

Abstract
Pyroptosis is a fulminant form of macrophage cell death, contributing to release of pro‐inflammatory cytokines. In humans, it depends on caspase 1/4‐activation of gasdermin D and is characterized by the release of cytoplasmic content. Pathogens apply strategies to avoid or antagonize this host response. We demonstrate here that a small accessory protein (PB1‐F2) of contemporary H5N1 and H3N2 influenza A viruses (IAV) curtails fulminant cell death of infected human macrophages. Infection of macrophages with a PB1‐F2‐deficient mutant of a contemporary IAV resulted in higher levels of caspase‐1 activation, cleavage of gasdermin D, and release of LDH and IL‐1β. Mechanistically, PB1‐F2 limits transition of NLRP3 from its auto‐repressed and closed confirmation into its active state. Consequently, interaction of a recently identified licensing kinase NEK7 with NLRP3 is diminished, which is required to initiate inflammasome assembly.
Keywords: influenza A virus, NEK7, NLRP3, PB1‐F2, pyroptosis
Subject Categories: Immunology; Microbiology, Virology & Host Pathogen Interaction
Contemporary influenza A viruses limit pyroptosis and IL‐1β production in human macrophages by the expression of PB1‐F2. Via its C‐terminus PB1‐F2 binds to the pyrin and LRR domains of NLRP3 and locks it in its closed confirmation.

Introduction
Infections with IAV are accompanied by a pronounced inflammatory host response largely responsible for the clinical manifestations experienced by human patients or infected model animals (Xu et al, 2006; Short et al, 2014; Liu et al, 2016). Productive viral replication takes place in pneumocytes, but other cell types, e.g., lung‐resident macrophages and inflammatory monocytes, are infected in an abortive fashion. Nevertheless, these abortive infections contribute significantly to the host response (Lin et al, 2008; van Riel et al, 2011; Friesenhagen et al, 2012). Pro‐inflammatory cytokines are key mediators in this response and are secreted by a number of cell types, foremost lung‐resident macrophages and infiltrating monocytes (Sprenger et al, 1996; Herold et al, 2006; Hogner et al, 2013). Inflammatory responses are especially pronounced in humans infected with zoonotic avian viruses, e.g., H5N1 or H7N9 subtypes (de Jong et al, 2006; Wang et al, 2014). One of the key cytokines in a pro‐inflammatory response is IL‐1β. Due to its possibly detrimental potential for the host organism, its production and secretion are regulated on multiple levels (Fenton et al, 1988; Kostura et al, 1989). Pro‐IL‐1β mRNA expression depends on recognition of pathogen‐associated molecular patterns (PAMP), e.g., by Toll‐like receptors (TLR) and downstream activation of NF‐κB signaling (Dinarello, 1998). Pro‐IL‐1β itself remains inactive in the cytoplasm until proteolytically activated and released. For this step, a second danger‐associated molecular pattern (DAMP), e.g., extracellular ATP or HMGB, needs to be detected (Hogquist et al, 1991; Chin & Kostura, 1993). Nod‐like receptors (NLR) were shown to respond to DAMP by forming large signaling platforms, which are in turn required to recruit and activate pro‐inflammatory caspases (Tschopp et al, 2003). NLR family, pyrin domain containing 3 (NLRP3) is known to respond to viral, bacterial and chemical DAMPs via the C‐terminal leucine‐rich repeat (LRR) domain (Cassel et al, 2009). It is believed that DAMP binding triggers NLRP3 transition from a closed auto‐repressed confirmation into an open, active conformation (Martinon & Tschopp, 2005). This change allows formation of homo‐oligomers via the central NACHT domain and recruitment of the downstream adapter ASC via the N‐terminal pyrin domain. In turn ASC forms CARD‐CARD interfaces with the executor caspases, performing pro‐IL‐1β cleavage (Tschopp & Schroder, 2010). Recently, it was demonstrated that caspase‐1 and caspase‐4 (caspase 11 in mice) also cleave and activate gasdermin D through non‐canonical inflammasome activation. The N‐terminus of gasdermin D forms pores in the plasma membrane, allowing free exchange of macromolecules with the extracellular space. Eventually, this uncontrolled leakage of cytoplasmic content results in a fulminant form of cell death termed pyroptosis (Kayagaki et al, 2015; Shi et al, 2015). In a feed‐forward loop, exposure of neighboring cells to cytoplasmic content of the dying cell is once more perceived as a danger event and provokes an amplification of the pro‐inflammatory tissue response (Aglietti & Dueber, 2017).
Secretion of IL‐1β accompanies IAV infections in vivo (Vacheron et al, 1990; Hennet et al, 1992). IL‐1β directly induces an antiviral transcription program in dendritic cells infected with the laboratory strain WSN (Aarreberg et al, 2018). Moreover, IL‐1β enhances CD4 T cell differentiation and expansion and promotes CD8 T cell expansion and effector function (Ben‐Sasson et al, 2009; Ben‐Sasson et al, 2013). IL1R−/− mice show reduced CD4+ T cell priming and reduced antiviral IgM production in IAV infection (Schmitz et al, 2005). In accordance knockout of NLRP3, ASC or IL1R results in reduced viral titers in vivo at late time points post‐infection and/or increased host susceptibility to succumb to IAV infection (Schmitz et al, 2005; Szretter et al, 2007; Allen et al, 2009; Ichinohe et al, 2009; Thomas et al, 2009; Chang et al, 2011). Chemical inhibition of NLRP3 pointed to a time dependent effect of NLRP3 (Tate et al, 2016), which might be a result of exacerbated inflammation leading to severe host immune pathology. Conversely, hyperactivation of the NLRP3 inflammasome restricts IAV replication in mice (Niu et al, 2019).
In light of these findings, one could assume that a successfully replicating virus has means to counteract pyroptotic host cell death. Different IAV proteins can interfere with antiviral signaling. Most of the research focused on the block of type I interferon (Schmolke & Garcia‐Sastre, 2010; Hsu, 2018), while inhibition of pro‐inflammatory signaling is less well understood. The accessory protein PB1‐F2 was previously shown in different model systems to act proapoptotic (McAuley et al, 2013; Pinar et al, 2017); here, we challenge these findings using contemporary IAV strains and their isogenic PB1‐F2 deficient mutants to infect human macrophages.
Results and Discussion
H5N1 and H3N2 PB1‐F2 expression reduces levels of secreted IL‐1β from infected macrophages
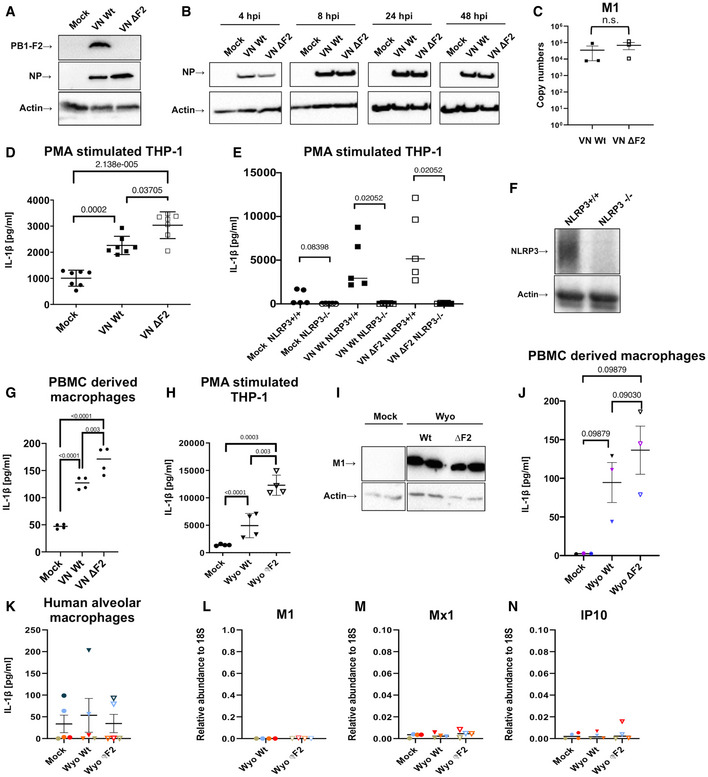
PB1‐F2 is a small accessory protein, predominantly expressed in avian, e.g., zoonotic H5N1 and H7N9 IAV, but also found in contemporary human H3N2 isolates (Zell et al, 2007; Kamal et al, 2017). Initial findings with laboratory‐adapted IAV strains identified PB1‐F2 as a pro‐apoptotic and pro‐inflammatory viral factor (Chen et al, 2001; McAuley et al, 2007). Using contemporary H5N1 and H3N2 IAV, we here challenge this idea. We first decided to study its function in context of a zoonotic infection scenario using a low‐pathogenic variant of a H5N1 subtype virus, A/Viet Nam/1203/2004 (VN). Deletion of PB1‐F2 was achieved by introduction of two premature stop codons by reverse genetics (Mazel‐Sanchez et al, 2018), without altering the start AUG, in order to avoid downstream effects on a second alternative gene product of segment 2, N40 (Wise et al, 2009; Tauber et al, 2012). We used THP‐1‐derived human macrophages as a first model to study the response of inflammatory monocyte derived macrophages to IAV infection. Low‐pathogenic IAV does not complete a full replication cycle in macrophages (van Riel et al, 2011; Friesenhagen et al, 2012). Total cell lysates of infected THP‐1‐derived human macrophages infected with VN Wt and VN ΔF2 virus confirmed absence of PB1‐F2 expression under comparable infection conditions as indicated by the presence of viral nucleoprotein (Fig 1A and B). Additionally, we verified that levels of viral M1 mRNA were similar at 16 h post‐infection (hpi) in infected THP‐1 (Fig 1C). These results ruled out discrepancies in viral infection between VN Wt and VN ΔF2 virus in macrophages, which might otherwise jeopardize downstream readouts, and further made presence of high amounts of defective particles in one of the stocks unlikely.
Figure 1. Secretion of IL‐1β upon infection with different IAV strains.
-
A, BTHP‐1‐derived macrophages were infected with the wild‐type (Wt) and ΔF2 H5N1 (VN) virus at MOI 10. Viral protein expression for PB1‐F2 (A) or NP (B) was detected by immunoblotting at 24 h post‐infection (hpi) or indicated time points, respectively. Equal loading was confirmed by detection of beta actin.
-
CTHP‐1‐derived macrophages were infected with the wild‐type (Wt) and ΔF2 H5N1 (VN) virus at MOI 10. Levels of viral M1 mRNA expression were detected by RT–qPCR at 16 hpi. The mean ± standard deviation of three independent biological samples is shown. Statistical analysis was performed by Student’s t‐test.
-
DTHP‐1‐derived macrophages were infected with VN and PR8 Wt and ΔF2 strains for 24 h at MOI 10. Levels of IL‐1β were quantified by ELISA from the supernatants. The mean ± standard deviation of seven independent experiments is shown. Statistical analysis was performed by one‐way ANOVA, and P‐values are indicated.
-
ETHP‐1 control and knockout cells for NLRP3 were mock treated or infected with the VN Wt and ΔF2 strains for 24 h at MOI 10 and IL‐1β levels were assessed by ELISA from the supernatants. The mean ± standard deviation of five independent experiments is shown. Statistical analysis was performed by one‐way ANOVA, and P‐values are indicated.
-
FImmunoblot detection for NLRP3 on control and NLRP3 knockout THP‐1 cells. Equal loading was confirmed by specific blotting against beta actin. Representative blot of three independent repeats is shown.
