

Abstract
Among the electroneutral Na+-dependent chloride transporters, NKCC1 had until now evaded identification as a protein causing human diseases. The closely related SLC12A transporters, NKCC2 and NCC have been identified some 25 years ago as responsible for Bartter and Gitelman syndromes: two renal-dependent salt wasting disorders. Absence of disease was most surprising since the NKCC1 knockout mouse was shown in 1999 to be viable, albeit with a wide range of deleterious phenotypes. Here we summarize the work of the past 5 years that introduced us to clinical cases involving NKCC1. The most striking cases are of 3 children with inherited mutations, who have complete absence of NKCC1 expression. These cases establish that lack of NKCC1 causes deafness; CFTR-like secretory defects with mucus accumulation in lung and intestine; severe xerostomia, hypotonia, dysmorphic facial features, and severe neurodevelopmental disorder. Another intriguing case is of a patient with a dominant deleterious SLC12A2 allele. This de novo mutation introduced a premature stop codon leading to a truncated protein. This mutant transporter seems to exert dominant-negative effect on wild-type transporter only in epithelial cells. The patient who suffers from lung, bladder, intestine, pancreas, and multiple endocrine abnormalities has, however, normal hearing and cognition. Finally, new reports substantiate the haploinsufficiency prediction of the SLC12A2 gene. Cases with single allele mutations in SLC12A2 have been linked to hearing loss and neurodevelopmental disorders.
Keywords: gastrointestinal tract, lung, salivary gland, nervous system, testis, sensorineural deafness, membrane trafficking, cystic fibrosis, CFTR, SLC12A2, exon 21
Graphical Abstract
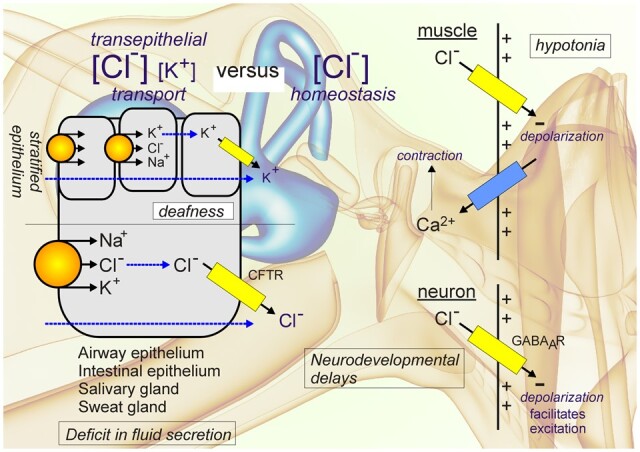
The Na-K-2Cl cotransporter-1, NKCC1, is encoded by the SLC12A2 gene. Transporters in the SLC12 family mediate the electroneutral transport of Cl− accompanied by Na+ and/or K+ across the plasma membrane of both epithelial and nonepithelial cells. The family is divided into two branches defined by Na+ transport: the Na+-dependent branch, which includes SLC12A1 (NKCC2), SLC12A2 (NKCC1) and SLC12A3 (NCC) and the Na+-independent branch which encodes SLC12A4 (KCC1), SLC12A5 (KCC2), SLC12A6 (KCC3) and SLC12A7 (KCC4). In addition, there are 2 orphan proteins; CCC9 (SLC12A8) and CIP (SLC12A9) whose molecular structure and function are yet to be characterized.1 The Na-(K)-Cl transporters are activated by phosphorylation and they mediate Na+-driven Cl− influx into cells, whereas K-Cl cotransporters are activated by dephosphorylation and they mediate K+-driven Cl− efflux. In many cell types, NKCC(s) and KCC(s) are reciprocally regulated by kinases and phosphatases and this balance is important to maintain intracellular Cl− and cell volume.2–4 Extensive work over the past 20 years has uncovered the kinases and phosphatases involved in the regulation of the cation-chloride cotransporters, including NKCC1. We know today that With-No-Lysine (WNK) kinases phosphorylate and activate STE20/SPS1-related proline-alanine-rich protein kinase (SPAK) and oxidative stress response 1 (OSR1), which in turn phosphorylate and activate NKCC1 to import Cl−, Na+ and K+ inside the cell.5–8 Dephosphorylation is mostly mediated by protein phosphatase 1 (PP1).9,10 Unlike renal-specific NCC and NKCC2 which are mainly expressed on the apical membrane of distal convoluted tubules (DCT) and thick ascending limb of Henle (TALH), respectively, NKCC1 has a broad expression pattern and is targeted to the basolateral membrane of Cl− secreting epithelia.
While mutations in some SLC12A genes have been linked to human Mendelian diseases, SLC12A2 has for the most part evaded identification as a disease-causing gene in the human population. There are several possible explanations for this lack of clinical evidence: 1) absence of transporter function is compensated by other transport mechanisms; 2) mutations in NKCC1 are not well-tolerated; or 3) the clinical presentation associated with mutations in NKCC1 is complex and has yet to be established. The second reason is very likely true, as the probability of intolerance for loss-of-function (pLI) index of SLC12A2 in the gnomAD database is 0.96, a value that establishes that the gene is extremely intolerant to loss of function mutations.11,12 As we will see in the next sections, the third explanation turns out to also be correct, SLC12A2 is a disease-causing gene. The gene had just to be identified as such for geneticists to start paying attention. As the transporter is expressed in almost all tissues, its dysfunction leads to complex symptoms. Some of these features are validated by mouse models, while others still need experimental support. In this review, we focus on the recent developments that establish SLC12A2 as a disease-causing gene in humans.
Gene, Expression, and Structure
The SLC12A2 gene is located on the long arm of chromosome 5 at 5q23.3 (GRCh38.p13, 5:128083766-128189677). The gene is highly conserved among species and more than 32 orthologues have now been described. Human NKCC1 is composed of 27 exons,13,14 with two splice variants identified: NKCC1a (full-length) and NKCC1b (lacking exon 21).13 Interestingly, exon 21 is nonexistent in SLC12A1, the gene encoding the kidney-specific isoform NKCC2. This exon has been postulated to play a role in targeting the cotransporter to the basolateral membrane,15 however, we recently showed that both NKCC1a and NKCC1b (Δ21) are targeted to the basolateral membrane of MDCK cells grown polarized in 3D Matrigel substrate.16 We found instead that a carboxyl-terminal dileucine motif (1087DLPPVLL1193) was responsible for targeting the cotransporter to the basolateral membrane. Substitution of the two leucine residues into alanine led to the mistrafficking of the transporter to the apical membrane.16 While NKCC1 is not in every cell, it is broadly expressed throughout the body.17 In chloride secreting epithelia such as salivary gland, sweat gland, lung, and intestine, the cotransporter is targeted to the basolateral membrane.17 This proximity to the serosal side allows NKCC1 to mediate the electroneutral movement of one Na+, one K+, and two Cl− ions from the basal interstitium into the cell. In the submandibular salivary gland, NKCC1 mediates 75%–80% of carbachol-induced Cl− uptake; which drives the bulk of transepithelial fluid secretion.18,19 Our 3D cell culture studies revealed that NKCC1 is first targeted to apical membrane initiation site and then subsequently targeted to the basolateral membrane upon epithelial cell polarization (Figure 1). Remarkably, the NKCC1 dileucine mutant or an NKCC1 with truncated C-terminus failed to relocate to the basolateral membrane upon formation of the 4-cell stage epithelium in vitro. This process may play a critical role during cell division of fast-dividing epithelia, such as in the small intestine since in polarized epithelium NKCC1 is directly targeted from the trans-Golgi network to the basolateral membrane. In some epithelia, NKCC1 is targeted to the apical membrane. This is the case of the olfactory epithelium,20 the retinal pigment epithelium,21 and choroid plexus epithelium.22,23 However, the precise mechanisms directing NKCC1 to the apical membrane in these distinct tissues are not well understood.
Figure 1.
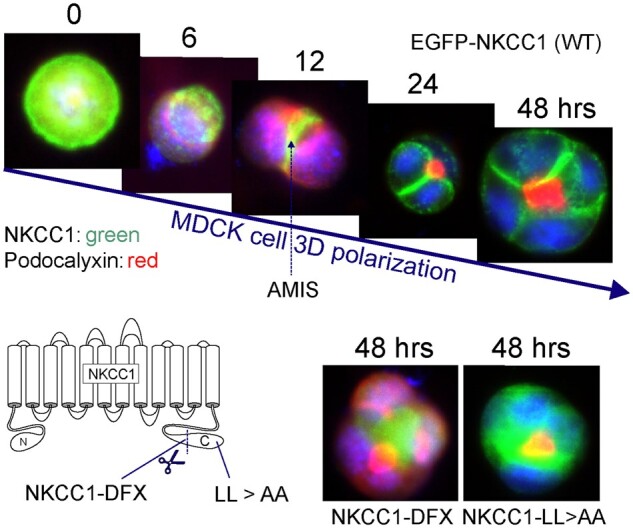
Membrane Localization of NKCC1 During Polarization of MDCK Cells in Matrigel. Top panels: MDCKII cells were transfected with EGFP-tagged NKCC1 and plated in Matrigel. As a single cell divide to create a polarized structure, NKCC1 which is first seen all around the cell now concentrates at the Apical Membrane Initiation Site prior to localizing at its final destination: the basolateral membrane. The process takes 24–48 h to create the first four cell polarized structure. Bottom panels: This process is prevented by truncating the carboxyl-terminus of NKCC1, as in the NKCC1-DFX patient or by mutating a dileucine motif located at the extreme carboxyl-terminal tail of the cotransporter. In each case, NKCC1 staining colocalizes with podocalyxin at the apical pole. NKCC1 is labeled in green; whereas the apical marker podocalyxin is labeled in red. Data were redrawn from Koumangoye et al.16
SLC12A2 encodes a 1205 amino acid transmembrane protein with 12 transmembrane domains (TMD1 to TMD12). NKCC1 has been shown to form functional dimers when expressed in oocytes or HEK293 cells.24,25 Significantly, Chew and colleagues have recently reported the structure of the zebrafish (Danio rerio) NKCC1 using cryo-electron microscopy.26 The 3D structure of NKCC1 features a domain-swap dimer architecture where the transmembrane domain (TMD) of one protomer is in close contact with the C-terminal domain (CTD) of the other protomer and these interactions are likely central to the function of the cotransporter. Similar overall architectures were observed with the structures of the related K-Cl cotransporters.27–29 As mentioned above, NKCC1 activity is regulated by the WNK-SPAK/OSR1 kinase pathway.30–32 SPAK and OSR1 activate the cotransporter by phosphorylation at key conserved threonine residues (Thr197, Thr201, and Thr206) located within the NH2-terminal domain of NKCC1.33 Note that Chew and colleagues were unable to elucidate the structure of the N-terminal domain because of its high degree of flexibility and lack of order.26 In fact, alignment of the N-terminus of NKCC1 from 12 species, ranging from shark to human, revealed overall poor conservation with the exception of the two SPAK binding domains5 and the small region containing the threonine residues.33 Of interest is the observation that the NKCC1 protein also contains a nucleolar localization signal (residues 1032-1055) which becomes active upon truncation of a portion of the carboxyl-terminus. The physiological significance of this site is still unknown.16
Pharmacology
Drugs such as loop diuretics, which target NKCC2, have been developed to lower blood pressure, minimize volume expansion, and prevent cardiovascular diseases.34,35 Furosemide (LasixR) and bumetanide (BumexR), however, inhibit both NKCC2 and NKCC1.36 The effective dose of bumetanide seems to be much lower (1/40) than furosemide in producing diuresis,37 likely due to the higher binding affinity of furosemide to serum albumin.38 The loop diuretics seem to have similar effect on NKCC1 and NKCC2 since the apparent affinity of bumetanide in isolated tissues or cells ranges from 10−8 to 10−6 M for each cotransporter.39–43 Recently, Hampel et al. described Azosemide as a more potent inhibitor of human NKCC1,44 and Savardi et al. identified a new compound, ARN23746, that modestly inhibits NKCC1 function (30% at 10 uM and 95% at 100 uM), but had no effect on NKCC2.45
NKCC1 Mouse Knockout Models
In 1999, two groups independently reported the creation NKCC1-KO mouse models46,47, and one group identified a radiation-induced mutant mouse at the Jackson Laboratory that also was a functional knockout of NKCC1.48 Two additional NKCC1 knockout mouse models were published the following year.49 As it will become clear in the next few paragraphs, the NKCC1 knockout mouse exhibits phenotypes that highlight a large number of tissues affected (Figure 2). In all cases, the NKCC1-null mice are smaller when compared to their wild-type counterparts, they are deaf and show imbalance characteristic of inner ear defects.46–49 In the cochlea, the cotransporter is highly expressed in the stria vascularis, a two-layer epithelium that produces the K+-rich endolymph.46 Loss of hearing due to absence of NKCC1 is also consistent with the known ototoxic effect of high doses of loop diuretics in the human pediatric population.50–52 The deficit in the vestibular system is rather debilitating for the mice, as it leads to constant spinning/circling and head-bobbing behavior. This phenotype is severe enough to interfere with the study of knockout animals, especially when neurobehavioral studies rely on locomotion. Because the deficit in endolymph K+ secretion leads to the death of hair cells, it is unlikely that this phenotype can be rescued. It is interesting, however, that short-term inhibition of NKCC1 with ethacrynic acid (another NKCC1 inhibitor) leads to the reversible elimination of auditory brain responses, while the inhibitor had no effect on vestibular gravity receptor function. This observation indicates that acute suppression of K+ secretion affects the cochlea to a greater extent than the vestibular system.53
Figure 2.
