

Abstract
Massively parallel reporter assays (MPRAs) functionally screen thousands of sequences for regulatory activity in parallel. To date, there has been no systematic comparison of differences in MPRA design. Here, we screen a library of 2,440 candidate liver enhancers and controls for regulatory activity in HepG2 cells using nine different MPRA designs. We identify subtle but significant differences that correlate with epigenetic and sequence-level features, as well as differences in dynamic range and reproducibility. We also validate en masse that enhancer activity is robustly independent of orientation, at least for our library and designs. Finally, with a new method, we assemble and test the same enhancers as 192-mers, 354-mers, and 678-mers, and observe surprisingly large differences. This work provides a framework for the experimental design of high-throughput reporter assays, suggesting that the extended sequence context of tested elements, and to a lesser degree the precise assay, influence MPRA results.
INTRODUCTION
Spatiotemporal control of gene expression is orchestrated in part by distally located DNA sequences known as enhancers. The first enhancers were identified by cloning fragments of DNA into a plasmid with a reporter gene and promoter1–4. Transcriptional enhancement in such reporter assays continues to be widely used for evaluating whether a putative regulatory element is a bona fide enhancer. However, conventional, one-at-a-time reporter assays are insufficiently scalable to test the >1 million putative enhancers in the human genome5–8.
Massively parallel reporter assays (MPRAs) modify in vitro reporter assays to facilitate simultaneous testing of thousands of putative regulatory elements9–11 per experiment. MPRAs characterize each element through sequencing-based quantification of transcribed, element-linked barcodes9–15. MPRAs have facilitated the scalable study of putative regulatory elements for goals including functional annotation16–18, variant effect prediction10–15,19 and evolutionary reconstruction20,21.
Over the past decade, diverse designs for enhancer-focused MPRAs have emerged. Major differences include whether the enhancer is upstream10,11 vs. within the 3′ UTR of the reporter16, and whether the construct remains episomal vs. integrated18. Additionally, most MPRAs test sequences in only one orientation, effectively assuming enhancer activity is orientation-independent. Finally, while sheared genomic DNA16,22, PCR amplicons12 or hybrid captured sequences23,24 have been used in MPRAs, most studies synthesize libraries of candidate enhancers on microarrays, generally limiting them to <200 bp.
Unfortunately, we have, as a field to date, largely failed to systematically evaluate how these design choices impact or bias the results of MPRAs; previous work in this vein is summarized in Supplemental Note 1. Particularly as efforts to validate a vast number of putative enhancers5–8 take shape, a clear-eyed understanding of the biases and tradeoffs introduced by MPRA experimental design choices is needed. We performed a systematic comparison by testing the same 2,440 sequences for regulatory activity using nine MPRA strategies, including conventional episomal, STARR-seq, and lentiviral designs. We further tested the same sequences in both orientations. Finally, we improved multiplex pairwise assembly25 and applied it to test differently sized versions of the same enhancers. Our results quantify the impact of MPRA experimental design choices and provide further insight into the nature of enhancers.
RESULTS
Implementation and testing of nine MPRA strategies
We sought to systematically compare nine MPRA strategies (Figure 1): (1) the “classic” MPRA, using the pGL4.23c vector, wherein the enhancer library resides upstream of a minimal promoter, and the associated barcodes reside in the 3′ UTR of the reporter gene (“pGL4”)10,26. (2, 3) Self-Transcribing Active Regulatory Region Sequencing (STARR-seq), wherein the enhancer library resides in the 3′ UTR of the reporter gene, either as originally described (“HSS”)16 or using the bacterial origin of replication for transcriptional initiation (“ORI”)22. In both cases, we introduce barcodes immediately adjacent to the enhancers in the 3’ UTR to facilitate consistent procedures with other assays. (4, 6, 8) LentiMPRA, wherein lentiviral integration is used to mitigate concerns about potential differences in chromatin between episomes vs. chromosomes, either with the enhancer library upstream of the minimal promoter and the associated barcodes in the 3′ UTR of the reporter (5′/3′ WT)18, the enhancer library upstream of the minimal promoter and the barcodes in the 5′ UTR of the reporter (5′/5′ WT), or with both the enhancer library and the barcodes in the 3′ UTR of the reporter (3′/3′ WT). The 5′/5′ WT design was developed to address distance-dependent template switching prior to lentiviral integration27,28, as it reduces the distance between the enhancer and barcode from 801 to 102 bp. The 3′/3′ WT design is analogous to STARR-seq, but integrated into the genome, and also addresses template switching by positioning the enhancer and barcode immediately adjacent to one another. (5, 7, 9) These designs are identical to the three lentiMPRA designs, except that the vector harbors a mutant integrase such that the constructs remain episomal (5′/3′ MT, 5′/5′ MT, 3′/3′ MT)18.
Figure 1. Nine MPRA strategies and experimental workflow.
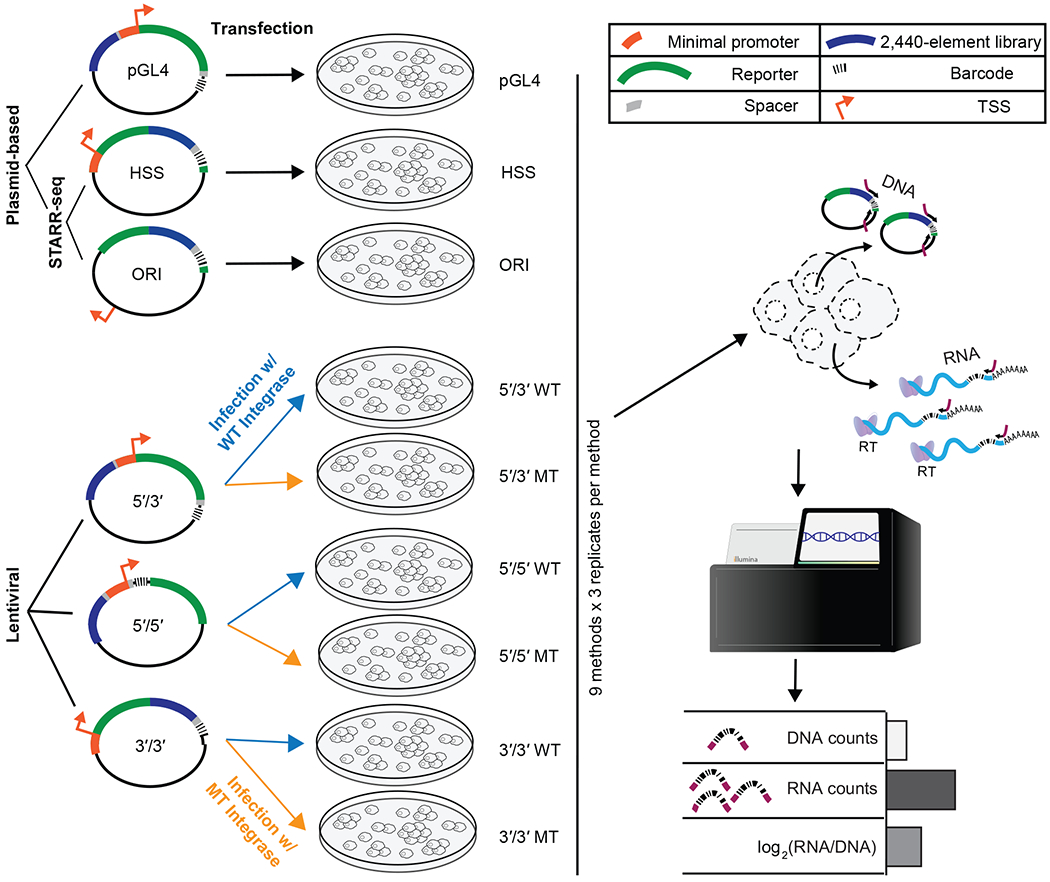
Nine different MPRA designs were tested. These are schematically represented on the left, and from top to bottom include pGL4.23c (pGL4); the original STARR-seq vector (HSS); STARR-seq with no minimal promoter (ORI); and lentiMPRAs with the enhancer library upstream of the minimal promoter and the associated barcodes in the 3′ UTR of the reporter gene (5′/3′), the enhancer library upstream of the minimal promoter and barcodes in the 5′ UTR of the reporter (5′/5′), or with both the enhancer library and the barcodes in the 3′ UTR of the reporter (3′/3′). The episomal designs (pGL4, HSS, ORI) were transfected into HepG2 cells, while 5′/5′, 5′/3′, and 3′/3′ were packaged with either wild type (WT) or mutant (MT) integrase and infected into HepG2 cells. DNA and RNA were extracted from the cells, and the enhancer-associated barcodes amplified and sequenced, and a normalized activity score for each element computed on the basis of the counts.
For a common set of sequences to test, we turned to a previously developed library18 consisting of 2,236 candidate enhancer sequences based on HepG2 ChIP-seq peaks, along with 204 controls (Supplemental Table 1). Of these, 281 overlap promoters (+/− 1 kilobase (Kb) of TSS of protein-coding gene). The controls consist of synthetically designed sequences that previously demonstrated enhancer MPRA activity (100 positives) or lack thereof (100 negatives)29 in HepG2 cells, along with 2 positive and 2 negative controls derived from endogenous sequences that were previously validated with luciferase assays18. All sequences were 171 base pairs (bp) and synthesized on a microarray together with common flanking sequences. A 15 bp degenerate barcode was appended during PCR amplification, and the amplicons cloned to the human STARR-seq (HSS) vector. The enhancer/barcode region of the HSS library was amplified and used for two purposes -- first, it was sequenced to link barcodes to enhancers; second, the amplicons were cloned at high complexity into other vectors to create libraries for the remaining eight MPRA designs (Supplemental Figure 1). As such, the relative abundances of enhancers and barcodes, as well as the enhancer-barcode associations, were consistent across all MPRA libraries. Cloning details and references for each of the nine assay designs are provided in the Methods.
Plasmid libraries were transfected into HepG2 cells in triplicate (three different days). LentiMPRA libraries were packaged with either wild-type (WT) or defective (MT) integrase lentivirus and infected into HepG2 in triplicate (three different days). We extracted DNA and RNA, amplified barcodes via PCR and RT-PCR respectively, and sequenced amplicons to generate barcode counts (Figure 1). An activity score for each element was calculated as the log2 of the normalized count of RNA molecules from all barcodes corresponding to the element, divided by the normalized number of DNA molecules from all barcodes corresponding to the element (Supplemental Table 2). For each of the 27 experiments (9 assays x 3 replicates), only barcodes observed in both RNA and DNA were considered. For 26 of 27 experiments (all but 3′/3′ MT replicate 1), the median number of barcode counts per element was greater than 100 (Supplemental Figure 2).
Different MPRA designs yield different results
We first sought to evaluate the technical reproducibility of each assay. Most assays were highly correlated between the three replicates. Specifically, the intra-assay Pearson correlations for pairwise comparisons of activity scores of replicates exceeded 0.90 for all assays except for the 5′/3′ MT (mean r = 0.87) and 3′/3′ MT (mean r = 0.54) assays (Figure 2A; Supplemental Figure 3A). We also confirmed correlations for 5′/3′ WT and 5′/3′ MT between this and our previous study18 (r = 0.92 for 5′/3′ WT and r = 0.81 for 5′/3′ MT; Supplemental Figure 3B).