-
GHuman primary macrophages differentiated from PBMCs from two healthy donors were infected with H5N1 Wt and ΔF2 for 5 h at MOI 10. Levels of IL‐1β were measured by ELISA in technical replicates. The mean of two independent experiments is shown. Statistical analysis was performed by one‐way ANOVA, and P‐values are indicated.
-
HTHP‐1‐derived macrophages were infected with a human H3N2 IAV strain (Wyo) Wt and ΔF2 for 24 h at MOI 4. Levels of IL‐1β were measured by ELISA from the supernatants. The mean ± standard deviation of four independent experiments is shown. Statistical analysis was performed by one‐way ANOVA, and P‐values are indicated.
-
IDetection of viral matrix protein M1 by immunoblotting in cells infected with the H3N2 Wt and ΔF2 strains.
-
JHuman primary macrophages isolated and differentiated from PBMCs from three healthy donors were infected with H3N2 Wt and ΔF2 for 24 h at MOI 4. Levels of IL‐1β were measured by ELISA. The mean ± standard deviation of three independent experiments is shown. Statistical analysis was performed by one‐way ANOVA, and P‐values are indicated.
-
K–NPrimary human alveolar macrophages from BALF of five pediatric patients (patients are color coded) were infected with the human H3N2 strain (Wyo) Wt and ΔF2 for 16 h at MOI 1. (K) Levels of IL‐1β were measured by ELISA from the supernatants. The mean ± standard deviation is shown. Levels of viral M1 mRNA (L), Mx1 mRNA (M), and IP10 mRNA (N) expression were detected by RT–qPCR. The mean ± standard deviation for cells from four patients are shown. Individual patients are color coded.
Source data are available online for this figure.
During IAV infection, macrophages are a major source of pro‐inflammatory cytokines such as IL‐1β (Peschke et al, 1993; Lehmann et al, 1996; Wang et al, 2012; Oshansky et al, 2014; Cole et al, 2017; Wong et al, 2018). In context of infected tissue, release of IL‐1β is facilitated by breach of plasma membrane integrity and uncontrolled release of cytoplasmic content, such as ATP (Piccini et al, 2008), which is perceived as a danger signal, e.g., by the NLRP3 pathway (Mariathasan et al, 2006). When testing the supernatants of infected macrophages, we found higher secretion of IL‐1β from VN ΔF2 infected cells (Fig 1D). Infection with a lower dose (5MOI) resulted in comparable differences (Fig EV1A) 24 h post‐infection. However, at 12 hpi PB1‐F2‐dependent differences were not yet visible (Fig EV1B). IL‐1β secretion strictly depended on NLRP3, as CRISPR/Cas9 generated NLRP3−/− THP1 (kindly provided by Dr. V. Hornung, LMU Munich) (Schmid‐Burgk et al, 2015) did not release any IL‐1β upon infection (Fig 1E and F). In order to confirm these data in a biologically more relevant system, we infected human PBMC‐derived macrophages from three independent healthy donors. As for THP‐1‐derived macrophages, we found increased IL‐1β secretion of cells infected with VN ΔF2 at 5 hpi (Fig 1G), supporting validity of our THP‐1 model. Full‐length open reading frames of PB1‐F2 are not only found in zoonotic avian IAV (e.g., H5N1 or H7N9) but also in contemporary human H3N2 isolates. Despite phyllogenetic distance between H5N1 and H3N2 PB1‐F2 on amino acid level (Appendix Fig S1), we confirmed increased IL‐1β secretion from THP‐1 or human PBMC‐derived macrophages infected with A/Wyoming/03/2003 (WYO/03) ΔF2 virus (Mazel‐Sanchez et al, 2018) at 24 hpi when compared to the corresponding Wt strain (Fig 1H and J). As for H5N1, the deletion of the PB1‐F2 ORF did not affect viral replication in human macrophages (Fig 1I).
Figure EV1. Contemporary IAV strains lacking PB1‐F2 do not display an in vivo phenotype in mice or murine cells.
-
ATHP‐1‐derived macrophages were infected with the wild‐type (Wt) and ΔF2 H5N1 (VN) virus at MOI 5 for 24 h. Levels of IL‐1β were measured by ELISA. The mean ± standard deviation of five independent experiments is shown. Statistical analysis was performed by one‐way ANOVA, and P‐values are indicated.
-
BTHP‐1‐derived macrophages were infected with the wild‐type (Wt) and ΔF2 H5N1 (VN) virus at MOI 10 for 12 h. Levels of IL‐1β were measured by ELISA. The mean ± standard deviation of three independent experiments is shown. Statistical analysis was performed by one‐way ANOVA, and P‐values are indicated.
-
CLung viral loads from 5 individual mice on day 3 post‐infection were assessed by plaque assay on MDCK cells. Median titers are indicated Statistical analysis was performed by Student’s t‐test as compared to mock samples (*P < 0.05, **P < 0.01).
-
DGroups of n = 5 mice were mock treated or infected with 40 pfu of the VN Wt or VN ΔF2 strains for 3 days. LDH assay was performed on BAL samples in technical duplicates (# P < 0.05 compared with wt **P < 0.01 compared with mock).
-
EELISA for IL‐1β in BALF of mock (n = 3), VN Wt (n = 5) or VN ΔF2 (n = 5) infected mice.
-
F–OGroups of mice were mock treated (n = 2) or infected with 40 pfu of the VN Wt (n = 5) or VN ΔF2 (n = 4–5, of note for F–H the staining for macrophages for the red sample failed due to technical problems and is not indicated) for 7 days. Flow cytometry data for indicated cell populations. Statistical analysis was performed by one‐way ANOVA, and P‐values are indicated. (*P < 0.05, **P < 0.01, ***P < 0.001).
-
PGroups of n = 3–5 mice were infected with 40 pfu of the VN Wt or VN ΔF2 strains for 8–11 days. Lung viral loads were assessed by plaque assay on MDCK cells.
-
QStreptococcus pneumoniae colonies in serial dilutions of lung homogenate are shown in trypcase soy agar +5% sheep blood plates. Representative results are shown in technical triplicates
-
RStreptococcus pneumoniae bacteria in the lungs (cfu/organ) 24 h post‐infection from mock treated, VN Wt or VN ΔF2 pre‐infected animals (mock n = 4, virus infected n = 5). Median values ± SD are indicated. Statistical analysis was performed by one‐way ANOVA P‐values toward mock samples are indicated. (**P < 0.01, ***P < 0.001).
-
SELISA for IL‐1β secretion in BMDMs (n = 3) mock treated of infected with VNWt and ΔF2 strains. Statistical analysis was performed by one‐way ANOVA, and P‐values are indicated. Median values ± SD are indicated. P‐values toward mock samples are indicated. (****P < 0.0001).
-
TImmunoblot of cell lysates from (S). For all panels, the median of the indicated independent experiments is shown.
Source data are available online for this figure.
Ex vivo infection of human lung tissue revealed that 4–11% of infected cells are alveolar macrophages (Weinheimer et al, 2012). Tissue‐resident alveolar macrophages originate from yolk sac and auto‐replenish in situ (Guilliams et al, 2013). Since origin and function of AMs during viral infection differ substantially from PBMC‐derived macrophages (Short et al, 2012), we tested a potential role of PB1‐F2 in inflammatory responses of IAV infected primary human alveolar macrophages (AM). AMs protect type I pneumocytes from IAV infection (Cardani et al, 2017). Direct infection of AMs in vivo is still under debate. Viral antigens were frequently found in AMs during IAV infection, but could stem from uptake of remnants of IAV‐positive epithelial cells.
Here, we isolated AM from bronchoalveolar lavage fluid (BALF) of five pediatric donors (see Appendix Table S1 for cohort description) were infected with WYO/03 Wt or ΔF2 and analyzed for secretion of IL‐1β. Baseline levels of IL‐1β varied substantially between donors and virus exposure did not increase IL‐1β secretion (Fig 1K). qPCR for viral M1 mRNA and the mRNA of two interferon stimulated genes (Mx1 and IP10) revealed no detectable viral transcription and no antiviral response in these cells (Fig 1L–N). These data suggest that tissue‐resident human AMs are not the primary target cell type for IAV and consequently do not substantially contribute to the host’s inflammatory response to IAV. AM and MDM infection largely depends on the virus subtype (reviewed in Lee et al, 2012). Recent data suggest that human AMs might be resistant to IAV infection with seasonal IAV strains (Ettensohn et al, 2016).
Enhanced NLRP3 inflammasome activation and secretion of IL‐1β, as seen in VN ΔF2 infected macrophages, could support an antiviral state in vivo. For IAV infection, mouse data show increased mortality in animals deficient for NLRP3 (Allen et al, 2009; Thomas et al, 2009). NLRP3 was found to be essential to mount effective adaptive immune responses against IAV (Ichinohe et al, 2009), albeit this finding was not confirmed by Thomas et al (2009) and Allen et al (2009). Experiments in IL1R−/− mice confirmed the importance of IL‐1β signaling for adaptive responses (CD4 priming and IgM induction) during IAV infection (Schmitz et al, 2005). Here however, we did not find differences in viral replication (Fig EV1C), IL‐1β levels in BALF (Fig EV1D) or influx of macrophages, leukocytes or lymphocytes (Fig EV1E–O) between VN Wt or VNΔF2‐infected mice. In line with this, we found no differences in viral clearance (Fig EV1P) or susceptibility to bacterial super infection with Streptococcus pneumoniae (Fig EV1Q and R). The discrepancy toward the findings in human cells might be explained by a species‐specific antagonism of IL‐1β production, since mouse BMDMs infected with VN ΔF2 did not show an increased IL‐1β secretion as compared to VN Wt infected cells (Fig EV1S and T). This points to a species‐specific effect of PB1‐F2 on IL‐1β secretion.
We thus decided to focus on the human cell system to address the PB1‐F2‐dependent inhibition of IL‐1β secretion in IAV infection.
Expression of H5N1 PB1‐F2 delays cell death of infected human macrophages
In virus‐infected tissue, IL‐1β production often is accompanied by substantial cell death. Onset of cell death (as indicated by annexin V staining) was more rapid (12 h post‐infection) for VN ΔF2‐infected THP1‐derived macrophages (Fig 2A and B). Following this process, these cells also lost the early cell death phenotype earlier (Fig 2C) and displayed signs of loss of membrane integrity 24 h post‐infection as VN Wt‐infected cells (as indicated by Zombie dye staining) (Figs 2D and EV2).
Figure 2. Expression of H5N1 PB1‐F2 delays pyroptotic cell death.
-
A–DTHP‐1 cells were infected with the wild‐type (Wt) and ΔF2 H5N1 (VN) virus at MOI 10. Flow cytometry results for annexin V single positive (A and C) and annexin V and zombie dye double positive (B and D) cells. The percentage of positive cells are depicted at 12 hpi or 24 hpi.
-
E, FTHP‐1 cells were infected with the wild‐type (Wt) and ΔF2 H5N1 (VN) virus at MOI 10. LDH release measured from supernatants of mock treated or infected cells at 12 hpi (E) or 24 hpi (F).
G THP‐1 Caspase 1−/− cells were infected with the wild‐type (Wt) and ΔF2 H5N1 (VN) virus at MOI 10. LDH release measured from supernatants of mock treated or infected cells at 12 hpi or 24 hpi. n = 3 for 12 hpi, n = 5 for 24 hpi. Medians are indicated, and statistical analysis was performed by one‐way ANOVA.
