

Abstract
Background
Regulator of G‐protein signaling 5 (RGS5) is a negative modulator of G‐protein–coupled receptors. The role of RGS5 in brain endothelial cells is not known. We hypothesized that RGS5 in brain microvascular endothelial cells may be an important mediator of blood‐brain barrier function and stroke severity after focal cerebral ischemia.
Methods and Results
Using a transient middle cerebral artery occlusion model, we found that mice with global and endothelial‐specific deletion of Rgs5 exhibited larger cerebral infarct size, greater neurological motor deficits, and increased brain edema. In our in vitro models, we observed increased Gq activity and elevated intracellular Ca2+ levels in brain endothelial cells. Furthermore, the loss of endothelial RGS5 leads to decreased endothelial NO synthase expression and phosphorylation, relocalization of endothelial tight junction proteins, and increased cell permeability. Indeed, RGS5 deficiency leads to increased Rho‐associated kinase and myosin light chain kinase activity, which were partially reversed in our in vitro model by pharmacological inhibition of Gq, metabotropic glutamate receptor 1, and ligand‐gated ionotropic glutamate receptor.
Conclusions
Our findings indicate that endothelial RGS5 plays a novel neuroprotective role in focal cerebral ischemia. Loss of endothelial RGS5 leads to hyperresponsiveness to glutamate signaling pathways, enhanced Rho‐associated kinase– and myosin light chain kinase–mediated actin‐cytoskeleton reorganization, endothelial dysfunction, tight junction protein relocalization, increased blood‐brain barrier permeability, and greater stroke severity. These findings suggest that preservation of endothelial RGS5 may be an important therapeutic strategy for maintaining blood‐brain barrier integrity and limiting the severity of ischemic stroke.
Keywords: blood‐brain barrier, cell signaling, ischemic stroke, vascular biology
Subject Categories: Blood-Brain Barrier, Cerebrovascular Disease/Stroke, Neuroprotectants, Vascular Disease
Nonstandard Abbreviations and Acronyms
- EC‐Rgs5–/– mice
endothelial‐specific RGS5‐deficient mice
- HBMEC
human brain microvascular endothelial cell
- MCAO
middle cerebral artery occlusion
- MLCK
myosin light chain kinase
- OGD
oxygen‐glucose deprivation
- Rgs5–/– mice
RGS5‐deficient mice
- RGS5
regulator of G‐protein signaling 5
- Rgs5flox/flox mice
conditional RGS5 mice
- ROCK
Rho‐associated kinase
Clinical Perspective
What Is New?
Our data suggest that global or brain microvascular endothelial loss of regulator of G‐protein signaling 5 (RGS5) leads to increased blood‐brain barrier permeability and stroke severity in a rodent model of transient focal cerebral ischemia.
RGS5 deletion in brain microvascular endothelial cells leads to hyperresponsiveness to glutamate signaling pathways, enhanced Rho‐associated kinase– and myosin light chain kinase–mediated actin‐cytoskeleton reorganization, decreased endothelial NO synthase phosphorylation and expression, and increased blood‐brain barrier permeability both in vitro and in vivo.
What Are the Clinical Implications?
Our findings indicate a novel neuroprotective role of endothelial RGS5 in focal cerebral ischemia.
These findings also suggest that preservation of RGS5 may be an important therapeutic strategy for maintaining blood‐brain barrier integrity and limiting the severity of ischemic stroke.
Potential therapeutic benefits of brain endothelial RGS5 upregulation as a therapeutic target should be experimentally and clinically evaluated.
G‐protein–coupled receptors are a large and diverse family of transmembrane receptors. G‐protein–coupled receptors play a central role in virtually every important physiological process, many of which affect human diseases. 1 G‐protein–coupled receptors are negatively regulated by a class of GTPases called regulator of G‐protein signaling (RGS). 2 RGS5 belongs to the R4 subfamily of RGS proteins and is a potent negative regulator of Gαq and Gαi. The R4 subfamily structurally consists of a conserved RGS domain that binds to the corresponding Gα subunit, dephosphorylating the active GTP‐bound Gα subunit through the GTPase‐stimulating activity of the RGS domain. RGS5 is highly abundant in pericytes, vascular endothelial cells, vascular smooth muscle cells, and some neurons. Furthermore, limited expression of RGS5 has been detected in the heart, liver, lung, brain, small intestine, placenta, and colon. 3
Recent studies indicate that RGS5 plays an important role in the cardiovascular system. 4 For example, RGS5 regulates systemic blood pressure, 5 , 6 affects the development of cardiac hypertrophy, 7 and stabilizes blood vessel formation. 8 In pregnant mice, RGS5 deficiency leads to hypertension and preeclampsia. 5 Indeed, several single‐nucleotide polymorphisms of RGS5 are associated with essential hypertension in Black and Chinese Han populations, although the precise mechanisms by which RGS5 affects systemic blood pressure are not well characterized. 9 , 10 In endothelial cells, RGS5 is hypoxia inducible and is highly upregulated during angiogenesis and after vascular injury. 11 In tumorigenesis, loss of RGS5 stabilizes newly formed vessels by recruiting pericytes, thereby reducing vessel leakiness and ischemia. 12
Because the blood‐brain barrier (BBB) is composed of cerebral microvascular endothelial cells, we hypothesized that RGS5 plays a critical role in regulating BBB function after ischemic stroke. The aim of this study, therefore, is to determine the role of RGS5 in ischemic stroke and to determine the mechanisms by which RGS5 regulates endothelial and BBB function during cerebral ischemia.
METHODS
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Animals
For in vivo experiments, we used 12‐ to 20‐week‐old, age‐matched, global (Rgs5 –/–; total of 41 animals) and endothelial cell–specific RGS5‐deficient (EC‐Rgs5–/–; total of 43 animals) mice with corresponding controls. To overcome the previously demonstrated intrinsic “ischemic protection” in young adult female mice, 13 we only used male mice in this study. All mice were congenic strains on C57Bl/6J background. To establish these mouse lines, we made a conditional Rgs5flox/flox mouse strain by introducing “flox” sequences that flanked the RGS5‐binding domain from exon 3 to 5 on chromosome 1. Global Rgs5–/– and EC‐Rgs5flox/flox mice were then generated by crossing the Rgs5flox/flox mouse strain with Pgk‐Cre and inducible Cre recombinase under the control of the vascular endothelial cadherin promoter mice, 14 respectively. To induce endothelial cell–specific Rgs5 deletion, EC‐Rgs5flox/flox mice were treated with tamoxifen (20 mg/kg per day, IP, 5 days) (Sigma‐Aldrich, St. Louis, MO). Experiments using these mice were performed at least 5 days after date of last tamoxifen injection. The controls for Rgs5–/– and EC‐Rgs5–/– mice were Pgk‐Cre (total of 41 animals), Cdh5‐CreERT2 (total of 29 animals with tamoxifen treatment), and EC‐Rgs5flox/flox (total of 37 animals without tamoxifen treatment). Mice were kept under 12:12‐hour light/dark schedule (light phase: white light 124 lx from 6 am to 6 pm; dark phase: red light <2 lx from 6 pm to 6 am). Food (Harlan 2918) and water were available ad libitum.
Invasive Blood Pressure Measurement
Mice were anesthetized with isoflurane (2% and 1.2% for the initial dose and during surgery, respectively). Body temperature was monitored with a rectal thermometer and maintained at 37°C ±0.5°C with a heating pad. The neck area was shaved, and the right carotid artery was isolated after middle neck incision. The distal end of the right carotid artery was tied, and its proximal end was temporarily closed. An intravascular catheter (1F Millar catheter, SPR‐1000; Millar Instruments, Inc, Houston, TX) was inserted into the carotid artery and advanced to the ascendant aorta. The catheter was secured with a suture, and the blood pressure and heart rate were recorded with a pressure control unit (PCU‐2000; ADInstruments, Colorado Springs, CO) and PowerLab 35 series data acquisition system with LabChart Pro (ADInstruments).
Transient Middle Cerebral Artery Occlusion
Transient middle cerebral artery (MCA) occlusion was used as a rodent model for ischemic stroke. Mice were anesthetized with isoflurane, and the body temperature was maintained as above. MCA was transiently occluded by the insertion of commercially available 6‐0 silicon sutures (Doccol Corp, Redlands, CA) into the internal carotid artery through a small incision on the external carotid artery and by the advancement of the sutures to the origin of MCA, ≈9 mm from the common carotid artery bifurcation. Successful MCA occlusion (MCAO) was confirmed by measuring the relative cerebral blood flow in MCA territory (≥80% reduction of relative cerebral blood flow from baseline) using transcranial laser Doppler flow (Moor VMS‐LDF; Moor Instruments, UK). The sutures were withdrawn after 30 minutes, and reperfusion was confirmed with relative cerebral blood flow (returning to >95% of baseline), indicating complete reperfusion without residual occlusion. For sham surgery, all animals underwent the same surgical procedure for the same period but without MCAO. After the procedure, mice were rehydrated with subcutaneous injection of 0.5 mL of saline and were treated with analgesic buprenorphine (0.1 mg/kg). Mortality rate following surgery was <10%. All mice were euthanized with CO2 asphyxiation after 24 hours of reperfusion. Cerebral infarct size (volume) was measured by triphenyltetrazolium chloride of 4 contiguous 2‐mm‐thick coronal brain sections.
Neurological Deficit Score
Neuromotor deficits were evaluated with a 6‐grade scoring scheme by investigators who were blinded to the mouse genotype. The neurological deficit score was determined by the following: 0 (no deficit), 1 (mild deficit, circling without inconsistent rotation), 2 (consistent circling), 3 (consistent strong circling and holding of a rotation position for >2 seconds), 4 (severe rotation and loss of righting reflex), and 5 (unresponsive, comatose). Animals with a score of 5 were excluded from the study and euthanized. To minimize potential bias, unresponsive and comatose animals were only excluded after recommendation from a credentialed veterinary technician. Sensorimotor function was evaluated using the adhesion tape test. In brief, mice were pretrained for 5 days before MCAO. A 2‐mm2 adhesive tape was applied to the contralateral forepaw, and the time to remove the tape was measured (maximum of 180 seconds).