Figure 2. Quantitative comparison of different MPRA strategies.
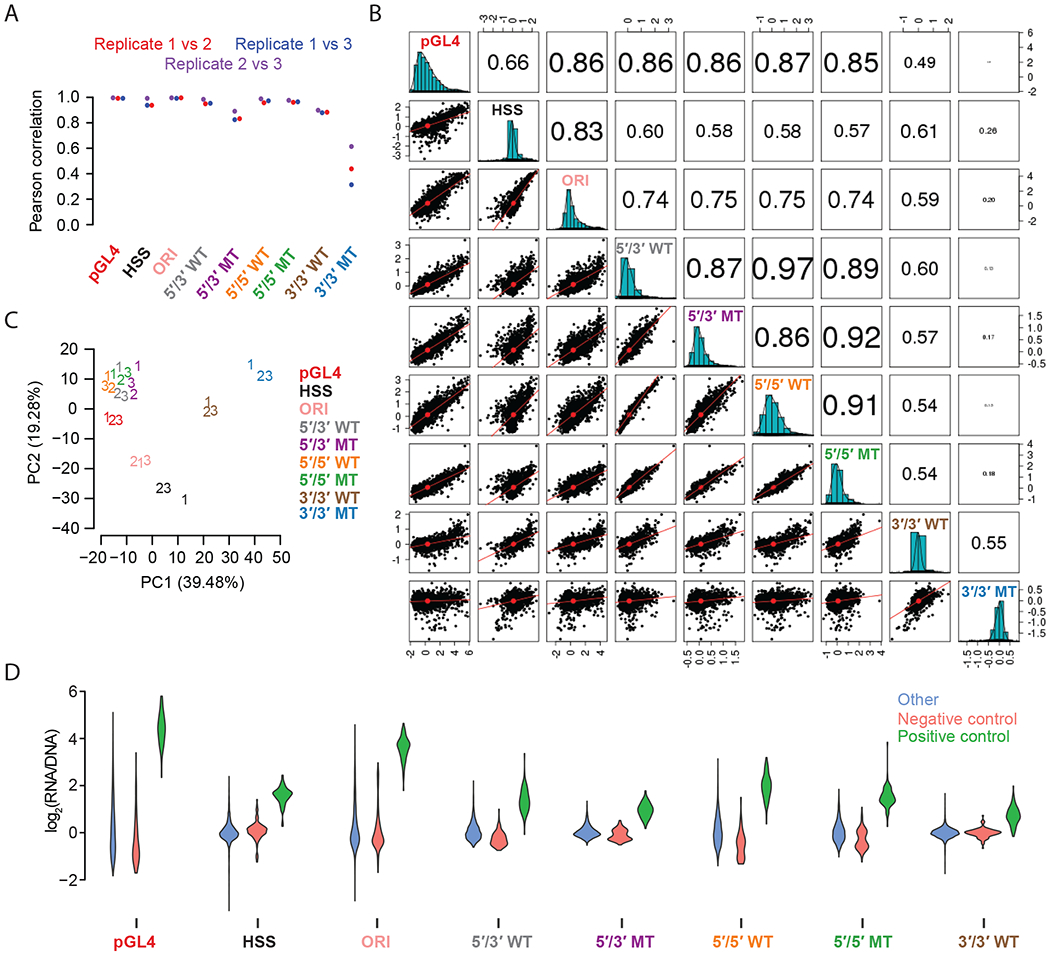
A) Beeswarm plot of the Pearson correlation values for each of the three possible pairwise comparisons among the replicates of each MPRA technique. B) Scatter matrix displaying scatter plots corresponding to each of the 36 pairs of possible inter-assay comparisons (lower diagonal elements). Shown on the diagonal is a histogram of the log2(RNA/DNA) ratios, averaged among replicate samples. Also shown are Pearson correlation values among each pair of comparisons, with the size of the text proportional to the magnitude of the correlation coefficient (upper diagonal elements). See Supplemental Figure 5 for equivalent but with Spearman correlations. C) PCA of 27 experiments, i.e. three replicates x nine different MPRA designs. Shown are the first two PCs that together explain over half of the variation. Slight jitter was added to each data point to enhance readability. D) Violin plots displaying the distribution of average log2(RNA/DNA) ratios across independent transfections for positive controls, negative controls, and putative enhancer sequences tested, for each of the nine assays.
We next sought to compare the results of the various assay designs to one another. We calculated the average activity score for each element across all technical replicates of a given assay (Supplemental Table 2), and then compared the assays to one another. Six of the nine assays demonstrated inter-assay Pearson and Spearman correlations of >0.7 with all other members of this group (Figure 2B, Supplemental Figure 4, Supplemental Figure 5). These were the ORI and pGL4, together with both WT and MT versions of the 5′/5′ and 5′/3′ assays. The remaining three assays (3’/3’ MT, 3’/3’ WT, HSS) did not show good agreement with the other six assays, nor with one another.
As a different approach to compare assays, we subjected activity scores from all 27 experiments (9 assays x 3 replicates) to Principal Components Analysis (PCA) (Figure 2C). The aforementioned six assays with inter-assay correlations of >0.7 clustered reasonably close to one another. Interestingly, PC1 tended to separate the assays wherein the enhancer resides upstream of the minimal promoter (5′/5′, 5′/3′, and pGL4) from those where it resides 3′ of the reporter gene (3′/3′, HSS, and ORI). In contrast, PC2 tended to separate lentiviral designs (5′/5′, 5′/3′, and 3′/3′) from plasmid-based designs (pGL4, HSS, and ORI). This suggests systematic differences in the enhancer activity measurements that relate to aspects of MPRA design. It also highlights that the location of the candidate enhancer on the plasmid backbone may play a larger role in differential activity than does the episomal vs. integrated aspect of the assay.
Next, we examined the dynamic range of activity scores (Figure 2D). The classic enhancer reporter vector (pGL4) and the promoter-less STARR-seq assay (ORI) exhibited the greatest dynamic range, with pGL4 showing the largest separation between positive and negative controls (two-sided t-statistic = 37.46). Among the lentiviral assays, the 5′/5′ WT design exhibited the greatest dynamic range and separation of controls (two-sided t-statistic = 30.92).
We generated lasso regression models based on 915 biochemical, evolutionary, and sequence-derived features (Supplemental Table 3, Supplemental Table 4) and 10-fold cross-validation (CV). We were able to best predict enhancer activities for the six aforementioned assays (Pearson r ranging from 0.59 for 5′/3′ WT to 0.71 for pGL4) (Supplemental Figure 6A–B). In general, strong enhancers tended to be underpredicted by the model while weak enhancers tend to be overpredicted.
Many of the top coefficients fit by these models correspond to ChIP-seq signal or sequence-based binding site predictions for transcriptional activators, coactivators, and repressors (Supplemental Figure 6C–D, Supplemental Table 5). We caution that the interpretation of feature selection and coefficient-based ranking is inherently limited by substantial multicollinearity among features (Supplemental Table 4), which in turn limits the determination of which features are mechanistically or causally involved. Potential reasons for inter-feature correlations are summarized in Supplemental Note 2.
We next sought to ask whether we could predict differences in enhancer activity between the assays, based on the same 915 features. For models predicting pairwise differences between the results of the pGL4, 5′/5′ WT, 3′/3′ WT, and ORI assays, we were able to achieve correlations of 0.4-0.5 (Figure 3A, Supplemental Figure 7A). We were particularly interested in whether features corresponding to RNA binding proteins and splicing factors would be especially predictive of promoterless STARR-seq (ORI) or 3′/3′ WT results, as in these assays the enhancer itself is included in the 3′ UTR. Indeed, SRSF1/2, BRUNOL4, PTBP1, PPRC1, KHDRBS2, SYNCRIP, and MBNL1, which are known to modulate mRNA stability and splicing, predict differences in measured activity in ORI or 3′/3′ WT vs. 5′/5′ WT or pGL4 (Figure 3B–C, Supplemental Figure 7B–C, Supplemental Table 5). Of note, SRSF1/2, PTBP1, PRPC1, SYNCRIP, and MBNL1 are all expressed in liver30, and could therefore influence MPRA results in HepG2. Additionally, several promoter-binding proteins (TEAD1, TEAD3, NRSF1, JUN, YY1), all expressed in the liver, favor pGL4 and 5’/5’ WT, while the CCAAT-enhancer-binding proteins favor HSS and ORI. This may correspond to a tradeoff wherein conventional MPRAs are biased towards testing for promoter-like activity, while STARR-seq MPRAs are biased by mRNA stability and splicing factors.
Figure 3. Predictive modeling of the ratios and differences between MPRA methods.
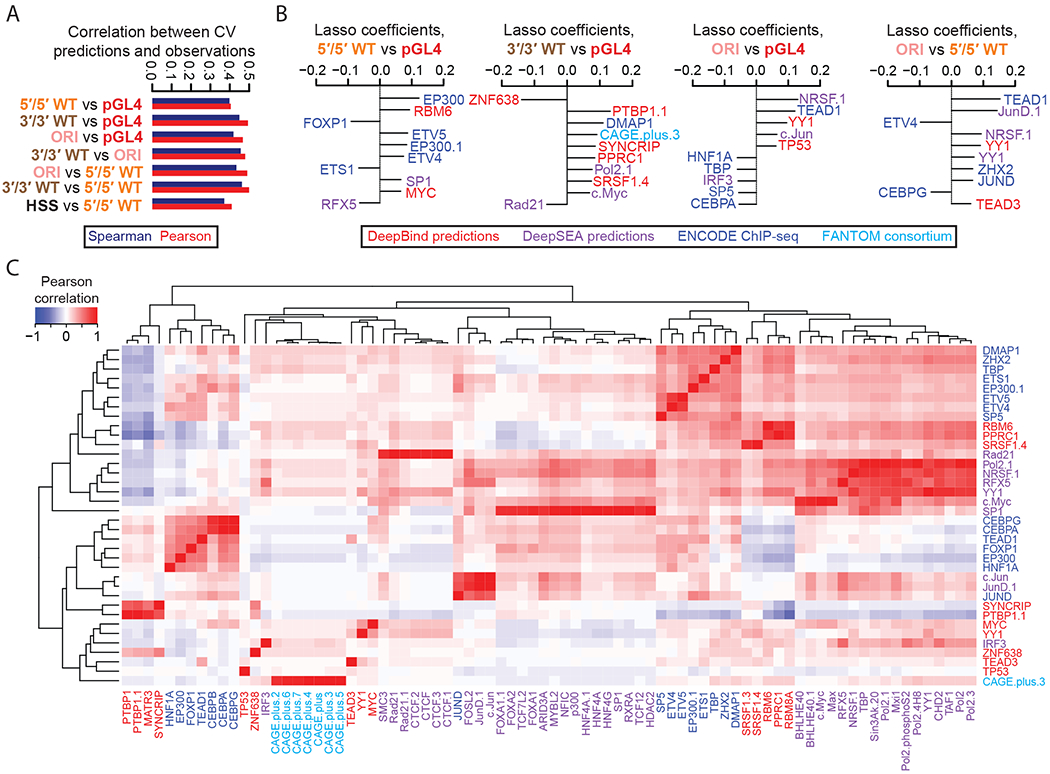
A) Pearson and Spearman correlation coefficients for 10-fold cross-validated predictions derived from lasso regression models and the observed RNA/DNA ratios, for each of the 7 indicated differential comparisons tested. Also indicated are the Pearson (r) and Spearman (rho) correlation values. B) The top 10 coefficients derived from lasso regression models trained on the full dataset to predict observed differences in the indicated pairs of MPRA methods. Features with the extension “.1”, “.2”, etc allude to redundant features or replicate samples. C) Pearson correlation matrix between the union of all top 10 features from (B), shown as rows, and other features sharing a Pearson correlation either ≤ −0.8 or ≥ 0.8, shown as columns. Feature names are colored according to the origin of the feature as shown in the boxed key above. Hierarchical clustering was used to group features exhibiting similar correlation patterns.
