

Abstract
PD‐1 is a highly glycosylated inhibitory receptor expressed mainly on T cells. Targeting of PD‐1 with monoclonal antibodies (MAbs) to block the interaction with its ligand PD‐L1 has been successful for the treatment of multiple tumors. However, polymorphisms at N‐glycosylation sites of PD‐1 exist in the human population that might affect antibody binding, and dysregulated glycosylation has been observed in the tumor microenvironment. Here, we demonstrate varied N‐glycan composition in PD‐1, and show that the binding affinity of camrelizumab, a recently approved PD‐1‐specific MAb, to non‐glycosylated PD‐1 proteins from E. coli is substantially decreased compared with glycosylated PD‐1. The structure of the camrelizumab/PD‐1 complex reveals that camrelizumab mainly utilizes its heavy chain to bind to PD‐1, while the light chain sterically inhibits the binding of PD‐L1 to PD‐1. Glycosylation of asparagine 58 (N58) promotes the interaction with camrelizumab, while the efficiency of camrelizumab to inhibit the binding of PD‐L1 is substantially reduced for glycosylation‐deficient PD‐1. These results increase our understanding of how glycosylation affects the activity of PD‐1‐specific MAbs during immune checkpoint therapy.
Keywords: camrelizumab, glycosylation, monoclonal antibody, PD‐1, structure
Subject Categories: Cancer; Immunology; Post-translational Modifications, Proteolysis & Proteomics
PD‐1 is a highly glycosylated receptor with a varied N‐glycan composition. The therapeutic antibody camrelizumab binds to the N58 glycan of PD‐1 and absent glycosylation results in decreased binding affinity and PD‐1/PD‐L1 blocking efficiency.
Introduction
Monoclonal antibody (MAb)‐based immune checkpoint therapy (ICT), which blocks programed death receptor 1 (PD‐1) inhibitory signaling pathway to restore tumor‐specific T‐cell responses, has been proven to be an unprecedented breakthrough in tumor therapy in recent years (Yu et al, 2019). Six anti‐PD‐1 MAbs have been approved worldwide, including nivolumab (2014, Bristol‐Myers Squibb), pembrolizumab (2014, Merck Sharp & Dohme), cemiplimab (2018, Sanofi and Regeneron), toripalimab (2018, Junshi), sintilimab (2018, Innovent), and camrelizumab (2019, HengRui). The clinical application of these MAbs has greatly improved both the overall survival rate and life quality of patients with multiple tumors (Nishino et al, 2017).
Glycosylation is a common modification of proteins in eukaryotic cells. Glycosylation changes, including abnormal core fucosylation and increased N‐glycan branching, have been observed in tumor cells (Radhakrishnan et al, 2014; Pinho & Reis, 2015; Schultz et al, 2016). Dysregulated glycosylation can alter protein function and play a critical role in cancer cell adhesion and migration, immune surveillance, cell signaling, and cellular metabolism (Newsom‐Davis et al, 2009; Wang et al, 2014; Munkley et al, 2015; Britain et al, 2018). The extracellular domain of PD‐1 (PD‐1‐ECD) contains an IgV domain with four potential N‐glycosylation sites; this domain interacts with the IgV domain of its ligands, PD‐L1 or PD‐L2 (Tan et al, 2016a,b). We have previously reported that the PD‐1‐ECD is highly glycosylated; its molecular weight substantially shifted from 14 kDa, when expressed in E. coli with no glycosylation, to about 35–40 kDa, when expressed in 293T cells with glycosylation similar to that in host cells (Tan et al, 2017). Additionally, SDS–PAGE analysis has revealed that when each of the four N‐glycosylation sites is mutated, PD‐1 exhibits substantially reduced molecular weight, indicating that all of the four N‐glycosylation sites are glycosylated (Tan et al, 2017). In addition to its expression on T cells, PD‐1 has been reported to be expressed in tumor cells, promoting their growth (Kleffel et al, 2015; Wang et al, 2020). Besides, among the four N‐glycosylation sites, nonsynonymous single nucleotide polymorphisms at N58 or N116 have been documented in the dbSNP database (N58D, rs1222512746; N116K, rs1256572186). Therefore, PD‐1 with varied glycan modifications may be present on T cells or tumor cells.
The complex structure of the interaction between PD‐1 and PD‐L1 has been reported, and analysis has revealed that all four N‐glycosylation sites are away from the PD‐1/PD‐L1‐binding interface (Zak et al, 2015). The reported complex structures of PD‐1 with the FDA‐approved MAbs, e.g., nivolumab, pembrolizuab, or toripalimab, have also indicated that the glycosylation sites of PD‐1 are not involved in the binding of these MAbs (Na et al, 2017; Tan et al, 2017; Chen et al, 2019; Liu et al, 2019). Nivolumab mainly binds to the N‐terminal loop of PD‐1, while pembrolizumab binds to the C'D loop, and toripalimab binds to the FG loop of PD‐1. However, the blocking efficacy of the MAbs involves not only direct binding to competitive binding surfaces with PD‐L1 but also conformational hindrance by the MAb. Although glycan modification is away from the binding interface of PD‐1/PD‐L1, there are still concerns that binding of MAbs to PD‐1 is affected by glycosylation. There are currently three additional FDA‐approved MAbs and few more are in various phases of clinical trials. Therefore, further studies are necessary to investigate the glycosylation‐dependent binding of these MAbs.
Camrelizumab is a humanized anti‐PD‐1 IgG4 MAb, also known as SHR‐1210, that has been approved by the National Medical Products Administration of China for the treatment of relapsed or refractory classical Hodgkin lymphoma in 2019 (Markham & Keam, 2019). Clinical investigations on camrelizumab have shown that it suppressed tumor growth, although unusual toxicity of capillary hemangioma occurred (Na et al, 2017; Chen et al, 2019). In this study, we analyzed the N‐glycan composition of PD‐1 and the dependency of camrelizumab binding on glycosylation. We found that the high binding affinity (K D) of camrelizumab to PD‐1 was substantially dependent on the glycosylation of PD‐1. These findings expand our knowledge regarding the binding mechanisms of camrelizumab in the context of tumor ICT.
Results and Discussion
Varied N‐glycan modifications of PD‐1 and binding of camrelizumab to PD‐1 produced in different expression systems
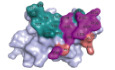
The N‐glycan composition of PD‐1 was analyzed with PD‐1‐ECD proteins that were expressed in 293T cells and treated with PNGase to obtain the N‐glycans present on PD‐1. The N‐glycans were then purified and analyzed with electrospray ionization mass spectrometry (ESI‐MS). Various N‐glycan compositions were identified, and most of the N‐glycans were fucosylated (Fig 1A). Glycan modifications have been found to be important for protein folding and stability (Xu & Ng, 2015). The influence of glycosylation on the conformational stability of PD‐1 was subsequently characterized with an all‐in‐one UNcle platform using PD‐1‐ECD proteins obtained from 293T cells or insect cells or refolded proteins expressed from E. coli cells; this enabled various levels of glycosylation modifications on PD‐1 proteins (Wang et al, 2017). The results indicated that the melting temperature (T m) was substantially decreased with PD‐1‐ECD proteins obtained from E. coli cells (no glycosylation; T m = 56.07°C), compared with PD‐1‐ECD proteins obtained from 293T cells or insect cells (T m = 63.78°C or 62.22°C) (Fig 1B). In addition, small sized, unfolded aggregates were observed with PD‐1 proteins expressed in E. coli cells by static light scattering (SLS) at 266 nm with an aggregation temperature (Tagg) of 39.87°C, while no substantial aggregates were observed with PD‐1 proteins obtained from 293T cells or insect cells (Fig 1C). These results suggest that the various glycosylations on PD‐1 may contribute to the maintenance of the conformational stability of PD‐1 and prevention of aggregation.
