

ABSTRACT
Background
Dietary carbohydrate type may influence cardiometabolic risk through alterations in the gut microbiome and microbial-derived metabolites, but evidence is limited.
Objectives
We explored the relative effects of an isocaloric exchange of dietary simple, refined, and unrefined carbohydrate on gut microbiota composition/function, and selected microbial metabolite concentrations.
Methods
Participants [n = 11; age: 65 ± 8 y; BMI (in kg/m2): 29.8 ± 3.2] were provided with each of 3 diets for 4.5 wk with 2-wk washout, according to a randomized, crossover design. Diets [60% of energy (%E) carbohydrate, 15%E protein, and 25%E fat] differed in type of carbohydrate. Fecal microbial composition, metatranscriptomics, and microbial-derived SCFA and secondary bile acid (SBA) concentrations were assessed at the end of each phase and associated with cardiometabolic risk factors (CMRFs).
Results
Roseburia abundance was higher (11% compared with 5%) and fecal SBA concentrations were lower (lithocolic acid –50% and deoxycholic acid –64%) after consumption of the unrefined carbohydrate diet relative to the simple carbohydrate diet [false discovery rate (FDR): all P < 0.05), whereas Anaerostipes abundance was higher (0.35% compared with 0.12%; FDR: P = 0.04) after the simple carbohydrate diet relative to the refined carbohydrate diet. Metatranscriptomics indicated upregulation of 2 cellular stress genes (FDR: P < 0.1) after the unrefined carbohydrate diet compared with the simple carbohydrate or refined carbohydrate diets. The microbial expression of 3 cellular/oxidative stress and immune response genes was higher (FDR: P < 0.1) after the simple carbohydrate diet relative to the refined carbohydrate diet. No significant diet effect was observed in fecal SCFA concentrations. Independent of diet, we observed 16 associations (all FDR: P < 0.1) of taxon abundance (15 phylum and 1 genera) with serum inflammatory markers and also with fecal SCFA and SBA concentrations.
Conclusions
Consuming an unrefined carbohydrate–rich diet had a modest effect on the gut microbiome and SBAs, resulting in favorable associations with selected CMRFs. Simple carbohydrate– and refined carbohydrate–rich diets have distinctive effects on the gut microbiome, suggesting differential mechanisms mediate their effects on cardiometabolic health. This trial was registered at clinicaltrials.gov as NCT01610661.
Keywords: carbohydrate quality, microbiome, metatranscriptomics, cardiometabolic risk factors, bile acids, randomized clinical trial
Introduction
Cardiovascular disease (CVD) affects 48% of adults in the United States and is closely associated with the clustering of key risk factors (abdominal obesity, insulin resistance, hyperglycemia, dyslipidemia, and hypertension) (1). Prospective cohort studies have consistently reported favorable associations between diets rich in unrefined carbohydrate (whole grains and fiber) and rates of CVD and total mortality (2, 3). In contrast, the consumption of simple and refined carbohydrate, as part of a Western diet, is generally associated with poor cardiometabolic health (4–7). Clinical trials indicate that the impact of carbohydrate quality on CVD is mediated in part via effects on cardiometabolic risk factors (CMRFs), including inflammatory markers, blood lipid, and lipoprotein profiles (8–10). We have previously documented that a diet enriched in refined compared with simple or unrefined carbohydrate resulted in higher fasting serum LDL and non-HDL cholesterol concentrations, suggesting that refined carbohydrate may have differential effects on CMRFs distinct from simple and unrefined carbohydrate (7).
The human gut microbiome and microbial-derived metabolites have emerged as important contributors to poor metabolic health and CVD development (11). Experimental evidence suggests that dietary carbohydrate quality and quantity can rapidly alter microbial composition and function (12, 13). Dietary fiber is a main fuel source for distinct microbial taxa (14, 15) and results in the production of SCFAs, which have a broad array of favorable physiological effects on the regulation of host metabolism, gut health, and immune responses (14, 15). Limited data suggest that dietary carbohydrate-induced changes in gut microbial composition affect the colonic bile acid pool and subsequently alter farnesoid X receptor and G protein-coupled bile acid receptor 1 (TGR5) signaling to influence host immune and metabolic signaling (16), as well as glucose homeostasis and inflammation (16, 17). Thus, modulation of gut microbiome composition and subsequent production of microbial-derived metabolites may be a potential mechanism mediating associations between carbohydrate quality and CMRFs.
In a secondary analysis of our prior work (7), we determined the effect of an isocaloric exchange of different types of carbohydrate (simple, refined, and unrefined) on gut microbial composition (16S sequencing) and function (metatranscriptomics) and concentrations of selected fecal microbial-derived metabolites [SCFAs and secondary bile acids (SBAs)]. In addition, we explored associations of microbial taxon with microbial-derived metabolites and serum CMRFs. We hypothesized that carbohydrate type would differentially affect gut microbial composition, function, and concentrations of fecal SCFAs and SBAs. In addition, we hypothesized that microbial composition would be associated with serum CMFRs.
