

Abstract
Vibrio parahaemolyticus is a Gram-negative, halophilic bacterium and opportunistic pathogen of humans and shrimp. Investigating the mechanisms of V. parahaemolyticus infection and the multifarious virulence factors it employs requires procedures for bacterial culture, genetic manipulation, and analysis of virulence phenotypes. Detailed protocols for growth assessment, generation of mutants, and phenotype assessment are included in this article.
Introduction:
Vibrio parahaemolyticus is a Gram-negative, halophilic gammaproteobacterial native to marine and estuarine environments. To date 49 pandemic serotypes have been identified, and isolates have been acquired from coastal waters worldwide, including those around North and South America, India, Southeast Asia, and Western Europe (CDC, 2019; Daniels et al., 2000; Han, Tang, Ren, Zhu, & Han, 2016). Although humans are not its primary reservoir, V. parahaemolyticus is nonetheless responsible for over 45000 cases of acute gastroenteritis annually in the United States (CDC, 2019). The life cycle of V. parahaemolyticus is facilitated by a vast array of virulence factors, including two type VI secretion systems (T6SS), two type III secretion systems (T3SS), adhesins, quorum sensing, type II secretion system (T2SS) secreted hemolysins, and a newly reported GDSL lipase/esterase (Chimalapati et al., 2020; Chimalapati et al., 2018; de Souza Santos, Salomon, Li, Krachler, & Orth, 2015; Zhang & Orth, 2013). Both the first and second T3SS (T3SS1 and T3SS2, respectively) induce cytotoxicity in vivo by mediating an extracellular and intracellular infection respectively, however, only the latter is responsible for the enterotoxicity that drives acute gastroenteritis in human hosts (Kodama et al., 2015; K.-S. Park et al., 2004; K. S. Park et al., 2004; Zhang et al., 2012). V. parahaemolyticus strains carrying the plasmid encoding the PirVP toxin are also capable of inducing acute hepatopancreatic necrosis disease (AHPND) in shrimp, a disease responsible for the widespread devastation of aquaculture worldwide, including over $20 billion in economic losses in Asia alone (Li et al., 2017; Undercurrentnews, 2016).
Given the clinical and economic impacts of V. parahaemolyticus infection, it is critical to understand the virulence factors the bacteria employ to facilitate their life cycle in and outside of the human host. Studying such a broad array of virulence factors requires a battery of experimental approaches. The most fundamental of these include V. parahaemolyticus culture techniques and maintenance, as well as evaluations of motility and growth rates under various culturing conditions. Cumulatively these techniques allow for the long-term propagation and storage of V. parahaemolyticus strains for study. In addition, they provide valuable insight into the impact of various nutritional sources, mutations, or other such stimuli on major cell-signaling pathways that govern swimming and swarming behavior, as well as on rates of replication.
Genetic manipulation represents a second core approach to investigating V. parahaemolyticus, as generating genomic knockouts and both chromosomal and plasmid complementation are critical for fulfilling molecular Koch’s postulates when investigating the roles of specific genes or virulence factors in the V. parahaemolyticus life cycle (Falkow, 2004). Assessing the virulence phenotypes associated with the genetic manipulations and various infection models are critical for understanding the role of specific factors in V. parahaemolyticus pathogenesis. Microscopy of the fixed and live cell imaging permit direct visualization of V. parahaemolyticus and host tissue culture cells at discrete stages of infection and can reveal major changes to pathogenesis resulting from mutations, while phenotypic assays such as gentamicin protection assays and lactate dehydrogenase assays provide more quantitative measures of intracellular bacteria and cytotoxicity in the host, respectively. Western blots and mass spectrometry are also valuable techniques for evaluating the secretion of different virulence factors. The protocols are designed for analyzing various virulence factors of V. parahaemolyticus strains.
Importantly, when assessing V. parahaemolyticus phenotypes, virulence factors from one system can mask the activity of another system, especially in cell culture assays. Therefore, when assessing the phenotypes associated with the virulence factors, it is necessary to delete one system to uncover the activity of another system. For example, as the T3SS1 kills within 2-3 hours in vitro, when studying the T3SS2 associated phenotypes, T3SS1 system must be inactivated so that the cytotoxicity of the T3SS1 does not mask the activities of the T3SS2 which can extend up to 7-8h during in vitro infections. In vivo, this is not as much a problem because of the different ways these two T3SSs are spatially activated.
In this article, Basic Protocol 1: Assessment of growth of V. parahaemolyticus; Basic Protocol 2: Swimming / Swarming motility assay; Basic Protocol 3: Genetic Manipulation; Basic Protocol 4: Virulence Factor and phenotype analyses are described in detail. Basic protocol 1 describes the growth curve protocol during which optical density of the culture is recorded at regular time intervals to assess the growth. The protocols for swimming and swarming motility patterns of Vibrio parahaemolyticus on motility agar plates are detailed in the basic protocol 2. Genetic manipulations such as generation of deletion and complementation mutants are described in the next section, basic protocol 3. Finally, for the assessment of various virulence factors and phenotype assessment, protocols for secretion assay, invasion assay, cytotoxicity assay and immunofluorescence detection of Vibrio parahaemolyticus are detailed in basic protocol 4.
Strategic planning:
Growth Conditions:
V. parahaemolyticus will grow under aerobic conditions between 18°C and 37°C, but does so best at 30°C. Its preferred broth growth medium is MLB (Luria-Bertani broth with 30g/L NaCl). For plating, MMM agar (Minimal Marine Medium, see Reagents and Solutions section for recipe) is standard, and plates should be pre-warmed, as V. parahaemolyticus will not grow after prolonged exposure to temperatures around 4°C. A neutral pH is recommended for standard growth conditions, though V. parahaemolyticus can survive across a broad pH range. Typically, V. parahaemolyticus will double every 20 minutes in rich growth media, and an overnight culture at stationary phase can be expected to yield > 1.5 X 109 CFU/mL. It is not recommended that ampicillin be used as for selection in V. parahaemolyticus, as most strains possess an innate resistance to the antibiotic.
Storage and Retrieval:
For storage, V. parahaemolyticus overnight cultures may be mixed 1:3 v/v with sterile 50% glycerol and stored at −80°C; V. parahaemolyticus may be streaked onto room temperature plates or inoculated in liquid media directly when cultured from storage, and will grow at 30°C within 8-12 hours. Generally, it is encouraged that a single colony be obtained by streaking out an inoculum from a frozen glycerol stock for inoculation of liquid culture. If small volumes (>500μL) of bacterial culture are needed, a 0.5mL MLB culture of V. parahaemolyticus inoculated from a −80°C freezer stock will reach logarithmic phase within three hours when incubated shaking at 30°C.
Induction of the T3SS:
The T3SS1 can be induced by incubating V. parahaemolyticus growing in logarithmic phase shaking at 37°C for one hour in a low calcium medium, such as high glucose DMEM (Sigma Aldrich, D5796) (Broberg, Zhang, Gonzalez, Laskowski-Arce, & Orth, 2010; "National Enteric Disease Surveillance: COVIS Annual Summary, 2014," 2016). MLB with 10mM sodium oxalate and 10mM MgCl2 can be used as an alternative to high glucose DMEM. The T3SS2 is induced in the presence of 0.05% bile salts (w/v prepared in deionized water) added to a logarithmic phase culture and incubation at 37°C for an hour (Kodama et al., 2015).
Basic Protocol 1: Assessment of growth of V. parahaemolyticus
In this protocol, V. parahaemolyticus is inoculated into growth medium at a very low density and growth pattern is followed by recording optical density at 600 nm (OD 600) at regular time intervals. V. parahaemolyticus follows a typical bacterial growth curve with an initial lag phase, followed by an exponential growth phase and then a stationary phase.
Note: V. parahaemolyticus is a Bio Safety Level 2 (BSL-2) pathogen and should be handled using appropriate safety precautions.
Materials
Sterile culture tubes
Sterile 125 ml Erlenmeyer flasks
Sterile Eppendorf tubes
Sterile MLB culture medium
Temperature controlled incubator with shaking
Benchtop centrifuge
Spectrophotometer
Prepare 3mL cultures in triplicate of the V. parahaemolyticus strain by inoculating from a single colony or from a frozen glycerol stock in MLB medium and incubate overnight (O/N) at 30°C with shaking at 200 rpm.