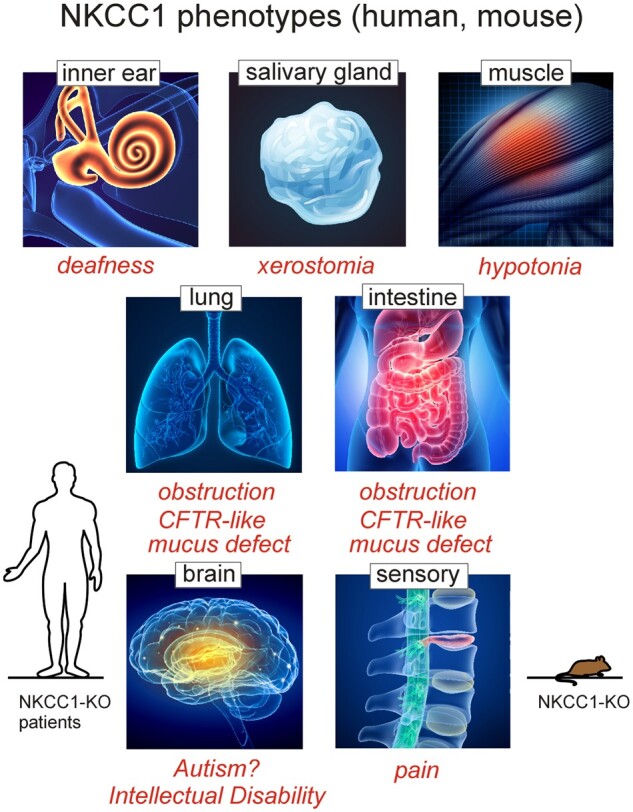
Phenotypes Associated with Loss of NKCC1 Expression in Humans and Mice. Inner ear, salivary gland, lung, and intestine are affected by lack of NKCC1-mediated transepithelial transport of ions (K+ and/or Cl−). Central neurons (brain), sensory neurons, and muscle cells are affected by lack of NKCC1-mediated intracellular Cl− accumulation. Resulting phenotypes are deafness, dry mouth, mucus obstruction in lung and intestine, neurodevelopmental delays, pain perception, and hypotonia. With the exception of autism, intellectual disability, and muscle weakness which have not been established in mouse, all other phenotypes are observed in both species and characteristic of the disorder associated with complete loss of NKCC1 function.
In one of the original models, peri-weaning lethality was observed and connected to intestinal obstruction and apparent bleeding in the cecum.47 Remarkably, this phenotype was not observed in another mouse model where all homozygote knockout mice survived past the peri-weaning period.46 This discrepancy was attributed to differences in mouse strain or background. The intestinal phenotype is consistent with NKCC1 playing a role in intestinal fluid secretion.54 Other tissues where NKCC1 plays a role in electrolyte and fluid secretion are the salivary gland and the lung. Deletion of NKCC1 in mice resulted in 70% decrease in saliva secretion18 and decreased fluid secretion in lung epithelium, although in this case, absence of airway disease in NKCC1 knockout animals suggests that the mice are able to compensate adequately for lack of NKCC1 function.55
Another phenotype that complicates the study of NKCC1 knockout mice is the male sterility. It prevents the breeding of homozygote animals which can then only be obtained from heterozygous crossings. The male sterility is due to the absence of spermatids in the lumen of the seminiferous tubules.56 Accumulation of spermatogonia (undifferentiated germ cells) is observed in the seminiferous tubules. These cells seem to be larger in size and not confined to the base of the tubules, as observed in wild-type mice.56,57 In addition, there is a greater number of cells undergoing apoptosis in the seminiferous tubules.57 While the level of testosterone in the testis was believed to be normal (the seminal vesicles were of similar sizes),56 blood levels of testosterone and luteinizing hormone were significantly lower in the knockout, indicating a possible neurological origin of the testis phenotype.57,58
Besides the stria vascularis in the inner ear, there are several other mouse tissues where NKCC1 is highly expressed. This is the case for sensory neurons in dorsal root ganglia and choroid plexus epithelial cells.22 In sensory neurons, the cotransporter accumulates Cl− above its electrochemical potential equilibrium,59,60 thereby facilitating GABA depolarization at the terminal ends of sensory fibers. In the knockout mouse, the GABA-mediated Cl− reversal potential shifts toward more negative (hyperpolarizing) values affecting presynaptic inhibition and filtering of sensory noise. As a consequence, NKCC1 mice are less sensitive to pain stimuli.2,61
The consequences of an absence of NKCC1 in choroid plexus are unknown. The cotransporter in this tissue is atypically located on the apical membrane.22,23,62 In 1972, it was shown that cerebrospinal fluid (CSF) secretion was inhibited by the application of bumetanide from the CSF side and not the blood side63; but for years that study was ignored and the transporter was wrongly assumed to be basolateral.64,65 In 1994, Richard Keep proposed a model placing the Na-K-2Cl cotransporter on the apical membrane,66 and this localization was later confirmed with the development of NKCC1-specific antibodies.22,23,62 A recent study showed that NKCC1 function and CSF secretion were both simultaneously stimulated following blood accumulation in the ventricles. Enhanced NKCC1 function was triggered by inflammatory response mediated by the activation of the TRL4 receptor, NFΚB, and the terminal kinase SPAK.67 Due to its location on the CSF-facing membrane, its role as a transporter facilitating CSF secretion is still controversial.68 Typically, the gradients for NKCC1-mediated transport are oriented inward and thus opposite to the direction of Na+ transport into the CSF. A recent paper from McCauley and colleagues argued that the gradients for Na-K-2Cl cotransporter in choroid plexus could in fact be reversed and outward facing.69 These data are, however, inconsistent with the fact that application of bumetanide leads to the shrinkage of choroid plexus cells23 and the observation that the epithelium is significantly shrunken in NKCC1 knockout mice, compared to wild-type mice.70
NKCC1 knockout mice are markedly hypotensive due to lower vascular tone.71 NKCC1 in vascular smooth muscle cells participate in depolarizing the membrane, thereby facilitating the depolarizing signals that lead to Ca2+ entry into the cell and muscle contraction.72 Studies have shown that the NKCC1 knockout kidney does not contribute to the hypotension as there was no defect that could be measured in the release of renin or in the ability of renal Na+ transporter expression to respond to increases in angiotensin II.73 Absence of renal contribution to the low blood pressure does not mean that the NKCC1 knockout mouse did not display a renal phenotype.
In the kidney, NKCC1 is mostly found in the renin-producing smooth muscle cells of the afferent arteriole, the intercalated cells type A in the outer medullary collecting duct,74 and in the inner medullary collecting duct.75 It is interesting that the signal in A-type intercalated cells was detected in rat but not mice, indicating possible physiological differences between the two models or species. Bumetanide-sensitive Na+ and K+-dependent secretion of Cl− was measured in collecting tubules from rats treated with mineralocorticoids.76 A functional NKCC1 transporter in this segment facilitates the secretion of Na+ and Cl− from blood to urine. Net reabsorption was observed upon application of bumetanide.77 Thus, detection of NKCC1 in the collecting duct could be dependent on the aldosterone status of the animal.
One striking renal phenotype of the NKCC1 knockout mouse was hyperkalemia.73 High plasma K+ levels could possibly originate from a deficit in muscle cells in loading and buffering plasma K+.78 But typically, increased plasma K+ leads to increased renal secretion to maintain homeostasis. In the NKCC1 knockout mouse, there was no increase in serum aldosterone and no increase in renal K+ secretion associated with the high K+ condition.73 NKCC1 mice also had reduced water excretion when consuming a salt (NaCl)-deficient diet.
NKCC1 “Knockout” Patients
The question as to whether a human with no cotransporter expression (NKCC1 knockout) is viable was answered recently. The case of a 5-year-old boy (Kilquist patient) with complete absence of NKCC1 expression was reported by Macnamara et al.79 The genetic of this patient is interesting as the defective allele inherited from his father has a large 22 kb deletion affecting exons 2–7. Unusual was the fact that the patient inherited two copies of chromosome 5 from his father (uniparental disomy) making him a homozygote knockout from one parent only. The proband suffers from a multitude of ailments including hearing loss, intellectual disability, respiratory weakness with mucus plugs, and gastrointestinal (GI) issues including malrotation, constipation, and blood in the GI tract. Some of his clinical presentations are similar to those observed in patients with cystic fibrosis, including inflammation, obstructive lung diseases, respiratory weakness, pancreatic exocrine dysfunction, dysmotility, intestinal obstruction, and growth disturbance (Table 1). In fact, the patient was originally tested for known diseases-causing mutations in CFTR and mitochondrial disease and all came back negative. The patient failed to thrive and necessitated the insertion of gastrotomy feeding tubes for nutrition. Whether the patients could have benefited from pancreatic enzymes replacement therapy earlier in life is unknown. In cystic fibrosis, morbidity is indeed often linked to pancreatic failure and deficit in bicarbonate secretion.82–85 Interestingly, the secretion of bicarbonate is affected by intracellular Cl−, and absence of NKCC1 on the basolateral membrane likely to affect not only Cl− secretion but also bicarbonate secretion. In addition, both the transports of Cl− and bicarbonate are regulated by the Cl− sensing kinases WNK, SPAK, and OSR1,86 and it was shown that WNK1 overexpression causes an increase in CFTR permeability (PHCO3/PCl) and conductance. This WNK1 effect was not dependent on SPAK and OSR1 and did not affect CFTR expression or trafficking, but affected the physical property of the CFTR channel.87 Finally, Hoegger and colleagues demonstrated that pharmacologic inhibition of NKCC1 in pig airway epithelial cell led to the secretion of a viscid mucus that remained attached to the epithelium in the absence of bicarbonate.88 These observations indicate that NKCC1 and CFTR dysfunction may share common underlining mechanisms that need to be further explored.