Brain Edema
Brain edema was measured with the wet‐dry method. 15 Brains were quickly removed and weighed. Each brain was separated into ipsilateral and contralateral hemispheres and dried overnight at 95°C. Water content was calculated as % H2O=100×(wet‐dry weight)/wet weight. Brain edema was also visually determined by extravasation of the injected Evans blue dye. BBB permeability was assessed after 2 hours of femoral vein injection of fluorescein sodium salt (Sigma‐Aldrich), 5 kDa dextran–Cascade Blue, and 40 kDa dextran–fluorescein isothiocyanate (FITC) (Thermo‐Fisher Scientific, Waltham, MA) in mice in deep isoflurane anesthesia 22 hours after reperfusion. Before dextran injection, mice were injected with saline. The brain hemispheres were quickly separated to measure fluorescence after extraction by a methanol dye. To visualize leakage of the injected dye, brains were transcardially perfused with saline, fixed in 4% paraformaldehyde, embedded in Tissue‐Tek (OCT compound; Sakura, Horgen, Switzerland), and imaged with a confocal laser‐scanning microscope (LSM 510; Zeiss, Germany).
4‐Amino‐5‐Methylamino‐2′,7′‐Difluorofluorescein Diacetate (DAF‐FM) Staining
Fresh brains were isolated, sliced into 2‐mm sections, and stained with 5 μmol/L DAF‐FM diacetate (Invitrogen, CA) in dark for 20 minutes. Sections were quickly washed in PBS and evaluated by fluorescent microscopy (BZ‐X700; Keyence, Itasca, IL) at 495/515 nm.
Cell Culture
Three identical genomic clones of primary isolated human brain microvascular endothelial cells (HBMECs) were purchased from ScienCell Research Laboratories (Carlsbad, CA), propagated, and used at passage 3. They were cultured at 37°C per manufacturer's instructions. RGS5 was knocked down by lentiviral‐containing RGS5 shRNA particles (Sigma‐Aldrich) at passage 3. For control, HBMECs were transduced with nonmammalian shRNA particles. Provided control GFP (green fluorescent protein) lentivirus construct was used as a visual confirmation of successful transfection and delivery. For isolation of mouse brain microvascular endothelial cells, we used 12‐ to 14‐week‐old mice. Mouse brains were isolated and gray matter was dissected and minced in Hanks balanced solution (Invitrogen Corp, Carlsbad, CA), then homogenized in a Dounce homogenizer. The microvessels were separated with a Percoll gradient (GE Healthcare Bio‐Science, Chicago, IL) and digested in 1 mg/mL collagenase/dispase (Roche, Indianapolis, IN) for 30 minutes at 37°C. Mouse brain endothelial cells were labeled with cluster of differentiation 31/cluster of differentiation 102 antibodies (BD Bioscience, San Jose, CA) and purified with magnetic beads (Dynabeads; Thermo‐Fisher, Waltham, MA). Endothelial cells were grown on plates coated with collagen IV (BD Bioscience, San Jose, CA) in endothelial cell medium (ScienCell Research Laboratories) at 37°C and 5% CO2.
Oxygen‐Glucose Deprivation With l‐Glutamate Treatment
HBMECs were subjected to oxygen‐glucose deprivation (OGD; glucose‐free DMEM in a gas mixture, 5% CO2/95% N2) with l‐glutamate treatment (1 mmol/L) for 3 hours at 37°C. Cells were then washed with PBS, cultured in DMEM with glucose supplementation, and placed in an incubator with 95% O2/5% CO2 at 37°C for 24 hours.
Western Blotting
Samples were lysed in a buffer (Cell Signaling Technology, Danvers, MA) containing phenylmethylsulfonyl fluoride (1 mmol/L). After brief sonication, samples were centrifuged (13 000g, 5 minutes) and supernatants were collected. Protein concentration was determined by the Bradford assay (Bio‐Rad Laboratories, Hercules, CA). Protein samples (25 μg) were separated on SDS‐PAGE and transferred to nitrocellulose membranes. After blocking with 5% BSA, membranes were immunoblotted with primary antibodies at 4 ºC overnight. For immunodetection, we used horseradish peroxidase–conjugated secondary antibodies (Tables S1 through S3), and detection was accomplished using chemiluminescence (ECL Substrate; Bio‐Rad Laboratories). Images were obtained and quantitated using the ChemiDoc MP System (Bio‐Rad Laboratories).
Real‐Time Quantitative Polymerase Chain Reaction
Total RNA was isolated using an RNA purification kit, according to manufacturer's instructions (Thermo‐Fisher, Waltham, MA). RNA (0.5 μg) was reverse transcribed into cDNA, and real‐time quantitative polymerase chain reaction was performed using the StepOnePlus Real‐Time PCR System (Thermo‐Fisher Scientific, Waltham, MA). Primer sets used were as follows: RGS5‐exon1 (forward, 5′‐GAT TAT TGA AGT TTC CAC AGA CG‐3′; and reverse, 5′‐GCC AGT CCC TTA CAC ATT T‐3′), RGS5‐exon2 (forward, 5′‐GTC AGC TGT TGA GAG GTT C‐3′; and reverse, 5′‐TTT CCA GGC ATG AGT GC‐3′), RGS5 (forward, 5′‐TCAAGA TCA AGT TGG GAA T‐3′; and reverse, 5′‐GAG AAT CCT TCT CCA TCA G‐3′), and GAPDH (forward, 5′‐GCA GTG GCA AAG TGG AGA TT‐3′; and reverse, 5′‐CAC ATT GGG GGT AGG AAC AC‐3′). PCR settings after initial denaturation (95°C for 30 seconds) were as follows (40 cycles): denaturation (95°C for 15 seconds), annealing/extension, and plate read (60°C for 60 seconds) (Applied Biosystems, StepOnePlus, Real‐Time PCR System, Foster City, CA). Expression level was normalized with GAPDH and quantified using the 2−ΔΔCt method. 16
Immunofluorescent Staining
For immunofluorescence staining, HBMECs were fixed with 4% paraformaldehyde, permeabilized with 0.5% Triton X for 5 minutes, and blocked with 1% normal goat serum for 1 hour. Cells were incubated with primary antibodies at 4°C overnight. Staining was visualized after 1‐hour incubation with fluorophore‐conjugated secondary antibodies (Tables S1 through S3) with a fluorescent microscope (BZ‐X700; Keyence, Itasca, IL). To visualize F‐actin formation, we used phalloidin staining according to manufacturer's recommendations (Thermo‐Fisher Scientific, Waltham, MA).
Measuring Gq Activity and Intracellular Calcium Concentration
To measure Gq activity, we used a commercially available assay for detecting its downstream metabolite, inositol monophosphate, according to manufacturer's recommendations (HTRF, IP‐one Assay; Cisbio Bioassays, Bedford, MA). For quantification of intracellular calcium concentration in HBMECs, we used a commercially available assay according to manufacturer's recommendations (calcium detection kit; Abcam, Cambridge, MA). To visualize intracellular calcium levels, we used Fura2‐AM staining according to manufacturer's recommendations (Abcam, Cambridge, MA).
Dextran Transwell Permeability Assay
HBMECs were grown on transwell polycarbonate membrane inserts (0.4‐μm pore size) at an initial density of 1×105 cells/mL for 7 days until confluency. Cells were then exposed to OGD with l‐glutamate treatment. After treatment, FITC‐labeled dextran (40 kDa) was added to the upper chamber (1 μg/mL) and supernatant from the lower chamber was collected after 24 hours. Fluorescence was measured (495/519 nm) according to a standard curve on a plate reader (Tecan 200, Mannedorf, Switzerland).
NO Assay
To measure total NO production in the brain tissue, we used a commercially available kit (Fluorometric Nitric Oxide Assay Kit; Abcam, Cambridge, MA) for detecting total nitrite concentration after conversion of nitrate to nitrite by nitrate reductase. Nitrite concentration was measured after reaction with the fluorescent probe DAN (2,3 diaminonaphthalene) (360/450 nm) according to a standard curve on a plate reader (Tecan 200, Mannedorf, Switzerland).
Statistical Analysis
All analyses were conducted with GraphPad Prism 8 (La Jolla, CA) or R version 3.5.2. The results with normal distribution are expressed as the mean±SEM, and data without normal distribution are shown as median±interquartile range (IQR). Normality of data was evaluated using Shapiro‐Wilk test. To assess for statistical significance of normal data, we used the Welch t tests for comparisons between 2 groups. In cases where the data were not normally distributed, we used the Mann‐Whitney U test for comparisons between 2 groups. In data sets where there were multiple comparisons, 1‐ or 2‐way ANOVA with post hoc Tukey test was conducted for normally distributed data sets and Kruskal‐Wallis tests were used for nonnormally distributed data sets. Statistical significance in data sets using count data was tested with the χ2 test. For nonnormally distributed paired data samples, we used the Wilcoxon signed‐rank test. All tests were 2 tailed, and P<0.05 was accepted as statistically significant.
Study Approval
All experimental animal protocols used in this study comply with the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee at the University of Chicago.