Measure the OD600 of the O/N cultures and calculate the amount of culture needed to obtain a final OD600 of 0.05 for 25mL culture medium using the formula 25X0.05 / OD600 and transfer this volume aseptically into Eppendorf tubes.
Centrifuge tubes at 16,000g for 5 min and wash the pellets once with 500 of sterile PBS.
Under aseptic conditions resuspend these pellets in 100 μl of MLB medium and transfer to 25 mL of sterile culture medium in sterile 125 ml Erlenmeyer flasks.
Immediately swirl the flasks and withdraw 1 ml sample from each flask.
Immediately Incubate the flasks at 30°C and 200 rpm.
Measure the OD600. of the 1 ml sample withdrawn to obtain T0 reading.
Aseptically withdraw 1 ml samples at 1 h intervals and measure OD600. When the OD reaches 0.6 and higher, accurate OD readings can be obtained by using a 10-fold dilution of the culture to obtain OD measurement. Continue with measurements of OD up to 8 h of growth.
Growth curves can be obtained by plotting OD600 readings against time intervals.
An example of V. parahaemolyticus CAB2 strain growth curve can be seen in Fig 1, with an initial lag phase up to 1 h, followed by exponential growth phase between 1-4h and an early stationary phase from 4-6h and late stationary phase between 6-8h. When the OD readings are taken using a 10-fold dilution, the reading should be multiplied by a factor of 10 to account for the dilution.
Fig 1.
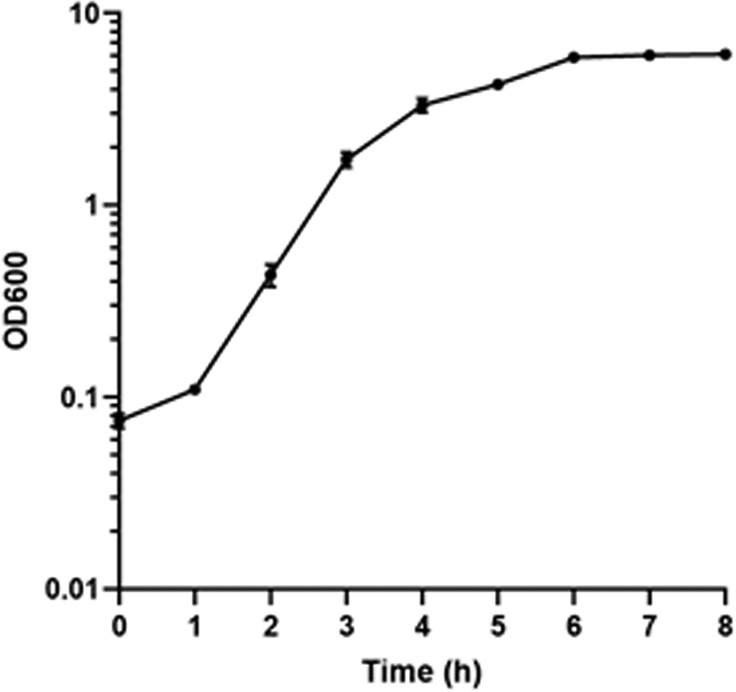
Growth curve for V. parahaemolyticus CAB2 strain in MLB medium (OD readings in triplicate).
Alternate Protocol 1: Assessment of growth of V. parahaemolyticus using a plate reader
The growth curves can also be obtained by using a plate reader for automated measurement.
Materials
Sterile culture tubes
Sterile Eppendorf tubes
Sterile culture medium (MLB /MMM)
Sterile 96 well flat bottom culture plate
Temperature controlled plate reader with provision for shaking
Benchtop centrifuge
Spectrophotometer
Prepare 3mL cultures in triplicate of the V. parahaemolyticus strain by inoculating from a single colony or from a frozen glycerol stock in MLB medium and incubate overnight (O/N) at 30°C with shaking at 200 rpm.
For each of the triplicate O/N cultures, measure the OD600 and calculate the amount of culture needed to obtain an OD600 of 0.05 for 1mL culture medium using the formula 1X0.05 / OD600 and transfer this volume aseptically into Eppendorf tubes.
Microcentrifuge tubes at 16,000g for 5 min and wash the pellets once with 500 of sterile PBS.
Under aseptic conditions, resuspend each of these pellets in 1 mL of MLB medium and aliquot 200 μl into 3 wells of a 96 well culture plate. Include 3 wells of 200 μl culture medium for blank measurement.
Immediately place the 96 well plate in the plate reader (set the plate reader temperature to 30°C with continuous shaking) and measure OD600.
Set the plate reader to measure OD600 at 1h time intervals up to 8h.
Growth curves can be obtained by plotting OD600 readings against time intervals.
Basic Protocol 2: Swimming / Swarming motility assay:
V. parahaemolyticus strains use their polar and lateral flagella for motility in swimming and swarming modes. The following protocols describe the assessment of motility phenotype.
Materials
Sterile culture tubes
Sterile Eppendorf tubes
Sterile swim and swarming agar plates (see ‘Reagents and Solution’ for recipes)
Sterile MLB
Benchtop centrifuge
Spectrophotometer
Temperature controlled incubator
Preparation of plates: Prepare swimming and swarming agar plates the day before the assay. (Follow the guidelines in the Reagents and Solutions to prepare these plates).
Use 150 mm plates so there is enough space to test multiple strains on the same plate.
Grow overnight cultures of bacteria in 3ml MLB as described in 1D.
Measure the OD600 of the O/N cultures and re-inoculate in 3ml MLB for a final OD of 0.2. Grow at 30°C and 200 RPM for 2 h to obtain the cultures in log phase.
Dry the swim /swarm plates for 2hrs at 37°C.
Spin 1 ml culture in a sterile 1.5 ml Eppendorf tubes and wash 3 times with sterile PBS and resuspend the pellet in 1 ml PBS and adjust OD600 to 0.1.
Spot inoculate 0.5 μl of each culture on the plates far enough from each other to swim / swarm. Up to 5 and 3 strains can be inoculated on 150 mm and 100mm plates respectively. Perform the spotting on 3 separate plates to obtain technical replicates.
Incubate plates upright at 30°C for 16h and measure the swimming / swarming diameter for each test strain.
An example of swimming and swarming motility of V. parahaemolyticus CAB2 strain can be seen in Fig 2. The diameter of the swimming and swarming zone may be determined using a ruler and used for comparison between strains.
Fig 2.

Examples of A) swimming and B) swarming patterns of V. parahaemolyticus CAB2 strain.
Basic Protocol 3: Genetic Manipulation:
Generation of V. parahaemolyticus mutants via genetic manipulation is described in the following protocols.
Generation of chromosomal Knockouts/ Knock-ins:
In this method an Ori6K/SacB suicide vector pDM4 is used to integrate required genomic change into the chromosome. The pDM4 vector containing the construct is introduced into V. parahaemolyticus via conjugation and, following recombination, the transconjugants are cured of the sacB containing pDM4 by counter selection on sucrose plates.
Materials
Standard molecular biology reagents for PCR and cloning
PCR Thermal Cycler
Sterile culture tubes
Temperature controlled incubator/shaker
Sterile PBS
LB – Chloramphenicol plates (LB Cm, refer to table 1 for concentration)
MMM – Chloramphenicol plates (MMM Cm, refer to table 1 for concentration)
MMM – Sucrose (15% w/v) plates
Heart Infusion No salts agar plates (HI NS- see ‘Reagents and Solution’ for recipe)
Table 1.
Generally used antibiotics and their concentrations for V. parahaemolyticus strainsa
| Antibiotic | Solvent | Stock concentration (mg/mL) |
Final concentration (μg/mL) |
|---|---|---|---|
| Spectinomycin | Deionized water | 50 | 50 |
| Chloramphenicol | 100% Ethanol | 25 | 25 |
| Kanamycin | Deionized water | 100 | 250 |
| Gentamicin | Deionized water | 50 | 100 |
All stock solutions should be sterilized by passing through 0.22 μM filters and stored at −20°C as aliquots.