Table 1.
Clinical Defects in NKCC1 Patients Compared to Cystic Fibrosis Patients
| Clinical presentation | Cystic fibrosis | p.Val1026Phefs∗2 | 22 kb deletion | c.1431delT |
|---|---|---|---|---|
| c.2006-1G>A | ||||
| Reference80 | Reference79 | Reference81 | ||
| Inflammation | + | + | + | + |
| Obstructive lung disease | + | + | + | + |
| Pancreatic exocrine dysfunction | + | + | + | ? |
| Dysmotility | + | + | + | + |
| Dysbiosis | + | ? | ? | ? |
| Intestinal obstruction | + | + | + | + |
| Deficiency in innate immunity | + | ? | ? | ? |
| Growth deficiency | + | + | + | + |
| Mucoviscidosis (viscid/sticky mucus) | + | + | + | + |
| Defective Cl−/ transport | +/+ | +/? | +/? | +/? |
The + sign means that the clinical presentation has been confirmed in the patient and for the last row that the deficit in Cl− secretion has been confirmed in all patients. The ? sign means that the clinical presentation has not been assessed or (for the last row) that HCO3− secretion has not been measured.
Stodberg et al. recently reported the case of a 9-year-old female patient with biallelic mutations in SLC12A2. The compound heterozygote patient carries a one base deletion (c.1431delT) inherited from her father, and a one base substitution (c.2006-1G>A) inherited from her mother. The one base pair deletion causes a frameshift in exon 8 which introduces an early stop codon, thereby prematurely terminating protein translation. The c.2006-1G>A mutation from the mother affects the splice acceptor site of exon 13, causing exon skipping and disruption of the NKCC1 open reading frame.81 Her clinical presentation mirrors the previously reported 5-year-old boy.79 She is deaf, presents dysmorphic facial features, suffers from severe intellectual disability, and cannot stand or walk on her own. She also presents deficits in tear, saliva, and sweat production. She has lung disease with dry airways that require frequent inhalations of hypertonic sodium chloride solution to prevent life-threatening mucus plugs. She also has multiple GI issues, including constipation and intestinal malrotation. The patient had an older sister, who died at 2 months of age; she carried the same mutations and presented similar symptoms.81 As with the Kilquist patient, the sisters were tested for cystic fibrosis and metabolic disease but no disease-causing mutations were found.
From these three cases, it is clear that individuals can live with complete absence of NKCC1 transport function. The clinical symptoms match closely some of the phenotypes that were observed in the NKCC1 knockout mouse models (Figure 2). These ailments are severe enough to cause one of the individuals to die 2 months after birth.
Patient with a C-Terminal Deletion in NKCC1
In 2016, we described the first patient carrying a loss of function mutation in NKCC1.80 The 14-year-old girl carried an 11 bp deletion in exon 22 of SLC12A2, resulting into a premature stop codon and truncation of the carboxyl-terminal tail of the cotransporter. Because the deletion led to a protein ending with an aspartic acid (D) followed by a phenylalanine (F) and a stop codon, we coined this mutant transporter: NKCC1-DFX. Because her father, mother, and three siblings did not carry the mutation, the mutation arose in germline or during early embryogenesis. Even in the presence of a wild-type (normal) allele, the mutation led to multiple organ deficits. Some of her clinical presentations, at the time of referral to the Undiagnosed Diseases Program at the National Institutes of Health, included autonomic bladder dysfunction, chronic pain, decreased energy, dietary intolerance, seizure-like episode, respiratory weakness, complete bladder failure, pancreatic insufficiency, and multiple endocrine abnormalities.80 The course of the disease revealed to be intriguing. Before the age of one year-old, the patient had mucus plugging of the lung and respiratory distress that led to her being tested for cystic fibrosis, for which the test came back negative. While this improved with age, she still needed to sleep starting at age 4 year-old with bi-level positive airway pressure (Bipap) therapy. It is worth noting that the lung of the NKCC1 knockout mouse showed a phenotype only during the neonatal period. The basal short-circuit current (Isc) of the tracheae in young NKCC1 mice was significantly reduced, compared to wild-type mice—a phenotype not observed in the adult mouse. When UTP was used to stimulate anion secretion in the wild-type mouse trachea, there was a sizable bumetanide-sensitive short circuit current at postnatal Days 4, 10, 15 which decreased by ages 21, 28, and 35 days and disappeared at P42.55 Thus, there seems to be a critical role of NKCC1 in the lung at early age which might be substituted by another mechanism later in life.
Her mother recalls that during the first few months of life, her daughter was sleeping far more than usual, and it was challenging to wake her up. Her pediatricians assumed that she had a severe metabolic phenotype and tested her for mitochondrial disease. While she clearly had abnormal mitochondrial DNA content and abnormal glycogen content,80 none of the tests pointed to specific diseases known to affect the mitochondria. Yet, when the patient fibroblasts were used to measure mitochondrial respiration, the rate of oxygen consumption was significantly higher in the patient fibroblasts, compared to fibroblasts isolated from healthy controls.89
Gastrointestinal function declined progressively. A gastric tube was placed at age 7 year-old to help her nutritional status and keep her out of metabolic crisis. This was followed by a jejunum tube and the feeding of elemental (predigested) food to try maintaining her nutritional needs. She then required total parenteral nutrition at the age of 11 year-old. Chronic infections of the GI tract necessitated a complete colectomy and partial resection of the small intestine. The resected colon pathology revealed abnormal mucus accumulation in the lumen of the colon, mucus that remained attached to the epithelium (Figure 3).90
Figure 3.

Role of NKCC1 in Fluid Secretion in the Intestine. Throughout the gastrointestinal tract, NKCC1 is expressed on the basolateral membrane of Cl− secreting epithelial cells. As seen by immunofluorescence, string NKCC1 signal (green) is observed at lateral membranes of mouse colonic epithelial cells, while some weaker signal is seen at the base. The hematoxylin/eosin panel located above shows a stained crypt lumen plugged with mucus in the colon of the NKCC1-DFX patient. The model depicts the pathway for Cl− movement through a basolateral NKCC1 and apical CFTR Cl− channel. CFTR is also permeant to bicarbonate. Na+ and water movement follows through the paracellular pathways. Na+ movement occurs through claudin-2, a protein of the junctional complex. Expression of claudin-2 is affected in the NKCC1-DFX and NKCC1 knockout mice. The tight junction protein is only observed at the base of the crypt and no longer observed at the epithelial cells along the side of the crypt.90
At age 13 year-old, she also had some pancreatic obstruction. Her common bile duct was enlarged and plugged with trapped bile which required the muscle of her biliary tree to be cut back and the placement of a pancreatic stent to help movement of the bile. At the age of 14, she lost complete function of her bladder.
The multiorgan involvement is consistent with NKCC1 being expressed in all affected tissues. The cotransporter certainly is involved in the fluid secretion characteristic of Cl− secreting epithelia (Figure 3). The multiorgan failure might also be indicative of a deficit of the autonomic nervous system. NKCC1 is highly expressed in the peripheral nervous system where, through Cl−, the transporter affects GABA-mediated synaptic transmission. As we mentioned in the NKCC1 mouse knockout section, the cotransporter is also involved in controlling the function of interstitial (Cajal) neurons which are found associated with all of the tissues affected in the patient. In addition to the interstitial neurons, most of these tissues also have smooth muscle cells that facilitate the movement of fluids. NKCC1 also serves a particular function in facilitating the depolarization that leads to Ca2+ entry and muscle contraction. Smooth muscle cells are found in the intestine, pancreatic duct, bladder, and vasculature as discussed in the mouse knockout section.
One aspect of this patient we have not yet addressed is the fact that she also carries a wild-type SLC12A2 allele. This allele should produce 50% of normal NKCC1 and provide enough function to avoid any overt or deleterious phenotype. In fact, absence of phenotype is established for the father of the Kilquist patient who carries only one copy of the abnormal allele and the parents of the Swedish sisters who also carry a wild-type allele alongside their null allele. What is special to the NKCC1-DFX allele that it causes such significant clinical features? A possible explanation is proposed in the following section.
Heterologous Expression Studies of the NKCC1-DFX Transporter
The first NKCC1-DFX experiment utilized fibroblasts isolated from the patient.80 K+ uptake experiments were performed in the absence and presence of ouabain, an inhibitor of the Na+/K+ pump, and in the presence or absence of bumetanide to inhibit the Na-K-2Cl cotransporter. The experiment was also done under control (isosmotic) or stimulated (hyperosmotic) conditions. Significant osmotically-induced, bumetanide-sensitive, K+ uptake was observed in cells from the patient, consistent with NKCC1 function. Because heterologous expression of the mutant transporter in Xenopus laevis oocytes failed to demonstrate transport function, the bumetanide-sensitive uptake in fibroblasts can only be attributed to the native transporter expressed from the wild-type allele. The level of NKCC1 function in the fibroblasts did not indicate a dominant-negative effect. Absence of dominant-negative effect was also confirmed by co-injecting equal amounts of NKCC1-DFX with wild-type NKCC1 cRNA into oocytes and comparing to equivalent amounts of NKCC1 alone.80
The situation was very different when fluorescently-tagged transporters were expressed in polarized epithelial cells.91 When MDCK cells were grown on permeabilized support, they formed a polarized cell layer on top of the polycarbonate membrane with tight junctions separating a well-delineated apical pole (accessible from top chamber) from a basolateral pole (at the membrane support and accessible from the bottom chamber). Expression of wild-type NKCC1 was observed at the basolateral membrane, whereas expression of the mutant transporter was observed at the apical membrane. When the same cells were grown on Matrigel (3D culture), they formed closed cysts with the epithelial layer separating the lumen or interior of the cyst from the external space. MDCK cells expressing wild-type EGFP-NKCC1 showed fluorescence signal at the basolateral pole, while cells expressing EGFP-NKCC1-DFX displayed most of the fluorescent signal at the apical pole indicating mistrafficking of the mutant transporter.91 The defect could be observed at very early stages of cyst formation in Matrigel. When cysts were at a stage of four cells (24–48 h post plating), signal from wild-type NKCC1 was clearly visible between the cells, whereas signal from NKCC1-DFX was predominantly observed at the apical pole (Figure 1). As discussed above, studies revealed that the loss of a carboxyl-terminal dileucine motif was sufficient to target the cells to the apical membrane.16 Intriguingly, the mutant transporter (through dimerization) was able to traffic some wild-type monomers to the apical membrane. This observation indicated to us that the mutant transporter exerts a significant dominant-negative effect on wild-type transporter, an effect that is notably limited to epithelial cells. Although the patient fibroblasts maintain 40% NKCC1 function, it is possible, because the truncated protein is translated and trafficked out of the ER and can dimerize with the wild-type protein, that the number of functional NKCC1 dimers could be lower in some specific cell types even if they are not epithelial cells (Figure 4).