RESULTS
Characterization of Global and Endothelial Cell–Specific RGS5 Knockout Mice
The Rgs5 gene is localized on chromosome 1 and consists of 5 exons. To generate Rgs5‐specific deletion, the genomic sequence encoding the entire RGS5‐binding domain (from exon 3 to 5) was replaced by a same domain flanked by loxP sites. The neomycin cassette, which was used for clonal selection, was flanked by flippase recognition target sites. The “floxed” Rgs5 mice were first bred with Flp mice to remove the neomycin cassette, and then bred with transgenic mice expressing Cre recombinase under the control of the phosphoglycerate kinase 1 promoter to generate mice with global deletion of Rgs5 (Rgs5–/– mice) (Figure S1A). Successful deletion was confirmed by Western blotting, showing loss of RGS5 in the brain, lung, heart, and liver of Rgs5–/– mice compared with control (Pgk‐Cre) mice (Figure 1A). Furthermore, efficient Rgs5 deletion was confirmed by Western blotting and immunostaining of sorted primary isolated brain endothelial cells after 2 cycles of sorting with anti–platelet endothelial cell adhesion molecule‐1 and anti–intercellular adhesion molecule‐2 antibody‐conjugated magnetic beads (Figure S1B and S1C). Real‐time quantitative polymerase chain reaction analysis of exon 1, exon 2, and the entire RGS5 mRNA confirmed complete Rgs5 deletion without the expression of truncated forms of exons 1 and 2 (data not shown). Rgs5–/– mice were fertile and have similar body weight up to 8 weeks as transgenic mice expressing Cre recombinase under the control of the phosphoglycerate kinase 1 promoter (Figure S1D). Using an invasive method for blood pressure measurement under anesthesia, we observed that Rgs5–/– mice have higher systolic (123.6±2.2 versus 113±0.9 mm Hg; n=5; P=0.016), diastolic (83.4±1.4 versus 75.6±0.8 mm Hg; n=5; P=0.015), and mean arterial blood pressures (97 [IQR, 98.5–95] versus 87 mm Hg [IQR, 89.5–87]; n=5; P<0.01) compared with that of transgenic mice expressing Cre recombinase under the control of the phosphoglycerate kinase 1 promoter. Heart rate did not differ between the 2 groups of mice (458.6±9 versus 448.6±6.6 bpm; n=5; P=0.39) (Figure 1B). To create EC‐specific RGS5 knockout mice (EC‐Rgs5–/–), we bred Rgs5flox/flox mice with tamoxifen‐inducible transgenic mice that express Cre under the control of the vascular endothelial‐cadherin (Cdh5) promoter (Cdh5‐CreERT2) (Figure S1E). Western blotting analyses of cell lysates from primary cultures of brain microvascular endothelial cells of EC‐Rgs5–/– mice (Figure 1C) and from whole brains, hearts, and lungs of EC‐Rgs5–/–mice (EC‐Rgs5flox/flox with tamoxifen) showed endothelial‐specific RGS5 deletion compared with that of control (EC‐Rgs5flox/flox without tamoxifen) (Figure 1D). The overall RGS5 expression in tissues was reduced in the whole brain (18.4±1.4%; n=5; P<0.01), whole heart (14.2±1.6%; n=5; P<0.01), and whole lung (29.4±2.1%; n=5; P<0.01) of EC‐Rgs5–/– mice compared with that of controls (Figure 1D). The blood pressure of endothelial‐specific Rgs5‐deficient mice (EC‐Rgs5flox/flox without tamoxifen) did not significantly differ from controls (systolic blood pressure, 113.3±2 versus 115±2.2 mm Hg [P=0.52]; diastolic blood pressure, 72.8±1.2 versus 75.2±2.4 mm Hg [P=0.36]; mean arterial blood pressure, 87 [IQR, 87–84.5] versus 88 mm Hg [IQR, 92.2–84.5] [P=0.61]) (Figure 1E). EC‐Rgs5–/– mice were fertile and had a similar body weight as control mice up to 8 weeks (Figure S1F).
Figure 1. Generation of global (Rgs5 −/−) and endothelial cell–specific regulator of G‐protein signaling 5 (RGS5) deficient mice (EC‐Rgs5 –/–).
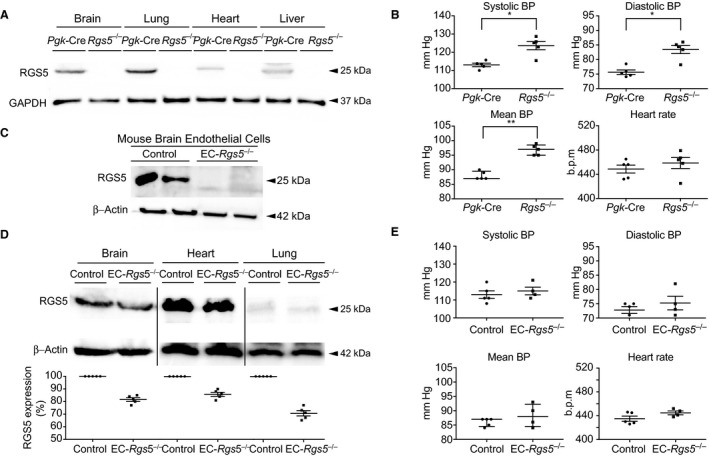
A, Western blotting of RGS5 in the brain, lung, heart, and liver of Rgs5 −/− mice compared with control, transgenic mice expressing Cre recombinase under the control of the phosphoglycerate kinase 1 promoter (Pgk‐Cre). B, Blood pressure and heart rate of Rgs5 −/− and Pgk‐Cre mice (n=5). Western blot analysis of RGS5 expression in primary brain endothelial cells (C) and brain, heart, and lung of EC‐Rgs5 –/– mice (D), with and without (Control) tamoxifen treatment (n=5). E, Blood pressure (BP) and heart rate of Control and EC‐Rgs5 –/– mice (n=5–4). The Welch t test was used for normally distributed data, and the Mann‐Whitney U test was used for all nonnormally distributed data set comparisons. *P<0.05, **P<0.01. B.p.m. indicates beats per minute.
Neuroprotective Effect of RGS5 After Focal Cerebral Ischemia
RGS5 expression is increased in the early period following the onset of ischemic injury, 11 suggesting a potential physiological role of RGS5 in focal cerebral ischemia. Indeed, triphenyltetrazolium chloride staining indicated Rgs5–/– mice had larger cerebral infarct sizes compared with control (Pgk‐Cre) mice (32.0±0.5% versus 15.9±0.9%; n=10; P<0.01) (Figure 2A). This was associated with more severe neurological motor deficits (n=10) (Figure 2B), greater loss of coordination, as determined by the adhesive tape removal test (122.4±6.7 versus 63.1±3.8 seconds; n=10; P<0.01) (Figure 2C), and increased brain edema formation (84.8±0.4% versus 81.9±0.3%; n=10; P<0.01) (Figure 2D) in Rgs5–/– mice. No differences in any parameters were observed between Rgs5–/– mice and transgenic mice expressing Cre recombinase under the control of the phosphoglycerate kinase 1 promoter after sham surgery. Compared with control mice, BBB permeability, as determined by leakage of Evan blue, was greater in Rgs5–/– mice (n=5) (Figure 2E). Similar findings of greater BBB permeability in Rgs5–/– mice were observed in the ipsilateral brain hemisphere after intravenous injection of 40 kDa FITC‐labeled dextran (18.3±0.45 versus 13.4±0.23 μg/g of tissue; n=6; P<0.01) (Figure 2F) and in coronal cryosections of brain peri‐infarct areas, as visualized by fluorescent microscopy (Figure 2G). There were no changes in BBB permeability with sodium‐FITC and 5 kDa dextran–Cascade Blue in sham operated animals (sodium‐FITC P=0.23, dextran–Cascade Blue P=0.69; n=5; Figure 2H).
Figure 2. Effect of global regulator of G‐protein signaling 5 (RGS5) deletion on cerebral injury after transient middle cerebral artery occlusion (MCAO).
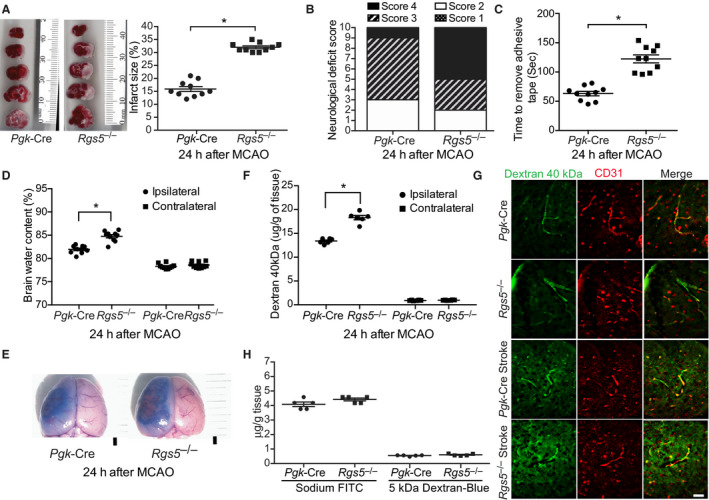
A, Coronal brain sections of control transgenic mice expressing Cre recombinase under the control of the phosphoglycerate kinase 1 promoter (Pgk‐Cre) and global Rgs5 deficient (Rgs5 −/−) mice stained with 2,3,5, triphenyltetrazolium chloride (left panel) with corresponding quantification of cerebral infarct volume (right panel) (n=10). Neurological deficit score (B), sensorimotor impairment (C), brain edema formation (n=10) (D), and Evans blue leakage (E) in Pgk‐Cre (Control) and global Rgs5 −/− mice 24 hours after MCAO (n=5). Bar=1 mm. Quantification of blood‐brain barrier (BBB) permeability (F) and microscopic visualization of fluorescein isothiocyanate (FITC)–labeled dextran 40 kDa leakage (G), 24 hours after MCAO in Pgk‐Cre (Control) and global Rgs5 −/− mice (n=6). Bar=50 μm. H, BBB permeability for sodium‐FITC and 5 kDa dextran–Cascade Blue of sham operated animals (n=5). For comparisons between 2 groups, the Welch t test was used. For brain edema formation, and BBB permeability data sets, 2‐way ANOVA (variables, ipsilateral vs contralateral and Pgk‐Cre vs Rgs5 –/–) was conducted followed by post hoc Tukey tests. The χ2 test was used for neurological deficit score data. *P<0.01.