Figure 1. N‐glycan profiles of PD‐1 and glycosylation‐binding dependency of camrelizumab.
-
AN‐glycan composition of PD‐1 protein expressed from 293T cells by ESI‐MS analysis. N‐glycans of representative peaks from ESI‐MS were identified by MS/MS analysis. H, mannose; N, N‐acetylglucosamine; F, fucose. Structural formulas: blue square, N‐acetylglucosamine; red triangle, fucose; green circle, mannose; yellow circle, galactose.
-
B, CAccelerated thermal stability of PD‐1 proteins obtained from 293T cells (green line), insect cells (pink line), or E. coli (blue line) is characterized in all‐in-one UNcle platform. Protein unfolding is observed as an increase in barycentric mean (B) while small particle formation of the samples is observed as an increase in SLS intensity at 266 nm (C).
-
DSPR analysis of the binding characteristics of camrelizumab with PD‐1 expressed in 293T cells, insect cells or E. coli cell, which enabled different levels of glycosylations. The binding affinity is presented as K D. The datum presented here is a representative of two independent experiments with similar results.
The binding of camrelizumab to PD‐1 proteins obtained from 293T cells, insect cells, or E. coli was analyzed by using surface plasmon resonance (SPR) assay. In this assay, full‐length camrelizumab was immobilized on the chip, and serially diluted PD‐1 proteins were then flowed through. The binding affinity of camrelizumab for PD‐1 expressed in E. coli cells (K D = 1.55 μM) was substantially lower than that to PD‐1 obtained from 293T cell (K D = 5.92 nM) or insect cell (K D = 6.73 nM) expression systems (Fig 1D and Appendix Table S1). The major differences among PD‐1 proteins expressed in different expression systems involve glycosylation modifications (Tan et al, 2017). Therefore, we hypothesized that the binding of camrelizumab to PD‐1 may be affected by the glycosylation modifications of PD‐1.
As a highly glycosylated protein, PD‐1 has four potential N‐linked glycosylation sites, and glycosylation at each of these sites has been reported in our previous studies (Tan et al, 2017). ESI‐MS analysis of the N‐glycoform composition indicated that the N‐glycosylation of PD‐1 is highly variable. Sun et al (2020) have recently reported that PD‐1 is extensively N‐glycosylated in T cells, and the intensities of its specific glycoforms are altered upon TCR activation. Decreased stability and increased aggregation potency of glycosylation‐deficient PD‐1 proteins indicate the importance of glycosylation in maintaining PD‐1 stability on the cell surface. Though glycosylation is not involved in PD‐1/PD‐L1 binding, there are still concerns that the binding of PD‐1‐specific MAbs with tumor suppression potency, which mainly function through the blocking of PD‐1/PD‐L1 interaction, may locate on these glycosylation sites.
Binding characteristics of camrelizumab for PD‐1/PD‐L1 blockade
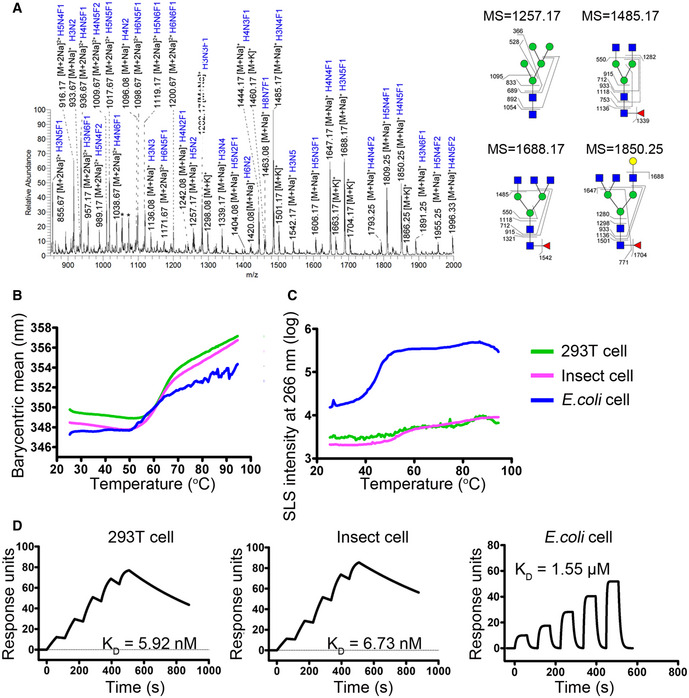
To elucidate the detailed binding characteristics of camrelizumab to PD‐1, the complex proteins of the single‐chain variable fragment (scFv) of camrelizumab and the fully glycosylated PD‐1‐ECD protein obtained from 293T cells were prepared for crystal screening (Fig 2A). The structure of the complex of camrelizumab‐scFv and PD‐1 was determined at a resolution of 2.8 Å (Appendix Table S2).
Figure 2. Overall structure of PD‐1 camrelizumab‐scFv complex.
- Gel filtration profiles of PD‐1 (orange), camrelizumab‐scFv (blue), and the PD‐1/camrelizumab‐scFv complex (red) are analyzed by size‐exclusion chromatography as indicated. The SDS–PAGE analyses are shown with peak 1 of camrelizumab‐scFv, peak 2 of camrelizumab‐scFv/PD‐1 complex, and peak 3 of PD‐1.
- The structure of camrelizumab‐scFv/PD‐1 complex. PD‐1 is shown as surface representation (white). Camrelizumab‐scFv is shown as cartoon representation. VL is colored in yellow and VH in deep salmon. The CDR1, CDR2, and CDR3 loops of the VL (LCDR1, LCDR2, and LCDR3) are colored in slate, warm pink, and violet purple, respectively. The CDR1, CDR2, and CDR3 loops of VH (HCDR1, HCDR2, and HCDR3) are colored in marine, orange, and cyan, respectively. Hydrogen bond interactions between PD‐1 and camrelizumab‐scFv are shown as sticks and labeled. Hydrogen bonds are shown as dashed red lines.
