

Abstract
Several human pathologies including neurological, cardiac, infectious, cancerous, and metabolic diseases have been associated with altered mitochondria morphodynamics. Here, we identify a small organic molecule, which we named Mito‐C. Mito‐C is targeted to mitochondria and rapidly provokes mitochondrial network fragmentation. Biochemical analyses reveal that Mito‐C is a member of a new class of heterocyclic compounds that target the NEET protein family, previously reported to regulate mitochondrial iron and ROS homeostasis. One of the NEET proteins, NAF‐1, is identified as an important regulator of mitochondria morphodynamics that facilitates recruitment of DRP1 to the ER–mitochondria interface. Consistent with the observation that certain viruses modulate mitochondrial morphogenesis as a necessary part of their replication cycle, Mito‐C counteracts dengue virus‐induced mitochondrial network hyperfusion and represses viral replication. The newly identified chemical class including Mito‐C is of therapeutic relevance for pathologies where altered mitochondria dynamics is part of disease etiology and NEET proteins are highlighted as important therapeutic targets in anti‐viral research.
Keywords: contact sites, mitochondria, morphodynamics, NEET proteins, virus
Subject Categories: Membrane & Intracellular Transport; Microbiology, Virology & Host Pathogen Interaction
This study identifies the small molecule Mito‐C as an activator of mitochondrial fragmentation. Mito‐C suppresses dengue virus replication by counteracting virus induced mitochondrial fusion.
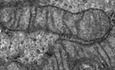
Introduction
Mitochondria are double membrane organelles essential for energy homeostasis in eukaryotes but also critical for regulating iron and calcium homeostasis, redox regulation, autophagy, innate immunity, and cell death (Galluzzi et al, 2012). A broad range of diseases, including viral infections, have been associated with irregular mitochondria morphology and dynamics, highlighting the central role of mitochondrial network dynamics in the maintenance of cellular homeostasis (Zemirli et al, 2018; Giacomello et al, 2020). Regulation of mitochondrial morphology is a consequence of a balance between fission and fusion events that maintain mitochondria number, size, and shape (Tilokani et al, 2018). This dynamic equilibrium enables adaptation to a wide range of stress situations. Moreover, mitochondria are physically bound to the endoplasmic reticulum (ER), via ER‐Mitochondria contact sites (ER‐MTcs). ER‐MTcs engage multiple proteins, from both ER and mitochondria, and are involved in a wide range of cellular functions, including autophagosome biogenesis and mitochondrial fission. A critical regulator of the mitochondrial fission process is the cytosolic GTPase dynamin‐related protein 1 (DRP1), which relocates to the mitochondrial surface to promote mitochondrial network fragmentation (Smirnova et al, 1998). Interestingly, DRP1 recruitment to ER‐MTcs defines the position of the mitochondrial division site (Friedman et al, 2011), although how this directed recruitment occurs still remains unclear.
In addition to proteins involved in fission/fusion regulation, many other proteins of critical importance to cell function such as the respiratory chain, innate immunity, redox regulation, and iron homeostasis are located on or in the mitochondria. Among these mitochondria‐associated proteins are members of the NEET family which regulate iron and reactive oxygen species (ROS) homeostasis in the mitochondria (Wiley et al, 2013; Tamir et al, 2015; Karmi et al, 2018; Mittler et al, 2019 the Unfolded Protein Response, and Ca2+ Homeostasis). There are three NEET proteins: MitoNEET, NAF‐1, and MiNT, and they are reported to localize at the mitochondria matrix (MiNT), the outer‐mitochondrial membrane (MitoNEET and NAF‐1), and the Endoplasmic Reticulum (NAF‐1). Their unique ability to reversibly bind the redox active [2Fe‐2S] cluster enables their function as iron–sulfur transfer proteins, transferring [2Fe‐2S] clusters out of the mitochondria (Tamir et al, 2015; Mittler et al., 2019). MitoNEET has been associated with the regulation of oxidative phosphorylation in mitochondria (Wang et al, 2017), and deletion of its gene (CISD1) alters the integrity of inter‐mitochondrial junctions (Vernay et al, 2017). In human, frame shift mutations in the CISD2 gene encoding NAF‐1 result in the autosomal recessive disorder Wolfram Syndrome Type 2 (WFS2), characterized at the cellular level by mitochondria dysfunctions, iron accumulation in mitochondria, increased autophagy, and cell death. In contrast to MitoNEET and NAF‐1, MiNT (encoded by CISD3 gene) is less characterized. Recently, the crystal structure of MiNT has been resolved and its contribution to tumorigenesis explored, especially in correlation with iron and ROS homeostasis in cancer cells (Lipper et al, 2018). Despite the numerous studies connecting NEET proteins to different mitochondrial disorders, no functional connection has been made to infectious diseases.
Here we find that Mito‐C, a compound not previously described, targets the NEET proteins family, modifies mitochondrial morphology and dynamics by stabilizing ER‐MTcs, and is able to counteract dengue virus‐induced mitochondria network hyperfusion and to strongly reduce Dengue virus replication. Furthermore, via pharmacological and genetic approaches we here also reveal the central role of NEET proteins in regulation of mitochondrial membrane dynamics via the ER‐mitochondria‐associated fission machinery.
Results and Discussion
We synthesized a novel small molecule, Mito‐C (2‐[(3,4‐Dimethoxybenzoyl)amino]‐6,6‐dimethyl‐5,7‐dihydro‐4H‐benzothiophene‐3‐carboxylic acid, Fig 1A) that was selected for its ability to affect the morphology of the mitochondrial network without adverse impact upon cell viability. The mitochondrial morphology modification was quantified using a phenotype‐based counting method, to compare elongated and fragmented mitochondria ratio. Mito‐C rapidly induces mitochondrial network fragmentation (Fig EV1A) in a dose‐dependent manner (Fig EV1B–D), with no evidence of cell death following much longer treatment (24 h) at higher concentrations (Appendix Fig S1A–C). The Mito‐C‐associated mitochondrial fragmentation observed was reversible, with cells recovering classical mitochondria following wash out and overnight culture (Appendix Fig S1D and E).
Figure 1. Mito‐C, a new chemical compound targeting NEETs proteins, induces mitochondrial network fragmentation.
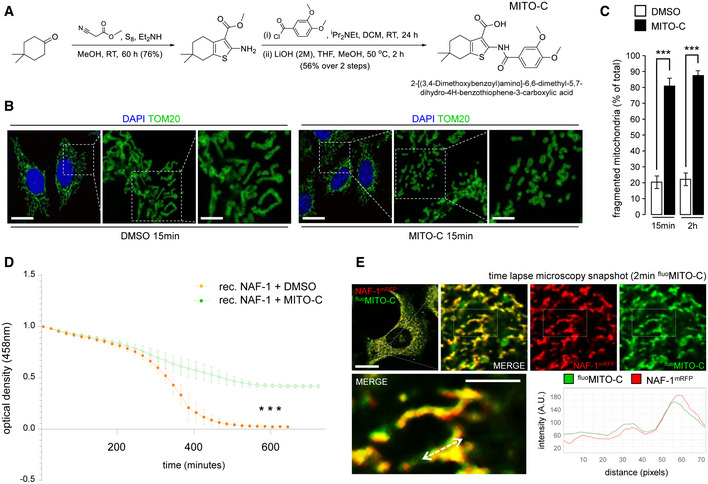
- Reaction scheme for synthesis of Mito‐C and structure of the compound.
- HeLa cells were treated with DMSO or 2 µM Mito‐C for 15 min and immunostained with anti‐TOM20 (green) antibody and DAPI; cropped areas show the mitochondria morphology changes.
- Quantification of mitochondria morphology (based on TOM20 immunostaining as shown in B) from HeLa cells treated with DMSO or 2 µM Mito‐C for 15 min or 2 h, expressed as ratio of fragmented mitochondria (percentage of total). Errors bars show the standard error of the mean (SEM) (n = 100, technical replicates).
- Profile of [2Fe–2S] cluster release from recombinant NAF‐1 (expressed and purified according to the procedure described in (Conlan et al, 2009)) was determined in untreated control (DMSO) or in presence of Mito‐C by monitoring absorbance at 458 nm as a function of time. Errors bars show the standard deviation (SD) of 3 independent experiments.
- Time‐lapse video‐microscopy snapshot on HeLa cells transfected with NAF‐1‐mRFP (red) and treated with fluoMito‐C (green). The distance/intensity fluorescence quantification graph illustrates the codistribution of fluoMito‐C and NAF‐1‐mRFP.
Data information: All scale bars = 10 µm, except magnifications areas (4 µm in B and 2 µm in E). For evaluating significance of differences observed in C, t‐test was used (*** indicates P < 0.0001); for differences observed in D, one‐way ANOVA followed by Dunn’s post‐test was used (*** indicates P < 0.0001).
Figure EV1. Time‐ and dose‐dependent mitochondrial fragmentation induced by Mito‐C.
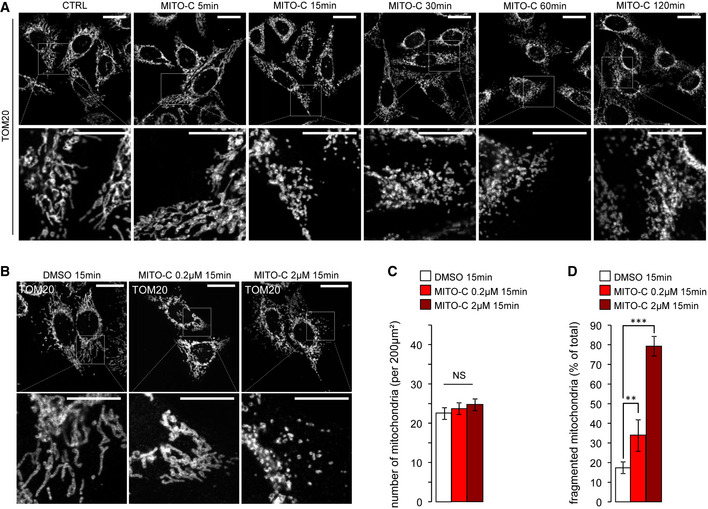
- HeLa cells were treated with 2 µM Mito‐C for the indicated times, fixed and immunostained for TOM20, and analyzed by light microscopy.