Neuroprotective Effect of Endothelial RGS5 After Focal Cerebral Ischemia
Next, we examined the role of endothelial RGS5 in focal cerebral ischemia. As with Rgs5–/– mice, EC‐Rgs5–/– mice also exhibited increased cerebral infarct size (24.5±0.7% versus 14.8±1.0% and 14.7±0.7%; n=5–6; P<0.01; Figure 3A), more severe motor neurological deficits (n=5; Figure 3B), poorer coordination, as measured by the adhesive tape removal test (81.4±2.3 versus 45.8±1.7 and 48±2.1 seconds; n=5; P<0.01; Figure 3C), and greater brain edema formation (83.8±0.3% versus 81.2±0.6% and 80.2±0.7%; n=5; P<0.01; Figure 3D) compared with control mice (EC‐Rgs5flox/flox mice without tamoxifen and inducible Cre recombinase under the control of the vascular endothelial cadherin promoter mice with tamoxifen). Furthermore, EC‐Rgs5–/– mice showed increased permeability of 40 kDa dextran‐FITC in the ipsilateral hemisphere compared with control mice (17±0.5 versus 13±0.3 μg/g of tissue; n=5; P<0.01) (Figure 3E), which was confirmed via visualization of coronal cryosections of brain peri‐infarct areas by fluorescent microscopy (Figure 3F). These findings indicate that endothelial‐specific RGS5 plays a major role in BBB integrity and limits the extent of injury following focal cerebral ischemia.
Figure 3. Effect of endothelial‐specific regulator of G‐protein signaling 5 deletion (EC‐Rgs5 –/–) on cerebral injury following transient middle cerebral artery occlusion (MCAO).
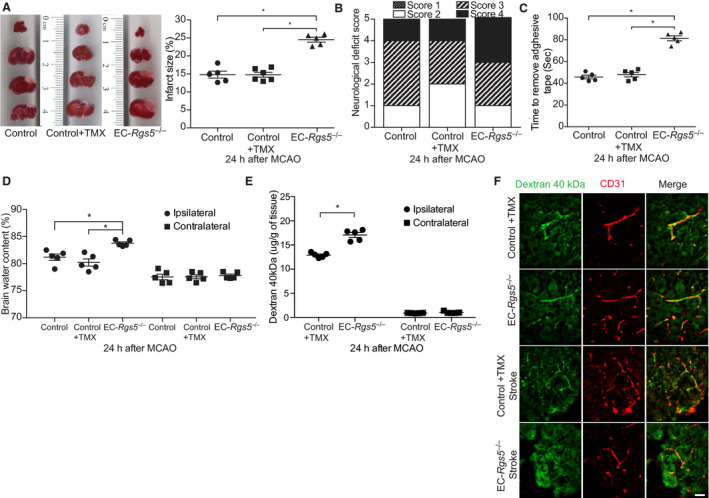
A, Coronal sections stained with 2,3,5, triphenyltetrazolium chloride (left panel) with quantification of cerebral infarct volume (right panel) (n=5 to 6). Evaluation of neurological deficit score (B) and sensorimotor impairment (C) in inducible Cre recombinase under the control of the vascular endothelial cadherin promoter mice (Cdh5‐CreERT2) tamoxifen treated (Control+TMX), and EC‐Rgs5 –/– with and without (Control) tamoxifen induction (n=5). D, Brain water content following MCAO in EC‐Rgs5 –/– and control mice (n=5). Quantification of blood‐brain barrier permeability (E) and microscopic visualization of fluorescein isothiocyanate–labeled dextran 40 kDa leakage (F), 24 hours after MCAO in EC‐Rgs5 –/– and Cdh5‐CreERT2 tamoxifen treated (Control+TMX) (n=5). Bar=50 μm. For all data sets, 1‐way ANOVA (A and C) or 2‐way ANOVA (D and E; variables, ipsilateral vs contralateral and type of mouse) was conducted followed by post hoc Tukey tests. The χ2 test was used for neurological deficit score data. *P<0.01.
RGS5 Deficiency Leads to Higher Gq Activity and Increased Intracellular Calcium Levels
RGS5 regulates the function of G‐protein–coupled receptors by enhancing Gαq GTPase activity and inhibiting Gαq signaling. 2 As Gαq mediates increases in intracellular calcium and calcium plays an important role in endothelial permeability, 17 we investigated whether the loss of function of RGS5 in HBMECs affects Gαq activity in vitro, with and without l‐glutamate stimulation, which mimics glutamate release during cerebral ischemia. Successful knockdown of RGS5 by lentiviral shRNA transduction was confirmed by Western blotting (Figure 4A). Furthermore, efficient transduction and delivery was confirmed by control GFP lentiviral transfection (Figure S1G). In RGS5 knockdown (RGS5‐KD) HBMECs, Gαq activity was higher under basal conditions and after l‐glutamate treatment in a concentration‐dependent manner compared with control cells (n=5; P<0.05; Figure 4B). Next, we examined the effect of RGS5 deficiency on intracellular calcium levels. In RGS5‐KD HBMECs, higher intracellular calcium concentration was observed under basal conditions (0.10±0.02 versus 0.04±0.01 μg/well; n=5; P=0.01; Figure 4C) and after l‐glutamate treatment (0.3±0.05 versus 0.15±0.01 μg/well; n=5; P=0.01; Figure 4C) compared with control cells. The increase in intracellular calcium in RGS5‐KD HBMECs was completely abolished by the cell‐permeable calcium chelator, 1,2‐bis(o‐aminophenoxy)ethane‐N,N,N′,N′‐tetraacetic acid‐acetoxylmethyl ester (BAPTA‐AM). Similarly, the increase in intracellular calcium concentration in RGS5‐KD HBMECs after l‐glutamate treatment was also observed with Fura‐2 staining (Figure 4D).
Figure 4. Effect of regulator of G‐protein signaling 5 knockdown (RGS5‐KD) on Gq activity, intracellular calcium levels, and myosin light chain kinase/Rho‐associated kinase (MLCK/ROCK) activity in human brain endothelial cells.
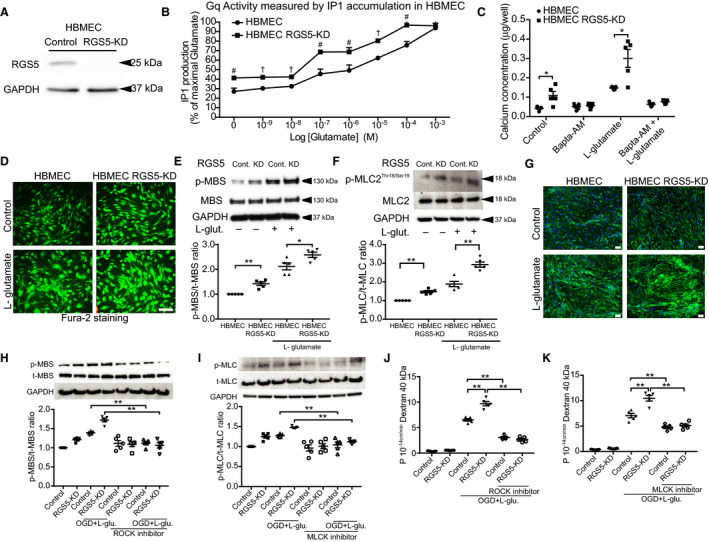
A, Western blotting evaluation of RGS5‐KD. B, Gq activity measured before and after treatment with l‐glutamate in a concentration‐dependent manner (# P<0.01, and † P<0.001; n=5). C, Quantification of intracellular calcium concentration after treatment with l‐glutamate and BAPTA‐AM (n=5). D, Fura‐2 staining following treatment with l‐glutamate. Western blotting showing phosphorylation of myosin phosphatase target subunit 1 (MBS) (E) and myosin light chain 2 (MLC2) (F), before and after treatment with l‐glutamate (n=5). G, Phalloidin staining showing increased F‐actin staining. Bar=50 μm. Western blotting analysis of ROCK activity (MBS phosphorylation) (H) and MLCK activity (MLC2 phosphorylation) (I) in control and RGS5‐KD, with or without oxygen glucose deprivation and l‐glutamate stimulation (OGD+l‐glutamate stimulation; 1 mmol/L for 3 hours), in the presence or absence of either ROCK (Y‐27632; 20 μmol/L) or MLCK (ML‐7; 10 μmol/L) pharmacological inhibition. Cell permeability, as determined by fluorescein isothiocyanate–labeled dextran 40 kDa of control or RGS5‐KD human brain microvascular endothelial cells (HBMECs), with or without OGD+l‐glutamate, in the presence or absence of ROCK inhibitor (Y‐27632; 20 μmol/L) (J) or MLCK inhibitor (ML‐7; 10 μmol/L) (K) (n=5). Wilcoxon signed‐rank test was used for the Gq activity data set. All other data sets used 2‐way ANOVA followed by post hoc Tukey tests (variables, treatment groups and control vs RGS5 knockdown). *P<0.05, **P<0.01.