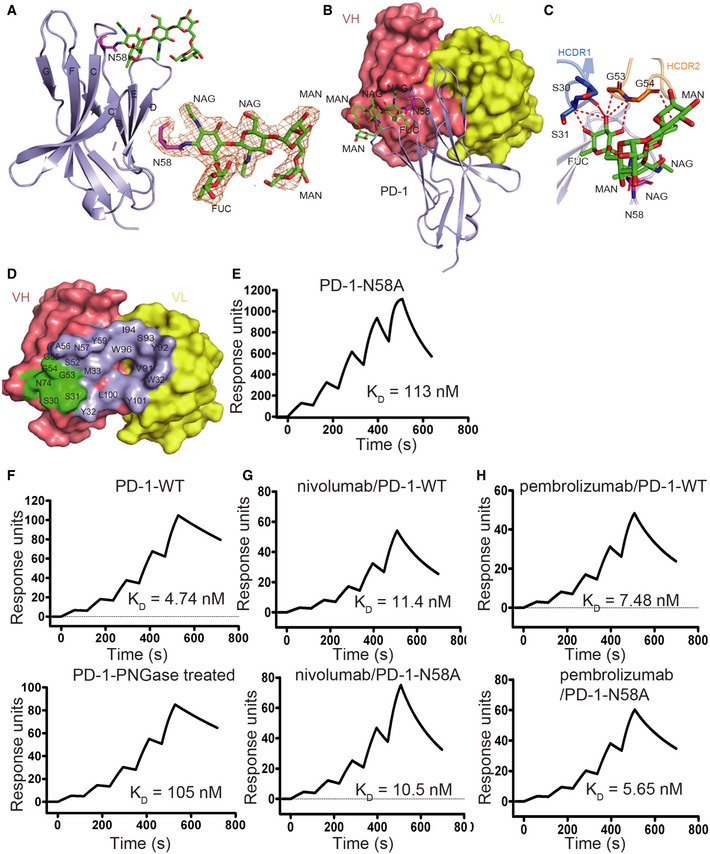
Overall, the structure of the camrelizumab‐scFv/PD‐1 complex includes two camrelizumab‐scFvs and two PD‐1 molecules (Appendix Fig S1). Camrelizumab binds to PD‐1 with a buried surface of 1966 Å2 (Fig 2B). Specifically, the heavy chain variable domain (VH) of camrelizumab was found to dominate the interaction with PD‐1 by all of the three CDRs, while light chain variable domain (VL) partially contributes to the binding with LCDR1 and LCDR3 loops (Fig 2B and Appendix Table S3). The camrelizumab binding areas on PD‐1 were predominantly occupied by the BC loop, C'D loop, and FG loop of PD‐1. Hydrogen bonds were formed between amino acids of the LCDR3 (Y92, I94) of camrelizumab and the FG loop of PD‐1 (P130, A132), as well as between HCDR2 (S52, A56, N57, and W59) of camrelizumab and the BC loop (E61 and S62) of PD‐1 (Fig 2B and Appendix Table S3). It is worth noting that the glycans on N58 of PD‐1 also interacted with amino acids from HCDR1 (S30, S31) and HCDR2 (G53, G54, and A56) of camrelizumab (Appendix Table S3).
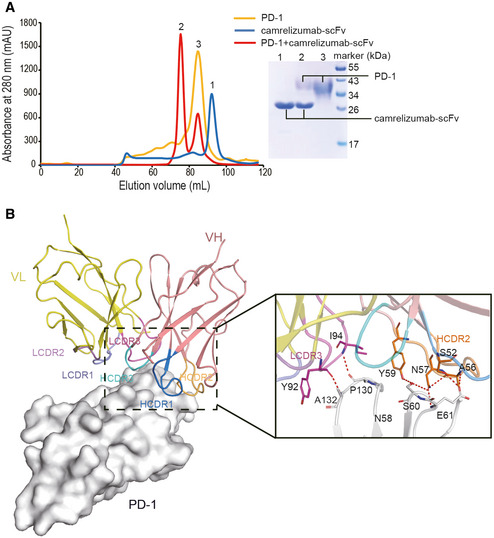
To analyze the mechanisms through which camrelizumab blocks the PD‐1/PD‐L1 interaction, the structure of the complex of camrelizumab/PD‐1 was superimposed with that of the PD‐1/PD‐L1 complex (PDB: 4ZQK). The binding of camrelizumab imposed a stereospecific hindrance to the binding of PD‐L1, which was mainly mediated by the VL domain of camrelizumab (Fig 3A). Additionally, there was a substantial overlap in the binding areas between camrelizumab and PD‐L1 on PD‐1 (Fig 3B). The overlapped binding areas were mainly located on the FG loop of PD‐1, which has also been shown to be occupied by nivolumab and toripalimab (Tan et al, 2016a; Liu et al, 2019). The FG loop has been suggested to be a hot spot for the binding of PD‐1/PD‐L1‐blocking MAbs, such as toripalimab, GY‐5, and GY‐14 (Chen et al, 2019; Liu et al, 2019). Unlike the FG loop that was substantially shifted upon binding of nivolumab or toripalimab, in the camrelizumab/PD‐1 complex, the FG loop maintained a conformation similar to that in the PD‐1/PD‐L1 complex (Appendix Fig S2). The binding surface of camrelizumab on PD‐1 was similar to that of toripalimab, nivolumab, and two other FG loop‐bound MAbs, GY‐5 and GY‐14, but substantially different from that of pembrolizumab (Fig 3C). In summary, the blocking mechanisms of camrelizumab mainly depend on its VL domain, which sterically inhibits the binding of PD‐L1 to PD‐1.
Figure 3. Structural basis of the competitive binding of camrelizumab with PD‐L1.
- Superimposition of camrelizumab/PD-1 with PD‐1/PD-L1 complex structure (PDB code: 4ZQK). PD‐1 is shown as surface format in light blue, while PD‐L1 is shown as deep teal cartoon and camrelizumab‐scFv is depicted as cartoon. VL of camrelizumab‐scFv is colored in yellow and VH in deep salmon.
- Competitive binding surfaces of camrelizumab with PD‐L1 on PD‐1. The residues contact with PD‐L1 alone are colored in deep teal while the residues bind to camrelizumab alone are colored in deep salmon. The residues contacted by both camrelizumab and PD‐L1 are colored by magenta.
- The binding surface of PD‐L1 and FDA‐approved MAbs on PD‐1 is presented in different colors. The binding area of PD‐L1 and the MAbs targeting PD‐1, camrelizumab, toripalimab, nivolumab, pembrolizumab, GY‐5, and GY‐14 are colored in deep teal, deep salmon, magenta, green, orange, light pink, and yellow, respectively.
Though the construction of camrelizumab‐scFv with a shorter GS linker has yielded an unexpected dimeric scFv, which binds to two PD‐1 molecules, it retains all of the structural features and binding properties (Appendix Fig S1). The binding regions on PD‐1 include the BC loop, FG loop, and part of the C'D loop, which is similar to the binding regions of toripalimab (Zak et al, 2015). The conformation of the FG loop upon binding to different MAbs varied substantially, and the shifted orientation upon binding to camrelizumab was similar to that of toripalimab. Unlike the dominant binding of toripalimab to the FG loop of PD‐1, the binding of camrelizumab to PD‐1 involved both the BC and FG loops. Although the FG loop also interacted with nivolumab, the orientation of FG loop upon binding to nivolumab is distinct from that to toripalimab and camrelizumab. We have reported that the FG loop was also the major targeted area that interacted with MAbs following screening of a panel of PD‐1/PD‐L1‐blocking MAbs, which showed tumor suppression potency (Chen et al, 2019).
