

Abstract
The function and maintenance of muscle stem cells (MuSCs) are tightly regulated by signals originating from their niche environment. Skeletal myofibers are a principle component of the MuSC niche and are in direct contact with the muscle stem cells. Here, we show that Myf6 establishes a ligand/receptor interaction between muscle stem cells and their associated muscle fibers. Our data show that Myf6 transcriptionally regulates a broad spectrum of myokines and muscle‐secreted proteins in skeletal myofibers, including EGF. EGFR signaling blocks p38 MAP kinase‐induced differentiation of muscle stem cells. Homozygous deletion of Myf6 causes a significant reduction in the ability of muscle to produce EGF, leading to a deregulation in EGFR signaling. Consequently, although Myf6‐knockout mice are born with a normal muscle stem cell compartment, they undergo a progressive reduction in their stem cell pool during postnatal life due to spontaneous exit from quiescence. Taken together, our data uncover a novel role for Myf6 in promoting the expression of key myokines, such as EGF, in the muscle fiber which prevents muscle stem cell exhaustion by blocking their premature differentiation.
Keywords: muscle stem cells, Myf6, myogenic factors, myokines, skeletal muscles
Subject Categories: Development & Differentiation, Regenerative Medicine
Skeletal muscle fibers are a key signaling component of the muscle stem cell niche. Myf6‐induced expression of myokines in skeletal muscle maintains myofiber‐associated muscle stem cell pools by blocking their premature differentiation in adult mice.

Introduction
In adult skeletal muscle, muscle stem cells (MuSCs) are spatially housed between the sarcolemma of the muscle fibers and the basal lamina, a layer of extracellular matrix proteins (ECM) that are closely juxtaposed with the myofibers. The physical enclosure of MuSCs in such an anatomical space ensures that they remain in close proximity to the myofiber in a mitotically quiescent state, under normal physiological conditions. Injury or trauma to the myofiber causes disruptions to the MuSC niche, during which they re‐enter cell cycle, transiently proliferate, and undergo myogenic differentiation to repair the injured muscle (Charge & Rudnicki, 2004; Lepper et al, 2011; Comai & Tajbakhsh, 2014). Induction of MuSC activation triggers a temporally regulated myogenic differentiation program orchestrated by four myogenic factors, namely Myf5, MyoD, Myogenin, and Myf6 (Braun et al, 1994; Berkes & Tapscott, 2005; Soleimani et al, 2012). MyoD and Myf5 transiently amplify MuSCs, but also induce differentiation by upregulating Myogenin in a context‐dependent manner (Soleimani et al, 2012; Siles et al, 2013). While Myf5, MyoD, and Myogenin are transiently expressed during myogenic commitment and differentiation of MuSCs; in the fully differentiated myofiber, Myf6 is the predominant myogenic factor that remains expressed under normal physiological conditions. Previous studies have implicated Myf6 involvement in myofiber maturation and in the maintenance of their differentiated state (Buckingham, 1994). Aside from its function in differentiation, Myf6 has also been implicated as a muscle determination factor in the absence of MyoD and Myf5 (Kassar‐Duchossoy et al, 2004).
While the epigenetic and transcriptional control of muscle formation has been extensively studied (Bentzinger et al, 2012; Buckingham & Rigby, 2014), less is known about the precise mechanisms by which the muscle stem cell pool is maintained in adult skeletal muscle. Maintenance of the MuSC pool crucially depends on a dynamic balance between quiescence and activation (Boonsanay et al, 2016); a process that is intricately dependent on the interaction of MuSCs with their niche microenvironment (Yin et al, 2013; Rayagiri et al, 2018; Verma et al, 2018). Many non‐myogenic cells have been shown to contribute to the niche milieu of MuSCs, such as endothelial cells under normal physiological conditions (Verma et al, 2018), fibroblasts under hypertrophy (Fry et al, 2017), and macrophages and neutrophils during muscle injury (Joe et al, 2010; Jin et al, 2018; Wang et al, 2018). The physical enclosure of MuSCs by the ECM creates an anatomically defined niche microenvironment where the muscle stem cell resides in a quiescent state. However, the manner in which the molecular communication between myofibers and their associated MuSCs regulates their fate remains unknown.
Beyond its contractile properties, skeletal muscle plays an important role as an endocrine organ. The secretome of skeletal muscle is composed of proteins and peptides known as myokines, which are thought to have system‐wide effects through autocrine, paracrine, and endocrine functions (Pedersen & Febbraio, 2012; Whitham et al, 2012). Exercise‐induced myokines such as Interleukin‐6 (IL6), Interleukin‐15 (IL15), and brain‐derived neurotrophic factor (BDNF) among others, have been studied in the context of their effect on inflammation, metabolic homeostasis, and lipid oxidation, respectively (Matthews et al, 2009; Munoz‐Canoves et al, 2013; Quinn et al, 2013).
Here, we show that Myf6 plays a key role in the endocrine function of skeletal muscle by transcriptionally regulating multiple myokines, such as epidermal growth factor (EGF) and vascular endothelial growth factor A (VEGFA). Our data show that the regulation of myokine expression by Myf6 is required to maintain the adult muscle stem cell pool. In the absence of Myf6, MuSCs break quiescence, upregulate the p38 MAPK signaling pathway, and undergo premature differentiation. Importantly, our data suggest that the loss of Myf6 does not confer a cell‐intrinsic defect in MuSCs, as shown by the lack of a deficit in their activation or their transient differentiation ex vivo. Collectively, our data suggest that Myf6 has a novel function as a myogenic niche regulator and plays a critical role in regulating the stem cell pool in adult skeletal muscle by transcriptionally regulating key secreted factors and myokines from the muscle fiber.
Results
Myf6/MRF4 cistrome and associated genetic networks in skeletal muscle
Among the four myogenic regulatory factors (MRFs), Myf6 is the only factor that is primarily expressed in fully differentiated muscle fibers (Figs 1A and EV1A). The remaining three MRFs, namely Myf5, MyoD, and Myogenin, are transiently expressed during muscle stem cell activation, commitment, and terminal differentiation. To identify genome‐wide Myf6 targets in primary myotubes, we first used Chromatin Tandem Affinity Purification Sequencing (ChTAP‐Seq), an ideal alternative to traditional ChIP‐Seq when a commercial ChIP‐grade antibody is not available. Here, we generated primary myoblast cell lines over‐expressing a C‐terminal Tandem Affinity Purification tag (Myf6‐CTAP), using a retroviral vector. We validated the expression, subcellular localization and functionality of the ectopic Myf6‐CTAP in muscle and non‐muscle cells using immunofluorescence (IF), Western blotting (WB), and Luciferase assays (Fig EV1B–F). Next, we performed ChTAP‐Seq on multinucleated primary myotubes formed after 5 days in differentiation media as described previously (Soleimani et al, 2013). Using MACS2 (Zhang et al, 2008) with a P‐value threshold of 10−5 and default parameters, we identified 12,885 binding sites for Myf6 in primary myotubes, using sequencing reads in the affinity‐matched empty vector control (EV‐ChTAP‐Seq) as background (Fig 1B). Motif analysis of the peak‐covered sequences from the total number of Myf6 peaks using MEME suite (Bailey et al, 2009) identified the top scoring canonical E‐box motif, centrally located and closely juxtaposed by the MEF2A binding motif located peripheral to the E‐box counterparts (Fig 1C). This is in accordance with the previous report of a physical interaction between Myf6 and MEF2 (Moretti et al, 2016). Comparative analysis between the densities of the E‐box motif distribution in the Myf6 ChIP‐Seq peaks compared with random genomic blocks showed significantly denser clustering of E‐boxes under Myf6 peaks (Fig 1E). Such patterns of motif distribution in Myf6 peaks mirror those of other myogenic factors such as MyoD and Myf5 (Soleimani et al, 2012). Within Myf6 peaks, highly enriched motifs for known MRF cooperative heterodimers such as E‐protein (E12/E47) and antagonistic interactors such as TWIST1 and ID4 followed similar distribution patterns as those of E‐box motifs (Fig 1D).
Figure 1. Genome‐wide binding sites of Myf6/MRF4 in primary myotubes.
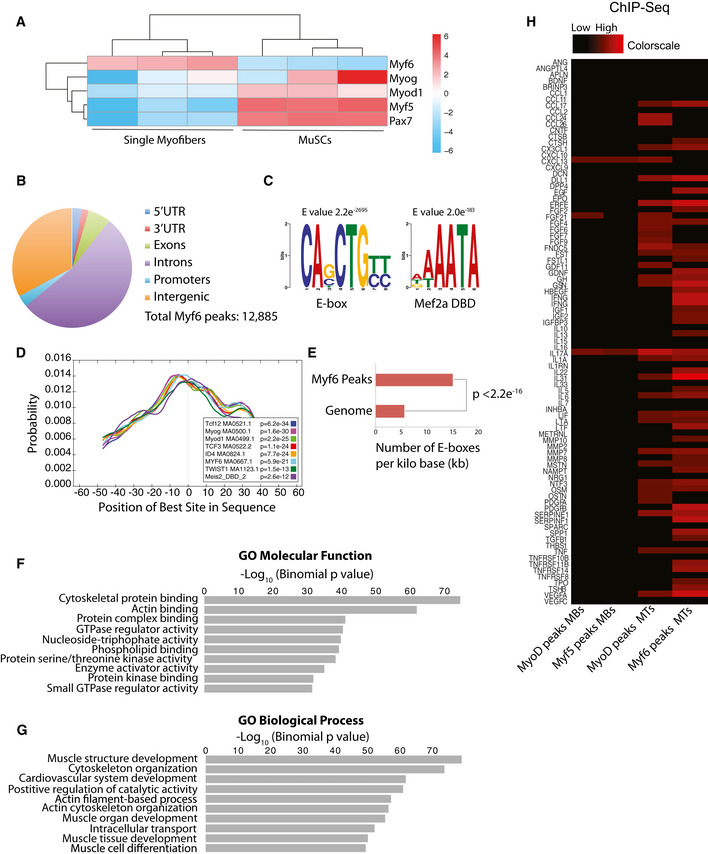
- Pie chart showing the distribution of Myf6 ChIP‐Seq peaks within various genomic features.
- Motif analysis using MEME suite (Bailey et al, 2009) identified canonical E‐box motif as the most highly enriched sequence among all sequences under Myf6 peaks, followed by the Mef2a DNA binding motif.
- Spatial distribution of transcription factor binding motifs within Myf6 ChIP‐Seq peaks using CentriMo available via MEME suite (Bailey et al, 2009).
- Normalized distribution of E‐box motifs that occur within Myf6 peaks compared with their occurrences in the genome normalized per kilo base of DNA (two‐tailed t‐test).
- Gene Ontology (GO Molecular Function) analysis of genes associated with Myf6 peaks based on association by proximity (single gene within 100 kb of peaks) analyzed by Genomic Regions Enrichment of Annotations Tool (GREAT) (McLean et al, 2010).
- Gene Ontology (GO Biological Process) analysis of genes associated with Myf6 peaks based on proximity similar to the analysis in (F).
- Colormap of Myf5, MyoD, and Myf6 peaks within 100 kb of the Transcription Start Sites (TSS) of myokines (Table EV3) ranging from zero (black) to six peaks (red) occupancy. The onset of differentiation coincides with increased binding of MRFs to the regulatory domains of the myokines genes.
Figure EV1. Functional validation Myf6‐CTAP construct used for ChIP‐seq analysis.
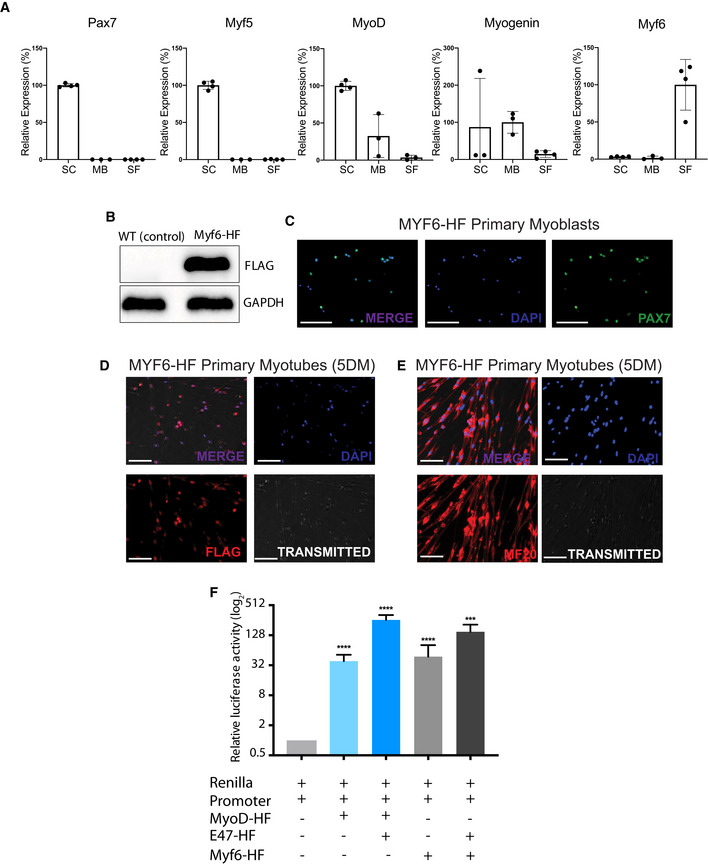
-
ARelative expression of Pax7, Myf5, MyoD, Myog, and Myf6 in mouse satellite cells (SC), primary myoblasts (MB), and single myofibers (SF) normalized to RPS2 as assayed by quantitative real‐time PCR (RT–qPCR). n = 3–4 biological replicates per group, two‐tailed t‐test, error bars = ± SD.
-
BWestern blot analysis of C2C12 myoblasts infected with a retrovirus containing Myf6‐CTAP.