RGS5 Regulates the Actin Cytoskeleton Through Inhibition of Rho‐Associated Kinase and Myosin Light Chain Kinase
Rho‐associated kinase (ROCK) and myosin light chain kinase (MLCK) are 2 important mediators of the actin cytoskeleton and, consequently, play central roles in endothelial barrier maintenance. 18 To determine the effects of RGS5 deficiency on ROCK and MLCK activity, we measured the downstream phosphorylation targets of ROCK (Thr853‐MBS) and MLCK (Ser/Thr‐MLC2). Compared with control cells, RGS5‐KD HBMECs exhibited higher ROCK activity (phospho‐myosin‐binding subunit/total‐myosin‐binding subunit, pMBS/tMBS) under basal conditions (1.42±0.07 versus 1.00±0.0; P<0.01; n=5) and after l‐glutamate treatment (2.58±0.10 versus 2.12±0.14; P=0.03; n=5) (Figure 4E). Similarly, RGS5‐KD HBMECs exhibited higher MLCK activity (phospho‐myosin light chain 2/total‐myosin light chain 2) under basal (1.47±0.06 versus 1.00±0.0; P<0.01; n=5) and l‐glutamate treatment conditions (2.93±0.14 versus 1.88±0.13; P<0.01; n=5) (Figure 4F). Indeed, RGS5‐KD HBMECs showed greater F‐actin staining under basal conditions and with l‐glutamate treatment compared with controls (Figure 4G).
An in vitro model of ischemia‐reperfusion injury consisting of exposing HBMECs to OGD with l‐glutamate (ischemia) followed by replenishment with oxygen and glucose but without l‐glutamate (reperfusion) was used to gain further mechanistic insight. Using this model, we found that ROCK and MLCK activities were both increased by OGD and l‐glutamate (Figure 4H and 4I). Cotreatment with Y‐27632, an inhibitor of ROCK, or ML‐7, an inhibitor of MLCK, prevented the increase in ROCK and MLCK activities in both control and HBMEC‐RGS5‐KD cells, respectively. This correlated with a reduction in OGD/l‐glutamate–induced RGS5‐KD HBMEC cell permeability with either ROCK inhibition (control, 6.43±0.34 versus 3.13±0.3, n=5, P<0.01; HBMEC‐RGS5‐KD, 9.86±0.73 versus 2.66±0.34, n=5, P<0.01; Figure 4J) or MLCK inhibition (control, 7.06±0.43 versus 4.78±0.25, n=5, P<0.01; HBMEC‐RGS5‐KD, 10.48±0.54 versus 5.12±0.3, n=5, P<0.01; Figure 4K). These findings indicate that RGS5 deficiency leads to activation of ROCK and MLCK, enhanced F‐actin staining, and increased cell permeability in a ROCK/MLCK‐dependent manner.
Inhibition of Gq‐Coupled Metabotropic Glutamate Receptor 1 and Ligand‐Gated Ionotropic Glutamate Receptor by RGS5 Mediates Changes in ROCK and MLCK Activity and Cell Permeability
Because l‐glutamate is known to activate Gαq‐coupled metabotropic glutamate receptor 1 (mGluR1) and ligand‐gated ionotropic glutamate receptor (NMDAR) in the brain, we investigated whether selective inhibitors of Gαq, mGluR1, and NMDAR could reverse the effects of OGD/l‐glutamate–induced ROCK/MLCK activity and cellular permeability in control and RGS5‐KD HBMECs. Cotreatment with the Gαq inhibitor, YM254980, blocked the increase in ROCK activity (0.96±0.06 versus 1.48±0.03; n=5; P<0.01) and MLCK activity (3.5 [IQR, 4–2.6] versus 5.34 [IQR, 5.6–4.3]; n=5; P=0.02) in RGS5‐KD HBMECs (Figure 5A). Similarly, cotreatment with the mGluR1 inhibitor, JNJ 16259685, prevented the upregulation of ROCK activity (0.86±0.08 versus 1.48±0.02; n=5; P<0.01) and MLCK activity (1.04±0.03 versus 2.05±0.05; n=5; P<0.01) in RGS5‐KD HBMECs (Figure 5B). Finally, cotreatment with the NMDAR inhibitor, MK‐801, also inhibited the increase in ROCK activity (1.12±0.04 versus 1.45±0.02; n=5; P<0.01) and MLCK activity (0.97 [IQR, 1.16–0.92] versus 1.54 [IQR, 1.57–1.33]; n=5; P<0.01) in RGS5‐KD HBMECs (Figure 5C). These inhibitory effects of YM254980, JNJ 16259685, and MK‐801 on ROCK and MLCK activities in RGS5‐KD HBMECs correlated with decreases in cell permeability, as measured by FITC‐labeled 40 kDa dextran (Gαq inhibition: 4.16±0.3 versus 10.08±0.75, n=5, P<0.01; mGluR1 inhibition: 3.58±0.2 versus 9.16±0.6, n=5, P<0.01; NMDR inhibition: 4.48±0.19 versus 9.62±0.43, n=5, P<0.01) (Figure 5D through 5F). These findings suggest that inhibition of Gαq, mGluR1, and NMDAR, and their downstream targets, ROCK and MLCK, by RGS5 in HBMECs is critical for maintaining endothelial cell integrity and limiting cell permeability in response to OGD/l‐glutamate.
Figure 5. Effect of regulator of G‐protein signaling 5 knockdown (RGS5‐KD) on actin cytoskeleton reorganization and cellular permeability during oxygen glucose deprivation and l‐glutamate stimulation (OGD+l‐glutamate; 1 mmol/L for 3 hours) and reperfusion treatment.
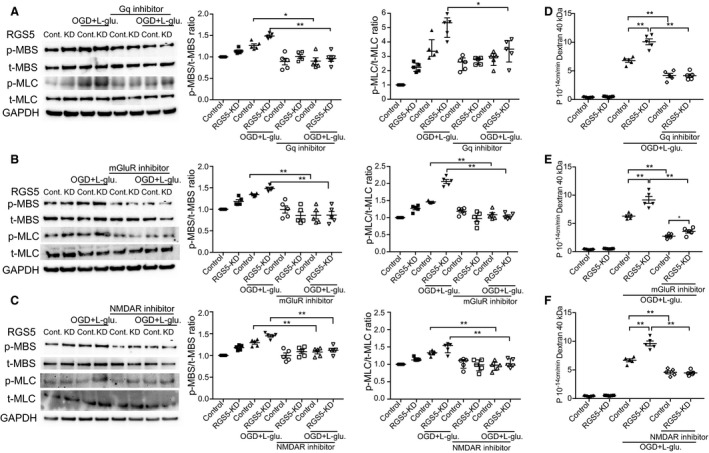
Western blotting analysis of myosin phosphatase target subunit 1 (MBS) and myosin light chain 2 (MLC2) phosphorylation, before and after treatment with Gq inhibitor (10 μmol/L; YM254980) (A), metabotropic glutamate receptor (mGluR) inhibitor (10 mmol/L; JNJ16259685) (B), and ligand‐gated ionotropic glutamate receptor (NMDAR) inhibitor (10 mmol/L; MK‐801) (C). Cell permeability, as determined by fluorescein isothiocyanate–labeled dextran 40 kDa in the presence or absence of Gq inhibitor (D), mGluR (E), or NMDAR inhibitor (F). n=5 for all above experiments. Normally distributed data sets used 2‐way ANOVA (variables, treatment groups and control vs RGS5‐KD) followed by post hoc Tukey tests, whereas nonnormally distributed data sets used Kruskal‐Wallis tests. *P<0.05, **P<0.01.
Maintenance of Endothelial Junctional Proteins and Endothelial NO Synthase Expression by RGS5
Loss of membrane junctional proteins through cellular relocalization is an important mechanism that leads to increased cell permeability in response to ischemia‐reperfusion injury. 19 Indeed, immunofluorescent staining for endothelial junctional proteins, including zonula occludens‐1, occludin, and claudin‐5, showed increases in membrane relocalization of these junction proteins in RGS5‐KD HBMECs after OGD/l‐glutamate treatment (Figure 6A through 6C; n=5). The relocalization of these junction proteins was prevented by inhibition of Rho/ROCK, MLCK, Gαq, mGluR1, and NMDAR. There were no differences in total expression of tight junction proteins before or after MCAO or OGD/l‐glutamate treatment in our in vivo and in vitro models of ischemic stroke, respectively (P>0.05; n=5–8) (Figure S2A and S2B).
Figure 6. Effect of regulator of G‐protein signaling 5 knockdown (RGS5‐KD) on brain endothelial tight junction proteins.
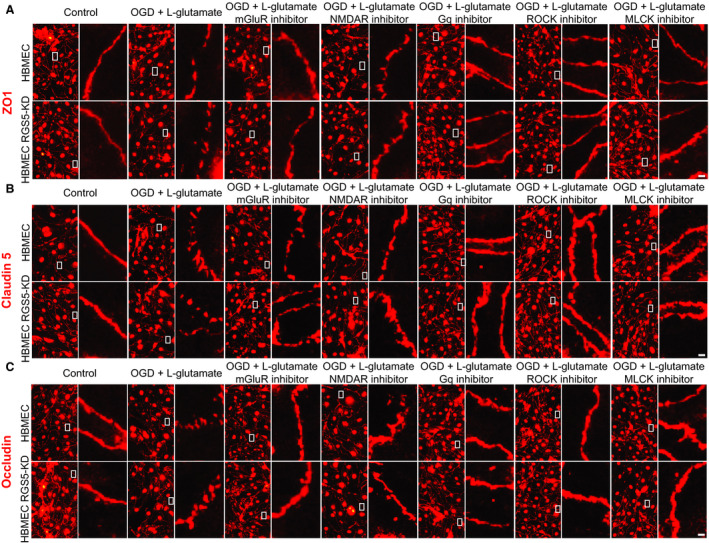
A through C, Immunofluorescent staining of tight junction proteins, zonula occludens 1 (ZO1), occluding, and claudin‐5 before after stimulation with oxygen glucose deprivation and l‐glutamate treatment (OGD+l‐glutamate; 1 mmol/L for 3 hours), with and without metabotropic glutamate receptor inhibitor (10 mmol/L; JNJ16259685), ligand‐gated ionotropic glutamate receptor inhibitor (10 mmol/L; MK‐801), Gq inhibitor (10 μmol/L; YM254980), Rho‐associated kinase (ROCK) inhibitor (Y‐27632; 20 μmol/L), or myosin light chain kinase (MLCK) inhibitor (ML‐7; 10 μmol/L) (n=5). Bar=10 μm.