Interaction between camrelizumab and N‐glycans at N58 of PD‐1
We next investigated the contribution of N‐glycosylation of PD‐1 to the binding of camrelizumab. In the structure of the camrelizumab and PD‐1 complex, the glycan modifications on all four N‐glycosylation sites can be observed (Appendix Fig S3). In particular, the glycan on N58 was in contact with the VH of camrelizumab, and the observed glycan structure included two N'acetylglucosamines (NAG), two mannose (MAN) residues, and a fucose (FUC) residue, which are conserved with some of the core glycan structures determined in ESI‐MS N‐glycan analysis (Figs 1A and 4A and B). The glycan chain of N58 interacted with HCDR1 and HCDR2 through Van der Waals forces between amino acids from HCDR1 (S30) and HCDR2 (G54, A56); the FUC and MAN also formed multiple hydrogen bonds with amino acids from HCDR1 (S31) and HCDR2 (G53) (Fig 4C and D, and Appendix Table S3). The binding of camrelizumab to N58A‐mutated PD‐1 protein obtained from 293T cells was examined by using the SPR assay. The binding affinity of camrelizumab for N58A‐mutated PD‐1 (K D = 113 nM) was substantially lower than that with wild‐type (WT) PD‐1 (K D = 5.92 nM) (Fig 4E). In addition, the Tm of N58A‐mutated PD‐1 (T m = 62.5°C) was lower compared with that of WT PD‐1‐ECD proteins obtained from 293T cells (T m = 63.78°C). No substantial aggregates were observed with N58A‐mutated PD‐1 proteins (Appendix Fig S4). Therefore, these results indicate that N58 glycosylation contributes to the high binding affinity of camrelizumab.
Figure 4. N‐glycan of N58 on PD‐1 is crucial for binding to camrelizumab.
-
AThe overall structure of N‐glycan on N58 of PD‐1 is presented with N‐glycans on N58 of PD‐1 highlighted in sticks in green. The 2 Fo‐Fc electron density map of the N58 N‐glycans contoured at 1.0 sigma is represented in orange on the right. The β strands were labeled according to canonical IgV‐strand designations.
-
BLocation of N58 glycosylation in the interaction with camrelizumab‐scFv. VL of camrelizumab‐scFv is colored in yellow and VH in deep salmon.
-
CDetailed interactions of N‐glycans on N58 of PD‐1 with camrelizumab‐scFv. The observed glycans on N58 of PD‐1 are highlighted in sticks and colored in green. Residues with hydrogen bond and van der Waals force interaction are shown as sticks and labeled while Hydrogen bond and van der Waals force interaction are shown as dashed red lines.
-
DBinding paratope on VH and VL of camrelizumab by PD‐1 is presented with residues in contact with glycan of N58 highlighted in green.
-
ESPR analysis of the binding of camrelizumab with N58A mutated PD‐1 expressed in 293T cells.
-
FThe SPR analysis of the binding of camrelizumab with WT (upper) or PNGase F treated PD‐1 proteins (lower) expressed in 293T cells.
-
G, HThe SPR analysis of the binding of nivolumab (G) with WT (upper) or N58A mutated (lower) PD‐1 expressed in 293T cells, and the binding of pembrolizumab (H) with WT (upper) or N58A‐mutated (lower) PD‐1 proteins.
To further verify the binding dependency of PD‐1 glycosylation, the binding of camrelizumab to PNGase F‐treated PD‐1 proteins was investigated, with PD‐1 proteins without PNGase F treatment tested in parallel as a control. SDS–PAGE analysis revealed that the molecular weight of PD‐1 protein was substantially reduced from ~35 kDa to ~20 kDa after treatment with PNGase F (Appendix Fig S5). The SPR analysis revealed that the binding affinity of camrelizumab to PD‐1 proteins treated with PNGase F has reduced to a K D of 105 nM, compared with the K D of 4.74 nM with PD‐1 proteins without PNGase F treatment (Fig 4F).
To investigate whether the N58 glycan affects the binding of the two widely used anti‐PD‐1 antibodies, nivolumab and pembrolizumab, the binding of WT or N58A‐mutated PD‐1 proteins to nivolumab or pembrolizumab was further analyzed by using SPR assay (Fig 4G and H). The binding affinities of nivolumab or pembrolizumab for WT and N58A‐mutated PD‐1 proteins were not substantially different. The previously reported structures of the complex of nivolumab or pembrolizumab with PD‐1 also suggest that N58 of PD‐1 is far from the binding area of these two MAbs (Tan et al, 2016a). This indicates that N58 glycan does not play a role in the binding of nivolumab or pembrolizumab to PD‐1.
As observed in the structure of the camrelizumab/PD‐1 complex, the binding of camrelizumab to PD‐1 mainly depends on its VH domain, while blockage of the PD‐1/PD‐L1 interaction is mainly mediated by its VL domain. Therefore, the binding region of PD‐1/PD‐L1‐blocking MAbs on PD‐1 is not necessarily identical to that of PD‐L1, as observed in the binding of camrelizumab to N58 glycosylation in the present study. Therefore, in addition to N58, N74 or N116, which are not on the opposite side of the PD‐1/PD‐L1 interface, could also be targeted by PD‐1/PD‐L1‐blocking MAbs.
Reduced blocking efficiency of camrelizumab against N58 glycosylation‐deficient PD‐1
The therapeutic potency of camrelizumab in tumor ICT mainly relies on its ability to block the PD‐1/PD‐L1 interaction. However, the binding affinity of PD‐1 for camrelizumab was dramatically reduced from about 1 000‐fold higher with WT PD‐1 (K D = 4.74 nM) compared with PD‐1/PD‐L1 (K D = 0.77–8.2 μM) to merely about 10‐fold higher (K D = 113 nM) with N58 glycosylation‐deficient PD‐1 (Tan et al, 2016a). The decreased binding affinity of camrelizumab for N58 glycosylation‐deficient PD‐1 prompted us to further evaluate the PD‐1/PD‐L1 blocking efficiency of camrelizumab.