-
CImmunofluorescent staining of primary myoblasts expressing Myf6‐HF for PAX7. Myf6‐HF cell lines were generated from Myf6‐null myoblasts infected with a retrovirus expressing Myf6‐HF (scale bar = 100 μm).
-
D, EImmunofluorescent staining of primary myotubes expressing Myf6‐HF for FLAG (D) and MF20 (E). Myf6‐HF myotubes were generated from Myf6‐null myoblasts infected with a retrovirus expressing Myf6‐HF, followed by 5 days of differentiation. (scale bar = 20 μm).
-
FDual luciferase assays showing the transcriptional activity of MYF6‐CTAP on a synthetic regulatory sequence containing a trimeric canonical E‐box motif (CAGCTG) spread over a 72 bp stretch of a synthetic DNA. The synthetic sequence was subcloned into the pGL4.23[luc2/minP] luciferase vector (Promega). Dual luciferase assays with Renilla/luciferase constructs were performed in Cos7 cells, and a vector containing the full‐length mouse MyoD (Soleimani et al, 2012) was used as a positive control. Relative transcriptional activity of the MyoD and the Myf6 constructs were measured with and without co‐transfection of Cos7 cells with the transcription factor E2‐alpha (E47), a heterodimer of the myogenic regulatory factors (MRFs). (n = 6 technical replicates), bars represent mean ± SD, two‐tailed t‐test, asterisks (*, **, ***, ****) correspond to P‐values of < 0.05, < 0.01, < 0.001, and < 0.0001, respectively.
Source data are available online for this figure.
To identify the genetic networks regulated by Myf6 in differentiated muscle cells, we first used Genomic Regions Enrichment of Annotations Tool (GREAT) (McLean et al, 2010) and performed Gene Ontology (GO) analysis using an association rule of single nearest genes (100 kb maximum extension). Within those regions, 12,270 out of 12,885 peaks (95.2%) were associated with a single gene and 610 peaks (4.73%) were not associated with any gene. Computation of GO terms using binomial tests, implemented in GREAT (McLean et al, 2010) showed that the majority of the Myf6 ChIP‐Seq peaks in the primary differentiated myotubes were within the regulatory domains of genes involved in regulation of muscle structure, muscle cell development and differentiation, and actin filament organization, as expected (Fig 1F and G). However, in addition to the expected differentiation genes, we identified a large set of genes for secreted proteins, including many known myokines and chemokines that are bound and transcriptionally regulated by Myf6 (Figs 1H and 2A–E, and EV2). When comparing transcription factor binding at the regulatory elements of myokine genes, there is a partial yet incomplete overlap between Myf6 and MyoD peaks in myotubes (Figs 1H and EV2B). Together, these data suggest that Myf6 but not MyoD or Myf5 is the predominant MRF which binds to the vicinity of various myokine genes.
Figure 2. Temporally regulated occupancy of myogenic regulatory factors (MRFs) to cis‐regulatory domains of myokine genes.

-
AGene expression analysis of myokines (Table EV3) during a 5 day time course of myogenic differentiation going from cycling myoblasts in growth media (Ham's F10 supplemented with 20% Fetal Bovine Serum, 1% penicillin/streptomycin, 2.5 ng/ml basic Fibroblast Growth Factor) to terminally differentiated myocytes (2 days in differentiation media, DMEM supplemented with 5% horse serum) to postmitotic multinucleated myotubes (5 days in differentiation media). Gene expression was assayed in biological triplicate by microarray (Soleimani et al, 2012, 2018). Expression of each gene was averaged across three replicates and normalized to mean zero and standard deviation of one. Expression values were then truncated at ± 3 for color display. Blue indicates lower than average expression, while red indicates higher than average expression.
-
BDistribution of Myf6 peaks within 100 kb of the TSS of the myokine genes (Table EV3), similar to the analysis shown in (A).
-
C–ESnapshots of the UCSC genome browser showing enrichment of Myf6 ChIP‐Seq reads compared with control around EGF (C), LIF and OSM (D), and VEGFA (E) loci superimposed on reads for Histone H3 lysine 4 mono methyl (H3K4me1), Histone H3 lysine 4 tri methyl (H3K4me3), and total H3 in myoblasts (pink) and myotubes (green) (GSE80588, Soleimani et al., 2018).
-
FSnapshots of representative IGV tracks displaying RNA‐Seq data from WT whole muscle (WM), single myofibers (SF), and satellite cells (SC) showing the expression of EGF, EGFR, and VEGFA. Ckm and Myf6 are used as positive controls for whole muscle and single fibers. PDGFRA, a marker of fibro/adipogenic progenitor cells, is used as a control to show the lack of niche cells in the single myofiber samples.
-
G, HRNA‐Sequencing Reads Per Million (RPM) for EGF (G) and EGFR (H) in satellite cells and single myofibers. RNA‐Sequencing libraries were prepared from 1,000 satellite cells freshly isolated by FACS or from a single myofiber as described in the Materials and Methods. (n = 3 biological replicates per group) bars = mean ± SD, two‐tailed t‐test).
-
IActivation of EGFR in MuSCs on EDL myofibers treated with EGF compared with untreated control. Myofibers were cultured for 48 h and were treated with 40 ng/ml of EGF for 10 min before fixing and staining with antibodies for PAX7 and phospho‐EGFR. Scale bar = 30 μm.
-
GActivation of EGFR signaling in myoblasts treated with recombinant EGF. Myoblasts were treated with 2 ng/ml of recombinant protein in growth media (Ham's F10 supplemented with 20% FBS, 2.5 ng/ml bFGF) for 15 min and lysed in RIPA lysis buffer. Western blots were performed with antibodies against EGFR and phospho‐EGFR (pEGFR‐Y1068).
-
HDepletion of Myf6 transcript by siRNAs in differentiated primary myotubes (5DM). (n = 3 biological replicates) Two‐tailed t‐test, bars = mean ± SD.
-
IDepletion of Myf6 expression by siRNAs leads to a significant reduction in the gene expression output of Epidermal Growth Factor (EGF). (n = 3 biological replicates) Two‐tailed t‐test, bars = mean ± SD.
Figure EV2. Myf6 and MyoD share a vast core myogenic genetic network.
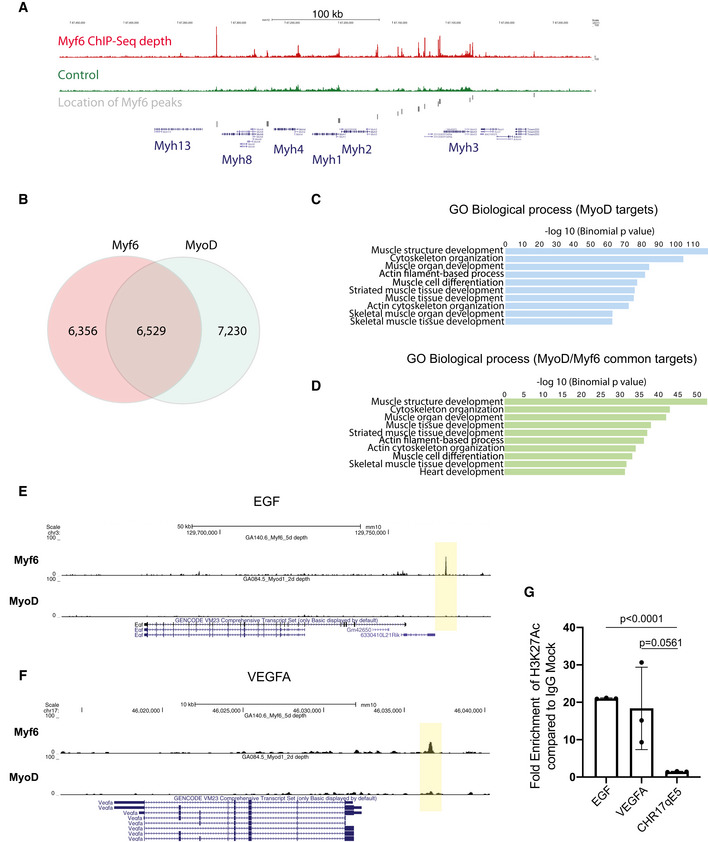
-
AEnrichment of Myf6 ChIP‐Seq signal on the myosin heavy chain gene cluster and location of Myf6 peaks.
-
BVenn diagram showing genome‐wide binding sites for MyoD and Myf6 and their overlap in differentiated muscle cells. The overlap is defined as any peaks that have any overlap between the two datasets.
- C
-
DSimilar analysis as in (C) for genes associated with MyoD/Myf6 common peaks. The association rule is based on single nearest gene within 300 kb of the peaks. Within that window the number of peaks associated with more than one gene is minimal for ChIP‐Seq datasets.
- E, F
-
GChIP‐qPCR for H3K27Ac compared with IgG control at the TSS of EGF, VEGFA, and a negative control region of Chromosome 17 qE5. (n = 3 technical replicates), two‐tailed t‐test, error bars = ± SD.
Myf6 occupies regulatory domains of a number of cytokine genes in differentiated muscle cells
To determine the role of Myf6 in the transcriptional regulation of myokines, we first analyzed Myf6 binding sites in their vicinity (Figs 1H and 2A and B). Notably, our ChIP‐Seq data show strong enrichment of Myf6 ChIP‐Seq reads, but not the reads from the affinity‐matched control ChIP‐Seq, on the enhancer elements in the vicinity of the TSS of EGF, LIF, OSM, and VEGFA in primary myotubes (Fig 2C–E). Next, we determined the pattern of regulatory histone marks including Histone H3 mono methyl lysine 4 (H3K4me1), a marker for enhancer elements and histone H3 trimethyl lysine 4 (H3K4me3), marking active/poised TSS in the vicinity of select cytokine genes (Fig 2C–E). Notably, our analysis of ChIP‐Seq data indicates that Myf6 binding sites overlap with H3K4me1 (Fig 2C–E). In primary myotubes, the presence of histone mark Histone 3 lysine 27 Acetyl (H3K27Ac) at the Myf6 binding site in the vicinity of the TSS of EGF and VEGFA further supports their active transcription (Fig EV2G). These data suggest that a novel function of Myf6 in adult skeletal muscle may be the establishment of a myokine‐mediated regulatory network. EGFR and STAT3 have recently been shown to play crucial roles in regulating muscle stem cell self‐renewal and expansion (Zhu et al, 2016; Wang et al, 2019). Both EGFR and STAT3 are readily activated in proliferating myoblasts (Figs 2J and EV3N) as well as in myofiber‐associated MuSCs (Figs 2I and EV3M) in response to exogenously applied ligands, indicating that these pathways are operationally active in muscle cells. Other myokines, such as vascular endothelial growth factor (VEGFA), are also transcriptionally regulated by Myf6 (Figs 2A, B, E and EV3B–C, and G). VEGFA plays an important role in maintaining MuSCs quiescence via recruitment of endothelial cells (Verma et al, 2018). In vitro depletion of Myf6 transcript by RNAi in differentiating primary myotubes shows that Myf6 is required for the transcriptional regulation of EGF and VEGFA (Figs 2K and L, and EV3G and H). While some ligands such as VEGFA are produced by both progenitors as well as differentiated myotubes (Fig 2A and F), others such as EGF are principally produced in differentiated myotubes and mature myofibers (Figs 2A, F, G and EV3F). This finding suggests that in the skeletal muscle EGFR signaling in satellite cells may be operationally dependent on the transcriptional regulation of its ligands by Myf6 in myofibers (Fig 2G and H). Together, these data indicate that the differentiation of muscle stem cells creates a physical niche whereby myokines (ligands) are produced in myofibers while their respective receptors are expressed in the associated MuSCs, suggesting the existence of a myokine‐mediated communication network between myofibers and MuSCs.
Figure EV3. Myf6 Regulates the Expression of Various Cytokine Genes.
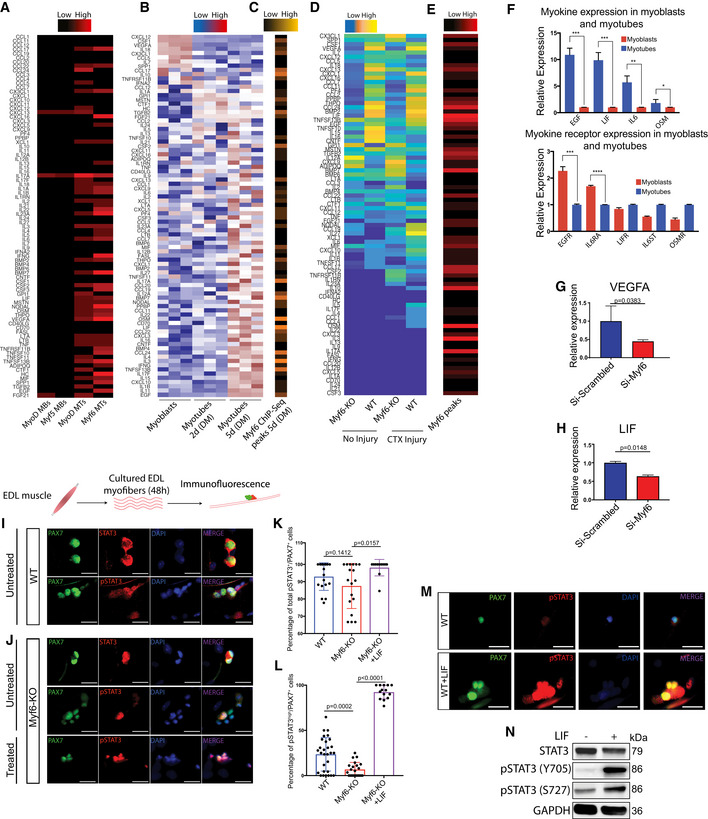
-
AColormap of Myf5, MyoD, and Myf6 peaks within 100 kb of the Transcription Start Sites (TSS) of cytokines ranging from zero (black) to six peaks (red) occupancy. Black indicates no binding (i.e., zero peaks), red indicates up to six ChIP‐Seq peaks. The onset of differentiation coincides with increased binding of MRFs to the regulatory domains of the cytokine genes.
