

Abstract
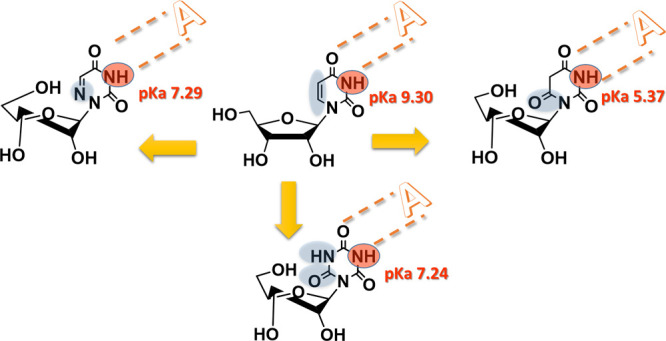
Uridine (U) mimetics are sought after as tools for biochemical and pharmacological studies. Previously, we have identified recognition patterns of U by proteins. Here, we targeted the characterization of uridine mimetics—cyanuryl-ribose (CR), barbituryl-ribose (BR), and 6-azauridine (AU)—with a view to identify analogs with potentially more binding interactions than U with target biomolecules. We found that CR, BR, and AU retain selective U’s natural H-bonds with adenosine vs guanosine. CR/AU and BR were 100- and 10,000-fold more acidic, respectively, than U. Under physiological pH, 54, 51, and 77% of CR, AU, and BR molecules, respectively, are ionized vs 13% for U. The electron-rich nature of CR and BR vs U was reflected by their 13C NMR chemical shifts and ε values. CR/AU and BR prefer N conformation (up to 73%) vs U (56%). Unlike U that prefers gg conformation around exocyclic methylol (48%), CR/AU and BR prefer both gt and gg rotamers. In conclusion, replacement of uridine’s C6 by N or carbonyl, or C5–C6 by an amide, results in significant changes in U’s ionization, electron density, conformation, base-stacking, etc., leading to potentially tighter binding than U with a target protein or nucleic acid and potential use for various biochemical and pharmacological applications.
Introduction
Over the years, many modifications of the uracil ring were reported, motivated by the need to develop better agonists or antagonists of various kinds of receptors (e.g., P2Y2/4/6-Rs), or inhibitors of enzymes (e.g., sorivudine, an inhibitor of a viral DNA polymerase),1 involved in a variety of pathophysiological conditions.2−11 These modifications of the uracil ring involved mostly substitution of various groups at the uracil C5 or C6 positions.3,4
Other pyrimidine-based antiviral and anticancer drugs, e.g., 6-azauracil, have been developed.5 In this type of uracil analog, the atoms constituting the ring have been replaced by others, e.g., a carbon atom was replaced by a nitrogen atom in 6-azauracil. Such changes affect chemical properties of the uracil ring, such as H-bonds, degree of aromaticity, and acid–base character. Another kind of uracil modification involves varying the size of the ring, e.g., a seven-membered ring analog of the uracil ring, azepine ring.6
The significance of the interaction of uracil or uridine or related nucleotides with target proteins in health and disease encouraged us to study binding patterns of uridine.7,8 Our data mining analysis of complexes of protein with uracil nucleosides/nucleotides provided insights regarding molecular recognition patterns. Thus, molecular recognition of the uracil moiety in uridine involves three types of interactions: π–cation interactions between the uracil moiety and positively charged residues in the proteins were detected in 25% of the complexes. π–π interactions between the uracil moiety and aromatic residues in the proteins were found in 59% of the inspected structures. Hydrogen bonds were the most dominant interaction between the uracil moiety and its binding proteins. The most abundant hydrogen bond interaction, present in 98% of the structures, was found between the uracil N3-H and a protein H-bond acceptor. The uracil O2 and O4 atoms act as H-bond acceptors in 80 and 93% of the cases, respectively (Figure 1).7
Figure 1.
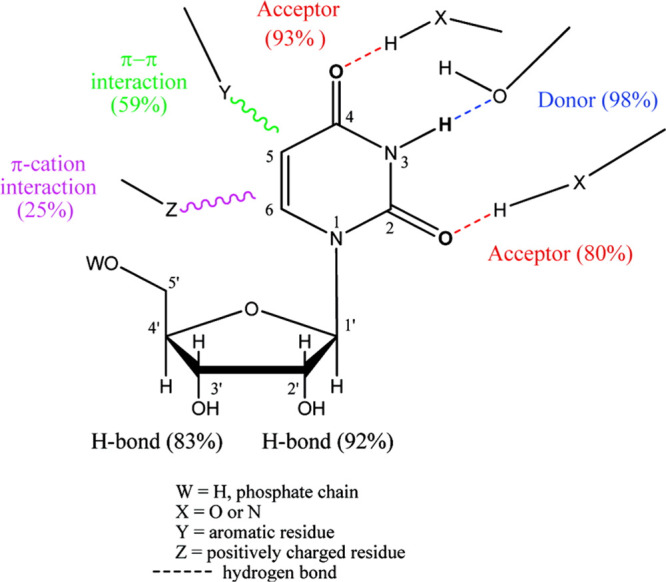
Recognition patterns in uracil-nucleos(t)ide binding proteins we previously reported.7 Numerical values in brackets represent percentages of occurrences of H-bonding\π–π interactions\π–cation interactions in the set of protein-uracil-nucleos(t)ide complexes studied.
Based on these findings, the potential number of H-bonds formed by uracil is expected to increase by replacing the C5 and/or C6 carbon atoms of the uracil ring by NH/carbonyl moieties.
Indeed, such
uridine mimetics, e.g., cyanuryl ribose (CR, 1), barbituryl
ribose (BR, 2), and 6-aza-uridine
(AU, 3), have been synthesized before.9,10 Uridine
mimetics, cyanuryl-2′-deoxy-riboside and 5-thiophene-6-aza-uridine,
have been incorporated in nucleic acids, and their effect on various
features of nucleic acids was studied.11−13
Here, we targeted the characterization of certain physico-chemical properties of CR, BR, and AU, as compared with uridine, with a view to identify the usefulness of the former as agents for biochemical and pharmacological applications. Specifically, we studied features of compounds 1–3 that are indicative of potential binding interactions of these analogs, such as acid–base equilibria, base pairing, base stacking, and nucleoside conformation, as compared to each other and to uridine.
Results and Discussion
Synthesis
For the improvement of the synthesis of CR14 (1), we employed the Vorbrüggen method, which is commonly used for the synthesis of nucleosides.15,16 However, the Vorbrüggen reaction suffers from unpredictable regioselectivity in some cases when heterocycles containing multiple basic sites are used, such as cyanuric acid.17,18
Indeed, when we treated silylated cyanuric acid with 1-O-acetyl-2,3,5-tri-O-benzoyl-β-d-ribofuranose at RT for 2 h, CR was obtained in only 2% yield together with disubstituted traizinone 6, obtained in 19% yield due to reaction of protected ribose 5, with two nitrogen atoms in silylated cyanuric acid (Scheme 1).
Scheme 1. Synthesis of CR.