Data information: Mean values of n = 3 independent experiments measured in technical duplicates are indicated (A, B, and E) or n = 3 independent experiments for (C and D) or n = 4 independent experiments for (F). Statistical analysis was performed by one‐way ANOVA, and P‐values are indicated.
Figure EV2. Representative FACS plots for Annexin V and Zombie‐Red staining.
Upper panels show representative panels for Fig 2A/B, lower panels show representative panels for Fig 2C/D.
Next, we quantified the presence of the cytoplasmic protein lactate dehydrogenase (LDH) in the supernatants of mock treated or infected macrophages. Release of LDH indicates compromised plasma membranes, a hallmark of fulminant forms of cell death. In line with the earlier onset of cell death, we also found higher levels of LDH in the supernatants of VN ΔF2‐infected macrophages compared with Wt‐infected macrophages after 12 h (Fig 2E) or 24 h post‐infection (Fig 2F). Pyroptotic cell death depends on caspase 1 activation (Fink & Cookson, 2005; Swanson & Molofsky, 2005). Notably, we did not detect virus induced LDH release after 12 h of infection in caspase 1 deficient THP‐1‐derived macrophages and after 24 h substantially less than in Wt THP‐1, with no differences between VN Wt and VN ΔF2 (Fig 2G). Overall, our data imply increased levels of pyroptotic cell death in VN ΔF2 infected macrophages as compared to the isogenic Wt strain.
PB1‐F2 was first described as a pro‐apoptotic pathogenicity factor using PR/8 as a model virus (Chen et al, 2001). A later study showed that this characteristic is strain specific to PR/8 using a limited panel of IAV strains (McAuley et al, 2010). Sequence variations in PB1‐F2 (see Fig EV1C) might explain the strain‐specific differences between former studies and the data for contemporary viruses presented here.
IAV PB1‐F2 does not interfere with signal one of NLRP3 inflammasome activation
NLRP3 inflammasome activation is thought to occur in a two‐step fashion. Priming of the inflammasome occurs in response to PRR‐dependent recognition of microbiological PAMPs. Experimentally, this step is often mimicked by LPS stimulation of macrophages. TLR4‐dependent recognition of LPS results in downstream activation of NF‐κB and transcriptional upregulation of inflammasome components, pro‐caspase‐1 and pro‐IL‐1β (Dinarello, 2009; Lee et al, 2015). Triggering of the inflammasome assembly requires a second danger signal, e.g., extracellular ATP. IAV infection provides both of these signals and activates the inflammasome independently of LPS priming (Kanneganti et al, 2006; Ichinohe et al, 2009); potentially, this is a consequence of detection of viral RNA, but also expression of the M2 ion channel was found to trigger inflammasome assembly (Ichinohe et al, 2010). Assembly of the NLRP3 inflammasome was found to be both detrimental and beneficial, depending on the infecting virus (reviewed in Ong et al, 2016). As described for other viruses (Shrivastava et al, 2016) IAV developed strategies to avoid or antagonize NLRP3 activation. The non‐structural protein NS1 inhibits NLRP3 signaling by binding via its TRIM25/RIG‐I interaction domain (Moriyama et al, 2016). NS1 would consequently limit step 1 of inflammasome activation. The same authors showed that expression of full‐length PB1‐F2 or an early‐truncated isoform differentially inhibited the NLRP3 inflammasome. In detail, HEK293T cells reconstituted with the NLRP3 inflammasome showed decreased Il‐1β secretion and ASC oligomerization in cells transfected with the full‐length PB1‐F2 compared with the control. This decrease was lost when cells were expressing the early‐truncated PB1‐F2 isoform (Yoshizumi et al, 2014).
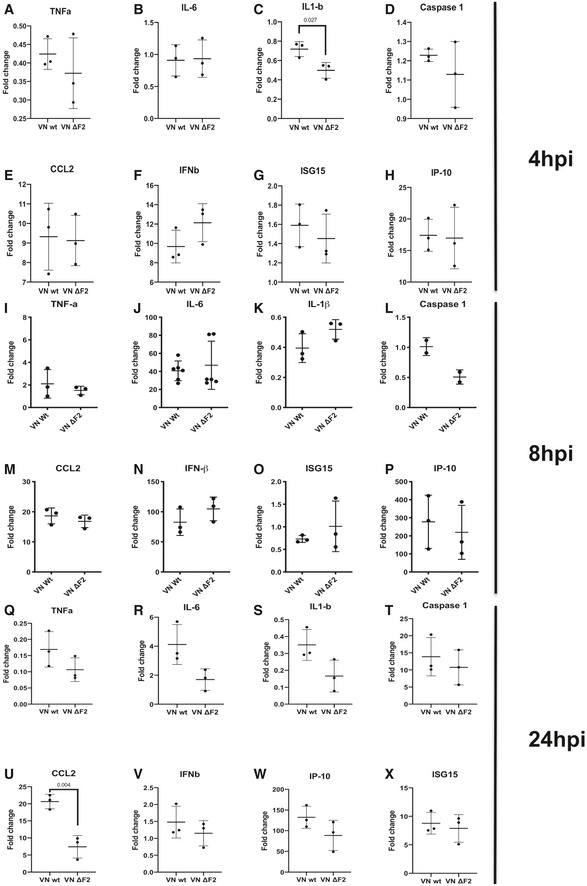
In order to understand, if PB1‐F2 interferes with signal 1 or 2 of the inflammasome activation, we first tested expression of classical NF‐κB‐dependent mRNA by RT–qPCR from total RNA of VN Wt‐ or VN ΔF2‐infected THP‐1‐derived macrophages 4, 8, 18, and 24 hpi. Despite upregulation of NF‐κB‐dependent marker mRNAs for TNFα or IL‐6, we found no increase in gene expression induction in VN ΔF2‐infected cells (Fig EV2A–X). Pro‐IL‐1β mRNA was surprisingly downregulated, however, to the same extent by both viruses (at 4 h post‐infection significantly more in VN ΔF2 infected THP‐1). It is currently unclear if this effect is based on a PB1‐F2‐independent shutoff mechanism by IAV, e.g., by NS1 (Pothlichet et al, 2013). For pro‐caspase‐1 mRNA detection, we observed no upregulation during infection at 4 an 8 hpi (Fig EV3D and L) and comparable induction after 24 h of infection (Fig EV3T). Equal expression of IFNβ and two ISGs (IP10 and ISG15) (Fig EV3F–H, N–P and V–X) further confirms no differences in the levels of defective interfering viral particles in the viral stocks used here, which could have been a source of aberrant inflammasome activation.
Figure EV3. Induction of NF‐κB‐dependent genes upon H5N1 infection.
-
A–XTHP‐1 cells were mock treated or infected with the VN Wt and ΔF2 strains for 4 h (A–H), 8 h (I–O), 18 h (P), and 24 h (Q–X) at MOI MOI 6 (I–O)‐10 (A–H and P–X). Relative expression levels normalized to the average of mock were determined by RT–qPCR for indicated mRNAs normalized to 18S rRNA and calibrated to mock. The mean ± standard deviation of 2–6 independent experiments with at least two technical replicates each is shown (each dot represents an independent biological sample). Statistical analysis was performed by paired two‐tailed Student’s t‐test, and P‐values are indicated.
Our data provide no evidence for differential transcriptional upregulation of NF‐κB‐dependent genes in absence of PB1‐F2 in macrophages infected with IAV. We thus ruled out a PB1‐F2‐dependent interception with signal 1 of NLRP3 inflammasome activation. Our data point to an interference with NLRP3 assembly or signaling.
PB1‐F2 interferes with inflammasome signaling at the level of or upstream of caspase‐1
We next aimed at understanding at which level PB1‐F2 might interfere with the classical inflammasome signaling pathway, starting from bottom to top.
Release of bioactive, cleaved IL‐1β requires caspase‐dependent activation of gasdermin D, which forms pores in the cytoplasmic membrane upon cleavage (Shi et al, 2015; Chen et al, 2016). By specific Western blot, we confirmed increased cleavage of full‐length gasdermin D after infection of macrophages with VN ΔF2 when compared to VN Wt infected cells at 24 hpi, but not at 12 hpi (Figs 3A and B, and EV4A). Increased levels of gasdermin cleavage products confirmed activation of this cell death executor (Fig 3C–E). This finding is in perfect agreement with the increased loss of membrane integrity and elevated LDH levels in the supernatant of VN ΔF2‐infected macrophages (Fig 2B and D–F).
Figure 3. The PB1‐F2 null mutation amplifies NLRP3‐dependent inflammasome activation in H5N1 infected cells.
- THP‐1 cells infected with VN Wt and ΔF2 virus at a MOI 10 for 24 h or treated with 6.7 μM of nigericin (nig) for 45 min were analyzed by immunoblotting. Detection of the full‐length gasdermin D. Equal loading was confirmed by beta actin. One representative blot of five independent repeats is shown.
- Quantification of band intensity for the gasdermin D full‐length isoform. The mean ± standard deviation five independent experiments is shown. Statistical analysis was performed by the paired two‐tailed Student’s t‐test. The asterisk (*) marks a significant difference when compared to the mock group **P < 0.01.
- THP‐1 cells infected with VN Wt and ΔF2 virus at a MOI 10 for 24 h or treated with 6.7 μM of nigericin (nig) for 45 min were analyzed by immunoblotting. Detection of cleaved gasdermin D N‐terminal and C‐terminal isoforms by immunoblot in supernatants; one representative experiment of three independent repeats is shown.
- Quantification of band intensity for N‐terminal gasdermin D isoform band. Three independent experiments are shown. Statistical analysis was performed by the paired two‐tailed Student’s t‐test. *P < 0.05.
- Quantification of band intensity for C‐terminal gasdermin D isoform band. Three independent experiments are shown. Statistical analysis was performed by the paired two‐tailed Student’s t‐test. *P < 0.05.
- THP‐1 cells infected with VN Wt and ΔF2 virus at a MOI 10 for 24 h or treated with 6.7 μM of nigericin (nig) for 45 min were analyzed by immunoblotting. Pro‐caspase‐1 and active caspase‐1 were detected by immunoblot in cell lysates (cell, upper panels) and supernatants (sup, lower panels) from the same experiment. One representative experiment of three independent repeats is shown.
- Quantification of band intensity for active caspase‐1 band. Five independent experiments are shown. Statistical analysis was performed by the paired two‐tailed Student’s t‐test. *P < 0.05.
- Levels of total pro‐caspase‐1 were quantified on pellets and respective supernatants. Three independent experiments are shown. Statistical analysis was performed by the paired two‐tailed Student’s t‐test.
Source data are available online for this figure.
Figure EV4. IAV dependent activation of the NLRP3 inflammasome and caspase 3 after 12 and 24 h.
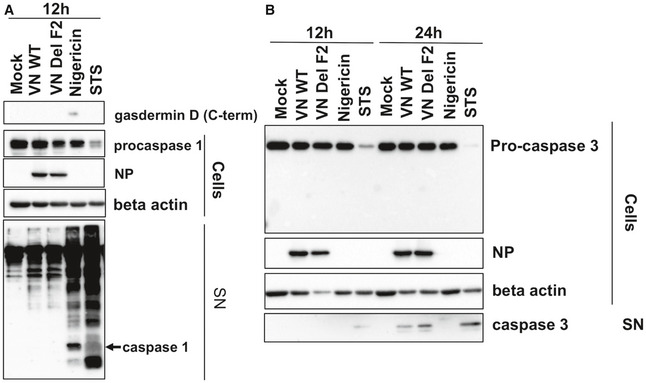
- Supernatants of THP‐1 cells infected with VN Wt and ΔF2 virus at a MOI 10 for 12 h or treated with 6.7 μM of nigericin for 45 min or treated with 1 µM of staurosporine for 12 h or 24 h were analyzed by immunoblotting against gasdermin D, caspase 1, NP, and beta actin (n = 3).