-
BGene expression analysis of cytokines during a 5 day time course of myogenic differentiation going from cycling myoblasts in growth media (Ham's F10 supplemented with 20% Fetal Bovine Serum, 1% penicillin/streptomycin, 2.5 ng/ml basic Fibroblast Growth Factor) to terminally differentiated myocytes (2 days in differentiation media, DMEM supplemented with 5% horse serum) to the postmitotic multinucleated myotubes (5 days in differentiation media). Gene expression was assayed in biological triplicate by microarray (Soleimani et al, 2012, 2018). Expression of each gene was averaged across three replicates and normalized to mean zero and standard deviation of one. Expression values were then truncated at ± 3 for color display. Blue indicates lower than average expression, while red indicates higher than average expression.
-
CDistribution of Myf6 peaks within 100 kb of the TSS of the cytokines genes, similar to the analysis shown in (A).
-
DColormap of genes that are up/downregulated in the whole muscle transcriptome (RNA‐Seq) of hindlimb muscles from Myf6‐KO and WT counterparts. Differential expression is Log2 transformed, and truncated at ± 3. Color scale goes from blue (−3 = lower than mean), to white (0 = mean) and finally to red (+3 = higher than mean).
-
EHeat map showing the distribution of Myf6 peaks within 100 kb of the TSS of the myokine genes ranging from zero (black) to six ChIP‐Seq peaks (red) occupancy.
-
FQuantitative Real‐Time PCR (RT–qPCR) analysis of a select set of myokine and myokine receptor genes involved in the activation of STAT3 and EGFR signaling pathways. RT–qPCR analysis was performed on the total RNA isolated from primary myoblasts and multinucleated primary myotubes derived from MuSCs isolated by fluorescence‐activated cell sorting (FACS, see the Extended Materials and Methods). Myotubes were obtained by feeding > 90% confluent primary myoblasts with differentiation media (DMEM supplemented with 5% horse serum) for 5 days. (n = 3 technical replicates), bars = mean ± SD, two‐tailed t‐test, asterisks (*, **, ***, ****) correspond to P‐values of < 0.05, < 0.01, < 0.001, and < 0.0001, respectively.
-
G, HRT–qPCR analysis of primary myotubes for that have been depleted of Myf6 expression by siRNAs compared to controls, for vascular endothelial growth factor A (VEGFA) (G) and leukemia inhibitory factor (LIF) (n = 3 biological replicates), Two‐tailed t‐test, bars represent mean ± SD (H).
-
I, JImmunofluorescence staining of WT (I) and Myf6‐KO (J) EDL myofibers stained with PAX7, STAT3 and phospho‐STAT3 antibodies. Live fibers were maintained in culture media for 44 h, followed by 4 h of serum starving in non‐supplemented DMEM. For treated samples, 1,000 U/ml of recombinant LIF was added to the non‐supplemented DMEM for 10 min before fixation. Activation status of STAT3 signaling pathways was assessed using antibodies against phospho‐STAT3 (pSTAT3‐Y705). Scale bar = 30 μm.
-
KQuantification of the percentage of all pSTAT3+/PAX7+ (defined as PAX7+ cells showing low or high pSTAT3 signal) cells on WT (n = 18 fibers from n = 3 mice), Myf6‐KO (n = 18 fibers from n = 3 mice), and Myf6‐KO EDL myofibers treated with LIF (n = 11 fibers from n = 3 mice). Two‐tailed t‐test, bars represent mean ± SD.
-
JQuantification of the percentage of pSTAT3high/PAX7+ cells (defined as PAX7+ cells showing high pSTAT3 signal) on WT (n = 29 fibers from n = 3 mice), Myf6‐KO (n = 22 fibers from n = 3 mice), and Myf6‐KO EDL myofibers treated with LIF (n = 14 fibers from n = 3 mice). Two‐tailed t‐test, bars represent mean ± SD.
-
KActivation of STAT3 signaling in MuSCs on EDL myofibers treated with LIF compared with untreated control. Myofibers were cultured for 48 h and were treated with 1,000 U/ml of recombinant LIF for 10 min before fixation and staining with antibodies for PAX7 and p‐STAT3. Scale bar = 30 μm.
-
LActivation of STAT3 signaling in myoblasts treated with recombinant LIF. Myoblasts were treated with 2 ng/ml of recombinant protein in growth media (Ham's F10 supplemented with 20% FBS, 2.5 ng/ml bFGF) for 15 min and lysed in RIPA lysis buffer. Western blots were performed with antibodies against STAT3 and phospho‐STAT3 (pSTAT3‐Y705).
Loss of Myf6 leads to impaired myokine production in adult mice
To determine the requirement of Myf6 in the regulation of myokine expression in vivo, we first analyzed the whole muscle transcriptome of Myf6‐knockout mice under normal physiological conditions and after cardiotoxin (CTX) injury by RNA‐Seq. For this, we used Myf6CE mice, in which a Cre‐ERT2 cassette is knocked into the first exon of the Myf6 gene, rendering it a null allele (Fig 3A; Southard et al, 2014). Mice with biallelic deletion of Myf6 were born with normal skeletal muscles and were indistinguishable from their WT littermates (Fig 3B). Quantitative Real‐Time PCR (RT–qPCR) of RNA extracted from hindlimb muscles showed efficient depletion of Myf6 transcript in homozygous Myf6‐knockout mice (Fig 3C). RNA‐Seq analysis showed that genetic deletion of Myf6 in skeletal muscles led to deregulation of a vast array of genes involved in myoblast differentiation, muscle development, muscle contraction, and pre‐mRNA splicing (Dataset EV1). However, in addition to this, our RNA‐Seq data also suggest that loss of Myf6 leads to the downregulation of the expression of key myokine genes, such as VEGFA and EGF in vivo (Figs 3D and EV3D). Analysis of Myf6 binding sites by ChIP‐Seq showed enrichment of Myf6 peaks around the transcription start sites (TSS) of the aforementioned genes (Figs 3E and EV3E). Next, to determine if loss of Myf6 leads to diminished myokine protein production in vivo, we isolated blood from Myf6‐KO and heterozygous counterparts and performed Enzyme‐Linked Immunosorbent Assay (ELISA) analysis on serum. Our data show that serum from Myf6‐KO mice has a significant reduction in key myokines, EGF, and VEGFA compared with controls (Fig 3F and G). In parallel, we also isolated MuSCs from Myf6‐KO and WT counterparts by fluorescent‐activated cell sorting (FACS) and differentiated fully confluent cultures of primary myoblasts for 7 days in differentiation media. Next, we collected media (secretome) from myotubes and performed ELISA followed by comparative analysis of protein expression by normalizing protein amount to the number of nuclei. Secretome from Myf6‐KO myotubes had significantly lower levels of EGF and VEGFA compared with their WT counterparts (Fig 3H and I). Importantly, reintroduction of Myf6 by retroviral infection into Myf6‐KO myotubes leads to rescue of EGF protein levels compared with uninfected Myf6‐KO myotube controls (Fig 3J). In accordance with this data, RT–qPCR analysis of Myf6‐KO myotubes transduced retrovirally by exogenous Myf6 also showed restoration of EGF and VEGFA levels (Fig 3K–M). Together, these data suggest that Myf6 is required for the production of multiple myokines, at both the transcript and protein level, in adult mice.
Figure 3. Genetic Deletion of Myf6 Impairs Myokine Expression in Adult Skeletal Muscles.

-
ASchematic drawing of the Myf6 locus showing insertion of Cre‐ERT2 sequence rendering the Myf6 locus a null allele (Southard et al, 2014).
-
BPicture of 8‐week-old Myf6‐KO and WT mice.
-
CRelative expression of Myf6 transcript in the hindlimb skeletal muscles of Myf6‐KO and WT mice measured by Quantitative Real‐Time PCR (RT–qPCR) (n = 4 mice). Boxes represent 1st and 3rd quartile, central line represents the median, and whiskers represent the range. Two‐tailed t‐test.
-
DColormap of genes that are up/downregulated in the whole muscle transcriptome (RNA‐Seq) of injured and uninjured hindlimb muscles from Myf6‐KO and WT counterparts. Differential expression is Log2 transformed and truncated at ± 3. Color scale goes from blue (−3 = lower than mean) to yellow (+3 = higher than mean).
-
EHeat map showing the distribution of Myf6 peaks within 100 kb of the TSS of the myokine genes ranging from zero (black) to six ChIP‐Seq peaks (red) occupancy.
-
FExpression of EGF protein in the blood serum from Myf6‐KO and heterozygous counterparts as measured by enzyme‐linked immunosorbent assay (ELISA). (n = 5 heterozygous biological replicates, n = 3 Myf6‐KO biological replicates) Two‐tailed t‐test, error bars = ± SD.
-
GExpression of VEGFA protein in the blood serum from Myf6‐KO and heterozygous counterparts as measured by enzyme‐linked immunosorbent assay (ELISA). (n = 5 heterozygous biological replicates, n = 3 Myf6‐KO biological replicates) Two‐tailed t‐test, error bars = ± SD.
-
HExpression of VEGFA protein in the secretome of Myf6‐KO and WT myotubes as measured by enzyme‐linked immunosorbent assay (ELISA). Primary myotubes were derived from MuSCs isolated by fluorescence‐activated cell sorting (FACS, see the Extended Materials and Methods) from Myf6‐KO and WT mice. Myotubes were obtained by feeding > 90% confluent primary myoblasts with differentiation media (DMEM supplemented with 5% horse serum) for 7 days. Media from the last 48 h of differentiation (days 5–7) were collected for ELISA secretome analysis. Protein readouts were normalized to the total number of nuclei in each plate. (n = 4 WT biological replicates, n = 3 Myf6‐KO biological replicates) Two‐tailed t‐test, error bars = ± SD.
-
IExpression of EGF protein in the secretome of Myf6‐KO and WT myotubes as measured by enzyme‐linked immunosorbent assay (ELISA). Primary myotubes were derived from MuSCs isolated by fluorescence‐activated cell sorting (FACS, see the Extended Materials and Methods) from Myf6‐KO and WT mice. Myotubes were obtained by feeding > 90% confluent primary myoblasts with differentiation media (DMEM supplemented with 5% horse serum) for 7 days. Media from the last 48 h of differentiation (days 5–7) were collected for ELISA secretome analysis. Protein readouts were normalized to the total number of nuclei in each plate. (n = 4 WT biological replicates, n = 3 Myf6‐KO biological replicates) Two‐tailed t‐test, error bars = ± SD.
-
JExpression of EGF protein in the secretome of Myf6‐HF expressing myotubes derived from Myf6‐KO myoblasts (rescue) compared with Myf6‐KO myotubes as measured by enzyme‐linked immunosorbent assay (ELISA). Primary myotubes were derived from MuSCs isolated by fluorescence‐activated cell sorting (FACS, see the Extended Materials and Methods) from Myf6‐KO mice. Myotubes were obtained by feeding > 90% confluent primary myoblasts with differentiation media (DMEM supplemented with 5% horse serum) for 5 days. Media from the last 48 h of differentiation (days 3–5) were collected for ELISA secretome analysis. Protein readouts were normalized to the total number of nuclei in each plate. (n = 3 Myf6‐KO technical replicates, n = 4 rescue technical replicates). Two‐tailed t‐test, error bars = ± SD.
-
KRT–qPCR of Myf6 expression in Myf6‐KO (n = 2 technical replicates), WT (n = 3 biological replicates), and Myf6‐HF (rescue) myotubes (n = 3 technical replicates) (5DM). “Rescue” myotubes were generated from Myf6‐KO myoblasts infected with a retrovirus containing Myf6‐HF. Two‐tailed t‐test, error bars = ± SD.
-
L, MRT–qPCR of EGF (L) and VEGFA (M) expression in Myf6‐KO (n = 5) and rescue (n = 3‐4) primary myotubes (5DM). Two‐tailed t‐test, error bars = ± SD.
-
N, OImmunofluorescence staining of WT (N) and Myf6‐KO EDL (O) myofibers stained with PAX7, EGFR, and phospho‐EGFR antibodies. Live fibers were maintained in culture media for 44 h, followed by 4 h of serum starving in non‐supplemented DMEM. For treated samples, 40 ng/ml of recombinant EGF was added to the non‐supplemented DMEM for 10 min before fixation. Activation status of EGFR was assessed using antibodies against phospho‐EGFR (pEGFR‐Y1068). Scale bar = 30 μm.
-
PQuantification of the percentage of all pEGFR+/PAX7+ cells (defined as PAX7+ cells showing low or high pEGFR signal) on WT (n = 24 fibers from n = 3 mice), Myf6‐KO (n = 22 fibers from n = 3 mice), and Myf6‐KO EDL myofibers treated with EGF (n = 14 fibers from n = 3 mice). Two‐tailed t‐test, error bars = ± SD.
-
OQuantification of the percentage of EGFRhigh/PAX7+ cells (defined as PAX7+ cells showing high pEGFR signal) on WT (n = 37 fibers from n = 3 mice), Myf6‐KO (n = 34 fibers from n = 3 mice), and Myf6‐KO EDL myofibers treated with EGF (n = 11 fibers from n = 3 mice). Two‐tailed t‐test, error bars = ± SD.