Figure 4.

Behavior of WT-NKCC1 and NKCC1-DFX Proteins in X. laevis Oocytes, HeLa Cells, and MDCK Cells. (A) Western blot analysis of cmyc-tagged wild-type and NKCC1-DFX protein expressed in X. laevis oocytes showing the mutant transporter predominantly as a dimer. (B) Similarly, NKCC1 transporters expressed at the surface of HeLa cells show as monomers and dimers for wild-type transporters but only as dimers for NKCC1-DFX transporters. tTomato-NKCC1 (red); EGFP-NKCC1-DFX (green); overlap (yellow). (C) In polarized MDCK cells grown on polycarbonate filters, wild-type NKCC1 is pulled-down from the basolateral membrane; whereas NKCC1-DFX is pulled down from both basolateral and apical membrane. (D) There are three possible dimer combinations originating from the wild-type and mutant SLC12A2 alleles: two wild-type NKCC1 monomers, one wild-type and one mutant monomer, and two mutant monomers. Expression of mutant monomers only results in a nonfunctional transporter. To date, in absence of direct testing, it is unclear if the heterodimer is functional and if that it is the case, it would lead to further reduction in NKCC1 function. T4: monoclonal anti-NKCC1 antibody. Data redrawn from Delpire et al.80 and Koumangoye et al.91
NKCC1-DFX Mouse Model
There is a high degree of exon/intron architecture and sequence conservation between human and mouse NKCC1. Therefore, a mouse model recapitulating the exact same 11 base pair deletion in exon 22 was engineered. This led to the expression in the mouse of the NKCC1-DFX protein observed in the patient.91 Immunofluorescence analysis of salivary gland and intestinal tissue from the NKCC1-DFX mice confirmed the abnormal expression of the cotransporter at the apical pole. As the NKCC1 antibody was directed against the carboxyl-terminus of the transporter, and thus did not recognize the mutant transporter, the apical signal observed was limited to mistrafficked wild-transporters. With a subset of transporters located on the wrong membrane in salivary gland, it was surprising that pilocarpine-induced saliva secretion was only reduced by 10% in heterozygous NKCC1-DFX mice compared to wild-type mice; a difference that did not reach statistical significance.91 The absence of a salivary gland phenotype contrasted with the NKCC1-DFX patient who suffers from dry mouth and who often chew ice to keep her mouth hydrated.80 Note that the contribution of NKCC1 to saliva production is also different in the different salivary glands, with highest contribution in the human submandibular gland, followed by the parotid gland, and then the sublingual gland.19 The disparity in phenotype might be due to differences in stimulated versus basal secretion or might reflect some species difference in contribution of NKCC1 in saliva production in each of the gland.
One tissue that clearly showed an abnormal phenotype in the NKCC1-DFX mouse was the intestine. NKCC1 plays a key role in producing fluid and electrolytes in the gut. Both NKCC1-DFX and NKCC1-KO mice demonstrated a 40%–50% decrease in fecal water content.90 This decreased was associated with decreased expression along the crypts of claudin-2, a paracellular Na+ and water junctional channel. Both the NKCC1-DFX and knockout mice also showed abnormal goblet cell mucus granules secretion. Typical mucus release involves the disintegration of the granule membrane and release of the mucus. In the NKCC1-DFX and NKCC1 knockout mice, entire groups of intact granules were released in the colon lumen. The mechanism behind the abnormal mucus release was not addressed in the study, but could be related to improper bicarbonate secretion and mucosal surface pH.88,92 Because of this abnormal mucus release, the inner mucus layer was much thinner and occasionally disrupted in the mutant mice compared to the wild-type mice.90 Consistent with a disrupted mucus barrier, commensal bacteria were seen in close contact with the intestinal epithelium. Immunoreactive CD3 signal also increased, indicating infiltration of immune cells deep into the epithelial layer. When mice were administered Citrobacter rodentium (a mild pathogen) through gavage, wild-type mice cleared the bacterial infection within 3–4 days, whereas NKCC1-DFX and NKCC1 knockout mice had not cleared the infection by Day 9.
Thus, the mouse studies determined that abnormal NKCC1 function leads not only to the secretion of viscid mucus, but also to the release of nondegranulated mucus granules which potentially cause more obstruction of the airway, pancreas, and intestinal epithelia. These studies also uncovered a novel mechanism by which NKCC1 protects against enteric infection and gastrointestinal inflammation.
Note that the inflammatory response could also be affected by the disruption in NKCC1 expression in immune cells. Intracellular Cl−, and particularly Cl− efflux, is a well-known trigger of neutrophil activation.93–96 A recent paper also demonstrated that NKCC1 function (through intracellular Cl− accumulation) inhibited efferocytosis, a process by which macrophages remove apoptotic corpses. Genetic deletion of Slc12a2, or pharmacologic inhibition of NKCC1, or manipulation of the upstream WNK-SPAK/OSR1 kinase pathway consistently promoted this process.97 More relevant was the observation that the loss of NKCC1 led to macrophage switching their program from anti-inflammatory to proinflammatory. Using a well-established apoptotic-cell-clearance model, the authors demonstrated that a low dose of LPS delivered intranasally in mice, caused neutrophil and monocyte infiltration in the lung with induction of proinflammatory cytokines. Animals administered with bumetanide, or a WNK inhibitor, exhibited enhanced levels of proinflammatory cytokines in the bronchoalveolar fluid, compared to vehicle-treated animals.97 NKCC1-deficient phagocytes also demonstrated increased amounts of reactive oxygen species (ROS) known to induce the production of proinflammatory cytokines. Remarkably, fibroblasts isolated from NKCC1 knockout mice also displayed significant increases in ROS, as established through increase in hydrogen peroxide production and peroxidase activity.89 These data indicate that the phagocyte phenotype switch might be extendable to other cell types.
SLC12A2 Haplotype Insufficiency and Hearing Loss
Two recent papers linked human NKCC1 mutations to hearing loss. Mutai et al. reported the cases of three SLC12A2 variants from three unrelated Japanese families (Table 2). The affected individuals presented with severe hearing loss.98 In the first family, a 7-month-old female infant and her twin brother shared a SLC12A2 c.2145C > G variant, resulting in p.Asp981Tyr mutation. In the second family, the affected individual carried a c.2930-2A > G mutation; which affects the splice acceptor site of exon 21. In the third family, the affected infant had a c.2962C > A variant; resulting in the substitution of a proline residue into threonine (p.Pro988Thr). Interestingly, all these variants were confined to exon 21 (Figure 5).98 In addition to the hearing loss, affected infants in two families had difficulties in holding their heads up, maintaining a sitting position, or walking, indicative of minor motor developmental delays. In a second study, we identified three individuals from two independent families with mutations in exon 21 associated with nonsyndromic bilateral sensorineural deafness (Table 2).99 In the first family, a 2-year-old boy had a p.Glu980Lys mutation, and in the second family, a 44-year-old father and his 5-year-old son carried a mutation of the preceding residue (p.Glu979Lys). Note that two additional patients with mutations in other parts of the transporter (p.Ala327Val and p.Trp892*) also demonstrated bilateral sensorineural hearing impairment.99
Table 2.
Disease-Causing SLC12A2 Variants
| Variant | Sex | Age | Origin | Exon | Prot. domain | Reference |
|---|---|---|---|---|---|---|
| Hearing | Dev. del. | Phenotype | Effect | |||
| p.Val1026Phefs∗2 | Girl | 14 year old | California | Exon 22 | C-terminus | Delpire et al. 80 |
| NKCC1-DFX | hetero | None | Lung, GI, bladder, endocrine | Dominant Negative | ||
| 22 kb deletion | Boy | 5 year old | Illinois | Exons 2–7 | TM1–TM5 | Macnamara et al.79 |
| Killquist | homo | Deaf | Brain, lung, GI, sweat | Knockout | ||
| c.1431delT c.2006-1G>A |
Girl | 8 year old | Sweden | Exon 8 Exon 13 |
Stodberg et al.81 | |
| comp | Deaf | Brain, lung, GI, sweat | Knockout | |||
| p. Asp981Tyr | Girl + Brother + 4 adults |
7 months | Japan | Exon 21 | C-terminus | Mutai et al.98 |
| Hetero | Loss | LOF | ||||
| p.Pro988Thr | Boy | 22 months | Japan | Exon 21 | C-terminus | Mutai et al.98 |
| Hetero | Loss | Minor motor developmental delay | LOF | |||
| c.2930AA>G | Girl | 17 months | Japan | Exon 21 | 3′ splicing | Mutai et al.98 |
| Hetero | Loss | Minor motor developmental delay | ||||
| p.Val327Ala | Boy | 1 year old | United Kingdom | Exon 4 | TM2 | McNeill et al.99 |
| Hetero | Loss | LOF | ||||
| p Arg410Gln | Boy | 9 year old | United Kingdom | Exon 6 | TM4 | McNeill et al.99 |
| Hetero | LOF | |||||
| p.Try892* | Girl | 15 year old | United Kingdom | Exon 18 | C-terminus | McNeill et al.99 |
| Hetero | Loss | LOF | ||||
| p.Asn376Ile | Girl | 3 year old | United Kingdom | Exon 5 | TM3 | McNeill et al.99 |
| Hetero | LOF | |||||
| p.His186Alafs*17 | Girl | 6 year old | United Kingdom | Exon 1 | N-terminus | McNeill et al.99 |
| Hetero | LOF | |||||
| p.Ala379Leu | Boy | 21 year old | Italy | Exon 5 | TM3 | McNeill et al.99 |
| Hetero | LOF | |||||
| p.Glu980Lys | Boy | 2 year old | United States | Exon 21 | C-terminus | McNeill et al.99 |
| Hetero | Loss | LOF | ||||
| p.Glu979Lys | Man | 44-year-old father | France | Exon 21 | C-terminus | McNeill et al.99 |
| Hetero | Loss | LOF | ||||
| p.Glu979Lys | Boy | 5-year-old son | France | Exon 21 | C-terminus | McNeill et al.99 |
| Hetero | Loss | LOF | ||||
| p. Tyr199Cys | Quebec | Exon 1 | N-terminus | Merner et al.100 | ||
| Hetero | Normal | Schizophrrenia | GOF? |
Comp, compound heterozyote; Hetero, heterozygote; Homo, homozygote; LOF, loss-of-function; GOF, gain-of--function; TM2, transmembrane domain 2; TM4, transmembrane domain 4; TM1, transmembrane domain 1; TM5, transmembrane domain 5.
Figure 5.