For gene deletion, as a first step, design PCR primers and amplify 1 kb upstream (−1kb) and downstream (+1kb) amplicons of the target gene such that 3’ of the upstream and 5’ of the downstream amplicons have same restriction endonuclease sites. Refer to Figure 3 for a schematic representation of these amplicons. Make sure that the amplicons do not contain the chosen restriction endonuclease sites, online tools such as NEB cutter may be used to visualize the restriction sites present in the amplicon. Complete genomic sequence of V. parahaemolyticus chromosome 1 and 2 are available via accession numbers NC_004613.1 and BA000032.2 respectively. Many online tools are available for designing primers such NCBI Primer Blast or Primer3. Following the standard molecular cloning techniques clone the (−1kb and +1kb) amplicons into pDM4 vector and transform into S17 (λpir) E. coli strain. Similarly, for a gene knock-in 1kb upstream and downstream homology arms are first fused to the amplicon containing the intended knock-in fragment as explained above. This construct is cloned into pDM4 using standard cloning techniques and transformed into S17 E. coli.
Day 1. Grow O/N cultures of the V. parahaemolyticus parental strain and the S17 E. coli strain harboring the amplicon containing pDM4 plasmid in 3mL of MLB and LB with chloramphenicol respectively.
Day 2. Transfer 100μl each of the above O/N cultures aseptically into a single sterile Eppendorf tube and mix by gentle vortexing.
Spot inoculate 10μl of the above mixture into 10 spots on to a single, pre warmed HI NS plate and incubate the plate at 37°C O/N taking care that the spots do not mix with each other (leave at room temperature until the spots are adsorbed on to the agar).
Day 3. Aseptically add 5mL of sterile PBS onto the O/N grown HI NS plate and gently scrape the growth into PBS using a sterile 1 mL pipette tip, re-suspend and collect the bacterial suspension into a sterile 15 ml culture tube. Gently vortex to break clumps of bacterial growth.
Aseptically transfer 10, 20 and 50 μl of the bacterial suspension onto 3 pre warmed MMM chloramphenicol plates containing a pool of 100 μl of sterile PBS and spread using sterile glass beads. Incubate the plates at 30°C for up to 48h until 1-1.5mm colonies become apparent.
Day 5. Aseptically patch 20 individual colonies on to MMM Cm plates using sterile toothpicks to confirm the integration of chloramphenicol marker in the V. parahaemolyticus parental strain. Incubate the plates at 30°c for 24 to 48h until 1-1.5mm colonies become apparent.
Day 6. Pick up to 8 colonies and streak aseptically on pre warmed MMM sucrose plates (up to 4 colonies can be streaked on to a single plate in a pie wedge format) in order to obtain individual colonies and incubate at 30°c for up to 48h until 1-1.5mm colonies become visible.
Grow O/N cultures in MLB of 2-3 colonies from each pie wedge and confirm the genomic edit by performing PCR and/or sequencing. Prepare glycerol stocks and store at −80°C.
Fig 3.

Schematic representation of upstream (−1kb) and downstream (+1kb) amplicons for the deletion of gene of interest.
Alternate protocol 2: Natural transformation.
In this protocol V. parahaemolyticus strain expressing the competence factor tfoX is used to increase the natural competence for taking up extracellular DNA. In this method the target gene for deletion is replaced by a selectable antibiotic marker.
Materials
Standard molecular biology reagents for PCR and cloning
PCR Thermal Cycler
Water bath
Sterile culture tubes
Temperature controlled incubator/shaker
Sterile PBS
MMM – Chloramphenicol plates (MMM Cm, refer to table 1 for concentration)
MMM – Sucrose (15% w/v) plates
Design primers and amplify 3kb PCR fragments upstream and downstream of the target gene and generate a single construct containing the 3kb upstream and downstream amplicons fused to either side (upstream amplicon 5’ and downstream amplicon on 3’) of an antibiotic resistance marker such as chloramphenicol following standard molecular biology PCR techniques. This construct is referred to as the transforming DNA (tDNA).
Day 1. Grow O/N culture of V. parahaemolyticus expressing pMMBsacBtfoX plasmid in 3 mL of MLB+ kanamycin. pMMBsacBtfoX is a bacterial expression vector that can be induced with Isopropyl-beta-D-thiogalactoside (IPTG) and it contains a sacB gene for curing the plasmid after homologous recombination and integration of the tDNA into the genome. tfoX is a Vibrio competence factor that is used to increase the natural competence of Vibrio strains for taking up extracellular DNA.
Day 2. For each transformation, transfer 3.5 μL of the overnight culture into an Eppendorf tube and dilute with 350 μL of 2X Instant Ocean (2XIO, see appendix for recipe) +100 μM IPTG (no antibiotic needed). Invert gently to mix. Include a “no DNA” control reaction.
Add tDNA (500 ng to 1μg) to each reaction and invert gently to mix. Incubate these reactions statically at 30°C for 4-6 hours.
Add 1 ml MLB to each reaction and incubate at 30°C with shaking for 2 hours.
Using glass beads spread plate all the reaction mixture on MMM antibiotic plates (MMM Cm plates if using chloramphenicol as selectable marker) from the tube containing the tDNA construct. Divide between 2-3 plates if necessary. For the control reaction 1 −2 plates are sufficient. Incubate the plates inverted at 30°C up to 2-3 days until colonies are visible.
Streak up to 10 colonies on MMM agar with 15% sucrose and antibiotic (MMM sucrose + Cm plates if using chloramphenicol as selectable marker) and incubate at 30°C up to 48 h or until isolated colonies appear.
Patch about 50 colonies on MMM plates with and without kanamycin (similar to replica plating, each colony will need to be patched onto MMM Kan plates first and then onto MMM plates). For each patched colony, select the colonies that grow on MMM but fail to grow on MMM Kan. These colonies denote V. parahaemolyticus strains that are cured of the pMMBSacBtfoX plasmid but have their target gene replaced by the antibiotic resistance marker.
Grow O/N cultures of 10 such colonies and confirm the gene deletion by performing PCR and/or sequencing. Prepare glycerol stocks and store at −80°C.
Basic Protocol 4: Secretion Assay and Sample Preparation for Mass Spectrometry Analysis
V. parahaemolyticus secretes either T3SS1 or T3SS2 mediated effectors depending on the type of induction. In this protocol, preparation of T3SS2 effectors secreted by V. parahaemolyticus CAB2 strain upon bile salts induction for mass spectrometry is described. T3SS1 effectors can be induced by growing V. parahaemolyticus in low calcium medium such as high glucose DMEM and incubation at 37°C. Wear gloves for handling of all reagents and materials as collagen contamination can mask signals in final sample.
Materials:
5mL overnight culture of V. parahaemolyticus CAB2 strain grown in MLB
Growth media – MLB
Sterile 50 mL conical tubes
Sterile Eppendorf tubes
Ice bucket
5% w/v Bile salts (Bile Bovine, Sigma Aldrich, B3883) solution
150mg/mL Sodium deoxycholate (DOC)
100% Trichloroacetic acid TCA (use within a month of preparation, as this reagent is labile)
100% Acetone
70% Methanol
From overnight cultures inoculate 50mL of growth media containing 0.05% bile salts to an OD600 = 0.2.
Incubate the cultures shaking at 37°C for 3–5h.
Measure the OD600 of all cultures. The final OD600 should be between 4 and 5, as all cultures should have reached stationary phase. Transfer cultures to a 50mL conical tube, normalizing the volumes of each culture so that an equivalent number of bacteria are added to each tube and all 50mL of the culture with the lowest OD600 is transferred.
Pellet for 15 minutes at 4°C at 3200g.
Filter the supernatants through a 0.22μm filter into a fresh 50mL conical tube.
Add 150mg/mL DOC to each supernatant to a final concentration of 150μg/mL; vortex and incubate on ice 15 minutes.
Add TCA to a final concentration of 8% (v/v) to each sample; vortex and incubate at 4°C overnight on ice in a cold room if possible.
Resuspend the precipitated protein and transfer the suspensions to Nalgene centrifuge tubes that have been rinsed with MeOH and dried to remove collagen contamination
Pellet the precipitated protein 1h at 4°C, 27000g.
Discard the supernatant; apply 4mL −20°C 100% acetone and spin 40 minutes at 4°C, 27000g to rinse; discard the supernatant and repeat.
Discard the supernatant and air dry each pellet 10 minutes.