We have previously shown that ROCK is an important negative regulator of endothelial NO synthase (eNOS) expression and activity. 20 , 21 Because RGS5 deletion/deficiency leads to increased ROCK activity, we first investigated the effects of RGS5 on eNOS expression and activity. RGS5‐KD HBMECs exhibited lower phosphorylation of eNOS at Ser1177 under basal conditions (0.8 [IQR, 0.84–0.65] versus 1.0; n=5; P<0.01) and after OGD/l‐glutamate treatment (0.95 [IQR, 0.84–0.65] versus 1.33 [IQR, 1.12–0.9]; n=5; P<0.01) (Figure 7A). Furthermore, eNOS expression was lower in RGS5‐KD HBMECs before (0.82 [IQR, 0.88–0.80] versus 1.0; n=5; P<0.01) and after OGD/l‐glutamate treatment (0.94 [IQR, 0.99–0.90] versus 1.16 [IQR, 1.24–1.14]; n=5; P<0.01) (Figure 7A).
Figure 7. Effect of regulator of G‐protein signaling 5 deficiency (RGS5) on endothelial NO synthase (eNOS) expression in human brain microvascular endothelial cells (HBMECs) and brain tissue before and after middle cerebral artery occlusion (MCAO).

A, Western blotting analysis and quantification of eNOS phosphorylation and expression in control and RGS5 knockdown (RGS5‐KD) HBMECs, with and without oxygen glucose deprivation and l‐glutamate treatment (OGD+l‐glutamate; 1 mmol/L for 3 hours) (n=5). NO production measured in whole brains before MCAO (B) and in contralateral and ipsilateral hemispheres after MCAO (C) of controls and endothelial Rgs5 deficient (EC‐Rgs5–/–) mice (n=5). Rho‐associated kinase (ROCK) activity, measured by myosin phosphatase target subunit 1 (MBS) phosphorylation (D) and eNOS phosphorylation (E) and expression in contralateral and ipsilateral hemisphere after MCAO of control and EC‐Rgs5 –/– mice. F, DAF‐FM staining of EC‐Rgs5–/– and control mice coronal brain sections 24 hours after MCAO (n=3). Bar=1 mm. For all data sets, 1‐way ANOVA (B) or 2‐way ANOVA (C through E; variables, ipsilateral vs contralateral and type of mice) followed by post hoc Tukey tests. Nonnormally distributed data sets were analyzed by Kruskal‐Wallis tests (A). *P<0.05, **P<0.01, #P<0.001. Control indicates EC‐Rgs5flox/flox without tamoxifen; Control+TMX, Cdh5‐CreERT2 with tamoxifen; and EC‐Rgs5–/–, endothelial cell–specific RGS5‐deficient mice.
Given that eNOS appears to be implicated in our in vitro data, we sought to evaluate the role of RGS5 in our in vivo model of ischemic stroke. Although basal NO production in whole brains of EC‐Rgs5–/– mice did not differ compared with that of control mice (P>0.05; n=5) (Figure 7B), EC‐Rgs5–/– mice showed decreased overall NO production in ischemic brain hemispheres following transient MCAO (64.4±4.6 versus 90.2±4.7 and 93.2±3.7; n=5; P<0.01) (Figure 7C). ROCK activity in the ipsilateral brain hemisphere was also higher in EC‐Rgs5–/– compared with controls (1.13±0.08 versus 1.75±0.25; n=5; P=0.04) (Figure 7D). This higher ROCK activity correlated with decreased eNOS expression and phosphorylation in the ipsilateral hemisphere (phosphorylated eNOS, 1.52±0.08 versus 1.25±0.03, n=8, P=0.015; eNOS, 1.41±0.04 versus 1.16±0.04, n=8, P<0.01) (Figure 7E). Furthermore, we visualized the decreased NO production in EC‐Rgs5–/–, compared with controls, by DAF‐FM staining following transient MCAO (Figure 7F). The fluorescent staining, corresponding to NO production, was greater in the infarct area of control mice (EC‐Rgs5flox/flox without tamoxifen and inducible Cre recombinase under the control of the vascular endothelial cadherin promoter mice with tamoxifen) compared with that of EC‐Rgs5–/– mice. These findings indicate that RGS5 is an important mediator of endothelial function, and maintains endothelial function under ischemic conditions.
DISCUSSION
We have shown that RGS5 stabilizes and maintains BBB after focal cerebral ischemia through inhibition of Gαq and its coupled receptors, mGluRs and NMDARs. This leads to attenuation of ROCK and MLCK signaling pathways, which affect actin cytoskeletal reorganization, endothelial tight junction, cell permeability, and stroke severity. Indeed, both global and endothelial RGS5 deficiency lead to increased BBB permeability, greater brain edema formation, larger cerebral infarct size, and worsened neurologic function. These findings suggest that RGS5 plays an important role in maintaining endothelial function and BBB integrity during focal cerebral ischemia.
The BBB is composed of endothelial cells, pericytes, and astrocyte end‐feet processes, and plays a critical role in maintaining brain homeostasis. 22 Thus, alteration of BBB can lead to increased brain edema, ion dysregulation, immune cell infiltration, entry of blood‐borne molecules, and energy disbalance, which can cause neuronal dysfunction and death. 23 Recent experimental and clinical studies support the notion that BBB dysfunction is an important contributor to the outcome of neurological diseases, such as ischemic stroke, brain trauma and tumorigenesis, multiple sclerosis, epilepsy, and Alzheimer disease. 23 However, the complex mechanisms by which BBB is regulated, both under homeostatic and pathophysiological conditions, are not well understood. Our findings implicate RGS5 as a novel regulator of BBB integrity, suggesting that RGS5 can be therapeutically exploited to attenuate the severity of neurological diseases affected by BBB alterations.
A new finding of this study is the effect of glutamate on nonneuronal cells, such as brain microvascular endothelial cells, as it pertains to BBB permeability and stroke severity. The release of glutamate, an important excitatory neurotransmitter, is an important mediator of rapid neuronal death, especially during ischemic stroke. Indeed, glutamate accumulation in neurons during ischemic stroke leads to early necrosis or delayed apoptosis. 24 Although the effects of glutamate on mGluRs and NMDARs are known in neuronal cells, the physiological role of these receptors in HBMECs, a vital component of the BBB, has not been fully characterized. 25 , 26 , 27 Some studies suggest that stimulation of NMDARs by glutamate may be the primary cause of increased endothelial permeability, whereas other studies have suggested that inhibitors of mGluRs may have neuroprotective effects after ischemic stroke. 28 , 29 , 30 The precise mechanisms by which glutamate receptors exert their BBB‐disruptive effects, however, are unknown. Because RGS5 is known to modulate the activities of Gαq and Gαi, 2 2 G‐protein subunits that are coupled to mGluR1, it is likely that RGS5 can negatively modulate mGluR1 signaling in brain endothelial cells during ischemic stroke. Indeed, inhibition of mGluR1 partially reversed the deleterious effects of RGS5 deficiency in brain microvascular endothelial cells by improving endothelial function and decreasing BBB permeability.
The expression of RGS5 is restricted to certain tissues. For example, RGS5 is abundantly expressed in vascular cells, such as endothelial cells, smooth muscle cells, and pericytes, but has limited expression in neurons. 31 , 32 Although our findings support endothelial RGS5 as an important mediator of BBB maintenance and stroke outcome, it is possible that RGS5 in other cell types, such as vascular smooth muscle, pericytes, and immune cells, also plays important roles in maintaining BBB integrity. 33 , 34 Indeed, we found greater BBB leakage and worse stroke outcomes in global Rgs5–/– mice compared with endothelial‐specific EC‐Rgs5–/– mice, suggesting that other cell types lacking RGS5 may also influence BBB stability and stroke outcome. Interestingly, a recent study showed that the loss of RGS5 affects pericyte coverage of endothelial cells, thus affecting BBB formation and integrity. 35 In contrast with our findings, their results do not show any differences in cerebral infarct size and brain edema formation between Rgs5 –/– and control mice following cerebral ischemia. It is uncertain why their results differ from ours, but a potential explanation could be the difference in the generation of the RGS5 knockout animals and the model of cerebral ischemia used. Özen et al 35 used mice created by replacing the in‐frame RGS domain of Rgs5 from part of exon 2 to exon 5 by a GFP reporter. It is possible, therefore, that the GFP cassette could introduce hypomorphic features in their mice, including a decrease in blood pressure. Furthermore, it is unclear whether their mice retained the expression of exon 1 of Rgs5, which could also produce a confounding phenotype. In contrast, we confirmed complete absence of the Rgs5 gene, including the lack of expression of truncated forms of exons 1 and 2. Another potential difference between their findings and ours is the difference in the model of cerebral ischemia used. Ozen et al performed permanent middle cerebral artery occlusion model by electrocoagulation of the distal part of the middle cerebral artery after craniectomy, whereas we used a transient, intraluminal filament model of middle cerebral artery occlusion.
Hypertension is associated with poor outcome and hemorrhagic transformation after ischemic stroke. 36 In concordance with the results of other studies, we show that global RGS5‐deficient mice have elevated blood pressures, which may potentially affect the outcome of acute ischemic stroke. 5 , 6 However, for the first time, we show that endothelial‐specific RGS5 deletion does not have a major impact on blood pressure. Consequently, worsened BBB stability and infarct size in ischemic stroke may be caused by endothelial dysfunction, as shown in this study, rather than an increase in blood pressures.