- HeLa cells were treated with increasing concentrations of Mito‐C for 15 min, fixed and immunostained for TOM20, and analyzed by light microscopy.
- Quantification of mitochondria number (based on TOM20 immunostaining as shown in b, n = 30). Errors bars show the standard error of the mean (SEM).
- Quantification of mitochondria phenotype morphology (based on TOM20 immunostaining as shown in b, n = 45–50). Errors bars show the standard error of the mean (SEM).
Data information: To evaluate significance of differences observed in C, one‐way ANOVA followed by Bonferroni’s post‐test was used (** indicates P < 0.001 and ***P < 0.0001). Scale bars = 10 µm, except 5 µm in magnification areas in A and B.
To determine the protein binding target of Mito‐C, we used a photo‐affinity labeling‐based method (Capture Compound Mass Spectrometry) to identify the cellular target(s) of Mito‐C. Mito‐C was synthesized with a photo‐reactive function for covalent cross‐linking to bound protein(s) and a sorting function for isolation of bound target and identification by mass spectrometric analysis (Köster et al, 2007). This approach revealed interaction of the capture compound with five proteins including the NEET protein MiNT (Appendix Fig S2A). MiNT is the only protein localized at the mitochondria and was thus selected for validation tests. Interestingly, capture experiment in the presence of an excess of “free” compound dramatically prevents MiNT pull down demonstrating the specificity of Mito‐C binding to MiNT (Appendix Fig S2B). To examine the functional effect of Mito‐C upon NEET proteins, possible effect upon the rate of release of the [2Fe‐2S] cluster from each purified NEET protein was tested with or without (DMSO) Mito‐C. As shown in Fig 1D (for NAF‐1) and Fig EV2 (for MitoNEET and MiNT) the presence of Mito‐C significantly enhanced the stability of NAF1, MitoNEET, and MiNT for their bound [2Fe‐2S] cluster thus in effect inhibiting their [2Fe‐2S] transfer function. Such a functional test confirms the physical binding of Mito‐C to NEET proteins.
Figure EV2. Mito‐C impacts Fe‐S clusters release from MitoNEET and MiNT proteins.
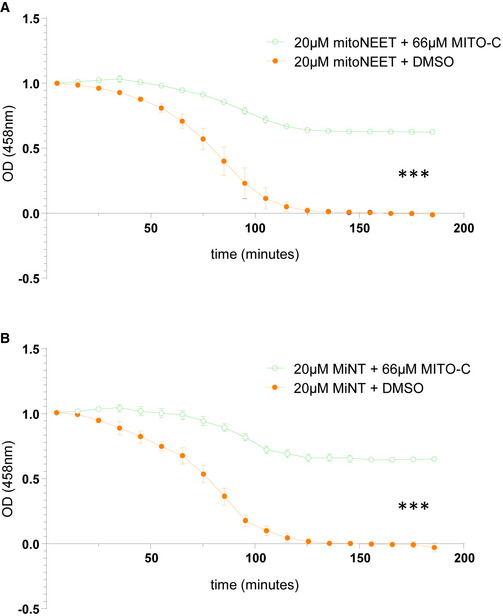
- Profile of [2Fe–2S] clusters release from purified recombinant MitoNEET and MiNT were determined in presence or absence (DMSO) of Mito‐C via monitoring their absorbance at 458 nm as a function of time. Errors bars show the standard deviation (SD) of 3 independent experiments.
Data information: For differences observed in both A and B, one‐way ANOVA followed by Dunn’s post‐test was used (N = 3, *** indicates P < 0.0001).
MiNT and MitoNEET have both been reported to localize exclusively to the mitochondrial matrix and outer membrane, respectively (Wiley et al, 2007). However, NAF1 has been reported to localize to the ER (Amr et al, 2007; Wiley et al., 2007; Appendix Fig S3A), to the mitochondria‐associated membranes (MAMs) and MT outer membranes (Chen et al, 2009) and Appendix Fig S3B). We now show that NAF‐1 is localized at ER and ER‐MT interface subdomains (Appendix Fig S3C) using PTPIP51 protein as a reporter of mitochondria membrane at the interface of the ER (Helle et al, 2013; Gomez‐Suaga et al, 2017). To strengthen the hypothesis that Mito‐C elicits its effects in cells by modulating NEET protein function(s), a fluorescently tagged version of Mito‐C (fluoMito‐C) was generated by linking the 7‐nitrobenzofurazan green fluorescent tag on the para position of the right hand phenol ring (see Fig 1A). Time‐lapse microscopy imaging revealed that fluoMito‐C targets both mitochondria (MT) and Endoplasmic Reticulum (ER) as illustrated by the colocalization between fluoMito‐C and the MT live probe MitoTracker RedOx or the ER marker Sec61β‐RFP (Appendix Fig S4A and B). The codistribution observed by time‐lapse microscopy between the NAF1‐RFP and fluoMito‐C further indicates that the NEET proteins and Mito‐C are localized to the same cellular compartments in living cells (Fig 1E). Altogether, our data indicate that Mito‐C targets NEET proteins by, at least, inhibiting their [2Fe‐2S] transfer function and initiates immediate and reversible MT network fragmentation.
To address whether the MT‐induced fragmentation correlates with impairment of the energetic capacity of MT, we measured the mitochondrial oxygen consumption rate (OCR), the MT transmembrane potential, and dynamic bioenergetics mitochondrial parameters by high‐resolution respirometry (HRR) on cells treated with Mito‐C. While we observed partial mitochondrial cristae disorganization in cells treated with Mito‐C (Fig EV3A), no significant changes in MT respiration or membrane potential were observed (Fig EV3B–D). Furthermore, no effect on MT mass was detected (Fig EV3E and F). Importantly, our results showed as well no change in the different bioenergetics parameters assessed by HRR, which included routine respiration, ATP‐linked respiration (oligomycin‐sensitive), uncoupled respiration (CCCP‐induced), and spare respiratory capacity (Fig EV3G). Furthermore, no change in cell viability was observed in galactose (glucose‐deprived) (Rossignol et al, 2004) medium (Fig EV3H), indicating a valid oxidative phosphorylation (OXPHOS) system. These findings suggest that Mito‐C is a novel not mitotoxic mitochondrial morphology modulator.
Figure EV3. Mitochondrial characteristics of Mito‐C treated cells.
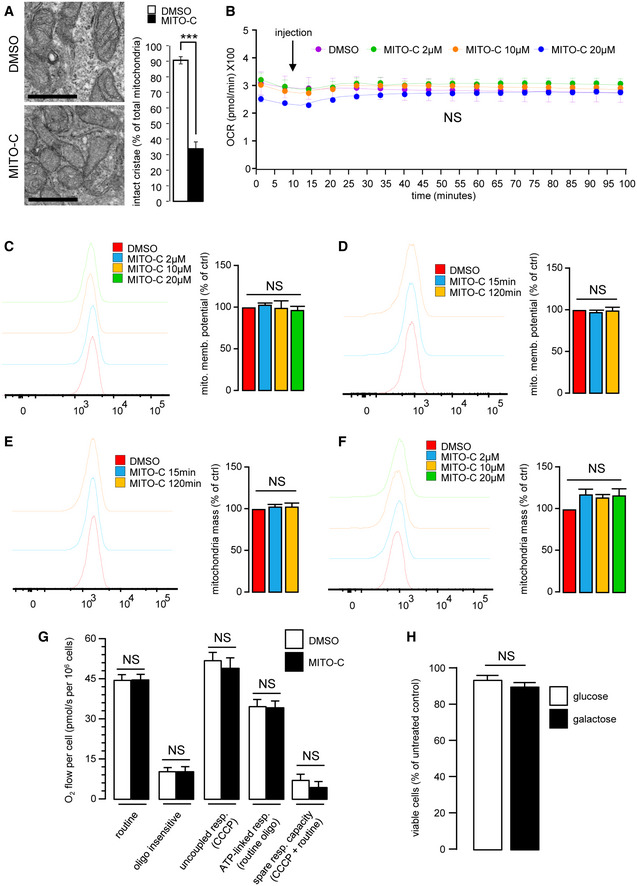
- HeLa cells were treated with 2 µM Mito‐C or DMSO for 24 h. Cells were fixed and prepared for electron microscopy analyses (left panel). Intact mitochondrial cristae were quantified (right panel). Errors bars show the standard deviation (SD) of 3 independent experiments.
- Oxygen consumption rate (Viard et al.) of cells treated with Mito‐C (2, 10 and 20µM) were measured by Seahorse© technique. Measurements start before starting the treatment, arrow indicates the Mito‐C injection. Graphs show the standard deviation (SD) of 3 independent experiments.
- To evaluate the mitochondrial potential, cells were treated with Mito‐C for 24 h at the indicated range of concentrations and stained with MitoTracker Redox and analyzed by cytometry. Graphs show the standard deviation (SD) of 3 independent experiments.
- To evaluate the mitochondrial potential, cells were treated with Mito‐C at 2 µM over a time course and then stained with MitoTracker Redox and analyzed by cytometry. Graphs show the standard deviation (SD) of 3 independent experiments.
- To evaluate the total mitochondrial mass over a time course of treatment, HeLa cells were treated with DMSO or Mito‐C at 2 µM for the time duration indicated, stained with MitoTracker green and analyzed by cytometry. Graphs show the standard deviation (SD) of 3 independent experiments.
- To evaluate the total mitochondrial mass at a fixed time point following treatment with an increasing range of concentrations of Mito‐C cells were treated with DMSO or Mito‐C for 24 h at concentration indicated, stained with MitoTracker green and analyzed by cytometry. Graphs show the standard deviation (SD) of 3 independent experiments.