Next, we looked for differences between episomal vs. integrated assays. We note that FOXP1 is more predictive of integrated activity, while ETS-variant TFs are more predictive of episomal activity, suggesting that these or correlated factors play a differential role in episomal vs. integrated contexts (Figure 3B–C, Supplemental Figure 7B–C).
Interestingly, general transcriptional activity, as measured by cap analysis gene expression (CAGE)31, was among the most predictive features of the 3′/3′ WT assay (Supplemental Figure 6C). As this is the only assay where the tested elements are both genomically integrated and distally located from the promoter, this observation suggests CAGE-based transcriptional activity may be a good predictor of distal enhancer activity32,33.
Enhancer activity is largely, but not completely, independent of sequence orientation
We next set out to test a key aspect of the canonical definition of enhancers, that they function independently of their orientation with respect to the promoter. We directionally cloned 2,336 sequences (the 2,236 candidates described above extended out to 192 bp genomic reference sequence, along with 50 positive and 50 negative controls from Vockley et al.12), in both orientations into the pGL4 vector, pooled these libraries together, and transfected HepG2 cells in quadruplicate (Figure 4A). The median number of barcode counts per element was >100 (Supplemental Figure 8) and the measured activities reproducible (Pearson r > 0.98; Figure 4B, Supplemental Figure 9, Supplemental Table 6). Interestingly, enhancer activities for the same elements cloned in forward vs. reverse orientation to the pGL4 vector were also highly correlated (mean r = 0.88) but less so than same-orientation comparisons (r > 0.98; Figure 4B). This suggests that enhancer activity in reporters is largely, but not completely, independent of orientation.
Figure 4. Enhancer activity is largely, but not completely, independent of sequence orientation.
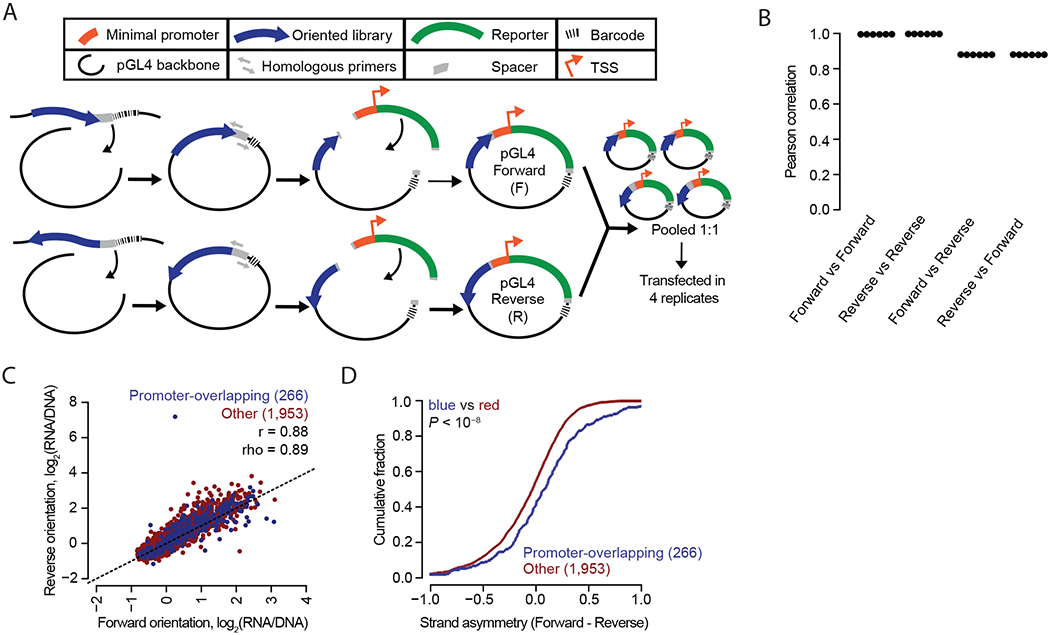
A) Workflow used to produce an MPRA library with each element in both orientations. The 2,336 element library was cloned into the pGL4 backbone in both orientations as two separate libraries. These were then pooled and transfected into HepG2 cells in quadruplicate. B) Beeswarm plot of the Pearson correlations corresponding to each of the six possible pairwise comparisons among the four replicates. The correlations are computed between observed enhancer activity values for elements positioned either in the same (Forward vs. Forward and Reverse vs. Reverse) or opposite (Forward vs. Reverse and Reverse vs. Forward) orientations. C) Scatter plots of the average activity score of each element in the Forward vs. Reverse orientation, split out by promoter-overlapping (blue; +/− 1 Kb of the TSS of a protein-coding gene) and other (red) elements. D) Cumulative distributions measuring strand asymmetry between promoter-overlapping elements and other elements. Here, “Forward” and “Reverse” were defined as “sense” and “antisense”, respectively, in relation to the orientation of the TSS for promoter-overlapping elements (n = 266); and were defined as “plus” and “minus”-stranded, respectively, in relation to the chromosome annotation for other elements (n = 1,953). Similarity of the blue distribution to that of the red was tested (one-sided Kolmogorov–Smirnov [K–S] test, P value).
In contrast with enhancers, promoters are established to be directional34,35. 266 of 281 promoter-overlapping elements were successfully measured in both orientations. We tested whether these behaved differently than 1,953 more distally located elements. Indeed, the promoter-overlapping sequences exhibited greater differences in activity between the two orientations than distal elements, supporting the conclusion that they contain signals to promote transcription in an asymmetric fashion (Figure 4C–D).
Appending sequence context leads to differences in the results of MPRAs.
Most MPRAs use array-synthesized libraries that are, for technical reasons, limited in length, typically to less than 200 bp. To evaluate the impact of this length restriction, we designed 192 bp (“short”), 354 bp (“medium”), and 678 bp (“long”) versions of our candidate enhancer library, centered at the same genomic position, and corresponding to the equivalent 2,236 candidate enhancers tested above (i.e., including more flanking sequence from reference genome; Supplemental Table 1). We also included 50 high and low-scoring putative elements from Vockley et al.12 in the short and medium libraries (excluded from long libraries because they were all shorter than 678 bp).
The 192 bp versions of these candidate enhancers were synthesized directly on a microarray; sequencing showed a 100% yield (2,336/2,336) and a 3.8-fold interquartile range (IQR) for relative abundance (Supplemental Figure 10A). To generate the 354 bp versions, we performed our previously published Multiplex Pairwise Assembly (MPA)25 on overlapping pairs of array-synthesized 192 bp fragments (95% yield (2,241/2,336); 4.9-fold IQR; Supplemental Figure 10A). Finally, to generate the 678 bp versions, we developed a “two-round” version of MPA, which we call Hierarchical Multiplex Pairwise Assembly (HMPA; Supplemental Figure 10B, Supplemental Figure 11). HMPA of overlapping pairs of array-synthesized 192 bp fragments yielded overlapping pairs of 354 bp fragments, which were further assembled to generate 678 bp fragments (84% yield (1,887/2,236); 27.9-fold IQR; Supplemental Figure 11A). We verified a subset of our long enhancers with PacBio sequencing (Supplemental Figure 11C–D; chimera rate of 16.5% ).
We cloned all three libraries into the pGL4 vector, then pooled and transfected them in quadruplicate to HepG2 cells (Figure 5A, Supplemental Table 7). Requiring each element to be detected with at least 10 unique barcodes, there were 651 candidate enhancers tested at all three lengths. Technical replicates within any given length class were highly reproducible, albeit modestly less so for long elements (mean Pearson r = 0.94, Figure 5B, Supplemental Figure 12–13). However, there was substantially less agreement for the same candidate enhancers tested at different lengths (short vs. medium, mean r = 0.78; medium vs. long, mean r = 0.67; short vs. long, mean r = 0.53; Figure 5B–C). Finally, we observed that the positive control sequences were significantly more active than the negative controls when tested as either 192 bp or 354 bp fragments (P < 0.01, Wilcoxon sign-rank test; Figure 5D).
Figure 5. Including additional sequence context around tested elements leads to differences in the results of MPRAs.
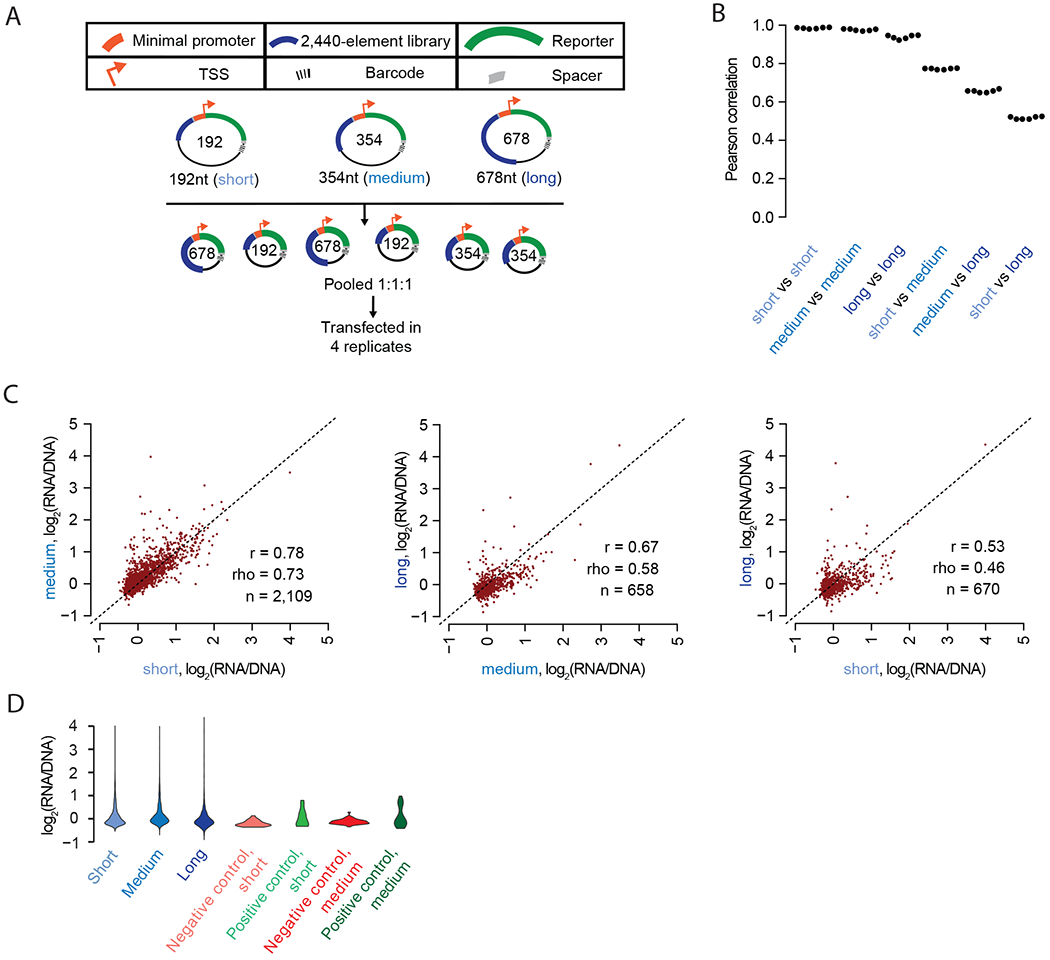
A) Experimental schematic. 192 bp, 354 bp, and 678 bp libraries were synthesized, assembled, and cloned into the pGL4 backbone. These were pooled and transfected into HepG2 cells in quadruplicate. B) Beeswarm plot of the Pearson correlation values corresponding to each of the six possible pairwise comparisons among the four replicates. The correlations are computed between observed enhancer activity values for elements measured in each of the three possible size classes. C) Scatter plots of the average activity score of each element, comparing short vs. medium, medium vs. long, and short vs. long versions of each element, and restricting to elements detected with at least 10 unique barcodes at both lengths (n). D) Violin plot displaying the distribution of average log2(RNA/DNA) ratios for short, medium, and long versions of the elements tested, as well as for positive and negative controls at short and medium lengths.