Resuspend each pellet in 1mL 10mM Tris pH 8 and transfer to a 1.5mL Eppendorf tube.
Add 150mg/mL DOC to each supernatant to a final concentration of 150μg/mL; vortex and incubate on ice 15 minutes.
Add TCA to a final concentration of 8% (v/v) to each sample; vortex and incubate at 4°C 1-5 h.
Pellet the precipitated protein 1hr at 4°C, 21000g in a microfuge.
Discard the supernatant and apply 400 μL −20°C 100% acetone and spin 20 minutes at 4°C, 21000g in a microfuge to rinse; discard the supernatant and repeat.
Discard the supernatant and air dry each pellet 10 minutes.
Suspend each pellet in 20 μL 5x Sodium Dodecyl Sulfate (SDS) sample buffer (more may be used if needed, though the volume must be kept consistent for each sample). Ensure that the pH of each sample does not exceed the buffer capacity of the sample buffer by adding 10M NaOH in 0.2μL increments as needed to keep sample neutral (blue).
Boil samples for 10 minutes.
Apply each sample to a MiniProtean Any kD precast SDS PAGE gel (Bio RAD, 4569033); run at 80V for 20 minutes or until the entire sample has permeated the gel
Excise each sample (~ 1 cm2) from the gel using the dye front as a guide and submit for mass spectrometry protein identification analysis.
Basic Protocol 5: Invasion assay (Gentamicin protection assay)
This protocol describes the HeLa cell invasion by V. parahaemolyticus strains and assessment of the invasion phenotype. When assessing V. parahaemolyticus invasion, the T3SS1 system must be genetically inactivated so that the cytotoxicity of the T3SS1 does not mask the invasion phenotype of the T3SS2.
Materials:
Tissue culture biosafety hood
24-well tissue culture plates
Sterile culture tubes
Spectrophotometer
Temperature controlled incubator / Shaker
CO2 incubator for tissue culture (37°C, 5% CO2)
HeLa cells
MLB
5% Bile salts solution (Bile Bovine (Sigma Aldrich, B3883) in in deionized water, w/v and filter sterilized)
DMEM complete medium (see ‘Reagents and Solution’ for recipe)
DMEM infection medium (see ‘Reagents and Solution’ for recipe)
Un-supplemented DMEM (see ‘Reagents and Solution’ for recipe)
Gentamicin (100mg/mL stock solution)
Sterile 0.5% Triton-X-100 prepared in PBS
Sterile PBS
Day 1. Using the tissue culture biosafety hood, plate HeLa cells in complete DMEM medium at a concentration of 7x104 cell /mL (1 mL/well, in triplicate for each V. parahaemolyticus strain) in 24 well tissue culture plates. Plate one 24 well for each time point.
Grow O/N cultures of V. parahaemolyticus strains in MLB at 30°C and 200 RPM.
Day 2. Measure the OD600 of diluted O/N grown cultures and dilute in 5 mL MLB containing 0.05% of bile salts solution to an initial OD600 of 0.3 and incubate the cultures at 37°C and 200 RPM for 1.5h to induce T3SS2.
Warm up the MMM plates to room temperature.
Pre warm the infection medium and plain DMEM at 37°C.
- Measure OD600 of the cultures (dilute if necessary for accuracy) after 1.5h growth at 37°C. 1 mL of culture with an OD 600 of 1 is equivalent to 5x108 bacteria. Calculate the volume of bacteria needed to infect HeLa cells for an MOI of 10, following the equation:
Prepare adequate volume of bacterial infection medium for each strain (based on time course of infection) by adding the required volume of bacteria to prewarmed infection medium as calculated above.
Gently wash HeLa cells plated in 24 wells with un-supplemented DMEM twice and start the infection process by adding 1 mL of bacterial infection medium to HeLa cells plated in each well. There will be 3 replicates in total for each strain for every timepoint.
Synchronize the infection by centrifuging the 24 well plates at 1000 RPM (~200x g) for 5 minutes and incubate the plates at 37°C in TC incubator for 2 h.
Wash HeLa cells twice with un-supplemented DMEM and add 1 mL of infection medium containing 100 μg/ml of gentamicin to each well.
Return the plates to the TC incubator and further incubate each plate for the required length of invasion process, typically for 1, 3, 5 and 7h.
At the end of each time point, wash wells with sterile PBS twice (1mL/well).
Lyse the cells by adding 1 mL/well of sterile 0.5% TX-100 prepared in PBS and incubate for 10 minutes with shaking at room temperature.
Pipette the cell lysates vigorously 10 times using a 1 mL pipette tip to break any clumps of cells and to obtain a uniform cell lysate.
From each lysate sample, prepare 10-fold serial dilutions up to 10−3 using sterile PBS.
Spot inoculate 10 μl of each dilution and undiluted lysate onto room temperature MMM plates. Leave at room temperature undisturbed until the spotted lysates are adsorbed onto the MMM plates and incubate them at 30°C O/N or until colonies become visible.
Count the colonies and multiply by 100 (include the dilution factor if counting colonies from diluted samples) to determine CFU/mL. Invasion phenotype can be assessed by plotting CFU/mL at various time points of invasion.
An example of invasion phenotype of V parahaemolyticus CAB2 strain during gentamicin protection assay has been shown in Fig 4. The number of intracellular bacteria during HeLa cell invasion assay increases with time up to 5 h which then reduces at 7h as the bacteria egress from the invaded HeLa cell and encounter the extracellular gentamicin.
Fig 4.
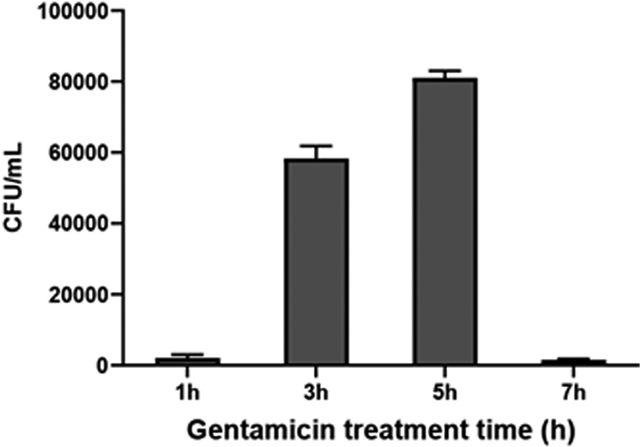
Invasion phenotype of V. parahaemolyticus CAB2 strain as assessed by gentamicin protection assay.
Basic Protocol 6: Immunofluorescence detection of intracellular V. parahaemolyticus
In this protocol, HeLa cells are infected with V. parahaemolyticus strains that have been induced with bile (an activator of the T3SS2) and are treated with cell impermeable gentamicin antibiotic to assess the behavior of intracellular bacteria. This protocol can be modified to evaluate other cell types, organelles, and host cell proteins of interest. When assessing V. parahaemolyticus invasion, the T3SS1 system must be inactivated so that the cytotoxicity of the T3SS1 does not mask the invasion of the T3SS2. It is recommended that any V. parahaemolyticus strain used constitutively express GFP to visualize intracellular bacteria. Alternatively, an anti-V. parahaemolyticus antibody may also be used to detect V. parahaemolyticus by immunofluorescence.
Materials:
Tissue culture biosafety hood
Glass coverslips
6-well tissue culture plate
DMEM complete medium (see ‘Reagents and Solution’ for recipe)
DMEM infection medium (see ‘Reagents and Solution’ for recipe)
Un-supplemented DMEM (see ‘Reagents and Solution’ for recipe) HeLa cells
CO2 incubator for tissue culture (37°C, 5% CO2)
GFP expressing V. parahaemolyticus glycerol stocks
Luria-Bertani (LB) broth, supplemented with 3% NaCl (MLB)
5% Bile salts solution (Bile Bovine (Sigma Aldrich, B3883) in in deionized water, w/v and filter sterilized)
30°C incubator
37°C incubator
Spectrophotometer
Gentamicin (100mg/mL stock solution)
1x phosphate buffered saline (PBS)
Fixative: 4% paraformaldehyde (PFA) in 1x PBS (v/v)
Permeabilization solution: 0.5% saponin in 1x PBS (w/v)
Blocking solution: 1% bovine serum albumin (BSA) and 0.1% saponin in 1x PBS (w/v)
Antibody dilution solution: 0.5% BSA and 0.1% saponin in 1x PBS (w/v)
Wash solution: 0.1% saponin with 1x PBS (w/v)
Hoechst 33342
ProLong Gold Antifade Mountant
Glass slides
Zeiss LSM confocal microscope
Day 1. UV sterilize glass coverslips and place into sterile 6-well tissue culture plate(s) in the tissue culture biosafety hood.