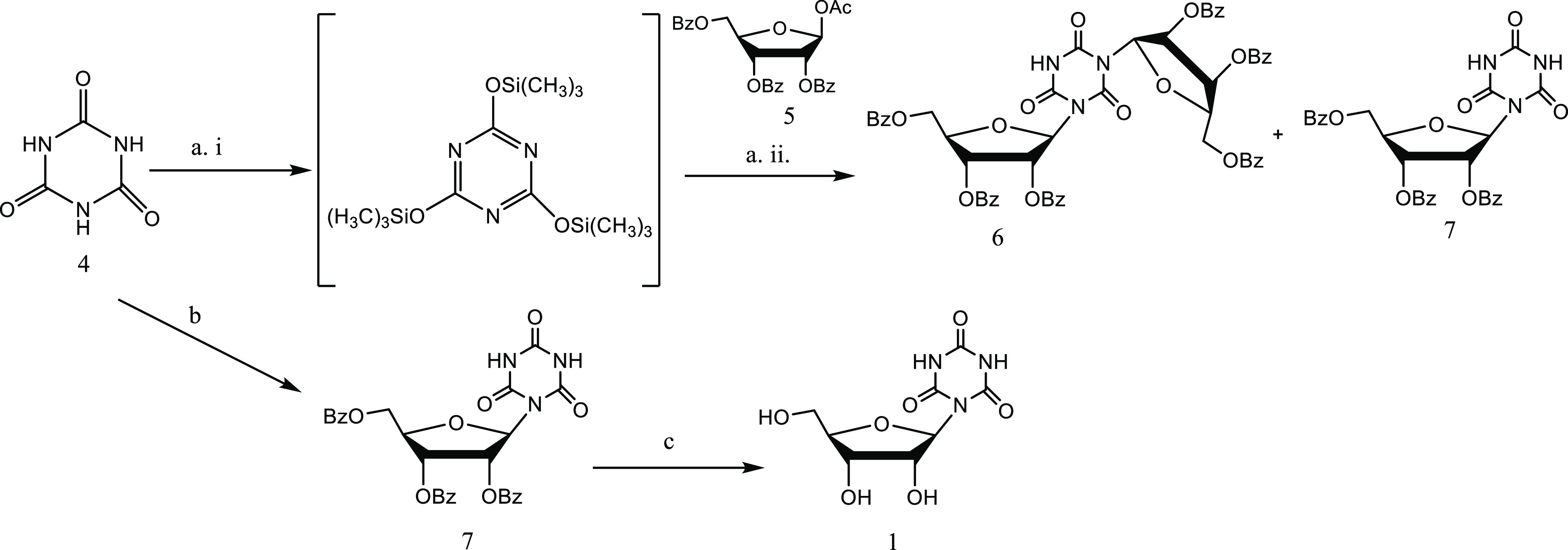
Reagents and conditions: (a) i: cyanuric acid, dry HMDS, reflux, 6 h. ii: dry CH3CN, protected ribose (5), SnCl4, 1 h at RT.18 (b) cyanuric acid, protected ribose (5), CH3CN, HMDS, TMSCl, SnCl4, 1 h at RT. (c) 1 M NaOH in MeOH.
Therefore, an improved synthesis was developed based on the Vorbrüggen reaction to prevent the formation of disubstituted traizinone 6.19 Specifically, we performed a one-pot reaction, where TMSCl (1 equiv) and SnCl4 (2 equiv) were added to an excess of cyanuric acid 4 (3 equiv) and 1-O-acetyl-2,3,5-tri-O-benzoyl-β-d-ribofuranose 5 (1 equiv) in HMDS and acetonitrile at RT. The turbid solution turned clear after about 1 h. Compound 7 was obtained in 82% yield. Finally, CR (1) was obtained quantitatively upon treatment of 7 with NaOH in MeOH at RT for 1 h.
Analogs 2 and 3 were commercially available.
Uridine Mimetics 1 and 2 Prefer Ribose N-Conformer and gg/gt-Rotamers
In the 70s, the conformation of various nucleotides and nucleosides was studied by NMR spectroscopy.9,20,21 NMR studies indicated that the ribose ring interconverts rapidly between N and S conformations with approximately equal residence. For example, uridine prefers N puckering (56%) over the S conformation (Figure 2). In addition, slight preference for the gauche–gauche rotamer of the exocyclic CH2OH group was indicated (Figure 3).22
Figure 2.

Ribose ring pucker: S and N conformations.
Figure 3.

Possible conformations of the exocyclic CH2OH group of nucleosides.
Conformational analysis of BR was not reported, while sugar puckering of CR and the conformation of its exocyclic CH2OH group have been studied only by low-field NMR.21,22 The ribose conformation of AU is known.10
Here, we employed detailed high-field 1H NMR and 13C NMR spectroscopy to analyze the solution conformation and to characterize physicochemical properties, such as base pairing, base stacking, and acid–base equilibrium of CR, BR, and AU.
Sugar Puckering
The chemical shifts and coupling constants for the sugar protons of CR, BR, and AU vs those of uridine are shown in Table S1 in the Supporting Information and were used for the determination of ribose puckering.
The chemical shifts for H-1′, 2′, and 3′ in CR, BR, and AU show a small degree of de-shielding relative to uridine, while the other three protons (H-4′, 5′, and 5″) are slightly shielded. This suggests that carbonyls C2/C6 of the cyanuryl and barbituryl moieties are above the ribose ring and close to H-4′, 5′, and 5″ (in the case of AU, an N atom replaces a C6 carbon atom) (Figure 4). This is consistent with the data shown below for the conformation of the exocyclic ribose methylol. The anti-glycosidic angle (−1 to 44° for the N conformer and 39–66° for the S conformer) observed for uridine23 is needed for removal of steric hindrance of the exocyclic ribose methylol with C2 carbonyl. Here, both the cyanuryl and burbituryl moieties possess two carbonyls (C2/C6), and therefore, steric hindrance is removed by rotation of C2/C6 carbonyl above the ribose ring and away from the 5′-hydroxymethylene group. In addition, this conformation is stabilized by an intramolecular H-bond between C2/C6 carbonyl and 5′-OH (Figure 4).
Figure 4.