- High‐resolution respirometry (HRR) evaluation of HeLa cells treated for 15 min with 2 µM Mito‐C. Different bioenergetics parameters were analyzed: routine respiration, oligomycin‐sensitive, and CCCP‐sensitive. The term "routine" respiration is defined as the respiratory rate of intact cells measured in 5 mM glucose DMEM under atmospheric conditions at 37°C and sensitive to 2.5 µM antimycin A inhibition. The term "oligo insensitive" respiration is the respiratory rate measured in the routine conditions after addition of the F1F0‐ATP synthase inhibitor oligomycin at 20 µg/ml. This "oligomycin" state of respiration does not depend on ADP phosphorylation. The ATP‐linked respiration is calculated as the difference between the routine and the oligomycin‐sensitive. The term "uncoupled" respiration defines the rate of respiration measured in the "oligo" conditions after addition of the uncoupler CCCP used at 2 µM. The "CCCP" state allows evaluating the maximal capacity of the respiratory chain in presence of energy substrates and oxygen concentration as defined in the "routine" conditions. The ATP‐linked respiration (routine‐oligo) indicates the part of respiration used for ATP synthesis and the spare respiratory capacity (CCCP‐routine) gives a measure of the capacity of the respiratory chain to be chemically uncoupled. It indicates how far from the maximal capacity the routine respiration operates. Bars show the standard deviation (SD) of 3 independent experiments.
- Comparison of cell viability from HeLa cells grown on high glucose (25 mM) (open bar) versus cells grown on galactose (10 mM) (solid bar) treated with 2 µM Mito‐C for 24 h. Data are expressed as mean ± SEM of N = 3 biological replicates.
Data information: For evaluating significance of differences observed in A, t‐test was used (*** indicates P < 0.0001). Scale bar in A = 200 nm.
To correlate the contribution of NEET proteins to the observed effects of Mito‐C, we analyzed the impact of siRNA‐mediated knockdown of each NEET protein transcript (Fig 2A–C) on MT morphology (Fig 2D). While knockdown of mitoNEET (siCISD1) protein does not induce significant alteration of MT morphology, reducing both NAF‐1 (siCISD2) or MiNT (siCISD3) protein expression caused a significant MT fragmentation (Fig 2E). To better assess the role of NAF‐1 protein in MT morphodynamics, we analyzed the MT network in cells stably expressing shRNA designed to reduce NAF‐1 expression (Fig 2G). Electron microscopy analysis shows an increase in MT fragmentation when NAF‐1 protein is reduced, and this effect is rescued upon restoration of NAF‐1 protein expression (Fig 2F–H).
Figure 2. Knocking down NEETs causes mitochondrial fragmentation.
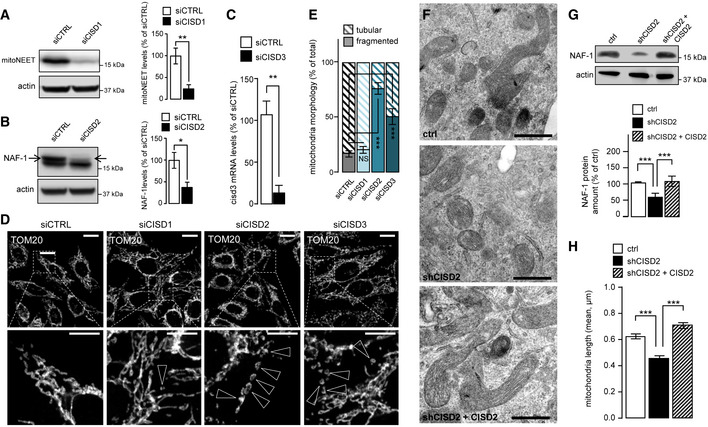
- Western blot analysis of MitoNEET expression in HeLa cells transfected with CISD1 siRNA; Bar chart (right panel) shows replicates quantification of MitoNEET expression. Errors bars show the standard deviation (SD) of 3 independent experiments.
- Western blot analysis of NAF‐1 expression in HeLa cells transfected with CISD2 siRNA; graph (right panel) shows the replicates quantification of NAF‐1 expression. Errors bars show the standard deviation (SD) of 3 independent experiments.
- RT–qPCR analysis of CISD3 mRNA levels from HeLa cells transfected with siRNA targeting CISD3. Errors bars show the standard deviation (SD) of 9 independent experiments.
- HeLa cells transfected with siRNA to reduce expression of MitoNEET (CISD1), NAF‐1 (CISD2), or MiNT (CISD3) and immunostained with anti TOM20 antibody. Arrowheads indicate fragmented mitochondria.
- Quantification of mitochondria morphology (based on TOM20 immunostaining as shown in D) from HeLa cells transfected with siRNA targeting mitoNEET (siCISD1), NAF1 (siCISD2), or MiNT (siCISD3). Quantification is expressed as ratio of tubular and fragmented mitochondria. Errors bars show the standard error of the mean (SEM). (n = 125–130 cells).
- Electron microscopy pictures from 150 INS‐1 cells transfected as described in G.
- Western blot analysis of NAF‐1 expression in cells stably transfected with control shRNA, shRNA‐targeting reduction in NAF‐1 protein expression (shCISD2), or shRNA‐targeting reduction in NAF‐1 protein expression complemented with plasmid derived expression of NAF‐1 (shCISD2 + CISD2); Western blot quantification of NAF‐1 is shown on the accompanying bar chart. Errors bars show the standard deviation (SD) of 3 independent experiments.
- Quantification of mitochondrial length in EM images from cells transfected as described in G. Errors bars show the standard error of the mean (SEM) (n = 260–270, technical replicates).
Data information: To evaluate significant differences observed in A, B, C, and G, a t‐test was used (for A ** indicates P < 0.001; for B *P is 0.045; C **P is 0.005; for G ***P is 0.0001); for differences observed in D and E, one‐way ANOVA followed by Bonferroni’s post‐test was used (*** indicates P < 0.0001); in H, one‐way ANOVA followed by Dunn’s post‐test was used (*** indicates P < 0.0001). Immunofluorescence scale bars = 10 µm, except 4 µm in magnification area in D. Scale bars from EM analyses = 0.5 µm.
During mitochondrial fission, the dynamin‐related protein DRP1 is recruited to ER‐MTcs to trigger the fission process (Smirnova et al, 1998; Chen et al., 2009), but the mechanisms that result in its enrichment and/or stability at mitochondria fission sites and at the ER‐MTcs remain poorly understood. Interestingly, NAF‐1 colocalizes partially with DRP1 at ER‐MT interface (as detected by presence of PTPIP51 protein, Fig EV4A). Interestingly, fluorescence image analysis shows that 15 min of Mito‐C treatment leads to increased recruitment of DRP1 at mitochondrial surface (Fig 3A and B). This observation was confirmed by subcellular fractionation experiments (Fig 3C and D), illustrating that a short‐time treatment of Mito‐C induces DRP1 accumulation at MT. Time course analysis of DRP1 expression following Mito‐C treatment also showed an increase in total DRP1 protein (Appendix Fig S5A and B), without any observed changes in phosphorylation at Serine 616 (DRP1S616), (Appendix Fig S5A and C) a post‐translational modification known to promote mitochondrial fission during mitosis (Taguchi et al, 2007; Liesa et al, 2009). To assess whether the effects of Mito‐C on mitochondrial morphology were directly connected to DRP1 mitochondrial recruitment and function, we co‐expressed a dominant negative version of DRP1 (drp1K38A) and mCherry, to discriminate between transfected and non‐transfected cells. Expression of drp1K38A is known to inhibit mitochondria fission (Smirnova et al, 1998; Zemirli et al, 2014). We observed that Mito‐C treatment causes the expected fragmented MT phenotype in non‐transfected cells (NT, Fig 3E and F), while this effect was no longer observed in cells expressing drp1K38A, which rather resulted in a hyperfused mitochondrial network both in the presence and absence of Mito‐C (Fig 3E and F). These data demonstrate that Mito‐C modifies MT fission in a manner dependent on DRP1 recruitment at mitochondrial membrane, presumably at ER‐MTcs subdomains.
Figure EV4. MITO‐C impact on NAF1 levels and subcellular localization.
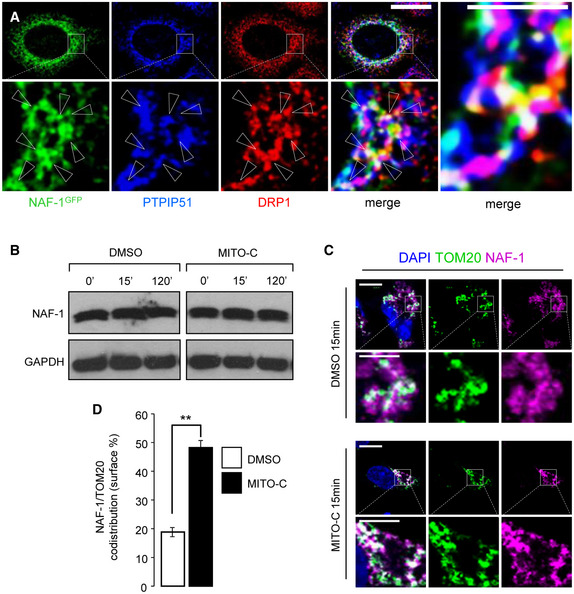
- HeLa cells were transfected with NAF‐1‐GFP plasmid (green) and immunostained with antibodies to PTPIP51 (Blue) and DRP1 (red); three color merged image is shown in the far‐right panel with arrowheads indicating the white triple colocalization domains.
- Western blot analysis of NAF‐1 and GAPDH proteins expression in total extracts from HeLa cells treated with 2 µM Mito‐C or DMSO for the indicated times.