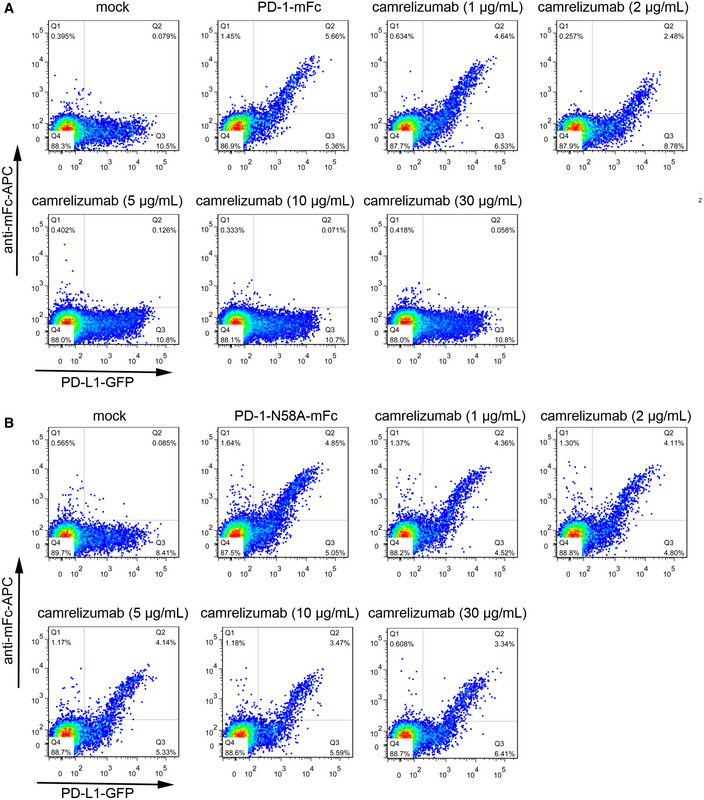
To investigate the blocking efficiency of camrelizumab for WT and N58 glycosylation‐deficient PD‐1, mFc tagged WT or N58A‐mutated PD‐1 recombinant proteins (PD‐1‐mFc) were prepared through transient expression in 293T cells. The binding of PD‐L1 to PD‐1 was assayed by flow cytometry following staining of 293T cells transiently expressing full‐length PD‐L1 on the cell surface with WT or N58A‐mutated PD‐1‐mFc proteins. The blocking of PD‐1/PD‐L1 interaction was examined using serial dilutions of camrelizumab pre‐incubated with 1 μg/ml WT or N58A‐mutated PD‐1‐mFc proteins and followed by staining PD‐L1‐expressing 293T cells. The frequency of WT PD‐1‐mFc‐positive 293T cells expressing PD‐L1 was substantially decreased from 51% of total PD‐L1‐GFP‐positive 293T cells to 22% in the presence of 2 μg/ml camrelizumab (Fig 5A). The binding of PD‐1‐mFc to PD‐L1‐expressing 293T cells was completely abolished in the presence of 5 μg/ml camrelizumab. However, the efficacy of camrelizumab in blocking the binding of N58A‐mutated PD‐1 to PD‐L1 was substantially reduced compared with WT PD‐1. N58A‐mutated PD‐1‐mFc positively stained cells remained 34% even at the high concentration of 30 μg/ml, compared with 49% positivity in the absence of camrelizumab (Fig 5B). Taken together, these results indicate that the efficiency of camrelizumab in blocking the interaction between N58 glycosylation‐deficient PD‐1 and PD‐L1 is substantially reduced.
Figure 5. Reduced blocking efficiency of camrelizumab to N58 glycosylation‐deficient PD‐1.
-
A, BThe blocking of the binding of WT PD‐1-mFc (A) or N58A‐mutated PD-1‐mFc (B) to PD‐L1 transiently expressed on the surface of 293T cells are analyzed with varied concentrations of camrelizumab. The PD‐L1 expressing 293T cells stained with PBS alone is used as negative controls, whereas the cells stained with WT or N58A‐mutated PD‐1-mFc proteins are used as a positive control. The frequency of PD‐1-mFc staining positive cells in PD‐L1-GFP‐positive cells is labeled in the upright corner. The reduced frequency of the PD‐L1-mFc staining positive subpopulation compared with positive control indicates the blockage of the PD-1/PD-L1 interaction. The density of events at a given position in the plot is color‐coded, with red representing the highest number of events at that point in the graph, while yellow, green, and blue represent progressively lower event densities. The results presented here are representative of three independent experiments with similar results.
Glycosylation‐dependent binding of camrelizumab may have a profound impact on both PD‐1/PD‐L1 blocking efficiency and binding specificity. Dysregulated glycosylation occurs in tumor cells; there are nonsynonymous single nucleotide polymorphisms at N58, with an estimated frequency of 8 per million as reported in TOPMed project reports (Pinho & Reis, 2015; Schultz et al, 2016). The binding affinity of camrelizumab for N58A‐mutated PD‐1 was substantially reduced to 113 nM, which is ~24‐fold lower than that of the WT PD‐1. The blocking efficiency of camrelizumab was not only dependent on the overlapping binding area of camrelizumab and PD‐L1 on PD‐1, but also on the overwhelming higher binding affinity of camrelizumab for PD‐1 compared with PD‐L1. Therefore, the substantially reduced binding affinity of camrelizumab for N58A‐mutated PD‐1 would result in attenuated efficiency in blocking the PD‐1/PD‐L1 interaction, which is about 0.77–8.2 μM (Tan et al, 2016a). As observed in the present study, the PD‐1/PD‐L1 blocking efficiency of camrelizumab is substantially attenuated by the N58A mutation, indicating the loss of T‐cell reactivation potency by camrelizumab. However, the functional consequences of PD‐1 glycosylation upon camrelizumab administration in primary T cells still need further investigation. Therefore, the function of camrelizumab may be affected in these cases. The structure of the camrelizumab/PD‐1 complex demonstrates that the glycosylation‐dependent high binding affinity of camrelizumab is mainly due to the N‐glycan chains in the core structure of N58 glycan, which is conserved in various N‐glycans, as was also observed in the N‐glycoform composition analysis in this study. Therefore, we hypothesize that the binding specificity of the MAbs which showed glycosylation‐binding dependency may be lower than those that bind only to amino acids of PD‐1. Finlay et al have reported that camrelizumab showed low‐affinity binding to other human receptors such as vascular endothelial growth factor receptor 2 (VEGFR2), frizzled class receptor 5, and UL16‐binding protein 2 (ULBP2; Finlay et al, 2019). Recently, other PD‐1 MAbs were also reported to recognize PD‐1 glycosylation, though the therapeutic potency and non‐specific binding to other human receptors remain less understood (Wang et al, 2019; Liu et al, 2020; Sun et al, 2020). The most common side effects reported in clinical trials with camrelizumab included cutaneous reactive capillary endothelial proliferation, which may be correlated with these low‐affinity off‐target bindings (Fang et al, 2018; Song et al, 2019). The results presented in this study suggest that the glycosylation‐dependent high‐affinity binding of camrelizumab may be responsible for these off‐target interactions. However, the difficulties in direct detection of N58 glycosylation deficiency on PD‐1 in clinical samples have impeded further evaluation of its clinical manifestations.
In summary, the variable N‐glycan composition of PD‐1 elucidated in the present study may play a critical role in maintaining the stability of PD‐1. We found that the binding of camrelizumab to PD‐1 substantially depends on the N‐glycans on N58 of PD‐1, and the PD‐1/PD‐L1 blocking efficiency of camrelizumab is substantially reduced with N58 glycosylation‐deficient PD‐1. These findings will further our understanding of the roles of N‐glycan of PD‐1 in the interaction with MAbs for tumor ICT and shed light for future design of next generation anti‐PD‐1 antibodies.
Materials and Methods
Plasmid construction and protein purification
The genes encoding the PD‐1‐ECD (UniProt: Q15116) expressed by different expression systems were constructed as previous reported (Tan et al, 2017). Briefly, for 293T cell expression, residues M1‐Q167, including signal peptide, of PD‐1 with six histidines at the C terminus of the sequence were cloned into pCAGGS vector (Addgene) (pCAGGS‐PD‐1). Plasmid pCAGGS‐PD‐1 was transiently transfected into 293T cells for protein expression. To allow expression of the PD‐1 N58A mutant proteins, the code substitutions of residues, N58A, was done by site‐directed mutagenesis using the overlapping extension PCR method and expressed in the same way in 293T cells. For Bac‐to‐Bac baculovirus expression system, residues N33‐R147 were inserted into pFastbac1 vector (Invitrogen) with an N‐terminal gp67 signal peptide for secretion and a C‐terminal hexa‐His tag for purification(Nanda et al, 2016). The recombinant baculovirus was then used to infect Hi5 cells to produce soluble PD‐1(Zhang et al, 2010; Song et al, 2016). In each case, the protein was purified by His‐Trap HP column (GE Healthcare) after the supernatant collected and filtration by 0.22 μm filter membrane. Then, PD‐1 protein was exchanged to 20 mM Tris (pH 8.0) and 150 mM NaCl buffer and further purified by Superdex 200 10/300 GL (GE Healthcare).