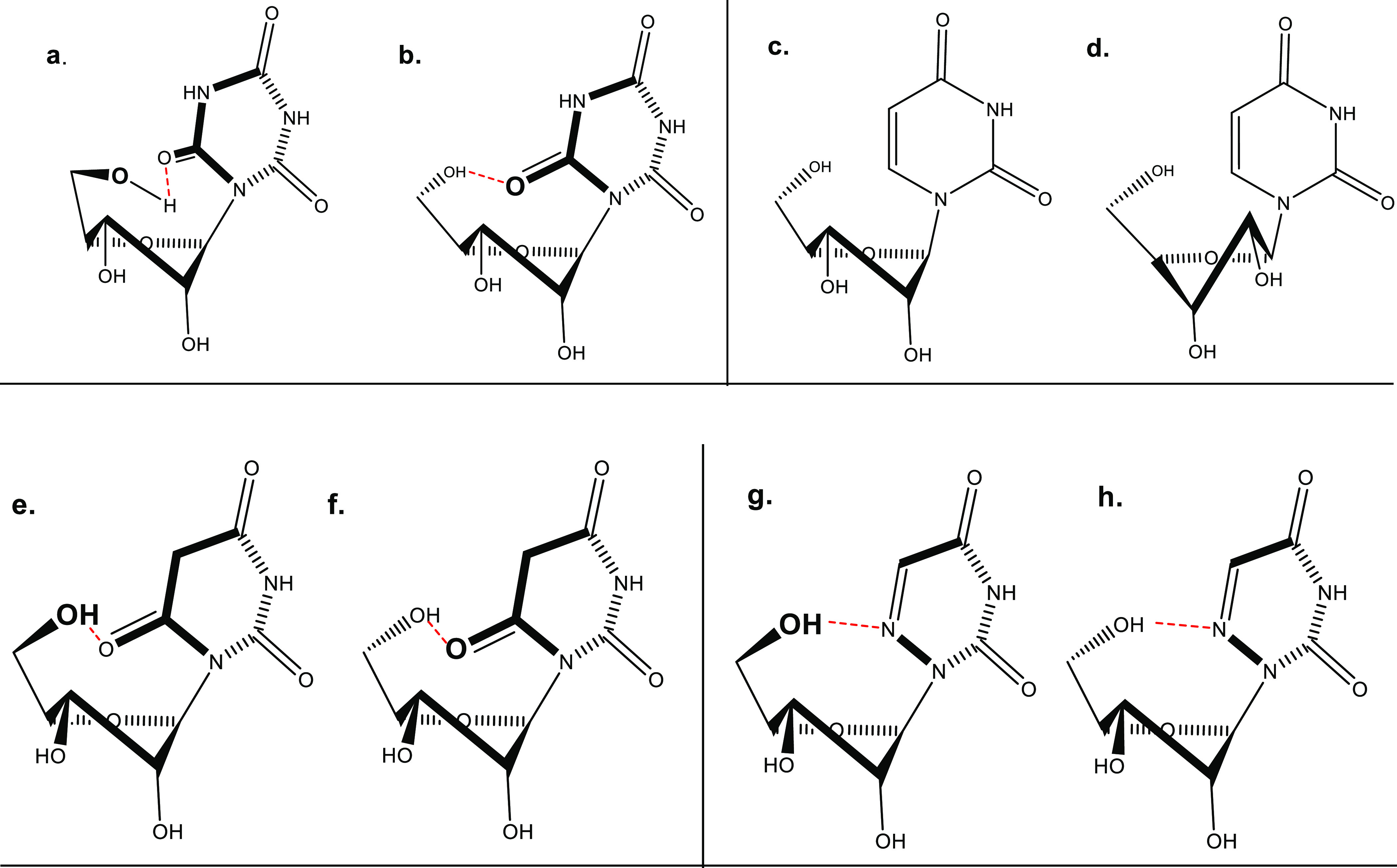
Suggested conformations of CR, BR, and AU as compared to that of uridine. (a) CR: exocyclic gt rotamer around the C4′–C5′ bond of N conformer (70%). (b) CR: exocyclic gg rotamer for the C4′–C5′ bond of N conformer. (c) Uridine: exocyclic gg around the C4′–C5′ bond of N (56%) conformer. (d) Uridine: exocyclic gg around the C4′–C5′ bond of S (44%) conformer. (e) BR: exocyclic gt rotamer for the C4′–C5′ bond of N conformer (73%). (f) BR: gg rotamer for the C4′–C5′ bond of N conformer. (g) AU: exocyclic gt rotamer around the C4′–C5′ bond of N conformer (60%). (h) AU: gg rotamer around the C4′–C5′ bond of N conformer.
The ribose moiety of nucleosides most often adopts either a C3′-endo (north, N) or a C2′-endo (south, S) orientation, where endo means the atoms located above the plane defined by C1, O1, and C4.24 The conformation of the ribose ring of nucleoside is analyzed in terms of a dynamic equilibrium between the north conformer and south conformer.23
To analyze the population of N and S conformers of uridine mimetics, we have extracted J1′2′ and J3′4′ coupling constants from their 1H NMR spectrum at 600 MHz at 300 K (Supporting Information, Table S1). The corresponding data for uridine are included for comparison. Peak assignments were confirmed by obtaining 1H × 1H correlations in COSY spectra.
Since the equilibration rate of the two conformers is fast in the NMR timescale, the observed vicinal couplings are weighted averages of their values in the two conformers. Thus, the mole fractions of conformers S and N were calculated directly from the observed values of J1′2′ and J3′4′ (see the Supporting Information, Si) and are shown in Table 1.
Table 1. Conformational Parameters of CR, BR, and AU in D2O Solution.
| conformer
population around C4′–C5′
bond |
||||
|---|---|---|---|---|
| compound | sugar puckering %N | %gg | %tg | %gt |
| CR | 70 | 46 | 10 | 44 |
| BR | 73 | 46 | 10 | 44 |
| AU | 60 | 48 | 14 | 38 |
| U25 | 56 | 58 | 20 | 22 |
Our NMR data are consistent with literature10,25 showing that the ribose ring is preferentially in the C3′-endo (N), which constitutes 70% of the total population for CR, 73% for BR, and 60% for 6-azauridine, whereas for the uridines, the value is 56% (Table 1).
Conformation of 5′-Hydroxymethylene Group (CH2OH)
In general, the nucleoside coupling constants J4′5′ and J4′5′ can be interpreted in terms of three classical staggered rotamers (gg, tg, and gt) with a preferred gauche–gauche (gg) conformation.26 The mole fractions of each staggered rotamer of C4′–C5′ were calculated as described in the Supporting Information (Sii).
The resulting percentages of populations of conformers gg, tg, and gt (Figure 3) calculated for uridine mimetics 1–3 are presented in Table 1. In contrast to uridine, 1–3 do not show preference for the gauche–gauche rotamer. However, major rotamers for 1–3 are both gauche–trans and gauche–gauche rotamers. This is apparently due to H-bonding between the 5′-hydroxyl and one of the carbonyls groups of the cyanuryl\barbituryl moiety and the N6 atom of 3, respectively, possible in those two rotamers. The conformations of the ribose ring of β-barbituryl ribose and β-cyanuryl ribose are similar, 70–73% N due to the steric hindrance between the carbonyl at position 6 and C2′. The percentage of N conformer of AU is lower than those of BR and CR but similar to that of uridine since there is no significant steric hindrance with C2′ position. The major rotamers of both CR and BR exo-cyclic methylol are gg and gt due to H-bonding between the 5′-hydroxyl and one of the carbonyls groups of the barbituryl and cyanuryl moiety. AU shows higher preference for gg and gt than uridine. This is apparently due to H-bonding between the 5′-hydroxyl and N6 nitrogen atom, possible in those two rotamers (Figure 4).
Uridine Mimetics 1–3 Preserve Uridine’s H-Bonding Pattern with Adenosine
The natural base pairing pattern of modified nucleic acids is useful for hybridization-based diagnostics, therapeutics, research, etc.27 Therefore, we investigated here the mode of base pairing between adenosine and CR, BR, and AU by 1H NMR-monitored titration (Figures 5 and 6). Specifically, 0.1 M CR, BR, and AU in DMSO-d6 were added to 0.1 M adenosine in DMSO-d6, and the NH-3/5 signal of the uridine mimetics, which may be involved in H-bond interactions with adenosine, was inspected (Figure 5).
Figure 5.