We chose ten MPRA-active candidate enhancers to test in individual luciferase assays, five that showed differential activity between their long and medium forms (cyan, Supplemental Figure 14A), and five that did not (green, Supplemental Figure 14A). Of the five that showed differential activity, three were active in the luciferase assay (2, 3, 4), all concordant with MPRA results (Supplemental Figure 14B–D). Of the five that did not show differential activity in the MPRA, all were active in the luciferase assay in at least one form and four had differential activity, possibly due to greater sensitivity of the luciferase assay or subtle differences between the constructs (Supplemental Figure 14B–D). We also tested versions of all ten of these MPRA-active candidates in their long form but with the middle 354bp deleted; all of these showed insignificant (n=8) or reduced (n=2) activity in the luciferase assay (Supplemental Figure 14B). Overall, these results highlight the relevance of the lengths and boundaries of elements tested in MPRAs in influencing measured activity.
We trained lasso regression models to predict activities using features which were re-computed for each of the three size classes (Figure 6A, Supplemental Figure 15, Supplemental Table 4). The lower performance of the model for the long element library is possibly consequent to its fewer sequences, lower technical reproducibility, or an increase in the effect of non-linear interactions between features that reduce predictive performance. Known predictors of enhancer activity were consistently present in the top coefficients, although their relative rankings differed depending on the size class being examined (Supplemental Figure 15C, Supplemental Table 5). Next, we sought to explicitly model how differences in predicted factor binding might explain differences in enhancer activity, as measured by different pairs of size classes. For example, in attempting to explain observed activity differences in long vs. short elements, we computed a set of features as the differences in predicted binding, or measured ChIP-seq signal, between the long element and corresponding short element (e.g., ∆ARID3A = ARID3Along – ARID3Ashort). Many of the top features originated from sequence-based differences in predicted binding in the extra genomic context surrounding the core element. Features consistently observed to explain activity differences in longer elements include RPC155, the catalytic core and largest component of RNA polymerase III; Jun and FOS, components of the AP-1 complex; ATF2, EZH2, and HDAC1/2, core histone-modifying enzymes; and the transcription factors ARID3A, DRAP1, and SP1/2/3 (Figure 6B–C, Supplemental Figure 16, Supplemental Table 5).
Figure 6. Predictive modeling of factors dependent on element size.
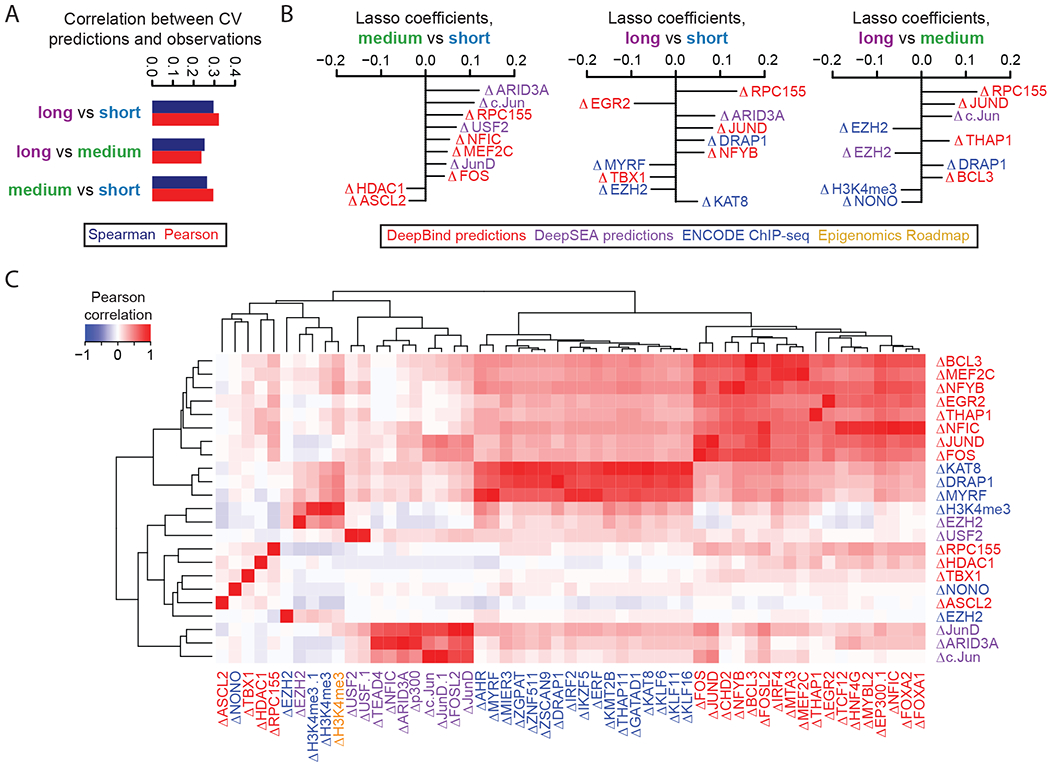
A) Pearson and Spearman values between the 10-fold cross-validated predictions and observed values for each of the three size classes tested. B) The top 10 coefficients derived from lasso regression models trained on the full dataset to predict observed values from the differential size comparisons indicated. Features with the extension “.1”, “.2”, etc allude to redundant features or replicate samples. C) Pearson correlation matrix between the union of all top 10 features from (B), shown as rows, and other features sharing a Pearson correlation either ≤ −0.8 or ≥ 0.8, shown as columns. Feature names are colored according to the origin of the feature as shown in the boxed key above. Hierarchical clustering was used to group features exhibiting similar correlation patterns.
DISCUSSION
Over the past decade, MPRAs have enabled researchers to functionally test large numbers of DNA sequences for regulatory activity and in the process address numerous biological questions. While different groups utilize various backbones and assay designs, there has been no systematic comparison of how these different strategies influence results.
Here, we have sought to perform a systematic comparison of all major MPRA strategies, and to concurrently investigate the consequences of other key design choices, i.e. orientation and length. We generally observe concordance between different MPRA designs, albeit to varying degrees. Six of the nine assays exhibited both technical reproducibility as well as reasonable agreement with one another (pGL4, ORI, 5′/5′ WT, 5′/5′ MT, 5′/3′ WT, 5′/3′ MT). Furthermore, as we previously showed for the 5′/3′ WT and 5′/3′ MT assays, enhancer activities as measured by MPRAs18 are reasonably well predicted by models based on primary sequence together with biochemical measurements at the corresponding genomic locations. Nonetheless, despite the general agreement, there are subtle but significant differences that are predictable by similar features, consistent with what we previously observed in comparing 5′/3′ WT vs. 5′/3′ MT MPRAs18. Taken together, our results support a view wherein diverse MPRAs are all measuring enhancer activity, but design differences (e.g. integrated vs. episomal; 5′ vs. 3′ location of the enhancer) influence the results to a modest degree. For example, features influencing mRNA stability and splicing favor assays with the enhancer transcribed in the 3’ UTR (ORI and 3’/3’ WT), while promoter-binding TFs favor assays with the enhancer upstream of the promoter (pGL4 and 5’/5’ WT).
Overall, our results support a preference for three of the nine MPRA designs evaluated here—pGL4, ORI, and 5′/5′ WT, which all had reasonable inter-assay correlations. The pGL4 assay has the advantage of representing the “classic” enhancer reporter assay design, had the greatest dynamic range, and was the most predictable with our lasso regression, but the disadvantages of being episomal rather than integrated and of confounding enhancer activity with possible effects from promoter-binding proteins. The ORI assay (i.e. promoterless STARR-seq) has the advantage of eliminating the need to associate barcodes, potentially allowing for greater library complexities, and has a large dynamic range, but the disadvantages of confounding enhancer activity with possible effects on mRNA splicing and/or stability, and also of being episomal rather than integrated. The 5′/5′ WT assay has the advantage of being integrated rather than episomal, and—amongst lentiviral assays—mitigates the template switching issue by minimizing the distance between the enhancer and barcode. However, template switching still occurs to some degree, the assay exhibits a lower dynamic range than pGL4 or ORI assays, and has similar potential for bias from promoter-binding proteins as pGL4.
A caveat of our HSS and ORI experiments is that by incorporating a barcode downstream of the enhancer, we introduced the possibility that barcode counts include short transcripts initiating within the candidate enhancer itself. Further exploration of this potential confounder, including additional experiments, is summarized in Supplemental Note 3.
Another key finding is our confirmation that the activity of enhancers, is largely, but not completely, independent of orientation, at least as measured for our subset of candidate enhancers tested. This is of course part of the original definition of enhancers1, but efforts to systematically test the validity of this assumption across a large number of sequences have been limited16,36,37. Previously, a subset of PIC-bound enhancers were shown to have strong orientation-dependent activity, highlighting that these trends may be influenced by the choice of elements tested38. Candidate enhancer sequences derived from the vicinity of TSSs exhibited greater directionality, consistent with a subset of these bearing features of oriented promoters.