Genetic deletion of Myf6 leads to exhaustion of the MuSC pool in adult skeletal muscle
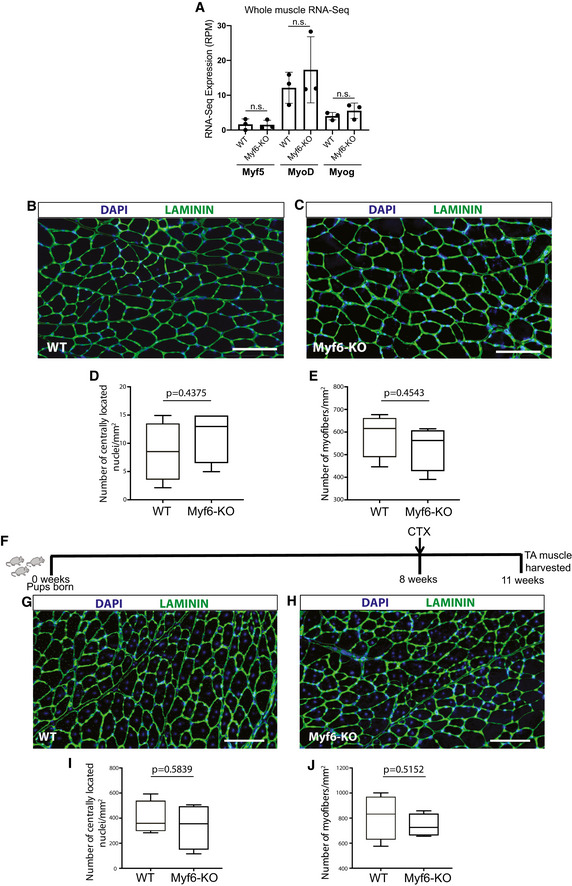
Genetic deletion of Myf6 led to the downregulation of many myokine genes including EGF, LIF, VEGFA, and Il‐15 (Figs 2 and 3). Given that EGF activates the EGFR signaling pathway (Fig 2I and J), we hypothesized that genetic deletion of Myf6 in the skeletal muscle myofiber can lead to defects in phosphorylation and subsequent activation of EGFR. To test this hypothesis, we isolated EDL myofibers from Myf6‐KO and WT animals and cultured them in growth media for 44 h. To eliminate the confounding effect of mitogens and growth factors in fiber culture, we switched fibers to unsupplemented DMEM for 4 h before fixing and staining. Immunofluorescence of Myf6‐KO EDL myofibers shows defects in phosphorylation of EGFR (pEGFR‐Y1068) (Fig 3N–Q). Importantly, treatment of EDL myofibers from Myf6‐KO animals with recombinant EGF rescues defects in EGFR phosphorylation (Fig 3N–Q). These data suggest that Myf6 is required to maintain functional signaling pathways like EGFR in the myofiber‐associated MuSCs (Figs 3N–Q and EV3I–L). Therefore, we next analyzed how the loss of Myf6 affects muscle regeneration and the stem cell pool. Immunofluorescent analysis of cross sections of TA muscles showed no significant difference in the number of regenerating fibers (i.e., myofibers with centrally located myonuclei), or the number of myofibers per unit area (mm2) between Myf6‐KO and WT mice (Fig EV4B–J). These data suggest that Myf6 is dispensable for muscle formation and MuSC differentiation during development. Next, we analyzed the effect of genetic deletion of Myf6 on the MuSC pool postnatally (P7, P21) and during adulthood (3 and 10 months of age) by first performing immunofluorescent analysis of the TA muscles from Myf6‐KO and WT animals with antibodies against Pax7 as a marker of MuSCs and Laminin to mark the periphery of myofibers (Table EV1). Our data indicate that loss of Myf6 has no significant effect on the establishment of the MuSC pool during development as the MuSC compartment remains unchanged at postnatal day 7 (P7) and 21 (P21) (Fig 4A–D). However, by the age of 3 months we observed a significant reduction in the number of Pax7+ cells in the Myf6‐KO TA muscles compared with their WT counterparts (Fig 4E and F). The reduction in the stem cell pool continues into late adulthood at 10 months of age (Fig 4G and H). Likewise, analysis of muscle stem cells associated with the Extensor Digitorum Longus (EDL) muscles stained with anti‐PAX7 antibody immediately after isolation (T = 0 h after isolation) showed a similar significant reduction in the number of MuSCs in Myf6‐KO myofibers (Fig 4I–L). Taken together, these data suggest that maintenance of the MuSC pool in adult mice is dependent on Myf6.
Figure EV4. Myf6 is dispensable for muscle development and regeneration in adult mice.
-
ATranscript expression (RPM) for myogenic factors Myf5, MyoD, and Myogenin from whole muscle RNA‐Sequencing of Myf6‐null compared with WT mice. (n = 3 biological replicates per group) Two‐tailed t‐test, bars represent mean ± SD.
-
B, CImmunofluorescent analysis of TA muscles from WT and Myf6‐KO counterparts stained with Laminin (LAMA1) and counterstained with DAPI. Scale bar = 100 μm.
-
DQuantification of the number of regenerating fibers (depicted by the presence of centrally located myonuclei) between the WT and the Myf6‐KO mice. Boxes represent 1st and 3rd quartile, central line represents the median, and whiskers represent the range. Two‐tailed t‐test.
-
DQuantification of the number of myofibers per unit area (mm2) of the TA muscles between WT and Myf6‐KO counterparts. (n = 4 mice per group). Boxes represent 1st and 3rd quartile, central line represents the median, and whiskers represent the range. Two‐tailed t‐test.
-
ESchematic timeline of cardiotoxin injury experiments.
-
G, HImmunofluorescent analysis of TA muscle from WT and Myf6‐KO animals 3 weeks after CTX injury (see extended Materials and Methods) stained with Laminin and counterstained with DAPI. Scale bar = 100 μm.
-
IQuantification of the number of regenerating myofibers (centrally located myonuclei) between WT and Myf6‐KO counterparts. Boxes represent 1st and 3rd quartile, central line represents the median, and whiskers represent the range. Two‐tailed t‐test.
-
HQuantification of the number of myofibers per unit area (mm2) between WT and Myf6‐KO counterparts. (n = 4 mice per group) Boxes represent 1st and 3rd quartile, central line represents the median, and whiskers represent the range. Two‐tailed t‐test.
Figure 4. Genetic Deletion of Myf6 Leads to Depletion of the Muscle Stem Cell Pool.
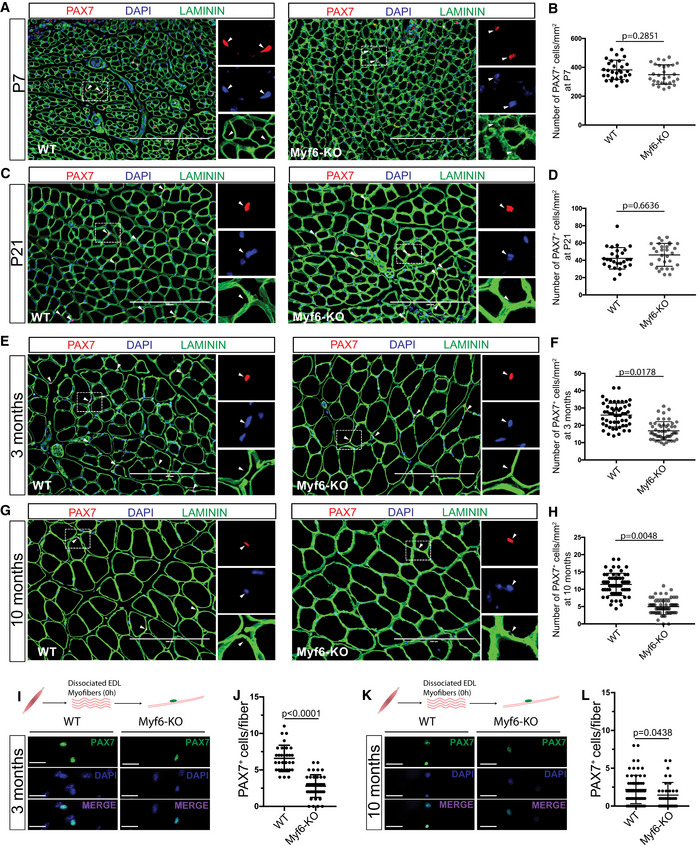
- Immunofluorescent analysis of TA muscle cross‐sections from Myf6‐KO and WT mice with PAX7 and Laminin (LAMA1) antibodies at postnatal day 7. Arrowheads indicate PAX7+ satellite cells. Scale bar = 200 μm.
- Quantification of the number of PAX7+ cells per unit area (mm2) in TA muscle cross‐sections from Myf6‐KO and WT counterparts at postnatal day 7. (n = 30 fields of view from n = 3 animals). Two‐tailed t‐test, error bars = ± SD.
- Immunofluorescent analysis of TA muscle cross‐sections from Myf6‐KO and WT mice with PAX7 and Laminin (LAMA1) antibodies at postnatal day 21. Arrowheads indicate PAX7+ satellite cells. Scale bar = 200 μm.
- Quantification of the number of PAX7+ cells per unit area (mm2) in TA muscle cross‐sections from Myf6‐KO and WT counterparts at postnatal day 21. (n = 27 fields of view from n = 3 animals). Two‐tailed t‐test, error bars = ± SD.
- Immunofluorescent analysis of TA muscle cross‐sections from Myf6‐KO and WT mice with PAX7 and Laminin (LAMA1) antibodies at 3 months of age. Arrowheads indicate PAX7+ satellite cells. Scale bar = 200 μm.
- Quantification of the number of PAX7+ cells per unit area (mm2) in TA muscle cross‐sections from Myf6‐KO and WT counterparts at 3 months of age. (n = 52 fields of view from n = 5 animals) Two‐tailed t‐test, error bars = ± SD.
- Immunofluorescent analysis of TA muscle cross‐sections from Myf6‐KO and WT mice with PAX7 and Laminin (LAMA1) antibodies at 10 months of age. Arrowheads indicate PAX7+ satellite cells. Scale bar = 200 μm.
- Quantification of the number of PAX7+ cells per unit area (mm2) in TA muscle cross‐sections from Myf6‐KO and WT counterparts at 10 months of age. (n = 60 fields of view from n = 3 animals). Two‐tailed t‐test, error bars = ± SD.
- Immunofluorescent analysis of myofiber‐associated MuSCs isolated from the EDL muscles of 3 month‐old Myf6‐KO and WT counterparts. Fibers were stained with PAX7 antibody and counterstained with DAPI at time T = 0 h after fiber isolation. Scale bar = 25 μm.
- Quantification of the number of MuSCs per fiber between 3‐month‐old Myf6‐KO (n = 41 fibers from n = 3 mice) and WT (n = 31 fibers from n = 3 mice) counterparts. Two‐tailed t‐test, error bars = ± SD.
- Immunofluorescent analysis of myofiber‐associated MuSCs isolated from the EDL muscles of 10‐month‐old Myf6‐KO and WT counterparts. Fibers were stained with PAX7 antibody and counterstained with DAPI at time T = 0 h after fiber isolation. Scale bar = 25 μm.
- Quantification of the number of MuSCs per fiber between 10‐month‐old Myf6‐KO (n = 38 fibers from n = 2 mice) and WT (n = 67 fibers from n = 3 mice) counterparts. Two‐tailed t‐test, error bars = ± SD.