- HeLa cells were treated with DMSO or 2 µM Mito‐C for 15 min and immunostained with anti‐TOM20 (green) and anti‐NAF‐1 (fuchsia) antibodies and DAPI (Blue).
- Quantification of NAF‐1 signal on TOM20 positive structures. Errors bars show the standard error of the mean (SEM) (n = 75 cells from 3 independent experiments).
Data information: In D, one‐way ANOVA followed by Dunn’s post‐test was used (** indicates P < 0.005). scale bars = 10 µm, except 4 µm in magnification area in A and in C.
Figure 3. Mito‐C causes DRP1‐dependent mitochondrial fission and ER‐mitochondria contact‐sites increase.
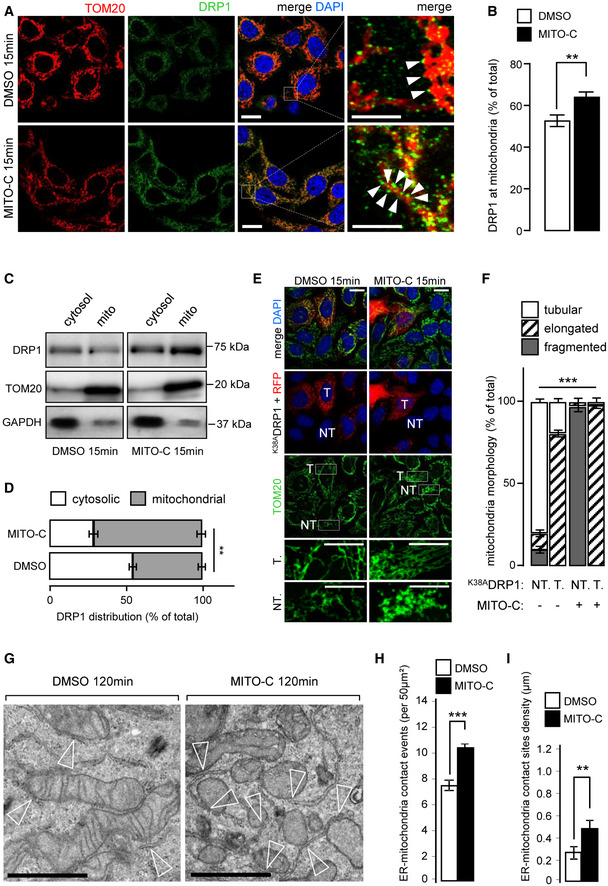
- HeLa cells treated or not, with 2 µM Mito‐C for 15 min were immunostained with anti‐TOM20 (red) and anti‐DRP1 (green); arrowheads in the far‐right panel indicate recruitment of DRP1 onto the mitochondrial surface (TOM20).
- Quantification of DRP1 signal on TOM20 positive structures. Errors bars show the standard error of the mean (SEM) (n = 45, technical replicates).
- Western blot analysis of DRP1, GAPDH (cytosolic marker), and TOM20 (mitochondrial marker) protein in cytosolic and mitochondrial fractions treated or not, with 2 µM Mito‐C as indicated.
- Quantification of Western blots showed in C and expressed as a distribution of DRP1 in the cytosolic and mitochondrial fractions. Errors bars show the standard deviation (SD) of 5 independent experiments.
- HeLa cells were transfected with DRP1K38A mutant and mCherry and treated or not, with Mito‐C (T for transfected, NT for not transfected).
- Quantification of the mitochondrial phenotypes observed in E. Errors bars show the standard error of the mean (SEM) (30–35 images each with an average of 15–20 cells from triplicate independent experiments were analyzed).
- Electron micrographs (EM) from HeLa cells treated with 2 µM Mito‐C or DMSO for 120 min. Empty arrowheads indicate ER‐mitochondria contact sites.
- Quantification of ER‐mitochondria membrane contact‐sites events (reported in 50 µm2 section; 50 images from an average of 30 cells from triplicate independent experiments were analyzed). Errors bars show the standard error of the mean (SEM).
- Quantification of ER‐mitochondria membrane contact‐sites density (in µm; 30 images from an average of 30 cells from triplicate independent experiments were analyzed). Errors bars show the standard error of the mean (SEM).
Data information: To evaluate significant differences observed in D, Mann–Whitney test was used (**P is 0.003); to evaluate statistical differences shown in B, F, H, and I, a one‐way ANOVA test was used (** indicates P < 0.001 *** indicates P < 0.0001). All scale bars = 10 µm, except magnifications areas (4 µm in A and 6 µm in E). Scale bars in G = 0.5 µm.
Because ER‐MTcs and DRP1 interplay is central in DRP1‐associated MT morphology regulation (Rowland et al, 2014), we analyzed the effect of Mito‐C on ER‐MTcs density by electron microscopy. As shown in Fig 3G, we observed an increase of the ER‐MT membrane tethering events in cells treated with Mito‐C compared with control conditions (Fig 3G–I). In parallel, we show that Mito‐C did not affect NAF1 cellular levels (Fig EV4B) but promoted an increased surface codistribution of NAF1 with mitochondria (Fig EV4C and D), highlighting the expanded membrane contacts between mitochondrial and ER membranes. Altogether, these data suggest that Mito‐C treatment alters ER‐mitochondria membrane interface by positively regulating the local contact sites, in agreement with the DRP1 mobilization we observed in Mito‐C‐treated cells.
Further investigation of the Mito‐C‐stimulated MT fragmentation phenotype examined the level of isoforms of the MT‐associated membrane dynamics regulator OPA1 (optic atrophy 1 protein). Long forms of OPA1 (L‐OPA1) are associated with fusion (Duvezin‐Caubet et al, 2006; MacVicar & Langer, 2016) while shorter forms (S‐OPA1) are considered fission mediators (Anand et al, 2014). Interestingly, Mito‐C treatment induces a time‐dependent decrease in L‐OPA1 and a concomitant accumulation of S‐OPA1 further confirming that Mito‐C and thereby NAF‐1 function shifts the mitochondrial dynamic equilibrium toward a fission state (Appendix Fig S5D–F).
Taken together, our data suggest that NAF1 functional alteration by Mito‐C is correlated with DRP1‐dependent MT fission via its increased mobilization at the ER and mitochondria interface.
Several viruses are known to modulate mitochondrial morphogenesis to promote their elongation, as a necessary part of the viral replication cycle (Moreno‐Altamirano et al, 2019). These include dengue virus, human immunodeficiency virus type 1, Sendai virus, and SARS coronavirus which have been shown to promote elongation of the MT network in infected cells (Castanier et al, 2010; Shi et al, 2014; Chatel‐Chaix et al, 2016; Fields et al, 2016). The effects of Mito‐C on MT morphodynamics thus motivated an investigation into the possible effect of Mito‐C upon the replication of specific viruses. We show that Mito‐C treatment during dengue virus infection significantly reduces the viral titer by more than one log (Fig 4A) and concomitantly inhibits dengue virus‐induced MT hyperfusion (Fig 4B and C). Our results suggest that Mito‐C treatment, through effects of MT fission, counteracts replication of viruses reliant upon elongated MT morphogenesis for successful replication. Conversely, hepatitis B virus (HBV) is described to induce mitochondrial fragmentation (Kim et al, 2013). Interestingly, treatment with Mito‐C failed to impair HBV replication cycle, as indicated by the quantification of the surface antigen (HBsAg) and relaxed circular DNA (rcDNA) in the supernatant of infected cells (Fig EV5) thus providing a clue on the specificity of Mito‐C anti‐viral properties, based on mitochondrial morphodynamics phenotypes. Altogether, our data suggest that Mito‐C possesses in vitro anti‐viral properties specifically against viruses that enhance mitochondrial fusion as a necessary part of their replication cycle.
Figure 4. Mito‐C counteracts dengue virus replication.
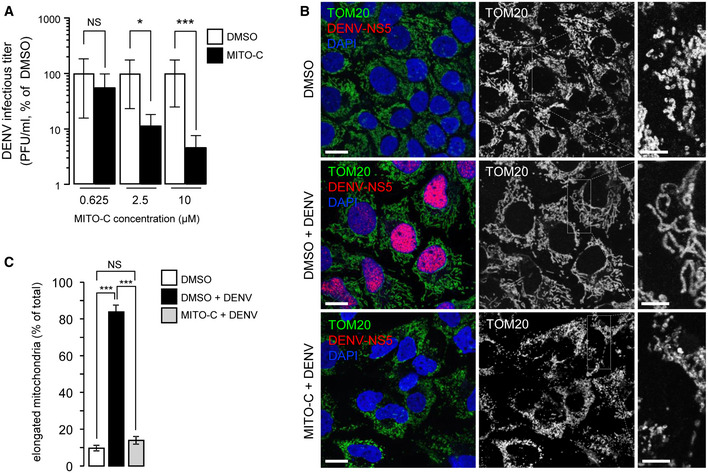
- Huh7 cells were treated with Mito‐C at the indicated concentrations and simultaneously infected with dengue virus (MOI = 5). Infectious titers are presented as % of DMSO in Mito‐C concentration of 0.625, 2.5 and 10 µM conditions, upon 48 h of viral infection. Errors bars show the standard error of the mean (SEM) (n = 3).
- Huh7 cells were treated with or without (DMSO) Mito‐C, infected with dengue virus and fixed after 48 h of infection. Cells were then immunostained for endogenous TOM20 (green channel), viral non‐structural protein NS5 (red) and DAPI (Blue). Cropped areas illustrate the mitochondrial morphology in described conditions.
- Quantification of the mitochondrial phenotypes observed in b. Errors bars show the standard error of the mean (SEM) (15–20 images each with an average of 20–25 cells from triplicate independent experiments were analyzed).
Data information: To evaluate significance of differences observed in A, we used one simple t‐test (theoretical mean of 100%, NS, for non‐significant, P = 0.0375 (> 0.2), *P = 0.0019 (< 0.01), ***P = 0.0002 (< 0.001)). To evaluate significance of differences observed in C, two‐way ANOVA followed by Bonferroni’s post‐test was used. (NS, for non‐significant, *** indicates P < 0.0001). Scale bars = 10 µm, except 4 µm in magnification area in B.