Finally, we developed improved methods to efficiently assemble longer DNA fragments from array-synthesized oligonucleotides, and applied them to evaluate the extent to which including additional sequence context around tested elements impacts MPRA results. We successfully assembled 95% of 2,336 x 354 bp targets using MPA, compared to just 71% of 2,271 x 192-252 bp targets in our original description of the method25. Moreover, our new hierarchical MPA (HMPA) is to our knowledge the first protocol to in vitro assemble thousands of sequences, each over 600 bp, as a single library. In this manuscript, we synthesized >600 elements, each 678bp, for 1-2% of what it would have cost from commercial vendors. Unlike potential alternatives, it does not require specialized equipment, making it more widely accessible39.
The sub-200 bp length of subsequences typically tested is a choice related to the technical limits of microarray-based synthesis. In the genome, there are no such limits, and it remains unclear what the appropriate “enhancer size” is to test in MPRAs and whether this choice matters. To evaluate this, we tested candidate enhancers at three different lengths. We observe correlations between the same elements tested at all lengths, but these correlations clearly drop off as a function of length differences. At the extreme, the activities of 678 bp vs. 192 bp versions of the same candidate enhancers were more poorly correlated than nearly all of our inter-assay comparisons (Pearson r = 0.53, Spearman rho = 0.46). Furthermore, these data suggest that the longer sequences are adding biologically relevant signal, as features corresponding to relevant TFs explain differences in activity of longer vs. shorter sequences. For example, a feature corresponding to RPC155, the catalytic subunit of RNA polymerase III, is the strongest coefficient separating the 678 bp constructs from the 192 and 354 bp constructs, and also one of the stronger coefficients separating the 354 bp from 192 bp constructs. Although it is challenging to offer strict guidance in the absence of in vivo ground truth, we recommend testing longer sequences when possible.
In conclusion, we set out to rank the relative contribution of assay design, orientation, and length on the results of MPRAs. We found that sequence length had the greatest effect, followed by assay design and finally orientation. Moreover, our results favor the use of one of three MPRA designs: pGL4, ORI, and 5′/5′ WT. Each of these assays exhibited strong technical reproducibility, reasonable interassay correlation with one another despite design differences, and reasonable predictability based on sequence and biochemical features of the corresponding genomic regions. Our results also suggest a degree of caution in interpreting the results of all MPRAs, as they are all subject to influence by aspects of the assay design. Finally, we conclude that whereas MPRA results are largely independent of the orientation for our tested elements, they are surprisingly dependent on the length of the elements tested—in other words, sequence context matters. Although MPRAs of high-complexity genome-wide fragment libraries are not length limited16,22, MPRAs of designed libraries largely still are. For designed libraries in particular, further work is necessary to develop or improve methods like HMPA to facilitate the construction of complex, uniform MPRA libraries of longer sequences, as well as to further explore the optimal parameters of element design (e.g. length, centering).
ONLINE METHODS
Design, barcoding, and cloning of the enhancer library into the human STARR-seq (HSS) vector
We used an existing array library from Inoue et al.18. This library consists of 2,440 unique 171 bp candidate enhancer sequences, based on ChIP-seq peaks in HepG2. Each sequence was flanked with a 15 bp sequence on the 5′ end (Original_Array_5adapter) and a 44 bp sequence on the 3′ end (Original_Array_3adapter) (Supplemental Table 8). More detail on enhancer design can be found in that manuscript18. We first amplified the library using the following primers: STARR-Seq-AG-f and spacer-AG-r (Supplemental Table 8). These amplify the library excluding the previously designed barcodes, while adding homology to the STARR-seq vector (Addgene ID:71509)16 on the 5′ end and a spacer sequence on the 3′ end that we use for all subsequent libraries. We amplified 10 ng of array oligos with KAPA HiFi 2x Readymix (Kapa Biosystems) with a 65 °C annealing temperature and 30 s extension, following the manufacturer’s protocol. We followed the reaction in real time using Sybr Green (Thermo fisher scientific), and stopped the reaction before plateauing, after 10 cycles. We then purified the PCR product with a 1.8x AMPure XP (Beckman coulter) cleanup and eluted in 50 μl of Qiagen Elution Buffer (EB), following the manufacturer’s protocol. We took 1 μL of purified PCR product and amplified in triplicate a second reaction using Kapa HiFi 2x Readymix using primers STARR-Seq-AG-f and STARR-BC-spacer-r with a 35 s extension time and 65 °C annealing temperature for eight cycles (Supplemental Table 8). This round of PCR added a 15 bp degenerate barcode on the 3′ end of the spacer as well as homology arms to the 3′ end of the human STARR-seq vector. We then pooled the three reactions together, ran on a 1.5% agarose gel, and gel extracted the amplicon using the QIAquick Gel Extraction Kit (Qiagen), following the manufacturer’s protocol, eluting in 17.5 μL of Qiagen EB. We then cloned a 2:1 molar excess of our gel-extracted insert into 100 ng of the human STARR-seq vector (linearized with AgeI and SalI) with the NEBuilder HiFi DNA Assembly Cloning Kit (NEB), following the manufacturer’s protocol. We transformed 10-beta electrocompetent cells (NEB C3020) with the plasmids in duplicate following the manufacturer’s protocol, along with a no insert negative control. We pooled the two transformations during recovery and plated 15 μL to estimate complexity. The following day, we estimated complexity as approximately 750,000, and grew a third of the transformation to represent a library of 250,000 in 100 mL of LB+Ampicillin, so that each candidate enhancer is expected to associate on average with 100 different barcodes. We extracted the plasmid using the ZymoPURE II Plasmid Midiprep Kit (Zymo Research).
Barcode association library for the 9 MPRA assays
We amplified 5 ng of the human STARR-seq (HSS) library with the following primers: P5-STARR-AG-ass-f and P7-STARR-ass-r (Supplemental Table 8). These primers add a sample-specific barcode and Illumina flow cell adapters. We then spiked the library into a NextSeq Mid 300 cycle kit with paired-end 149 bp reads and a 20 bp index read (which captured the 15 bp barcode as well as 5 bp of extra sequence to help filter for read quality), using the following custom primers: Read1 as STARR-AG-seq-R1, Read2 as spacer-seq-R2, Index1 as pLSmp-ass-seq-ind1 and Index2 as STARR-AG-ind2 (Supplemental Table 8).
Library cloning
From HSS to ORI vector:
We amplified 5 ng of the HSS library with the following primers: STARR-Seq-AG-f and STARR-Seq-AG-r (Supplemental Table 8) using KAPA HiFi 2x Readymix (Kapa Biosystems) with a 65 °C annealing temperature and 30 s extension. These primers amplify both candidate enhancers and previously assigned degenerate barcodes and add homology arms to the ORI vector (Addgene 99296)22. We followed the reaction in real time with Sybr Green (Thermo fisher scientific) and stopped the reaction before plateauing, at 13 cycles. We gel extracted the amplicon on a 1.5% agarose gel as described above. We then cloned the library in a 2:1 molar excess into 100 ng of the hSTARR-seq_ORI vector (addgene ID:99296), linearized with AgeI and SalI, using the NEBuilder HiFi DNA Assembly Cloning Kit (NEB), following the manufacturer’s protocol. We then transformed 10-beta electrocompetent cells (NEB C3020) with the plasmids in duplicate following the manufacturer’s protocol, along with a no insert negative control. We pooled the two positive transformations during recovery, plated 15 μL to estimate complexity and grew the remainder of the culture in 100 mL LB+Amp. The following day, we estimated the complexity as >500K and extracted the plasmid using the ZymoPURE II Plasmid Midiprep Kit (Zymo Research).
From human STARR-seq to pGL4.23c MPRA vector:
As described above, we amplified 5 ng of the human STARR-seq library with the following primers: pGL423c-AG-1f and pGL423c-AG-1r (Supplemental Table 8). These primers amplify both candidate enhancers and previously assigned degenerate barcodes and add homology arms to the pGL4.23c MPRA vector (GenBank MK484105). We stopped the reaction before plateauing, at 18 cycles. We linearized the pGL4.23c MPRA backbone, while removing the minimal promoter and reporter using HindIII and XbaI. We treated the linearized plasmid with antarctic phosphatase (NEB) following the manufacturer’s protocol, and then gel extracted the plasmid on a 1% agarose gel, as described above. We then cloned our insert into the linearized backbone, transformed, and extracted DNA as described above. We then relinearized the pGL4.23c backbone, containing our enhancer library with SbfI and EcoRI, gel extracted, and inserted our minimal promoter + GFP cassette, which contains overlaps for SbfI and EcoRI.
From human STARR-seq to lentiMPRA 5′/5′:
We used similar methods as in the pGL4.23c library cloning with the following changes. The human STARR-seq library was amplified with pLSmP-AG-2f and pLSmP-AG-5r (Supplemental Table 8) for 17 cycles. After gel extraction, we cloned the insert into the pLS-mP (Addgene 81225)18, which had been linearized with SbfI and AgeI and treated with antarctic phosphatase. The resulting library was recut with SbfI and AgeI, residing between the designed candidate enhancer and barcode, and the minimal promoter was ligated in. We generated the minimal promoter with oligos minP_F and minP_R (Supplemental Table 8), which provide overlaps for SbfI and AgeI. The minimal promoter oligos were phosphorylated and annealed using T4 Ligation Buffer and T4 Polynucleotide Kinase (NEB) at 37 °C for 30 minutes followed by 95 °C for five minutes, ramping down to 25 °C at 5 °C/min. We then diluted the annealed oligos at 1:200 and cloned into the linearized pLS-mP backbone with our enhancer library at a 2:1 molar excess.
From human STARR-seq to lentiMPRA 5′/3′:
We used similar methods as for the pGL4.23c library with the following changes. The human STARR-seq library was amplified with pLSmP-AG-2f and pLSmP-AG-3r (Supplemental Table 8) for 17 cycles. After gel extraction, we cloned the insert into the pLS-mP backbone (Addgene 81225)18, which had been linearized with SbfI and EcoRI, and treated with antarctic phosphatase. Similar to the pGL4.23c library, the resulting library was recut with SbfI and EcoRI again, and we inserted our minimal promoter + GFP cassette, containing overlaps for SbfI and EcoRI.
From human STARR-seq to lentiMPRA 3′/3′:
We used similar methods as for the pGL4.23c library with the following changes. The human STARR-seq library was amplified with pLSmP-AG-3f and pLSmP-AG-3R (Supplemental Table 8) for 13 cycles. After gel extraction, we cloned the insert into the pLS-mP backbone (Addgene 81225)18, which had been linearized with EcoRI only and treated with antarctic phosphatase.