Residues L25‐R147 of PD‐1 were cloned into the pET‐21a expression vector (Novagen). Camrelizumab‐scFv was constructed as VL‐GGGGS‐VH and cloned into the pET21a expression vector. Plasmids of PD‐1 or camrelizumab‐scFv were then transformed into E. coli strain BL21 (DE3) cells and overexpressed as inclusion bodies. The inclusion bodies of PD‐1 were purified and then refolded as previous described(Liu et al, 2017; Zhao et al, 2018; Lu et al, 2019; Zhu et al, 2019). After refolding, we concentrated and exchanged PD‐1 proteins into a buffer containing 20 mM Tris–HCl (pH 8.0) and 150 mM NaCl, and purified by using a Superdex™ 200 Increase 10/300 GL column (GE Healthcare).
The full‐length heavy chain and light chain gene of camrelizumab were cloned into the pCAGGS vector individually with Eco RI and Xho I sites and then co‐transfected into 293T cells. The culture medium was changed to fresh DMEM without FBS 6 h after transfection. The supernatants were collected on the 3rd day and 7th day after transfection, and the proteins were purified with protein A column (GE Healthcare). Nivolumab was prepared as previously described (Tan et al, 2017), while pembrolizumab was a gift from Junshi Pharma.
Release of N‐Glycans and N‐glycan MS analysis
This experiment was performed as previously reported (Li et al, 2016, 2019). Briefly, 250 μl of water and 200 μl of protein denaturation solution were added to 2 mg of PD‐1 proteins obtained from 293T cells, prior to heating at 100°C for 10 min. The sample was then cooled to room temperature (25°C), and 50 μl of sodium phosphate buffer (1 M, pH 7.5), 50 μl of 10% NP‐40 aqueous solution, and 1 μl (500 U) of PNGase F solution were successively added. After incubation at 37°C for 24 h, the N‐glycans were purified using a C18 column and graphite carbon columns (Sigma‐Aldrich) and were retained in the inner tube. The derived PD‐1 N‐glycans were collected for further analysis.
N‐Glycan MS was conducted on an LTQ XL linear ion trap mass spectrometer coupled to an electrospray ion source and an HPLC system (Thermo Scientific, San Diego, CA, USA) using the parameters reported for ESI‐MS by (Li et al, 2016). A 50% methanol stream was passed through the Rheodyne loop at a flow rate of 20 μl/min. The spray voltage was set at 4 kV, the sheath gas (N2) flow rate was 20 arb, the auxiliary gas (nitrogen) flow rate was 10.0 arb, the capillary voltage was 37 V, the tube lens voltage was 250 V, and the capillary temperature was 300°C.
Online LC‐UV‐ESI‐MS/MS analysis was performed on a high‐performance liquid chromatography (HPLC)‐ESI‐MS system (Thermo Scientific, San Jose, CA, USA) using a 4.6 mm × 250 mm TSKgel Amide‐80 column at room temperature as previously reported (Li et al, 2016). Data acquisition was performed using Xcalibur software (Thermo Scientific).
Tm and Tagg characterization
The stability of PD‐1 proteins including unfolding and aggregation was evaluated with an all‐in‐one UNcle stability platform (Unchained Labs, Norton, MA) with PD‐1 proteins obtained from 239T cell, insect cell, or E. coli as previously reported (Nanda et al, 2016). The barycentric mean (BCM) and static light scattering (SLS) at 266 nm were used to monitor the evolution of protein structures upon temperature increasing at a rate of 2°C/min from 20°C to 95°C. The label‐free fluorescence from intrinsic aromatic amino acid residues excited by the inset excitation wavelength of 266 nm was collected for melting temperature (the midpoint of unfolding event, T m) calculation, while SLS at 266 nm was monitored, respectively, to determine the temperature where PD‐1 protein started to aggregate (the starting point of aggregation event, Tagg).
Totally, 9 μl of PD‐1 (PD‐1 expressed in 239T cell was 1 mg/ml, insect cell 1.7 mg/ml or E. coli 1.67 mg/ml) in PBST buffer of pH 7.4 was loaded into each sample well for the SLS and intrinsic fluorescence test with duplicates.
Camrelizumab‐scFv/PD‐1 complex preparation and crystallization
To get the complex of camrelizumab‐scFv/PD‐1 proteins, purified 293T cell expressed PD‐1 and camrelizumab‐scFv were mixed together at a molar ration of 1:1. After incubating on ice for 30 min, the mixture was purified by Superdex™ 200 10/300 GL (GE Healthcare) to exclude the unbound proteins. Purified camrelizumab‐scFv/PD‐1 proteins were then concentrated to 10 mg/ml. Then, 1 μl of protein solution was mixed with 1 μl of crystal growing reservoir solution. The resulting solution was sealed and equilibrated against 100 μl of reservoir solution at 4 or 18°C. Crystals of camrelizumab‐scFv/PD‐1 complex were grown in 0.1 M cadmium chloride, 0.1 M Na acetate, pH 4.6, 30% v/v PEG 400 a month later.
Data collection and structure determination
Crystals were flash‐cooled in liquid nitrogen after incubating in antifreezing buffer (reservoir solution containing 20% (v/v) glycerol). Diffraction data of crystals were collected at Shanghai Synchrotron Radiation Facility (SSRF) BL19U. All data were processed with HKL2000 after collection (Otwinowski & Minor, 1997). The complex structure was solved by molecular replacement using phase with the reported PD‐1 structure (PDB: 5WT9) and antibody structure (PDB: 5WT9) as the search models(Collaborative Computational Project, 1994; Read, 2001). Subsequent model building and refinement were performed using coot and phenix to refine the results, respectively (Emsley & Cowtan, 2004; Adams et al, 2010). The stereochemical qualities of the final model were assessed with MolProbity(Chen et al, 2010). The detail information of data collection and refinement statistics are summarized in Table S2. All structural figures were generated by Pymol (http://www.pymol.org).
SPR analysis
All the SPR measurements were done by BIAcore8000 system (GE healthcare) with CM5 chips (GE Healthcare). For all measurements, PBST was used as running buffer. To detect the camrelizumab binding to different forms of PD‐1, 669 response units camrelizumab full‐length MAb was immobilized on the chip and a blank channel was used as the negative control. Mammalian cell‐expressed PD‐1 and Bac‐to‐Bac baculovirus‐expressed PD‐1 was serially diluted to concentrations ranging from 2.5 nM to 40 nM. PD‐1 N58A mutant protein and E. coli expressed PD‐1 was serially diluted to concentrations ranging from 0.25 to 4 μM. Different PD‐1 proteins were then flowed over the chip surface one by one. After each cycle, the sensor surface was regenerated with pH 1.7 glycine.