For each well/coverslip, seed 1x105 HeLa cells in 2mL of complete DMEM. Incubate HeLa cell plate in CO2 incubator at 37 °C with 5% CO2.
Inoculate 5 mL of MLB with V. parahaemolyticus strains from glycerol stocks. Add spectinomycin at 50μg/mL to maintain expression of GFP and add other antibiotics if necessary. Incubate overnight at 30 °C and 200 rpm. If GFP expressing V. parahaemolyticus strains are not available, a commercially available anti V. parahaemolyticus O3 antibody (Abcam 78751) can be used to visualize the bacteria as detailed later in the protocol.
Day 2. Measure the OD600 of the overnight cultures.
Start fresh cultures in 5 mL MLB for each strain from the overnight Vibrio cultures for an OD600 of 0.3. Add spectinomycin at 50μg/mL to maintain expression of GFP and add other antibiotics if necessary.
To induce expression of the T3SS2, add 50 μL of 5% bile salt solution for a final concentration of 0.05%. Then incubate the cultures at 37 °C for 90 minutes at 200rpm.
After the incubation, check the OD600 of each culture. 1 mL of culture with an OD 600 of 1 is equivalent to 5x108 bacteria.
- Using the OD600, calculate the volume of each bacterial sample needed for an approximate MOI of 10 according to the following equation:
Create the infection solution by adding calculated volume of bacterial cultures to pre-warmed DMEM infection media.
Wash HeLa cells 2x with un-supplemented DMEM.
Infect cells by adding 1mL of the infection solution to each well of HeLa cells.
Centrifuge the 6-well plate(s) at 1000 rpm (~200x g) for 5 min to synchronize the infection by forcing contact of the bacteria with the HeLa cells.
Incubate plate(s) in the CO2 incubator at 37 °C with 5% CO2 for 2 hours.
Wash cells 2x with un-supplemented DMEM.
Make a solution by adding gentamicin (100 μg/mL final concentration) to fresh DMEM infection media. Add 1 mL into each well and incubate the plate at 37 °C with 5% CO2 for each time point (usually between 1-7h).
At the end of each time point, remove the 6-well plate from the incubator and protect from light for following steps.
Wash the coverslips with 1mL of 1x PBS and then fix the cells on the coverslip by adding 1mL of 1x PBS with 4% PFA for 10 minutes at room temperature.
Wash the coverslips three times with 1mL of 1x PBS for each well. If required 6 well plates containing coverslips may be stored at 4°C covered with aluminum foil up to 24 h.
If using anti V. parahaemolyticus antibody to detect extracellular bacteria, incubate the coverslips with diluted antibody (1: 500 in 1x PBS) and incubate at room temperature for 1 hour.
Wash the coverslips three times for 5 minutes with 1x PBS.
Incubate the coverslips with a fluorescent secondary antibody diluted in 1x PBS (see step 37 for fluorescent secondary antibody options) and incubate at room temperature for 1 hour.
Wash the coverslips three times for 5 minutes with 1x PBS.
Permeabilize the cells on the coverslips with 1mL of 1x PBS with 0.5% saponin for each well and incubate at room temperature for 10 minutes.
Wash the coverslips one time with 1mL of 1x PBS for each well.
If desiring to image other cellular proteins and markers, then block the cells on the coverslips using 1x PBS with 1% bovine serum albumin (BSA) and 0.1% saponin for 30 min at room temperature. If only assessing the bacterium, then skip to step 30.
Incubate the coverslips with primary antibody diluted in 1x PBS containing 0.5% BSA and 0.1% saponin at an appropriate dilution and incubate at room temperature for 1 hour.
Wash the coverslips three times for 5 minutes with 1x PBS containing 0.1% saponin.
Incubate the coverslips with secondary antibody diluted in 1x PBS containing 0.5% BSA and 0.1% saponin at an appropriate dilution and incubate at room temperature for 1 hour.
Wash the coverslips three times for 5 minutes with 1x PBS containing 0.1% saponin.
To visualize nuclei, incubate coverslips with 1x PBS containing 1μg/mL Hoechst 33342 for 10 minutes at room temperature.
To visualize actin, incubate coverslips with 1x PBS containing Rhodamine Phalloidin Reagent (Abcam ab235138) to a final concentration of 1:1000.
Wash the coverslips two times with 1x PBS.
Mount the coverslips on to glass slides with ProLong Gold Antifade Mountant.
Cure the slides overnight.
Day 3. Seal the edges of the coverslips with clear nail polish.
Image on Zeiss LSM confocal microscope.
Some common antibodies and dyes and their excitation / emission wavelength parameters used for imaging intracellular V. parahaemolyticus are described below.
Phalloidin binds F-actin and can be used to visualize the effects of the bacterium on the host actin cytoskeleton. This dye can be used to stain the coverslip when incubating with the Hoechst stain. 10 minutes at room temperature is sufficient for staining.
- Fluorescent phalloidin is commercially available and examples are listed below:
- Alexa Fluor™ 488 phalloidin (green, excitation: 495nm, emission: 518nm)
- Rhodamine-phalloidin (red, excitation: 540nm, emission: 565nm)
- Alexa Fluor™ 647 phalloidin (red, excitation: 650nm, emission: 668nm)
Hoechst 33342 is cell permeable and can be used to stain the nucleus of host cells as well as bacterial DNA. (Blue, excitation: 361nm, emission: 497nm)
Cellular compartments can be visualized by staining with primary antibodies following secondary antibodies conjugated to different fluorophores (e.g. Alexa Fluor 488 - green, Alexa Fluor 555 - red, and Alexa Fluor 680 – far red). Examples of common markers for cellular compartments are EEA-1 (early endosome), Lamp-1 (late endosome/lysosome), KDEL and Calnexin (ER), and COX-4 (mitochondria).
Examples of confocal micrographs of HeLa cells invaded by V. parahaemolyticus GFP expressing CAB2 strain are given in Fig 5. A, B, C and D denote HeLa cells containing intra cellular bacteria at 1, 3, 5 and 7h post gentamicin treatment respectively during a gentamicin protection assay. HeLa cell actin was stained with rhodamine phalloidin (red) and DNA was stained with Hoechst (blue). At 1 h, post gentamicin treatment V. parahaemolyticus are typically restricted to endosomes, hence their appearance is in small puncta. At 3h post gentamicin treatment, the bacteria that escaped from the endosome can be visualized in the cytoplasm where they replicate. This process typically continues up to 5 to 6h post gentamicin treatment. At 7h post gentamicin treatment, most bacteria have egressed from the host cell into the extracellular medium where they are killed by extracellular gentamicin.
Fig 5.

Confocal micrographs of HeLa cells invaded by V. parahaemolyticus GFP expressing CAB2 strain at A)1, B)3, C)5 and D)7h post gentamicin treatment during a gentamicin protection assay.
HeLa cell actin was stained with rhodamine phalloidin (red) and DNA was stained with Hoechst (blue). Scale bars = 10 μm.
Basic Protocol 7: Cytotoxicity Assay for the T3SS2
Lactate dehydrogenase (LDH) is an abundant metabolic enzyme found in nearly all living cells. When host cells undergo lysis and the plasma membrane is damaged, this enzyme is released into the extracellular environment with other intracellular contents. The amount of LDH released from a host cell population correlates with the proportion of lysed cells and thus serves as an indirect assessment of cytotoxicity. When induced, the type III secretion system 2 (T3SS2) of V. parahaemolyticus and its effectors cause nearly 100% HeLa cell lysis by 5-6 hours. By contrast, when the T3SS1 system is induced cytolysis can be detected in as little as two hours. Thus, when assessing V. parahaemolyticus invasion, the T3SS1 system must be genetically inactivated so that the cytotoxicity of the T3SS1 does not mask the invasion of the T3SS2. This protocol can be modified to test other cell types and other bacterial virulence factors.