Cell culture, lentivirus packaging, and titration
HEK293T and HepG2 cell culture, lentivirus packaging and titration were performed as previously described with modifications18. Briefly, twelve million HEK293T cells were seeded in 15 cm dishes and cultured for 48 hours. To generate wild-type lentiviral libraries (5′/5′ WT, 5′/3′ WT, and 3′/3′ WT), the cells were co-transfected with 5.5 μg of lentiMPRA libraries, 1.85 μg of pMD2.G (Addgene 12259) and 3.65 μg of psPAX2 (Addgene 12260), which encodes a wild type pol, using EndoFectin Lenti transfection reagent (GeneCopoeia) according to the manufacturer’s instruction. To generate non-integrating lentiviral libraries (5′/5′ MT, 5′/3′ MT, and 3′/3′ MT), pLV-HELP (InvivoGen) that encodes a mutant pol was used instead of psPAX2. After 18 hours, cell culture media was refreshed and TiterBoost reagent (GeneCopoeia) was added. The transfected cells were cultured for 2 days and lentivirus harvested and concentrated using the Lenti-X concentrator (Takara) according to the manufacturer’s protocol. To measure DNA titer for the lentiviral libraries, HepG2 cells were plated at 1x105 cells/well in 24-well plates and incubated for 24 hours. Serial volume (0, 4, 8, 16 μL) of the lentivirus was added with 8 μg/ml polybrene, to increase infection efficiency. The infected cells were cultured for three days and then washed with PBS three times. Genomic DNA was extracted using the Wizard SV genomic DNA purification kit (Promega). Multiplicity of infection (MOI) was measured as relative amount of viral DNA (WPRE region, WPRE_F and WPRE_F) over that of genomic DNA [intronic region of LIPC gene, LIPC_F and LIPC_R (Supplemental Table 8)] by qPCR using SsoFast EvaGreen Supermix (BioRad), according to the manufacturer’s protocol.
Transient transfections and lentiviral infections
HepG2 cells were seeded in 10 cm dishes (2.4 million cells per dish) and incubated for 24 hours. For plasmid-based MPRA, the cells were transfected with 10 μg of the plasmid libraries (HSS, ORI, and pGL4) using X-tremeGENE HP (Roche) according to the manufacturer’s protocol. The X-tremeGENE:DNA ratio was 2:1. For the lentiMPRA, the cells were infected with the lentiviral libraries (5′/5′ WT/MT, 5′/3′ WT/MT, and 3′/3′ WT/MT) along with 8 μg/ml polybrene, with the estimated MOI of 50 for wild-type and 100 for mutant libraries. The cells were incubated for 3 days, washed with PBS three times, and genomic DNA and total RNA was extracted using AllPrep DNA/RNA mini kit (Qiagen). mRNA was purified from the total RNA using Oligotex mRNA mini kit (Qiagen). All experiments for nine libraries were carried out simultaneously to minimize batch effect. Three independent replicate cultures were transfected or infected on different days.
RT-PCR, amplification, and sequencing of RNA and DNA
DNA for all experiments was quantified using the Qubit dsDNA Broad Range Assay kit (Thermo Fisher Scientific). For all samples, a total of 12 μg of DNA was split into 24 50 μL PCR reactions (each with 500 ng of input DNA) with KAPA2G Robust HostStart ReadyMix (Kapa Biosystems) for three cycles with a 65 °C annealing and 40 s extension, using an indexed P5 primer and a unique molecular identifier (UMI)-containing P7 primer (Supplemental Table 9). After three cycles, reactions were pooled and purified with a 1.8x AMPure cleanup, following the manufacturer’s instructions, and eluted in a total of 344 μL of Qiagen EB. The entire purified product was then used for a second round of PCR, split into 16 x 50 μL reactions each, with primers P5 and P7 (Supplemental Table 8). The reaction was followed in real time with Sybr Green (Thermo Fisher Scientific) and stopped before plateauing. PCRs were then pooled and 100 μL of the pooled PCR products was purified with a 0.9x AMPure cleanup and eluted in 30 μL for sequencing.
mRNA for all experiments was treated with Turbo DNase (Thermo Fisher Scientific) following the manufacturer’s instructions and then quantified using the Qubit RNA Assay kit (Thermo Fisher Scientific). For all samples, we performed three 20 μL reverse transcription reactions, each with one third of the sample (up to 500 ng of mRNA). RT was performed using SuperScript IV (Thermo Fisher Scientific) and a gene-specific primer, which attached a UMI (Supplemental Table 9), following the manufacturer’s instructions.
cDNA for each sample was split into eight 50 μL PCRs using an indexed P5 primer and P7 (Supplemental Table 8) for three cycles. Reactions were then pooled together and purified with a 1.5x AMPure reaction and eluted in 129 μL of Qiagen EB. The purified PCR product was then split into six 50 uL PCRs with P5 and P7 following in real time with Sybr Green and stopped before plateauing. PCRs were then pooled and 100 μL of the pooled PCR products was purified with a 0.9-1.8x AMPure cleanup depending on background banding, and eluted in 30 μL for sequencing.
Two experiments at a time (each with three DNA replicates and three RNA replicates) were run on a 75 cycle NextSeq 550 v2 High-Output kit with custom primers for each assay (Supplemental Table 8).
MPRA to evaluate the impact of enhancer orientation
To test enhancers in both orientations relative to the promoter (in the “forward” and “reverse” orientations), we synthesized the same 2,236 genomic sequences tested above18, along with 100 controls previously tested in STARR-seq, which are described below in the Length section below12. These sequences were synthesized as 192 bp fragments with HSS-F-ATGC and HSS-R (Supplemental Table 8). The forward orientation was amplified in a 50 μL PCR reaction using KAPA HiFi 2x Readymix (Kapa Biosystems) and primers “HSS_pGL4_F” and “HSS_pGL4_R1”; the PCR for the reverse orientation used the primers “HSS_pGL4_F_orr2” and “HSSpGL4_1_orr2” (Supplemental Table 8). PCRs were followed in real time with Sybr Green, stopped before plateauing (7 cycles), and purified in a 1X AMPure reaction, eluting in 25 μL of Qiagen EB. 1 μL of the purified products were put into a second PCR reaction, which added 15 bp of barcode sequence and homology to the pGL4.23c vector; the forward orientation used primers HSS_pGL4_F and HSS_pGL4_R2, and the reverse orientation used primers HSS_pGL4_F_orr2 and HSS_pGL4_R2 (Supplemental Table 8).
We linearized the pGL4.23c MPRA backbone with HindIII and XbaI (removing the minimal promoter and reporter), and gel extracted the backbone and insert PCR products. Inserts were cloned into the pGL4.23c plasmid using NEBuilder HiFi DNA Assembly Cloning Kit (NEB), following the manufacturer’s protocol. We transformed 10-beta electrocompetent cells (NEB C3020) with the plasmids, grew up transformations in 100 mL of LB+Amp, and extracted plasmid libraries using a ZymoPure II Plasmid Midiprep Kit (Zymo Research).
To clone in the minimal promoter and GFP for the forward orientation, 20 ng of the forward backbone was amplified with Len_lib_linF and Len_lib_linR (Supplemental Table 8) using NEBNext High-Fidelity 2X PCR Master Mix (NEB); the minimal promoter and GFP was amplified from 10 ng of the pLS-mP plasmid using minGFP_Len_HAF and minGFP_Len_HAR (Supplemental Table 8). For the reverse orientation, 20 ng of the backbone was linearized with Len_lib_linF and Rorr_R2_LinR (Supplemental Table 8); for the reverse orientation insert, previously gel extracted minimal promoter and GFP from pLS-mP was amplified using minPGFP_Revorr_Len_HA_F and Len_lib_linR (Supplemental Table 8). Both backbones were treated with antarctic phosphatase, following the manufacturer’s protocol. All backbones and inserts were gel extracted, with the exception of the reverse orientation insert, which we purified in a 1.8x AMPure reaction. Plasmid libraries were cloned and extracted as previously described.
Transfections (4 independent transfections), DNA/RNA extractions, reverse transcription of mRNA, and qPCRs to amplify barcodes for sequencing were all performed as previously described for the enhancer-length experiments. The final PCRs for the DNA samples were purified in a 1.5X AMPure reaction, using 50 μL of PCR reaction, and eluting in 15 μL of Qiagen EB; cDNA PCRs were gel purified. Libraries were separately denatured and pooled, pooling twice as much of the RNA samples as the DNA samples. Samples were loaded at a final concentration of 1.8 pM on a 75 Cycle NextSeq v2 High-Output kit.
MPRA to evaluate the impact of including additional sequence context at tested elements
Design of enhancer length libraries for array synthesis:
We chose to synthesize the same 2,236 genomic sequences tested above18. We also included the top 50 and bottom 50 haplotypes, averaging 409 bp, from a screen conducted in the STARR-seq vector12 and designed libraries of 192 bp and 354 bp sequences, centered at the position of the previously tested design. We also designed a library of 678 bp sequences for the 2,236 genomic sequences above. We extracted genomic sequence using bedtools getfasta40. To the 192 bp library, we added the HSS-F-ATGC sequence to the 5′ end and the HSS-R-clon sequence to the 3′ end (Supplemental Table 8).
For the 354 bp library, we split each sequence into two overlapping fragments, A and B. Fragment A included positions 1-190 and fragment B included positions 161-354. To fragment A, we appended the HSS-F-ATGC adapter to the 5′ end and the DO_15R_Adapter to the 3′ end. To fragment B, we appended the DO_5F_Adapter to the 5′ end and the HSS-R-clon adapter to the 3′ end (Supplemental Table 8).
For the 678 bp library, we only designed the 2,236 sequences from Inoue et al.18. We split the sequences into 13 different sets of 172 sequences each. We then split each sequence into four fragments. Fragment A included positions 1-190, fragment B included positions 161-352, fragment C included positions 323-514, and fragment D included positions 485-678. Adapters and primers used for the 13 sets of HMPA are included in Supplemental Table 10.
Amplification of the 192 bp library:
All 192 bp enhancers were amplified from the array using HSSF-ATGC and HSS-R-clon (Supplemental Table 8) with KAPA HiFi HotStart Uracil+ ReadyMix PCR Kit (Kapa Biosystems) with SYBR Green (Thermo Fisher Scientific) on a MiniOpticon Real-Time PCR system (Bio-Rad), and stopped before plateauing.
Multiplex pairwise assembly for 354 bp library:
All 5′ fragments were amplified off the array using HSSF-ATGC and DO_15R_PU (Supplemental Table 8) with KAPA HiFi HotStart Uracil+ ReadyMix PCR Kit (Kapa Biosystems) and stopped before plateauing. All 3′ fragments were amplified off the array using DO_5F_PU and HSS-95R (Supplemental Table 8). Both were purified using a 1.8x AMPure cleanup and eluted in 20 μL Qiagen EB. 2 μL of USER enzyme (NEB) was added directly to each purified PCR product, and incubated for 15 minutes at 37 °C followed by 15 minutes at room temperature. Reactions were then treated with the NEBNext End Repair Module (NEB) following the manufacturer’s protocol, and purified using the DNA Clean and Concentrator 5 (Zymo Research) and eluted in 12 μL EB, following the manufacturer’s protocol. We then quantified DNA concentrations for both treated samples using a Qubit and diluted samples to 0.75 ng/uL. We then assembled the 5′ and 3′ fragments as described previously25. Briefly, fragments were allowed to anneal and extend for 5 cycles with KAPA HiFi 2X HotStart Readymix (Kapa Biosystems) before primers HSSF-ATGC and DO_95R were added for amplification (Supplemental Table 8).