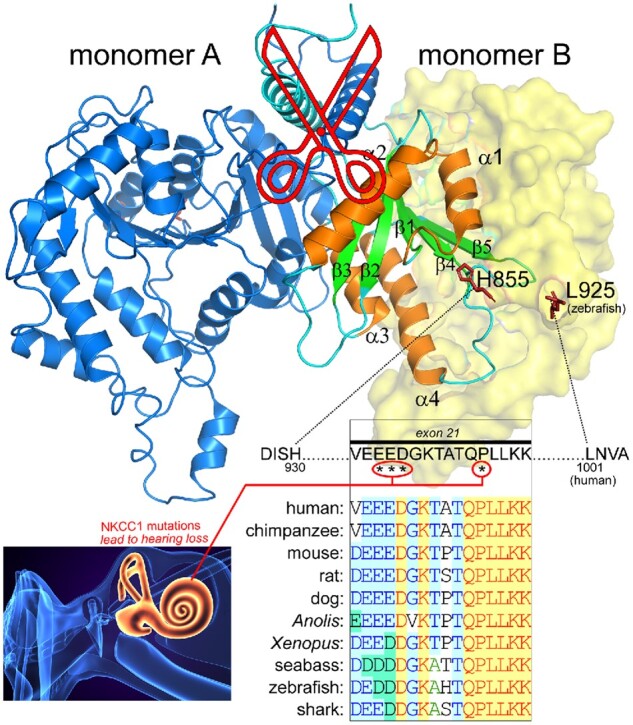
Structure of the Carboxyl-Terminus of NKCC1 Highlighting the Absence of a Peptide Containing Exon 21. Note that monomer A sits under the transmembrane core of monomer B and vice versa, with two helices crossing in scissor shape. The α-helices 1–4 and β-sheet 1–5 of monomer B are highlighted in orange and green colors, respectively. Residues between His855 and L925 (zebrafish) were nor resolved and therefore missing from the structure. Note that they are oriented in opposite directions in the two monomers making them unlikely to interact. The 69 stretch of missing residues in the structure includes the 16 amino acid encoded by exon 21. Four residues in this exon (human sequence, labeled by stars), including those creating an acidic motif, are involved in loss of hearing. These residues are highly conserved among vertebrate species. The D. rerio (zebrafish) NKCC1 structure26 was drawn using Pymol Molecular Graphic System (version 2.3.1, Schrodinger). The sequence alignment was created using VectorNti (version 6, Informax).
The NKCC1-DFX patient, the father of the Kilquist patient, and the father and mother of the sisters from Sweden all carry a wild-type SLC12A2 allele alongside an abnormal SLC12A2 allele, and they have no signs of hearing deficit or loss. Thus, 50% expression of the cotransporter in the inner ear is sufficient to support proper or adequate hearing. This observation does not exclude the possibility that they will experience accelerated age-related hearing loss.101 Why is exon 21 so significant to the function of the inner ear? From these observations, it is evident that the NKCC1 protein expressed in the stria vascularis must contain exon 21. In fact, this was addressed in the Dixon et al. study which established that the exon 21-containing splice form was the only transcript detected in the cochlea.48 Because the lack of exon 21 (splicing mutations) or single amino acid substitutions in this exon lead to hearing loss, it is possible that the 16 amino acids peptide encoded by this exon is part of a protein-protein interaction that is specific to the inner ear. What would cause a defective exon 21 to exert a dominant-negative effect specifically in the inner ear is currently unknown and should be investigated. Note that the structure of the zebrafish NKCC1 did not resolve the region of the carboxyl-terminus containing this unique exon (Figure 5).
SLC12A2 Haplotype Insufficiency and Neurodevelopmental Disorders
The study of McNeill and colleagues reported the cases of 6 children with de novo mutations in SLC12A2 linked to neurodevelopmental disorders such as autism spectrum disorders (ASDs), spastic dysplasia, developmental delays, and spastic quadriparesis. Two of these patients have been already mentioned since they had bilateral sensorineural hearing loss. Remarkably, neurodevelopmental disorders such as attention-deficit/hyperactivity disorder and anxiety-related disorders occur at rates 2–3 times higher in deaf children compared with children with normal hearing.102 While nonbiologic factors such as social interaction might be involved, mouse models of inner ear dysfunction support the link between vestibular dysfunction and hyperactivity and anxiety. Similarly, autistic patients can develop symptoms related to abnormal modulation of vestibular input.103
Additional human SLC12A2 variants have been linked to intellectual disability,104 macrocephaly and epilepsy associated with ASDs105 and schizophrenia.100
Variants in the Vanderbilt BioVU Database
With the increase of genetic data available, we wanted to query a database existing in our institution and test the hypothesis that variants in SLC12A2 can be identified in patients with CFTR-like symptoms. BioVU is Vanderbilt Medical Center’s biorepository of DNA extracted from discarded blood collected during routine clinical testing and linked to de-identified medical records in the Synthetic Derivative. To date, BioVU contains over 250,000 samples from both adult and pediatric populations. We queried two BioVU cohorts to identify individuals with rare exonic mutations in SLC12A2. The exome cohort contained a set of 35,842 individuals and the MEGA cohort contained a set of 94,489 individuals. In total, we identified 9 rare coding variants in SLC12A2 with data available in both Exome and MEGA cohorts (Table 3). On the Exome Beadchip, 38 individuals were heterozygotes for one of these variants and 2 homozygous. A phenome-wide association study (PheWAS) was conducted on the 36 SLC12A2 rare variant heterozygotes against all wild-type individuals. In total, we tested 1426 phenotypes with at least 100 cases; and excluded pregnancy-related codes. Only one association exceeded the Bonferroni correction (phecode 686.1—carbuncle and furuncle). Several associations related to what was observed in the NKCC1 patients were found to be nominally significant (P < 0.05) in this analysis, including dyshidrosis, chronic bronchitis, and intestinal obstruction (see Table 4). These data highlight the need for clinicians and geneticists to pay attention to deleterious mutations in the Na-K-2Cl cotransporter-1 gene when lung and/or intestinal obstruction symptoms are associated.
Table 3.
Rare SLC12A2 Coding Variants Identified in BioVU
| dbSNP ID | Coding | Protein | # Patients |
|---|---|---|---|
| rs115472345 | c.G2987A, c.G3035A | p.S996N, p.S1012N | 3 |
| rs142251614 | c.A770G | p.D257G | 1 |
| rs146381223 | c.A839G | p.K280R | 15 |
| rs147627093 | c.A1204G | p.M402V | 1 |
| rs116191759 | c.A2380G | p.M794V | 14 |
| rs142105778 | c.G2402A | p.R801H | 1 |
| rs114752904 | c.A3400G, c.A3448G | p.I1134V, p.I1150V | 1 |
One individual who was homozygous for a rare variant in SLC12A2 (p.M794V) was a pediatric patient seen at VUMC between the ages of 12 and 16 years. Using phecodes, we determined that this patient was diagnosed with abdominal pain and constipation, chronic sinusitis, and migraines. Upon further chart review we found that, despite extensive workup, the underlying cause of this patient GI complaints was never identified.
Table 4.
PheWas Data from BioVU Query of SLC12A2 Variants
| Phecode | Description | P-value | OR (L95–U95) |
|---|---|---|---|
| 686.1 | Carbuncle and furuncle | 0.0000075** | 8.97 (3.43–23.43) |
| 705.1 | Dishydrosis | 0.0003* | 9.26 (2.74–31.2) |
| 705 | Sweat gland disorders | 0.0012* | 5.73 (1.99–16.50) |
| 496.2 | Chronic bronchitis | 0.0018* | 4.35 (1.73–10.94) |
| 772.1 | Muscular wasting | 0.0086* | 6.96 (1.64–29.64) |
| 560.4 | Other intestinal obstruction | 0.0123* | 3.37 (1.30–8.73) |
| 578 | Gastrointestinal hemorrhage | 0.0205* | 2.32 (1.14–4.74) |
| 496.21 | Obstructive chronic bronchitis | 0.0212* | 3.59 (1.21–10.66) |
| 771.1 | Swelling of limb | 0.0539 | 2.12 (0.99–4.57) |
| 497 | Bronchitis | 0.0569 | 2.77 (0.97–7.94) |
| 496 | Chronic airway obstruction | 0.0650 | 2.11 (0.95–4.66) |
Cases were defined using phecodes (ICD-10 codes), requiring only one billing code. No exclude ranges were used. Association results were generated using logistic regression, with sex, race, and age at last visit as covariates. The Bonferroni correction for this analysis was calculated as 0.05/1426. We also analyzed single SLC12A2 homozygote individually, using phecodes followed up by chart review. All analyses were performed in R statistical software. OR, odds ratio (confidence interval). Use of the BioVU and Synthetic Derivative databases was approved by our Institutional Review Board.
P < Bonferroni correction value.
P < 0.05.
NKCC1, Bumetanide, and Autism
ASDs are a group of neurodevelopmental disorders in children that affect their ability to socially interact and communicate with peers, parents, and others. The condition is also characterized by restricted and repetitive pattern of behaviors and limited interests.106 Hundreds of genes have been linked to ASD, and there is no definitive etiology or diagnosis established. In some cases, children with similar genetic variants present with drastically different phenotypes.107 ASD is thought to be caused by a complex interaction between genetic variations and environmental factors. Although the molecular mechanisms that cause ASD are not fully understood, recent studies suggest that genes and pathways involved in neuronal excitability and GABAergic signals are involved in the pathophysiology of ASD.108–110 NKCC1 and KCC2 play key roles in modulating GABA-mediated synaptic transmission as they control the level of intracellular Cl− concentration (Figure 6).111–114 In newborn mice, NKCC1 expression is high, while KCC2 expression is low. The resulting effect is an elevated intracellular Cl− concentration. Consequently, when GABA binds to its ligand-activated Cl− channel, it causes net Cl− efflux that leads to membrane depolarization and excitation. This is the basis for the giant depolarizing potentials (GDPs) observed in young rat brains.115 In the more mature brain, NKCC1 expression is lower, while expression of KCC2 is high leading to a low intracellular Cl− concentration. When GABA binds, the opening of the Cl− conductance leads to an influx of the anion into the cell, further hyperpolarizing the plasma membrane.3,116 Thus, the cotransporters contribute to the developmental shift in GABA-mediated synaptic responses from depolarizing to hyperpolarizing (Figure 6). In human brain development, this switch is believed to occur prenatally toward term or 40 weeks of gestation.117
Figure 6.

Regulation of Intracellular [Cl−] in Neurons and Effect on GABA Receptor Function. (A) In immature central nervous system (CNS) neurons and in peripheral nervous system (PNS) neurons, the Na-K-2Cl cotransporter, NKCC1, is highly expressed, whereas the K-Cl cotransporter, KCC2, is minimally expressed (as in the CNS) or not expressed (as in the PNS). The activity of NKCC1 results in accumulation of intracellular Cl−. [Cl−] values range from 30 to 60 mM. As GABA binds to its receptor (GABAAR), it triggers the opening of a Cl− conductance and Cl− follows its electrochemical driving force leaving the cell and leading to depolarization of the neuronal membrane. The membrane potential is created by the leak of K+ through specific K+ channels. (B) In mature CNS neurons, NKCC1 expression or activity is low whereas expression and activity of KCC2 high. This results into low intracellular Cl− concentrations (<10 mM). When GABA binds, Cl− enters the cell and hyperpolarizes the membrane.
In rodents, birth constitutes an important event that is critical for brain development. During delivery, the release of oxytocin produces a signal that leads to GABA inhibition.118 This is at a time when intracellular Cl− in neurons is still elevated and GABA release produces these giant depolarizing potentials. During delivery, however, [Cl−]i drops precipitously, in so doing producing GABA inhibition and sedation.118 Importantly, this signal is abolished in two rodent models of autism. The likely explanation is that in these models, at P0, NKCC1 activity remains high and intracellular Cl− remains elevated. This hypothesis was confirmed with the addition of bumetanide on hippocampal slices, as well as through pretreatment of the dams with bumetanide before delivery. In addition, blocking oxytocin signaling in wild-type mice, produced offspring having electrophysiological and behavioral autistic-like features.118 This study supports the idea of a link between NKCC1 function, GABA signaling, and autism.
