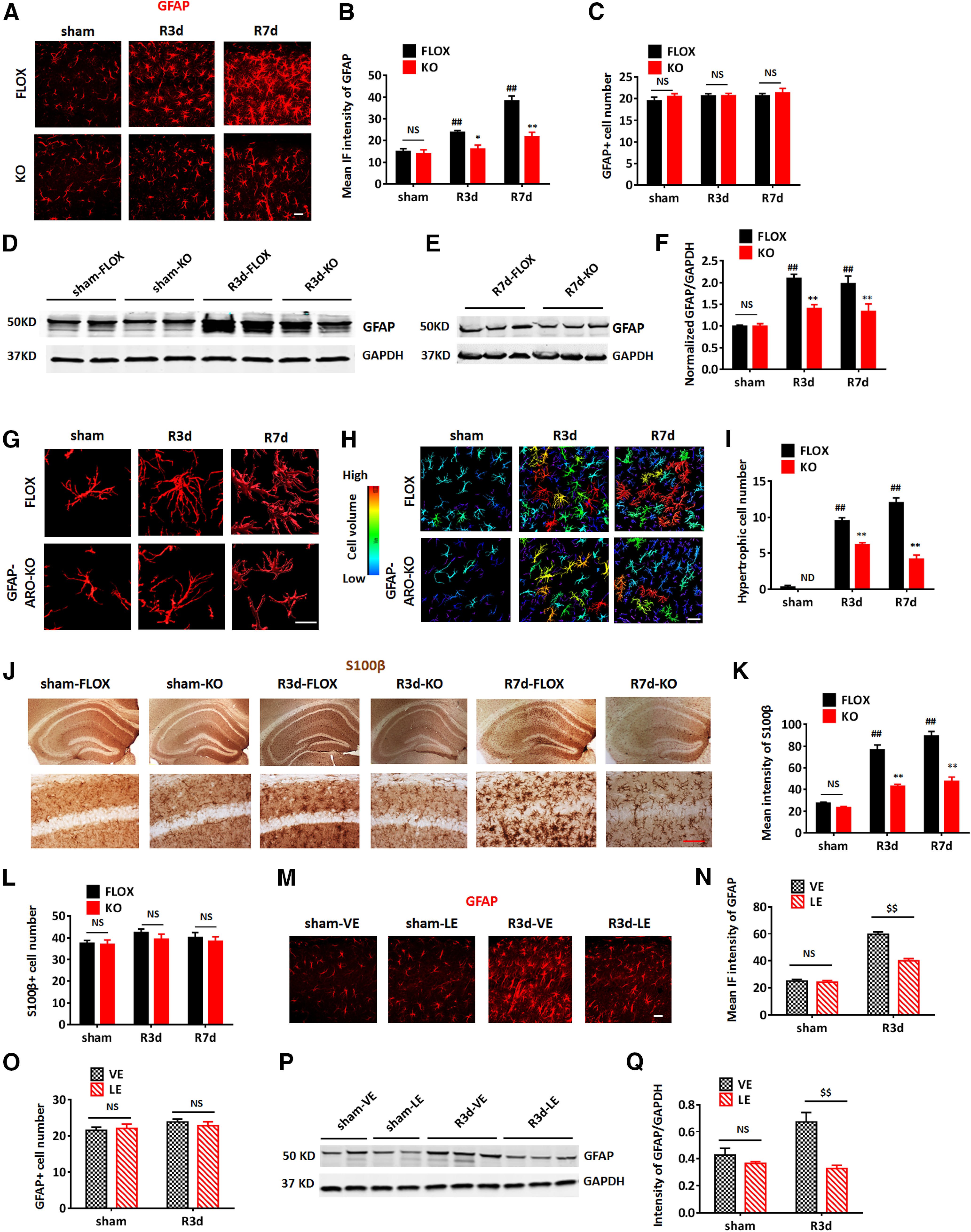
Depletion of astrocyte-derived E2 in male mice leads to a decrease in reactive astrogliosis after GCI. A–C, Representative images (A) and quantification of mean IF intensity of GFAP (B) and GFAP+ cell number (C) in the hippocampal CA1 region of sham, R3d, and R7d FLOX and GFAP-ARO-KO (KO) mice. Scale bar: 20 μm. D–F, Western blot analysis and quantification of GFAP expression in hippocampal tissue samples collected from male FLOX and KO animals. G–I, Representative 3-D. Reconstructions of single astrocyte (G) and astrocytes in the hippocampal CA1 region (H). Scale bars: 20 μm. I, Cell volume was quantified based on 3-D reconstruction. Cells with volume >750,000 units (yellow, orange, and red cells) were counted as hypertrophic. J–L, Representative images (J) and quantification of mean intensity of S100β (K) and s100β+ cell number (L) in the hippocampal CA1 region. Scale bar: 20 μm. M–O, Representative images (M) and quantification of mean IF intensity of GFAP (N) and GFAP+ cell number (O) in the hippocampal CA1 region of the vehicle (VE)-treated and letrozole (LE)-treated mouse brain. Scale bars: 20 μm. P, Q, Western blot analysis and quantification of GFAP expression in hippocampal tissue samples collected from the male vehicle-treated and letrozole-treated animals. Data are mean ± SEM; n = 6 biologically independent animals; ##p < 0.01 compared with sham, two-way ANOVA followed by post hoc tests; *p < 0.05, **p < 0.01 compared with FLOX, two-way ANOVA; $$p < 0.01 compare to vehicle two-way ANOVA. NS, not significant. ND, not detectable.
Next, we sought confirmation of our KO mouse results on reactive astrogliosis by performing acute pharmacological aromatase inhibition in WT sham and GCI mice with the aromatase inhibitor, letrozole. Letrozole (10 mg/kg/d, i.p.) was administered to three-month-old WT male mice beginning 1 d before GCI or sham surgery, and continued daily until reperfusion day 3. As shown in Figure 3M–Q, similar to our results in KO mice, GFAP immunostaining intensity and expression levels were significantly decreased in the hippocampus of letrozole-treated mice at R3d, as compared with the vehicle-treated animals measured by IF intensity (n = 6, F(1,32) = 38.31, p < 0.01, two-way ANOVA followed by Sidak's test; Fig. 3M,N) and Western blot analysis (n = 4, F(1,8) = 12.73, p < 0.01, two-way ANOVA followed by Sidak's test; Fig. 3P,Q). Note that letrozole treatment did not have a significant effect on the morphology and GFAP expression in sham astrocytes (n = 6, p > 0.05, two-way ANOVA followed by Sidak's test; Fig. 3M,N,P,Q). Also similar to KO mice, quantification of GFAP+ cell number demonstrated that letrozole treatment did not change the number of GFAP+ astrocytes in the hippocampus either in sham or R3d groups (n = 6, F(1,44) = 0.6146, p = 0.4373, two-way ANOVA; Fig. 3O), indicating that changes in GFAP reflect decreased activation of astrocytes after GCI rather than a decrease in astrocyte cell number. These findings demonstrate that aromatase deletion by GFAP-cre or acute aromatase inhibition by pharmacological treatment results in decreased reactive astrogliosis in response to GCI.
Male GFAP-ARO-KO mice exhibit greater neuronal damage and increased hypertrophic microglia after GCI
We next investigated neuronal damage in the hippocampus of FLOX and KO male mice at 3 and 7 d after GCI reperfusion. Representative photomicrographs for NeuN staining are shown in Figure 4A. Quantification of NeuN+ cell number in the hippocampal CA1 region revealed that the neuron number displayed a progressive decrease at R3d and R7d in FLOX after GCI, as compared with FLOX-sham (n = 6, R3d, p < 0.05; R7d, p < 0.01, two-way ANOVA followed by Tukey's test; Fig. 4A,E). Moreover, the neuron number in the KO-GCI hippocampal CA1 region was significantly less than FLOX-GCI at both R3d and R7d (n = 6, F(2,30) = 31.32, p < 0.01, two-way ANOVA followed by Sidak's test; Fig. 4A,E). In addition, immunostaining and fluorescent-intensity analysis showed that the expression of the apoptosis marker, cleaved caspase-3 in the hippocampal CA1 region was significantly increased in KO-GCI as compared with FLOX-GCI at R3d and R7d (n = 6, F(2,18) = 9.307, p < 0.01, two-way ANOVA followed by Sidak's test; Fig. 4A,F). Western blot analysis further confirmed that the expression of cleaved caspase-3 was significantly higher in hippocampal tissue samples from the KO-GCI group, as compared with the FLOX-GCI group at both R3d and R7d (n = 6, F(2,16) = 4.776, p < 0.05, two-way ANOVA followed by Sidak's test; Fig. 4B–D). These data indicate that loss of astrocyte-derived E2 results in increased neuronal damage and more neuronal loss in the KO hippocampus after GCI, as compared with FLOX controls.
Figure 4.
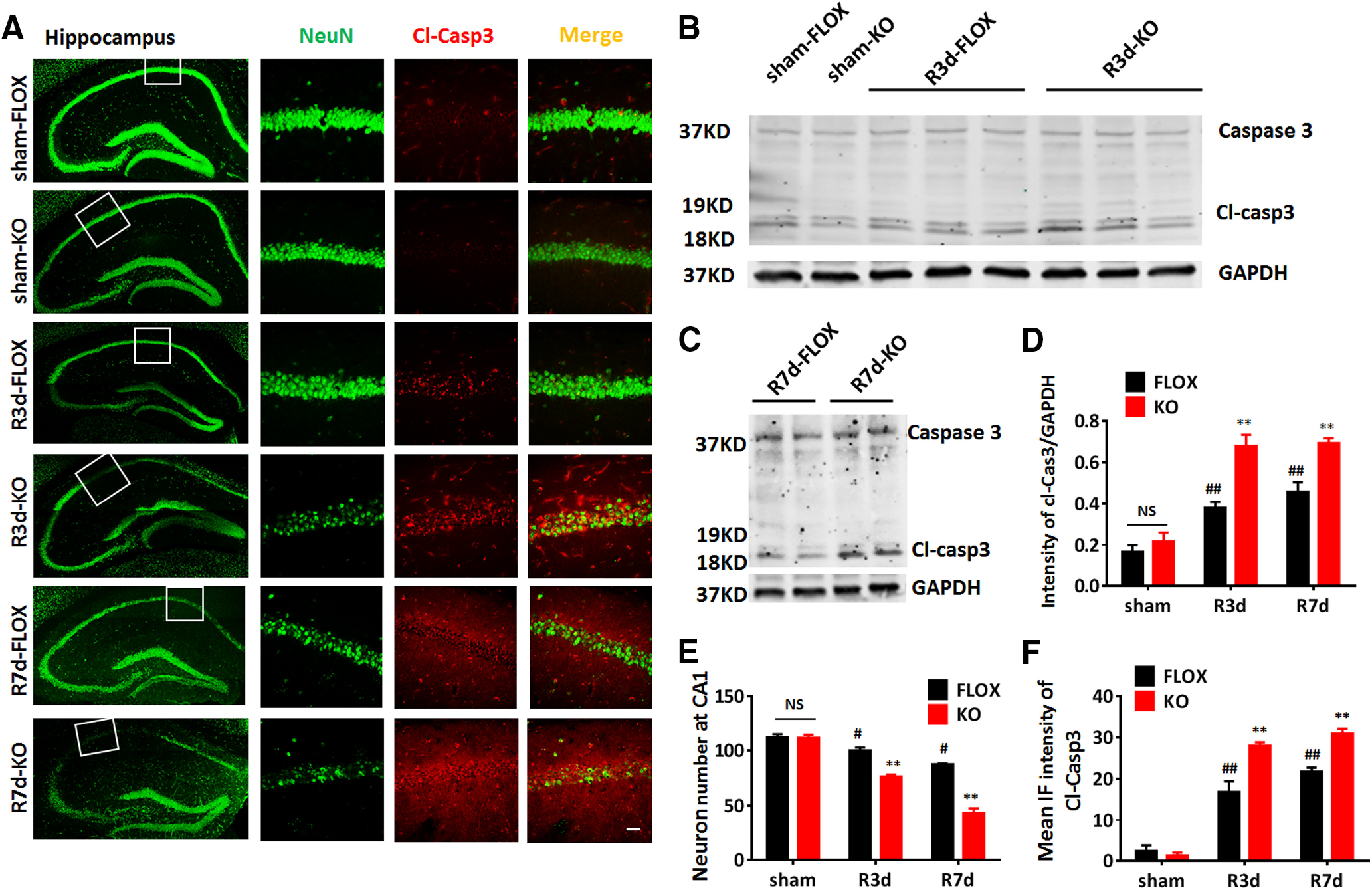
Increased neuronal damage in male GFAP-ARO-KO mice after GCI. A, Representative images of NeuN and cleaved caspase-3 (Cl-Casp3) immunostaining in the sham R3d, or R7d hippocampus and CA1 region of FLOX and GFAP-ARO-KO (KO) mice. B–D, Western blot analysis and quantification of relative Cl-Casp3 expression in the hippocampus of sham, R3d, and R7d animals. E, Quantification of NeuN+ cell number in hippocampal CA1 region in sham, R3d, and R7d animals. F, Quantification of mean IF intensity of Cl-Casp3. Data are mean ± SEM; n = 6 mice. Scale bar: 20 μm; #p < 0.05, ##p < 0.01 compared with sham, two-way ANOVA; **p < 0.01 compared with FLOX, two-way ANOVA followed by post hoc tests. NS, not significant.