Materials:
Tissue culture biosafety hood
DMEM complete medium (see ‘Reagents and Solution’ for recipe)
DMEM High glucose without phenol red (Sigma Aldrich)
HeLa cells
CO2 incubator for tissue culture (37 °C, 5% CO2)
Vibrio parahaemolyticus glycerol stocks
Luria-Bertani (LB) broth, supplemented with 3% NaCl (MLB)
5% Bile salts solution (Bile Bovine (Sigma Aldrich B3883) in in deionized water, w/v and filter sterilized)
30°C incubator
37°C incubator
24-well tissue culture plates
96-well clear bottom plates
OD600 Spectrophotometer
Multichannel pipet
LDH Cytotoxicity Detection Kit (Takara Bio)
10% Triton X-100 solution
Day 1. In the tissue culture biosafety hood, seed 7x104 HeLa cells in 1 mL complete DMEM for each well of the 24-well plate. For each sample at each time point, cells should be plated in triplicate. In addition to experimental strains, include a Vibrio parahaemolyticus strain with intact T3SS2 as positive control and a Vibrio parahaemolyticus strain with inactivated T3SS2 as negative control.
Separate plates should be used for each time point and each plate should include 3 extra wells of HeLa cells (total lysis control).
Inoculate 5 mL of MLB with V. parahaemolyticus strains from glycerol stocks. Add antibiotics if necessary. Incubate overnight at 30 °C and 200rpm.
Day 2. Measure the OD600 of the overnight cultures.
Start fresh cultures in 5 mL MLB for each strain and from the overnight Vibrio cultures for an OD600 of 0.3. Add antibiotics if necessary.
To induce expression of the T3SS2, add 50 μL of 5% bile salt solution for a final concentration of 0.05%. Then incubate the cultures at 37 °C for 90 minutes at 200rpm.
After the incubation, check the OD600 of each culture. 1 mL of culture with an OD 600 of 1 is equivalent to 5x108 bacteria.
- Using the OD600, calculate the volume of each bacterial sample needed for an approximate MOI of 10. For 1 mL of cells:
1mL of media is required for each well of HeLa cells. Make the infection solution by adding the calculated volume of bacteria to pre-warmed DMEM without phenol red.
Wash HeLa cells twice with DMEM without phenol red to remove residual phenol red and antibiotics.
To each well of HeLa cells, add 1 mL/well of the bacteria infection solution. For the lysis control wells, add 1mL/well of the DMEM without phenol red (no bacteria). Add 1 mL/well of DMEM without phenol red to three empty wells as blanks.
Centrifuge the plate(s) at 1000 rpm (~200x g) for 5 minutes to synchronize the infection by forcing contact of the bacteria with the HeLa cells.
Incubate plate(s) in the CO2 incubator at 37 °C with 5% CO2.
10 minutes prior to each time point, remove 500 μL from the total lysis control wells and add 500 μL of DMEM without phenol red + 2% Triton X-100.
At the end of each time point, remove 200 μL of media in triplicate from each well and transfer to a 96-well plate. Spin this plate down for 5 min at 1000 rpm to remove any debris.
Carefully take out 100 μL of supernatant and transfer to a new 96-well plate. Place the new plate at 4 °C.
LDH can be measured by kits from many manufacturers. Here, we use the LDH Cytotoxicity Detection Kit from Takara Bio.
For 50 reactions/wells, prepare the working solution by mixing 125 μL of the catalyst solution + 5.625 mL of the dye solution. Scale up as necessary.
To each well of the 96 well plate(s), add 100 μL of the working solution with a multichannel pipettor.
If desired, add 50 μL of 1N HCl to stop the reaction (final concentration = 0.2N HCl). If not, the reaction product can be measured without a stop solution.
Measure the absorbance at 490nm using a 96 well plate reader.
- Determine the average absorbance of technical replicates and then calculate the percent of lysis by the equation below:
Results can be plotted as a percentage of cytotoxicity relative to the total lysis control.
Representative results of LDH assay are presented in Fig 5, from an infection of HeLa cells with Vibrio parahaemolyticus strain CAB2 (T3SS2 only). LDH levels were measured at 1.5, 3, 4.5, and 6 hours post infection with a MOI of 10. The percent cytotoxicity is calculated by comparing the LDH levels in the sample wells to the total lysis control. Cytotoxicity is used as a proxy for infection-mediated cell death, and is measured based levels LDH, a stable enzyme released into the media upon disruption of tissue culture cells’ plasma membranes. LDH reduces NAD+ to NADH/H+ in the process of converting lactic acid to pyruvate. Cell-free supernatant from the infection at each time point is mixed with a lactic acid reaction mixture provided by the LDH assay kit for a set period of time, and the resulting accumulated NADH/H+ is then allowed to react with a colorimetric dye that turns red when reduced by NADH/H+. More cytotoxicity results in increased release of LDH into the media, which when applied to the kit produces more pyruvate and NADH/H+, in turn reacting with more of the dye to produce a stronger absorbance measurement in the red spectrum. Absorbance levels recorded experimentally are normalized to an LDH standard to generate the percent cytotoxicity shown on the Y-axis. The cytotoxicity level increases during the infection of HeLa cells with Vibrio parahaemolyticus CAB2 strain and reaches maximal levels between 5 and 6h of infection as depicted in Fig 6.
Fig 6.
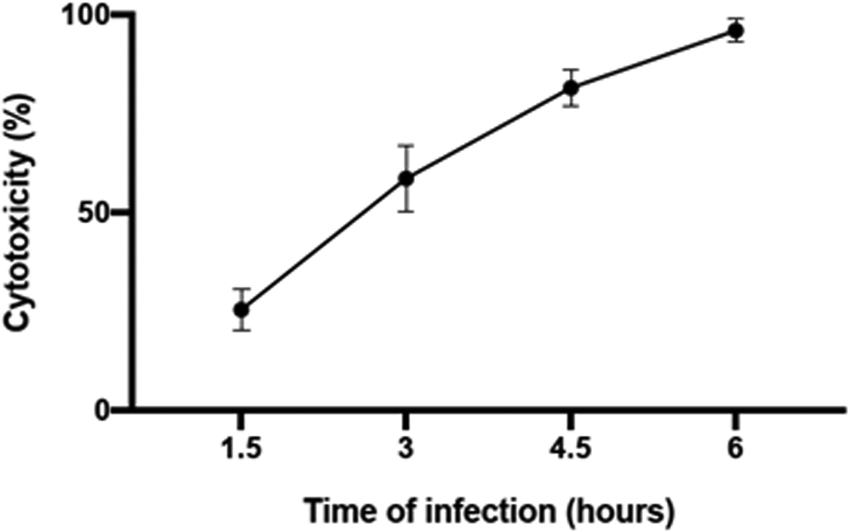
Representative LDH assay results from infection with Vibrio parahaemolyticus strain CAB2 (T3SS2 only). LDH levels were measured at 1.5, 3, 4.5, and 6 hours post infection with a MOI of 10. The percent cytotoxicity is calculated by comparing the LDH levels in the sample wells to the total lysis control.
Reagents and Solutions:
MLB (LB broth with 3% NaCl (w/v)): Tryptone 10g, Yeast extract 5g, NaCl 30g / liter of Milli-Q deionized (Mq) H2O. Adjust pH to 7.4 with NaOH and sterilize by autoclaving at 15 psi for 20 min.
Swimming agar plates: Tryptone 10g, NaCl 20g, Agar 3.25g / liter of MqH2O, pH 7.4. Autoclave at 15 psi for 20 min. Do not invert plates, store them upright.
Swarming agar plates: HI (Heart infusion broth) 25g, NaCl 20g, Agar 15g / liter of MqH2O, pH 7.4. Autoclave at 15 psi for 20 min. These plates can be inverted.
MMM agar: NaCl 20g, Agar 15g / 925 mL of Mq H2O, Autoclave at 15 psi for 20 min and cool to 55°C. Add 50 mL sterile 20X Salts, 20 mL sterile 20% galactose, 5 mL sterile 1 M MgSO4. Mix well and pour plates.
20X Salts: K2SO4 20g, K2HPO4 270g, KH2PO4 94g, NH4Cl 22g / Liter of Mq H2O. Autoclave at 15 psi for 20 min.