Methods
Study design and participants
The current study is a secondary analysis of a randomized clinical trial designed to examine the effect of carbohydrate type on CMRFs. Results from the primary study have been published, including details of the study design, inclusion/exclusion criteria, recruitment, and power calculations for the primary outcomes (7). The randomization sequence for each participant was generated by the statistician before the start of the study according to a block design, and assignment was based on enrollment date and time. Investigators and laboratory personnel were blinded to the random order. The study was conducted between 2012 and 2015, in accordance with the Declaration of Helsinki guidelines and with approval of the Institutional Review Board of Tufts University and Tufts Medical Center, and registered at clinicaltrials.gov as NCT01610661 on November 7, 2011.
Briefly, 11 participants (7 postmenopausal women and 4 men; 64% Caucasian) met all recruitment criteria and completed all 3 diet phases, following a randomized crossover design (Supplemental Figure 1). Blood samples were collected at the end of each diet phase. Three samples were collected following a 12-h fast, and 1 postprandial sample was collected 4 h following the consumption of a meal consistent with the respective diet phase.(7) Weight was maintained constant throughout the study. Average ± SD of baseline characteristics were as follows: age: 65 ± 8 y; systolic blood pressure: 123 ± 10 mm Hg; diastolic blood pressure: 71 ± 9 mm Hg; BMI (in kg/m2): 29.8 ± 3.2; glucose: 5.6 ± 0.6 mmol/L; and LDL-C: 3.5 ± 0.7 mmol/L (7).
Study diets
Participants consumed each of 3 diets enriched in simple, refined, or unrefined carbohydrate-containing foods for 4.5 wk, with a minimum 2-wk washout period between each diet (Supplementary Figure 1). Each participant visited the Metabolic Research Unit 3 times per week to consume 1 study meal on site and received additional meals for consumption offsite. Diets for each carbohydrate phase were isocaloric, matched for macronutrients (60% energy total carbohydrate, 15% energy protein, and 25% energy total fat), and differed only in carbohydrate type. The simple carbohydrate diet contained a higher proportion of foods containing sucrose and/or high-fructose corn syrup. The refined grain carbohydrate diet included a higher proportion of foods made from refined grains, such as white rice, white bread, and white pasta. The unrefined grain carbohydrate diet contained a higher proportion of foods made from whole grains. Detailed sample study menus have previously been described (7).
Stool collection
Study participants were given stool sample collection kits after the end of the third week of each dietary intervention phase along with instructions for collection and storage during week 4 of the intervention. Stool collection kits contained an ice pack for transporting samples back to the Metabolic Research Unit at the Jean Mayer USDA Human Nutrition Research Center on Aging within 18 h of production. Samples were aliquoted upon receipt, with 1 aliquot immediately processed with a PowerMicrobiome RNA Extraction kit from MoBio in the Phoenix laboratory at Tufts Medical Center. Remaining aliquots were kept at −80°C until the extraction of bacterial DNA using Qiagen's QIAmp DNA Stool minikit and for SCFA analysis. Another aliquot was freeze-dried for subsequent bile acid analysis.
Sample processing
16S amplification and sequencing
We used the 515F/806R primers described by Caporaso et al. (18) (forward primer sequence of GTGCCAGCMGCCGCGGTAA and reverse primer sequence of GGACTACHVGGGTATCTAAT) to isolate and amplify the V4 hypervariable region of the bacterial 16S ribosomal subunit gene in DNA extracted from stool samples. V4 16S amplicon molecules were sequenced using an Illumina MiSeq machine at the Tufts University Core Facility. Reads were automatically demultiplexed into individual paired forward and reverse FASTq files for each sample. Sequences with ambiguous, low-quality, mismatched, or unknown barcodes were discarded.
Metatranscriptomics ribosomal RNA-depleted RNA sequencing
We used MoBio's PowerMicrobome RNA Extraction kit to isolate RNA from aliquots from each stool sample, and we used Illumina's TruSeq Stranded Total RNA kit with Ribo-Zero to deplete ribosomal RNA (rRNA). These rRNA-depleted complementary DNA libraries were barcoded, pooled, and run through an Illumina HiSeq 2500 with 6 samples per lane. To remove lingering rRNA and human RNA contamination, we aligned all reads to hg19 and rRNA reference libraries using Bowtie2 and removed all hits.
Fecal bile acids
We measured fecal concentrations of bile acids in freeze-dried stool samples as described previously (19). Extraction and purification were completed using a chloroform methanol solution followed by a multistage sample purification process (19). SBAs were quantified by isotopic dilution using LC–quadrupole time-of-flight MS. Data were normalized by the dry weight of fecal samples and are expressed as micrograms per milligram.