Based on these studies, bumetanide is being tested in clinical trials as a potential therapeutic agent to treat patients with ASDs.119–123 While these early trials are encouraging, this work is ongoing and no final conclusions can be made at this time regarding the effectiveness of the treatment. Bumetanide was also proposed as a potential therapeutic agent for the treatment of neonatal seizures.124 In a clinical trial, the diuretic however failed to treat hypoxic-ischemic encephalopathy-induced seizures in newborn babies.125
We should also mention the existence of an extensive literature linking glial cell and blood-brain barrier NKCC1 function to ischemic brain injury. During stroke and/or cerebral ischemia, there is release of K+ in the extracellular space from neurons and glial cells.126 High K+, in turn, leads to NKCC1-mediated astrocyte swelling and the release of excitatory amino acids.127,128 In addition, the K+-induced increased in NKCC1 function leads to the accumulation of Na+, the activation of the Na+/Ca2+ exchanger (NCX1), and Ca2+ overloading in mitochondria.129 Administration of bumetanide in rats was shown to reduce edema and neuronal damage following occlusion of the left middle cerebral artery.130 Similarly, the extent of damage caused by focal cerebral edema was significantly reduced in NKCC1 knockout mice.131 Thus, NKCC1 function seems to be detrimental following injury and therefore a reduced cotransporter function might be protective in the setting of stroke or ischemic injury.
Conclusions
The data and literature reviewed here provide evidence that disruption of human SLC12A2 (NKCC1) results in severe dysfunction involving multiple organs and systems. The spectrum of disorders caused by mutations in SLC12A2 is vast because the cotransporter is expressed in so many tissues and its specific function cannot be compensated by other transporters. With the exception of the kidney which abundantly expresses the Na-K-2Cl cotransporter-2, NKCC1 is the only isoform of the Na-K-2Cl cotransporter that is found in all other tissues. During late embryonic–early postnatal development, NKCC1 function in the nervous system seems to be tightly controlled and cotransporter dysfunction might be involved in autism, neonatal seizures, and schizophrenia. Additional studies are also required to better understand the molecular underlying mechanisms by which a loss of NKCC1 function leads to intestinal and pulmonary obstruction, pancreatic insufficiency, chronic GI, and lung infections, constipation, intestinal dysmotility, or malrotation, failure to grow and maintain body weight, muscle weakness, fatigue, chronic pain, and sensorineural hearing loss. This review and its recent primary sources now better identify NKCC1 as a disease-causing transporter. Ultimately, it is hoped that information here should prompt genetic testing when early symptoms of deafness, hypotonia, lung, and intestinal obstruction are encountered; and the study of additional cases can be translated to a more personalized and scientifically-driven approach for treating these ailments.
Funding
This work was supported by National Institute of Diabetes and Digestive and Kidney Diseases Grant DK093501 and by Leducq Foundation grant 17CVD05.
Conflict of interest statement
The authors declare that there is no conflict of interest.
References
- 1. Gamba G. Molecular physiology and pathophysiology of electroneutral cation-chloride cotransporters. Physiol Rev 2005;85:423–493. [DOI] [PubMed] [Google Scholar]
- 2. Sung K-W, Kirby M, McDonald MP, Lovinger DM, Delpire E.. Abnormal GABAA-receptor mediated currents in dorsal root ganglion neurons isolated from Na-K-2Cl cotransporter null mice. J Neurosci 2000;20:7531–7538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zhu L, Lovinger D, Delpire E.. Cortical neurons lacking KCC2 expression show impaired regulation of intracellular chloride. J Neurophysiol 2005;93:1557–68. [DOI] [PubMed] [Google Scholar]
- 4. Delpire E, Gagnon KB.. Water homeostasis and cell volume maintenance and regulation. Curr Top Membr 2018;81:3–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Piechotta K, Lu J, Delpire E.. Cation-chloride cotransporters interact with the stress-related kinases SPAK and OSR1. J Biol Chem 2002;277:50812–50819. [DOI] [PubMed] [Google Scholar]
- 6. Dowd BF, Forbush B.. PASK (proline-alanine-rich STE20-related kinase), a regulatory kinase of the Na-K-Cl cotransporter (NKCC1). J Biol Chem 2003;278:27347–27353. [DOI] [PubMed] [Google Scholar]
- 7. Vitari AC, Thastrup J, Rafiqi FH, et al. Functional interactions of the SPAK/OSR1 kinases with their upstream activator WNK1 and downstream substrate NKCC1. Biochem J 2006;397:223–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gagnon KB, England R, Delpire E.. Volume sensitivity of cation-chloride cotransporters is modulated by the interaction of two kinases: SPAK and WNK4. Am J Physiol Cell Physiol 2006;290:C134–C142. [DOI] [PubMed] [Google Scholar]
- 9. Darman RB, Flemmer A, Forbush BI.. Modulation of ion transport by direct targeting of PP1 to the Na-K-Cl cotransporter. J Biol Chem 2001;276:34359–34362. [DOI] [PubMed] [Google Scholar]
- 10. Gagnon KB, Delpire E.. Multiple pathways for protein phosphatase 1 (PP1) regulation of Na-K-2Cl cotransporter (NKCC1) function. The N-terminal tail of the Na-K-2Cl cotransporter serves as a regulatory scaffold for Ste20-related proline/alanine-rich kinase (SPAK) and PP1. J Biol Chem 2010;285:14115–14121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lek M, Karczewski KJ, Minikel EV, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016;536:285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Karczewski KJ, Francioli LC, Tiao G, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020;581:434–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Randall J, Thorne T, Delpire E.. Partial cloning and characterization of Slc12a2: the gene encoding the secretory Na+-K+-2Cl- cotransporter. Am J Physiol Cell Physiol 1997;273:C1267–C1277. [DOI] [PubMed] [Google Scholar]
- 14. Morita Y, Callicott JH, Testa LR, et al. Characteristics of the cation cotransporter NKCC1 in human brain: alternate transcripts, expression in development, and potential relationships to brain function and schizophrenia. J Neurosci 2014;34:4929–4940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Carmosino M, Giménez I, Caplan M, Forbush B.. Exon loss accounts for differential sorting of Na-K-Cl cotransporters in polarized epithelial cells. Mol Biol Cell 2008;19:4341–4351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Koumangoye R, Omer S, Delpire E.. A dileucine motif in the C-terminal domain of NKCC1 targets the cotransporter to the plasma membrane. Am J Physiol Cell Physiol 2019;316:C545–C558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Delpire E, Gagnon KB.. Na+-K+-2Cl- cotransporter (NKCC) physiological function in nonpolarized cells and transporting epithelia. Compr Physiol 2018;8:871–901. [DOI] [PubMed] [Google Scholar]
- 18. Evans RL, Park K, Turner RJ, et al. Severe impairment of salivation in Na+/K+/2Cl- cotransporter (NKCC1)-deficient mice. J Biol Chem 2000;275:26720–26726. [DOI] [PubMed] [Google Scholar]
- 19. Kondo Y, Nakamoto T, Jaramillo Y, Choi S, Catalan MA, Melvin JE.. Functional differences in the acinar cells of the murine major salivary glands. J Dent Res 2015;94:715–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hengl T, Kaneko H, Dauner K, Vocke K, Frings S, Mohrlen F.. Molecular components of signal amplification in olfactory sensory cilia. Proc Natl Acad Sci USA 2010;107:6052–6057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Milićević N, Duursma A, Ten Asbroek A, Felder-Schmittbuhl MP, Bergen AA.. Does the circadian clock make RPE-mediated ion transport "tick" via SLC12A2 (NKCC1)? Chronobiol Int 2019;36:1592–1598. [DOI] [PubMed] [Google Scholar]
- 22. Plotkin MD, Kaplan MR, Peterson LN, Gullans SR, Hebert SC, Delpire E.. Expression of the Na+-K+-2Cl- cotransporter BSC2 in the nervous system. Am J Physiol Cell Physiol 1997;272:C173–C183. [DOI] [PubMed] [Google Scholar]
- 23. Wu Q, Delpire E, Hebert SC, Strange K.. Functional demonstration of Na-K-2Cl cotransporter activity in isolated, polarized choroid plexus cells. Am J Physiol Cell Physiol 1998;275:C1565–C1572. [DOI] [PubMed] [Google Scholar]
- 24. Parvin MN, Gerelsaikhan T, Turner RJ.. Regions in the cytosolic C-terminus of the secretory Na(+)-K(+)-2Cl(-) cotransporter NKCC1 are required for its homodimerization. Biochemistry 2007;46:9630–9637. [DOI] [PubMed] [Google Scholar]
- 25. Parvin MN, James Turner RJ.. Identification of key residues involved in the dimerization of the secretory Na+–K+–2Cl– cotransporter NKCC1. Biochemistry 2011;50:9857–9864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chew TA, Orlando BJ, Zhang J, et al. Structure and mechanism of the cation-chloride cotransporter NKCC1. Nature 2019;572:488–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Liu S, Chang S, Han B, et al. Cryo-EM structures of the human cation-chloride cotransporter KCC1. Science 2019;366:505–508. [DOI] [PubMed] [Google Scholar]
- 28. Reid MS, Kern DM, Brohawn SG.. Cryo-EM structure of the potassium-chloride cotransporter KCC4 in lipid nanodiscs. Elife 2020;9:e52505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Xie Y, Chang S, Zhao C, et al. Structures and an activation mechanism of human potassium-chloride cotransporters. Science Adv 2020;6:eabc5883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gagnon KB, Delpire E.. Molecular physiology of SPAK and OSR1: two Ste20-related protein kinases regulating ion transport. Physiol Rev 2012;92:1577–1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Piala AT, Moon TM, Akella R, He H, Cobb MH, Goldsmith EJ.. Chloride sensing by WNK1 involves inhibition of autophosphorylation. Sci Signal 2014;7:ra41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sun Q, Wu Y, Jonusaite S, et al. Intracellular chloride and scaffold protein Mo25 cooperatively regulate transepithelial ion transport through WNK signaling in the malpighian tubule. J Am Soc Nephrol 2018;29:1449–1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gagnon KB, England R, Delpire E.. A single binding motif is required for SPAK activation of the Na-K-2Cl cotransporter. Cell Physiol Biochem 2007;20:131–142. [DOI] [PubMed] [Google Scholar]
- 34. Chobanian AV, Bakris GL, Black HR, et al. The Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure: the JNC 7 report. JAMA 2003;289:2560–2572. [DOI] [PubMed] [Google Scholar]