Microglia activation is also an important component of the neuroinflammatory response after cerebral ischemia (Zhang, 2019). We thus examined whether microglia activation is altered in GFAP-ARO-KO male mice following GCI. Expression and intensity of the microglia marker, Iba1, was used as an indicator of microglial activation. Compared with sham, both male FLOX and KO hippocampus exhibited a significantly increased intensity of Iba1 immunostaining at R3d and R7d (n = 6, p < 0.01, two-way ANOVA followed by Tukey's test; Fig. 5A,B). Interestingly, significantly higher Iba1 immunostaining intensity was detected in the KO-GCI hippocampus as compared with the FLOX-GCI at R3d and R7d (n = 6, F(2,28) = 10.54, R3d, p < 0.05; R7d, p < 0.01, two-way ANOVA followed by Sidak's test; Fig. 5A,B), indicating greater microglial activation in KO mice after GCI. 3-D reconstruction of the confocal imaging of Iba1 immunostaining showed that microglia from the KO hippocampus displayed a more enlarged cell soma and enriched branches at the single-cell level (Fig. 5C). Cell volume measurement also found that the number of hypertrophic microglia defined by the Imaris software based on the cell volume pixel (>750,000 units; Fig. 5D, yellow, orange, and red cells) was significantly increased in the KO-GCI group, as compared with the FLOX-GCI group at R3d and R7d (n = 6, F(2,20) = 32.84, p < 0.01, two-way ANOVA followed by Sidak's test; Fig. 5D,E). Western blot analysis further confirmed a significant increase of Iba1 expression in the KO-GCI hippocampus, as compared with the FLOX-GCI group at R3d and R7d (n = 6, F(2,18) = 7.315, R3d, p < 0.05, R7d, p < 0.01, two-way ANOVA followed by Sidak's test; Fig. 5F–H). These results demonstrate microglial activation is enhanced in the KO hippocampus, as compared with FLOX hippocampus at R3d and R7d.
Figure 5.
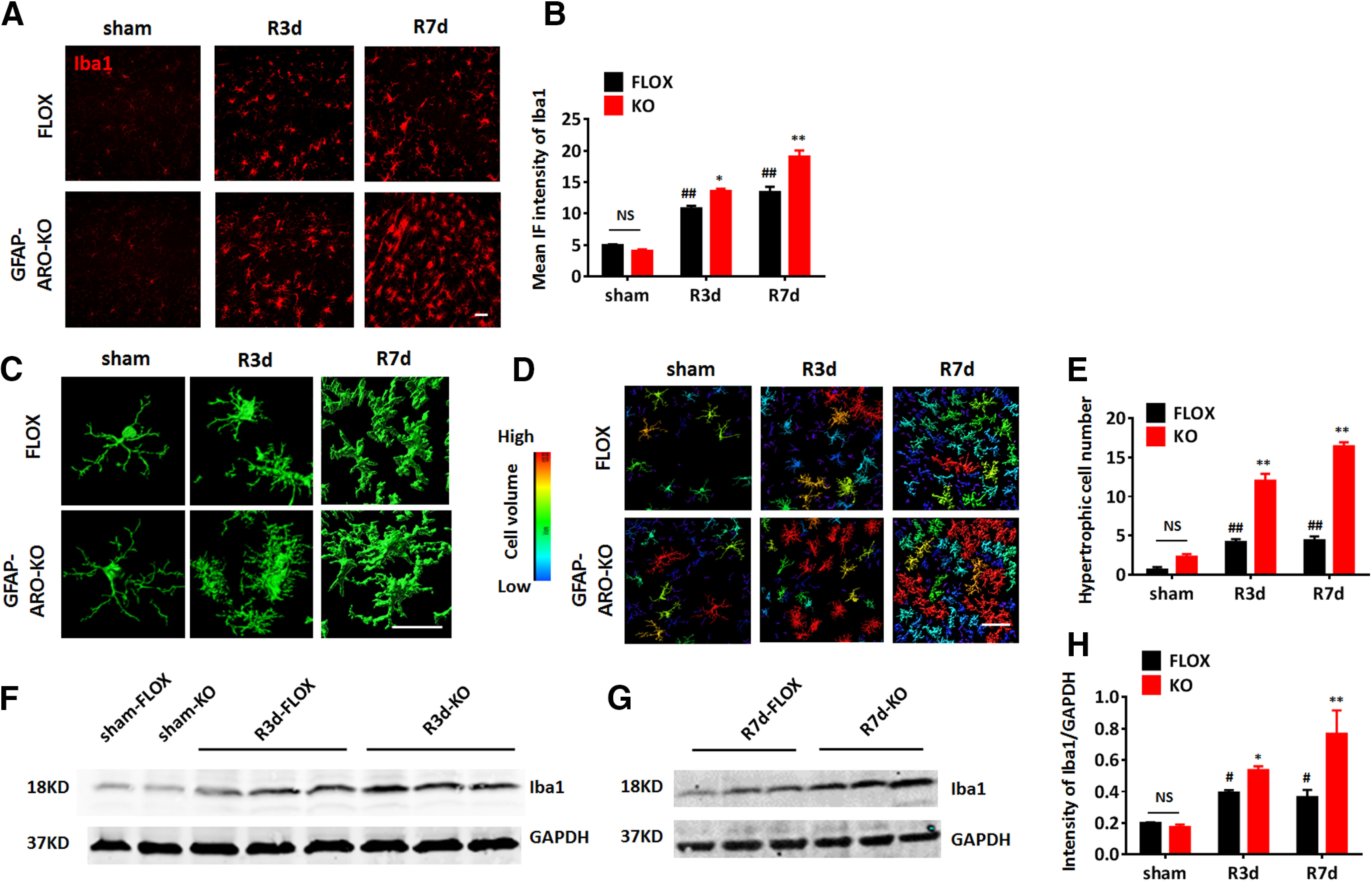
Increased microglia activation in the hippocampus of male GFAP-ARO-KO mice after GCI. A, B, Representative images (A) and quantification of mean IF intensity (B) of Iba1 immunostaining in the hippocampal CA1 region of sham, R3d, and R7d FLOX and GFAP-ARO-KO (KO) mice. C–E, Representative 3-D. Reconstructions of single microglia (C) and microglia in hippocampal CA1 region (D). Cell volume was quantified based on 3-D reconstruction. E, Cells with volume >750,000 units (yellow, orange, and red cells) were counted as hypertrophic and quantified. F–H, Western blot analysis and quantification of relative Iba1 expression in the hippocampus of FLOX and KO animals. Scale bars: 20 μm; #p < 0.05, ##p < 0.01 compared with sham, two-way ANOVA followed by post hoc; *p < 0.05, **p < 0.01 compared with FLOX, two-way ANOVA followed by post hoc. NS, not significant.
Increased cognitive deficits in male GFAP-ARO-KO mice after GCI
GCI, which occurs during cardiac arrest, hypotensive shock, and asphyxia, can lead to significant cognitive decline (Neumann et al., 2013). The cognitive decline after GCI is associated with neuronal damage and loss in the hippocampal CA1 region following the brain ischemia and reperfusion process. Next, we sought to examine whether the depletion of astrocyte-derived E2 affected cognitive outcome after GCI. Toward this end, cognitive behavioral tests were performed on male FLOX and KO mice at 7d after sham or GCI/reperfusion surgery (sham and R7d groups). The results revealed that the KO-sham mice did not show any significant difference as compared with FLOX-sham mice in cognitive and motor activity behavioral analysis, as determined by the Barnes maze test, novel object recognition test, and open field test (n = 9 mice/group, p > 0.05 two-way ANOVA followed by Sidak's test; Fig. 6A–C). In contrast, after ischemia/reperfusion, FLOX-GCI mice showed a significant increase of the latency to first entry on day 3 in the training trial of the Barnes maze test (n = 9, sham-day 3 vs R7d-day 3, p < 0.05, two-way ANOVA followed by Sidak's test; Fig. 6A(a),A(b)), and a significant decrease of quadrant occupancy in the probe trial of the Barnes maze test (n = 9, p < 0.01 compared with sham, two-way ANOVA followed by Sidak's test; Fig. 6A(c),A(d)). This result indicates cognitive impairments were present in FLOX mice at R7d after GCI. Interestingly, KO mice at R7d had even more significant cognitive defects than FLOX-R7d mice, as evidenced by a significant increase of the latency to first entry on days 2 and 3 in the Barnes maze training trial (n = 10, F(2,48) = 9.485, day 2, p < 0.05; day 3, p < 0.01; two-way ANOVA followed by Sidak's test; Fig. 6A(a),A(b)), and a decrease of quadrant occupancy in the probe trial of Barnes maze test (n = 10, F(1,36) = 4.28, p < 0.05 compared with FLOX-R7d, two-way ANOVA followed by Sidak's test; Fig. 6A(c),A(d)).
Figure 6.

Increased cognitive deficits in male GFAP-ARO-KO mice after GCI. A, Barnes maze tests. Representative tracking plots on the third day of the training trial are shown in A(a), and the representative tracking plots for the probe trial are shown in A(c). The escape latency in training trial A(b) and the quadrant occupancy A(d) in probe trial were recorded and statistically analyzed. B, Representative tracking plots of open field tests are shown in B(a), and the total distance moved were recorded and statistically analyzed B(b). C, Novel object recognition test. Representative tracking plots on the choice day are shown in C(a), the discrimination index of the sampling day C(b), and choice day C(c) were analyzed. Data are mean ± SEM; n = 9; #p < 0.05, ##p < 0.01 compared with sham, two-way ANOVA followed by post hoc tests; *p < 0.05, **p < 0.01 compared with FLOX-R7d, two-way ANOVA followed by post hoc tests. NS, not significant.
In the novel object recognition test, which tests hippocampal-dependent recognition memory, both FLOX-sham and KO-sham demonstrated a normal novel object discrimination capacity (discrimination index: sham-FLOX, 0.20 ± 0.05 vs sham-KO, 0.19 ± 0.05; Fig. 6C). In contrast, FLOX-R7d mice displayed a significant decrease in novel object discrimination capacity (n = 8–10, p < 0.05 compared with FLOX-sham, two-way ANOVA followed by Sidak's test; Fig. 6C(c)). Furthermore, KO-R7d mice exhibited an even greater decrease in novel object discrimination capacity as compared with FLOX-R7d mice (n = 8–10, F(1,38) = 4.392, p < 0.01, two-way ANOVA followed by Sidak's test; Fig. 6C(c)), indicating that KO mice had a greater deficit in hippocampal-dependent recognition memory after GCI as compared with FLOX-R7d mice. No significant differences were observed in the discrimination capacity between FLOX and KO mice at sampling stage (n = 8–10, F(1,28) = 0.0002, p = 0.9888, two-way ANOVA; Fig. 6C(b)). Finally, performance of the open field test demonstrated that there was no significant difference between FLOX and KO mice in total travel distance in both sham and R7d groups (n = 8–10, F(1,28) = 0.6872, p = 0.4141, two-way ANOVA followed by Sidak's test; Fig. 6B), indicating the differences demonstrated in the Barnes maze tests are not because of motor functional disability. These results indicate that after brain ischemia, loss of astrocyte-derived E2 leads to more significant cognitive deficits in GFAP-ARO-KO mice, which is consistent with the increased neuronal damage in the hippocampal CA1 region of KO mice after GCI.