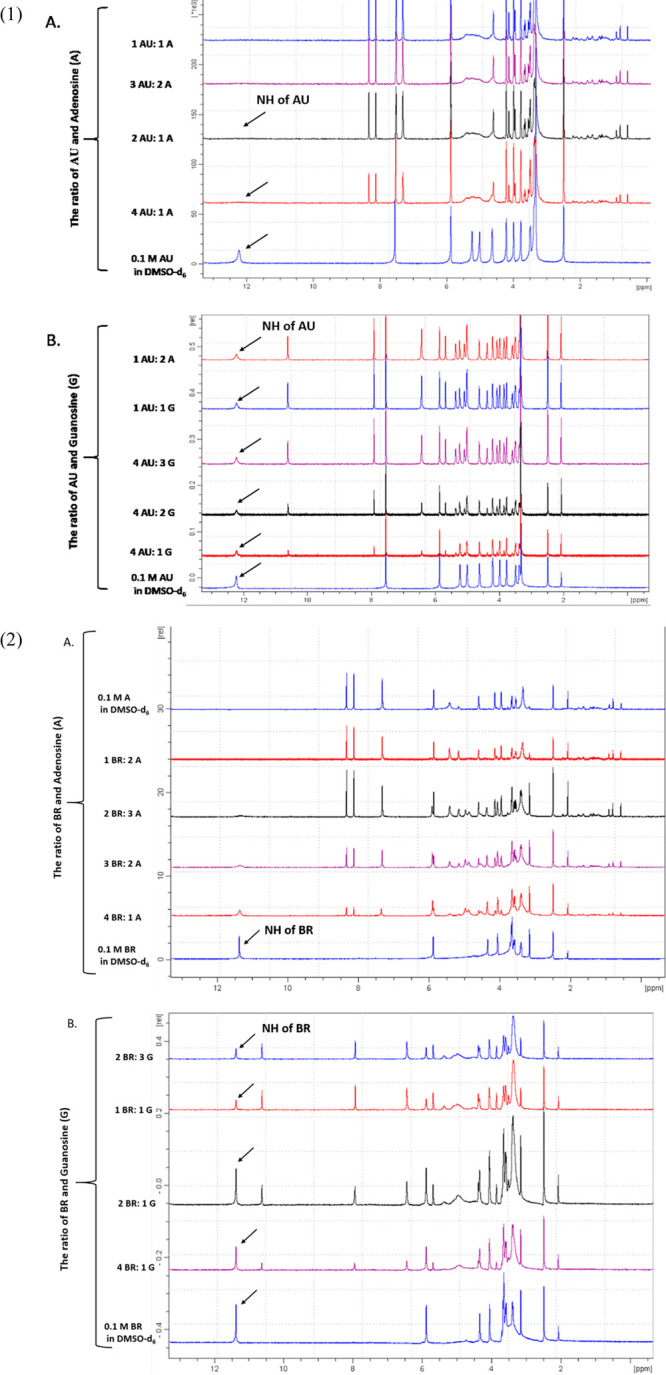
Changes in 1H NMR spectra (600 MHz, 300 K) of (1) 0.1 M AU upon addition of (A) 0.1 M adenosine in DMSO-d6 solution and (b) 0.1 M guanosine in DMSO-d6 solution and (2) 0.1 M BR upon addition of (a) 0.1 M adenosine in DMSO-d6 solution and (b) 0.1 M guanosine in DMSO-d6 solution.
Figure 6.
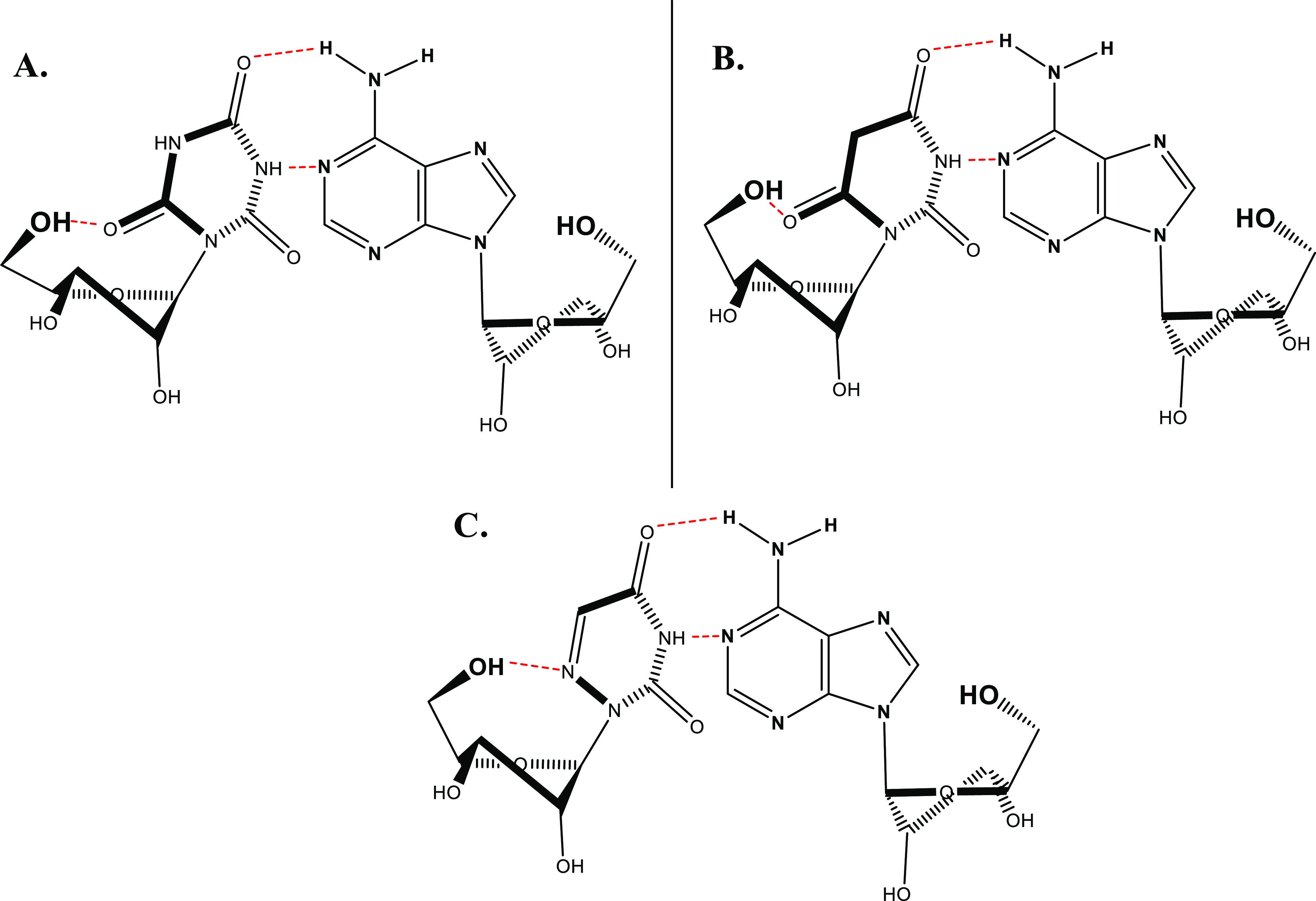
Schematic representation of H-bonded base pairing between (A) CR, (B) BR, and (C) AU and adenosine.
We observed a decrease in the signals of NH protons of the uracil-mimetic moiety, until they disappeared. This indicates the formation of H-bonding between the uracil-mimetic moiety and adenine moiety. Unexpectedly, de-shielding of the protons of the adenosine’s exocyclic amine was not significant. This is presumably due to the averaging of both signals of the N6-amine protons.