- 35. Group AaL-LTtPHATCR. Diuretic versus alpha-blocker as first-step antihypertensive therapy: final results from the Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial (ALLHAT). Hypertension 2003;42:239–246. [DOI] [PubMed] [Google Scholar]
- 36. Hannaert P, Alvarez-Guerra M, Pirot D, Nazaret C, Garay RP.. Rat NKCC2/NKCC1 cotransporter selectivity for loop diuretic drugs. Naunyn-Schmiedeberg's Arch Pharmacol 2002;365:193–199. [DOI] [PubMed] [Google Scholar]
- 37. Asbury MJ, Gatenby PB, O'Sullivan S, Bourke E.. Bumetanide: potent new "loop" diuretic. Br Med J 1972;1:211–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Goto S, Yoshitomi H, Miyamoto A, Inoue K, Nakano M.. Binding of several loop diuretics to serum albumin and human serum from patients with renal failure and liver disease. J Pharmacobiodyn 1980;3:667–676. [DOI] [PubMed] [Google Scholar]
- 39. Forbush BI, Haas M, Lytle C.. Na-K-Cl cotransport in the shark rectal gland. I. Regulation in the intact perfused gland. Am J Physiol Cell Physiol 1992;262:C1000–C1008. [DOI] [PubMed] [Google Scholar]
- 40. Franklin CC, Turner JT, Kim HD.. Regulation of Na+/K+/Cl- cotransport and [3H]bumetanide binding site density by phorbol esters in HT29 cells. J Biol Chem 1989;264:6667–6673. [PubMed] [Google Scholar]
- 41. Haas M, Forbush B. 3rd. Photolabeling of a 150-kDa (Na + K + Cl) cotransport protein from dog kidney with a bumetanide analogue. Am J Physiol 1987;253:C243–C252. [DOI] [PubMed] [Google Scholar]
- 42. Kaji D. Na+/K+/2Cl- cotransport in medullary thick ascending limb cells: kinetics and bumetanide binding. Biochim Biophys Acta 1993;1152:289–299. [DOI] [PubMed] [Google Scholar]
- 43. Suvitayavat W, Palfrey HC, Haas M, Dunham PB, Kalmar F, Rao MC.. Characterization of the endogenous Na(+)-K(+)-2Cl- cotransporter in Xenopus oocytes. Am J Physiol Cell Physiol 1994;266:C284–C292. [DOI] [PubMed] [Google Scholar]
- 44. Hampel P, Römermann K, MacAulay N, Löscher W.. Azosemide is more potent than bumetanide and various other loop diuretics to inhibit the sodium-potassium-chloride-cotransporter human variants hNKCC1A and hNKCC1B. Sci Rep 2018;8:9877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Savardi A, Borgogno M, Narducci R, et al. Discovery of a small molecule drug candidate for selective NKCC1 inhibition in brain disorders. Chem 2020;6:2073–2096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Delpire E, Lu J, England R, Dull C, Thorne T.. Deafness and imbalance associated with inactivation of the secretory Na-K-2Cl co-transporter. Nat Genet 1999;22:192–19. [DOI] [PubMed] [Google Scholar]
- 47. Flagella M, Clarke LL, Miller ML, et al. Mice lacking the basolateral Na-K-2Cl cotransporter have impaired epithelial chloride secretion and are profoundly deaf. J Biol Chem 1999;274:26946–269. [DOI] [PubMed] [Google Scholar]
- 48. Dixon MJ, Gazzard J, Chaudhry SS, Sampson N, Schulte BA, Steel KP.. Mutation of the Na-K-Cl co-transporter gene Slc12a2 results in deafness in mice. Hum Mol Genet 1999;8:1579–1584. [DOI] [PubMed] [Google Scholar]
- 49. Pace AJ, Madden VJ, Henson OWJ, Koller BH, Henson MM.. Ultrastructure of the inner ear of NKCC1-deficient mice. Hear Res 2001;156:17–30. [DOI] [PubMed] [Google Scholar]
- 50. Mathog RH, Matz GJ.. Ototoxic effects of ethacrynic acid. Ann Otol Rhinol Laryngol 1972;81:871–875. [DOI] [PubMed] [Google Scholar]
- 51. Rybak LP. Ototoxicity of loop diuretics. Otolaryngol Clin North Am 1993;26:829–844. [PubMed] [Google Scholar]
- 52. Ikeda K, Oshima T, Hidaka H, Takasaka T.. Molecular and clinical implications of loop diuretic ototoxicity. Hear Res 1997;107:1–8. [DOI] [PubMed] [Google Scholar]
- 53. Lee C, Jones TA.. Acute blockade of inner ear marginal and dark cell K(+) secretion: effects on gravity receptor function. Hear Res 2018;361:152–156. [DOI] [PubMed] [Google Scholar]
- 54. Grubb BR, Lee E, Pace AJ, Koller BH, Boucher RC.. Intestinal ion transport in NKCC1-deficient mice. Am J Physiol Gastrointest Liver Physiol 2000;279:G707–G718. [DOI] [PubMed] [Google Scholar]
- 55. Grubb BR, Pace AJ, Lee E, Koller BH, Boucher RC.. Alterations in airway ion transport in NKCC1-deficient mice. Am J Physiol Cell Physiol 2001;281:C615–C623. [DOI] [PubMed] [Google Scholar]
- 56. Pace AJ, Lee E, Athirakul K, Coffman TM, O’Brien DA, Koller BH.. Failure of spermatogenesis in mouse lines deficient in the Na+-K+-2Cl- cotransporter. J Clin Invest 2000;105:441–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Gagnon KB, Delpire E.. Physiology of SLC12 transporters: lessons from inherited human genetic mutations and genetically-engineered mouse knockouts. Am J Physiol Cell Physiol 2013;304:C693–C714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Moenter SM, DeFazio RA.. Endogenous gamma-aminobutyric acid can excite gonadotropin-releasing hormone neurons. Endocrinology 2005;146:5374–5379. [DOI] [PubMed] [Google Scholar]
- 59. Alvarez-Leefmans FJ, Gamiño SM, Giraldez F, Nogueron I.. Intracellular chloride regulation in amphibian dorsal root ganglion neurons studied with ion-selective microelectrodes. J Physiol 1988;406:225–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Alvarez-Leefmans FJ, Delpire E. Thermodynamics and kinetics of chloride transport in neurons: An outline. In: Alvarez-Leefmans FJ, Delpire E, eds. Physiology and Pathology of Chloride Transporter and Channels in the Nervous System: From Molecules to Diseases. London: Academic Press, 2009:81–108. [Google Scholar]
- 61. Laird JM, Garcia-Nicas E, Delpire EJ, Cervero F.. Presynaptic inhibition and spinal pain processing in mice: a possible role of the NKCC1 cation-chloride co-transporter in hyperalgesia. Neurosci Lett 2004;361:200–203. [DOI] [PubMed] [Google Scholar]
- 62. Plotkin MD, Snyder EY, Hebert SC, Delpire E.. Expression of the Na-K-2Cl cotransporter is developmentally regulated in postnatal rat brains: a possible mechanism underlying GABA's excitatory role in immature brain. J Neurobiol 1997;33:781–795. [DOI] [PubMed] [Google Scholar]
- 63. Buhrley LE, Reed DJ.. The effect of furosemide on sodium-22 uptake into cerebrospinal fluid and brain. Exp Brain Res 1972;14:503–510. [DOI] [PubMed] [Google Scholar]
- 64. Saito Y, Wright EM.. Bicarbonate transport across the frog choroid plexus and its control by cyclic nucleotides. J Physiol 1983;336:635–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Johanson CE, Murphy VA, Dyas M.. Ethacrynic acid and furosemide alter Cl, K, and Na distribution between blood, choroid plexus, CSF, and brain. Neurochem Res 1992;17:1079–1085. [DOI] [PubMed] [Google Scholar]
- 66. Keep RF, Xiang J, Betz AL.. Potassium cotransport at the rat choroid plexus. Am J Physiol Cell Physiol 1994;267:C1616–C1622. [DOI] [PubMed] [Google Scholar]
- 67. Karimy JK, Zhang J, Kurland DB, et al. Inflammation-dependent cerebrospinal fluid hypersecretion by the choroid plexus epithelium in posthemorrhagic hydrocephalus. Nature Med 2017;23:997–1003. [DOI] [PubMed] [Google Scholar]
- 68. Delpire E, Gagnon KB.. Elusive role of the Na-K-2Cl cotransporter in the choroid plexus. Am J Physiol Cell Physiol 2019;316:C522–C524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Steffensen AB, Oernbo EK, Stoica A, et al. Cotransporter-mediated water transport underlying cerebrospinal fluid formation. Nature Commun 2018;9:2167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Gregoriades JMC, Madaris A, Francisco J,, Alvarez FJ, Alvarez-Leefmans FJ.. Genetic and pharmacologic inactivation of apical NKCC1 in choroid plexus epithelial cells reveals the physiological function of the cotransporter Am J Physiol Cell Physiol 2019;316:C525–C544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Meyer JW, Flagella M, Sutliff RL, et al. Decreased blood pressure and vascular smooth muscle tone in mice lacking basolateral Na(+)-K(+)-2Cl(-) cotransporter. Am J Physiol Heart Circ Physiol 2002;283:H1846–H1855. [DOI] [PubMed] [Google Scholar]
- 72. Garg P, Martin CF, Elms SC, et al. Effect of the Na-K-2Cl cotransporter NKCC1 on systemic blood pressure and smooth muscle tone. Am J Physiol Heart Circ Physiol 2007;292:H2100–H2105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Wall SM, Knepper MA, Hassell KA, et al. Hypotension in NKCC1 null mice: role of the kidneys. Am J Physiol Renal Physiol 2006;290:F409–F416. [DOI] [PubMed] [Google Scholar]
- 74. Ginns SM, Knepper MA, Ecelbarger CA, et al. Immunolocalization of the secretory isoform of Na-K-Cl cotransporter in rat renal intercalated cells. J Am Soc Nephrol 1996;7:2533–2542. [DOI] [PubMed] [Google Scholar]
- 75. Kaplan MR, Plotkin MD, Brown D, Hebert SC, Delpire E.. Expression of the mouse Na-K-2Cl cotransporter, mBSC2, in the terminal IMCD, the glomerular and extraglomerular mesangium and the glomerular afferent arteriole. J Clin Invest 1996;98:723–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Wall SM, Fischer MP, Mehta P, Hassell KA, Park SJ.. Contribution of the Na+-K+-2Cl- cotransporter NKCC1 to Cl- secretion in rat OMCD. Am J Physiol Renal Physiol 2001;280:F913–F921. [DOI] [PubMed] [Google Scholar]
- 77. Wall SM, Fischer MP.. Contribution of the Na(+)-K(+)-2Cl(-) cotransporter (NKCC1) to transepithelial transport of H(+), NH(4)(+), K(+), and Na(+) in rat outer medullary collecting duct. J Am Soc Nephrol 2002;13:827–835. [DOI] [PubMed] [Google Scholar]
- 78. Gosmanov AR, Lindinger MI, Thomason DB.. Riding the tides: K+ concentration and volume regulation by muscle Na+-K+-2Cl- cotransport activity. News Physiol Sci 2003;18:196–200. [DOI] [PubMed] [Google Scholar]
- 79. Macnamara EF, Koehler AE, D'Souza P, et al. Kilquist syndrome: a novel syndromic hearing loss disorder caused by homozygous deletion of SLC12A2. Hum Mut 2019;40:532–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Delpire E, Wolfe L, Flores B, et al. A patient with multisystem dysfunction carries a truncation mutation in human SLC12A2, the gene encoding the Na-K-2Cl cotransporter, NKCC1. Cold Spring Harb Mol Case Studies 2016;2:a001289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Stodberg T, Magnusson M, Lesko N, et al. SLC12A2 mutations cause NKCC1 deficiency with encephalopathy and impaired secretory epithelia. Neurol Genet 2020;6:e478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Quinton PM. Cystic fibrosis: lessons from the sweat gland. Physiology (Bethesda) 2007;22:212–225. [DOI] [PubMed] [Google Scholar]