Decreased A2 astrocyte signature genes in GFAP-ARO-KO mice after GCI
To identify the potential genes regulated by astrocyte-derived E2 after GCI, we performed global transcriptome analysis. RNA samples were isolated from ovx-female FLOX and KO hippocampal tissue at 24 h after GCI reperfusion (R24h) and were subjected to RNA-seq. Among the 773 identified DEGs (>2-fold, adjusted p < 0.05), 369 genes were upregulated (KO/FLOX fold change >2), while 404 genes were downregulated (KO/FLOX fold change <0.5) in KO versus FLOX mice at 24 h after GCI (Fig. 7A). Next, the DEGs were subjected to GO analysis (Fig. 7B). Pathways analysis, in terms of either biological processes or molecular functions revealed that the DEGs were involved in cytokine, chemokine and inflammatory response signaling pathways (Fig. 7B). Pathway analysis using IPA demonstrated that the IL-6 signaling was among the top regulated canonical pathways with –log 10 (p value) 1.86. Further GSEA revealed that these gene sets of cytokine, chemokine, and inflammatory response signaling pathways, such as IL-6/JAK/STAT3 signaling (Normalized Enrichment Score (NES) = −2.24, FDR q value = 0.000), cytokine mediated signaling pathway (NES = −2.23, FDR q value = 0.000), positive regulation of inflammatory response (NES = −2.40, FDR q value = 0.000), and chemokine-signaling pathway(NES = −2.20, FDR q value = 0.000), were negatively correlated with GFAP-ARO-KO GCI modulated genes as compared with the FLOX-GCI modulated genes (Fig. 7C(a)–7(d)). In addition, GSEA also revealed that E2F target genes signatures showed a positive correlation (NES = 2.11, FDR q value = 0.000) with the GFAP-ARO-KO-GCI modulated genes (Fig. 7C(e)). Since the E2F pathway is known to induce apoptosis, this data mirrors the results of increased hippocampal neuronal apoptosis detected by IHC and Western blot analysis (Fig. 4) in KO-GCI mice as compared with FLOX-GCI mice.
Figure 7.
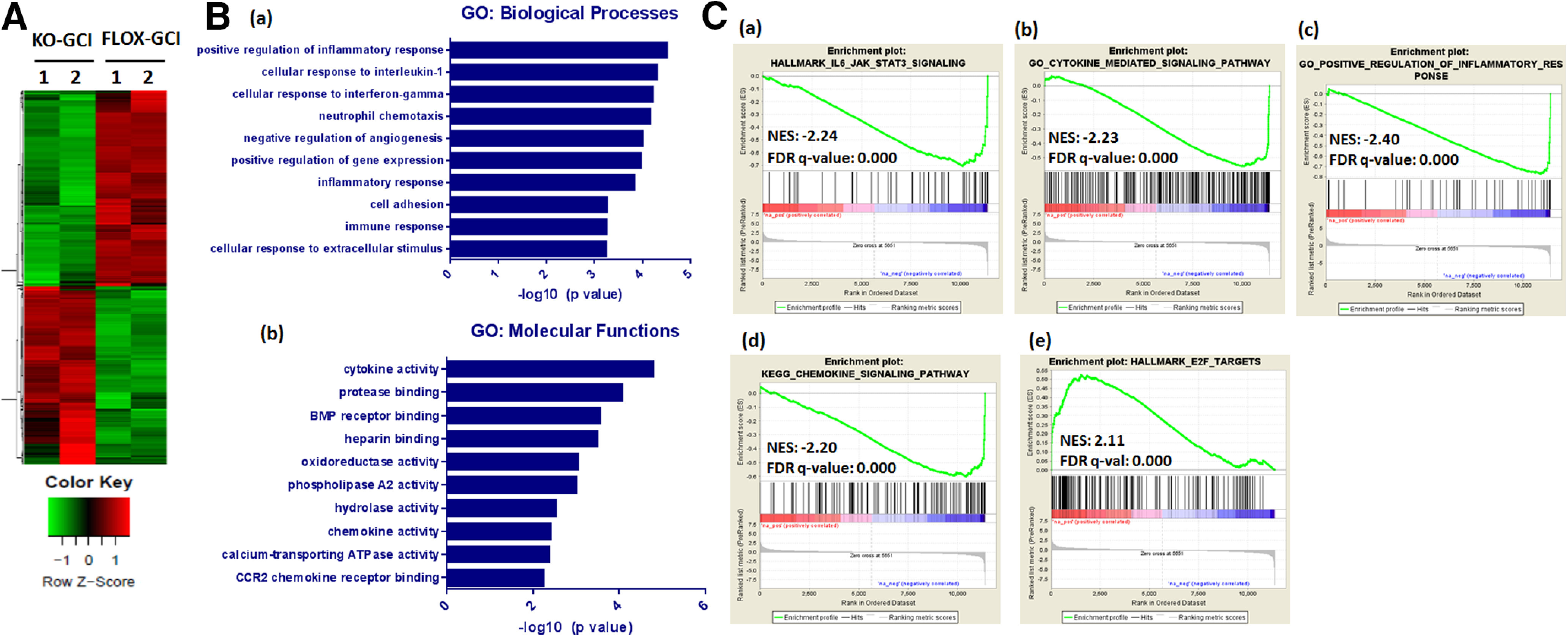
RNA-seq analysis of hippocampal transcriptome alterations in ovx-female GFAP-ARO-KO mice after GCI. A, Heat map of DEGs between FLOX-GCI and KO-GCI groups at 24 h after GCI is shown (n = 2, >2-fold, p < 0.05). B, GO analysis of DEGs in terms of biological processes [B(a)] or molecular functions [B(b)] was examined using DAVID software and the top ten significantly altered pathways are shown. C, GSEA revealed that the IL-6/JAK/STAT3 signaling [C(a)], cytokine mediated signaling pathway [C(b)], positive regulation of inflammatory response [C(c)], and chemokine-signaling pathway gene signatures [C(d)] showed negative correlation with genes altered in KO-GCI-R24h; E2F-targets [C(e)] showed positive correlation with genes altered in KO-GCI-R24h. NES, normalized enrichment score.
Glial cells, including astrocytes and microglia, play an active role and can mediate cytokine, chemokine, and inflammatory response signaling pathways in the brain after ischemic injury (Choudhury and Ding, 2016). Based on the gene profile study of astrocytes in response to specific injuries, reactive astrocytes have recently been proposed to be classified into at least two main types, called A1 and A2 (Zamanian et al., 2012). A1 astrocytes are induced in lipopolysaccharides insults as well as chronic neuroinflammatory situations, and are proposed to mediate pro-inflammatory and neurotoxic effects (Liddelow et al., 2017), while A2 astrocytes are induced acutely by ischemia and have been proposed to exert neuroprotective effects (Zamanian et al., 2012). We next studied the astrocyte transcriptome profiles by doing expression analysis (log2 fold change, GFAP-ARO-KO over FLOX, n = 2; Fig. 8A) of the PAN-specific, A1-specific, and A2-specific reactive gene sets based on the RNA-seq data. The results revealed that six of the A2-specific genes (Tgm1, Ptx3, S100a10, Ptgs2, Tm4sf1, and Cd14) were significantly decreased in KO-GCI mice (p < 0.05, multiple t tests) compared with FLOX-GCI; five of the PAN-reactive panel genes (S1pr3, Timp1, Cd44, Osmr, and GFAP) were significantly decreased (p < 0.05, multiple t tests). Among the A1-specific genes, Psmb8 was significantly decreased (p < 0.05, multiple t tests), and Fkbp5 was significantly increased (p < 0.05, multiple t tests) in the KO mice at GCI-R24h. To verify the RNA-seq data, we performed qRT-PCR to examine the expression level of astrocyte profile genes in the hippocampal RNA samples from five biologically independent animals. The results showed a significantly decreased (p < 0.05, unpaired t test) expression level in five of the Pan-reactive genes (S1pr3, Timp1, Cxcl10, Cd44, and GFAP), seven of A2-specific genes (Clcf1, Tgm1, Ptx3, S100a10, Ptgs2, Tm4sf1, and Cd14) as compared with the FLOX-GCI mice. In addition, four of the A1-specific genes (H2-T23, Serping1, Fbln5 and Fkbp5) were significantly increased (p < 0.01, unpaired t test) in KO-GCI, as compared with the FLOX-GCI (Fig. 8B). These results demonstrate that the ischemia-induced astrocyte reactive genes (PAN and A2-specific) were predominantly reduced in the KO hippocampus after GCI; while in contrast, several of the inflammation-induced astrocyte reactive (A1-specific) genes were increased in the KO-GCI hippocampus as compared with FLOX-GCI.
Figure 8.
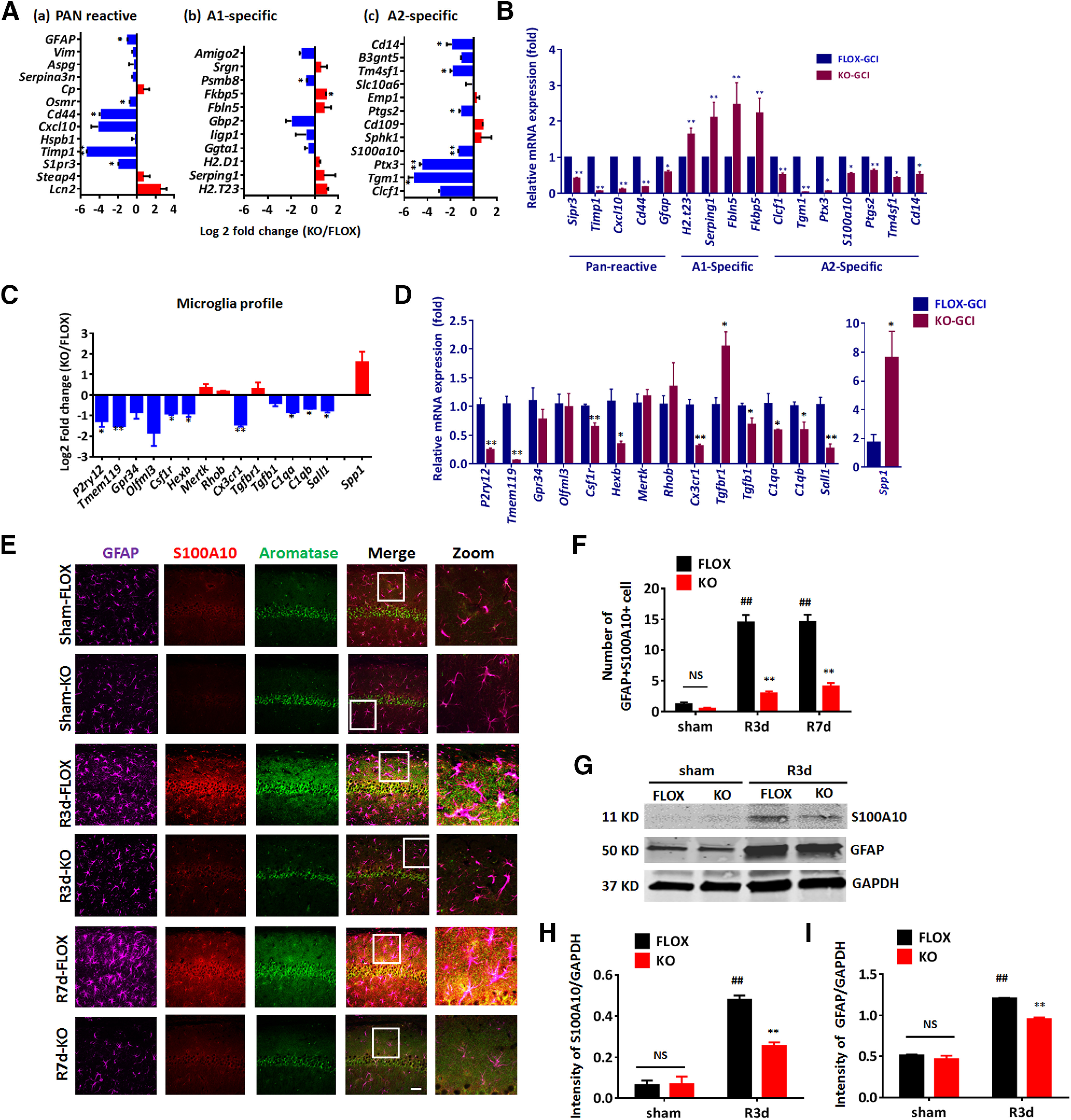
A2-type reactive astrocytes are decreased in male GFAP-ARO-KO mice after GCI. A, Log2 fold change of PAN/A1/A2 astrocyte genes between the FLOX-GCI-R24h and KO-GCI-R24h samples. B, PAN/A1/A2 astrocyte genes expression profile changes between FLOX-GCI and KO-GCI groups were verified using qRT-PCR (n = 5 biologically independent mice). C, Log2 fold change of microglia-specific signature genes between the FLOX-GCI-R24h and KO-GCI-R24h samples. D, Microglia-specific signature genes changes between FLOX-GCI and KO-GCI groups were verified using qRT-PCR (n = 5 biologically independent mice). E, Representative images of immunostaining for GFAP, S100A10, and aromatase in hippocampal CA1 region. Scale bar: 20 μm. F, Quantification of GFAP-S100A100 double-positive cell number in hippocampal CA1 region. G, Western blot analysis of S100A10 on protein lysates of ACSA2+ astrocytes isolated from the hemispheres of sham and R3d mice. H, I, Densitometry analysis of Western blot analysis results by ImageJ. Intensities of the S100A10 (H) and GFAP (I) band were normalized to the intensity of GAPDH from two independent experiments. Samples from three mice were pooled for one collection for each experiment. Data are mean ± SEM; #p < 0.05, ##p < 0.01 versus sham, two-way ANOVA followed by post hoc; *p < 0.05, **p < 0.01 versus KO with FLOX, (A–D) multiple t test; (F–I) two-way ANOVA followed by post hoc. NS, not significant.