- Supernatants and cell lysates of THP‐1 cells infected with VN Wt and ΔF2 virus at a MOI 10 for 12 h or treated with 6.7 μM of nigericin for 45 min or treated with 1 µM of staurosporin for 12 h were analyzed by immunoblotting against caspase 1, NP, and beta actin (n = 3).
Source data are available online for this figure.
Similarly, we observed more active caspase‐1 in the supernatants of VN ΔF2 infected macrophages at 24 h post‐infection (Fig 3F and G), while both viruses did not cause release of caspase 1 after 12 h of infection (Fig EV4B). Of note, pro‐caspase‐1 levels in the supernatants of infected cells fluctuated substantially but did not show a clear pattern over several experiments (Fig 3H). Potentially, this is a consequence of slightly different timing in gasdermin D pore formation in the independent experiments we performed. Detection of active caspase 1 from the cell lysates was not successful, potentially due to ongoing release into the supernatant. To address an involvement of classical apoptosis, we determined the cleavage of pro‐caspase 3. In cell pellets, no differences were found in pro‐caspase 3 cleavage between mock treated and infected THP‐1‐derived macrophages, while staurosporine clearly diminished the levels of pro‐caspase 3 (Fig 3C). Active caspase 3 could also not be detected in the cell pellets. However, we found active caspase 3 in the supernatants of infected THP‐1 [only at 24 h post‐infection, (Fig EV4C)]. VN ΔF2 caused slightly higher levels of caspase 3 in the supernatants. It is currently unclear, if this is solely a consequence of increased pore formation or a sign of classical apoptosis occurring in parallel.
PB1‐F2 binds to the pyrin and LRR domain of NLRP3 and prevents NEK7 binding
So far our data indicate that PB1‐F2‐dependent inhibition interferes above the level of pro‐caspase 1 cleavage. The simplest model of PB1‐F2 interfering with this step of the NLRP3 signaling pathway would involve a direct interaction of PB1‐F2 with NLRP3. We thus performed pull‐down experiments of overexpressed full‐length N‐terminally flag tagged PB1‐F2s of VN (H5N1), PR8 (H1N1), Wyo (H3N2), and A/Shanghai/1/2013 (H7N9) with V5‐tagged NLRP3. We found robust pull‐down of NLRP3 with the VN and Wyo PB1‐F2 and slightly reduced pull‐down was observed with the H7N9 PB1‐F2 (Fig 4A and B). Of course, pull‐down of endogenous NLRP3 with PB‐F2 from IAV‐infected macrophages would be important to demonstrate, but currently anti‐PB1‐F2 antibodies of high quality are not available. The H1N1 strain PR8 PB1‐F2 did not interact with NLRP3, which could be a consequence of multiple amino acid exchanges as compared to the three binding PB1‐F2s (Fig 4C). Intriguingly, earlier work by McAuley and colleagues indicated that PB1‐F2 of PR/8 might actively induce the NLRP3 inflammasome in a mouse model (McAuley et al, 2013). Using exogenous application of PR8 PB1‐F2‐derived peptides in cell culture and mouse models, they showed increased secretion of Il‐1β after peptide treatment. Similar results were obtained with H7N9 PB1‐F2‐derived peptides (Pinar et al, 2017). It remains currently elusive, if the capacity of PB1‐F2 derived peptides to form large agglomerates (Vidic et al, 2016) contributes to the activation of NLRP3 inflammasomes (McAuley et al, 2013; Pinar et al, 2017). We cannot rule out strain‐specific effects explaining the opposite phenotype as described for PR/8 PB1‐F2 considering the distant phylogenetic relation with contemporary PB1‐F2s (see Appendix Fig S1 and Fig 4C). A set of four amino acids was previously indentified in PR/8 PB1‐F2 to be the molecular determinant of its immunopathogenic function (Alymova et al, 2011); however, these appear to be conserved among the four strains we compared for PB1‐F2‐NLRP3 interaction.
Figure 4. PB1‐F2 interacted with NLRP3 in a strain‐dependent manner.
- Western blot analysis of whole cell lysate (input) and Flag‐specific immunoprecipitation (IP) from 293T cells co‐transfected with expression plasmids encoding V5‐NLRP3 and Flag ZsGreen‐PB1‐F2 from indicated viral strains for 24 h. Representative blot of three independent repeats is shown.
- Quantification of the NLRP3 IP band was normalized to the respective NLRP3 input band (n = 3 independent biological replicates, normalized to the respective input). Mean values ± SD are indicated.
- Alignment of PB1‐F2 sequence from two avian IAV strains (VN (H5N1) and Sha (H7N9)) and two mammalian/human IAV strains (PR8 (H1N1) and Wyo (H3N2)) (Combet et al, 2000). * above the sequence shows a conserved amino acid between the four strains while ↑ on the bottom shows a different amino acid for PR8 in comparison with the other strains.
- Schematic diagrams showing the truncated versions of Flag ZsGreen PB1 F2 used in (E) (not to scale).
- 293T cells co‐transfected with expression plasmids encoding different truncation mutants for Flag Zsgreen‐PB1‐F2 from VN (H5N1) and V5‐NLRP3 were processed as described in (A). Representative blot of three independent repeats is shown.
Source data are available online for this figure.
In our hands, PB1‐F2 of H5N1, H7N9, and human H3N2 viruses binds to NLRP3 and accordingly PB1‐F2 deficient H5N1 and H3N2 viruses induced more Il‐1β in infected human macrophages (both THP1 or PBMC‐derived). This implicates a broader biological function of PB1‐F2 beyond zoonotically infecting viruses. Interestingly, PB1 segments of avian origin were introduced into the pandemic H2N2 viruses (1957) and H3N2 viruses (1968), which could argue for an early function of PB1‐F2 in human hosts. It currently remains elusive, why H1N1 viruses tend to lose a full‐length PB1‐F2 in mammals. Potentially, the NLRP3 inhibition by NS1 could provide a redundant system to control pro‐inflammatory cytokine production (Moriyama et al, 2016).
We next addressed, which domain of PB1‐F2 binds to NLRP3 by using two overlapping truncated versions (aa 1–50 and aa 40–90) of PB1‐F2 in pull‐down experiments (Fig 4D). Only the C‐terminus of VN PB1‐F2 pulled down NLRP3 in these settings, locating the interaction domain to the terminal 40 aa of PB1‐F2 (Fig 4E), which is overall more conserved than the N‐terminus (Fig 4C).
Next, we aimed at narrowing down the interaction domain in NLRP3 by expression of previously defined functional domains (Fig 5A; Yu et al, 2017). Full‐length PB1‐F2 of VN interacts with both the N‐terminal pyrin and the C‐terminal LRR domain of NLRP3 (Fig 5B), but not with the central NACHT domain. PB1‐F2 did not prevent NLRP3 from binding to ASC in a co‐expression system (Fig 5C) confirming the mechanism of action at the level of NLRP3.
Figure 5. Mechanism for PB1‐F2 interaction with NLRP3 hence preventing Nek7 binding.
- Schematic diagrams showing the NLRP3 constructs used in (B).
- 293T cells were co‐transfected with expression plasmids encoding for the indicated V5‐tagged domains of NLRP3 for 24 h. Whole cell lysate (left blot) and flag‐specific IP (right blot) were analyzed by immunoblotting. Equal loading was confirmed by blotting against tubulin. Representative blots of two independent experiments are shown.
- 293T cells were co‐transfected with expression plasmids encoding Flag‐NLRP3, V5‐PB1‐F2, and V5‐ASC for 24 h. Whole cell lysate and flag‐specific IP were analyzed by immunoblotting for indicated antigens. Equal loading of whole cell lysate was assured by blotting against beta actin. Representative blot of two independent repeats is shown.
- 293T cells were co‐transfected with expression plasmids encoding for Flag‐NLRP3, PB1‐F2 or NEK7 or combinations thereof as indicated. Whole cell lysate and Flag IP were analyzed by immunoblotting. Equal loading was confirmed by blotting against beta actin. Representative blots of three independent experiments are shown. Right panel: Quantification of IP bands’ intensity relative to their input. The mean ± standard deviation of three independent experiments is shown. Statistical analysis was performed by paired two‐tailed Student’s t‐test (*P < 0.05).
- As in (D): 293T cells were co‐transfected with expression plasmids encoding for Flag‐NLRP3‐LRR, PB1‐F2 or NEK7 or combinations thereof as indicated. Whole cell lysate and NLRP3‐LRR IP were analyzed by immunoblotting. Equal loading was confirmed by blotting against beta actin. Representative blots of three independent experiments are shown.
- Quantification of IP bands’ intensity relative to their input. The mean ± standard deviation of three independent experiments is shown. Statistical analysis was performed by paired two‐tailed Student’s t‐test (*P < 0.05).
- As in (D and E): 293T cells were co‐transfected with expression plasmids encoding for Flag‐NLRP3‐LRR, PB1‐F2, or V5‐NLRP3‐Pyrin or combinations thereof as indicated. Whole cell lysate and NLRP3‐LRR IP were analyzed by immunoblotting. Equal loading was confirmed by blotting against tubulin. Representative blots of three independent experiments are shown.
Source data are available online for this figure.
Recently, PB1‐F2 was shown to interfere with IFNβ by interaction with the NLR family protein NLRX1 (Jaworska et al, 2014). While a fine‐mapping of the interaction site has not been performed by the authors, it is of note that NLRP3 and NLRX1 share common functional domains; i.e., the NACHT and the LRR domain. Although experimental evidence is missing, this could implicate that PB1‐F2 is generally capable of binding NLRs potentially by interaction through their LRR domain.
NLRP3 was proposed to rest in its auto‐repressed state in non‐activated macrophages. In this conformation the LRR domain makes contact with the NACHT and Pyrin domain, thus preventing auto‐oligomerization of NLRP3‐NACHT domains and NLRP3‐ASC interaction through the pyrin domain (Martinon & Tschopp, 2005). It is conceivable that PB1‐F2 maintains NLRP3 in its auto‐repressed state, thus preventing activation of the downstream signaling events leading to pyroptosis. In order to corroborate this hypothesis, we took advantage of a recently identified host kinase NEK7, binding only to the open/active conformation of NLRP3 (He et al, 2016; Schmid‐Burgk et al, 2016; Shi et al, 2016). We hypothesized that NEK7 should bind less efficiently to NLRP3 in presence of PB1‐F2. Indeed, overexpression of PB1‐F2 substantially reduces NLRP3‐NEK7 interaction (Fig 5D). Recently, published cryo‐EM data revealed in detail the structural nature of the NLRP3‐NEK7 complex (Sharif et al, 2019). NEK7 was proposed to license NLRP3 activation partially by binding to its LRR domain. More specifically, NEK7 is required to bridge the LRRs neighboring NLRP3 monomers, thus allowing efficient oligomerization, which is the structural basis for inflammasome activation. Our data so far did not exclude a direct competition of PB1‐F2 and NEK7 for the same binding site on the LRR of NLRP3. We thus repeated competition IPs using only the LRR domain and NEK7 in the presence or absence of PB1‐F2. Here, PB1‐F2 did not affect the co‐precipitation of NEK7 with the NLRP3‐LRR (Fig 5E and F). Since PB1‐F2 binds both the pyrin and the LRR domain, we hypothesized that it might “lock” NLRP3 in an inactive closed conformation. In absence of PB1‐F2, we did not detect substantial co‐precipitation of LRR and pyrin domain, when the LRR domain was used as bait. Importantly co‐expression of IAV PB1‐F2 enforced co‐precipitation of pyrin and LRR domain of NLRP3 (Fig 5G). These data confirm that PB1‐F2 could stabilize the auto‐repressed and closed confirmation of NLRP3 through interaction with LRR and pyrin domain. Intriguingly, a similar conformational lock was recently described as mechanism of action of the specific NLRP3 inhibitor MCC950 (Tapia‐Abellan et al, 2019), which is proposed to target the NACHT domain.