Figure EV5. Absence of MITO‐C effect on Hepatitis B virus replication.
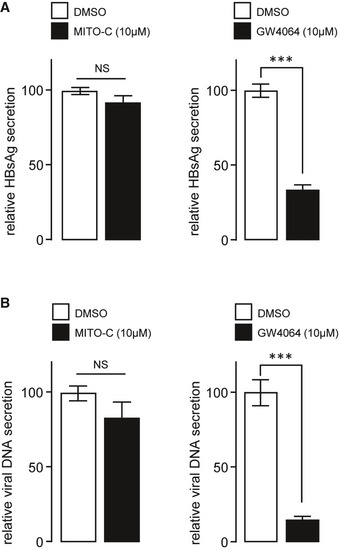
- Relative secretion of HBsAg (from Hepatitis B virus (HBV)) in dHepaRG cells treated post‐infection with 10 µM of Mito‐C or the FXR agonist GW4064. Bars show the standard deviation (SD) of 3 independent experiments.
- Relative secretion of HBV viral DNA in dHepaRG cells treated post‐infection with 10 µM of Mito‐C or GW4064 and quantified by quantitative PCR. Bars show the standard deviation (SD) of 3 independent experiments.
Data information: In A and B, one‐way ANOVA followed by Dunn’s post‐test was used (*** indicates P < 0.0001).
Here, we report a novel small molecule (Mito‐C) that targets the NEET protein family, presumably via their [2Fe‐2S] lability/cluster transfer function. Mito‐C stimulates rapid and reversible mitochondrial fragmentation and alters mitochondria cristae, without altering mitochondrial respiration and cell viability. The importance of the NEET protein NAF‐1 in Mito‐C mechanism of action is demonstrated by the fact that NAF‐1 protein expression reduction recapitulates the fragmented mitochondrial phenotype. NAF‐1 colocalizes with DRP1 at the ER‐MT contact sites, and the effect of Mito‐C on mitochondrial morphodynamics is dependent upon rapid DRP‐1 recruitment to the surface of mitochondria. Mito‐C also stimulates MT fragmentation concomitant with accumulation of S‐OPA1, a cleavage product of the fusion mediator OPA1 known to mediate MT fission. The therapeutic relevance of these observations is illustrated by the effect of Mito‐C against replication of Dengue virus reliant upon altered MT morphogenesis for successful infection.
Our present data suggest that beside mitochondrial iron level modulation, the NEET family NAF‐1 protein can participate in the regulation of DRP1 recruitment to ER/ MT interface. If and how NAF‐1 [2Fe‐2S] cluster lability/ transfer at the ER‐MT contact sites regulates mitochondrial morphogenesis remains to be elucidated. However, it is interesting to note that NEET proteins have been associated with different pathological conditions in which MT morphology as well as ER‐MT contact‐sites alterations appear to be a frequent signature: metabolic disorders, neurological, cancer (Erpapazoglou et al, 2017; Naia et al, 2017; Tubbs & Rieusset, 2017; Doghman‐Bouguerra & Lalli, 2019), and now infections. Importantly, we show here that chemical‐mediated alteration of NEET proteins' biological function via Mito‐C increases the ER‐mitochondria contact sites, a situation previously reported in patients with a mutated form of NAF‐1 (Rouzier et al, 2017) and suffering from Wolfram syndrome (Pallota et al, 2019).
Interestingly enough, NAF‐1 has also been shown to physically interact with Beclin 1, a key protein of the class III PI3K required for autophagosome biogenesis (Chang et al, 2010). Autophagosomes have been proposed to form at the ER‐MT contact sites (Hamasaki et al, 2013), and several pathological conditions, including infections, activate the autophagic pathway (Shi et al, 2014; Chatel‐Chaix et al., 2016). The chemical compound Mito‐C allows us to highlight a function of NEET proteins at the sites of MT fission machinery recruitment suggesting that these proteins may participate in different processes reliant upon ER‐MT contact‐sites machinery.
A role for mitochondrial morphodynamics, as well as ER‐MT interface regulation, in viral replication and/or host response is highlighted here by the actions of Mito‐C upon dengue virus replication, while the lack of efficacy against hepatitis B virus does not preclude the activity against a diverse array of viruses, and the observed inability of the virus to stimulate mitochondrial network elongation in treated cells. Eliciting an anti‐viral effect by targeting human NEET protein‐specific functions at mitochondrial membrane interface opens important new perspectives in anti‐viral research. Interestingly, ER‐MT contact‐sites membrane domains are described to mediate MAVS‐dependent innate immune signaling (Horner et al, 2011), thus opening directions for future exploration of Mito‐C impact on viruses that are sensed by RIG‐like receptors.
Our study points to the ER‐MT contact sites as regions of intracellular communication integrating stress conditions such as viral infection and highlights NEET proteins as an important therapeutic target in diseases where altered mitochondria dynamics is implicated in disease etiology. The novel chemical class to which Mito‐C belongs may prove a powerful tool to modulate mitochondrial dynamics in such pathological situations.
Materials and Methods
Cell culture, transfection, and treatment with molecules
HeLa cells (ATCC) were grown in Minimum Essential Medium (MEM, Gibco), supplemented with GlutaMAX and 10% FCS at 37°C and 5% CO2. Huh7 cell were maintained in Dulbecco’s modified Eagle's medium (DMEM) supplemented with 2 mM l‐glutamine, 1× non‐essential amino acids, 100 U/ml penicillin, 100 µg/ml streptomycin (all from GIBCO, Life Technologies), and 10% fetal calf serum (Capricorn). INS‐1E‐cells were grown in RPMI 1640 with 11.1 M d‐glucose supplemented with 10% heat‐inactivated fetal bovine serum, 100 U/ml penicillin, 100 g/ml streptomycin, 10 M HEPES, 2 M l‐glutamine, 1 M sodium pyruvate, and 50 M mercapto‐ethanol. INS‐1E‐knockdown cell lines were generated as described previously (Sohn et al, 2013) and maintained in the same medium as control cells with Puromycin (1 g/ml). Human HepaRG cells were grown in William's E Medium supplemented with 50 IU/ml penicillin, 50 μg/ml streptomycin, 2 mM l‐glutamine, 5 μg/ml insulin, 25 μg/ml hydrocortisone hemisuccinate, and 10% FCS at 37°C and 5% CO2. Plasmid transfections in HeLa cells were performed using FuGENE® HD Transfection Reagent (Promega) following the manufacturer’s instructions. Analyses were performed 24 h after transfection. Constructs used include NAF‐1‐GFP and NAF‐1‐RFP which were generated by cloning the ORF sequence NM_001008388.4 (CISD2) into pcDNA3.1(+)‐C‐eGFP and pmCherry‐N1 Vector by GeneScript; the dominant mutant K38A DRP1 was used as previously described (Zemirli et al, 2014); Sec61β‐RFP was a kind gift from T. Rapoport (Harvard University, Cambridge, MA, USA). siRNA transfections were performed using Lipofectamine RNAiMAX (Invitrogen) following the manufacturer’s guidelines. Analyses were performed 72 h after transfection. siRNAs used were siCTRL (Qiagen, AllStars Negative Control siRNASI03650318); siCISD1 (Qiagen, SI04758194); siCISD2 (Qiagen, si04985855); and siCISD3 (Qiagen, SI04758187). Cells were treated with Mito‐C and fluoMito‐C compounds at the indicated concentrations diluted directly in culture media, as negative control the same amount of DMSO was used.
Protein extracts, membrane fractionation and Western blot analyses
For total protein extracts, cells were washed twice with 1XPBS and then directly lysed on ice with 1× Laemmli buffer (60 mM Tris–HCl pH = 6.8, 2% SDS, 10% Glycerol, bromophenol blue, supplemented with fresh added 100 mM DTT final), and lysates were incubated for 10 min at 90°C. For mitochondrial and cytosolic fraction preparations, after treatment HeLa cells were washed twice and gently scraped in cold PBS. Cell pellets were recovered via centrifugation at 600 g for 5 min. Cells were gently re‐suspended in M buffer (440 mM mannitol, 140 mM sucrose, 40 mM HEPES, 1 mM EDTA, 2 mg/ml fatty acid‐free BSA, and protease/phosphatase inhibitor cocktail) and placed on ice for 10 min. After homogenization with Dounce homogenizer (20 stroke), the lysate was centrifuged at 600 g for 5 min to pellet nuclei and recover the post nuclear supernatant (PNS). The PNS was than centrifuged at 7,200 g for 15 min at 4°C to collect the mitochondria‐enriched fraction and the supernatant (cytosolic fraction). For routine SDS–PAGE, precast gradient gels (4–20% Tris‐Glycine, Invitrogen) were used and home‐made 7.5% Tris‐Glycine gels for OPA‐1 detection. Separated proteins were transferred onto PVDF membranes. Membranes were blocked with BSA 3%/ TBS/0.1% Tween for 1 h and incubated with primary antibodies overnight at 4°C in 2% BSA/TBS/0.1% Tween. Immunoblot analysis was performed by chemiluminescence (Millipore) in a ChemiDoc MP Imaging System (Bio‐Rad). Quantification of band intensities was carried using ImageJ software.