To evaluate microglia transcriptome profile changes in the hippocampus of KO-GCI mice, we conducted expression analysis on microglia homeostatic genes, inflammatory molecular genes (Krasemann et al., 2017), and disease-associated microglia (DAM) signature genes (Deczkowska et al., 2018). Among these three groups of genes, we observed that eight out of the 14 microglia homeostatic genes (P2ry12, Tmem119, Csf1r, Hexb, Cx3cr1, C1qa, C1qb, and Sall1) were significantly decreased in KO-GCI (p < 0.05, unpaired t test, compared with FLOX-GCI; Fig. 8C). We also noticed one of the inflammatory molecular genes, spp1, was significantly increased in KO-GCI (p < 0.05, unpaired t test, compared with FLOX-GCI; Fig. 8C). No significant change in the DAM signature gene was detected between the FLOX-GCI and KO-GCI. To verify these RNA-seq results, we next performed qRT-PCR using biologically independent samples. qRT-PCR confirmed that nine of the 14 microglia homeostatic genes (P2ry12, Tmem119, Csf1r, Hexb, Cx3cr1, Tgfb1, C1qa, C1qb, and Sall1) were significantly decreased, while the inflammatory molecular gene Spp1 was significantly increased in KO-GCI (n = 5 mice, p < 0.05, unpaired t test, compared with FLOX-GCI; Fig. 8D).
Following the transcriptome analysis, we next examined the expression of an A2 astrocyte marker, S100A10, and aromatase at the protein level in the sham R3d and R7d hippocampal astrocytes of male mice. In this study, triple-immunostaining was performed in male hippocampal CA1 region for GFAP (astrocyte marker), S100A10, and aromatase. The results demonstrated that aromatase expression was highly induced in FLOX-GCI S100A10+astrocytes, but not in KO-GCI S100A10+astrocytes at R3d and R7d (Fig. 8E). The number of GFAP+S100A10+ cells was significantly increased in FLOX-R3d and R7d as compared with FLOX-sham mice (n = 6, p < 0.01, two-way ANOVA followed by Sidak's test; Fig. 8F). In contrast, the number of GFAP+S100A10+ cells was significantly decreased in KO-GCI mice, as compared with FLOX-GCI mice at R3d and R7d (n = 6, F(2,41) = 35.68, p < 0.01, two-way ANOVA; Fig. 8F). To further confirm these results, we also performed Western blot analysis on lysates from purified astrocytes collected from the brain hemispheres to detect the expression of GFAP, and the A2 astrocyte marker S100A10 (Fig. 8G–I). The results showed that the expression of S100A10 was highly increased in FLOX-GCI astrocytes, as compared with expression in FLOX-sham astrocytes at R3d (n = 6 mice, p < 0.05, two-way ANOVA followed by Sidak's test; Fig. 8H). Furthermore, the expression of S100A10 in KO-GCI astrocytes was significantly reduced as compared with FLOX-GCI astrocytes (n = 6 mice, F(1,4) = 20.61, p < 0.05, two-way ANOVA followed by Sidak's test; Fig. 8H). Expression of GFAP in the purified KO astrocytes was also significantly reduced compared with that in the FLOX astrocytes after GCI (n = 6 mice, F(1,4) = 17.25, p < 0.05, two-way ANOVA followed by Sidak's test; Fig. 8I). These findings are consistent with the expression analysis results of the RNA-seq data, and support that astrocyte-derived E2 has a key role in enhancing the A2-type astrocyte activation following GCI.
Astrocyte-derived E2 in male mice regulates LIF/STAT3 signaling in astrocytes after GCI
We next explored the potential mechanism for the reduced astrocyte activation in male KO mice after GCI. Based on our RNA-seq results, LIF was among the top differentially regulated cytokine activity genes in FLOX and KO hippocampus after GCI. Interestingly, LIF belongs to the IL-6 cytokine family, which can induce activation of the downstream STAT3 signaling pathway (Murakami et al., 2019). STAT3 is known to be a critical regulator of reactive astrogliosis in response to CNS injury, and STAT3 deletion in GFAP+ cells diminishes the neuroprotective activity of reactive astrocytes (Herrmann et al., 2008). Therefore, we next sought confirmation of changes in the LIF/IL-6-STAT3 signaling pathway at the protein level by examining the cytokine levels of LIF and IL-6, as well as the activation of STAT3 in male FLOX and KO hippocampal tissues after GCI. ELISA analysis of the cytokine LIF in the hippocampal tissue lysate detected a significant increase of LIF levels in FLOX-R3d and FLOX-R7d, as compared with the FLOX-sham group (n = 6, p < 0.01, two-way ANOVA followed by Tukey's test; Fig. 9A). In contrast, LIF levels in the KO-GCI hippocampus were significantly lower than the levels observed in the FLOX-GCI hippocampus at R3d and R7d (n = 6, F(2,28) = 7.230, R3d, p < 0.05; R7d, p < 0.05, two-way ANOVA followed by Sidak's test; Fig. 9A). Meanwhile, ELISA analysis found that IL-6 levels in the hippocampus were not significantly different in FLOX-GCI mice, as compared with the sham group (n = 6, p = 0.4866, two-way ANOVA followed by Tukey's test; Fig. 9B). Furthermore, IL-6 levels in KO-GCI hippocampus did not show a significant difference as compared with FLOX-GCI (n = 6, F(2,27) = 0.1496, p = 0.8617, two-way ANOVA followed by Sidak's test; Fig. 9B). Finally, co-immunostaining of LIF with GFAP in hippocampal slices showed an upregulated LIF expression in astrocytes in FLOX-GCI mice compared with FLOX-sham (n = 6, p < 0.01, two-way ANOVA followed by Tukey's test; Fig. 9C,E), and the IF intensity of LIF in KO-GCI hippocampus is significantly decreased compared with FLOX-GCI (n = 6, F(2,24) = 9.649, p < 0.01, two-way ANOVA followed by Sidak's test; Fig. 9C,E).
Figure 9.
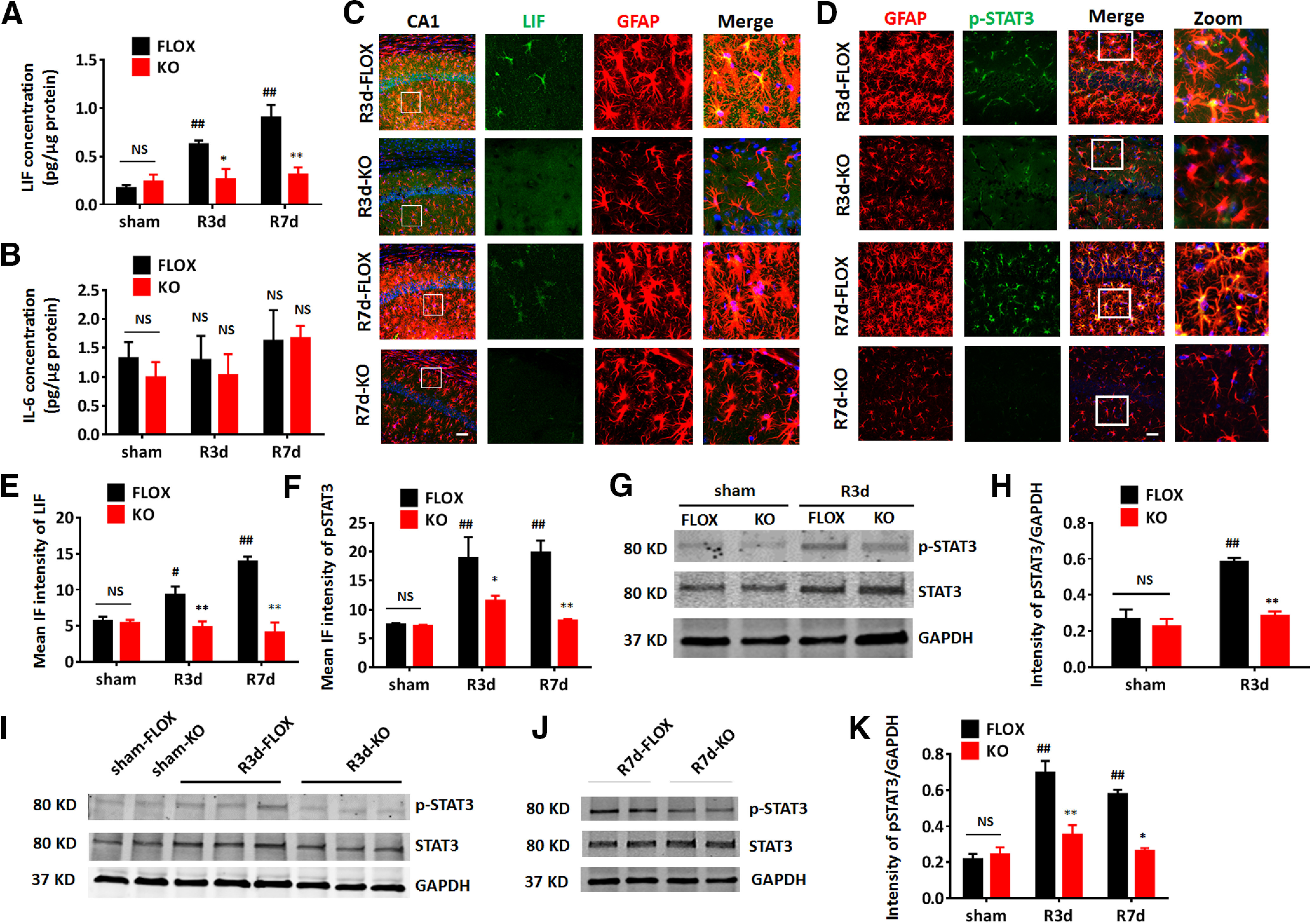
Astrocyte-derived E2 regulates the LIF-STAT3 pathway in astrocytes of male mice after GCI. A, B, ELISA to measure cytokine LIF (A) and IL-6 (B) levels in the hippocampus. C, D, Representative images of immunostaining for GFAP and LIF (C)/p-STAT3 (D) in hippocampal CA1 region at R3d and R7d. Scale bar: 20 μm. E, F, IF quantification of LIF (E) and pSTAT3 (F) intensity in hippocampal CA1. G, H, Western blot analysis and quantification of p-STAT3, and STAT3 on protein lysates from purified ACSA2+ astrocytes isolated from the hemispheres of sham and R3d mice. Intensities of the p-STAT3 band were normalized to the intensity of GAPDH from two independent experiments. Samples from three mice were pooled for one collection for each experiment. I–K, Western blot analysis and quantification of relative p-STAT3 expression in hippocampus of sham, R3d, and R7d animals. Data are mean ± SEM; #p < 0.05, ##p < 0.01 versus sham, two-way ANOVA followed by post hoc; *p < 0.05, **p < 0.01 versus KO with FLOX, two-way ANOVA followed by post hoc. NS, not significant.