In the simplest model supported by our data, PB1‐F2 prevents conformational changes of NLRP3 after upstream activation. This conformational arrest prevents a recently identified critical interaction with NEK7 and with it, activation of the downstream signaling cascade funneling into pyroptosis.
A logical next step to our study would be a follow‐up in a suitable, confirmatory in vivo model. Using macrophages (primary and immortalized), we demonstrated the NLRP3‐limiting effect of PB1‐F2 knockout in the human system. Obviously, it would be of great interest to understand the consequences of this viral strategy to antagonize the innate immune system. Out data in mouse macrophages suggest a species dependent effect of PB1‐F2. The origin of this effect is currently unknown, but might originate from alternate interaction of PB1‐F2 with NLRP3 in the two species. Alternative suitable models are ferret and guinea pig; however, for both we currently lack efficient genetic tools to achieve targeted suppression of the NLRP3 inflammasome system. The pathological consequences of NLRP3 activation in presence or absence of a functional PB1‐F2 could be addressed in a causal fashion in such a system.
Materials and Methods
Plasmids
pCR3.V62‐Met‐Flag‐NLRP3 was kindly provided by Dr. F. Martinon, University of Lausanne, Switzerland. The cDNAs for NLRP3 and its functional domains, for PB1‐F2 isoforms and for Nek7 coupled to Flag ZsGreen or V5 tag positioned n‐terminally were amplified by standard PCR and inserted into a pCAGGS expression plasmid using standard restriction enzyme based cloning techniques (Niwa et al, 1991). pDONR223‐NEK7 was a gift from Dr. W. Hahn & Dr. D. Root (Addgene plasmid # (Schmolke et al, 2011) 23395) (Johannessen et al, 2010). A list of primers for cloning and qPCR is provided in Appendix Table S2.
Antibodies
Antibodies used for immunoblotting included mouse monoclonal anti‐actin‐HRP (Sigma‐Aldrich, Cat# A5441), mouse monoclonal anti‐tubulin‐HRP (Cell signaling, Cat# 12351), rabbit monoclonal anti‐NEK7 (Abcam, Cat# ab133514), rabbit monoclonal anti‐caspase‐1 (Cell signaling, Cat# 3866S), mouse monoclonal anti‐caspase‐1 (Adipogen, Cat# AG‐20B‐0048) rabbit monoclonal anti‐ASC (Cell signaling, Cat# 13833), rabbit monoclonal anti‐NLRP3 (Cell signaling, Cat# 15101), rabbit polyclonal anti‐Gasdermin D (Novus, NBP2‐33422), rabbit monoclonal anti‐Gasdermin D (Abcam, ab210070) mouse anti‐Flag‐HRP (Sigma; Cat# A8592), mouse anti‐V5 (Invitrogen; R960‐25), goat polyclonal anti‐mouse‐HRP (Sigma‐Aldrich Cat# A5278), goat polyclonal anti‐rabbit‐HRP (Sigma‐Aldrich, Cat# A0545), rabbit polyclonal anti‐influenza virus NP (Invitrogen, Cat# PA5‐32242), mouse monoclonal antibody anti‐influenza virus M1 (Bio‐Rad; Cat# MCA40), and rabbit polyclonal anti‐H5N1 PB1‐F2 clone 9947, described elsewhere (Schmolke et al, 2011).
Viruses
Low‐pathogenicity H5N1 virus [A/Viet Nam/1203/04, HAlo (Steel et al, 2009)], H1N1 virus (A/Puerto Rico/8/1934), and human H3N2 virus (A/Wyoming/03/2003) and corresponding PB1‐F2 mutants of these viruses were generated by reverse genetics as described in (Mazel‐Sanchez et al, 2018). Reverse genetics systems were kindly provided by Dr. A. García‐Saste and Dr. P. Palese, Icahn School of Medicine at Mount Sinai, New York, USA. H5N1 (VN) and H1N1 (PR8) strains were expanded in embryonated chicken eggs (Eisfeld et al, 2014), and stocks of the H3N2 (Wyo) virus strain were propagated in MDCK cells (Anchisi et al, 2018).
PB1‐F2 derived peptides
Peptide sequences of A/Viet Nam/1203/2003 PB1‐F2 C‐terminus (aa 60–90) and a matching scrambled peptide are indicated in Appendix Fig S1. Peptides were synthesized at the Peptide Synthesis Platform (Faculty of Medicine, University of Geneva) using a standard Fmoc solid phase peptide synthesis strategy on a MultiPep Rsi synthesizer (Intavis AG, Germany), with the 5(6)‐Carboxyfluorescein manually coupled to the N‐terminal amine of the peptide. After resin cleavage, crude peptides were purified by reverse‐phase HPLC. Purified peptides were assessed for purity using analytical reverse‐phase HPLC (> 90%) and authenticated by mass spectrometry. THP‐1‐derived macrophages were exposed to 200 μg/ml of peptide for indicated time points. LDH release was measured from supernatants as described below.
Ethics statement
All human donors of monocytes provided informed consent in written form. All animal procedures were in accordance with federal regulations of the Bundesamt für Lebensmittelsicherheit und Veterenärwesen (BLV), Switzerland (Tierschutzgesetz), and approved by direction de l'expérimentation animale and the cantonal authorities of the canton Geneve (license number GE/44/17). Embryonated chicken eggs were obtained from the University of Geneva Animalerie d'Arare and infected on day 10 of embryonic development.
Mice
C57BL/6J age matched, 8‐ to 10‐week‐old female mice were purchased from Charles River Laboratories (France) and housed under SPF/BSL2 conditions. Animals were housed in enriched environments under a strict 12‐h day/night cycle and fed ad libitum.
For infection with H5N1 Wt and ΔF2 strains, mice were first injected intraperitoneally with a mix of ketamin/xylazine (100 and 5 mg/kg, respectively) in 100 µl of sterile PBS. Upon reaching deep anesthesia, mice were inoculated with 40 µl of PBS or 40 PFU of virus via the intranasal route. Body weights were measured daily during the light phase. Animals were euthanized using controlled CO2 exposure. BAL sampling was performed as previously described (Sun et al, 2017). Tools were changed in between experimental groups to avoid cross‐contamination. Lung tissue was collected for viral titers analyses. Viral titers in the lungs were determined by standard plaque assay on MDCK cells as described before (Mazel‐Sanchez et al, 2018). Supernatants from BAL samples were stored at −80°C until being processed.
For FACS analysis of invading immune cells, total lungs were harvested, cut in small pieces, and digested in 3 ml of digestion mix (HBSS, collagenase D at 1 mg/ml (Roche), DNase I at 10 μg/ml (Roche) and FBS 1%) for 40 min with one step of mechanical dissociation (resuspension up and down with a pipette) after 20 min. After the digestion step, samples were filtered on 70 μm cell strainer. Cells were then separated by centrifugation (1,030 g for 30 min, 18°C) on a discontinuous isotonic Percoll gradient containing 70 and 30% layers. Mononuclear cells at the 70–30% interface were collected and washed extensively in FACS buffer.
Study population for primary human alveolar macrophages
Bronchoalveolar lavage (BAL) was performed in five children aged 1–16 years (see Appendix Table S2) free from infection undergoing bronchoscopy after written informed consent of parents was obtained. BAL was performed with three aliquots of NaCl 0.9% (1 ml/kg per aliquot, maximum 20 ml each) from the right middle pulmonary lobe. BAL portions 1 and 2 were analyzed for presence of bacterial pathogens per microbial culture and respiratory viruses per PCR (all negative in all children). BAL portion 3 was analyzed for cell distribution (normal in all five children with alveolar macrophages 91–97%, lymphocytes 2–7%, neutrophils 1–3%), and remnants were used for AM cultivation. Reasons for bronchoscopy were as follows: 1 after suspected foreign body aspiration ultimately retrieved from upper esophagus, 3 during surveillance bronchoscopy 2–9 months post‐lung transplantation (LTx), 1 early (2 weeks) post‐LTx. Patients after lung transplantation were treated with immunosuppressants at the time of BAL (prednisolone 0.1–0.6 mg/kg/day, mycophenolate mofetil 33–45 mg/kg/day, tacrolimus with trough serum levels of 4.6–24 µg/l). Lung function tests were stable in all patients.
Cell culture and infection conditions of human alveolar macrophages
Human AMs were obtained by seeding the cells of the 3rd cell fraction of freshly obtained BALs at a concentration of 1 × 106/ml in tissue culture dishes in RPMI 1640 supplemented with 1% glutamine, 1% penicillin–streptomycin, and 10% human AB plasma. After overnight culture, non‐adherent cells were removed and adherent AMs infected for 16 h with human H3N2 virus (A/Wyoming/03/2003) and the corresponding PB1‐F2 mutant. Control AMs were treated for 16 h with PBS. Supernatants were harvested for subsequent cytokine analyses, and cells were harvested for total RNA isolation and subsequent gene expression analyses.
PBMC‐derived macrophages
Peripheral blood buffy coats were obtained from healthy blood donors provided by the Centre de Transfusion Sanguine, Hôpitaux Universitaires Genève. Mononuclear cells were selected by density centrifugation with Ficoll (Lymphoprep™, AXIS‐SHIELD), and monocyte isolation was performed by magnetic labeling using CD14 Microbeads (Miltenyi Biotec), according to the manufacturer’s instructions. Isolated monocytic cells were differentiated into macrophages by exposure to 2 μg/ml of GM‐CSF during 7 days.
Cell lines
HEK 293T cells (Human embryonic kidney; ATCC) and MDCK (Madin‐Darby canine kidney; ATCC) were cultured in DMEM (Dulbecco's modified Eagle medium, Gibco). Cell culture media were supplemented with 10% heat‐inactivated fetal bovine serum (FBS) (Gibco) and 1% penicillin–streptomycin (Pen‐Strep) (Sigma‐Aldrich). Human monocytic THP‐1 (ATCC) cells were cultured in RPMI 1640 media (Gibco) complemented with 10% FBS, 1% Pen‐Strep and 0.05 mM of 2‐mercaptoethanol (Sigma‐Aldrich). CRISPR/Cas‐modified THP‐1 (kindly provided by Dr. Veit Hornung LMU, Munich, Germany) (Schmid‐Burgk et al, 2015) was cultured in RPMI 1640 media complemented with 10% FBS, 1% Pen‐Strep and 1% sodium pyruvate (Gibco). All cells were incubated at 37°C with 5% CO2 and 90% humidity.