Hierarchical multiplex pairwise assembly for 678 bp library:
All libraries were amplified off the array using the primers indicated in Supplemental Table 10 with KAPA HiFi HotStart Uracil+ ReadyMix PCR Kit (Kapa Biosystems) as described above. During the first round of assembly, fragments A and B were assembled with HSSF-ATGC and DO_31R_PU and fragments C and D were assembled with DO_8F_PU and HSS_R (Supplemental Table 10). Assembled libraries were then purified with a 0.65x Ampure cleanup following the manufacturer’s protocol, and eluted in 20 μl. 2 μl of USER enzyme (NEB) was added to the purified assembly reactions and incubated at 37 °C for 15 minutes followed by 15 minutes at room temperature, and then repaired using the NEBNext End Repair Module (NEB), following the manufacturer’s protocol, and purified using the DNA Clean and Concentrator 5 (Zymo Research) and eluted in 10 μL EB. All libraries were then quantified using the Qubit dsDNA HS Assay kit (Thermo Fisher Scientific) and eluted to 0.75 ng/ul. Assemblies AB and CD were then assembled together following the multiplex pairwise assembly protocol25. After the second assembly, libraries were purified using a 0.6x AMPure cleanup and eluted in 30 μL EB. We then amplified 1 uL of each assembly with HSS-F-ATGC-pu1F and HSS-R-clon-pu1R to add flow cell adapters and indexes (Supplemental Table 8). We performed the assembly for each set of 172 sequences separately, as well as for different combinations of sets, up to all 2,236 sequences at once41.
Sequence validation of assembled libraries:
Before cloning, we verified assembly and uniformity of our libraries. The multiplex pairwise assembly library (2,336 354mers) was sequenced on a Miseq v3 600 cycle kit with paired-end 305 bp reads. Reads were merged with PEAR v0.9.542 and aligned to a reference fasta file with BWA mem v0.7.10-r78943. Each of the 13 hierarchical pairwise assembly sub-libraries (172 678mers) as well as different complexities (344, 688, 1032, 1376, 1720, 2064, 2236) were sequenced on a Miseq v3 600 cycle kit with paired-end 300 bp reads. Paired end reads were aligned to a reference fasta file with BWA mem v0.7.10-r78943. As our HMPA library was longer than the maximum Illumina sequencing length (600 bp), we prepared our HMPA sub-library 3 (172 678mers) for sequencing on the PacBioSequel System using V2.1 chemistry (Pacific Biosciences). The library was amplified with pu1L and pu1R and sent to the University of Washington PacBio Sequencing Services for library preparation and sequencing. We obtained 312,277 productive ZMWs with an average Pol Read length of 30,806 bp. After generating circular consensus sequences, we obtained 218,240 CCS reads with a mean read length of 882 bp.
Barcoding and cloning of length libraries into pGL4.23c:
We performed a two-step PCR to add barcodes and cloning adapters for pGL4.23c onto our three different libraries. For the 192mer and 354mer library, we amplified 20 ng of the library with HSS-pGL4_F and HSS-pGL4_R1 (Supplemental Table 8) using NEBNext High-Fidelity 2X PCR Master Mix (NEB) for 16 cycles. For the 678mer libraries, we pooled all 13 sub-libraries at equal concentrations, and then amplified 20 ng with the same primers above and conditions above. All PCR products were purified with a 1.5x AMPure cleanup following the manufacturer’s instructions and eluted in 50 μL. We then used 1μL of each purified reaction for a second PCR to append the 15 bp degenerate barcodes and cloning adapters. For the second reaction, we used HSS-pGL4_F and HSS_pGL4_R2 (Supplemental Table 8).
We linearized the pGL4.23c MPRA backbone, while removing the minimal promoter and reporter using HindIII and XbaI. We treated the linearized plasmid with antarctic phosphatase following the manufacturer’s protocol, and then gel extracted the plasmid on a 1% agarose gel. We then cloned all three libraries into the pGL4.23c plasmid using the NEBuilder HiFi DNA Assembly Cloning Kit (NEB), following the manufacturer’s protocol. The library was then transformed into 10-beta electrocompetent cells (NEB C3020), grown in 100 mL of LB+Amp, and extracted using the ZymoPure II Plasmid Midiprep Kit (Zymo Research). We then relinearized each library with Len_lib_linF and Len_lib_linR and amplified the minimal promoter and GFP from 10 ng of the pLSMP plasmid using minGFP_Len_HAF and minGFP_Len_HAR (Supplemental Table 8). We then gel extracted all linearized libraries and the minimal promoter + GFP insert on a 1% agarose gel. We inserted the minimal promoter and GFP using the NEBuilder HiFi DNA Assembly Cloning Kit (NEB) as described above.
MPRA of all enhancer length libraries:
The day before transfection, we seeded HepG2 cells in five 10 cm dishes. Day of transfection, we combined the 192, 354, and 678 pGL4.23c libraries at a 1:1:1 molar ratio and transfected 21 μg of pooled libraries into each 10 cm dish using Lipofectamine 3000 (Thermo Fisher Scientific), following the manufacturer’s protocol. 48 hours post transfection, we extracted DNA and RNA from each replicate using the AllPrep DNA/RNA Mini Kit (Qiagen), following the manufacturer’s instructions.
We added UMIs to a total of 4 μg of DNA from each replicate split across eight reactions with KAPA2G Robust HotStart ReadyMix (Kapa Biosystems) for three cycles with a 65 °C annealing and 40 s extension, using P5-pLSmP-5bc-idx and P7-pGL4.23c-UMI (Supplemental Table 8). After three cycles, reactions were pooled and purified with a 1.8x AMPure cleanup, following the manufacturer’s instructions, and eluted in a total of 87 μL of Qiagen EB. The entire purified product was then used for a second round of PCR, split into 6 50 μL reactions each, with primers P5 and P7. The reaction was followed in real time with Sybr Green and stopped before plateauing. PCRs were then pooled and 100 μL of the pooled PCR products was purified with a 0.9x AMPure cleanup and eluted in 30 μL for sequencing.
RNA for each replicate was treated with Turbo DNase (Thermo Fisher Scientific) following the manufacturer’s protocol and then quantified using the Qubit RNA Assay kit (Thermo Fisher Scientific). For all samples, we performed two 15 μL reverse transcription reactions, using a total of 15.75 μL RNA (½ total). RT was performed using Thermo Fisher SuperScript IV (Thermo Fisher Scientific) and a gene-specific primer (P7-pGL4.23c-UMI), which attached a UMI, following the manufacturer’s instructions. cDNA for each sample was split into four 50 μL PCRs using P5-pLSmP-5bc-idx and P7 for three cycles. Reactions were then pooled together and purified with a 1.5x AMPure reaction and eluted in 64.5 μL of Qiagen EB. The purified PCR product was then split into three 50 μL PCRs with P5 and P7, followed in real time with Sybr Green (Thermo Fisher Scientific), and stopped before plateauing (11 cycles). PCR products were purified with a 1.5x AMPure reaction before sequencing on a 75 cycle NextSeq 550 v2 High-Output kit.
For barcode associations, we amplified 5 ng of each library with P5_pGL4_Idx_assF and P7-pGL4-ass-R (Supplemental Table 8), following in real time with Sybr Green for 14-15 cycles. PCR products were purified with a 1x AMPure cleanup and eluted in 20 μL of Qiagen EB for sequencing. Libraries were separately denatured and pooled to account for part of the clustering bias on the NextSeq. We brought the 192 library to a final concentration of 1.65 pM, the 354 library to a final concentration of 2.15 pM, and the 678 library to a final concencentration of 2.9 pM. We then pooled an equal volume of each library and loaded on a 300 cycle NextSeq 550 v2 Mid-Output kit with an 80 bp read 1 and 213 bp read 2 (in order to sequence part of contributing oligos A, C, and D).
MPRA processing pipeline
Reproducible MPRA analysis pipeline implementation.
We developed and utilized a fully reproducible processing pipeline to process the raw MPRA data. The sections below document the various components of the pipeline, which borrow heavily from our earlier work18, and were implemented into a reproducible Nextflow-based codebase named MPRAflow44.
Associating barcodes to designed elements.
For each of the barcode association libraries, we generated Fastq files with bcl2fastq v2.18 (Illumina Inc.), splitting the sequencing data into an index file delineating the barcode and two paired-end read files delineating the corresponding element linked to the barcode. If the paired-end reads overlapped in sequence, they were merged into one and aligned using BWA mem v0.7.10-r78943 to a reference Fasta file comprised of the designed elements (Supplemental Table 2). We carried forward the subset of merged reads whose mapped length corresponded to the expected length of the designed element ± 5 bp (i.e., 171 ± 5, 192 ± 5, 354 ± 5, and 678 ± 5, depending on the element size), allowing indels or mismatches. To minimize the impact of sequencing errors, we associated a barcode to an element if: i) the barcode:element pair was sequenced at least three independent times, and ii) ≥90% of the barcode mapped to a single element. These barcode associations were then used as a dictionary to match barcodes detected in the RNA and DNA sequencing libraries in different MPRA designs.
Replicates, normalization, and RNA/DNA activity scores:
Barcodes were counted for RNA and DNA samples for each MPRA experiment, using UMIs to collapse barcodes derived from the same molecule during PCR, and mapped to the element they were linked to, as identified by the dictionary of barcode:element associations. To normalize RNA and DNA for different sequencing depths in each sample, we followed a nearly identical scheme as one we had previously devised18. Briefly, for each replicate of each MPRA design, we first considered the subset of barcodes that were observed for both the RNA and DNA samples of the replicate. We then summed up the counts of all barcodes contributing to each element and computed the normalized counts as the counts per million (cpm) sequenced reads of that library. Finally, we computed enhancer activity scores as log2(RNA [cpm]/DNA [cpm]). To account for the differential scale among replicates of each experiment, we divided the RNA/DNA ratios by the median across the replicate value before averaging them. Due to low counts in the initial round of sequencing and poor sample quality, the three replicates from the 5′/3′ MT and 3′/3′ MT were re-sequenced, and the data from each pair of technical replicates was pooled together across the two independent sequencing runs. Even after pooling, the first replicates of these two assays exhibited poorer inter-replicate concordance than the other replicates (Figure 2A, Supplemental Figure 3), and thus were excluded during replicate averaging (Supplemental Table 2). In practice, this decision very modestly altered the numerical results, and did not change the study’s conclusions.
Modeling and Analyses
Features considered:
For each candidate enhancer, we computed a total of 915 features derived from either: i) the sequence itself, or ii) experimentally measured information, computed as a mean signal extracted from the corresponding region of the human genome (Supplemental Table 3, with a full list of features and data sources: Supplemental Table 4). The sequence-based features represent the conservation of the sequence, general G/C content, predicted chromatin state, and likelihood of binding to an assortment of transcription factors and RNA binding proteins. In contrast, the experimentally derived features represent empirical measurements of chromatin/epigenetic state, binding to transcription factors, or transcriptional activity. The features were derived from custom Perl scripts, the UCSC genome browser45, DeepSEA v0.9446, DeepBind v0.1147, with epigenomic data derived from the Epigenomics Roadmap Consortium48, CAGE data from the FANTOM Consortium33, and ChIP-seq data from the ENCODE Consortium7.