We next examined STAT3 activation by performing double IHC co-localization of phosphorylated-STAT3 (the activated form of STAT3, Tyr705; p-STAT3) with GFAP in the male FLOX and KO hippocampus. IF analysis showed a significant co-localization of p-STAT3 with GFAP in FLOX-GCI but not in KO-GCI hippocampal CA1 region at R3d and R7d (n = 6, F(2,23) = 3.528, R3d, p < 0.05; R7d, p < 0.01, two-way ANOVA followed by Sidak's test; Fig. 9D,F). Western blot analysis on purified astrocytes collected from FLOX and KO brain hemispheres at 3 d after GCI reperfusion also detected an increase of p-STAT3 in FLOX-GCI astrocytes compared with FLOX-sham astrocytes (n = 6 animals, p < 0.01, two-way ANOVA followed by Sidak's test; Fig. 9G,H), and the p-STAT3 level in KO-GCI astrocytes was significantly decreased as compared with FLOX-GCI astrocytes (n = 6 animals, F(1,4) = 11.75, p < 0.05, two-way ANOVA; Fig. 9G,H). Furthermore, Western blot analysis of hippocampal tissue lysate detected a significant increase of p-STAT3 levels in FLOX-GCI as compared with sham at R3d and R7d (n = 6, p < 0.01, two-way ANOVA followed by Sidak's test; Fig. 9I–K). Interestingly, the p-STAT3 levels in the KO-GCI hippocampal samples were significantly lower than in FLOX-GCI hippocampal samples (n = 6, F(2,12) = 8.088, R3d, p < 0.01; R7d, p < 0.05, two-way ANOVA followed by Sidak's test; Fig. 9I–K). Altogether, these data mirror the RNA-seq analysis results and demonstrate that the LIF/STAT3 signaling is highly induced in FLOX astrocytes but not in KO astrocytes after GCI. These findings suggest that astrocyte-derived E2 is crucial for ischemia-induced astrocyte activation, and that effect may involve modulation of the LIF/STAT3 signaling pathway.
Astrocyte aromatase deletion in the intact female brain induces a similar phenotype as in the male after GCI
Similar to males, aromatase is also strongly expressed in the forebrain of female mice (Lu et al., 2019). Thus, we next examined whether astrocyte aromatase deletion in the female brain induces a similar phenotype in female mice as in males. Three-month-old intact female FLOX and KO mice were examined in sham and R3d groups for neuronal damage, glial activation, and LIF/STAT3 signaling pathway activation. Immunostaining of NeuN and quantification on the CA1 neuron number revealed that a 20-min ischemia resulted in a mild but not significant decrease of the CA1 neurons in the female FLOX mice (n = 6, p > 0.05, two-way ANOVA followed by Sidak's test; Fig. 10B). However, the intact female KO mouse exhibited a significant decrease of CA1 neurons as compared with FLOX after GCI (n = 6, F(1,15) = 23.13, p < 0.01, two-way ANOVA followed by Sidak's test; Fig. 10A,B), indicating the loss of astrocyte-derived E2 leads to increased susceptibility to neuronal damage in female mouse brain after GCI. Following GCI, an enhanced astrocyte activation was detected in the FLOX-GCI hippocampus, as demonstrated by a significant increase in GFAP IF intensity compared with FLOX-sham (n = 6, p < 0.01, two-way ANOVA followed by Sidak's test; Fig. 10A,C). Similar to the case in male mice, astrocyte activation was significantly decreased in the hippocampus of intact female KO mice after GCI, as compared with intact female FLOX-GCI mice (n = 6, F(1,20) = 9.963, p < 0.01, two-way ANOVA followed by Sidak's test; Fig. 10A,C). We next examined microglia activation in the intact female mice. Quantification of IF intensity revealed an enhanced Iba1 intensity in both FLOX-GCI and KO-GCI hippocampus, as compared with sham controls (n = 6, p < 0.01, two-way ANOVA followed by Sidak's test; Fig. 10D,E). Moreover, the Iba1 intensity in the intact female KO-GCI hippocampus is significantly higher than that in the intact female FLOX-GCI hippocampus at R3d (n = 6, F(1,20) = 25.16, p < 0.01, two-way ANOVA followed by Sidak's test; Fig. 10D,E). Next, we sought to further confirm the IF intensity data for GFAP and Iba1 using Western blot analysis, which confirmed a reduced GFAP expression (n = 4, F(1,12) = 13.98, p < 0.01, two-way ANOVA followed by Sidak's testl Fig. 10F–H) and enhanced Iba1 expression (n = 4, F(1,12) = 4.900, p < 0.05; two-way ANOVA followed by Sidak's test; Fig. 10F–H) in the intact female KO-GCI hippocampal tissue, as compared with intact female FLOX-GCI at R3d. Finally, we also evaluated the activation of the LIF/STAT3 signaling pathway in the intact female mice after GCI. IHC and IF intensity quantification revealed an upregulation of LIF expression in astrocytes of intact female FLOX-GCI mice but not in intact female KO-GCI mice at R3d (n = 6, F(1,20) = 95.22, p < 0.01, two-way ANOVA followed by Sidak's test; Fig. 10I,J). Western blot analysis similarly confirmed an enhanced activation of p-STAT3 in intact female FLOX-GCI hippocampus, as compared with intact female FLOX-sham at R3d (n = 4, p < 0.01, two-way ANOVA followed by Sidak's test; Fig. 10K,L). Furthermore, the p-STAT3 level was significantly lower in intact female KO-GCI hippocampus, as compared with intact female FLOX-GCI at R3d (n = 4, F(1,8) = 7.864, p < 0.05, two-way ANOVA; Fig. 10K,L). In summary, these results demonstrate that astrocyte aromatase deletion in the intact female mouse results in increased neuronal damage, decreased astrocyte activation, and enhanced microglia activation with compromised LIF/STAT3 signaling pathway after GCI, similar to the phenotype observed in male KO mice.
Figure 10.
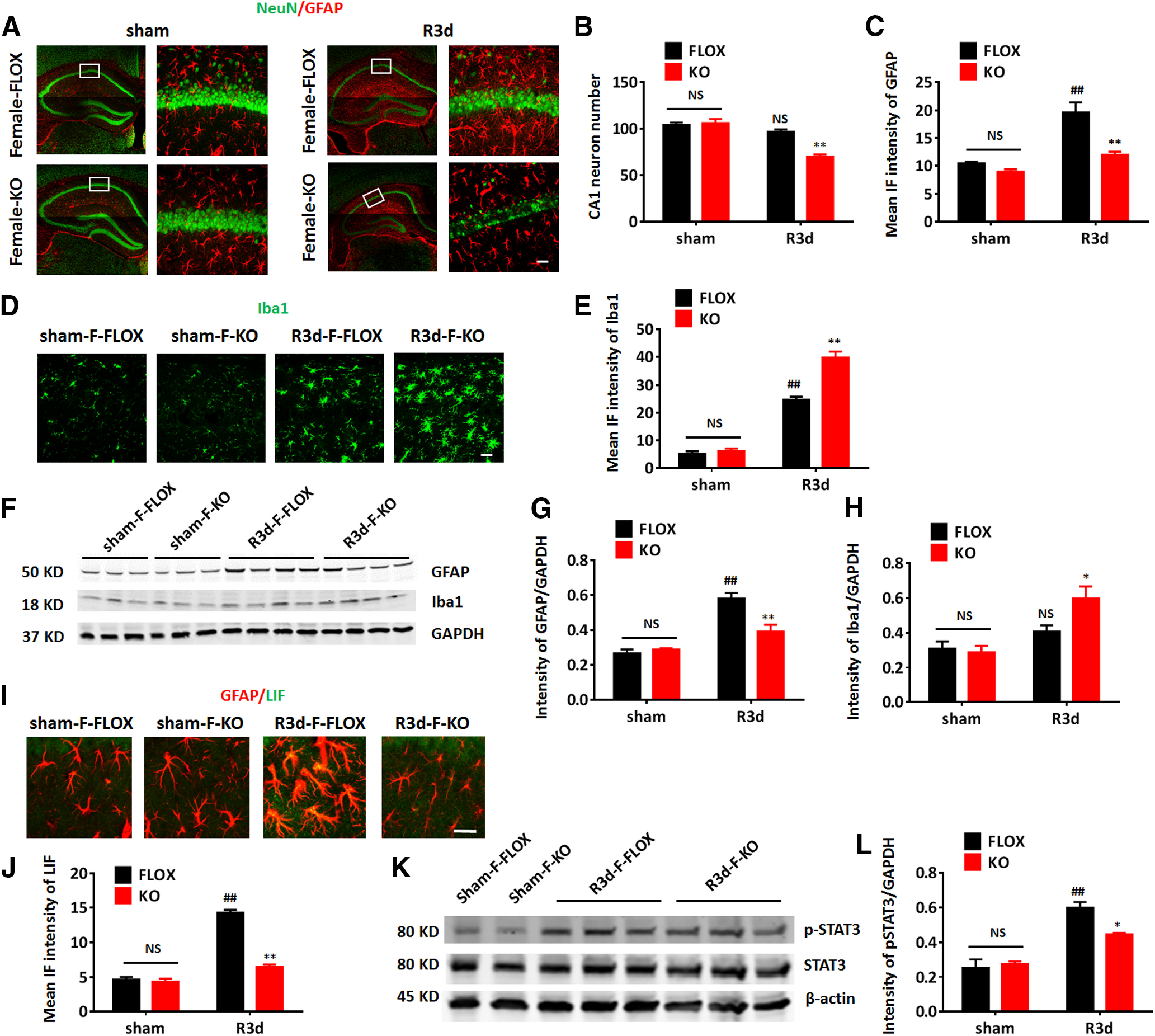
Astrocyte-derived E2 deletion in the intact female brain induces a similar phenotype as in the male after GCI. A–C, Representative images of immunostaining for GFAP and NeuN (A) and quantification of the CA1 neuron number (B) and the mean IF intensity of GFAP (C) in the intact female hippocampus. D, E, Representative images of immunostaining for Iba1 (D) and quantification of mean IF intensity (E) of Iba1 the intact female hippocampal CA1. F–H, Western blot analysis (F) and quantification of GFAP (G) and Iba1 (H) expression in hippocampal tissue samples collected from intact female mice. I, J, Representative images of immunostaining for GFAP and LIF (I) and quantification of the mean IF intensity of LIF (J) in the intact female hippocampal CA1. K, L, Western blot analysis (K) and quantification of p-STAT3 in hippocampal tissue samples collected from intact female mice. Data are mean ± SEM; ##p < 0.01 versus sham, two-way ANOVA followed by post hoc; *p < 0.05, **p < 0.01 versus KO with FLOX, two-way ANOVA followed by post hoc. NS, not significant. Scale bars: 20 μm.