Cell differentiation and stimulation
Monocytic THP‐1 cells were stimulated to differentiate into adherent macrophage‐like cells by treatment with 0.5 μM of PMA (Phorbol 12‐myristate 13‐acetate) (Sigma‐Aldrich) for 3 h at 37°C, washed once in PBS (Gibco), and seeded at 2 × 106 cells/ml in complete media overnight. As a positive control for cell death, differentiated THP‐1 cells were treated with 1 μM of staurosporine (STS) (Sigma‐Aldrich) for 12 or 24 h. As a positive control for inflammasome activation, differentiated THP‐1 cells were treated with 6.7 μM of nigericin (Tocris) for 45 min. Primary human monocytes were induced into macrophage differentiation by treatment with 100 ng/ml of GM‐CSF (Sigma‐Aldrich) for 7 days in RPMI supplemented with 10% FBS.
PMA‐differentiated THP‐1 cells were seeded in 12‐well plates at 2 × 106 cells/ml for infection 16 h post‐seeding. Cells were washed once with PBS followed by incubation with each indicated virus in OPTI‐MEM (Thermo Fischer) at a MOI of 10 (unless stated otherwise in the figure legend). After adsorption for 40 min at 37°C, the infection media were removed and 500 μl of OPTI‐MEM was added to the cells and cells were kept in culture for the times indicated in figure legends.
For primary human macrophages, cells were treated as described in the “Cell differentiation and stimulation” section above and afterward infected in the same manner as for THP‐1 cells.
Infections and handling of infected cells was carried out in a biosafety cabinet under BSL2 conditions.
Flow cytometry assay
THP‐1 cells were infected at a MOI of 10 for 12 h or 24 h. At the mentioned time points, the cells were harvested using trypsin‐EDTA (Sigma‐Aldrich) and washed with PBS. Centrifugations were performed at 400 rcf. The staining protocol was performed as described in (Mazel‐Sanchez et al, 2018). Briefly, after washing, the cells were incubated with Zombie Red fixable viability dye (Bio‐legend) 1/100 in PBS, in the dark for 30 min at 4°C. Then, the cells were washed once in PBS‐2% BSA and once with annexin V binding buffer (Bio‐legend) followed by incubation with annexin V (Bio‐legend) 5/100 for 15 min at room temperature (RT). After incubation, cells were washed once with annexin V binding buffer and once with PBS‐Ca‐Mg. Cells were later fixed in 2% formaldehyde diluted in PBS‐Ca‐Mg for 20 min at RT. Cells were washed again and resuspended in PBS‐Ca‐Mg. Samples were then analyzed by flow cytometry using a Gallios 4 (Beckman Coulter), and results were analyzed with the Kaluza Analysis 1.5a software (Beckman).
For differential staining of immune cells invading the lung during infection, we used antibodies from BD biosciences: TCR‐BV711 (H57‐597), CD4‐BUV737 (GK1.5), Invitrogen/ebiosciences: CD11c‐Pecy7 (N418), F4/80‐PE‐e610 (BM8), CD8‐A700 (53‐6.7), IFNγ‐PE (XMG1.2), and biolegend: CD11b‐BV605 (M1/70), CD19‐BV785 (6D5), CD45‐A647 (30‐F11), CD45‐BV510 (30‐F11), GranzymeB‐Pacific blue (GB11).
For viability staining, cells were stained for 15 min at 4°C with fixable viability dye eFluor 780 in PBS (Ebiosciences). For surface markers, cells were stained for 30 min at 4°C with fluorophore‐conjugated antibodies in FACS buffer (BSA 0.05%, 0.05 mM EDTA, PBS). For intracellular stainings, cells were fixed after surface staining for 15 min at room temperature and thereafter permeabilized using the Intracellular Fixation & Permeabilization buffer set (eBioscience). For IFN‐γ, IL‐17, and GM‐CSF staining, cells were first re‐stimulated in complete RPMI containing PMA/ionomycin and Golgi plug (BD Biosciences) and incubated 4 h at 37°C, 5% CO2. Samples were acquired on the BD Biosciences Fortessa device and analyzed using FlowJo software (FlowJo company).
Immunoblotting
Cell lysates were collected in RIPA buffer, sonicated, and boiled for 5 min at 85°C. After BCA protein quantification (Pierce, Thermo scientific), the same protein amount for each condition was electrophoretically separated in denaturing SDS‐polyacrylamide gels, transferred to nitrocellulose membranes (GE Healthcare), blocked with 5% skim milk in TBS containing 0.1% Tween 20 (Applichem), and incubated with respective primary antibodies (described above), overnight at 4°C Secondary anti‐mouse or anti‐rabbit conjugated to HRP (when necessary) were incubated for 1 h and developed using the ECL chemiluminescence reagent (Witec AG).
ELISA
Human IL‐1β was quantified in the supernatants by ELISA. Assays were performed on clear Nunc‐Immuno plates (Thermo scientific) according to the manufacturer's instructions (BD Biosciences). TMB solution (Sigma‐Aldrich) was added to the plates followed by 1 M of H3PO4 (Fischer scientific) to stop the reaction. Captured cytokines were detected by reading absorbance values in a Bio‐Rad 680 microplate reader. Analysis was done using the four‐parameter logistic ELISA curve fitting available on https://elisaanalysis.com/.
LDH assay
THP‐1 were plated overnight and infected with VN Wt or ΔF2 for 12 h with 10 MOI as described above. After 12 h, the supernatants were collected for LDH release assay. LDH present in the supernatants was measured using the Cytotoxicity Detection Kit PLUS (Sigma‐Aldrich, Cat# 04 744 926 001) according to the manufacturer’s instructions.
For LDH measurements in the bronchoalveolar lavage (BAL), PBS aspirated after injection into the lungs of mice mock treated or infected with VN Wt or ΔF2 for 3 d was exposed to the substrate and treated as described for THP‐1 cells supernatants. Each BAL sample from 5 mice per condition was analyzed twice.
Immunoprecipitation assay
80% sub‐confluent HEK293T cells (35 mm dish) were co‐transfected with varying amounts of DNA (1 μg per expression plasmid using 2 μl/μg DNA of Trans‐IT LT1 (Mirus)) according to the manufacturer’s protocol. 24 h post‐transfection, cells were washed in cold PBS and cross‐linked with 1% formaldehyde in PBS for 10 min (for NLRP3 and Nek7 interaction studies only). The fixing solution was removed, and 125 mM of glycine in PBS was added for another 5 min, followed by washing with PBS. Cells were lysed on ice with 300 μl of lysis buffer [50 mM Tris–HCl (pH 7.5), 150 mM NaCl, 0.5 mM EDTA, 0.5% (vol/vol) NP‐40], containing a protease and phosphatase inhibitor cocktail (Pierce, Cat# 88266 and Sigma‐Aldrich Cat# 4906845001, respectively). Samples were sonicated and 100 μl of the supernatants obtained after centrifugation of the cell lysates was bound to anti‐Flag M2 affinity gel agarose beads (Sigma‐Aldrich, Cat# A2220) and incubated overnight at 4°C, while 50 μl was mixed with sample buffer to serve as input control. The beads were washed extensively in lysis buffer, and the samples were analyzed by immunoblotting (described above) with the indicated antibodies.
RT–qPCR
Total RNA was extracted from infected or mock treated cells using the EZNA®Total RNA Kit I (Omega Bio‐tek) or the NucleoSpin® kit (Machery&Nagel), according to the manufacturer’s instructions. Total RNA was quantified from the samples using the NanoDrop 2000 (Thermo Scientific), and 500 ng per sample was used for reverse transcription reaction, performed with SuperScript™ II Reverse Transcriptase (Invitrogen). Primers for 18S or respective cytokine mRNA (see Appendix Table S2) were used, and the ΔΔC t values were calculated (Livak & Schmittgen, 2001).
Phyllogenetic analysis of PB1‐F2 sequences
Phylogenetic tree obtained from of PB1‐F2 protein sequences of Influenza A virus selected from various epidemic periods, after removal of partial sequences. The multiple sequence alignment and NJ were performed in MAFFT (Kuraku et al, 2013) using default parameters. The Newick resulting tree was formatted in MEGA7 (Molecular Evolutionary Genetics Analysis) (Kumar et al, 2016). Sequences obtained for human H3N2 isolates formed a large cluster colored in blue. The tree is not rooted and drawn to scale, with branch lengths in the same units as those of the evolutionary distances used to infer the phylogenetic tree.
Statistical analysis
All statistical analysis was performed in Prism 7 (GraphPad Software, version 7.0d). The number of independent experiments is indicated in each figure legend. Statistical tests and significance are indicated in each figure legend.
Author contributions
IB‐C, BM‐S, SY, FS, CN, NW, JPPBL, JS, LG, and SH performed experiments and/or analyzed data; LG and SH analyzed cell influx into the lungs of infected mice; PF established phyllogenetic analysis of human IAV PB1‐F2; JC isolated AM from the lung of human patients; DV. NS, and JS isolated and infected primary human alveolar macrophages; IB‐C, DV, SH, and MS designed experiments; IB‐C designed the figures; IB‐C and MS conceptualized the project; IB‐C, BM‐S, and MS wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Review Process File
Expanded View Figures PDF
Source Data for Expanded View
Appendix
Source Data for Figure 1
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Acknowledgements
This work was supported by the Boninchi Foundation and the Swiss National Fund (SNF 310030_155949, SNF 310030_155949/2, and SNF 310030_182475/1) granted to MS and SNF310030‐185255 granted to SH). This work was supported by grants to D.V. from the DFG under Germany’s Excellence Strategy—EXC 2155‐390874280 (RESIST). We are grateful for the excellent service provided by the CMU animal facility, the Bio‐imaging platform, and the Centre de Transfusion Sanguine (HUG). We are grateful for technical advice from Dr. Fabio Martinon.
EMBO Reports (2020) 21: e50421.
Data availability
No large data sets where generated in this study.