Immunofluorescence, live, and confocal microscopy
For immunofluorescence analyses, cells were plated on 12‐mm glass coverslips and fixed with 4% paraformaldehyde in PBS for 20 min at room temperature. After 3 washes of 20 min each in PBS, cells were blocked with fetal calf serum (10%) in PBS for 30 min. Incubation with primary antibodies was performed in permeabilization buffer (0.05% saponin in blocking buffer). Coverslips were mounted on microscope slides using home‐made Mowiol mounting medium with or without DAPI. Images were obtained either using a 63× oil immersion objective with a Zeiss Apotome‐2 fluorescence wide‐field microscope or using a 63× oil immersion objective with Leica TCS SP5 confocal microscope, using a 405 nm diode laser line exciting DAPI, a 488 nm argon laser line exciting Alexa Fluor 488, a 561 nm diode laser line for Alexa Fluor 546 and laser He/Ne 633 nm for Alexa 647. Acquisitions were done in sequential mode and fluorescence acquired in separated channels. For some experiments, optical sections were acquired with a 63×/1.4 Oil immersion objective using the LAS‐X software and fluorescent pictures were collected with a PMTs GaAsP hybride camera (Hamamatsu). For confocal imaging of living cells, cells seeded onto 25‐mm round glass coverslips, treated as indicated, were placed on the stage of a Leica SP5 inverted microscope at 37°C and under 5% CO2 atmosphere. For mitochondria analysis in live microscopy, we used MitoTracker Red CMXRos (Thermo Fisher, M7512).
Image analysis and statistics of data
Image analysis was performed using ImageJ software or manually by quantification of mitochondria phenotypes. For mounting representative images, background was reduced using brightness and contrast adjustments applied to the whole image. For analyzing the recruitment of DRP1 on mitochondria, a mask on the fluorescence in TOM20 channel was used to define the region to measure the total intensity of the DRP1 signal in the DRP1 channel. For statistics of data, we used GraphPad Prism software. After evaluation of mean, standard deviation, and standard errors, we evaluate data distributions using normality test: KS normality test, D'Agostino & Pearson omnibus normality test, and Shapiro–Wilk normality test. Data were processed as normally distributed when at least 2 out of 3 test resulted positive, otherwise they were processed as non‐parametric distributions. Gaussian distributions were analyzed with the more appropriate t‐test or ANOVA and non‐Gaussian distributed sets of data were evaluated with the more appropriate non‐parametric test, Mann–Whitney, or ANOVA, as specified in figure legends.
Antibodies used and dilutions
The primary antibodies used are the following: mouse anti‐TOMM20 (BD Biosciences 612278,1:1,000 for WB, 1:400 for immunofluorescence); rabbit anti‐MitoNEET (ProteinTech 16006‐1‐AP, 1:3,000); rabbit anti‐NAF‐1 (ProteinTech 13318‐1‐AP, 1:3,000); mouse anti‐DRP1(BD Biosciences 611112, 1:1,000); rabbit anti‐DRP1 P616 (Cell signaling 3455, 1:1,000); rabbit anti‐PTPIP51 (Novus biological NBP1‐84738, 1:1,000 for WB, 1:200 for immunofluorescence); mouse anti‐OPA‐1 (BD Biosciences 612607, 1:1,000); rabbit anti‐GAPDH (Sigma G9545, 1:5,000 for WB); mouse anti beta‐actin (Abcam ab8226, 1:1,000); and rabbit anti‐Dengue virus NS5 protein (GeneTex GTX124253, 1:1,000). For secondary antibodies, we used for immunofluorescence an Alexa 488‐conjugated donkey anti‐mouse (Invitrogen A21202, 1:800), Alexa 488‐conjugated donkey anti‐rabbit (Invitrogen A21206, 1:800), Alexa 647‐conjugated donkey anti‐mouse (Invitrogen A31571, 1:800), Alexa 647 conjugated donkey anti‐rabbit (Invitrogen A31573, 1:800). For Western blotting, we used Secondary HRP‐conjugated anti‐rabbit IgG (GE Healthcare, 1:10,000) and HRP‐conjugated anti‐mouse IgG (Bio‐Rad 1:10,000).
RNA extraction and quantification
RNA was extracted from cells using NucleoSpin RNA kit (Macherey‐Nagel, 740984.50) according to the manufacturer’s instructions. cDNA was made from 1 μg of RNA using M‐MLV reverse transcriptase (Life Technologies, 28025013). Detection of MiNT transcript was performed by qPCR with iTAQ universal SYBR Green supermix (Applied Biosystem) using the following primers for 5′‐GCAGGGAAAACCTACAGGTG‐3′ and 5′‐TGAGTGGAGATAGGCCAGTG‐3′ (Eurofins) in a qTOWER machine (Analytik Jena).
Cell proliferation assay (BrdU)
Cells were treated with Mito‐C or DMSO at the indicated concentrations diluted directly in culture media. Cell proliferation assay was performed following manufacturer’s protocol (Cell Proliferation ELISA, BrdU Kit (colorimetric), Sigma). Briefly, Huh7 cells were cultured and treated with Mito‐C or DMSO in 96‐well plates. BrdU labeling solution was added 4 h before incubation with FixDenat solution. After incubation with Anti‐BrdU POD, washing steps and subtract addition, the reaction was stopped with H2SO4 (1 M). Absorbance was recorded using TECAN Spark 20M at wavelength of 450 nm.
Flow cytometry analyses
Cells were treated as indicated in each figure legend. After treatment, cell death was quantified using Annexin V‐FITC/Propidium Iodide (PI) assay according to the manufacturer’s protocol (Annexin V‐FITC Apoptosis Detection Kit II, 556570, BD PharmingenTM). For mitochondria analysis, we used MitoTracker Red CMXRos (Thermo Fisher, M7512) and MitoTracker Green FM (Thermo Fisher, M7514). Data were analyzed by a LSRFortessaTM flow cytometer (BD Biosciences, San Jose, CA) and processed using Cell Quest software (BD Biosciences) and FlowJo software (FLOWJO, LLC).
Oxygen consumption rate and mitochondrial activity measurements
For measurements of mitochondria activity, Hela cells were seeded at a density of 6,000/well in a XFe96 cell culture microplate. 16 h later cells were balanced for 1 h in un‐buffered XF assay media (Agilent Technologies) supplemented with 2 mM Glutamine, 10 mM Glucose, and 1 mM Sodium Pyruvate, and then oxygen consumption rate were measured by Seahorse bio analyzer. Values were taken every 3 min and Mito‐C was injected during the assay at reading time of 10 min (indicated by the arrow in the Appendix Fig S7A) at the final concentrations of 2, 10 or 20 µM. The data were normalized to protein content measured in each well using BCA assay (Thermo Fisher Scientific) according to manufacturer’s instructions. High‐Resolution Respirometry analysis (HRR) of Hela cells treated with Mito‐C. High‐resolution respirometry using the Oroboros O2k apparatus was used for measuring precisely mitochondrial respiration on HeLa cells treated 15 min with 2 µM of Mito‐C compound. Mitochondrial respiration was evaluated at 37°C using the O2k system (Oroboros Instruments, Austria). The respiratory medium consisted of 5 mM Glucose DMEM supplemented with 10% FCS. To assess the ATP‐linked respiration, 20 µg/µl oligomycin (Sigma‐Aldrich) was used. The "uncoupled" respiration was measured in the "oligo" conditions after addition of the uncoupler CCCP used at 2 µM. Last, to exclude non‐mitochondrial respiration 2 mM KCN was used. The respiratory rates were expressed as pmol/s O2 per 106 cells.
Mito‐C compound synthesis
Mito‐C (2‐[(3,4‐Dimethoxybenzoyl)amino]‐6,6‐dimethyl‐5,7‐dihydro‐4H‐benzothiophene‐3‐carboxylic acid) was synthetized by Charles River company as following: a reaction vessel was charged 4,4‐dimethylcyclohexanone (CAS: 4255‐62‐3, 5.00 g, 39.6 mmol), methyl cyanoacetate (CAS: 105‐34‐0, 3.8 ml, 43.6 mmol), diethylamine (CAS: 109‐89‐7, 2.0 ml, 19.8 mmol), and sulfur (CAS: 7704‐34‐9, 1.52 g, 47.5 mmol). The reaction was solvated in methanol (25 ml) and set to stir at RT. The reaction mixture was stirred at room temperature for 60 h. The volatiles were removed under reduced pressure and the residue was purified by flash chromatography on silica gel (eluting with 0–20% EtOAc in isohexane) which afforded methyl 2‐amino‐6,6‐dimethyl‐4,5,6,7‐ tetrahydrobenzo[b] thiophene‐3‐carboxylate as a pale yellow solid (7.22 g, yield 76%). 1H NMR (CDCl3, 400 MHz): δ = 5.92 (s, 2H), 3.79 (s, 3H), 2.69 (t, J = 6.4 Hz, 2H), 2.27 (s, 2H), 1.48 (t, J = 6.4 Hz, 2H), 0.98 (s, 6H). To a solution of methyl 2‐amino‐6,6‐dimethyl‐4,5,6,7‐tetrahydrobenzo[b]thiophene‐3‐carboxylate (Preparation #1, 200 mg, 0.84 mmol) in DCM (5.0 ml) was added DIPEA (CAS: 7087‐68‐5, 220 µl, 1.25 mmol) and 3,4‐dimethoxybenzoyl chloride (CAS: 3535‐37‐3, 120 µl, 1.00 mmol). The reaction mixture was stirred at RT overnight. The resulting mixture was diluted with DCM and water. The two phases were separated. The organic layer was passed through a phase separator, and the solvent was removed under reduced pressure. The residue was dissolved in THF (4.0 ml) and MeOH (2.0 ml). To the solution was added LiOH aq. (CAS: 1310‐66‐3, 2.0 M, 1.7 ml, 3.36 mmol). The reaction mixture was stirred at 50°C for 2 h. The mixture was allowed to cool to RT and acidified with 1 N aqueous HCl solution. The reaction mixture was then extracted with EtOAc. The organic phase was washed with brine and dried over Na2SO4. The solvent was removed under reduced pressure. Purification by RP‐HPLC. RP‐HPLC purification condition: Column XSELECT CSH Prep C18 19 × 250 mm, 5 µm. Mobile phase: MeCN in water (0.1% HCOOH), Flow rate: 20 ml/min; Wavelength: 210–260 nm DAD. Sample injected in DMSO (+ optional formic acid and water), 22 min non‐linear gradient from 10 to 95% MeCN, centered on a specific focused gradient. 1H NMR (DMSO‐d6, 400MHz): δ = 13.34 (br s, 1H), 12.36 (s, 1H), 7.49–7.45 (m, 2H), 7.18 (d, J = 8.4 Hz, 1H), 3.86 (s, 3H), 3.85 (s, 3H), 2.75 (t, J = 5.9 Hz, 2H), 2.42 (s, 2H), 1.50 (t, J = 6.3 Hz, 2H), 0.98 (s, 6H). LC/MS (Appendix Fig S3A, Method A) Rt = 5.31 min; MS m/z: 390 [M + H]+. Purification by RP‐HPLC afforded the title compound in 56% yield over 2 steps. Compounds were > 95% pure, reconstituted in 100% DMSO (Sigma) and diluted in cell culture medium for assays.