Decreased astrocyte activation in GFAP-ARO-KO mice can be rescued by exogenous E2 administration
Similar to the results obtained in intact male and intact female KO mice, we found that ovx-KO mice also had increased neuronal damage, decreased astrocyte activation, decreased LIF-STAT3 signaling, and enhanced microglial hypertrophy, as compared with ovx-FLOX mice after GCI (n = 6, p < 0.05, two-way ANOVA followed by Sidak's tests; Fig. 11). Using the ovx-female mice, we next examined whether these effects could be rescued by exogenous E2 administration. Exogenous E2 or placebo (20% β-cyclodextrin) was administered by minipumps to ovx-KO mice subcutaneously. Seven days after E2 or placebo administration, the mice were subjected to GCI, and hippocampal samples were collected for analysis at R3d. E2 measurement by ELISA showed that exogenous E2 administration could restore the local E2 level in the KO-R3d hippocampus back to levels observed in the FLOX-R3d hippocampus (n = 6, F(2,30) = 42.26, p < 0.01, one-way ANOVA followed by Tukey's test; Fig. 11A). Immunostaining demonstrated that the total intensity of GFAP in the ovx-KO hippocampus was significantly enhanced at R3d in E2-treated but not in placebo-treated animals, compared with the ovx-KO-R3d (n = 6, F(3,18) = 20.44, p < 0.01, one-way ANOVA followed by Tukey's test; Fig. 11B,C). Western blot analysis confirmed that GFAP levels in the hippocampus at R3d were increased in ovx-KO mice after E2 treatment, but not in placebo-treated mice (n = 6, F(3,7) = 22.38, p < 0.05, one-way ANOVA followed by Tukey's test; Fig. 11D,F). Next, immunostaining for NeuN, a neuronal marker, was used to determine neuron number. The results revealed that exogenous E2, but not placebo treatment, significantly increased the number of surviving neurons in the ovx-KO hippocampal CA1 region at R3d (n = 6, F(3,24) = 14.56, p < 0.01, one-way ANOVA followed by Tukey's test; Fig. 11H,I). Immunostaining assay also found that the intensity of the microglia marker, Iba1 in the hippocampal CA1 region was significantly decreased in E2-treated, but not placebo-treated mice (n = 6, F(3,24) = 20.74, p < 0.01, one-way ANOVA followed by Tukey's test; Fig. 11H,J). Western blot analysis further confirmed a decrease in Iba1 levels in the hippocampus of ovx-KO-E2-treated mice, as compared with the ovx-KO-R3d group (n = 6, F(3,6) = 21.71, p < 0.05, one-way ANOVA followed by Turley's test; Fig. 11E,G). Moreover, exogenous E2 infusion also rescued the LIF level in the KO hippocampus (n = 6, F(3,19) = 5.202, p < 0.05, one-way ANOVA followed by Tukey's test; Fig. 11K) as measured by ELISA. Finally, Western blot analysis demonstrated that p-STAT3 levels in the hippocampus at R3d were significantly rescued in ovx-KO mice by exogenous E2 treatment (F(3,5) = 8.842, p < 0.05, one-way ANOVA followed by Tukey's test; Fig. 11L,M).
Figure 11.
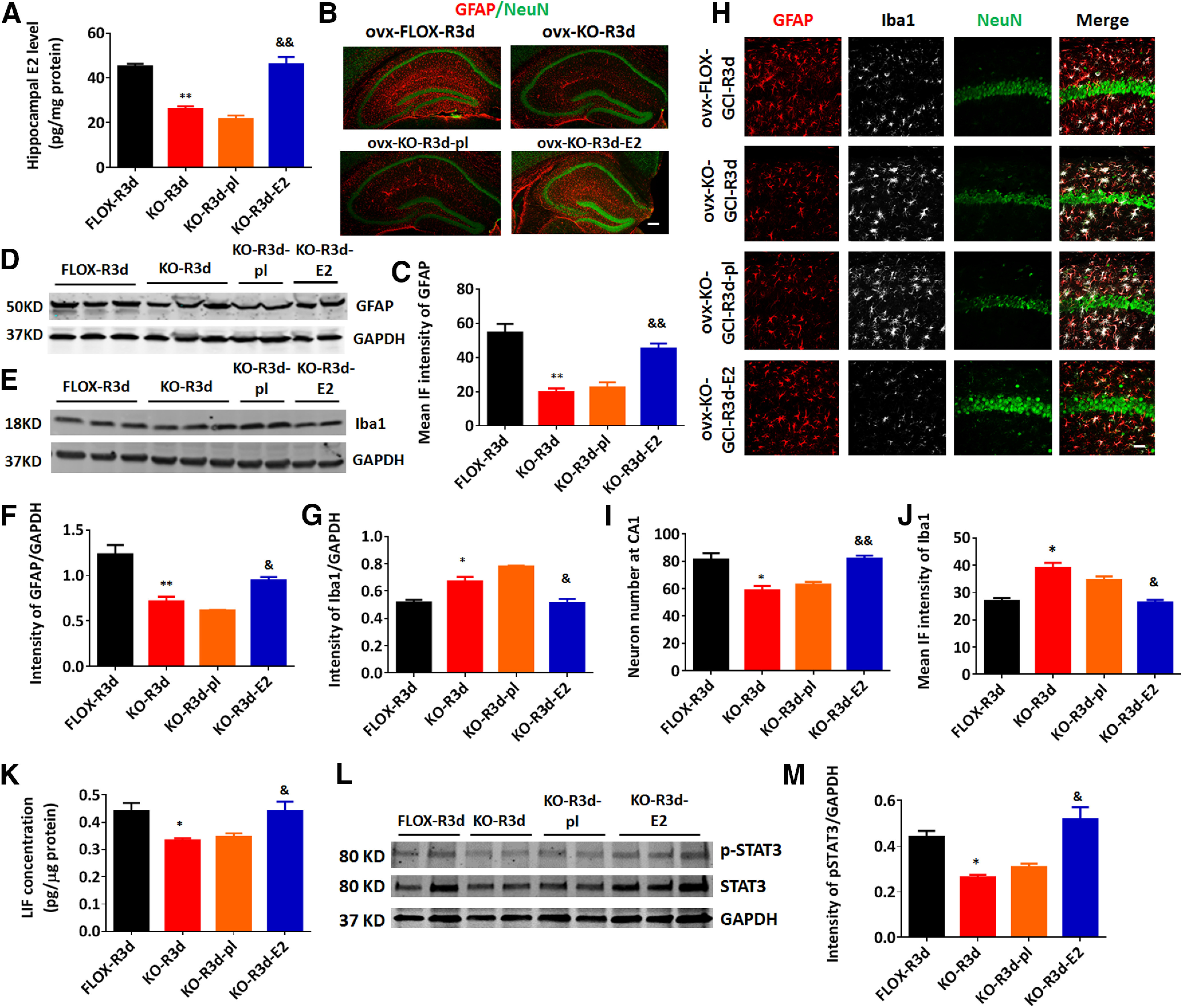
Reinstatement of forebrain E2 levels by exogenous E2 administration rescues reactive astrogliosis, reverses the increase of microglia activation, and is neuroprotective in the ovx-female GFAP-ARO-KO mice. A, E2 levels in hippocampal tissue samples were measured by ELISA at R3d in ovx-female animals. B, C, Representative images of immunostaining for GFAP and NeuN (B) and quantification of the mean IF intensity of GFAP (C) in the ovx-female hippocampus. Scale bar: 50 μm. D–G, Western blot analysis and quantification of GFAP (D, F) and Iba1 (E, G) expression in hippocampal tissue samples collected from ovx-female animals. H–J, Representative images of immunostaining for GFAP, Iba1, and NeuN (H) in the hippocampal CA1 region of ovx-female mice, and quantification of the neuron number in the hippocampal CA1 region (I) and the mean IF intensity of Iba1 (J) in H. Scale bar: 20 μm. K, LIF levels in hippocampal tissue samples were measured by ELISA at R3d in ovx-female animals. L, M, Western blot analysis and quantification of p-STAT3 level in hippocampal tissue samples collected from ovx-female animals. Data are mean ± SEM; n = 6 biologically independent animals; *p < 0.05, **p < 0.01 compared with FLOX-R3d, two-way ANOVA; &p < 0.05, &&p < 0.01 compared with KO-R3d, one-way ANOVA followed by post hoc tests.
To establish whether exogenous E2 treatment could rescue the suppressed reactive astrogliosis gene expression in KO mice after GCI, we measured the mRNA expression of the reactive astrocyte genes in the hippocampal tissue of placebo-treated or E2-treated ovx-female mice. Compared with FLOX-R3d-pl mice, KO-GCI-pl mice had a significant decrease in the expression of nine of the PAN (Steap4, Serpina3n, Timp1, Cxcl10, Cd44, Osmr, Aspg, Vimentin, and Gfap) and 8 of the A2 (Clcf1, Tgm1, Ptx3, S100a10, Emp1, Slc10a6, Tm4sf1, and Cd14) reactivate astrocyte genes (n = 3–5, compared with FLOX-GCI-pl, p < 0.05, one-way ANOVA followed by Tukey's test; Fig. 12A,B). Intriguingly, exogenous E2 treatment was able to significantly rescue the mRNA level of these genes in KO mice (n = 3–5, compared with KO-GCI-pl, p < 0.05, one-way ANOVA followed by Tukey's test; Fig. 12A,B). Collectively, these data demonstrate that reinstating forebrain E2 levels rescues the defects observed in the KO mouse.
Figure 12.
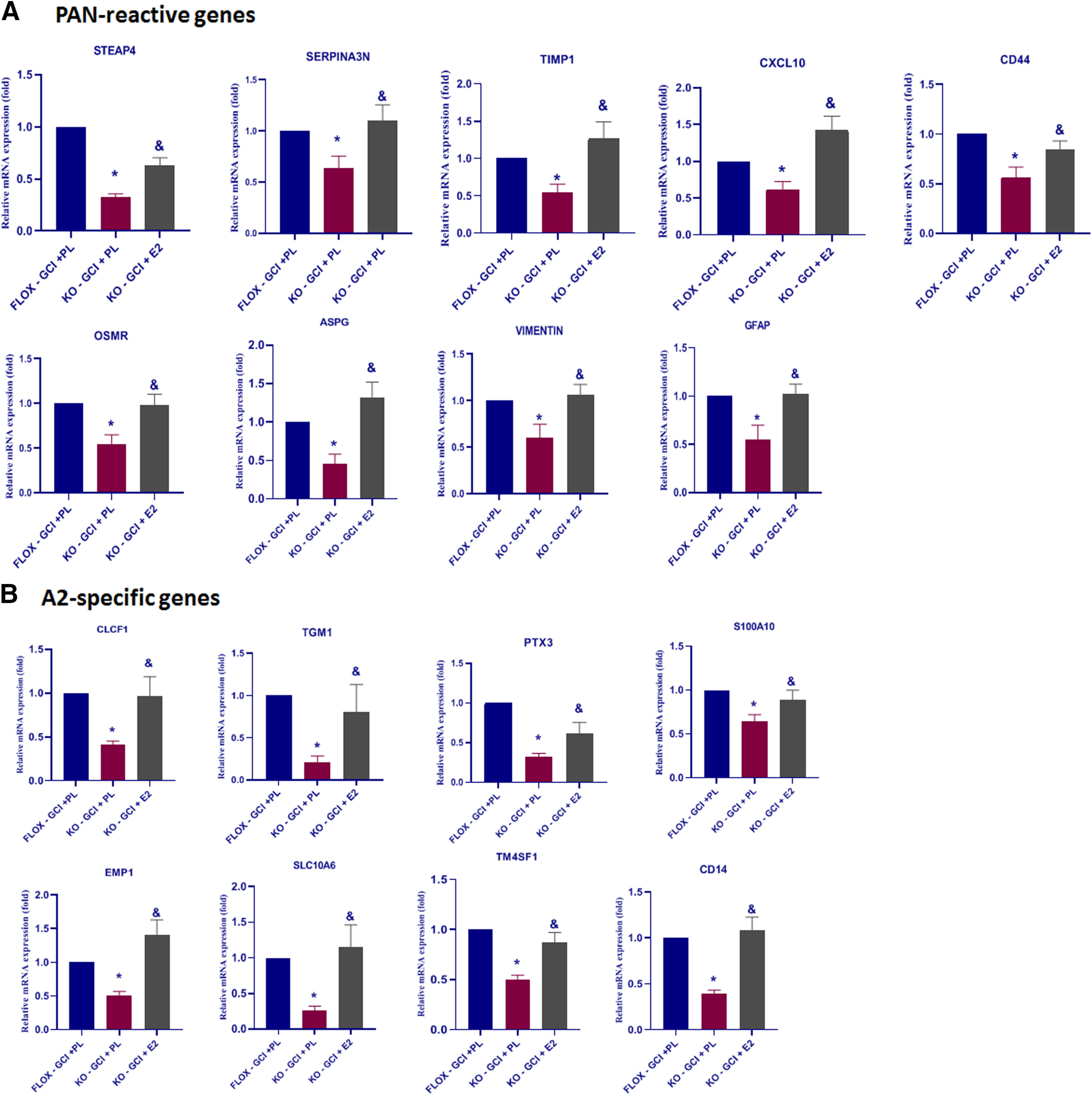
Reinstatement of forebrain E2 levels by exogenous E2 administration can rescue the suppressed reactive astrocyte genes in the ovx-female GFAP-ARO-KO hippocampus after GCI. qRT-PCR analysis for PAN-specific (A) and A2-specific (B) reactive astrocyte genes expression level in FLOX-GCI-placebo, KO-GCI-placebo, and KO-GCI-E2 hippocampus at R3d. Data are mean ± SEM; n = 3–5; *p < 0.05 versus FLOX-GCI +PL, &p < 0.05 versus KO – GCI + PL, one-way ANOVA followed by post hoc tests.