2XIO (2X Instant Ocean): Instant Ocean (Instant Ocean Sea Salts for aquarium) 28g / Liter of Mq H2O. Autoclave at 15 psi for 20 min.
SDS Sample Buffer: 250mM Tris pH6.8, 50% glycerol v/v, 5% SDS, 0.25% Bromophenol blue m/v, 1:1000 2-mercaptoethanol.
DMEM complete medium: High glucose DMEM with phenol red (Sigma Aldrich, D5796) Add the following supplements to the final concentrations as listed. Fetal bovine serum (FBS): 10% FBS, Sodium pyruvate (100mM, Sigma Aldrich S8636): 1mM, Penicillin/streptomycin/l-glutamine (Sigma Aldrich G1146): 1%.
DMEM infection medium: High glucose DMEM with phenol red (Sigma Aldrich, D5796) Add the following supplements to the final concentrations as listed. Fetal bovine serum (FBS): 10% FBS, Sodium pyruvate (100mM, Sigma Aldrich S8636): 1mM.
Un-supplemented DMEM: High glucose DMEM with phenol red (Sigma Aldrich, D5796), without any additives.
5% Bile salts solution: Dissolve 0.5 g of Bile Bovine (Sigma Aldrich B3883) in 10 mL deionized water, and filter sterilize by passing through 0.22 μm filters. This solution can be stored at room temperature and should be used within one month.
Commentary
Background information
V. parahaemolyticus was first isolated from a food poisoning outbreak in Japan in 1950 and was originally categorized as Pasteurella parahaemolytica, which was later re-classified into the genus Vibrio based on advances in taxonomy and after its halophilic nature was discovered in 1955 (Shinoda, 2011). Prior to the emergence of the pandemic strain O3:K6 serotype in 1996 from India, V. parahaemolyticus was generally confined to localized outbreaks and linked to various serotypes such as O2:K3, O3:K6 and O4:K8 (Okuda et al., 1997; Wong et al., 2000). The alphabets O and K refer to the somatic and capsular antigens of V. parahaemolyticus respectively. The pandemic O3:K6 strain quickly spread globally and lead to several outbreaks throughout the world. Recently 22 new serotypes (e.g. O4:K68, O1:K25, O1:KUT (untypable)) have been discovered with genotyping profiles identical to O3:K6 and referred to as serovariants of the pandemic strain O3:K6. Based on MLST (multilocus sequence typing), a recent study reported a total of 49 pandemic serotypes that grouped into 14 sequence types (Han et al., 2016).
Multiple laboratory strains have been developed to study the virulence factors of V. parahaemolyticus. The clinical isolate RIMD 2210633 was collected from a patient from Osaka, Japan in 1996 and is the parent strain of many such laboratory mutants, most notably the CAB and POR strains (Makino et al., 2003; Ono, Park, Ueta, Iida, & Honda, 2006). POR1 features a deletion of the two hemolysin-encoding genes tdhA and tdhS and is typically utilized as a wild type control in T3SS studies because it permits study of the cytotoxic effects of both T3SS without interference from the cytotoxic properties of the hemolysins. From this strain were derived additional POR strains, POR2, which lacks a functional T3SS1 due to deletion of export apparatus vcrD1, POR3, which lacks a functional T3SS2 due to deletion of export apparatus gene vcrD2, and POR4, which contains both mutations and no functional T3SS (De Nisco et al., 2017; K. S. Park et al., 2004). Also derived from POR1 were the CAB strains – CAB2, which lacks a functional T3SS1 due to a deletion of the transcription regulatory gene exsA, CAB3, which lacks a functional T3SS2 due to a deletion of the transcription regulatory gene vtrA, and CAB4, which lacks any functional T3SS due to deletions in both aforementioned transcriptional regulatory genes (Calder et al., 2014; Zhang et al., 2012).
Critical parameters and trouble shooting
Genetics:
If no or slow growth is observed in liquid cultures inoculated from freezer stocks, it is possible that too much bacteria has been used for inoculation in limited volume; it is recommended that only very small volume of glycerol stocks be used for inoculation for optimal growth. Moreover, if no colonies appear after transconjugation matings after 72 hours of incubation, adjusting the ratio of bacteria from 1:1 to 1:2 Vibrio to E. coli donor/helper strains may rectify the issue.
For natural transformations, issues associated with the ligation of fragments via PCR may be rectified by shortening the homologous amplicons from 3kb up and downstream of the target gene to 2kb, resulting in a shorter overall nucleotide strand that is easier to amplify (note however that shortening the regions of homology may impact the efficiency of recombination). Gibson assembly may also be used to increase the efficiency of ligating PCR amplicons to the antibiotic marker gene.
Proteomics:
Whenever analyzing secretion profiles for mass spectrometry, it is critical that gloves be worn at all times and that all tubes be washed with 70% methanol to minimize collagen contamination, as this can significantly decrease the sensitivity of the assay. Use of a positive pressure hood for sample preparation is recommended when possible.
Cell biology:
For visualization of V. parahaemolyticus via confocal microscopy, it is important to minimize exposure of slides to light even before staining if the bacteria express GFP, as the GFP signal is often weak in comparison to that of many phalloidin stains, and photobleaching will further compromise the resolution of images taken. If you use a fluorescent reporter to label the bacteria (e.g. GFP), be aware that using methanol for tissue fixation may result in loss of signal and may require an anti-GFP antibody for visualization. If there is difficulty seeing the bacteria through the microscope lens due to dim signal from the antibody staining/fluorescent reporter, it is possible to find them with the Hoechst stain with the blue channel (Hoechst will stain the bacterial DNA). Different antibody markers may necessitate testing different types of fixation and permeabilization methods for the best staining. A list of some more common problems encountered during these assays and possible remedies are presented in Table 2.
Table 2.
List of commonly encountered problems and possible remedies
| Problem | Possible reason | Possible remedy |
|---|---|---|
| Low or no growth in overnight Vibrio cultures and during regular growth cycle | High inoculum from the frozen glycerol stocks; Contaminated culture tubes; Low ratio of growth flask to culture | Inoculate very low volume from frozen glycerol stocks, make sure to use clean and sterile culture tubes; for growth curves, make sure that the ratio of culture / vessel volume is at least 1:5. |
| Contamination in motility agar plates | Moisture accumulation | Use freshly prepared plates, as the swimming plates cannot be stored inverted causing moisture-based contamination issues. Make sure to dry the plates at 37°C prior to inoculation. |
| No colonies with the intended genetic change (gene deletion or complementation) after genetic manipulation, only wild type colonies | Lack of homologous recombination | For streaking colonies on to MMM sucrose plates, use a very low initial inoculum from the MMM Cm plates. Giving enough time (24-48h) to obtain well, isolated colonies from low inoculum is crucial thus encouraging double crossover and homologous recombination providing the intended mutant colonies. |
| No colonies after gentamicin protection assay/ No intracellular bacteria in invasion assay / No cytotoxicity in LDH assay | T3SS2 not induced | Use freshly prepared bile salts solution and make sure the Vibrio cultures are grown at 37°C which is crucial for T3SS2 induction. It is also important to use fresh MMM plates (use within a month of preparation) as the salts precipitate in the agar after prolonged storage at 4°C. |
| Emulsion/ brown coloration in sample buffer after attempted resuspension of precipitated protein | Too much NaOH | Be mindful of 10M NaOH droplets that cling to the outside of the pipette tip before adding NaOH, as the base is highly concentrated and too much can prevent the protein precipitate from properly resuspending. Supplementation of 10M HCl in 0.2μL increments or increasing the volume of SDS sample buffer may help, but often it is better to retry the experiment. Diluting NaOH to 5M in MilliQ deionized water may help prevent this outcome as well. |
| Low hit count of proteins on mass-spectrometry analysis of secreted proteins | Collagen contamination, Not enough precipitated protein | Use clean PPE, rinse gloves and all tubes with 70% MeOH before use, and prepare samples in a positive pressure hood; Increase induction culture volume to 75mL or greater as needed |
Understanding Results:
See the figures and text within each protocol for expected results and interpretation. Expected results for each assay are shown at the end of the respective protocol.