Dengue virus infection and estimation of virus titers upon Mito‐C treatment
Dengue virus serotype 2 strain New Guinea C was produced as previously described and infectious titers determined by limiting dilution assay using Huh7 cells (Roth et al, 2017). Huh7 cells (1 × 105) were infected with dengue virus at a multiplicity of infection (MOI) of 5 TCID50 per cell and simultaneously treated with indicated concentrations of Mito‐C for 48 h. Supernatants were harvested, filtered through 0.45 µM pore size membrane, and titrated by plaque assays. Briefly, Vero E6 (2.5 × 105) cells were infected with a serial dilution of the harvested supernatants. Two hours post‐infection inoculum was replaced by serum‐free MEM medium (Gibco, Life Technologies) containing 1.5% carboxymethyl cellulose (Sigma‐Aldrich). At day 8 post‐infection, cells were fixed by addition of formaldehyde to a final concentration of 5%. Cells were stained with crystal violet solution (1% crystal violet, 10% ethanol in H2O) for 30 min at room temperature and rinsed extensively with H2O. Infectious titers were calculated considering the corresponding dilution factor. For immunofluorescence assays, Huh7 cells (1 × 105) were infected with dengue virus at a MOI of 0.5 TCID50 per cell and simultaneously treated with 10 µM of Mito‐C for 48 h.
HBV infection and viral growth estimation under Mito‐C or GW4064 treatment
Hepatitis B virus (HBV) inoculum was prepared from stably transfected HepG2.2.15 cell line as previously described (Kim et al, 2013). HepaRG cell inoculation was performed with 100 genome‐equivalent per cell in culture medium that contained 4%v/v PEG8000 (Sigma‐Aldrich) for 24 h at 37°C. At day 2 post‐infection, cultures were treated for 10 days with the following compounds Mito‐C or GW4064 at indicated concentrations. HBs antigen (HBsAg) secreted into cell supernatants were quantified on the miniVIDAS apparatus using the VIDAS Ultra tests (BioMérieux, Marcy‐l’Etoile, France). Viral HBV DNA was extracted from cell supernatant using the QIAAmp MinElute virus spin kit (Qiagen, Courtaboeuf, France) on a QIAcube apparatus (Qiagen). Extractions were carried out following the manufacturer’s recommendations. Eluates were directly used for quantification of secreted viral DNA by quantitative PCR experiments using primers for rcHBV DNA: forward 5′‐GGGGAGGAGATTAGGTTAAAGGTC‐3′, reverse 5′‐CACAGCTTGGAGGCTTGAACAGTGG‐3′ and the QuantiFast SYBR green PCR kit (Qiagen) on a LightCycler 480 II (Roche Applied Science).
Transmission electron microscopy
Cells were fixed for 2 h at room temperature with 2% glutaraldehyde in 0.1 µM sodium cacodylate buffer at pH of 7.4. Samples were then rinsed three times of 10 min each in 0.1 µM sodium cacodylate buffer and post‐fixed with 1% osmium tetroxide in 0.1 µM sodium cacodylate buffer for 30 min. Contrast was done in uranyl acetate 1% for 30 min, and then, the samples were dehydrated in a graded series of ethanol ending with 100% ethanol. Samples were then embedded in EPON. About 70 nm sections were prepared with a Leica Ultracut 7 ultramicrotome. The sections were observed using a Philips CM120 electron microscope operating at 120 kV. Images were captured using a GATAN Orius 200 camera.
Fe‐S cluster transfer assay
The stability of NEET protein binding for its [2Fe–2S] cluster was determined from monitoring their characteristic absorbance at 458 nm as a function of time with 66 µM Mito‐C (or DMSO) and 20 µM of purified mitoNEET, NAF‐1 or MiNT proteins in a 20 mM Tris–HCl pH 5.5, 100 mM NaCl buffer on a Synergy 2 microplate reader (BioTek Instruments). NEET proteins were expressed and purified as previously described (Zuris et al, 2011).
Author contributions
Project conception: DM, IP‐C, BC, EMo; Design, Biochemical and cell biology experiments, Data organization, Statistical analysis, Manuscript writing: DM. Some of the biochemical and cell biology experiments, Experiment analysis, Electron microscopy experiments, Research design, Manuscript writing: IP‐C; Electron microscopy, Iron–sulfur cluster release experiments: H‐BM; Respiratory‐related experiments: NDA, RR; Some of the contact site‐related experiments: LR; Generation and characterization of stable shCISD2/NAF‐1 cell lines: OK, Y‐SS; Cytometry experiments and analyses: AH; Iron–sulfur cluster release experiments: LL; HBV experiments: PR, SJ; Some biochemical experiments, Research design, Manuscript editing: DA; Dengue infection experiments, Research design, Manuscript editing: KK, AR; Research design: JV; Research design, Manuscript writing: PC, FG, EMe, PM, RN; Study co‐supervision, Research design, Microscopy experiments, Light and electron microscopy experiment analysis, Manuscript writing: BC and EMo.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Review Process File
Acknowledgements
We thank Dr. Rappaport, Dr. Schmidt‐Chanasit and Dr. Mikaelian for kindly sharing reagents and advices with us. We thank our team of colleagues as well at ENYO Pharma and at INEM for fruitful discussions and constant support. We also acknowledge the INEM‐associated imaging, metabolic platform, and FACS facilities (SFR Necker INSERM US24, CNRS UMS 3633). We thank Ivan Nemazanyy at INEM for its help with designing and performing the Seahorse© essays. The present work has benefited from Imagerie‐Gif core facility supported by I’Agence Nationale de la Recherche (ANR‐11‐EQPX‐0029, ANR‐10‐INBS‐04, ANR‐11‐IDEX‐0003‐02). F.G and L.R. were supported by ANR Jeune Chercheur (ANR0015TD), ATIP‐Avenir Program. Work in A.R.’s laboratory was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation)—Projektnummer 240245660—SFB 1129 TP13. RN acknowledges BSF (Binational Science Foundation (BSF) Grant No. 2015831). This work was supported by institutional funding from INSERM (U1151SE19KA and U115120K), University de Paris (16CONV30UPDE and L17V22CONV02) and grants from ANR (ANR‐17‐CE14‐0030‐02 and ANR‐17‐CE13‐0015‐003), to P.C. and E.Mo.
EMBO Reports (2020) 21: e49019.
Contributor Information
Benoit de Chassey, Email: bdc@enyopharma.com.
Etienne Morel, Email: etienne.morel@inserm.fr.
Data availability
The authors declare that no data were deposited in a public database.