Furthermore, we tested the selectivity of molecular recognition of adenosine vs guanosine by CR, BR, and AU. For this purpose, we have added gradually 0.1 M CR, BR, and AU in DMSO-d6 to 0.1 M guanosine in DMSO-d6 (Figure 5, CR titrations shown in Supporting Information, Figure S5). No change in the base NH proton signal was observed.
Base-Stacking Was Observed for CR but Not for BR and AU
Next, we investigated by NMR the possibility of self-base stacking interactions of the cyanuryl, barbituryl, and 6-azauracil moiety of compounds 1–3 at high concentrations. We monitored the shift of chemical shifts of H5 and H1′ of the ribose proton in AU and BR. In CR, we monitored just the ribose H1′ proton, which indicates stacking interactions between the base moieties, due to the lack of CH group in the cyanuryl moiety.
Table 2 shows the 1H chemical shifts of protons of the sugar moiety of CR and of uridine in dilute solution (0.005 M). These chemical shifts represent the monomeric forms of 1–3 and uridine (δ0). In addition, we determined the asymptotic chemical shifts (δ∞) of the stacked heterocyclic moieties obtained at high concentration (0.4 M).
Table 2. Chemical Shifts (ppm) for Monomeric (δ0) and Self-Stacked (δ∞) CR, BR, and AU and Related Data for Uridine from the Literature28.
| H5 |
H6 |
H1′ |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| compound | δ0 | δ∞ | Δδ | δ0 | δ∞ | Δδ | δ0 | δ∞ | Δδ |
| U | 5.90 | 5.79 | 0.11 | 7.87 | 7.82 | 0.05 | 5.92 | 5.81 | 0.11 |
| AU | 7.56 | 7.05 | 0.05 | 6.04 | 6.05 | 0.01 | |||
| CR | 5.89 | 6.01 | –0.12 | ||||||
| BR | 3.28 | 3.27 | 0.01 | 6.03 | 6.04 | 0.01 | |||
For uridine, as for other nucleosides, the protons of the sugar moiety are deshielded as the concentration is increased due to base stacking interactions, e.g., positive Δδ values for H5, H6, and H1′.28 This observation is not surprising because in solution, nucleosides assume the anti-conformation with respect to the glycosidic bond.29
Unexpectedly, the data for CR were different. The chemical shifts of the protons in the ribose moiety of CR were shielded as concentration increased, resulting in a negative Δδ with an absolute value similar to that of uridine. This phenomenon may be due to the different stacking interactions of β-cyanuryl vs uracil moieties. Presumably, the cyanuryl moieties form H-bonds (similar to H-bonds in β-sheets of proteins) rather than stacking like aromatic purine and pyrimidines bases. The latter tend to have smaller upfield shifts because their ring currents are smaller than those in purines.30
In a previous report, uridine showed an upfield shift at an elevated concentration28 (Table 2); the same was observed for AU, for the H5 proton, but to a minute degree. There was no change in the chemical shift of AU’s H1′ proton. In contrast to CR, the chemical shifts of the protons in the ribose moiety of BR ribose did not shift at a high concentration. These findings imply that there is no base-stacking in BR and AU. It is noteworthy that pyrimidine derivatives are expected to produce smaller upfield shifts than purines because their ring currents are smaller.30
CR/AU and BR Are 100- and 10,000-fold More Acidic Than U, Respectively
We studied the effect of replacement of C5–C6 double bond in uridine by an amide moiety or additional N atom at position C6 in CR, BR, and AU on the uridine’s acid–base equilibria. Specifically, we established the pKa values of these analogs by pH titration monitored by 13C NMR.
A drastic downfield shift of the signals of carbonyls C2, C6, and C4 of the cyanuryl ring was observed (Figure 7). While the ribose signals only slightly shifted, the C4 chemical shift was highly affected by increasing pH and shifted downfield up to 22 ppm. C2 and C6 signals were shifted by 12 ppm, as expected.
Figure 7.

pH dependence of 13C chemical shifts of CR (1), BR (2), and AU (3). (1) 0.9 M CR solution was titrated with diluted HCl and NaOH, and the changes in 13C NMR spectra (at 600 MHz) were monitored as a function of pH. (A) Chemical shifts of C2 and C6 in a pH range of 3–9.5. (B) Chemical shifts of C2 and C6 in a pH range of 10–13.1. (C) Chemical shifts of C4 in a pH range of 3–9.5. (D) Chemical shifts of C4 in a pH range of 10–13.1. (2) 0.9 M BR solution was titrated with diluted HCl and NaOH, and the changes in 13C NMR spectra (at 600 MHz) were monitored as a function of pH. (A) Chemical shifts of C2 and C6 in a pH range of 2–7. (B) Chemical shifts of C2 and C6 in a pH range of 8–13.26. (C) Chemical shifts of C4 in a pH range of 2–7. (D) Chemical shifts of C4 in a pH range of 8–13.26. (3) 0.9 M AU solution was titrated with diluted HCl and NaOH, and the changes in 13C NMR spectra (at 600 MHz) were monitored as a function of pH. (A) Chemical shifts of C2 in a pH range of 2–12.7. (B) Chemical shifts of C4 in a pH range of 2–12.7.
At high pH, downfield shifts of the signals of carbonyls C2, C4, and C6 of BR ring and C2 and C4 of AU were observed, while at low pH, the downfield shifts were moderated (Figure 7). In contrast to CR where the shift of C4 was up to 22 ppm, the signals of C2, C4, and C6 in both BR and AU were affected by increasing pH and shifted downfield 10 ppm on average.
These shifts reflect the electron-rich nature of the deprotonated ring systems and indicate the existence of singly and doubly deprotonated N3 and N5 NH groups in CR, BR, and AU.
In contrast to uridine, CR and BR have two acidic protons. Two inflection points were observed in the sigmoid graph (Figure 7). Calculated pKa1 and pKa2 values for CR and for BR were 7.24 and 12.73 and 5.37 and 11.95, respectively. One inflection point was observed in the sigmoid graph for AU (Figure 7), and the calculated pKa was 7.29 vs 9.30 for uridine.28
The 100-fold higher acidity of CR vs uridine is due to resonance stabilization of the anion by the oxygen atom on C4 (Supporting Information, Figure S6). Therefore, the signal of C4 is highly affected and drastically shifted downfield.
Like CR, AU is 100-fold more acidic than uridine due to additional resonance stabilization involving the N6 nitrogen atom.
Remarkably, BR is 10,000-fold more acidic than uridine due to the aromatic resonance stabilization of the anion, which resembles that of barbituric acid (pKa, 4.01).31
The di-anion form of both CR and BR is stabilized by all carbonyls, although the C4 signal is most affected as compared to C2 and C6 signals. This is because of the interception of averaging of two peaks of NH obtained as one peak.
BR Anion Strongly Absorbs Light
CR, BR, and AU exist as several tautomeric forms including an aromatic tautomer (Supporting Information, Figure S7).32 To learn about the electron density of CR, BR, and AU vs uridine, we measured the UV spectra of all of them vs uridine33,34 under several conditions.