- 83. Ratjen F, Bell SC, Rowe SM, Goss CH, Quittner AL, Bush A.. Cystic fibrosis. Nat Rev Dis Primers 2015;1:15010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Quinton PM. Cystic fibrosis: impaired bicarbonate secretion and mucoviscidosis. Lancet (London, England) 2008;372:415–417. [DOI] [PubMed] [Google Scholar]
- 85. Borowitz D. CFTR, bicarbonate, and the pathophysiology of cystic fibrosis. Pediatr Pulmonol 2015;50(Suppl 40):S24–S30. [DOI] [PubMed] [Google Scholar]
- 86. Park HW, Nam JH, Kim JY, et al. Dynamic regulation of CFTR bicarbonate permeability by [Cl-]i and its role in pancreatic bicarbonate secretion. Gastroenterology 2010;139:620–631. [DOI] [PubMed] [Google Scholar]
- 87. Kim Y, Jun I, Shin DH, et al. Regulation of CFTR bicarbonate channel activity by WNK1: implications for pancreatitis and CFTR-related disorders. Cell Mol Gastroenterol Hepatol 2020;9:79–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Hoegger MJ, Fischer AJ, McMenimen JD, et al. Impaired mucus detachment disrupts mucociliary transport in a piglet model of cystic fibrosis. Science 2014;345:818–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Omer S, Koumangoye R, Delpire E.. A mutation in the Na-K-2Cl cotransporter-1 leads to changes in cellular metabolism. J Cell Physiol 2020;235:7239–7250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Koumangoye R, Omer S, Kabeer MH, Delpire E.. Novel human NKCC1 mutations cause defects in goblet cells mucus secretion and chronic inflammation. Cell Mol Gastroenterol Hepatol 2020;9:239–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Koumangoye R, Omer S, Delpire E.. Mistargeting of a truncated Na-K-2Cl cotransporter in epithelial cells. Am J Physiol Cell Physiol 2018;315:C258–C276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Garcia MA, Yang N, Quinton PM.. Normal mouse intestinal mucus release requires cystic fibrosis transmembrane regulator-dependent bicarbonate secretion. J Clin Invest 2009;119:2613–2622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Menegazzi R, Busetto S, Dri P, Cramer R, Patriarca P.. Chloride ion efflux regulates adherence, spreading, and respiratory burst of neutrophils stimulated by tumor necrosis factor-alpha (TNF) on biologic surfaces. J Cell Biol 1996;135:511–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Tager AM, Wu J, Vermeulen MW.. The effect of chloride concentration on human neutrophil functions: potential relevance to cystic fibrosis. Am J Respir Cell Mol Biol 1998;19:643–652. [DOI] [PubMed] [Google Scholar]
- 95. Menegazzi R, Busetto S, Decleva E, Cramer R, Dri P, Patriarca P.. Triggering of chloride ion efflux from human neutrophils as a novel function of leukocyte beta 2 integrins: relationship with spreading and activation of the respiratory burst. J Immunol 1999;162:423–434. [PubMed] [Google Scholar]
- 96. Busetto S, Trevisan E, Decleva E, Dri P, Menegazzi R.. Chloride movements in human neutrophils during phagocytosis: characterization and relationship to granule release. J Immunol 2007;179:4110–4124. [DOI] [PubMed] [Google Scholar]
- 97. Perry JSA, Morioka S, Medina CB, et al. Interpreting an apoptotic corpse as anti-inflammatory involves a chloride sensing pathway. Nature Cell Biol 2019;21:1532–1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Mutai H, Wasano K, Momozawa Y, et al. Variants encoding a restricted carboxy-terminal domain of SLC12A2 cause hereditary hearing loss in humans. PLoS Genet 2020;16:e1008643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. McNeill A, Lovino E, Bedoukian E, et al. SLC12A2 variants cause a neurodevelopmental disorder or cochleovestibular defect. Brain 2020;143:2380–2387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Merner ND, Mercado A, Khanna AR, et al. Gain-of-function missense variant in SLC12A2, encoding the bumetanide-sensitive NKCC1 cotransporter, identified in human schizophrenia. J Psychiatr Res 2016;77:22–26. [DOI] [PubMed] [Google Scholar]
- 101. Liu Y, Chu H, Chen J, et al. Age-related change in the expression of NKCC1 in the cochlear lateral wall of C57BL/6J mice. Acta Otolaryngol 2014;134:1047–1051. [DOI] [PubMed] [Google Scholar]
- 102. Antoine MW, Vijayakumar S, McKeehan N, Jones SM, Hébert JM.. The severity of vestibular dysfunction in deafness as a determinant of comorbid hyperactivity or anxiety. J Neurosci 2017;37:5144–5154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Leekam SR, Nieto C, Libby SJ, Wing L, Gould J.. Describing the sensory abnormalities of children and adults with autism. J Autism Dev Disord 2007;37:894–910. [DOI] [PubMed] [Google Scholar]
- 104. Anazi S, Maddirevula S, Salpietro V, et al. Expanding the genetic heterogeneity of intellectual disability. Hum Genet 2017;136:1419–1429. [DOI] [PubMed] [Google Scholar]
- 105. Marchese M, Valvo G, Moro F, Sicca F, Santorelli FM.. Targeted gene resequencing (astrochip) to explore the tripartite synapse in autism-epilepsy phenotype with macrocephaly. Neuromolecular Med 2016;18:69–80. [DOI] [PubMed] [Google Scholar]
- 106. Maenner MJ, Shaw KA, Baio J, et al. Prevalence of autism spectrum disorder among children aged 8 years - autism and developmental disabilities monitoring network, 11 sites, United States, 2016. MMWR Morb Mortal Wkly Rep 2020;69:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Rylaarsdam L, Guemez-Gamboa A.. Genetic causes and modifiers of autism spectrum disorder. Front Cell Neurosci 2019;13:385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Menold MM, Shao Y, Wolpert CM, et al. Association analysis of chromosome 15 gabaa receptor subunit genes in autistic disorder. J Neurogenet 2001;15:245–259. [DOI] [PubMed] [Google Scholar]
- 109. Dykens EM, Sutcliffe JS, Levitt P.. Autism and 15q11-q13 disorders: behavioral, genetic, and pathophysiological issues. Ment Retard Dev Disabil Res Rev 2004;10:284–291. [DOI] [PubMed] [Google Scholar]
- 110. Ma DQ, Whitehead PL, Menold MM, et al. Identification of significant association and gene-gene interaction of GABA receptor subunit genes in autism. Am J Hum Genet 2005;77:377–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Rivera C, Voipio J, Payne JA, et al. The K+/Cl- co-transporter KCC2 renders GABA hyperpolarizing during neuronal maturation. Nature 1999;397:251–255. [DOI] [PubMed] [Google Scholar]
- 112. Delpire E. Cation-chloride cotransporters in neuronal communication. NIPS 2000;15:309–312. [DOI] [PubMed] [Google Scholar]
- 113. Dzhala VI, Talos DM, Sdrulla DA, et al. NKCC1 transporter facilitates seizures in the developing brain. Nat Med 2005;11:1205–1213. [DOI] [PubMed] [Google Scholar]
- 114. Nardou R, Yamamoto S, Chazal G, et al. Neuronal chloride accumulation and excitatory GABA underlie aggravation of neonatal epileptiform activities by phenobarbital. Brain 2011;134:987–1002. [DOI] [PubMed] [Google Scholar]
- 115. Ben-Ari Y, Cherubini E, Corradetti R, Gaiarsa JL.. Giant synaptic potentials in immature rat CA3 hippocampal neurones. J Physiol (London) 1989;416:303–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Thompson SM, Deisz RA, Prince DA.. Outward chloride/cation co-transport in mammalian cortical neurons. Neurosci Lett 1988;89:49–54. [DOI] [PubMed] [Google Scholar]
- 117. Vanhatalo S, Palva JM, Andersson S, Rivera C, Voipio J, Kaila K.. Slow endogenous activity transients and developmental expression of K+-Cl- cotransporter 2 in the immature human cortex. Eur J Neurosci 2005;22:2799–2804. [DOI] [PubMed] [Google Scholar]
- 118. Tyzio R, Nardou R, Ferrari DC, et al. Oxytocin-mediated GABA inhibition during delivery attenuates autism pathogenesis in rodent offspring. Science 2014;343:675–679. [DOI] [PubMed] [Google Scholar]
- 119. Lemonnier E, Ben-Ari Y.. The diuretic bumetanide decreases autistic behaviour in five infants treated during 3 months with no side effects. Acta Paediatr 2010;99:1885–1888. [DOI] [PubMed] [Google Scholar]
- 120. Lemonnier E, Degrez C, Phelep M, et al. A randomised controlled trial of bumetanide in the treatment of autism in children. Transl Psychiatry 2012;2:e202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Lemonnier E, Villeneuve N, Sonie S, et al. Effects of bumetanide on neurobehavioral function in children and adolescents with autism spectrum disorders. Transl Psychiatry 2017;7:e1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. van Andel DM, Sprengers JJ, Oranje B, Scheepers FE, Jansen FE, Bruining H.. Effects of bumetanide on neurodevelopmental impairments in patients with tuberous sclerosis complex: an open-label pilot study. Mol Autism 2020;11:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Zhang L, Huang CC, Dai Y, et al. Symptom improvement in children with autism spectrum disorder following bumetanide administration is associated with decreased GABA/glutamate ratios. Transl Psychiatry 2020;10:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Kahle KT, Staley KJ.. The bumetanide-sensitive Na-K-2Cl cotransporter NKCC1 as a potential target of a novel mechanism-based treatment strategy for neonatal seizures. Neurosurg Focus 2008;25:E22. [DOI] [PubMed] [Google Scholar]
- 125. Ben-Ari Y, Damier P, Lemonnier E.. Failure of the Nemo trial: bumetanide is a promising agent to treat many brain disorders but not newborn seizures. Front Cell Neurosci 2016;10:90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Leis JA, Bekar LK, Walz W.. Potassium homeostasis in the ischemic brain. Glia 2005;50:407–416. [DOI] [PubMed] [Google Scholar]
- 127. Su G, Kintner DB, Sun D.. Contribution of Na(+)-K(+)-Cl(-) cotransporter to high-[K(+)](o)- induced swelling and EAA release in astrocytes. Am J Physiol Cell Physiol 2002;282:C1136–C1146. [DOI] [PubMed] [Google Scholar]
- 128. Su G, Kintner DB, Flagella M, Shull GE, Sun D.. Astrocytes from Na(+)-K(+)-Cl(-) cotransporter-null mice exhibit absence of swelling and decrease in EAA release. Am J Physiol Cell Physiol 2002;282:C1147–C1160. [DOI] [PubMed] [Google Scholar]
- 129. Lenart B, Kintner DB, Shull GE, Sun D.. Na-K-Cl cotransporter-mediated intracellular Na+ accumulation affects Ca2+ signaling in astrocytes in an in vitro ischemic model. J Neurosci 2004;24:9585–9587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Yan Y, Dempsey RY, Flemmer A, Forbush B, Sun D.. Inhibition of Na(+)-K(+)-Cl(-) cotransporter during focal cerebral ischemia decreases edema and neuronal damage. Brain Res 2003;961:22–31. [DOI] [PubMed] [Google Scholar]
- 131. Chen H, Luo J, Kintner DB, Shull GE, Sun D.. Na(+)-dependent chloride transporter (NKCC1)-null mice exhibit less gray and white matter damage after focal cerebral ischemia. J Cereb Blood Flow Metab 2005;25:54–66. [DOI] [PubMed] [Google Scholar]