References
- Aarreberg LD, Wilkins C, Ramos HJ, Green R, Davis MA, Chow K, Gale M Jr (2018) Interleukin‐1beta signaling in dendritic cells induces antiviral interferon responses. MBio 9: e00342‐18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aglietti RA, Dueber EC (2017) Recent insights into the molecular mechanisms underlying pyroptosis and gasdermin family functions. Trends Immunol 38: 261–271 [DOI] [PubMed] [Google Scholar]
- Allen IC, Scull MA, Moore CB, Holl EK, McElvania‐TeKippe E, Taxman DJ, Guthrie EH, Pickles RJ, Ting JP (2009) The NLRP3 inflammasome mediates in vivo innate immunity to influenza A virus through recognition of viral RNA. Immunity 30: 556–565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alymova IV, Green AM, van de Velde N, McAuley JL, Boyd KL, Ghoneim HE, McCullers JA (2011) Immunopathogenic and antibacterial effects of H3N2 influenza A virus PB1‐F2 map to amino acid residues 62, 75, 79, and 82. J Virol 85: 12324–12333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anchisi S, Goncalves AR, Mazel‐Sanchez B, Cordey S, Schmolke M (2018) Influenza A virus genetic tools: from clinical sample to molecular clone. Methods Mol Biol 1836: 33–58 [DOI] [PubMed] [Google Scholar]
- Ben‐Sasson SZ, Hu‐Li J, Quiel J, Cauchetaux S, Ratner M, Shapira I, Dinarello CA, Paul WE (2009) IL‐1 acts directly on CD4 T cells to enhance their antigen‐driven expansion and differentiation. Proc Natl Acad Sci USA 106: 7119–7124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben‐Sasson SZ, Hogg A, Hu‐Li J, Wingfield P, Chen X, Crank M, Caucheteux S, Ratner‐Hurevich M, Berzofsky JA, Nir‐Paz R et al (2013) IL‐1 enhances expansion, effector function, tissue localization, and memory response of antigen‐specific CD8 T cells. J Exp Med 210: 491–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardani A, Boulton A, Kim TS, Braciale TJ (2017) Alveolar macrophages prevent lethal influenza pneumonia by inhibiting infection of type‐1 alveolar epithelial cells. PLoS Pathog 13: e1006140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassel SL, Joly S, Sutterwala FS (2009) The NLRP3 inflammasome: a sensor of immune danger signals. Semin Immunol 21: 194–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang YJ, Kim HY, Albacker LA, Baumgarth N, McKenzie AN, Smith DE, Dekruyff RH, Umetsu DT (2011) Innate lymphoid cells mediate influenza‐induced airway hyper‐reactivity independently of adaptive immunity. Nat Immunol 12: 631–638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, Calvo PA, Malide D, Gibbs J, Schubert U, Bacik I, Basta S, O'Neill R, Schickli J, Palese P et al (2001) A novel influenza A virus mitochondrial protein that induces cell death. Nat Med 7: 1306–1312 [DOI] [PubMed] [Google Scholar]
- Chen X, He WT, Hu L, Li J, Fang Y, Wang X, Xu X, Wang Z, Huang K, Han J (2016) Pyroptosis is driven by non‐selective gasdermin‐D pore and its morphology is different from MLKL channel‐mediated necroptosis. Cell Res 26: 1007–1020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin J, Kostura MJ (1993) Dissociation of IL‐1 beta synthesis and secretion in human blood monocytes stimulated with bacterial cell wall products. J Immunol 151: 5574–5585 [PubMed] [Google Scholar]
- Cole SL, Dunning J, Kok WL, Benam KH, Benlahrech A, Repapi E, Martinez FO, Drumright L, Powell TJ, Bennett M et al (2017) M1‐like monocytes are a major immunological determinant of severity in previously healthy adults with life‐threatening influenza. JCI Insight 2: e91868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Combet C, Blanchet C, Geourjon C, Deleage G (2000) NPS@: network protein sequence analysis. Trends Biochem Sci 25: 147–150 [DOI] [PubMed] [Google Scholar]
- de Jong MD, Simmons CP, Thanh TT, Hien VM, Smith GJ, Chau TN, Hoang DM, Chau NV, Khanh TH, Dong VC et al (2006) Fatal outcome of human influenza A (H5N1) is associated with high viral load and hypercytokinemia. Nat Med 12: 1203–1207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinarello CA (1998) Interleukin‐1 beta, interleukin‐18, and the interleukin‐1 beta converting enzyme. Ann N Y Acad Sci 856: 1–11 [DOI] [PubMed] [Google Scholar]
- Dinarello CA (2009) Immunological and inflammatory functions of the interleukin‐1 family. Annu Rev Immunol 27: 519–550 [DOI] [PubMed] [Google Scholar]
- Eisfeld AJ, Neumann G, Kawaoka Y (2014) Influenza A virus isolation, culture and identification. Nat Protoc 9: 2663–2681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ettensohn DB, Frampton MW, Nichols JE, Roberts NJ Jr (2016) Human alveolar macrophages may not be susceptible to direct infection by a human influenza virus. J Infect Dis 214: 1658–1665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenton MJ, Vermeulen MW, Clark BD, Webb AC, Auron PE (1988) Human pro‐IL‐1 beta gene expression in monocytic cells is regulated by two distinct pathways. J Immunol 140: 2267–2273 [PubMed] [Google Scholar]
- Fink SL, Cookson BT (2005) Apoptosis, pyroptosis, and necrosis: mechanistic description of dead and dying eukaryotic cells. Infect Immun 73: 1907–1916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friesenhagen J, Boergeling Y, Hrincius E, Ludwig S, Roth J, Viemann D (2012) Highly pathogenic avian influenza viruses inhibit effective immune responses of human blood‐derived macrophages. J Leukoc Biol 92: 11–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guilliams M, De Kleer I, Henri S, Post S, Vanhoutte L, De Prijck S, Deswarte K, Malissen B, Hammad H, Lambrecht BN (2013) Alveolar macrophages develop from fetal monocytes that differentiate into long‐lived cells in the first week of life via GM‐CSF. J Exp Med 210: 1977–1992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y, Zeng MY, Yang D, Motro B, Nunez G (2016) NEK7 is an essential mediator of NLRP3 activation downstream of potassium efflux. Nature 530: 354–357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hennet T, Ziltener HJ, Frei K, Peterhans E (1992) A kinetic study of immune mediators in the lungs of mice infected with influenza A virus. J Immunol 149: 932–939 [PubMed] [Google Scholar]
- Herold S, von Wulffen W, Steinmueller M, Pleschka S, Kuziel WA, Mack M, Srivastava M, Seeger W, Maus UA, Lohmeyer J (2006) Alveolar epithelial cells direct monocyte transepithelial migration upon influenza virus infection: impact of chemokines and adhesion molecules. J Immunol 177: 1817–1824 [DOI] [PubMed] [Google Scholar]
- Hogner K, Wolff T, Pleschka S, Plog S, Gruber AD, Kalinke U, Walmrath HD, Bodner J, Gattenlohner S, Lewe‐Schlosser P et al (2013) Macrophage‐expressed IFN‐beta contributes to apoptotic alveolar epithelial cell injury in severe influenza virus pneumonia. PLoS Pathog 9: e1003188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogquist KA, Nett MA, Unanue ER, Chaplin DD (1991) Interleukin 1 is processed and released during apoptosis. Proc Natl Acad Sci USA 88: 8485–8489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu AC (2018) Influenza virus: a master tactician in innate immune evasion and novel therapeutic interventions. Front Immunol 9: 743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichinohe T, Lee HK, Ogura Y, Flavell R, Iwasaki A (2009) Inflammasome recognition of influenza virus is essential for adaptive immune responses. J Exp Med 206: 79–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichinohe T, Pang IK, Iwasaki A (2010) Influenza virus activates inflammasomes via its intracellular M2 ion channel. Nat Immunol 11: 404–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaworska J, Coulombe F, Downey J, Tzelepis F, Shalaby K, Tattoli I, Berube J, Rousseau S, Martin JG, Girardin SE et al (2014) NLRX1 prevents mitochondrial induced apoptosis and enhances macrophage antiviral immunity by interacting with influenza virus PB1‐F2 protein. Proc Natl Acad Sci USA 111: E2110–2119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johannessen CM, Boehm JS, Kim SY, Thomas SR, Wardwell L, Johnson LA, Emery CM, Stransky N, Cogdill AP, Barretina J et al (2010) COT drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature 468: 968–972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamal RP, Alymova IV, York IA (2018) Evolution and virulence of influenza A virus protein PB1‐F2. Int J Mol Sci 19: 96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanneganti TD, Body‐Malapel M, Amer A, Park JH, Whitfield J, Franchi L, Taraporewala ZF, Miller D, Patton JT, Inohara N et al (2006) Critical role for Cryopyrin/Nalp3 in activation of caspase‐1 in response to viral infection and double‐stranded RNA. J Biol Chem 281: 36560–36568 [DOI] [PubMed] [Google Scholar]
- Kayagaki N, Stowe IB, Lee BL, O'Rourke K, Anderson K, Warming S, Cuellar T, Haley B, Roose‐Girma M, Phung QT et al (2015) Caspase‐11 cleaves gasdermin D for non‐canonical inflammasome signalling. Nature 526: 666–671 [DOI] [PubMed] [Google Scholar]
- Kostura MJ, Tocci MJ, Limjuco G, Chin J, Cameron P, Hillman AG, Chartrain NA, Schmidt JA (1989) Identification of a monocyte specific pre‐interleukin 1 beta convertase activity. Proc Natl Acad Sci USA 86: 5227–5231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Stecher G, Tamura K (2016) MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol 33: 1870–1874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuraku S, Zmasek CM, Nishimura O, Katoh K (2013) aLeaves facilitates on‐demand exploration of metazoan gene family trees on MAFFT sequence alignment server with enhanced interactivity. Nucleic Acids Res 41: W22–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SM, Dutry I, Peiris JS (2012) Editorial: macrophage heterogeneity and responses to influenza virus infection. J Leukoc Biol 92: 1–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee DJ, Du F, Chen SW, Nakasaki M, Rana I, Shih VF, Hoffmann A, Jamora C (2015) Regulation and function of the Caspase‐1 in an inflammatory microenvironment. J Invest Dermatol 135: 2012–2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann C, Sprenger H, Nain M, Bacher M, Gemsa D (1996) Infection of macrophages by influenza A virus: characteristics of tumour necrosis factor‐alpha (TNF alpha) gene expression. Res Virol 147: 123–130 [DOI] [PubMed] [Google Scholar]
- Lin KL, Suzuki Y, Nakano H, Ramsburg E, Gunn MD (2008) CCR2+ monocyte‐derived dendritic cells and exudate macrophages produce influenza‐induced pulmonary immune pathology and mortality. J Immunol 180: 2562–2572 [DOI] [PubMed] [Google Scholar]
- Liu Q, Zhou YH, Yang ZQ (2016) The cytokine storm of severe influenza and development of immunomodulatory therapy. Cell Mol Immunol 13: 3–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real‐time quantitative PCR and the 2(‐Delta Delta C(T)) Method. Methods 25: 402–408 [DOI] [PubMed] [Google Scholar]
- Mariathasan S, Weiss DS, Newton K, McBride J, O'Rourke K, Roose‐Girma M, Lee WP, Weinrauch Y, Monack DM, Dixit VM (2006) Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 440: 228–232 [DOI] [PubMed] [Google Scholar]
- Martinon F, Tschopp J (2005) NLRs join TLRs as innate sensors of pathogens. Trends Immunol 26: 447–454 [DOI] [PubMed] [Google Scholar]