References
- Amr S, Heisey C, Zhang M, Xia X‐J, Shows KH, Ajlouni K, Pandya A, Satin LS, El‐Shanti H, Shiang R (2007) A homozygous mutation in a novel zinc‐finger protein, ERIS, is responsible for Wolfram syndrome 2. Am J Hum Genet 81: 673–683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anand R, Wai T, Baker MJ, Kladt N, Schauss AC, Rugarli E, Langer T (2014) The i‐AAA protease YME1L and OMA1 cleave OPA1 to balance mitochondrial fusion and fission. J Cell Biol 204: 919–929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castanier C, Garcin D, Vazquez A, Arnoult D (2010) Mitochondrial dynamics regulate the RIG‐I‐like receptor antiviral pathway. EMBO Rep 11: 133–138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang NC, Nguyen M, Germain M, Shore GC (2010) Antagonism of Beclin 1‐dependent autophagy by BCL‐2 at the endoplasmic reticulum requires NAF‐1. EMBO J 29: 606–618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatel‐Chaix L, Cortese M, Romero‐Brey I, Bender S, Neufeldt CJ, Fischl W, Scaturro P, Schieber N, Schwab Y, Fischer B et al (2016) Dengue virus perturbs mitochondrial morphodynamics to dampen innate immune responses. Cell Host Microbe 20: 342–356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y‐F, Kao C‐H, Chen Y‐T, Wang C‐H, Wu C‐Y, Tsai C‐Y, Liu F‐C, Yang C‐W, Wei Y‐H, Hsu M‐T et al (2009) Cisd2 deficiency drives premature aging and causes mitochondria‐mediated defects in mice. Genes Dev 23: 1183–1194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conlan AR, Axelrod HL, Cohen AE, Abresch EC, Zuris J, Yee D, Nechushtai R, Jennings PA, Paddock ML (2009) Crystal structure of Miner1: the redox‐active 2Fe‐2S protein causative in Wolfram syndrome 2. J Mol Biol 392: 143–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doghman‐Bouguerra M, Lalli E (2019) ER‐mitochondria interactions: both strength and weakness within cancer cells. Biochim Biophys Acta 1866: 650–662 [DOI] [PubMed] [Google Scholar]
- Duvezin‐Caubet S, Jagasia R, Wagener J, Hofmann S, Trifunovic A, Hansson A, Chomyn A, Bauer MF, Attardi G, Larsson N‐G et al (2006) Proteolytic processing of OPA1 links mitochondrial dysfunction to alterations in mitochondrial morphology. J Biol Chem 281: 37972–37979 [DOI] [PubMed] [Google Scholar]
- Erpapazoglou Z, Mouton‐Liger F, Corti O (2017) From dysfunctional endoplasmic reticulum‐mitochondria coupling to neurodegeneration. Neurochem Int 109: 171–183 [DOI] [PubMed] [Google Scholar]
- Fields JA, Serger E, Campos S, Divakaruni AS, Kim C, Smith K, Trejo M, Adame A, Spencer B, Rockenstein E et al (2016) HIV alters neuronal mitochondrial fission/fusion in the brain during HIV‐associated neurocognitive disorders. Neurobiol Dis 86: 154–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman JR, Lackner LL, West M, DiBenedetto JR, Nunnari J, Voeltz GK (2011) ER tubules mark sites of mitochondrial division. Science 334: 358–362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galluzzi L, Kepp O, Kroemer G (2012) Mitochondria: master regulators of danger signalling. Nat Rev Mol Cell Biol 13: 780–788 [DOI] [PubMed] [Google Scholar]
- Gomez‐Suaga P, Paillusson S, Stoica R, Noble W, Hanger DP, Miller CCJ (2017) The ER‐mitochondria tethering complex VAPB‐PTPIP51 regulates autophagy. Curr Biol 27: 371–385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamasaki M, Furuta N, Matsuda A, Nezu A, Yamamoto A, Fujita N, Oomori H, Noda T, Haraguchi T, Hiraoka Y et al (2013) Autophagosomes form at ER‐mitochondria contact sites. Nature 495: 389–393 [DOI] [PubMed] [Google Scholar]
- Helle SCJ, Kanfer G, Kolar K, Lang A, Michel AH, Kornmann B (2013) Organization and function of membrane contact sites. Biochim Biophys Acta Mol Cell Res 1833: 2526–2541 [DOI] [PubMed] [Google Scholar]
- Horner SM, Liu HM, Park HS, Briley J, Gale M (2011) Mitochondrial‐associated endoplasmic reticulum membranes (MAM) form innate immune synapses and are targeted by hepatitis C virus. Proc Natl Acad Sci USA 108: 14590–14595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karmi O, Marjault H‐B, Pesce L, Carloni P, Onuchic JN, Jennings PA, Mittler R, Nechushtai R (2018) The unique fold and lability of the [2Fe‐2S] clusters of NEET proteins mediate their key functions in health and disease. J Biol Inorg Chem 23: 599–612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S‐J, Khan M, Quan J, Till A, Subramani S, Siddiqui A (2013) Hepatitis B virus disrupts mitochondrial dynamics: induces fission and mitophagy to attenuate apoptosis. PLoS Pathog 9: e1003722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köster H, Little DP, Luan P, Muller R, Siddiqi SM, Marappan S, Yip P (2007) Capture compound mass spectrometry: a technology for the investigation of small molecule protein interactions. Assay Drug Dev Technol 5: 381–390 [DOI] [PubMed] [Google Scholar]
- Liesa M, Palacín M, Zorzano A (2009) Mitochondrial dynamics in mammalian health and disease. Physiol Rev 89: 799–845 [DOI] [PubMed] [Google Scholar]
- Lipper CH, Karmi O, Sohn YS, Darash‐Yahana M, Lammert H, Song L, Liu A, Mittler R, Nechushtai R, Onuchic JN et al (2018) Structure of the human monomeric NEET protein MiNT and its role in regulating iron and reactive oxygen species in cancer cells. Proc Natl Acad Sci USA 115: 272–277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giacomello M, Pyakurel A, Glytsou C, Scorrano L (2020) The cell biology of mitochondrial membrane dynamics. Nat Rev Mol Cell Biol 21: 204–224 [DOI] [PubMed] [Google Scholar]
- MacVicar T, Langer T (2016) OPA1 processing in cell death and disease ‐ the long and short of it. J Cell Sci 129: 2297–2306 [DOI] [PubMed] [Google Scholar]
- Mittler R, Darash‐Yahana M, Sohn YS, Bai F, Song L, Cabantchik IZ, Jennings PA, Onuchic JN, Nechushtai R (2019) NEET Proteins: a new link between iron metabolism, reactive oxygen species, and cancer. Antioxid Redox Signal 30: 1083–1095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno‐Altamirano MMB, Kolstoe SE, Sánchez‐García FJ (2019) Virus control of cell metabolism for replication and evasion of host immune responses. Front Cell Infect Microbiol 9: 95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pallotta MT, Tascini G, Crispoldi R, Orabona C, Mondanelli G, Grohmann U, Esposito S (2019) Wolfram syndrome, a rare neurodegenerative disease: from pathogenesis to future treatment perspectives. J Transl Med 17: 238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naia L, Ferreira IL, Ferreiro E, Rego AC (2017) Mitochondrial Ca2+ handling in Huntington's and Alzheimer's diseases ‐ Role of ER‐mitochondria crosstalk. Biochem Biophys Res Comm 483: 1069–1077 [DOI] [PubMed] [Google Scholar]
- Rossignol R, Gilkerson R, Aggeler R, Yamagata K, Remington SJ, Capaldi RA (2004) Energy substrate modulates mitochondrial structure and oxidative capacity in cancer cells. Can Res 64: 985–993 [DOI] [PubMed] [Google Scholar]
- Roth H, Magg V, Uch F, Mutz P, Klein P, Haneke K, Lohmann V, Bartenschlager R, Fackler OT, Locker N et al (2017) Flavivirus infection uncouples translation suppression from cellular stress responses. MBio 8: e02150‐16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouzier C, Moore D, Delorme C, Lacas‐Gervais S, Ait‐El‐Mkadem S, Fragaki K, Burté F, Serre V, Bannwarth S, Chaussenot A et al (2017) A novel CISD2 mutation associated with a classical Wolfram syndrome phenotype alters Ca2+ homeostasis and ER‐mitochondria interactions. Hum Mol Genet 26: 1599–1611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowland AA, Chitwood PJ, Phillips MJ, Voeltz GK (2014) Article ER contact sites define the position and timing of endosome fission. Cell 159: 1027–1041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi C‐S, Qi H‐Y, Boularan C, Huang N‐N, Abu‐Asab M, Shelhamer JH, Kehrl JH (2014) SARS‐coronavirus open reading frame‐9b suppresses innate immunity by targeting mitochondria and the MAVS/TRAF3/TRAF6 signalosome. J Immunol 193: 3080–3089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smirnova E, Shurland DL, Ryazantsev SN, van der Bliek AM (1998) A human dynamin‐related protein controls the distribution of mitochondria. J Cell Biol 143: 351–358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohn Y‐S, Tamir S, Song L, Michaeli D, Matouk I, Conlan AR, Harir Y, Holt SH, Shulaev V, Paddock ML et al (2013) NAF‐1 and mitoNEET are central to human breast cancer proliferation by maintaining mitochondrial homeostasis and promoting tumor growth. Proc Natl Acad Sci USA 110: 14676–14681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taguchi N, Ishihara N, Jofuku A, Oka T, Mihara K (2007) Mitotic phosphorylation of dynamin‐related GTPase Drp1 participates in mitochondrial fission. J Biol Chem 282: 11521–11529 [DOI] [PubMed] [Google Scholar]
- Tamir S, Paddock ML, Darash‐Yahana‐Baram M, Holt SH, Sohn YS, Agranat L, Michaeli D, Stofleth JT, Lipper CH, Morcos F et al (2015) Structure–function analysis of NEET proteins uncovers their role as key regulators of iron and ROS homeostasis in health and disease. Biochem Biophys Acta 1853: 1294–1315 [DOI] [PubMed] [Google Scholar]
- Tilokani L, Nagashima S, Paupe V, Prudent J (2018) Mitochondrial dynamics: overview of molecular mechanisms. Essays Biochem 62: 341–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tubbs E, Rieusset J (2017) Metabolic signaling functions of ER‐mitochondria contact sites: role in metabolic diseases. J Mol Endocrinol 58: R87–R106 [DOI] [PubMed] [Google Scholar]
- Vernay A, Marchetti A, Sabra A, Jauslin TN, Rosselin M, Scherer PE, Demaurex N, Orci L, Cosson P (2017) MitoNEET‐dependent formation of intermitochondrial junctions. Proc Natl Acad Sci USA 114: 8277–8282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Landry AP, Ding H (2017) The mitochondrial outer membrane protein mitoNEET is a redox enzyme catalyzing electron transfer from FMNH2 to oxygen or ubiquinone. J Biol Chem 292: 10061–10067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiley SE, Andreyev AY, Divakaruni AS, Karisch R, Perkins G, Wall EA, van der Geer P, Chen Y‐F, Tsai T‐F, Simon MI et al (2013) Wolfram syndrome protein, Miner1, regulates sulphydryl redox status, the unfolded protein response, and Ca2+ homeostasis. EMBO Mol Med 5: 904–918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiley SE, Murphy AN, Ross SA, van der Geer P, Dixon JE (2007) MitoNEET is an iron‐containing outer mitochondrial membrane protein that regulates oxidative capacity. Proc Natl Acad Sci USA 104: 5318–5323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zemirli N, Morel E, Molino D (2018) Mitochondrial dynamics in basal and stressful conditions. Int J Mol Sci 19: 564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zemirli N, Pourcelot M, Ambroise G, Hatchi E, Vazquez A, Arnoult D (2014) Mitochondrial hyperfusion promotes NF‐κB activation via the mitochondrial E3 ligase MULAN. FEBS J 281: 3095–3112 [DOI] [PubMed] [Google Scholar]
- Zuris JA, Harir Y, Conlan AR, Shvartsman M, Michaeli D, Tamir S, Paddock ML, Onuchic JN, Mittler R, Cabantchik ZI et al (2011) Facile transfer of [2Fe‐2S] clusters from the diabetes drug target mitoNEET to an apo‐acceptor protein. Proc Natl Acad Sci USA 108: 13047–13052 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Review Process File
Data Availability Statement
The authors declare that no data were deposited in a public database.