Feature pre-processing:
Right-skewed data such as ChIP-seq and CAGE signal were log-transformed to approximate a normal distribution, and each feature was then z-score normalized to scale the features similarly. This enabled a direct comparison of coefficients among features derived from the resulting linear models.
Model training:
As described before18, we trained a lasso regression model on each of 10 folds of the data, selecting enhancers which were measured with at least 10 independent barcodes to reduce the impact of measurement noise in the assessment of model quality. A lasso regression model was chosen specifically because it employs an L1 regularization penalty, which leads to the selection of the fewest features that maximally explain the data. The strength of the regularization was controlled by a single λ parameter, which was optimized using 10-fold cross-validation on the entire dataset. To evaluate the most relevant features selected, we trained a lasso regression model on the full dataset and visualize the top 10-30 coefficients with the greatest magnitude. A full table of the selected features and their coefficients are provided (Supplemental Table 5). To compare differential enhancer activity between a pair of assays, we fit a loess (“locally estimated scatterplot smoothing”) regression between one assay relative to the other and computed residuals from this fit, using the “loess” function in R. We then fit lasso regression models to explain these residuals, based upon the aforementioned procedure.
Luciferase assays
The “medium”, “long” and “deleted” versions of ten enhancers (total 30 sequences), APOE enhancer (positive control), and neg2 sequence (negative control) were synthesized along with minimal promoter and adaptor sequences (Supplemental Table 8) and cloned into the BglII and NcoI site of the pGL4.23c vector by Twist. These were selected based on highest differential activities, reproducibility and base balance (for synthesis). As two of them (chr2:106744003-106744357_medium and chr10:114391246-114391924_del) failed the cloning, these sequences were synthesized by Twist and manually cloned into the BglII and NcoI site of the pGL4.23c vector using NEBuilder HiFi DNA Assembly Cloning Kit (NEB). The plasmid sequences were confirmed by Sanger sequencing. All the 32 plasmids and empty pGL4.23c were individually transfected along with pGL4.74 (Promega) into 1x104 HepG2 cells, as previously described18. Four independent replicate cultures were transfected. Firefly and Renilla luciferase activities were measured on a Glomax microplate reader (Promega) using the Dual-Luciferase Reporter Assay System (Promega). Enhancer activity was measured as the fold change of each plasmid’s firefly luciferase activity normalized to Renilla luciferase activity.
DATA AVAILABILITY
We developed a fully reproducible MPRA processing pipeline available to process the data into final enhancer activity scores. Raw and processed data has been deposited in the Gene Expression Omnibus (GEO) at accession number GSE142696.
CODE AVAILABILITY
A reproducible processing pipeline for MPRA data is available as a Nextflow-based MPRA processing pipeline named MPRAflow (https://github.com/shendurelab/MPRAflow)44.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Seungsoo Kim and other members of the Shendure and Ahituv laboratories for general advice and critical feedback on the manuscript. This work was supported by the National Human Genome Research Institute grants 1UM1HG009408 (N.A. and J.S.), 5R01HG009136 (J.S.), 1R21HG010065 (N.A.), and 1R21HG010683 (N.A.), 5F30HG009479 (J.K.); National Institute of Mental Health grants 1R01MH109907 (N.A.) and 1U01MH116438 (N.A.), NRSA NIH fellowship 5T32HL007093 (V.A.) and the Uehara Memorial Foundation (F.I.). J.S. is an investigator of the Howard Hughes Medical Institute.
Footnotes
Competing interests
The authors declare no competing interests.
REFERENCES
- 1.Banerji J, Rusconi S & Schaffner W Expression of a beta-globin gene is enhanced by remote SV40 DNA sequences. Cell 27, 299–308 (1981). [DOI] [PubMed] [Google Scholar]
- 2.Moreau P et al. The SV40 72 base repair repeat has a striking effect on gene expression both in SV40 and other chimeric recombinants. Nucleic Acids Res. 9, 6047–6068 (1981). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Banerji J, Olson L & Schaffner W A lymphocyte-specific cellular enhancer is located downstream of the joining region in immunoglobulin heavy chain genes. Cell 33, 729–740 (1983). [DOI] [PubMed] [Google Scholar]
- 4.Neuberger MS Expression and regulation of immunoglobulin heavy chain gene transfected into lymphoid cells. EMBO J. 2, 1373–1378 (1983). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bernstein BE et al. The NIH Roadmap Epigenomics Mapping Consortium. Nat. Biotechnol. 28, 1045–1048 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kawaji H, Kasukawa T, Forrest A, Carninci P & Hayashizaki Y The FANTOM5 collection, a data series underpinning mammalian transcriptome atlases in diverse cell types. Sci Data 4, 170113 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.The ENCODE Project Consortium. A User’s Guide to the Encyclopedia of DNA Elements (ENCODE). PLoS Biol. 9, e1001046 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Patwardhan RP et al. High-resolution analysis of DNA regulatory elements by synthetic saturation mutagenesis. Nat. Biotechnol. 27, 1173–1175 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Patwardhan RP et al. Massively parallel functional dissection of mammalian enhancers in vivo. Nat. Biotechnol. 30, 265–270 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Melnikov A et al. Systematic dissection and optimization of inducible enhancers in human cells using a massively parallel reporter assay. Nat. Biotechnol. 30, 271 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vockley CM et al. Massively parallel quantification of the regulatory effects of noncoding genetic variation in a human cohort. Genome Res. 25, 1206–1214 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tewhey R et al. Direct Identification of Hundreds of Expression-Modulating Variants using a Multiplexed Reporter Assay. Cell 172, 1132–1134 (2018). [DOI] [PubMed] [Google Scholar]
- 14.Ulirsch JC et al. Systematic Functional Dissection of Common Genetic Variation Affecting Red Blood Cell Traits. Cell 165, 1530–1545 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu S et al. Systematic identification of regulatory variants associated with cancer risk. Genome Biol. 18, 194 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Arnold CD et al. Genome-wide quantitative enhancer activity maps identified by STARR-seq. Science 339, 1074–1077 (2013). [DOI] [PubMed] [Google Scholar]
- 17.Kwasnieski JC, Fiore C, Chaudhari HG & Cohen BA High-throughput functional testing of ENCODE segmentation predictions. Genome Res. 24, 1595–1602 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Inoue F et al. A systematic comparison reveals substantial differences in chromosomal versus episomal encoding of enhancer activity. Genome Res. 27, 38–52 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Klein JC et al. Functional testing of thousands of osteoarthritis-associated variants for regulatory activity. Nat. Commun. 10, 2434 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Arnold CD et al. Quantitative genome-wide enhancer activity maps for five Drosophila species show functional enhancer conservation and turnover during cis-regulatory evolution. Nat. Genet. 46, 685–692 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Klein JC, Keith A, Agarwal V, Durham T & Shendure J Functional Characterization of Enhancer Evolution in the Primate Lineage. bioRxiv 283168 (2018) doi: 10.1101/283168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Muerdter F et al. Resolving systematic errors in widely used enhancer activity assays in human cells. Nat. Methods 15, 141–149 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vanhille L et al. High-throughput and quantitative assessment of enhancer activity in mammals by CapStarr-seq. Nat. Commun. 6, 6905 (2015). [DOI] [PubMed] [Google Scholar]
- 24.Wang X et al. High-resolution genome-wide functional dissection of transcriptional regulatory regions and nucleotides in human. Nat. Commun. 9, 5380 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Klein JC et al. Multiplex pairwise assembly of array-derived DNA oligonucleotides. Nucleic Acids Res. 44, e43 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kircher M et al. Saturation mutagenesis of disease-associated regulatory elements: Supplementary Information. (2018) doi: 10.1101/505362. [DOI] [Google Scholar]
- 27.Hill AJ et al. On the design of CRISPR-based single-cell molecular screens. Nat. Methods 15, 271–274 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sack LM, Davoli T, Xu Q, Li MZ & Elledge SJ Sources of Error in Mammalian Genetic Screens. G3 6, 2781–2790 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Smith RP et al. Massively parallel decoding of mammalian regulatory sequences supports a flexible organizational model. Nat. Genet. 45, 1021–1028 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Consortium GTEx. Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science 348, 648–660 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shiraki T et al. Cap analysis gene expression for high-throughput analysis of transcriptional starting point and identification of promoter usage. Proc. Natl. Acad. Sci. U. S. A. 100, 15776–15781 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.FANTOM. Transcribed enhancers leadwaves of coordinated transcription in transitioning mammalian cells. Science (2015) doi: 10.6084/m9.figshare.1288777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Andersson R et al. An atlas of active enhancers across human cell types and tissues. Nature 507, 455–461 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Engreitz JM et al. Local regulation of gene expression by lncRNA promoters, transcription and splicing. Nature 539, 452–455 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.van Arensbergen J et al. Genome-wide mapping of autonomous promoter activity in human cells. Nat. Biotechnol. (2016) doi: 10.1038/nbt.3754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kvon EZ, Stampfel G, Yáñez-Cuna JO, Dickson BJ & Stark A HOT regions function as patterned developmental enhancers and have a distinct cis-regulatory signature. Genes Dev. 26, 908–913 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mikhaylichenko O et al. The degree of enhancer or promoter activity is reflected by the levels and directionality of eRNA transcription. Genes Dev. 32, 42–57 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Weingarten-Gabbay S et al. Systematic interrogation of human promoters. Genome Res. 29, 171–183 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Plesa C, Sidore AM, Lubock NB, Zhang D & Kosuri S Multiplexed gene synthesis in emulsions for exploring protein functional landscapes. Science 359, 343–347 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
METHODS-ONLY REFERENCES
- 40.Quinlan AR & Hall IM BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Klein JC, Agarwal V, Inoue F, Keith A, Martin B, Kircher M, Ahituv N, Shendure J. A systematic evaluation of the design, orientation, and sequence context dependencies of massively parallel reporter assays. Protocol Exchange (2020) doi: 10.21203/rs.3.pex-1065/v1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang J, Kobert K, Flouri T & Stamatakis A PEAR: a fast and accurate Illumina Paired-End reAd mergeR. Bioinformatics 30, 614–620 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li H Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv [q-bio.GN] (2013). [Google Scholar]
- 44.Gordon MG et al. lentiMPRA and MPRAflow for high-throughput functional characterization of gene regulatory elements. Nat. Protoc. 15, 2387–2412 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Karolchik D et al. The UCSC Genome Browser database: 2014 update. Nucleic Acids Res. 42, D764–70 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhou J & Troyanskaya OG Predicting effects of noncoding variants with deep learning-based sequence model. Nat. Methods 12, 931–934 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Alipanahi B, Delong A, Weirauch MT & Frey BJ Predicting the sequence specificities of DNA- and RNA-binding proteins by deep learning. Nat. Biotechnol. 33, 831–838 (2015). [DOI] [PubMed] [Google Scholar]
- 48.Roadmap Epigenomics, Consortium et al. Integrative analysis of 111 reference human epigenomes. Nature 518, 317–330 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
We developed a fully reproducible MPRA processing pipeline available to process the data into final enhancer activity scores. Raw and processed data has been deposited in the Gene Expression Omnibus (GEO) at accession number GSE142696.