At pH 2.7 and 5.5, maximum absorption of CR appeared at 214 nm, and extinction coefficients (ε) were 2900 and 3300 M–1 cm–1, respectively (Supporting Information, Figure S2). However, under pH 12, ε drastically increased to 12,900 M–1 cm–1 (λmax, 220 nm). In contrast, the absorption of uridine (260 nm) is pH non-dependent. Under both pH 11 and 14, uridine exhibits an ε value of 9800 M–1 cm–1 (Table 3).32,35
Table 3. Absorption and Extinction Coefficients of AU, CR, and BR vs Uridine.
| absorption
λ (nm) |
extinction coefficient
ε (cm–1 M–1) |
|||||||
|---|---|---|---|---|---|---|---|---|
| pH | uridine | AU | CR | BR | uridine | AU | CR | BR |
| 11.9 | 261 | 257 | 220 | 262 | 9900 | 7400 | 12,900 | 46,400 |
| 7.4 | 262 | 256 | 217 | 261 | 9820 | 5500 | 13,500 | 16,600 |
| 5.5 | 262 | 255 | 214 | 261 | 9980 | 2300 | 3300 | 14,900 |
| 2.7 | 262 | 263 | 213 | NA | 9930 | 6900 | 2900 | NAa |
NA = not available
Generally, absorption maxima and extinction coefficients are highly affected by an electronic-rich or electron-poor nature of the system. With more conjugated systems, the absorption wavelengths tend to shift toward the long wavelength region. The larger the molar extinction coefficient is, the more strongly light is absorbed. Thus, at pH 12, the CR anion species are dominant and exist as a fully conjugated structure, unlike CR at pH 2.7, resulting in a significantly larger molar extinction coefficient: ε 13,900 vs 2900 M–1 cm–1, respectively. The former ε value reflects the electron-rich nature of the deprotonated ring system.
Due to its high acidity, BR significantly absorbs light (ε 14,900 M–1 cm–1) at pH 5.5. Under pH 12, the BR di-anion strongly absorbs (ε 46,400 M–1 cm–1).
Conclusions
Our goal here was to identify uridine mimetics that potentially form more binding interactions with a potential protein partner, or nucleic acid, than those formed with uridine.
Indeed, based on the above NMR studies, we found that CR and BR as well as AU retain specific uridine’s H-bonds with adenosine but not guanosine. In particular, CR, BR, and AU keep uridine’s natural H-bonding involving N3-H and C4-carbonyl.
Replacement of uridine’s C5–C6 double bond by an amide moiety changes dramatically the acid–base character of the molecule. Thus, CR/AU and BR are 100- and 10,000-fold more acidic than uridine, respectively. Namely, under physiological pH, 54% of CR, 51% of AU, and 77% of BR molecules are ionized vs only 13% ionization for uridine, thus indicating potentially tighter binding of those uridine mimetics vs uridine to any protein partner via ionic interactions.
Furthermore, the electron-rich nature of CR and BR vs uridine under physiological pH was reflected by their 13C NMR chemical shifts and ε values vs that of uridine (13,500 and 16,600 vs 9820 M–1 cm–1), respectively. Thus, π-interactions with target biomolecules are expected to be significant for CR and BR.
In addition, we studied the conformation of the ribose ring in compounds 1–3 vs uridine. Our findings indicate that all compounds prefer N conformation, up to 73% vs 56% for uridine. Unlike uridine that prefers gg conformation (48%), for compounds 1–3, both major rotamers are gt and gg. Preference for both N conformation and gt and gg rotamers is more significant for 1–3 vs uridine due to the greater probability of H-bonding between the 5′-hydroxyl and C2/C6-carbonyl groups of the heterocyclic moiety. This conformational change of 1–3 vs uridine may also affect the molecular recognition of the former structures by biopolymers.
In conclusion, replacement of uridine’s C6 by N or carbonyl, or C5–C6 by an amide, results in significant changes in U’s ionization, electron density, conformation, base-stacking, etc., leading to potentially tighter binding than U with a target protein or nucleic acid and potential use for various biochemical and pharmacological applications.
Experimental Section
Synthesis of β-Cyanuryl Ribose
Below is an improved procedure for the synthesis of β-cyanuryl ribose based on literature.36 The original procedure involves several steps. We combined them to a one-pot reaction that affords the product in high yield. To cyanuric acid (4) (0.018 mol, 2.30 g, 3 equiv) and 1-O-acetyl-2,3,5-tri-O-benzoyl-β-d-ribofuranose (5) (6 mmol, 3 g, 1 equiv) in 50 mL acetonitrile hexamethyldisilazane (HMDS) (0.018 mmol, 3.76 mL, 3 equiv), tetramethylsilyl chloride (TMSCl) (6 mmol, 0.8 mL, 1 equiv) and tin tetrachloride (10 mmol, 1.2 mL, 2 equiv) were added consecutively. After few minutes, the clear solution became turbid. The mixture was stirred at room temperature for another 1 h. The progress of the reaction was monitored by TLC (ethyl acetate/toluene, 1:4). At the end of the reaction, two spots were observed on the TLC plate (Rf 0.58, Rf 0.17). Then, MeOH was added and the mixture was centrifuged for 10 min. After evaporation, the residue was dissolved in toluene and the organic phase was concentrated and purified by flash column chromatography (toluene/ethylacetate, 4:1). The fractions containing the product were evaporated, affording CR (7) (2.8 g, 83% yield). 1H NMR (400 MHz, CDCl3): δ 9.67 (s, 2H, NH) 8.04–7.87 (3d, 6H, Bz), 7.50–7.20 (m, 9H, Bz), 6.17 (dd, 1H, H-2), 6.08 (t, 1H, H-3), 4.80–4.65 (m, 3H, H-4, H-5) ppm. 13C NMR (100 MHz, CDCl3) 166.6, 165.9, 165.5 (CO), 148.4 (CO) 133.4, 129.6, 128.9, 128.8, 128.5 (C-Ar), 87.4 (C-1), 79.4 (C-4), 74.1 (C-2), 71.2 (C-3), 64.2 (C-5) ppm. HRMS m/s: 591.1703 [M + NH4]+ [C29H23N3O10]. β-d-Ribofuranose 1-cyanuryl 2,3,5-tribenzoate (7) (4 mmol, 2.3 g, 1 equiv) was added to a solution of 2 M NaOH (4 equiv) in MeOH (40 mL), and the resulting mixture was stirred at room temperature for 0.5 h. Then, the reaction was quenched by 10% HCl till pH 3 was obtained and extracted with water/chloroform in order to remove benzoic acid.18 The aqueous phase was freeze-dried, affording a white solid CR (1, 1.1 g, 100% yield). 1H NMR (D2O, 400 MHz): δ 6.10 (d, 1H, H-1′), 4.40 (dd, 1H, H-2′), 4.00 (t, 1H, H-3′), 3.90 (dt, 1H, H-4′), 3.72 (dd, 1H, H-5′) ppm. 13C NMR (D2O, 100 MHz) 149.5 (CO), 89.71 (C-1), 84.17 (C-4), 72.70 (C-2), 89.9 (C-3), 62.6 (C-5) ppm. HRMS m/s 260.05307 [M – H]− [C8H11N3O7].
NMR Measurements for Conformational Analysis of the Compounds 1–3
1H NMR spectra of 1 M CR, BR, and AU were measured in D2O at pD 7.44 at 700 MHz and 300 K on a Bruker Avance – III instrument (700.5 MHz).