Deletion of Myf6 leads to spontaneous break from quiescence and entry of MuSCs into differentiation
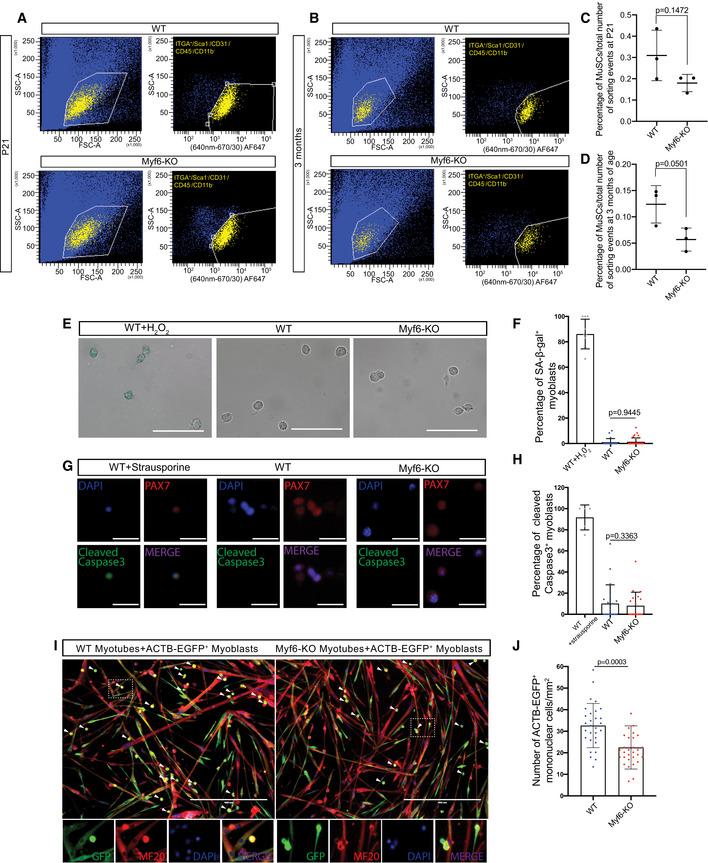
To determine the mechanism underlying the exhaustion of the muscle stem cell pool in Myf6‐KO adult mice, we first examined the quiescent state and the size of the muscle stem cell pool during three postnatal time points. By the third week of postnatal life (P21), we observe significantly fewer quiescent MuSCs in Myf6‐KO compared with WT mice, as shown by an increased number of PAX7+ cells that also express KI67, a marker for proliferating cells (Fig 5A–F; Table EV1). The spontaneous break of MuSCs from a quiescent state continues postnatally, and by 3 months of age there is a significant reduction in the MuSC pool of Myf6‐KO animals compared with their WT counterparts (Figs 4E and F, and 5E and F). Next, we isolated MuSCs from the Myf6‐KO and their WT counterpart by FACS and performed in vitro analysis of these MuSCs for differentiation potential, senescence, and apoptosis. Our data indicate that loss of Myf6 has no significant effect on the propensity of MuSCs to undergo apoptosis or senescence (Fig EV5E–H), suggesting that neither of these mechanisms is the cause of MuSC exhaustion in Myf6‐KO muscle. Next, we performed immunofluorescence analysis of myofiber‐associated MuSCs from the EDL myofibers at T0 after fiber isolation. At T0, WT MuSCs normally express PAX7 but not MYOD. However, in Myf6‐KO EDL myofibers we observed a significant number of MuSCs that express both PAX7 and MYOD, suggesting that a significant portion of MuSCs from the Myf6‐KO mice have exited quiescence (Fig 5K–M). These data are consistent with our earlier analysis showing that by 3 weeks of age, MuSCs from Myf6‐KO mice break quiescence and express KI67 (Fig 5C–F). Next, we performed RNA‐Seq analysis of freshly sorted MuSCs from Myf6‐KO and WT mice. To overcome the limitation in the number of MuSCs that can be isolated from each animal and, by proxy, the limited amount of total RNA, we used Switching Mechanisms at 5′ end of RNA Template (SMART technology, Clontech, Table EV1). Using an adjusted P‐value of < 0.01, we identified 754 genes that are significantly up‐ or downregulated in Myf6‐KO compared with their WT counterparts (Dataset EV2). Analysis of RNA‐Seq data shows that the p38 MAPK pathway is significantly upregulated in MuSCs of the Myf6‐KO mice compared with their WT counterparts (Fig 5G and H). Upregulation of p38 MAPK was further validated by immunofluorescence analysis of EDL myofiber‐associated MuSCs (Fig 5N–P). p38 MAPK plays a critical role in MuSC activation and exits from cell cycle and terminal differentiation (Segalés et al, 2016). Notably, our data show that progenitors derived from MuSCs from Myf6‐KO mice readily undergo myogenic differentiation with a fusion index approaching 100% as compared to their WT counterparts which exhibit a fusion index of approximately 60% (Fig 5I and J). Using tg‐ACTB‐EGFP+ myoblasts to trace the fate of these cells when cultured with either the WT or the Myf6‐KO myotubes, our data show that the tg‐ACTB‐EGFP+ cells fuse with their neighboring myotubes, as shown by the presence of MF20+/GFP+ multinucleated myotubes (Fig EV5I and J). Additionally, the lower number of mononuclear ACTB‐EGFP+ cells remaining when cultured with Myf6‐KO myotubes suggests that the enhanced fusion index seen in Myf6‐KO myotubes is due to the increased fusion of reserve cells as opposed to apoptosis or senescence (Fig EV5E–J). Furthermore, treatment of differentiating C2C12 myoblasts with recombinant EGF or LIF blocks p38 MAPK‐induced differentiation, as shown by the reduced expression of phospho‐p38 as well as the weakened expression of differentiation markers Myogenin and MF20 (Fig 5Q–T). Likewise, treatment of cultured EDL muscle fibers with EGF protein leads to the expansion of the PAX7+ satellite cell pool (Fig 5U and V). Taken together, these data suggest that loss of Myf6 in skeletal muscle leads to precocious activation of the differentiation program by dampening the EGFR pathway along with concomitant activation of p38 MAPK (Figs 4 and 5) and MyoD expression (Fig 5K–M).
Figure 5. MuSCs from Myf6‐KO Mice Exhibit Precocious Exit from Quiescence.

- Immunofluorescent analysis of TA muscle cross‐sections from Myf6‐KO and WT mice with PAX7 and KI67 antibodies at postnatal day 7. Scale bar = 100 μm. Arrowheads highlight select MuSC.
- Quantification of the percentage of KI67+ over PAX7+ MuSCs between Myf6‐KO and WT mice at postnatal day 7. (n = 15 fields of view from n = 3 animals) Two‐tailed t‐test, error bars = ± SD.
- Immunofluorescent analysis of TA muscle cross‐sections from Myf6‐KO and WT mice with PAX7 and KI67 antibodies at postnatal day 21. Scale bar = 100 μm. Arrowheads highlight select MuSC.
- Quantification of the percentage of KI67+ over PAX7+ MuSCs between Myf6‐KO (n = 28 fields of view from n = 3 animals) and WT mice (n = 31 fields of view from n = 3 animals) at postnatal day 21. Two‐tailed t‐test, error bars = ± SD.
- Immunofluorescent analysis of TA muscle cross‐sections from Myf6‐KO and WT mice with PAX7 and KI67 antibodies at 3 months of age. Scale bar = 100 μm. Arrowheads highlight select MuSC.
- Quantification of the percentage of KI67+ over PAX7+ MuSCs between Myf6‐KO and WT mice at 3 months of age (n = 30 fields of view from n = 5 mice). Two‐tailed t‐test, error bars = ± SD.
- Schematic drawing of the strategy for FACS isolation of Myf6‐KO and WT satellite cells for RNA‐Seq library preparation or for culture and differentiation.
- (Left) Heat Map of up‐ and downregulated genes in the p38 MAPK pathway (WP400) in Myf6‐KO and WT satellite cells. (Right) Gene set enrichment analysis of p38 MAPK pathway genes. In each plot, the blue curve represents the gene set enrichment score profile (Subramanian et al, 2005) across p38 MAPK pathway genes that are ranked by their differential expression. The gray curve represents the null expectation. P‐values are based on permutation test (10,000 permutations).
- Immunofluorescent analysis of WT and Myf6‐KO primary myotubes cultured in differentiation media for 7 days and stained with MF20 and DAPI. Scale bar = 200 μm.
- Quantification of the fusion index between Myf6‐KO (n = 54 fields of view from n = 3 mice) and WT (n = 81 fields of view from n = 4 mice) 7DM myotubes. Fusion index is calculated as the percentage of myonuclei out of the total number of nuclei per field of view. Two‐tailed t‐test, error bars = ± SD.
- EDL myofibers isolated from WT and Myf6‐KO mice fixed at time T = 0 and stained with PAX7, MYOD, and DAPI. Scale bar = 30 μm.
- Quantification of the percentage of MYOD+/PAX7+ satellite cells per fiber in Myf6‐KO (n = 88 myofibers) vs. WT (n = 86 myofibers) myofibers. Two‐tailed t‐test, error bars = ± SD.
- Quantification of the average percentage of MYOD+/PAX7+ satellite cells per mouse in Myf6‐KO and WT myofibers (n = 3 mice). Two‐tailed t‐test, error bars = ± SD.
- Staining for phospho‐p38 MAPK on EDL myofibers cultured for 48 h from Myf6‐KO and WT mice. Scale bar = 30 μm.
- Quantification of the percentage of phospho‐p38+/PAX7+ satellite cells per fiber in Myf6‐KO (n = 56 myofibers) vs WT (n = 67 myofibers) myofibers. Two‐tailed t‐test, error bars = ± SD.
- Quantification of the average percentage of phospho‐p38+/PAX7+ satellite cells per mouse in Myf6‐KO vs. WT myofibers (n = 3 mice). Two‐tailed t‐test, error bars = ± SD.
- Western blot analysis of C2C12 myoblasts treated with EGF, LIF, or EGF + LIF while in differentiation media and stained with phosphor‐p38, total p38, myogenin, MF20, and GAPDH. Cells were treated with EGF (50 ng/ml), LIF (1,000 U/ml) or EGF + LIF after 48 h in differentiation media, and every 6 h following initial treatment, followed by cell lysis at 72 h.
- Phospho‐p38 Western blot signal intensity normalized to total p38 signal intensity in control, EGF‐treated, LIF‐treated and EGF + LIF‐treated C2C12s. Signal intensity was measured by placing ROIs over Western blot bands using LI‐COR Image Studio. (n = 3 technical replicates) Two‐tailed t‐test, error bars = ± SD.
- Myogenin Western blot signal intensity normalized to GAPDH signal intensity in control, EGF‐treated, LIF‐treated and EGF + LIF‐treated C2C12s. Signal intensity was measured by placing ROIs over Western blot bands using LI‐COR Image Studio. (n = 3 technical replicates) Two‐tailed t‐test, error bars = ± SD.
- MF20 Western blot signal intensity normalized to GAPDH signal intensity in control, EGF‐treated, LIF‐treated, and EGF + LIF‐treated C2C12s. Signal intensity was measured by placing ROIs over Western blot bands using LI‐COR Image Studio. (n = 3 technical replicates) Two‐tailed t‐test, error bars = ± SD.
- EDL myofibers treated repeatedly with recombinant EGF protein (100 ng/ml) at 0 h, 24 h, 32 h, 40 h, and 48 h in culture and fixed at 48 h, compared with untreated controls.
- Quantification of the number of PAX7+ cells per fiber in EGF‐treated (n = 19 fibers from n = 1 mouse) and untreated control EDL myofibers (n = 16 fibers from n = 1 mouse) after 48 h in culture. Two‐tailed t‐test, error bars = ± SD.
Figure EV5. Myf6‐knockout mice show a reduced MuSC pool.
-
ARepresentative FACS plots for the isolation of MuSCs from Myf6‐KO and WT mice at P21. The yellow population corresponds to the ITGA7+/CD31−/SCA1−/CD45−/CD11b− population of MuSCs.
-
BRepresentative FACS plots for the isolation of MuSCs from Myf6‐KO and WT mice at 3 months. The yellow population corresponds to the ITGA7+/CD31−/SCA1−/CD45−/CD11b− population of MuSCs.
-
CQuantification of the percentage of total FACS sorting events corresponding to the MuSC population (number of ITGA+/ITGA7+/CD31−/SCA1−/CD45−/CD11b− events over the total number of FACS events) at P21 (n = 3 mice per group). Two‐tailed t‐test, error bars = ± SD.
-
DQuantification of the percentage of total FACS sorting events corresponding to the MuSC population (number of ITGA+/ITGA7+/CD31−/SCA1−/CD45−/CD11b− events over the total number of FACS events) at 3 months (n = 3 mice per group). Two‐tailed t‐test, bars represent mean ± SD.
-
ERepresentative photographs of SA‐β‐galactosidase staining in Myf6‐KO, WT, and WT H2O2‐treated cultured primary myoblasts. As a positive control, primary myoblasts from WT mice were cultured and treated with 20 μM H2O2 for 15 min, and left in fresh media for 48 h before fixation Scale bar = 100 μm.
-
FQuantification of the percentage of SA‐β‐Gal+ cells in Myf6‐KO (n = 33 fields of view, myoblasts from n = 2 mice), WT (n = 44 fields of view, myoblasts from n = 2 mice), and WT H2O2‐treated myoblasts. Two‐tailed t‐test, bars represent mean ± SD.
-
GRepresentative photographs of staining for cleaved caspase 3 in Myf6‐KO, WT, and WT strausporine‐treated cultured primary myoblasts. Primary myoblasts from WT mice were cultured and treated with 3 μM strausporine for 3 h and then fixed. Scale bar = 50 μm.
-
HQuantification of the percentage of cleaved caspase3+ cells in Myf6‐KO (n = 20 fields of view, myoblasts from n = 2 mice), WT (n = 20 fields of view, myoblasts from n = 2 mice), and WT strausporine‐treated myoblasts. Two‐tailed t‐test, error bars = ± SD.
-
IRepresentative images of a co‐culture assay of WT or Myf6‐KO myotubes with ACTB‐EGFP+ myoblasts. Briefly, WT and Myf6‐KO myoblasts were differentiated for 2 days before adding equal numbers of ACTB‐EGFP myoblasts for an additional 2 days, followed by staining for GFP, MF20, and DAPI. Arrows indicate mononuclear unfused cells. Arrowheads indicate unfused myoblasts. Scale bar = 400 μm.
-
JQuantification of the number of mononuclear cells remaining after 2 days in co‐culture with WT or Myf6‐KO 2DM myotubes (n = 30 fields of view, myoblasts from n = 2 mice per group). Two‐tailed t‐test, bars represent mean ± SD.