The increased Gαq activation and intracellular calcium concentration in brain microvascular endothelial cells, both in basal conditions and in response to glutamate, can likely be attributed to the lack of Gαq inhibition with RGS5 deficiency. The activation of Gαq‐coupled mGluR1 by glutamate leads to the upregulation of phospholipase C and hydrolysis of phosphatidylinositol 4,5‐biphosphate to diacylglycerol and inositol triphosphate. 37 Inositol triphosphate induces the release of calcium from the endoplasmic reticulum, thereby increasing intracellular calcium concentration. Similarly, the activation of NMDARs by glutamate could also directly lead to increases in intracellular calcium. Because MLCK is a calcium‐calmodulin binding protein, an increase in intracellular calcium would lead to MLCK activation and MLC phosphorylation. 38 , 39 MLC phosphorylation is a prerequisite for actomyosin formation, cellular contraction, relocalization of tight junction proteins, and cell permeability. 40 In addition, the activation of Gαi‐ and Gαq‐coupled receptors in RGS5‐deficient endothelial cells may also lead to activation of the Rho/ROCK signaling pathway, which could likewise maintain MLC phosphorylation through the inhibition of myosin light chain phosphatase or by direct phosphorylation of MLC. 41 Thus, the combined activation of ROCK and MLCK by glutamate through mGluR1s and NMDARs may potently stimulate endothelial cell contraction and BBB permeability. The mechanisms of Gαq activation of Rho/ROCK are not fully understood. According to previously published studies, Gαq can promote Rho/ROCK activity directly by activating RhoA. Gαq can also activate other Rho family members, such as Rac and Cdc42, which lead to activation of their downstream effectors rather than through RhoA activation. The existence of a large number of Rhoguanine nucleotide exchange factors (GEFs) may give us new candidates that can mediate Gαq activation of Rho, including p63RhoGEF and Trio, in addition to the RGS domain containing RhoGEFs that are activated by Gα12 and Gα13. 41 , 42 , 43
Although there is some evidence for cross talk between mGluR and NMDAR pathways, the potential regulation of NMDARs by G‐protein–coupled pathways, such as by RGS5, requires further evaluation. 44 , 45 The activation of Src‐family tyrosine kinase by certain Gαq coupled receptors might potentially explain the cross talk of RGS5 with NMDARs. 46 Nevertheless, in our study, we find that inhibition of Gαq, mGluR1s, or NMDARs leads to reversal of ROCK/MLCK activation and cellular permeability in RGS5‐deficient brain microvascular endothelial cells.
Our findings indicate that glutamate and RGS5 play important roles in ROCK activation in brain endothelial cells in a model of ischemic stroke. Because ROCK also plays an important role in the pathogenesis of endothelial dysfunction, hypertension, vasospasm, cardiac hypertrophy, heart failure, and stroke, 47 , 48 , 49 , 50 RGS5 may play a broader role in cardiovascular disease through its inhibitory effects on ROCK. Indeed, RGS5‐deficient brain microvascular endothelial cells exhibit lower eNOS expression and activity, likely mediated by the increased ROCK activity in these cells. 20 This is consistent with lower NO synthesis in the brain of EC‐Rgs5–/– mice, although the decrease in NO could also be because of lower activities of inducible NO synthase and neuronal NO synthase. Because RGS5 deficiency leads to impaired NO synthesis and more severe stroke outcomes, the observed decrease in NO production in the brains of EC‐Rgs5–/– mice is most likely caused by the downregulation of eNOS rather than downregulation of inducible NO synthase or neuronal NO synthase, which tend to have neurotoxic effects. 51 Indeed, we found that the deletion of RGS5 in mice leads to higher systolic and diastolic blood pressures. Again, there are conflicting reports on the role of RGS5 in blood pressure regulation, 52 , 53 , 54 perhaps because of different approaches in the generation of Rgs5 –/– mice. The finding that our Rgs5 –/– mice have higher blood pressure is consistent with the loss of RGS5 in gestational hypertension. 5 Similarly, different single‐nucleotide polymorphism variants of RGS5 have been linked to essential hypertension in Black and Chinese Han populations. 9 , 10 Our finding that RGS5 is an important positive regulator of eNOS and endothelial function may provide the basis for the observed blood pressure elevation in Rgs5 –/– mice.
There are a few limitations to our study. First, we decided to use primary HBMECs to evaluate the translatability of our findings in mice. It is possible that the data from HBMECs after OGD/l‐glutamate treatment may not fully encapsulate the molecular signaling pathways underling the phenotypic differences in our murine model of MCAO. Furthermore, as we implicate ROCK in RGS5 deficiency, it is likely that alternative mechanisms aside from tight junction relocalization are involved in BBB disruption. For instance, pinocytosis after transient focal ischemia, as regulated by ROCK, may be involved in disruption of the BBB, and was not evaluated in this article. Finally, as animals with a score of 5 on the neurological deficit score were excluded, survivorship bias may lead to the animals used in the study providing more optimistic parameters.
Nonetheless, we have identified endothelial RGS5 as an important regulator of mGluR1 and NMDAR signaling through its inhibition of Gαq. Global or endothelial‐specific loss of function of RGS5 leads to increased BBB permeability and stroke severity. It remains to be determined, however, whether RGS5 is a clinically useful therapeutic target for stroke and cardiovascular disease.
Sources of Funding
This study was supported by grants from the National Institutes of Health (NS070001 and HL052233; Liao) and the American Heart Association (17POST33650097; Sladojevic).
Disclosures
None.
Supporting information
Tables S1–S3
Figures S1–S2
Acknowledgments
We are grateful to Yoshiaki Kubota (Keio University, Japan) for providing the Cre recombinase under the control of the vascular endothelial cadherin promoter transgenic mice.
(J Am Heart Assoc. 2020;9:e017533 DOI: 10.1161/JAHA.120.017533.)
For Sources of Funding and Disclosures, see page 15.
References
- 1. Hilger D, Masureel M, Kobilka BK. Structure and dynamics of GPCR signaling complexes. Nat Struct Mol Biol. 2018;25:4–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Fu Y, Zhong H, Nanamori M, Mortensen RM, Huang X, Lan K, Neubig RR. RGS‐insensitive G‐protein mutations to study the role of endogenous RGS proteins. Methods Enzymol. 2004;389:229–243. [DOI] [PubMed] [Google Scholar]
- 3. Seki N, Sugano S, Suzuki Y, Nakagawara A, Ohira M, Muramatsu M, Saito T, Hori T. Isolation, tissue expression, and chromosomal assignment of human RGS5, a novel G‐protein signaling regulator gene. J Hum Genet. 1998;43:202–205. [DOI] [PubMed] [Google Scholar]
- 4. Ganss R. Keeping the balance right: regulator of G protein signaling 5 in vascular physiology and pathology. Prog Mol Biol Transl Sci. 2015;133:93–121. [DOI] [PubMed] [Google Scholar]
- 5. Holobotovskyy V, Chong YS, Burchell J, He B, Phillips M, Leader L, Murphy TV, Sandow SL, McKitrick DJ, Charles AK, et al. Regulator of G protein signaling 5 is a determinant of gestational hypertension and preeclampsia. Sci Transl Med. 2015;7:290ra288. [DOI] [PubMed] [Google Scholar]
- 6. Holobotovskyy V, Manzur M, Tare M, Burchell J, Bolitho E, Viola H, Hool LC, Arnolda LF, McKitrick DJ, Ganss R. Regulator of G‐protein signaling 5 controls blood pressure homeostasis and vessel wall remodeling. Circ Res. 2013;112:781–791. [DOI] [PubMed] [Google Scholar]
- 7. Li H, He C, Feng J, Zhang Y, Tang Q, Bian Z, Bai X, Zhou H, Jiang H, Heximer SP, et al. Regulator of G protein signaling 5 protects against cardiac hypertrophy and fibrosis during biomechanical stress of pressure overload. Proc Natl Acad Sci USA. 2010;107:13818–13823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Manzur M, Hamzah J, Ganss R. Modulation of the "blood‐tumor" barrier improves immunotherapy. Cell Cycle. 2008;7:2452–2455. [DOI] [PubMed] [Google Scholar]
- 9. Faruque MU, Chen G, Doumatey A, Huang H, Zhou J, Dunston GM, Rotimi CN, Adeyemo AA. Association of ATP1B1, RGS5 and SELE polymorphisms with hypertension and blood pressure in African–Americans. J Hypertens. 2011;29:1906–1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chang PY, Qin L, Zhao P, Liu ZY. Association of regulator of g protein signaling (RGS5) gene variants and essential hypertension in Mongolian and Han populations. Genet Mol Res. 2015;14:17641–17650. [DOI] [PubMed] [Google Scholar]
- 11. Jin Y, An X, Ye Z, Cully B, Wu J, Li J. RGS5, a hypoxia‐inducible apoptotic stimulator in endothelial cells. J Biol Chem. 2009;284:23436–23443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hamzah J, Jugold M, Kiessling F, Rigby P, Manzur M, Marti HH, Rabie T, Kaden S, Grone HJ, Hammerling GJ, et al. Vascular normalization in Rgs5‐deficient tumours promotes immune destruction. Nature. 2008;453:410–414. [DOI] [PubMed] [Google Scholar]
- 13. Manwani B, McCullough LD. Sexual dimorphism in ischemic stroke: lessons from the laboratory. Womens Health (Lond). 2011;7:319–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Okabe K, Kobayashi S, Yamada T, Kurihara T, Tai‐Nagara I, Miyamoto T, Mukouyama YS, Sato TN, Suda T, Ema M, et al. Neurons limit angiogenesis by titrating VEGF in retina. Cell. 2014;159:584–596. [DOI] [PubMed] [Google Scholar]