For the detection of direct binding of camrelizumab to N‐glycans from 239T cell‐expressed PD‐1, 8590 response unit full‐length camrelizumab protein was immobilized on the chip. The purified N‐glycans from 6 mg PD‐1 was dissolved in 1.5 ml PBST buffer, and six twofold serially diluted samples were prepared. The diluted N‐glycans were then flowed through the chip, and the response units were measured. The binding of nivolumab or pembrolizumab to WT or N58A mutated PD‐1 proteins were detected with SPR similarly as camrelizumab. Briefly, 694 response units nivolumab‐Fab and 1,431 units pembrolizumab full‐length MAb were immobilized on the chip, respectively. 293T cell expressed PD‐1 and PD‐1 N58A was serially diluted to concentrations ranging from 2.5 nM to 40 nM. Different PD‐1 proteins were then flowed over the chip surface one by one.
To remove the N‐glycan on PD‐1, 1 mg of PD‐1 proteins obtained from 293T cells were added 30 μg PNGase F. After incubation at 37°C for 24 h, the PD‐1 proteins were purified by Superdex™ 200 Increase 10/300 GL column (GE Healthcare) and then test the purification of PD‐1 by SDS–PAGE. The 3,167 response unit camrelizumab full‐length MAb were immobilized on the chip. The enzymolysis PD‐1 serially diluted to concentrations ranging from 62.5 nM–1 μM.
The binding kinetics were all analyzed with the software of Biacore™ Insight evaluation software (GE healthcare) using a 1:1 Langmuir binding model.
Flow cytometry based PD‐1/PD‐L1 blocking assay
The full‐length PD‐L1 was cloned into pEGFP‐N1 vector (Clontech). HEK 293 T cells were transiently transfected with this recombinant plasmid for 24 h and resuspended in PBS at 1 × 107 cells/ml. WT or N58A mutated PD‐1‐mFc protein at a concentration of 1 μg/ml and camrelizumab at different concentrations were pre‐incubated, respectively, at room temperature for 30 min. Then, these mixtures were used to incubate with the 293T cells expressing PD‐L1 fused EGFP protein at room temperature for further 30 min. After washing with PBS for three times, the cells were stained with secondary APC goat anti‐mouse secondary IgG antibody (CAT: 405308; Lot B30147; BioLegend) for another 30 min and analyzed using flow cytometry (BD FACS Canto Flow Cytometer). The figures were generated with FlowJo 7.6.
Author contributions
KL, JG, and QW performed experiments; ST, KL, CD, and GFG analyzed the data and wrote the paper; ST and KL designed the experiments; YC and JQ solved the structure; WJ and ZW analyzed the glycosylation composition; JY and HS analyzed the data.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Review Process File
Acknowledgements
This study was supported by National Major Science & Technology Major Project (2018ZX10302302‐001‐002, 2018ZX10101004‐001‐003 and 2018ZX09711003‐002‐001), Strategic Priority Research Program of Chinese Academy of Sciences (CAS) (XDB29040201), and the Chair Professor Grant (CPG) 2019‐00019‐FHS. We thank the staff of BL17U and BL19U beamline at the Shanghai Synchrotron Radiation Facility for assistance with data collection. We also thank Yuanyuan Chen, Bingxue Zhou, and Zhenwei Yang from the Institute of Biophysics, CAS, for their technical support in the SPR assay.
EMBO Reports (2020) 21: e51444
Contributor Information
Shuguang Tan, Email: tansg@im.ac.cn.
Chuxia Deng, Email: cxdeng@umac.mo.
George F Gao, Email: gaof@im.ac.cn.
Data availability
Atomic coordinates have been deposited in the Protein Data Bank (PDB, http://www.rcsb.org/pdb) under the accession code 7CU5.
References
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse‐Kunstleve RW et al (2010) PHENIX: a comprehensive Python‐based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66: 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Britain CM, Holdbrooks AT, Anderson JC, Willey CD, Bellis SL (2018) Sialylation of EGFR by the ST6Gal‐I sialyltransferase promotes EGFR activation and resistance to gefitinib‐mediated cell death. J Ovarian Res 11: 12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen VB, Arendall WB III, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC (2010) MolProbity: all‐atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr 66: 12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D, Tan S, Zhang H, Wang H, He W, Shi R, Tong Z, Zhu J, Cheng H, Gao S et al (2019) The FG loop of PD‐1 serves as a “hotspot” for therapeutic monoclonal antibodies in tumor immune checkpoint therapy. iScience 14: 113–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collaborative Computational Project N (1994) The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr 50: 760–763 [DOI] [PubMed] [Google Scholar]
- Emsley P, Cowtan K (2004) Coot: model‐building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60: 2126–2132 [DOI] [PubMed] [Google Scholar]
- Fang W, Yang Y, Ma Y, Hong S, Lin L, He X, Xiong J, Li P, Zhao H, Huang Y et al (2018) Camrelizumab (SHR‐1210) alone or in combination with gemcitabine plus cisplatin for nasopharyngeal carcinoma: results from two single‐arm, phase 1 trials. Lancet Oncol 19: 1338–1350 [DOI] [PubMed] [Google Scholar]
- Finlay WJJ, Coleman JE, Edwards JS, Johnson KS (2019) Anti‐PD1 ‘SHR‐1210’ aberrantly targets pro‐angiogenic receptors and this polyspecificity can be ablated by paratope refinement. mAbs 11: 26–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleffel S, Posch C, Barthel SR, Mueller H, Schlapbach C, Guenova E, Elco CP, Lee N, Juneja VR, Zhan Q et al (2015) Melanoma cell‐intrinsic PD‐1 receptor functions promote tumor growth. Cell 162: 1242–1256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Wang C, Qiang S, Zhao J, Song S, Jin W, Wang B, Zhang Y, Huang L, Wang Z (2016) Mass Spectrometric analysis of N‐glycoforms of soybean allergenic glycoproteins separated by SDS‐PAGE. J Proteome Res 64: 7367–7376 [DOI] [PubMed] [Google Scholar]
- Li C, Lu Y, Chen X, Yang M, Zou Z, Han J, Gao X, Tang R, Wang C, Huang L et al (2019) Analysis of the N‐glycoforms and immunoactivity of Chinese yam (Dioscorea opposita Thunb.) glycoprotein 30CYGP. J Proteome Res 19: 28–35 [DOI] [PubMed] [Google Scholar]
- Liu WJ, Lan J, Liu K, Deng Y, Yao Y, Wu S, Chen H, Bao L, Zhang H, Zhao M et al (2017) Protective T cell responses featured by concordant recognition of middle East respiratory syndrome coronavirus‐derived CD8+ T cell epitopes and host MHC. J Immunol 198: 873–882 [DOI] [PubMed] [Google Scholar]
- Liu H, Guo L, Zhang J, Zhou Y, Zhou J, Yao J, Wu H, Yao S, Chen B, Chai Y et al (2019) Glycosylation‐independent binding of monoclonal antibody toripalimab to FG loop of PD‐1 for tumor immune checkpoint therapy. mAbs 11: 681–690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu JX, Wang GQ, Liu L, Wu RJ, Wu Y, Fang C, Zhou XH, Jiao J, Gu Y, Zhou H et al (2020) Study of the interactions of a novel monoclonal antibody, mAb059c, with the hPD‐1 receptor. Sci Rep 9: 17830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu D, Liu K, Zhang D, Yue C, Lu Q, Cheng H, Wang L, Chai Y, Qi J, Wang LF et al (2019) Peptide presentation by bat MHC class I provides new insight into the antiviral immunity of bats. PLoS Biol 17: e3000436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markham A, Keam SJ (2019) Camrelizumab: first global approval. Drugs 79: 1355–1361 [DOI] [PubMed] [Google Scholar]