Discussion
While it is well accepted that astrocytes express aromatase after brain injury, the precise functions of astrocyte-derived E2 are not well understood. Using a novel GFAP-ARO-KO mouse model, the current study sheds new light on this issue by demonstrating that astrocyte-derived E2 has several key roles following cerebral ischemia, including (1) being critical for reactive astrogliosis in the hippocampal CA1 region; (2) inducing JAK-STAT3 signaling in astrocytes; (3) facilitating induction of the A2 panel of reactive astrocyte genes; (4) suppressing microglia activation; and (5) exerting neuroprotection and preserving hippocampal-dependent cognitive functions. It should be noted that while depletion of astrocytic aromatase leads to a loss of GCI-induced E2 in the KO forebrain, it can also produce an increase in androgen precursors. We believe the defects in the KO mouse are primarily because of the loss of E2, as reinstating forebrain E2 levels by exogenous E2 replacement was able to rescue the defects in KO mice after GCI.
Further characterization of the KO mice revealed that they had normal brain structure and development and exhibited no gross abnormalities or cognitive defects basally. This is perhaps not unexpected, as astrocytes express little to no aromatase basally, with significant induction of aromatase only observed after ischemia or injury (Peterson et al., 2001; Garcia-Segura et al., 2003; Zhang et al., 2014). Indeed, we observed a significant ischemia-induced expression of aromatase in astrocytes in FLOX mice, with a corresponding elevation of hippocampal E2 levels after GCI, an effect that was abolished in KO mice. Further examination of aromatase expression in purified astrocytes from FLOX and KO mice at R3d confirmed the ischemia-induced elevation of aromatase in FLOX astrocytes, and its abolishment in KO astrocytes. While ischemia-induced aromatase expression was abolished in KO astrocytes, constitutive basal aromatase expression in hippocampal neurons, as well as basal hippocampal E2 levels, were not significantly different in FLOX versus KO mice, indicating that the KO of aromatase was specific for astrocytes.
Astrocyte-derived E2 regulates reactive astrogliosis after ischemic injury
It is well known that following brain injury and neurodegenerative disorders, astrocytes highly express the intermediate filament, GFAP, and exhibit robust cellular hypertrophy, a process termed reactive astrogliosis (Takano et al., 2009). Although well characterized, the process and factors that regulate reactive astrogliosis after ischemia are not fully understood. The results of our current study enhance understanding in this area by demonstrating that astrocyte-derived E2 is critical for reactive astrogliosis following cerebral ischemia. Following GCI reperfusion injury, we observed that reactive astrogliosis in KO mice was significantly decreased compared with FLOX mice. This result was further confirmed using a pharmacological approach, in which we administered the aromatase inhibitor, letrozole to WT mice, and demonstrated a similar significant attenuation of reactive astrogliosis after GCI, as compared with placebo-treated mice. The decreased reactive astrogliosis in KO mice may explain, at least in part, the increased neuronal damage and cognitive deficits observed in KO mice after GCI. In support of this suggestion, there is growing evidence that reactive astrogliosis plays a key role in limiting tissue damage and contributes to the restoration of homeostasis following cerebral ischemia (Sofroniew and Vinters, 2010; Choudhury and Ding, 2016; Becerra-Calixto and Cardona-Gómez, 2017). For instance, GFAP−/−Vm−/− mice exhibit attenuated reactive astrogliosis and increased neuronal damage after cerebral ischemia (de Pablo et al., 2013). Likewise, inhibition of reactive astrocytes with fluorocitrate has been shown to attenuate neurovascular remodeling and recovery in mice after cerebral ischemia (Hayakawa et al., 2010). Recent work using transcriptome analysis has suggested that there are at least two major classes of astrocytes, an A1 “proinflammatory” astrocyte that is induced in chronic neurodegenerative disorders, and an A2 “neuroprotective” astrocyte, which is induced acutely following cerebral ischemia (Zamanian et al., 2012). In our study, RNA-Seq analysis revealed that many of the A2-reactive astrocyte genes are significantly lower in the KO mice, as compared with FLOX mice after GCI. IHC for the A2-selective marker, S100A10, confirmed a significant decrease in induction of A2 astrocytes in KO mice after GCI. Because of the proposed neuroprotective role of A2 astrocytes, it is possible that the reduced A2 astrocyte phenotype could also contribute to the increased neuronal damage and cognitive deficits in KO mice after GCI. However, definitive proof of a role of A2 astrocytes in neuroprotection in GCI will have to await the development of tools that allow specific blocking or ablation of A2 astrocytes in the brain.
Astrocyte-derived E2 regulates JAK-STAT3 signaling in astrocytes after GCI
Previous work has revealed that the JAK-STAT3 pathway plays a key role in mediating the activation of reactive astrocytes after ischemia and injury (Ceyzériat et al., 2016; Liddelow and Barres, 2017). This is intriguing as GSEA of our RNA-seq data revealed that the IL-6/JAK/STAT3 signaling pathway is negatively correlated with GFAP-ARO-KO after GCI. Our study also demonstrated that the STAT3 activation level (p-STAT3 upregulation) is significantly suppressed in KO astrocytes, as compared with FLOX astrocytes after GCI. The loss of STAT3 activation could explain the reduced reactive astrogliosis in our KO mice after GCI, as previous work showed that conditional deletion of STAT3 in astrocytes results in failure of reactive astrogliosis in a spinal cord injury model (Okada et al., 2006; Herrmann et al., 2008). Interestingly, mice with conditional STAT3 deletion in astrocytes displayed increased lesion volume and attenuated functional recovery, similar to our GFAP-ARO-KO mice. In further support of a key role for astrocyte-derived E2 in regulating the IL-6/JAK/STAT3 signaling pathway, we found that LIF, a member of the IL-6 cytokine family and an upstream regulator of STAT3 (Murakami et al., 2019), was significantly upregulated in the FLOX-R3d hippocampus, but not in the KO-R3d hippocampus after GCI, as measured by IHC and ELISA. Thus, the attenuated STAT3 activation in our KO mice after GCI may be because of loss of LIF induction. In further potential support of this possibility, we found that reinstatement of forebrain-E2 levels in KO mice rescued LIF expression and STAT3 activation in KO astrocytes, an effect that was associated with a rescue of astrocyte activation in KO mice.
It should be mentioned that the results of our study found similar defects in male, intact female, and ovx-female KO mice, with all showing a similar increased neuronal damage, decreased astrocyte activation, decreased LIF-STAT3 signaling, and enhanced microglial hypertrophy after GCI, as compared with FLOX controls. Since we focused primarily on the LIF-STAT3 signaling pathway, we cannot rule out that gender differences could exist in other pathways or mechanisms not examined in our study. For instance, previous studies have provided evidence that gender differences exist in cell death signaling, immune response, and recovery following cerebral ischemia (Spychala et al., 2017). Thus, more detailed studies on these pathways and processes may yet reveal gender differences in the KO mice.
Astrocyte-derived E2 regulates microglia activation after GCI
Microglia activation is well known to be an indicator of an inflammatory response and neuronal damage following ischemic brain injury (Patel et al., 2013). In our study, we observed enhanced hypertrophy of microglia, increased expression of Iba1, and suppressed microglial homeostatic gene profile in the hippocampus of KO mice after GCI, indicating that microglia in KO-GCI mice have increased microglia activation and compromised homeostasis maintenance function, as compared with microglia in FLOX-GCI mice. In a previous study in the rat, we found that antisense oligonucleotide knock-down of aromatase also increased microglia activation, although it was unclear whether this was because of loss of neuronal-derived or astrocyte-derived E2, as antisense oligonucleotide knock-down decreases aromatase in both cell types (Zhang et al., 2014). The current study, which depleted aromatase specifically in astrocytes, indicates that astrocyte-derived E2 has an important role in restraining microglia activation after GCI. This effect could be mediated directly on the microglia via estrogen receptors, as a number of studies have shown that microglia express estrogen receptors and that E2 can act directly on microglia to suppress their pro-inflammatory activation (Vegeto et al., 2003; Lee et al., 2018; Thakkar et al., 2018). Alternatively, enhanced neuronal damage in the KO-GCI hippocampus could also contribute to the increased microglia activation, as microglia can be activated by danger signals from damaged neurons (Podbielska et al., 2016; Gülke et al., 2018). Finally, whether the increased microglia activation contributes to the increased neuronal damage and astrocytic phenotype changes observed in our KO mice after GCI is unclear. Interpretation on this issue is complicated, as previous work has shown that reactive microglia display diverse polarization phenotypes in response to brain injury, and activated microglia can play both beneficial and detrimental roles after cerebral ischemia (Patel et al., 2013). Hence, further study will be needed to address this question.
In conclusion, the current study provides evidence that astrocyte-derived E2 has a critical role in regulating reactive astrogliosis, microglia activation and neuroprotection in the ischemic brain. Astrocyte-derived E2 is also critical for the activation of LIF/JAK/STAT3 signaling in astrocytes, which is a key regulatory pathway responsible for astrocyte activation. As a whole, the study provides novel genetic evidence of the multiple beneficial roles of astrocyte-derived E2 in the ischemic brain.
Footnotes
This work was supported by the National Institutes of Neurological Disorders and Stroke, National Institutes of Health Research Grant R01NS088058.
The authors declare no competing financial interests.