- Mazel‐Sanchez B, Boal Carvalho I, Silva F, Dijkman R, Schmolke M (2018) H5N1 influenza A virus PB1‐F2 relieves HAX‐1‐mediated restriction of avian virus polymerase PA in human lung cells. J Virol 92: e00425‐18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAuley JL, Hornung F, Boyd KL, Smith AM, McKeon R, Bennink J, Yewdell JW, McCullers JA (2007) Expression of the 1918 influenza A virus PB1‐F2 enhances the pathogenesis of viral and secondary bacterial pneumonia. Cell Host Microbe 2: 240–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAuley JL, Chipuk JE, Boyd KL, Van De Velde N, Green DR, McCullers JA (2010) PB1‐F2 proteins from H5N1 and 20 century pandemic influenza viruses cause immunopathology. PLoS Pathog 6: e1001014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAuley JL, Tate MD, MacKenzie‐Kludas CJ, Pinar A, Zeng W, Stutz A, Latz E, Brown LE, Mansell A (2013) Activation of the NLRP3 inflammasome by IAV virulence protein PB1‐F2 contributes to severe pathophysiology and disease. PLoS Pathog 9: e1003392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriyama M, Chen IY, Kawaguchi A, Koshiba T, Nagata K, Takeyama H, Hasegawa H, Ichinohe T (2016) The RNA‐ and TRIM25‐binding domains of influenza virus NS1 protein are essential for suppression of NLRP3 inflammasome‐mediated interleukin‐1beta secretion. J Virol 90: 4105–4114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu J, Wu S, Chen M, Xu K, Guo Q, Lu A, Zhao L, Sun B, Meng G (2019) Hyperactivation of the NLRP3 inflammasome protects mice against influenza A virus infection via IL‐1beta mediated neutrophil recruitment. Cytokine 120: 115–124 [DOI] [PubMed] [Google Scholar]
- Niwa H, Yamamura K, Miyazaki J (1991) Efficient selection for high‐expression transfectants with a novel eukaryotic vector. Gene 108: 193–199 [DOI] [PubMed] [Google Scholar]
- Ong JD, Mansell A, Tate MD (2016) Hero turned villain: NLRP3 inflammasome‐induced inflammation during influenza A virus infection. J Leukoc Biol 101: 863–874 [DOI] [PubMed] [Google Scholar]
- Oshansky CM, Gartland AJ, Wong SS, Jeevan T, Wang D, Roddam PL, Caniza MA, Hertz T, DeVincenzo JP, Webby RJ et al (2014) Mucosal immune responses predict clinical outcomes during influenza infection independently of age and viral load. Am J Resp Crit Care 189: 449–462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peschke T, Bender A, Nain M, Gemsa D (1993) Role of macrophage cytokines in influenza A virus infections. Immunobiology 189: 340–355 [DOI] [PubMed] [Google Scholar]
- Piccini A, Carta S, Tassi S, Lasiglie D, Fossati G, Rubartelli A (2008) ATP is released by monocytes stimulated with pathogen‐sensing receptor ligands and induces IL‐1beta and IL‐18 secretion in an autocrine way. Proc Natl Acad Sci USA 105: 8067–8072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinar A, Dowling JK, Bitto NJ, Robertson AA, Latz E, Stewart CR, Drummond GR, Cooper MA, McAuley JL, Tate MD et al (2017) PB1‐F2 peptide derived from avian influenza A virus H7N9 induces inflammation via activation of the NLRP3 inflammasome. J Biol Chem 292: 826–836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pothlichet J, Meunier I, Davis BK, Ting JP, Skamene E, von Messling V, Vidal SM (2013) Type I IFN triggers RIG‐I/TLR3/NLRP3‐dependent inflammasome activation in influenza A virus infected cells. PLoS Pathog 9: e1003256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid‐Burgk JL, Gaidt MM, Schmidt T, Ebert TS, Bartok E, Hornung V (2015) Caspase‐4 mediates non‐canonical activation of the NLRP3 inflammasome in human myeloid cells. Eur J Immunol 45: 2911–2917 [DOI] [PubMed] [Google Scholar]
- Schmid‐Burgk JL, Chauhan D, Schmidt T, Ebert TS, Reinhardt J, Endl E, Hornung V (2016) A genome‐wide CRISPR (clustered regularly interspaced short palindromic repeats) screen identifies NEK7 as an essential component of NLRP3 inflammasome activation. J Biol Chem 291: 103–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz N, Kurrer M, Bachmann MF, Kopf M (2005) Interleukin‐1 is responsible for acute lung immunopathology but increases survival of respiratory influenza virus infection. J Virol 79: 6441–6448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmolke M, Garcia‐Sastre A (2010) Evasion of innate and adaptive immune responses by influenza A virus. Cell Microbiol 12: 873–880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmolke M, Manicassamy B, Pena L, Sutton T, Hai R, Varga ZT, Hale BG, Steel J, Perez DR, Garcia‐Sastre A (2011) Differential contribution of PB1‐F2 to the virulence of highly pathogenic H5N1 influenza A virus in mammalian and avian species. PLoS Pathog 7: e1002186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharif H, Wang L, Wang WL, Magupalli VG, Andreeva L, Qiao Q, Hauenstein AV, Wu Z, Nunez G, Mao Y et al (2019) Structural mechanism for NEK7‐licensed activation of NLRP3 inflammasome. Nature 570: 338–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi H, Wang Y, Li X, Zhan X, Tang M, Fina M, Su L, Pratt D, Bu CH, Hildebrand S et al (2016) NLRP3 activation and mitosis are mutually exclusive events coordinated by NEK7, a new inflammasome component. Nat Immunol 17: 250–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, Zhuang Y, Cai T, Wang F, Shao F (2015) Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526: 660–665 [DOI] [PubMed] [Google Scholar]
- Short KR, Brooks AG, Reading PC, Londrigan SL (2012) The fate of influenza A virus after infection of human macrophages and dendritic cells. J Gen Virol 93: 2315–2325 [DOI] [PubMed] [Google Scholar]
- Short KR, Kroeze E, Fouchier RAM, Kuiken T (2014) Pathogenesis of influenza‐induced acute respiratory distress syndrome. Lancet Infect Dis 14: 57–69 [DOI] [PubMed] [Google Scholar]
- Shrivastava G, Leon‐Juarez M, Garcia‐Cordero J, Meza‐Sanchez DE, Cedillo‐Barron L (2016) Inflammasomes and its importance in viral infections. Immunol Res 64: 1101–1117 [DOI] [PubMed] [Google Scholar]
- Sprenger H, Meyer RG, Kaufmann A, Bussfeld D, Rischkowsky E, Gemsa D (1996) Selective induction of monocyte and not neutrophil‐attracting chemokines after influenza A virus infection. J Exp Med 184: 1191–1196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steel J, Lowen AC, Pena L, Angel M, Solorzano A, Albrecht R, Perez DR, Garcia‐Sastre A, Palese P (2009) Live attenuated influenza viruses containing NS1 truncations as vaccine candidates against H5N1 highly pathogenic avian influenza. J Virol 83: 1742–1753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun F, Xiao G, Qu Z (2017) Murine bronchoalveolar lavage. Bio Protoc 7: e2287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson MS, Molofsky AB (2005) Autophagy and inflammatory cell death, partners of innate immunity. Autophagy 1: 174–176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szretter KJ, Gangappa S, Lu X, Smith C, Shieh WJ, Zaki SR, Sambhara S, Tumpey TM, Katz JM (2007) Role of host cytokine responses in the pathogenesis of avian H5N1 influenza viruses in mice. J Virol 81: 2736–2744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapia‐Abellan A, Angosto‐Bazarra D, Martinez‐Banaclocha H, de Torre‐Minguela C, Ceron‐Carrasco JP, Perez‐Sanchez H, Arostegui JI, Pelegrin P (2019) MCC950 closes the active conformation of NLRP3 to an inactive state. Nat Chem Biol 15: 560–564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tate MD, Ong JD, Dowling JK, McAuley JL, Robertson AB, Latz E, Drummond GR, Cooper MA, Hertzog PJ, Mansell A (2016) Reassessing the role of the NLRP3 inflammasome during pathogenic influenza A virus infection via temporal inhibition. Sci Rep 6: 27912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tauber S, Ligertwood Y, Quigg‐Nicol M, Dutia BM, Elliott RM (2012) Behaviour of influenza A viruses differentially expressing segment 2 gene products in vitro and in vivo . J Gen Virol 93: 840–849 [DOI] [PubMed] [Google Scholar]
- Thomas PG, Dash P, Aldridge JR Jr, Ellebedy AH, Reynolds C, Funk AJ, Martin WJ, Lamkanfi M, Webby RJ, Boyd KL et al (2009) The intracellular sensor NLRP3 mediates key innate and healing responses to influenza A virus via the regulation of caspase‐1. Immunity 30: 566–575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tschopp J, Martinon F, Burns K (2003) NALPs: a novel protein family involved in inflammation. Nat Rev Mol Cell Biol 4: 95–104 [DOI] [PubMed] [Google Scholar]
- Tschopp J, Schroder K (2010) NLRP3 inflammasome activation: the convergence of multiple signalling pathways on ROS production? Nat Rev Immunol 10: 210–215 [DOI] [PubMed] [Google Scholar]
- Vacheron F, Rudent A, Perin S, Labarre C, Quero AM, Guenounou M (1990) Production of interleukin 1 and tumour necrosis factor activities in bronchoalveolar washings following infection of mice by influenza virus. J Gen Virol 71(Pt 2): 477–479 [DOI] [PubMed] [Google Scholar]
- van Riel D, Leijten LM, van der Eerden M, Hoogsteden HC, Boven LA, Lambrecht BN, Osterhaus AD, Kuiken T (2011) Highly pathogenic avian influenza virus H5N1 infects alveolar macrophages without virus production or excessive TNF‐alpha induction. PLoS Pathog 7: e1002099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidic J, Richard CA, Pechoux C, Da Costa B, Bertho N, Mazerat S, Delmas B, Chevalier C (2016) Amyloid assemblies of influenza A virus PB1‐F2 protein damage membrane and induce cytotoxicity. J Biol Chem 291: 739–751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Nikrad MP, Travanty EA, Zhou B, Phang T, Gao B, Alford T, Ito Y, Nahreini P, Hartshorn K et al (2012) Innate immune response of human alveolar macrophages during influenza A infection. PLoS One 7: e29879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Zhang A, Wan Y, Liu X, Qiu C, Xi X, Ren Y, Wang J, Dong Y, Bao M et al (2014) Early hypercytokinemia is associated with interferon‐induced transmembrane protein‐3 dysfunction and predictive of fatal H7N9 infection. Proc Natl Acad Sci USA 111: 769–774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinheimer VK, Becher A, Tonnies M, Holland G, Knepper J, Bauer TT, Schneider P, Neudecker J, Ruckert JC, Szymanski K et al (2012) Influenza A viruses target type II pneumocytes in the human lung. J Infect Dis 206: 1685–1694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wise HM, Foeglein A, Sun J, Dalton RM, Patel S, Howard W, Anderson EC, Barclay WS, Digard P (2009) A complicated message: identification of a novel PB1‐related protein translated from influenza A virus segment 2 mRNA. J Virol 83: 8021–8031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong SS, Oshansky CM, Guo XJ, Ralston J, Wood T, Seeds R, Newbern C, Waite B, Reynolds G, Widdowson MA et al (2018) Severe influenza is characterized by prolonged immune activation: results from the SHIVERS cohort study. J Infect Dis 217: 245–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu T, Qiao J, Zhao L, Wang G, He G, Li K, Tian Y, Gao M, Wang J, Wang H et al (2006) Acute respiratory distress syndrome induced by avian influenza A (H5N1) virus in mice. Am J Respir Crit Care Med 174: 1011–1017 [DOI] [PubMed] [Google Scholar]
- Yoshizumi T, Ichinohe T, Sasaki O, Otera H, Kawabata SI, Mihara K, Koshiba T (2014) Influenza A virus protein PB1‐F2 translocates into mitochondria via Tom40 channels and impairs innate immunity. Nat Commun 5: 4713 [DOI] [PubMed] [Google Scholar]
- Yu J, Wu Y, Wang J (2017) Activation and role of NACHT, LRR, and PYD domains‐containing protein 3 inflammasome in RNA viral infection. Front Immunol 8: 1420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zell R, Krumbholz A, Eitner A, Krieg R, Halbhuber KJ, Wutzler P (2007) Prevalence of PB1‐F2 of influenza A viruses. J Gen Virol 88: 536–546 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Review Process File
Expanded View Figures PDF
Source Data for Expanded View
Appendix
Source Data for Figure 1
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Data Availability Statement
No large data sets where generated in this study.