- Munkley J, Oltean S, Vodak D, Wilson BT, Livermore KE, Zhou Y, Star E, Floros VI, Johannessen B, Knight B et al (2015) The androgen receptor controls expression of the cancer‐associated sTn antigen and cell adhesion through induction of ST6GalNAc1 in prostate cancer. Oncotarget 6: 34358–34374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Na Z, Yeo SP, Bharath SR, Bowler MW, Balikci E, Wang CI, Song H (2017) Structural basis for blocking PD‐1‐mediated immune suppression by therapeutic antibody pembrolizumab. Cell Res 27: 147–150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nanda R, Chow LQ, Dees EC, Berger R, Gupta S, Geva R, Pusztai L, Pathiraja K, Aktan G, Cheng JD et al (2016) Pembrolizumab in patients with advanced triple‐negative breast cancer: phase Ib KEYNOTE‐012 study. J Clin Oncol 34: 2460–2467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newsom‐Davis TE, Wang D, Steinman L, Chen PF, Wang LX, Simon AK, Screaton GR (2009) Enhanced immune recognition of cryptic glycan markers in human tumors. Cancer Res 69: 2018–2025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishino M, Ramaiya NH, Hatabu H, Hodi FS (2017) Monitoring immune‐checkpoint blockade: response evaluation and biomarker development. Nat Rev Clin Oncol 14: 655–668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W (1997) Processing of X‐ray diffraction data collected in oscillation mode. Methods Enzymol 276: 307–326 [DOI] [PubMed] [Google Scholar]
- Pinho SS, Reis CA (2015) Glycosylation in cancer: mechanisms and clinical implications. Nat Rev Cancer 15: 540–555 [DOI] [PubMed] [Google Scholar]
- Radhakrishnan P, Dabelsteen S, Madsen FB, Francavilla C, Kopp KL, Steentoft C, Vakhrushev SY, Olsen JV, Hansen L, Bennett EP et al (2014) Immature truncated O‐glycophenotype of cancer directly induces oncogenic features. Proc Natl Acad Sci USA 111: E4066–E4075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Read RJ (2001) Pushing the boundaries of molecular replacement with maximum likelihood. Acta crystallogr D Biol Crystallogr 57: 1373–1382 [DOI] [PubMed] [Google Scholar]
- Schultz MJ, Holdbrooks AT, Chakraborty A, Grizzle WE, Landen CN, Buchsbaum DJ, Conner MG, Arend RC, Yoon KJ, Klug CA et al (2016) The tumor‐associated glycosyltransferase ST6Gal‐I regulates stem cell transcription factors and confers a cancer stem cell phenotype. Cancer Res 76: 3978–3988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song H, Qi J, Khedri Z, Diaz S, Yu H, Chen X, Varki A, Shi Y, Gao GF (2016) An open receptor‐binding cavity of hemagglutinin‐esterase‐fusion glycoprotein from newly‐identified Influenza D virus: basis for its broad cell tropism. PLoS Pathog 12: e1005411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Y, Wu J, Chen X, Lin T, Cao J, Liu Y, Zhao Y, Jin J, Huang H, Hu J et al (2019) A single‐arm, multicenter, phase II study of camrelizumab in relapsed or refractory classical Hodgkin lymphoma. Clin Cancer Res 25: 7363–7369 [DOI] [PubMed] [Google Scholar]
- Sun L, Li CW, Chung EM, Yang R, Kim YS, Park AH, Lai YJ, Yang Y, Wang YH, Liu J et al (2020) Targeting Glycosylated pd‐1 induces potent antitumor immunity. Cancer Res 80: 2298–2310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan S, Chen D, Liu K, He M, Song H, Shi Y, Liu J, Zhang CW, Qi J, Yan J et al (2016a) Crystal clear: visualizing the intervention mechanism of the PD‐1/PD‐L1 interaction by two cancer therapeutic monoclonal antibodies. Protein Cell 7: 866–877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan S, Zhang CW, Gao GF (2016b) Seeing is believing: anti‐PD‐1/PD‐L1 monoclonal antibodies in action for checkpoint blockade tumor immunotherapy. Signal Transduct Target Ther 1: 16029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan S, Zhang H, Chai Y, Song H, Tong Z, Wang Q, Qi J, Wong G, Zhu X, Liu WJ et al (2017) An unexpected N‐terminal loop in PD‐1 dominates binding by nivolumab. Nat Commun 8: 14369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Chen J, Li QK, Peskoe SB, Zhang B, Choi C, Platz EA, Zhang H (2014) Overexpression of alpha (1,6) fucosyltransferase associated with aggressive prostate cancer. Glycobiology 24: 935–944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Zhang X, Wu G, Tian Z, Qian F (2017) Optimization of high‐concentration endostatin formulation: harmonization of excipients’ contributions on colloidal and conformational stabilities. Inter J Pharm 530: 173–186 [DOI] [PubMed] [Google Scholar]
- Wang M, Wang J, Wang R, Jiao S, Wang S, Zhang S, Zhang M (2019) Identification of a monoclonal antibody that targets PD‐1 in a manner requiring PD‐1 Asn58 glycosylation. Commun Biol 2: 392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Yang X, Zhang C, Wang Y, Cheng T, Duan L, Tong Z, Tan S, Zhang H, Saw PE et al (2020) Tumor cell‐intrinsic PD‐1 receptor is a tumor suppressor and mediates resistance to PD‐1 blockade therapy. Proc Natl Acad Sci USA 117: 6640–6650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu C, Ng DT (2015) Glycosylation‐directed quality control of protein folding. Nat Rev Mol Cell Biol 16: 742–752 [DOI] [PubMed] [Google Scholar]
- Yu J, Hubbard‐Lucey VM, Tang J (2019) Immuno‐oncology drug development goes global. Nat Rev Drug Discov 18: 899–900 [DOI] [PubMed] [Google Scholar]
- Zak KM, Kitel R, Przetocka S, Golik P, Guzik K, Musielak B, Domling A, Dubin G, Holak TA (2015) Structure of the complex of human programmed death 1, PD‐1, and its ligand PD‐L1. Structure 23: 2341–2348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Qi J, Shi Y, Li Q, Gao F, Sun Y, Lu X, Lu Q, Vavricka CJ, Liu D et al (2010) Crystal structure of the swine‐origin A (H1N1)‐2009 influenza A virus hemagglutinin (HA) reveals similar antigenicity to that of the 1918 pandemic virus. Protein Cell 1: 459–467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao M, Liu K, Luo J, Tan S, Quan C, Zhang S, Chai Y, Qi J, Li Y, Bi Y et al (2018) Heterosubtypic protections against human‐infecting Avian Influenza viruses correlate to biased cross‐t‐cell responses. mBio 9: e01408–e01418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu S, Liu K, Chai Y, Wu Y, Lu D, Xiao W, Cheng H, Zhao Y, Ding C, Lyu J et al (2019) Divergent peptide presentations of HLA‐A(*)30 alleles revealed by structures with pathogen peptides. Front Immunol 10: 1709 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Review Process File
Data Availability Statement
Atomic coordinates have been deposited in the Protein Data Bank (PDB, http://www.rcsb.org/pdb) under the accession code 7CU5.