Myf6 is required for the establishment of functional niche environment in skeletal muscle
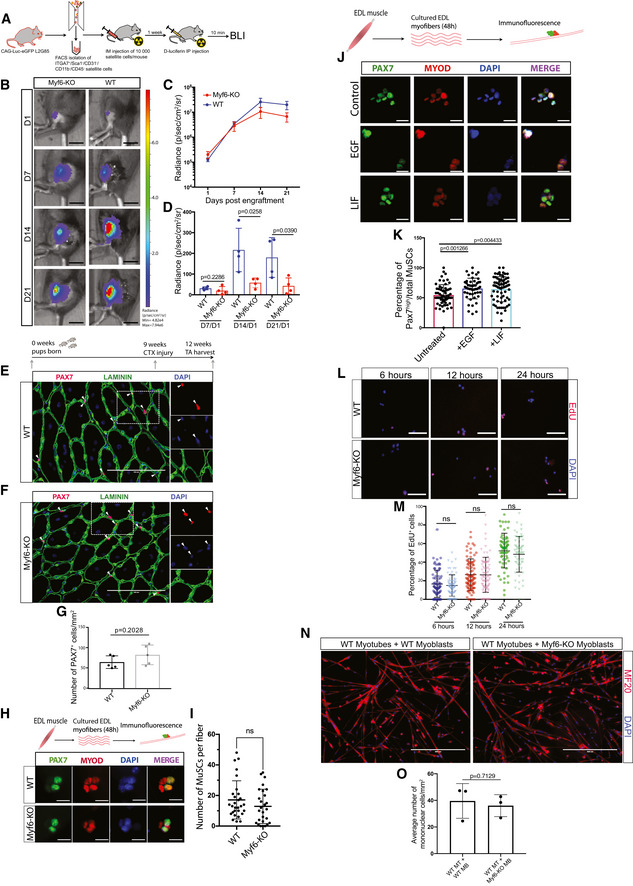
Loss of Myf6 led to a reduced MuSC pool in adult mice (Fig 4A–L) through spontaneous MuSC break from quiescence (Fig 5A–F). MuSCs from Myf6‐KO mice upregulate the p38 MAPK pathway, express MyoD (Fig 5K–P), and enter into myogenic differentiation (Fig 5G–J). Therefore, we next investigated whether defects in the stem cell compartment in Myf6‐KO mice are the result of a defective niche microenvironment. Since our earlier analysis established the requirement for Myf6 to regulate the expression of key myokines (Figs 2 and 3) that are required for the activation of EGFR (Fig 3), we hypothesized that genetic deletion of Myf6 leads to a defective niche microenvironment which is pro differentiation. To test this hypothesis, we isolated MuSCs from a luciferase mouse (B6;FVB‐Ptprc a Tg (CAG‐luc,‐GFP)L2G85Chco Thy1 a/J) (see Materials and Methods) and transplanted ten thousand satellite cells into either homozygous Myf6‐knockout or WT control mice. Next, we monitored the engraftment efficiency by bioluminescence imaging at day 1, 7, 14, and 21. Notably, in the Myf6‐knockout animals there is significantly less retention of donor MuSCs (Fig 6A–D), suggesting that the main effect of the loss of Myf6 on the MuSC compartment is due to a defective niche microenvironment. To eliminate the possibility that defects in the MuSC compartment in Myf6‐KO animals might be due to a cell‐intrinsic defect, we performed acute muscle regeneration assay using CTX injury. Importantly, following CTX‐induced injury, the MuSC pool in Myf6‐KO mice is reconstituted back to the level seen in their WT counterparts (Fig 6E–G), suggesting that exposure of muscle stem cells to injury‐induced sources of cytokines can rescue defects in the MuSC compartment. Similarly, continuous exposure of culturing EDL myofibers with chick embryo extract expands the number of MuSCs associated with EDL myofibers (Fig 6H and I). Furthermore, exposure of myofiber‐associated MuSCs to ligands, such as EGF and LIF, significantly increases the number of high Pax7 expressing MuSCs (Fig 6J–K). Our EdU incorporation data also show that cultured MuSCs from Myf6‐KO mice have a similar growth rate as that of their WT counterparts (Fig 6L–M). Lastly, when cultured in equal numbers alongside WT myotubes, Myf6‐KO and WT myoblasts differentiate to a similar extent, as shown by their equivalent number of reserve cells remaining after 2 days of co‐culture (Fig 6N and O). Taken together, our data indicate that the defect in the stem cell compartment of the Myf6‐KO animals is not due to cell‐intrinsic defects but is primarily due to an impairment in the niche microenvironment, in which depletion of myokine expression from the myofiber leads to activation of a signaling cascade that breaks MuSC quiescence, activates p38 MAPK signaling and induces precocious differentiation. This perturbation in the niche environment leads to a gradual reduction in the number of MuSCs in adult mice.
Figure 6. Exposure of MuSCs from Myf6‐KO mice to exogenous sources of cytokines rescues their self‐renewal defects.
-
ASchematic drawing of MuSC transplantation and in vivo bioluminescence experiments. Briefly, 10,000 MuSCs isolated by FACS from CAG‐Luc-eGFP mice were injected into the irradiated hindlimb of immunocompromised WT or Myf6‐KO mice. Bioluminescent imaging was performed at day 1, 7, 14, and 21 after donor MuSC injections.
-
BRepresentative images of bioluminescent signal from hindlimbs of Myf6‐KO and WT counterparts at day 1, 7, 14, and 21 after transplantation. Scale bar = 1 cm.
-
CTime course of average bioluminescent signal (total flux in radiance) in Myf6‐KO and WT mice at 1, 7, 14, and 21 days after MuSC transplantation showing engraftment dynamics of donor MuSCs (mean across 4–5 animals per group ± SEM at each time point, two‐tailed t‐test).
-
DBioluminescent signal (radiance) in Myf6‐KO and WT mice at each time point normalized to the bioluminescent signal at day 1 after MuSC transplantation. (n = 4–5 animals per group ± SD) Two‐tailed t‐test, bars represent mean ± SD.
-
E, FImmunostaining of TA cross‐sections of injured muscle from 3‐month‐old WT (E) and Myf6‐KO (F) mice stained with laminin (LAMA1) and PAX7 antibodies. Acute muscle injury was performed by injection of Cardiotoxin (CTX) into the TA muscle of Myf6‐KO and WT mice (n = 5 animals per group). Arrowheads indicate PAX7+ MuSCs. Scale bar = 100 μm.
-
FQuantification of the number of PAX7+ cells per unit area of TA muscle cross‐sections between Myf6‐KO and WT mice. (n = 5 animals per group) Two‐tailed t‐test, bars represent mean ± SD.
-
GImmunofluorescent analysis of Myf6‐KO and WT EDL myofibers cultured in growth media for 48 h and stained with PAX7 and MYOD antibodies. Scale bar = 20 μm.
-
HQuantification of the number of PAX7+ MuSCs per fiber in Myf6‐KO (n = 27 fibers from n = 3 mice) and WT (n = 29 fibers from n = 3 mice) mice after 48 h in culture. Two‐tailed t‐test, error bars = ± SD.
-
IImmunofluorescence staining of WT EDL myofibers cultured in growth media for 48 h and treated with LIF (1,000 U/ml) or EGF (80 ng/ml) every 8 h. Fibers were stained with antibodies against PAX7 and MyoD. Scale bar = 30 μm.
-
JQuantification of the percentage of PAX7high cells on the EDL myofiber treated with recombinant LIF (n = 80 fibers), EGF (n = 50 fibers) or vehicle control (n = 61 fibers). Two‐tailed t‐test, bars represent mean ± SD.
-
KEdU staining of cultured primary myoblasts isolated by FACS (see Materials and Methods) from Myf6‐KO and WT mice. Staining was analyzed after 6, 12, and 24 h of EdU incorporation. Scale bar = 100 μm.
-
LQuantification of the percentage of EdU+ myoblasts after 6 h (WT n = 75 fields of view from n = 3 mice, Myf6‐KO n = 72 fields of view from n = 3 mice), 12 h (WT n = 82 fields of view from n = 3 mice, Myf6‐KOn = 82 fields of view from n = 3 mice), and 24 h (WT n = 70 fields of view from n = 3 mice, Myf6‐KO n = 65 fields of view from n = 3 mice) of EdU incorporation. Two‐tailed t‐test, bars represent mean ± SD.
-
MRepresentative images of a co‐culture assay between WT myotubes and either WT or Myf6‐KO myoblasts. Briefly, WT myoblasts were differentiated for 2 days before adding equal numbers of either WT or Myf6‐KO myoblasts for an additional 2 days, followed by immunostaining for MF20 and DAPI. Scale bar = 400 μm.
-
NQuantification of the average number of myoblasts per mm2 that have remained mononuclear after their addition to 2DM WT myotubes, for 2 additional days (n = 3 biological replicates). Two‐tailed t‐test, bars represent mean ± SD.
Discussion
Skeletal muscle has an inherent endocrine function in addition to its primary roles of producing force and regulating energy metabolism (Pedersen & Febbraio, 2012; Pal et al, 2014; Nadeau & Aguer, 2019). However, mechanistic insights into how myokines are regulated and whether or not they function as part of the MuSC niche milieu remains largely unexplored. Here, we report that Myf6 is critical to maintain a myokine signaling loop where the muscle fiber acts as a source of ligands to signal to their associated satellite cells, which express the corresponding receptors. In other words, our data identify a novel function for Myf6 as a myogenic regulator of niche factors to maintain the muscle stem cell pool (Figs 4 and EV5A–D).
In accordance with previous work, the differentiation function of Myf6 in myogenesis is dispensable, likely because of the large overlap in the regulatory function of myogenic factors (Fig EV2; Olson et al, 1996). Consistent with this notion, comparative ChIP‐Seq analysis in differentiating myotubes shows that MyoD is fully capable of binding to a very similar set of genes, sharing a 50% overlap with Myf6 targets (Figs 1H and EV2, and Fig EV3A). Myf6‐knockout mice were born with no observable physical abnormalities. Moretti et al (2016) previously reported that treatment of skeletal muscles with Myf6 shRNA led to myofiber hypertrophy. Similarly, we noticed a slight trend toward larger fiber size in adult mice with a homozygous Myf6 deletion, despite there being no distinct difference in fiber morphology (Fig EV4B–J). Also consistent with previous reports, we noticed an upregulation of muscle‐specific genes in Myf6‐KO muscle (Moretti et al, 2016; Table EV3). Therefore, the differentiation function of Myf6 is likely compensated redundantly by other myogenic factors, i.e., MyoD and/or Myogenin during the transient differentiation of muscle stem cells (Figs EV2 and EV4B–J). However, once the muscle fiber is fully formed, MyoD and Myogenin are no longer largely expressed in WT or in Myf6‐null animals, and therefore cannot counteract the lack of Myf6 in the context of myokine production (Figs 1A and EV1A, and EV4A). Accordingly, using both siRNA to specifically knockdown Myf6 in myotubes (Figs 2K and L, and EV3G and H) and in vivo genetic deletion of Myf6 in skeletal muscle followed by whole muscle RNA‐Seq analysis (Figs 3D and EV3D), we show that the unique function of Myf6 in regulating myokine expression is not compensated by other myogenic factors, such as MyoD and Myogenin. The unanticipated function of Myf6 in regulating the MuSC pool via myokine expression is primarily due to its temporal position in the hierarchy of the muscle differentiation program. While Myf5, MyoD, and Myogenin are transiently expressed during MuSC differentiation, in mature skeletal muscle fibers, Myf6 is the predominantly expressed myogenic factor (Figs 1A and EV1A).
RNA‐sequencing of single myofibers clearly reveals that the primary source of EGF and VEGFA in the muscle is from fully differentiated myofibers (Fig 2F and G). Our ChIP‐Seq (Fig 1), gene expression analysis (Fig 2), and ELISA experiments (Fig 3) point to a role for Myf6 in the production of multiple regulatory myokines, such as EGF and VEGFA. Our striking observation that loss of Myf6 leads to a significant reduction of myokines, not only in the secretome of differentiating myotubes (Fig 3H and I) but also in serum (Fig 3F and G) suggests that the regulatory function of Myf6 may extend beyond skeletal muscle.
The impairment in the ability of Myf6‐KO MuSCs to activate pathways such as EGFR, followed by subsequent rescue (Fig 3N–Q), strongly suggests that Myf6 regulates MuSC activity by myokine signaling. A critical factor that is required for the maintenance of the MuSC pool throughout adult life is the ability of stem cells to remain quiescent under homeostatic conditions. In Myf6‐KO animals, muscle stem cells break quiescence and spontaneously enter into the differentiation program by upregulating the p38 MAPK pathway and expressing MyoD (Fig 5). Importantly, Myf6‐regulated myokines, namely EGF, have the ability to block p38 MAPK‐induced differentiation (Fig 5Q–T), instead promoting the expansion of the PAX7+ cell pool (Fig 5U and V). Therefore, the spontaneous exit from quiescence and entry into myogenic differentiation seen in Myf6‐KO animals results from the abrogation of myokine‐mediated communication between myofibers and their associated stem cells. The inability of donor stem cells to be efficiently retained in the skeletal muscles of Myf6‐KO mice further supports our conclusion that loss of Myf6 leads to defective communication between MuSCs and their niche microenvironment (Fig 6A–D). Our in vivo and in vitro analyses uncover a novel Myf6‐mediated myokine signaling pathway that establishes a direct communication between myofibers and their associated stem cells. Therefore, we propose that Myf6 is a myogenic niche regulator which acts upstream of EGFR and p38 MAPK signaling pathways to maintain the muscle stem cell pool.
Materials and Methods
Generation of Myf6‐CTAP retrovirus
Full open reading frame (FL‐ORF) of the mouse Myf6/MRF4 was subcloned into pBRIT retroviral backbone (Soleimani et al, 2012). The construct was verified by sequencing, and its functionality was confirmed using multiple functional assays (Fig EV1). Retroviral particles containing Myf6‐CTAP were generated by transfection of Phoenix helper‐free retrovirus producer lines, a kind gift from the Nolan laboratory (https://web.stanford.edu/group/nolan/_OldWebsite/retroviral_systems/phx.html) with Polyethylenimine (PEI, Polysciences Cat. 23966‐2). Viral supernatant was collected 48 h after transfection.
Generation of primary myoblasts over‐expressing Myf6‐CTAP
Muscle stem cells were isolated from 8‐week‐old WT C57BL/6 mice by FACS and were propagated and maintained in growth media (HAM's F10 supplemented with 20% FBS, 1% penicillin/streptomycin and 2.5 ng/ml basic Fibroblast Growth Factor, bFGF) for up to three passages. The resulting myoblasts were then infected with a retroviral cocktail containing Myf6‐CTAP virus and polybrene (Millipore) for 8 h. Cells were subsequently washed with 1 × PBS and fed with normal growth media. The selection of transduced cells was carried out by puromycin selection (2.5 μg/ml) in primary myoblast growth media for up to 1 week. Subsequently, stably transduced primary cells were maintained in low puromycin selection media (HAM's F10 supplemented with 20% FBS, 2.5 ng/ml bFGF, 1% penicillin/streptomycin, and 1.25 μg/ml puromycin). Differentiated multinucleated myotubes were obtained by placing > 90% confluent myoblasts in differentiation media (DMEM supplemented with 5% horse serum) for 5 days.