References
- Becerra-Calixto A, Cardona-Gómez GP (2017) The role of astrocytes in neuroprotection after brain stroke: potential in cell therapy. Front Mol Neurosci 10:88. 10.3389/fnmol.2017.00088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brann DW, Dhandapani K, Wakade C, Mahesh VB, Khan MM (2007) Neurotrophic and neuroprotective actions of estrogen: basic mechanisms and clinical implications. Steroids 72:381–405. 10.1016/j.steroids.2007.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceyzériat K, Abjean L, Carrillo-de Sauvage MA, Ben Haim L, Escartin C (2016) The complex STATes of astrocyte reactivity: how are they controlled by the JAK-STAT3 pathway? Neuroscience 330:205–218. 10.1016/j.neuroscience.2016.05.043 [DOI] [PubMed] [Google Scholar]
- Choudhury GR, Ding S (2016) Reactive astrocytes and therapeutic potential in focal ischemic stroke. Neurobiol Dis 85:234–244. 10.1016/j.nbd.2015.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deczkowska A, Keren-Shaul H, Weiner A, Colonna M, Schwartz M, Amit I (2018) Disease-associated microglia: a universal immune sensor of neurodegeneration. Cell 173:1073–1081. 10.1016/j.cell.2018.05.003 [DOI] [PubMed] [Google Scholar]
- de Pablo Y, Nilsson M, Pekna M, Pekny M (2013) Intermediate filaments are important for astrocyte response to oxidative stress induced by oxygen-glucose deprivation and reperfusion. Histochem Cell Biol 140:81–91. 10.1007/s00418-013-1110-0 [DOI] [PubMed] [Google Scholar]
- Duan S, Anderson CM, Stein BA, Swanson RA (1999) Glutamate induces rapid upregulation of astrocyte glutamate transport and cell-surface expression of GLAST. J Neurosci 19:10193–10200. 10.1523/JNEUROSCI.19-23-10193.1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan KA, Walters BJ, Saldanha CJ (2013) Traumatized and inflamed–but resilient: glial aromatization and the avian brain. Horm Behav 63:208–215. 10.1016/j.yhbeh.2012.02.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ennaceur A, Delacour J (1988) A new one-trial test for neurobiological studies of memory in rats. 1: behavioral data. Behav Brain Res 31:47–59. 10.1016/0166-4328(88)90157-x [DOI] [PubMed] [Google Scholar]
- Fester L, Prange-Kiel J, Jarry H, Rune GM (2011) Estrogen synthesis in the hippocampus. Cell Tissue Res 345:285–294. 10.1007/s00441-011-1221-7 [DOI] [PubMed] [Google Scholar]
- Garcia-Segura LM, Wozniak A, Azcoitia I, Rodriguez JR, Hutchison RE, Hutchison JB (1999) Aromatase expression by astrocytes after brain injury: implications for local estrogen formation in brain repair. Neuroscience 89:567–578. 10.1016/s0306-4522(98)00340-6 [DOI] [PubMed] [Google Scholar]
- Garcia-Segura LM, Veiga S, Sierra A, Melcangi RC, Azcoitia I (2003) Aromatase: a neuroprotective enzyme. Prog Neurobiol 71:31–41. 10.1016/j.pneurobio.2003.09.005 [DOI] [PubMed] [Google Scholar]
- Gülke E, Gelderblom M, Magnus T (2018) Danger signals in stroke and their role on microglia activation after ischemia. Ther Adv Neurol Disord 11:1756286418774254. 10.1177/1756286418774254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayakawa K, Nakano T, Irie K, Higuchi S, Fujioka M, Orito K, Iwasaki K, Jin G, Lo EH, Mishima K, Fujiwara M (2010) Inhibition of reactive astrocytes with fluorocitrate retards neurovascular remodeling and recovery after focal cerebral ischemia in mice. J Cereb Blood Flow Metab 30:871–882. 10.1038/jcbfm.2009.257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrmann JE, Imura T, Song B, Qi J, Ao Y, Nguyen TK, Korsak RA, Takeda K, Akira S, Sofroniew MV (2008) STAT3 is a critical regulator of astrogliosis and scar formation after spinal cord injury. J Neurosci 28:7231–7243. 10.1523/JNEUROSCI.1709-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hojo Y, Hattori TA, Enami T, Furukawa A, Suzuki K, Ishii HT, Mukai H, Morrison JH, Janssen WG, Kominami S, Harada N, Kimoto T, Kawato S (2004) Adult male rat hippocampus synthesizes estradiol from pregnenolone by cytochromes P45017alpha and P450 aromatase localized in neurons. Proc Natl Acad Sci USA 101:865–870. 10.1073/pnas.2630225100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishihara Y, Itoh K, Tanaka M, Tsuji M, Kawamoto T, Kawato S, Vogel CFA, Yamazaki T (2017) Potentiation of 17β-estradiol synthesis in the brain and elongation of seizure latency through dietary supplementation with docosahexaenoic acid. Sci Rep 7:6268. 10.1038/s41598-017-06630-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krasemann S, Madore C, Cialic R, Baufeld C, Calcagno N, El Fatimy R, Beckers L, O'Loughlin E, Xu Y, Fanek Z, Greco DJ, Smith ST, Tweet G, Humulock Z, Zrzavy T, Conde-Sanroman P, Gacias M, Weng Z, Chen H, Tjon E, et al. (2017) The TREM2-APOE pathway drives the transcriptional phenotype of dysfunctional microglia in neurodegenerative diseases. Immunity 47:566–581.e9. 10.1016/j.immuni.2017.08.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Lee SO, Kim GL, Rhee DK (2018) Estrogen receptor-β of microglia underlies sexual differentiation of neuronal protection via ginsenosides in mice brain. CNS Neurosci Ther 24:930–939. 10.1111/cns.12842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liddelow SA, Barres BA (2017) Reactive astrocytes: production, function, and therapeutic potential. Immunity 46:957–967. 10.1016/j.immuni.2017.06.006 [DOI] [PubMed] [Google Scholar]
- Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, Bennett ML, Münch AE, Chung WS, Peterson TC, Wilton DK, Frouin A, Napier BA, Panicker N, Kumar M, Buckwalter MS, Rowitch DH, Dawson VL, Dawson TM, Stevens B, et al. (2017) Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541:481–487. 10.1038/nature21029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, Sareddy GR, Wang J, Wang R, Li Y, Dong Y, Zhang Q, Liu J, O'Connor JC, Xu J, Vadlamudi RK, Brann DW (2019) Neuron-derived estrogen regulates synaptic plasticity and memory. J Neurosci 39:2792–2809. 10.1523/JNEUROSCI.1970-18.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami M, Kamimura D, Hirano T (2019) Pleiotropy and specificity: insights from the interleukin 6 family of cytokines. Immunity 50:812–831. 10.1016/j.immuni.2019.03.027 [DOI] [PubMed] [Google Scholar]
- Neumann JT, Cohan CH, Dave KR, Wright CB, Perez-Pinzon MA (2013) Global cerebral ischemia: synaptic and cognitive dysfunction. Curr Drug Targets 14:20–35. 10.2174/138945013804806514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada S, Nakamura M, Katoh H, Miyao T, Shimazaki T, Ishii K, Yamane J, Yoshimura A, Iwamoto Y, Toyama Y, Okano H (2006) Conditional ablation of Stat3 or Socs3 discloses a dual role for reactive astrocytes after spinal cord injury. Nat Med 12:829–834. 10.1038/nm1425 [DOI] [PubMed] [Google Scholar]
- Patel AR, Ritzel R, McCullough LD, Liu F (2013) Microglia and ischemic stroke: a double-edged sword Int J Physiol Pathophysiol Pharmacol 5:73–90. [PMC free article] [PubMed] [Google Scholar]
- Peterson RS, Saldanha CJ, Schlinger BA (2001) Rapid upregulation of aromatase mRNA and protein following neural injury in the zebra finch (Taeniopygia guttata). J Neuroendocrinol 13:317–323. 10.1046/j.1365-2826.2001.00647.x [DOI] [PubMed] [Google Scholar]
- Podbielska M, Das A, Smith AW, Chauhan A, Ray SK, Inoue J, Azuma M, Nozaki K, Hogan EL, Banik NL (2016) Neuron-microglia interaction induced bi-directional cytotoxicity associated with calpain activation. J Neurochem 139:440–455. 10.1111/jnc.13774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roselli CE, Liu M, Hurn PD (2009) Brain aromatization: classic roles and new perspectives. Semin Reprod Med 27:207–217. 10.1055/s-0029-1216274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sareddy GR, Zhang Q, Wang R, Scott E, Zou Y, O'Connor JC, Chen Y, Dong Y, Vadlamudi RK, Brann D (2015) Proline-, glutamic acid-, and leucine-rich protein 1 mediates estrogen rapid signaling and neuroprotection in the brain. Proc Natl Acad Sci USA 112:E6673–E6682. 10.1073/pnas.1516729112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlinger BA, Amur-Umarjee S, Shen P, Campagnoni AT, Arnold AP (1994) Neuronal and non-neuronal aromatase in primary cultures of developing zebra finch telencephalon. J Neurosci 14:7541–7552. 10.1523/JNEUROSCI.14-12-07541.1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpkins JW, Yang SH, Wen Y, Singh M (2005) Estrogens, progestins, menopause and neurodegeneration: basic and clinical studies. Cell Mol Life Sci 62:271–280. 10.1007/s00018-004-4382-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sofroniew MV, Vinters HV (2010) Astrocytes: biology and pathology. Acta Neuropathol 119:7–35. 10.1007/s00401-009-0619-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spychala MS, Honarpisheh P, McCullough LD (2017) Sex differences in neuroinflammation and neuroprotection in ischemic stroke. J Neurosci Res 95:462–471. 10.1002/jnr.23962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takano T, Oberheim N, Cotrina ML, Nedergaard M (2009) Astrocytes and ischemic injury. Stroke 40:S8–S12. 10.1161/STROKEAHA.108.533166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang FL, Zhao L, Zhao Y, Sun D, Zhu XJ, Mei L, Xiong WC (2020) Coupling of terminal differentiation deficit with neurodegenerative pathology in Vps35-deficient pyramidal neurons. Cell Death Differ 27:2099–2116. 10.1038/s41418-019-0487-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thakkar R, Wang R, Wang J, Vadlamudi RK, Brann DW (2018) 17β-estradiol regulates microglia activation and polarization in the hippocampus following global cerebral ischemia. Oxid Med Cell Longev 2018:4248526. 10.1155/2018/4248526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vegeto E, Belcredito S, Etteri S, Ghisletti S, Brusadelli A, Meda C, Krust A, Dupont S, Ciana P, Chambon P, Maggi A (2003) Estrogen receptor-alpha mediates the brain antiinflammatory activity of estradiol. Proc Natl Acad Sci USA 100:9614–9619. 10.1073/pnas.1531957100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Yu RK (2013) Interaction of ganglioside GD3 with an EGF receptor sustains the self-renewal ability of mouse neural stem cells in vitro. Proc Natl Acad Sci USA 110:19137–19142. 10.1073/pnas.1307224110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Ma MW, Dhandapani KM, Brann DW (2017) Regulatory role of NADPH oxidase 2 in the polarization dynamics and neurotoxicity of microglia/macrophages after traumatic brain injury. Free Radic Biol Med 113:119–131. 10.1016/j.freeradbiomed.2017.09.017 [DOI] [PubMed] [Google Scholar]
- Zamanian JL, Xu L, Foo LC, Nouri N, Zhou L, Giffard RG, Barres BA (2012) Genomic analysis of reactive astrogliosis. J Neurosci 32:6391–6410. 10.1523/JNEUROSCI.6221-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang QG, Wang R, Khan M, Mahesh V, Brann DW (2008) Role of Dickkopf-1, an antagonist of the Wnt/beta-catenin signaling pathway, in estrogen-induced neuroprotection and attenuation of tau phosphorylation. J Neurosci 28:8430–8441. 10.1523/JNEUROSCI.2752-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang QG, Wang R, Tang H, Dong Y, Chan A, Sareddy GR, Vadlamudi RK, Brann DW (2014) Brain-derived estrogen exerts anti-inflammatory and neuroprotective actions in the rat hippocampus. Mol Cell Endocrinol 389:84–91. 10.1016/j.mce.2013.12.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S. (2019) Microglial activation after ischaemic stroke. Stroke Vasc Neurol 4:71–74. 10.1136/svn-2018-000196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwain IH, Yen SS, Cheng CY (1997) Astrocytes cultured in vitro produce estradiol-17beta and express aromatase cytochrome P-450 (P-450 AROM) mRNA. Biochim Biophys Acta 1334:338–348. 10.1016/s0304-4165(96)00115-8 [DOI] [PubMed] [Google Scholar]