- 15. Pasban E, Panahpour H, Vahdati A. Early oxygen therapy does not protect the brain from vasogenic edema following acute ischemic stroke in adult male rats. Sci Rep. 2017;7:3221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real‐time quantitative PCR and the 2(‐delta delta C(T)) method. Methods. 2001;25:402–408. [DOI] [PubMed] [Google Scholar]
- 17. Tiruppathi C, Minshall RD, Paria BC, Vogel SM, Malik AB. Role of Ca2+ signaling in the regulation of endothelial permeability. Vascul Pharmacol. 2002;39:173–185. [DOI] [PubMed] [Google Scholar]
- 18. van Nieuw Amerongen GP, Beckers CM, Achekar ID, Zeeman S, Musters RJ, van Hinsbergh VW. Involvement of Rho kinase in endothelial barrier maintenance. Arterioscler Thromb Vasc Biol. 2007;27:2332–2339. [DOI] [PubMed] [Google Scholar]
- 19. Sandoval KE, Witt KA. Blood‐brain barrier tight junction permeability and ischemic stroke. Neurobiol Dis. 2008;32:200–219. [DOI] [PubMed] [Google Scholar]
- 20. Hiroi Y, Noma K, Kim HH, Sladojevic N, Tabit CE, Li Y, Soydan G, Salomone S, Moskowitz MA, Liao JK. Neuroprotection mediated by upregulation of endothelial nitric oxide synthase in Rho‐associated, coiled‐coil‐containing kinase 2 deficient mice. Circ J. 2018;82:1195–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wolfrum S, Dendorfer A, Rikitake Y, Stalker TJ, Gong Y, Scalia R, Dominiak P, Liao JK. Inhibition of Rho‐kinase leads to rapid activation of phosphatidylinositol 3‐kinase/protein kinase Akt and cardiovascular protection. Arterioscler Thromb Vasc Biol. 2004;24:1842–1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhao Z, Nelson AR, Betsholtz C, Zlokovic BV. Establishment and dysfunction of the blood‐brain barrier. Cell. 2015;163:1064–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Daneman R, Prat A. The blood–brain barrier. Cold Spring Harb Perspect Biol. 2015;7:a020412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lai TW, Zhang S, Wang YT. Excitotoxicity and stroke: identifying novel targets for neuroprotection. Prog Neurogibol. 2014;115:157–188. [DOI] [PubMed] [Google Scholar]
- 25. Krizbai IA, Deli MA, Pestenacz A, Siklos L, Szabo CA, Andras I, Joo F. Expression of glutamate receptors on cultured cerebral endothelial cells. J Neurosci Res. 1998;54:814–819. [DOI] [PubMed] [Google Scholar]
- 26. Macrez R, Ortega MC, Bardou I, Mehra A, Fournier A, Van der Pol SM, Haelewyn B, Maubert E, Lesept F, Chevilley A, et al. Neuroendothelial NMDA receptors as therapeutic targets in experimental autoimmune encephalomyelitis. Brain. 2016;139:2406–2419. [DOI] [PubMed] [Google Scholar]
- 27. Collard CD, Park KA, Montalto MC, Alapati S, Buras JA, Stahl GL, Colgan SP. Neutrophil‐derived glutamate regulates vascular endothelial barrier function. J Biol Chem. 2002;277:14801–14811. [DOI] [PubMed] [Google Scholar]
- 28. Andras IE, Deli MA, Veszelka S, Hayashi K, Hennig B, Toborek M. The NMDA and AMPA/KA receptors are involved in glutamate‐induced alterations of occludin expression and phosphorylation in brain endothelial cells. J Cereb Blood Flow Metab. 2007;27:1431–1443. [DOI] [PubMed] [Google Scholar]
- 29. Xhima K, Weber‐Adrian D, Silburt J. Glutamate induces blood‐brain barrier permeability through activation of N‐methyl‐D‐aspartate receptors. J Neurosci. 2016;36:12296–12298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kohara A, Takahashi M, Yatsugi S, Tamura S, Shitaka Y, Hayashibe S, Kawabata S, Okada M. Neuroprotective effects of the selective type 1 metabotropic glutamate receptor antagonist YM‐202074 in rat stroke models. Brain Res. 2008;1191:168–179. [DOI] [PubMed] [Google Scholar]
- 31. Silini A, Ghilardi C, Figini S, Sangalli F, Fruscio R, Dahse R, Pedley RB, Giavazzi R, Bani M. Regulator of G‐protein signaling 5 (RGS5) protein: a novel marker of cancer vasculature elicited and sustained by the tumor's proangiogenic microenvironment. Cell Mol Life Sci. 2012;69:1167–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Liu C, Hu Q, Jing J, Zhang Y, Jin J, Zhang L, Mu L, Liu Y, Sun B, Zhang T, et al. Regulator of G protein signaling 5 (RGS5) inhibits sonic hedgehog function in mouse cortical neurons. Mol Cell Neurosci. 2017;83:65–73. [DOI] [PubMed] [Google Scholar]
- 33. Yang S, Jin H, Zhu Y, Wan Y, Opoku EN, Zhu L, Hu B. Diverse functions and mechanisms of pericytes in ischemic stroke. Curr Neuropharmacol. 2017;15:892–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Yu Q, Tao H, Wang X, Li M. Targeting brain microvascular endothelial cells: a therapeutic approach to neuroprotection against stroke. Neural Regen Res. 2015;10:1882–1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Özen I, Roth M, Barbariga M, Gaceb A, Deierborg T, Genové G, Paul G. Loss of regulator of G‐protein signaling 5 leads to neurovascular protection in stroke. Stroke. 2018;49:2182–2190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. McManus M, Liebeskind DS. Blood pressure in acute ischemic stroke. J Clin Neurol. 2016;12:137–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Willard SS, Koochekpour S. Glutamate, glutamate receptors, and downstream signaling pathways. Int J Biol Sci. 2013;9:948–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Legros H, Launay S, Roussel BD, Marcou‐Labarre A, Calbo S, Catteau J, Leroux P, Boyer O, Ali C, Marret S, et al. Newborn‐ and adult‐derived brain microvascular endothelial cells show age‐related differences in phenotype and glutamate‐evoked protease release. J Cereb Blood Flow Metab. 2009;29:1146–1158. [DOI] [PubMed] [Google Scholar]
- 39. Speyer CL, Hachem AH, Assi AA, Johnson JS, DeVries JA, Gorski DH. Metabotropic glutamate receptor‐1 as a novel target for the antiangiogenic treatment of breast cancer. PLoS One. 2014;9:e88830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Afonso PV, Ozden S, Prevost MC, Schmitt C, Seilhean D, Weksler B, Couraud PO, Gessain A, Romero IA, Ceccaldi PE. Human blood‐brain barrier disruption by retroviral‐infected lymphocytes: role of myosin light chain kinase in endothelial tight‐junction disorganization. J Immunol. 2007;179:2576–2583. [DOI] [PubMed] [Google Scholar]
- 41. Chikumi H, Vazquez‐Prado J, Servitja JM, Miyazaki H, Gutkind JS. Potent activation of RhoA by Galpha q and Gq‐coupled receptors. J Biol Chem. 2002;277:27130–27134. [DOI] [PubMed] [Google Scholar]
- 42. Fukuhara S, Chikumi H, Gutkind JS. RGS‐containing RhoGEFs: the missing link between transforming G proteins and Rho? Oncogene. 2001;20:1661–1668. [DOI] [PubMed] [Google Scholar]
- 43. Rojas RJ, Yohe ME, Gershburg S, Kawano T, Kozasa T, Sondek J. Galphaq directly activates p63RhoGEF and Trio via a conserved extension of the Dbl homology‐associated pleckstrin homology domain. J Biol Chem. 2007;282:29201–29210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yang K, Jackson MF, MacDonald JF. Recent progress in understanding subtype specific regulation of NMDA receptors by G protein coupled receptors (GPCRs). Int J Mol Sci. 2014;15:3003–3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Tian M, Xu J, Lei G, Lombroso PJ, Jackson MF, MacDonald JF. STEP activation by Galphaq coupled GPCRs opposes Src regulation of NMDA receptors containing the GluN2A subunit. Sci Rep. 2016;6:36684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Salter MW, Kalia LV. Src kinases: a hub for NMDA receptor regulation. Nat Rev Neurosci. 2004;5:317–328. [DOI] [PubMed] [Google Scholar]
- 47. Sladojevic N, Yu B, Liao JK. ROCK as a therapeutic target for ischemic stroke. Expert Rev Neurother. 2017;17:1167–1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Masumoto A, Mohri M, Shimokawa H, Urakami L, Usui M, Takeshita A. Suppression of coronary artery spasm by the Rho‐kinase inhibitor fasudil in patients with vasospastic angina. Circulation. 2002;105:1545–1547. [DOI] [PubMed] [Google Scholar]
- 49. Shimizu T, Narang N, Chen P, Yu B, Knapp M, Janardanan J, Blair J, Liao JK. Fibroblast deletion of ROCK2 attenuates cardiac hypertrophy, fibrosis, and diastolic dysfunction. JCI Insight. 2017;2:e93187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Dong M, Liao JK, Fang F, Lee APW, Yan BPY, Liu M, Yu CM. Increased rho kinase activity in congestive heart failure. Eur J Heart Fail. 2012;14:965–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Chen ZQ, Mou RT, Feng DX, Wang Z, Chen G. The role of nitric oxide in stroke. Med Gas Res. 2017;7:194–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Cho H, Park C, Hwang IY, Han SB, Schimel D, Despres D, Kehrl JH. Rgs5 targeting leads to chronic low blood pressure and a lean body habitus. Mol Cell Biol. 2008;28:2590–2597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Nisancioglu MH, Mahoney WM Jr, Kimmel DD, Schwartz SM, Betsholtz C, Genove G. Generation and characterization of rgs5 mutant mice. Mol Cell Biol. 2008;28:2324–2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Arnold C, Demirel E, Feldner A, Genove G, Zhang H, Sticht C, Wieland T, Hecker M, Heximer S, Korff T. Hypertension‐evoked RhoA activity in vascular smooth muscle cells requires RGS5. FASEB J. 2018;32:2021–2035. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Tables S1–S3
Figures S1–S2