Tandem affinity chromatin immunoprecipitation sequencing (ChTAP‐Seq)
ChTAP‐Seq was carried out as described previously (Soleimani et al, 2013). Briefly, multi‐nucleated myotubes over‐expressing Myf6‐CTAP or empty vector‐ (EV‐CTAP) were harvested and 20 mg of chromatin was used as input for ChIP. ChIP was performed in tandem using anti FLAG antibody conjugated to agarose beads (Sigma). Chromatin was then eluted from the beads by competition with FLAG peptide (Sigma) and cleavage by TEV protease (New England Biolabs). The eluted chromatin was then used for a second purification step using Ni‐NTA Agarose with nickel‐charged affinity resin (Thermo Fisher), as described previously (Soleimani et al, 2013). Finally, 10 ng of ChIP DNA was used to construct ChIP‐Seq libraries using TruSeq ChIP Library Preparation Kit (Illumina). Reads from the Myf6 ChTAP‐ and the EV‐ChTAP‐Seq were mapped to the mouse mm10/GRCm38 mouse genome assembly. Peak calling was carried out using MACS2.0 (Zhang et al, 2008) with P‐value threshold of 10−5 using default parameters.
Luciferase assays
Luciferase assays were carried out as described previously (Soleimani et al, 2018). Briefly, Cos7 cells were co transected with retroviral vectors harboring MRFs (MyoD or Myf6) with and without mouse E47 expression vector together with the luciferase vector (pGL4.23 [luc2/minP]) containing a multimerized E‐box motif with three spaced out CAGCTG motifs, as described previously (Soleimani et al, 2018). Dual luciferase assay was performed using Promega Dual luciferase assay kit. Relative luciferase expressions were normalized to renilla and plotted relative to the promoter readout.
Generation of a stable cell line of primary myoblasts over‐expressing Myf6
Stable cell lines of Myf6‐KO primary myoblasts over‐expressing Myf6 were generated as previously described (Soleimani et al, 2012). Briefly, retrovirus particles containing Myf6‐Flag plasmid were generated using Phoenix helper‐free retrovirus producer lines with lipofectamine and chloroquine. Viral supernatant was collected 48 h after transfection and applied to primary myoblasts for 8 h. Cells were then washed twice with 1 × PBS and grown for 48 h in normal growth media. The cells were then treated with puromycin selection media (HAM's F10 supplemented with 20% FBS, 2.5 ng/ml bFGF, 1% penicillin/streptomycin, and 2.5 μg/ml puromycin) for 1 week. Afterward the cells were maintained in low puromycin maintenance media (HAM's F10 supplemented with 20% FBS, 2.5 ng/ml bFGF, 1% penicillin/streptomycin, and 1.25 μg/ml puromycin).
Immunofluorescent analysis of the tibialis anterior (TA) muscles
TA muscle sections were fixed in 4% paraformaldehyde (PFA) in PBS for 10 min on ice and washed three times for 5 min each in PBS. Permeabilization was done in 0.3% Triton‐X100 and 0.1 M glycine in PBS for 20 min on a shaker at room temperature, followed by washing once in PBT (0.05% Triton‐X100 in PBS, v/v).
Muscle cross‐sections were first blocked in Mouse blocking reagent (M.O.M reagents Vector Laboratories) for 1 h and subsequently in 10% goat serum (Thermo Fisher) supplemented with 3% bovine serum albumin (BSA) in a humid chamber at room temperature for 1 h. Subsequently, muscle sections were incubated with the primary antibodies (anti PAX7, concentrated DSHB 1:100 dilution; anti‐Laminin (LAMA1), Sigma 1:750 dilution) in goat serum with BSA blocking solution overnight in a humid chamber at 4°C. The next day, slides were washed twice for 10 min each time in PBT (0.05% Triton‐X100 in PBS, v/v), and once for 10 min in PBS. Samples were then incubated with the secondary antibodies (goat anti‐mouse IgG1, Alexa 568 1:1,000 dilution; goat anti‐rabbit IgG Alexa 488 1:500 dilution) diluted in goat serum in BSA blocking solution for 1 h at room temperature in a humid chamber protected from light. Sections were washed three times for 10 min each time in PBT followed by mounting with ProLong Gold AntiFade plus DAPI solution (Invitrogen P36935), and coverslips were sealed using clear nail polish.
Skeletal muscle injury and regeneration
Injury‐induced regeneration experiments were performed via intra‐muscular injection of 50 μl cardiotoxin (CTX, 5 μM, Sigma) into the TA muscle of 3‐4‐months old Myf6‐KO mutant and WT control animals. Prior to injury, mice were anaesthetized by intraperitoneal injection of Avertin (2,2,2‐tribromoethanol from TCI America at 15 μl per gram body weight of 20 mg per ml solution), or by the administration of Isoflurane. The right injured and the left uninjured TA muscles were harvested via dissection 21 days after CTX injury. Muscles were immediately mounted in tragacanth (Alfar Aesar) on cork and snap‐frozen in liquid nitrogen‐chilled isopentane (Sigma).
Isolation and immunofluorescent analysis of EDL myofibers
Briefly, the skin from the hindlimb of the mouse was removed and the TA muscle was dissected to gain access to the extensor digitorum longus (EDL). Excess tissue was removed around the knee to reveal the proximal tendon of the EDL muscle. The EDL muscle was dissected by cutting the distal tendon, gently peeling the muscle off the leg to reach the proximal tendon and severed that as well. The EDL was placed in a 1.5 ml tube with 1,000 U/ml collagenase in Dulbecco's Modified Eagle's Medium (DMEM) for 1 h at 37°C, 5% CO2. The EDL muscle was then transferred to a 6‐well plate containing unsupplemented DMEM that had previously been coated with 10% horse serum (HS) plus DMEM for 30 min. The fibers were released from the EDL muscle by using a large bore glass pipette with blunted edges at the nozzle, coated with HS, to gently pipette the muscle up and down.
Isolated fibers were transferred to a single well of a 12‐well plate that was coated with 10% HS plus DMEM for 30 min. Excess media were removed, and fibers were fixed with 4% PFA in PBS for 10 min at room temperature. After PFA removal, fibers were washed 3× with 0.1% Triton X100 in PBS. Fibers were then permeabilized with 0.1% Triton X100, 0.1 M glycine in PBS for 15 min at room temperature (RT). After removal of permeabilization buffer, fibers were washed 3× with 0.1% Triton X100 in PBS and blocked with a blocking solution composed of 5% HS, 2% BSA, 1% Triton X100 in PBS for at least 1 h at RT. Subsequently, blocking solution was removed and fibers were stained with appropriate primary antibodies diluted in the blocking solution overnight at 4°C. The next day, the primary antibody solution was removed and fibers were washed 3× with 0.1% Triton X100 in PBS. Subsequently, the secondary antibodies were diluted in the blocking solution and added to fibers at RT for at least 1 h. Lastly, antibody solution was removed and fibers were washed 3× with 0.1% Triton X100 in PBS and placed on a glass slide with mounting solution containing DAPI for microscopic analysis.
Immunofluorescence of cultured fibers was essentially the same as above with the exception that the fibers were placed in growth media [DMEM/sodium pyruvate (110 mg/l), supplemented with 10% FBS, 1% penicillin/streptomycin, 1% chick embryo extract, 2.5 ng/ml bFGF] and incubated at 37°C and 5% CO2 for 48 h prior to fixation.
Immunofluorescence imaging
Imaging was performed using an EVOS FL Imaging System (Thermo Fischer, AMF4300) equipped with GFP, Texas Red and DAPI filters, and 4×, 10×, 20×, 40× objectives. To determine the numbers of PAX7high and PAX7low cells when imaging EDL myofibers, images were captured at 60% signal intensity. Once all channels were overlaid, the GFP channel for PAX7 staining was set to 40% intensity. If no signal was visible at that threshold, cells were counted as PAX7low. If a signal was visible at that threshold, cells were counted as PAX7high. The imaging threshold was determined by focusing on a field of view containing cells with varying signal intensities, and adjusting the settings such that the brightest cells are visible after image capture whereas the dimmer cells are not. A similar method was used to determine numbers of pEGFRhigh and pSTAT3high cells. Equal imaging parameters were used between experimental and control sample groups.
Quantitative real‐time PCR
qPCR analysis was performed using SYBR Green qPCR Master Mix containing buffer, dNTPs and Thermo stable hot‐start DNA polymerase (Thermo Fisher). Gene‐specific primers (Table EV2) were designed using GenScript software. (https://www.genscript.com/tools/real-time-pcr-tagman-primer-design-tool) crossing exon junctions for specified genes. The reactions were run on a QuantStudio 7 Flex Real‐Time PCR System (Applied Biosystems – Thermo Fisher). Relative expression values were calculated based on the ΔΔCT method using RPS2 or GAPDH as housekeeping genes.
Isolation of muscle stem cells by fluorescence‐activated cell sorting
Hindlimb muscles from Myf6‐KO and WT mice were minced and digested in FACS digestion media, composed of HAM's F10 with 2.4 U/ml Collagenase D, 2.4 U/ml Dispase II and 0.5 mM CaCl2, for 60 min at 37°C in a 5% CO2 Tissue Culture (TC) incubator with intermittent trituration. The digested muscle slurry was supplemented with 9 ml of FBS and filtered through a 40 μm cell strainer, and cells were pelleted by centrifugation at 500 g, relative centrifugation force (RCF). The cell pellet was resuspended in 500 μl of 2% FBS: PBS (v/v) and incubated with antibodies and live cell stain (Alexa647‐conjugated to anti‐mouse ITGA7, PE‐CD31, PE‐CD45, PE‐CD11b, PE‐SCA1 and Hoechst 33342) for 15 min at room temperature. Cells were washed in 2% FBS:PBS (v/v) solution and sorted for the ITGA7+/Hoechst+/CD31−/CD45−/CD11b−/SCA1− population as described previously (Pasut et al, 2012) on a FACSAria Fusion system (Beckman‐Dickinson).
Isolated cells were sorted directly into lysis buffer for SMART RNA‐Seq library preparation or were cultured in growth media (Ham's F10 supplemented with 20% FBS, 1% penicillin/streptomycin and 2.5 ng/ml bFGF) and passaged after reaching 60% confluency.
SMART RNA‐Seq library preparation of satellite cells
1,000–3,000 quiescent satellite cells were lysed after FACS by sorting directly into a 0.2 ml tube containing 1 μl SMART‐Seq Reaction Buffer (95% SMART‐Seq 10 × lysis buffer containing 5% SMART‐Seq RNAse Inhibitor, Takara Bioscience, Cat. 634890) in 8 μl ddH20. RNA was reverse transcribed using SMART‐Seq Reverse Transcriptase followed by 11 cycles of PCR amplification (SMART‐Seq v4 kit, Takara Bioscience, Cat. 634890). Purification and size selection of DNA were carried out using Ampure XP beads (Beckman Coulter, Cat. A63880). Total cDNA was quantified using Quant‐iT PicoGreen dsDNA Assay Kit (Thermo Fisher). Sequencing libraries were created with the Nextera XT kit (Illumina Cat. FC‐131‐1024 and FC‐131‐2001), using 150 pg of total cDNA as input for tagmentation. Illumina sequencing adapters were added by PCR using 12 cycles of amplification (Illumina Cat. FC‐131‐1024 and FC‐131‐2001). Final sequencing libraries were purified and size selected using Ampure XP beads (Beckman Coulter, Cat. A63880).
Single myofiber RNA‐sequencing
Extensor Digitorum Longus myofibers were isolated from mouse hindlimbs as previously described under “Isolation and Immunofluorescent Analysis of EDL Myofibers”. To strip the myofibers of their associated satellite cells, trypsin was added to the digestion buffer at a final concentration of 0.25%. A single myofiber was then transferred to a 0.2 ml PCR tube, followed by visualization under microscope to confirm the presence of a single fiber. Fibers were resuspended in 9 μl of nuclease‐free H2O containing 1 μl of SMART‐Seq 10 × Reaction Buffer (95% SMART‐Seq 10 × lysis buffer containing 5% SMART‐Seq RNAse Inhibitor, Takara Bioscience, Cat. 634890). Sequencing‐ready libraries were prepared as previously described (Blackburn et al, 2019), using 250 pg of cDNA as input for Nextera XT (Illumina Cat. FC‐131‐1024 and FC‐131‐2001) library preparation.
Satellite cell transplantation
Hindlimbs of WT and Myf6‐KO mice were irradiated with 18 Gy using a MultiRad 225 irradiator and were administered a subcutaneous injection of immunosuppressant FK506 (2.5 mg/kg) the day before satellite cell transplantation. Luciferase‐expressing satellite cells were FACS sorted from B6;FVB‐Ptprc a Tg (CAG‐luc,‐GFP)L2G85Chco Thy1 a/J mice into 2%FBS:PBS (v/v), and 10,000 cells were injected into the hindlimb of WT and Myf6‐KO mice using a Hamilton syringe. FK506 injections (2.5 mg/kg) were maintained every 48 h throughout the bioluminescence imaging time course.
In vivo bioluminescence imaging
Mice were anesthetized and given two 100 μl contralateral intraperitoneal injections of D‐luciferin (Gold Biotechnology) diluted to 15 mg/ml in PBS. Bioluminescence imaging was performed 11 min after luciferin injections using an IVIS Spectrum machine (Perkin Elmer). Bioluminescence signal was measured using region of interest (ROI) placed over each hindlimb. Imaging was performed 1, 7, 14, and 21 days after donor cell transplantation.
Whole muscle RNA‐seq preparation
Total RNA was isolated from the hindlimb skeletal muscles of 4‐month‐old Myf6‐KO and WT mice using TRIzol reagent (Thermo Fisher). RNA quality and quantity were assessed by the Agilent Bio analyzer and 1.0 μg of RNA was used to construct RNA‐Seq libraries. cDNA libraries were constructed from total RNA with the TruSeq RNA v2 kit (Illumina). The size distribution of the libraries was analyzed with the Fragment Analyzer High Sensitivity NGS assay (AATI). DNA concentration was measured with the Qubit High Sensitivity DNA assay (Thermo Fisher).