UV Measurements
Absorption spectra of CR, BR, AU, and uridine were determined in aqueous solutions at a pH range of 2–12. Samples were measured at room temperature in a 10 mm quartz cell with a 1 cm path length. Absorption spectra were measured on a UV-2401PC UV–VIS recording spectrophotometer (Shimadzu, Kyoto, Japan). Extinction coefficients of those compounds were determined using samples at a concentration range of 10–100 μM.
Evaluation of Base-Pairing of CR, BR, and AU with Purine Nucleosides
CR (14 mg), BR (27 mg), AU (24.5 mg), adenosine (26.7 mg), and guanosine (28.3 mg) in volumetric flasks were stored under vacuum overnight to remove absorbed water. 1H NMR spectra were measured in dry DMSO-d6 at 600 MHz. The data were collected at 300 K. 1H NMR spectra were obtained at a range of concentrations of CR, BR, and AU (0.025, 0.05, 0.1, and 0.15 M) titrated with 0.1 M adenosine in dry DMSO-d6 solution. Adenosine solution (0.1 M, 400 μL) was titrated with 0.1 M compounds 1–3 starting at ratio 4:1 (adenosine/uridine mimetic) until 2:3, respectively. The final volume of the solution in the tube was 1 mL. This protocol was repeated for guanosine.
Base-Stacking Experiments
CR (50 mg), AU (39 mg), and BR (43.5 mg) in volumetric flasks were dried under vacuum overnight. 1H NMR spectra were measured in D2O at 600 MHz. The data were collected at 300 K. 1H NMR spectra were obtained at a range of concentrations of compounds 1–3 (0.003, 0.025, 0.04, 0.05, 0.25, and 0.4 M). NaNO3 was added to increase the ionic strength to 0.1 M.
pKa Measurements
Dilute DCl and NaOD solutions were added to 0.1 M CR, BR, and AU in D2O to reach the following pH values: for CR, 3.4, 4.1, 4.4, 4.8, 5.0, 5.3, 5.7, 6.1, 7.6, 8.9, 10.0, 10.3, 11.0, 11.7, 12.1, 12.7, and 13.1; for BR, 2.32, 2.69, 3.22, 3.54, 4.55, 5.32, 5.51, 5.92, 6.39, 8.34, 9.45, 9.95, 11.49, 12.14, 12.34, 12.59, 12.72, and 12.85; and for AU, 2.32, 2.82, 4.32, 5.52, 6.15, 6.61, 8.37, 9.53, 10.03, 11.48, and 12.29. Apparent pD values were measured with a Hanna instruments pH meter equipped with an electrode. pH is estimated from the pD meter measurement (apparent reading from the pH meter) as 0.41. 13C NMR spectra were measured in D2O at 150 MHz. The data were collected at 300 K. 13C NMR chemical shifts of the bases’ carbonyls were monitored as a function of pD.
The chemical shifts of C2, C4, and C6 were plotted vs pH, and a sigmoid curve was obtained. The pKa values were obtained from the inflection points, which are determined by the second derivative of the fitted sigmoid function using Mathlab. A five-parameter sigmoid function was fitted to the data using SigmaPlot.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.0c04788.
1H and 13C NMR spectra, UV–vis spectra, chemical shifts and coupling constants, equations of population, resonance, and tautomeric forms (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Ginsburg-Shmuel T.; Haas M.; Grbic D.; Arguin G.; Nadel Y.; Gendron F.-P.; Reiser G.; Fischer B. UDP made a highly promising stable, potent, and selective P2Y6-receptor agonist upon introduction of a boranophosphate moiety. J. Med. Chem. 2012, 20, 5483–5495. 10.1016/j.bmc.2012.07.042. [DOI] [PubMed] [Google Scholar]
- Brunschweiger A.; Muller C. E. P2 receptors activated by uracil nucleotides-an update. Med. Chem. 2006, 13, 289–312. 10.2174/092986706775476052. [DOI] [PubMed] [Google Scholar]
- Saladino R.; Crestini C.; Palamara A. T.; Danti M. C.; Manetti F.; Corelli F.; Garaci E.; Botta M. Synthesis, Biological Evaluation, and Pharmacophore Generation of Uracil, 4 (3 H)-Pyrimidinone, and Uridine Derivatives as Potent and Selective Inhibitors of Parainfluenza 1 (Sendai) Virus. J. Med. Chem. 2001, 44, 4554–4562. 10.1021/jm010938i. [DOI] [PubMed] [Google Scholar]
- Jacobson K. A.; Costanzi S.; Ivanov A. A.; Tchilibon S.; Besada P.; Gao Z.-G.; Maddileti S.; Harden T. K. Structure activity and molecular modeling analyses of ribose-and base-modified uridine 5′-triphosphate analogues at the human P2Y2 and P2Y4 receptors. Biochem. Pharmacol. 2006, 71, 540–549. 10.1016/j.bcp.2005.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connolly G. P.; Duley J. A. Uridine and its nucleotides: biological actions, therapeutic potentials. Trends Pharmacol. Sci. 1999, 20, 218–225. 10.1016/S0165-6147(99)01298-5. [DOI] [PubMed] [Google Scholar]
- Kunieda T.; Witkop B. Preparation and Photochemistry of 5, 6-Cyclopropyluridines and of Bicyclic Isomers of Thymines. J. Am. Chem. Soc. 1969, 91, 7751–7752. 10.1021/ja50001a043. [DOI] [PubMed] [Google Scholar]
- Ginsburg-Shmuel T.; Haas M.; Schumann M.; Reiser G.; Kalid O.; Stern N.; Fischer B. 5-OMe-UDP is a potent and selective P2Y6-receptor agonist. J. Med. Chem. 2010, 53, 1673–1685. 10.1021/jm901450d. [DOI] [PubMed] [Google Scholar]
- Codacci-Pisanelli G.; Kralovanszky J.; Van der Wilt C.; Noordhuis P.; Colofiore J. R.; Martin D. S.; Franchi F.; Peters G. J. Modulation of 5-fluorouracil in mice using uridine diphosphoglucose. Clin. Cancer Res. 1997, 3, 309–315. [PubMed] [Google Scholar]
- Jones A. J.; Winkley M. W.; Grant D. M.; Robins R. K. Carbon-13 nuclear magnetic resonance: naturally occurring nucleosides. Proc. Natl. Acad. Sci. U. S. A. 1970, 65, 27–30. 10.1073/pnas.65.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammerbacher K.; Görtemaker K.; Knies C.; Bender E.; Bonaterra G. A.; Rosemeyer H.; Kinscherf R. Combinatorial Synthesis of New Pyrimidine-and Purine-β-d-Ribonucleoside Nucleolipids: Their Distribution Between Aqueous and Organic Phases and Their In Vitro Activity Against Human-and Rat Glioblastoma Cells In Vitro. Chem. Biodiversity 2018, 15, e1800173. 10.1002/cbdv.201800173. [DOI] [PubMed] [Google Scholar]
- Beigelman L.; Karpeisky A.; Usman N. Synthesis of 6-Aza-& 6-methyl-pyrimidine ribonucleoside phosphoramidites and their incorporation in hammerhead ribozymes. Nucleosides, Nucleotides Nucleic Acids 1995, 14, 895–899. 10.1080/15257779508012497. [DOI] [Google Scholar]
- Cipriani F.; Gigli G.; Cingolani R.; Favaretto L.; Zambianchi M.; Sotgiu G.; Barbarella G.; Citro G.. Functionalized thiophene oligomers and their use as fluorescent markers. U.S. Patent No. US6841669B2, 2015.