Time Considerations:
Bacterial growth:
Bacterial cultures typically require 8 hours to reach stationary phase and are best prepared the night before they are needed. Inoculation generally takes at most 1-2 minutes, though additional time should be allocated based on the number of cultures needed. For transconjugation matings, colonies will typically appear within 2-3 days after plating on selection media, though up to 4 may be necessary depending on the gene being modified.
Phenotype assays:
During gentamicin protection assay or invasion assay, particularly when extended time points are required e.g. 7h and later, It is recommended to perform the 7h time point of infection first followed by 5, 3 and 1h time points to complete the experiment in a timely manner. it is recommended that infections be conducted in reverse chronological order. Application of bacteria to HeLa cells during infection should be carried out in pairs to ensure that the centrifuge is balanced during centrifugation to synchronize infections. Also, one should allot 5 minutes for washing and infecting each pair of tissue culture plates in addition to 5 minutes required for centrifugation, culminating in 40-50 minutes of prep for a standard 1/3/5/7-hour gentamicin protection or invasion assay.
Acknowledgements:
We thank current and past members of the Orth lab for their valuable contributions in designing these protocols. This work was funded NIH grants R01 GM115188 (KO), 5 T32 AI007520-20 (AEL), 5 T32 GM008014 (LC), Once Upon a Time…Foundation and the Welch Foundation I-1561. Dr. Kim Orth is a Burroughs Welcome Investigator, a Beckman Young Investigator, and a W. W. Caruth, Jr., Biomedical Scholar with an Earl A. Forsythe Chair in Biomedical Science.
Literature Cited:
- Broberg CA, Zhang L, Gonzalez H, Laskowski-Arce MA, & Orth K (2010). A <em>Vibrio</em> Effector Protein Is an Inositol Phosphatase and Disrupts Host Cell Membrane Integrity. Science, 329(5999), 1660. doi: 10.1126/science.1192850 [DOI] [PubMed] [Google Scholar]
- Calder T, de Souza Santos M, Attah V, Klimko J, Fernandez J, Salomon D, … Orth K (2014). Structural and regulatory mutations in Vibrio parahaemolyticus type III secretion systems display variable effects on virulence. FEMS microbiology letters, 361(2), 107–114. doi: 10.1111/1574-6968.12619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- CDC. (2019). Vibriosis Prevention Questions and Answers. Centers for Disease Control and Prevention. Retrieved from https://www.cdc.gov/vibrio/faq.html [Google Scholar]
- Chimalapati S, de Souza Santos M, Lafrance AE, Ray A, Lee WR, Rivera-Cancel G, … Orth K (2020). Vibrio deploys type 2 secreted lipase to esterify cholesterol with host fatty acids and mediate cell egress. Elife, 9. doi: 10.7554/eLife.58057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chimalapati S, de Souza Santos M, Servage K, De Nisco NJ, Dalia AB, & Orth K (2018). Natural Transformation in Vibrio parahaemolyticus: a Rapid Method To Create Genetic Deletions. J Bacteriol, 200(15). doi: 10.1128/JB.00032-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels NA, MacKinnon L, Bishop R, Altekruse S, Ray B, Hammond RM, … Slutsker L (2000). Vibrio parahaemolyticus Infections in the United States, 1973–1998. The Journal of Infectious Diseases, 181(5), 1661–1666. doi: 10.1086/315459 [DOI] [PubMed] [Google Scholar]
- De Nisco NJ, Kanchwala M, Li P, Fernandez J, Xing C, & Orth K (2017). The cytotoxic type 3 secretion system 1 of Vibrio rewires host gene expression to subvert cell death and activate cell survival pathways. Sci Signal, 10(479). doi: 10.1126/scisignal.aal4501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Souza Santos M, Salomon D, Li P, Krachler A-M, & Orth K (2015). 8 - Vibrio parahaemolyticus virulence determinants In Alouf J, Ladant D, & Popoff MR (Eds.), The Comprehensive Sourcebook of Bacterial Protein Toxins (Fourth Edition) (pp. 230–260). Boston: Academic Press. [Google Scholar]
- Falkow S (2004). Molecular Koch’s postulates applied to bacterial pathogenicity — a personal recollection 15 years later. Nature Reviews Microbiology, 2(1), 67–72. doi: 10.1038/nrmicro799 [DOI] [PubMed] [Google Scholar]
- Han C, Tang H, Ren C, Zhu X, & Han D (2016). Sero-Prevalence and Genetic Diversity of Pandemic V. parahaemolyticus Strains Occurring at a Global Scale. Frontiers in Microbiology, 7, 567. doi: 10.3389/fmicb.2016.00567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kodama T, Hiyoshi H, Okada R, Matsuda S, Gotoh K, & Iida T (2015). Regulation of Vibrio parahaemolyticus T3SS2 gene expression and function of T3SS2 effectors that modulate actin cytoskeleton. Cellular Microbiology, 17(2), 183–190. doi: 10.1111/cmi.12408 [DOI] [PubMed] [Google Scholar]
- Li P, Kinch LN, Ray A, Dalia AB, Cong Q, Nunan LM, … Orth K (2017). Acute Hepatopancreatic Necrosis Disease-Causing Vibrio parahaemolyticus Strains Maintain an Antibacterial Type VI Secretion System with Versatile Effector Repertoires. Appl Environ Microbiol, 83(13). doi: 10.1128/AEM.00737-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makino K, Oshima K, Kurokawa K, Yokoyama K, Uda T, Tagomori K, … Iida T (2003). Genome sequence of Vibrio parahaemolyticus: a pathogenic mechanism distinct from that of V cholerae. The Lancet, 361(9359), 743–749. doi: 10.1016/S0140-6736(03)12659-1 [DOI] [PubMed] [Google Scholar]
- National Enteric Disease Surveillance: COVIS Annual Summary, 2014. (2016). 11. [Google Scholar]
- Okuda J, Ishibashi M, Hayakawa E, Nishino T, Takeda Y, Mukhopadhyay AK, … Nishibuchi M (1997). Emergence of a unique O3:K6 clone of Vibrio parahaemolyticus in Calcutta, India, and isolation of strains from the same clonal group from Southeast Asian travelers arriving in Japan. J Clin Microbiol, 35(12), 3150–3155. doi: 10.1128/jcm.35.12.3150-3155.1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono T, Park KS, Ueta M, Iida T, & Honda T (2006). Identification of proteins secreted via Vibrio parahaemolyticus type III secretion system 1. Infection and Immunity, 74(2), 1032–1042. doi: 10.1128/iai.74.2.1032-1042.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park K-S, Ono T, Rokuda M, Jang M-H, Okada K, Iida T, & Honda T (2004). Functional Characterization of Two Type III Secretion Systems of Vibrio parahaemolyticus. Infection and Immunity, 72(11), 6659–6665. doi: 10.1128/IAI.72.11.6659-6665.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park KS, Ono T, Rokuda M, Jang MH, Okada K, Iida T, & Honda T (2004). Functional characterization of two type III secretion systems of Vibrio parahaemolyticus. Infection and Immunity, 72(11), 6659–6665. doi: 10.1128/IAI.72.11.6659-6665.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinoda S (2011). Sixty Years from the Discovery of Vibrio parahaemolyticus and Some Recollections. Biocontrol Science, 16(4), 129–137. doi: 10.4265/bio.16.129 [DOI] [PubMed] [Google Scholar]
- Undercurrentnews. (2016). Disease has cost Asia shrimp sector over $20bn… Retrieved from https://www.undercurrentnews.com/2016/09/09/disease-has-cost-asia-shrimp-sector-over-20bn/
- Wong HC, Liu SH, Wang TK, Lee CL, Chiou CS, Liu DP, … Lee BK (2000). Characteristics of Vibrio parahaemolyticus O3:K6 from Asia. Appl Environ Microbiol, 66(9), 3981–3986. doi: 10.1128/aem.66.9.3981-3986.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Krachler Anne M., Broberg Christopher A., Li Y, Mirzaei H, Gilpin Christopher J., & Orth K (2012). Type III Effector VopC Mediates Invasion for Vibrio Species. Cell Reports, 1(5), 453–460. doi: 10.1016/j.celrep.2012.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, & Orth K (2013). Virulence determinants for Vibrio parahaemolyticus infection. Current Opinion in Microbiology, 16(1), 70–77. doi: 10.1016/j.mib.2013.02.002 [DOI] [PubMed] [Google Scholar]