RNA‐seq analysis
Sequencing was performed on Illumina NextSeq 500 High Output Flow Cell. Sequenced reads were mapped to mouse mm10 genome assembly using HISAT2 (Kim et al, 2015). We used FeatureCounts to quantify gene expression using GENCODE gene definitions (Liao et al, 2014). We then normalized expression by adding one count to every gene (to avoid zeros) and converted gene expression into reads per million (RPM). Differentially expressed genes were identified using the R package DESeq2 (Love et al, 2014). P‐values were corrected by independent hypothesis weighting (Ignatiadis et al, 2016).
GSEA pathway analysis
Genes for p38 MAPK (WP400) pathways were used to perform gene set enrichment analysis (Tanabe & Kanehisa, 2012; Fabregat et al, 2018; Slenter et al, 2018) using a weighted Kolmogorov–Smirnov test. In each test, genes in the pathway were weighted by its Wald‐test statistic of differential expression. The null distribution of enrichment scores was calculated by permuting the gene labels 10,000 times, the enrichment significance (P‐value) was calculated by the comparison of the enrichment score of the original labels with the null distribution (Subramanian et al, 2005).
Experimental model and subject details
Animal procedures
All animal procedures were approved by the McGill University Animal Care Committee (UACC).
Cell isolation and culture
Muscle stem cells were isolated from the hindlimb muscles of 8‐week‐old mice by fluorescence‐activated cell sorting, as described in the Method Details. Sorted cells were cultured on collagen‐coated plates in growth media (Ham's F10 supplemented with 20% FBS, 1% penicillin/streptomycin and 2.5 ng/ml bFGF) and passaged after reaching 60% confluency.
Quantification and statistical analysis
Statistical analysis was performed using Prism7 (Graphpad). An unpaired two‐tailed t‐test was used for comparison between experimental and control groups. Error bars are representations of the standard deviation unless otherwise stated. Asterisks (*, **, ***, ****) correspond to P‐values of < 0.05, < 0.01, < 0.001, and < 0.0001, respectively.
Contact for reagent and resource sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Vahab Soleimani (vahab.soleimani@mcgill.ca).
Author contributions
Conceived the idea, VDS; performed experiments, VDS, FL, DMB, KS, NK, AJ‐A, AS, DN, and CL; Analyzed data, VDS, FL, DMB, TJP, HSN, and AHC; wrote the manuscript; FL and VDS.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Dataset EV1
Dataset EV2
Source Data for Expanded View
Review Process File
Acknowledgements
We thank Christian Young at the Lady Davis Institute for Medical Research—Jewish General Hospital—core facility for help with fluorescence‐activated cell sorting (FACS). We thank Dr. Michael A. Rudnicki at the Ottawa Hospital Research Institute—University of Ottawa for kindly providing us with crucial reagents. This work was supported by grants from the Canadian Institute of Health Research (CIHR) project grant PJT‐156087 to VDS, and NSERC discovery grant RGPIN‐2015‐06774 to VDS.
EMBO Reports (2020) 21: e49499
Data availability
The primary ChIP‐Seq and RNA‐Seq data reported in this paper are deposited in NCBI and available through Geo Accession numbers GSE123836, GSE80588, GSE158487, and GSE133505.
References
- Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L, Ren J, Li WW, Noble WS (2009) MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res 37: W202–W208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentzinger CF, Wang YX, Rudnicki MA (2012) Building muscle: molecular regulation of myogenesis. Cold Spring Harb Perspect Biol 4: a008342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkes CA, Tapscott SJ (2005) MyoD and the transcriptional control of myogenesis. Semin Cell Dev Biol 16: 585–595 [DOI] [PubMed] [Google Scholar]
- Blackburn DM, Lazure F, Corchado AH, Perkins TJ, Najafabadi HS, Soleimani VD (2019) High‐resolution genome‐wide expression analysis of single myofibers using SMART‐Seq. J Biol Chem 294: 20097–20108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boonsanay V, Zhang T, Georgieva A, Kostin S, Qi H, Yuan X, Zhou Y, Braun T (2016) Regulation of skeletal muscle stem cell quiescence by Suv4‐20 h1‐dependent facultative heterochromatin formation. Cell Stem Cell 18: 229–242 [DOI] [PubMed] [Google Scholar]
- Braun T, Bober E, Rudnicki MA, Jaenisch R, Arnold HH (1994) MyoD expression marks the onset of skeletal myogenesis in Myf‐5 mutant mice. Development 120: 3083–3092 [DOI] [PubMed] [Google Scholar]
- Buckingham M (1994) Muscle differentiation. Which myogenic factors make muscle? Curr Biol 4: 61–63 [DOI] [PubMed] [Google Scholar]
- Buckingham M, Rigby PW (2014) Gene regulatory networks and transcriptional mechanisms that control myogenesis. Dev Cell 28: 225–238 [DOI] [PubMed] [Google Scholar]
- Charge SB, Rudnicki MA (2004) Cellular and molecular regulation of muscle regeneration. Physiol Rev 84: 209–238 [DOI] [PubMed] [Google Scholar]
- Comai G, Tajbakhsh S (2014) Molecular and cellular regulation of skeletal myogenesis. Curr Top Dev Biol 110: 1–73 [DOI] [PubMed] [Google Scholar]
- Fabregat A, Jupe S, Matthews L, Sidiropoulos K, Gillespie M, Garapati P, Haw R, Jassal B, Korninger F, May B et al (2018) The reactome pathway knowledgebase. Nucleic Acids Res 46: D649–D655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fry CS, Kirby TJ, Kosmac K, McCarthy JJ, Peterson CA (2017) Myogenic progenitor cells control extracellular matrix production by fibroblasts during skeletal muscle hypertrophy. Cell Stem Cell 20: 56–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ignatiadis N, Klaus B, Zaugg JB, Huber W (2016) Data‐driven hypothesis weighting increases detection power in genome‐scale multiple testing. Nat Methods 13: 577–580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin RM, Warunek J, Wohlfert EA (2018) Chronic infection stunts macrophage heterogeneity and disrupts immune‐mediated myogenesis. JCI Insight 3: e121549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joe AW, Yi L, Natarajan A, Le Grand F, So L, Wang J, Rudnicki MA, Rossi FM (2010) Muscle injury activates resident fibro/adipogenic progenitors that facilitate myogenesis. Nat Cell Biol 12: 153–163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kassar‐Duchossoy L, Gayraud‐Morel B, Gomes D, Rocancourt D, Buckingham M, Shinin V, Tajbakhsh S (2004) Mrf4 determines skeletal muscle identity in Myf5: myod double‐mutant mice. Nature 431: 466–471 [DOI] [PubMed] [Google Scholar]
- Kim D, Langmead B, Salzberg SL (2015) HISAT: a fast spliced aligner with low memory requirements. Nat Methods 12: 357–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lepper C, Partridge TA, Fan CM (2011) An absolute requirement for Pax7‐positive satellite cells in acute injury‐induced skeletal muscle regeneration. Development 138: 3639–3646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, Shi W (2014) featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30: 923–930 [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biol 15: 550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews VB, Astrom MB, Chan MH, Bruce CR, Krabbe KS, Prelovsek O, Akerstrom T, Yfanti C, Broholm C, Mortensen OH et al (2009) Brain‐derived neurotrophic factor is produced by skeletal muscle cells in response to contraction and enhances fat oxidation via activation of AMP‐activated protein kinase. Diabetologia 52: 1409–1418 [DOI] [PubMed] [Google Scholar]
- McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, Wenger AM, Bejerano G (2010) GREAT improves functional interpretation of cis‐regulatory regions. Nat Biotechnol 28: 495–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moretti I, Ciciliot S, Dyar KA, Abraham R, Murgia M, Agatea L, Akimoto T, Bicciato S, Forcato M, Pierre P et al (2016) MRF4 negatively regulates adult skeletal muscle growth by repressing MEF2 activity. Nat Commun 7: 12397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz‐Canoves P, Scheele C, Pedersen BK, Serrano AL (2013) Interleukin‐6 myokine signaling in skeletal muscle: a double‐edged sword? FEBS J 280: 4131–4148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadeau L, Aguer C (2019) Interleukin‐15 as a myokine: mechanistic insight into its effect on skeletal muscle metabolism. Appl Physiol Nutr Metab 44: 229–238 [DOI] [PubMed] [Google Scholar]
- Olson EN, Arnold HH, Rigby PW, Wold BJ (1996) Know your neighbors: three phenotypes in null mutants of the myogenic bHLH gene MRF4. Cell 85: 1–4 [DOI] [PubMed] [Google Scholar]
- Pal M, Febbraio MA, Whitham M (2014) From cytokine to myokine: the emerging role of interleukin‐6 in metabolic regulation. Immunol Cell Biol 92: 331–339 [DOI] [PubMed] [Google Scholar]
- Pasut A, Oleynik P, Rudnicki MA (2012) Isolation of muscle stem cells by fluorescence activated cell sorting cytometry. Methods Mol Biol 798: 53–64 [DOI] [PubMed] [Google Scholar]
- Pedersen BK, Febbraio MA (2012) Muscles, exercise and obesity: skeletal muscle as a secretory organ. Nat Rev Endocrinol 8: 457–465 [DOI] [PubMed] [Google Scholar]
- Quinn LS, Anderson BG, Conner JD, Wolden‐Hanson T (2013) IL‐15 overexpression promotes endurance, oxidative energy metabolism, and muscle PPARdelta, SIRT1, PGC‐1alpha, and PGC‐1beta expression in male mice. Endocrinology 154: 232–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rayagiri SS, Ranaldi D, Raven A, Mohamad Azhar NIF, Lefebvre O, Zammit PS, Borycki AG (2018) Basal lamina remodeling at the skeletal muscle stem cell niche mediates stem cell self‐renewal. Nat Commun 9: 1075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segalés J, Perdiguero E, Muñoz‐Cánoves P (2016) Regulation of muscle stem cell functions: a focus on the p38 MAPK signaling pathway. Front Cell Dev Biol 4: 91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siles L, Sanchez‐Tillo E, Lim JW, Darling DS, Kroll KL, Postigo A (2013) ZEB1 imposes a temporary stage‐dependent inhibition of muscle gene expression and differentiation via CtBP‐mediated transcriptional repression. Mol Cell Biol 33: 1368–1382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slenter DN, Kutmon M, Hanspers K, Riutta A, Windsor J, Nunes N, Melius J, Cirillo E, Coort SL, Digles D et al (2018) WikiPathways: a multifaceted pathway database bridging metabolomics to other omics research. Nucleic Acids Res 46: D661–D667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soleimani VD, Yin H, Jahani‐Asl A, Ming H, Kockx CE, van Ijcken WF, Grosveld F, Rudnicki MA (2012) Snail regulates MyoD binding‐site occupancy to direct enhancer switching and differentiation‐specific transcription in myogenesis. Mol Cell 47: 457–468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soleimani VD, Palidwor GA, Ramachandran P, Perkins TJ, Rudnicki MA (2013) Chromatin tandem affinity purification sequencing. Nat Protoc 8: 1525–1534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soleimani VD, Nguyen D, Ramachandran P, Palidwor GA, Porter CJ, Yin H, Perkins TJ, Rudnicki MA (2018) Cis‐regulatory determinants of MyoD function. Nucleic Acids Res 46: 7221–7235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southard S, Low S, Li L, Rozo M, Harvey T, Fan CM, Lepper C (2014) A series of Cre‐ER(T2) drivers for manipulation of the skeletal muscle lineage. Genesis 52: 759–770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES et al (2005) Gene set enrichment analysis: a knowledge‐based approach for interpreting genome‐wide expression profiles. Proc Natl Acad Sci USA 102: 15545–15550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanabe M, Kanehisa M (2012) Using the KEGG database resource. Curr Protoc Bioinformatics 10.1002/0471250953.bi0112s38 [DOI] [PubMed] [Google Scholar]
- Verma M, Asakura Y, Murakonda BSR, Pengo T, Latroche C, Chazaud B, McLoon LK, Asakura A (2018) Muscle satellite cell cross‐talk with a vascular niche maintains quiescence via VEGF and notch signaling. Cell Stem Cell 23: 530–543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Zhao W, Ransohoff RM, Zhou L (2018) Infiltrating macrophages are broadly activated at the early stage to support acute skeletal muscle injury repair. J Neuroimmunol 317: 55–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YX, Feige P, Brun CE, Hekmatnejad B, Dumont NA, Renaud JM, Faulkes S, Guindon DE, Rudnicki MA (2019) EGFR‐aurka signaling rescues polarity and regeneration defects in dystrophin‐deficient muscle stem cells by increasing asymmetric divisions. Cell Stem Cell 24: 419–432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitham M, Chan MH, Pal M, Matthews VB, Prelovsek O, Lunke S, El‐Osta A, Broenneke H, Alber J, Bruning JC et al (2012) Contraction‐induced interleukin‐6 gene transcription in skeletal muscle is regulated by c‐Jun terminal kinase/activator protein‐1. J Biol Chem 287: 10771–10779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin H, Price F, Rudnicki MA (2013) Satellite cells and the muscle stem cell niche. Physiol Rev 93: 23–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W et al (2008) Model‐based analysis of ChIP‐Seq (MACS). Genome Biol 9: R137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu H, Xiao F, Wang G, Wei X, Jiang L, Chen Y, Zhu L, Wang H, Diao Y, Wang H et al (2016) STAT3 regulates self‐renewal of adult muscle satellite cells during injury‐induced muscle regeneration. Cell Rep 16: 2102–2115 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Dataset EV1
Dataset EV2
Source Data for Expanded View
Review Process File
Data Availability Statement
The primary ChIP‐Seq and RNA‐Seq data reported in this paper are deposited in NCBI and available through Geo Accession numbers GSE123836, GSE80588, GSE158487, and GSE133505.