- Fallon H. J.; Frei E. III; Block J.; Seegmiller J. E. The uricosuria and orotic aciduria induced by 6-azauridine. J. Clin. Invest. 1961, 40, 1906–1914. 10.1172/JCI104415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vorbrüggen H.; Ruh-Pohlenz C. Synthesis of nucleosides. Org. React. 2004, 55, 1–630. 10.1002/0471264180.or055.01. [DOI] [Google Scholar]
- Wittenburg E. Eine neue Synthese von Nucleosiden. Z. Chem. 1964, 4, 303–304. 10.1002/zfch.19640040806. [DOI] [Google Scholar]
- Choi W. B.; Wilson L. J.; Yeola S.; Liotta D. C.; Schinazi R. F. In situ complexation directs the stereochemistry of N-glycosylation in the synthesis of thialanyl and dioxolanyl nucleoside analogs. J. Am. Chem. Soc. 1991, 113, 9377–9379. 10.1021/ja00024a058. [DOI] [Google Scholar]
- Niedballa U.; Vorbrüggen H. Synthesis of nucleosides. 12. General synthesis of N-glycosides. Iv. Synthesis of nucleosides of hydroxy and mercapto nitrogen heterocycles. J. Org. Chem. 1974, 39, 3668–3671. 10.1021/jo00939a011. [DOI] [PubMed] [Google Scholar]
- Khaled A.; Ivannikova T.; Augé C. Synthesis of unnatural sugar nucleotides and their evaluation as donor substrates in glycosyltransferase-catalyzed reactions. Carbohydr. Res. 2004, 339, 2641–2649. 10.1016/j.carres.2004.09.002. [DOI] [PubMed] [Google Scholar]
- Vorbrüggen H.; Bennua B. Nucleoside syntheses, XXV1 A new simplified nucleoside synthesis. Chem. Ber. 1981, 114, 1279–1286. 10.1002/cber.19811140407. [DOI] [Google Scholar]
- Schweizer M. P.; Witkowski J. T.; Robins R. K. Nuclear magnetic resonance determination of syn and anti conformations in pyrimidine nucleosides. J. Am. Chem. Soc. 1971, 93, 277–279. 10.1021/ja00730a062. [DOI] [PubMed] [Google Scholar]
- Hruska F. E.; Grey A. A.; Smith I. C. P. Nuclear magnetic resonance study of the molecular conformation of. beta.-pseudouridine in aqueous solution. J. Am. Chem. Soc. 1970, 92, 4088–4094. 10.1021/ja00716a043. [DOI] [PubMed] [Google Scholar]
- Smith I. C. P.; Dugas H.; Blackburn B. J.; Robins R. K.; Deslauriers R. Nuclear magnetic resonance study of the conformation of. beta.-cyanuric acid riboside. Further evidence for the anti rotamer in pyrimidine nucleosides. J. Am. Chem. Soc. 1971, 93, 3468–3470. 10.1021/ja00743a029. [DOI] [PubMed] [Google Scholar]
- Altona C.; Sundaralingam M. Conformational analysis of the sugar ring in nucleosides and nucleotides. New description using the concept of pseudorotation. J. Am. Chem. Soc. 1972, 94, 8205–8212. 10.1021/ja00778a043. [DOI] [PubMed] [Google Scholar]
- Prado F. R.; Giessner-Prettre C.; Pullman B. On the conformational dependence of the proton chemical shifts in nucleosides and nucleotides III. Proton chemical shifts of 5′-nucleotides as a function of different conformational parameters. J. Theor. Biol. 1978, 74, 259–277. 10.1016/0022-5193(78)90075-9. [DOI] [PubMed] [Google Scholar]
- Remin M. Determination of the complete correlation between the sugar ring puckers and 5′-exocyclic group rotamers in conformationally-flexible nucleic acid components from NMR study. J. Biomol. Struct. Dyn. 1984, 2, 211–220. 10.1080/07391102.1984.10507558. [DOI] [PubMed] [Google Scholar]
- Blackburn B. J.; Grey A. A.; Smith I. C. P.; Hruska F. E. Determination of the molecular conformation of uridine in aqueous solution by proton magnetic resonance spectroscopy. Comparison with β-pseudouridine. Can. J. Chem. 1970, 48, 2866–2870. 10.1139/v70-482. [DOI] [Google Scholar]
- Sawada T.; Fujita M. A single watson– crick G·C base pair in water: aqueous hydrogen bonds in hydrophobic cavities. J. Am. Chem. Soc. 2010, 132, 7194–7201. 10.1021/ja101718c. [DOI] [PubMed] [Google Scholar]
- Scheller K. H.; Sigel H. A proton nuclear magnetic resonance study of purine and pyrimidine nucleoside 5′-diphosphates. Extent of macrochelate formation in monomeric metal ion complexes and promotion of self-stacking by metal ions. J. Am. Chem. Soc. 1983, 105, 5891–5900. 10.1021/ja00356a029. [DOI] [Google Scholar]
- Marzilli L. G. Metal-Ion Interactions with Nucleic Acids and Nucleic Acid Derivatives. Prog. Inorg. Chem. 1977, 255–378. [Google Scholar]
- Prado F. R.; Giessner-Prettre C. Parameters for the calculation of the ring current and atomic magnetic anisotropy contributions to magnetic shielding constants: Nucleic acid bases and intercalating agents. J. Mol. Struct. 1981, 76, 81–92. 10.1016/0166-1280(81)85115-9. [DOI] [Google Scholar]
- Villemin D.; Labiad B. Clay catalysis: dry condensation of barbituric acid with aldehydes under microwave irradiation. Synth. Commun. 1990, 20, 3333–3337. 10.1080/00397919008051567. [DOI] [Google Scholar]
- Ploeser J. M.; Loring H. S. The ultraviolet absorption spectra of the pyrimidine ribonucleosides and ribonucleotides. J. Biol. Chem. 1949, 178, 431–437. [PubMed] [Google Scholar]
- Clark L. B.; Tinoco I. Jr. Correlations in the Ultraviolet Spectra of the Purine and Pyrimidine Bases1. J. Am. Chem. Soc. 1965, 87, 11–15. 10.1021/ja01079a003. [DOI] [Google Scholar]
- Cavalieri L. F.; Bendich A.; Tinker J. F.; Brown G. B. Ultraviolet Absorption Spectra of Purines, Pyrimidines and Triazolopyrimidines1. J. Am. Chem. Soc. 1948, 70, 3875–3880. 10.1021/ja01191a102. [DOI] [PubMed] [Google Scholar]
- Miyaki M.; Shimizu B. N→N alkyl and glycosyl migration of purines and pyrimidines. III. N→N alkyl and glycosyl migration of purine derivatives. Chem. Pharm. Bull. 1970, 18, 1446–1456. 10.1248/cpb.18.1446. [DOI] [Google Scholar]
- Vorbrüggen H.; Bennua B. New simplified nucleoside synthesis. Tetrahedron Lett. 1978, 19, 1339–1342. 10.1016/0040-4039(78)80123-3. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.