Fecal SCFAs
Fecal SCFAs were analyzed in frozen stool samples using the method described previously (20). Samples underwent a derivatization with 3-nitrophenylhydrazine, and SCFAs were quantified using quadrupole ion trap 5500 LC-MS/MS. The intra-assay CVs were <12%.
Serum cardiometabolic risk factors
We have previously reported assay details and the results of dietary carbohydrate type on serum concentrations of CMRFs, including lipids and lipoproteins (total cholesterol, LDL, HDL, non-HDL, VLDL, triglycerides, and nonesterified fatty acids), glycemic markers (insulin, glycated hemoglobin, and HOMA-IR), and inflammatory markers [IL-6 and high-sensitivity C-reactive protein (hsCRP)] (7). For the current analysis, the CMRF data were used to explore associations with microbial taxon.
Bioinformatics analyses
16S amplicon sequencing data analysis
We processed demultiplexed 16S reads using Qiime2 (version 2018.4) (21). We converted the raw demultiplexed FASTq files into Qiime artifact format. We then denoised and grouped our reads into amplicon sequence variants (ASVs) using the DADA2 algorithm (built into Qiime2) (22) and selected parameters to trim 5 bp from the start of each read, but without truncation. Taxonomy was assigned to each ASV using a naive Bayes classifier built from the Greengenes database (release 13.8) (23). ASVs that the classifier failed to identify were given unique labels, marking them as unassigned but retaining them for analysis.
We performed paired differential analyses (for taxa between each pair of diets at the phylum and genus taxonomic levels) using the R package DESeq2 from Bioconductor (24), which applies a Wald test to a negative binomial generalized linear model. For microbiome data, DESeq2 has been shown to return lower false discovery rates (FDRs) compared with other methods (25), and it is particularly well suited for smaller data sets such as ours (26). Read counts were normalized using the “estimateSizeFactors()” function from DESeq2. Models to derive statistical significance included the host participant as a covariate. We performed an FDR correction, using the Benjamini–Hochberg procedure (27), on all P values within each taxonomic level independently. We considered an FDR P ≤ 0.1 as statistically significant.
Although rarefaction of metagenomic data is not ideal for many types of analyses, it is still a useful step in generating meaningful diversity metrics and is used in Qiime2’s standard operating procedures. For α- and β-diversity calculations only, we rarefied our data to a depth of 80,000 reads, a depth chosen based on DADA2’s output table in order to ensure all of our samples would be included in our diversity analyses with minimal information loss. We used Qiime2’s α-rarefaction tool to estimate the α-diversity of each sample using the Shannon index as our metric, iterating 10 times and taking the average value of all iterations. We used mafft, fasttree, and Qiime2’s alignment mask and phylogeny midpoint-root algorithms to construct a phylogenetic tree from the ASVs generated by DADA2, which we then used to calculate the β-diversity between each pair of samples, reported as weighted UniFrac distances. We performed a principal coordinate analysis on the UniFrac distance matrix. For all statistical tests involving microbial diversity (α and β), we considered a P value ≤ 0.05 as statistically significant.
Metatranscriptomic data analysis
To address limitations in functional annotation of microbial genes, we examined microbial gene families identified de novo by clustering genes closely related in both sequence and function into a single feature. This reduces the effective number of features under analysis (improving statistical power) and more clearly identifies changes in community function instead of community composition. All RNA-sequencing (RNA-seq) reads (after human and rRNA filtering) were pooled into a single FASTq file, which was submitted as an input to the Trinity de novo transcript assembler (28). Trinity builds contigs from input sequences and then clusters those contigs into putative genes. The filtered RNA-seq reads from each sample were aligned to the Trinity-derived transcripts using Bowtie2 (29), and gene and isoform expression values were calculated using RSEM (30). We used RSEM's expected counts output for each assembled transcript and each sample to derive an expression matrix for differential expression analysis. All differential expression calculations were performed with DESeq2 using the same models as described previously (16S amplicon sequencing data analysis). We performed pairwise differential expression between diets, and we report only data where any 1 of the 3 pairwise comparisons passed an FDR P-threshold ≤ 0.10.
Additional statistical analysis
We used a repeated-measures ANOVA model (PROC MIXED), built in SAS for Windows (version 9.4; SAS Institute), to test potential differences in fecal SBAs and SCFA concentrations between diet phases. The model used study participants as a random effect, with main effects including diet phase, diet sequence, age, BMI, and sex. We applied the Tukey–Kramer method as a post hoc analysis whenever our ANOVA model resulted in P values ≤ 0.05. All SCFA and SBA concentrations were log-transformed to normalize their distributions prior to analyses. We considered a P value ≤ 0.05 as statistically significant.
In an exploratory analysis, we assessed the associations of relative taxon abundances with concentrations of fecal SBAs, fecal SCFAs, and serum CMRF markers. Due to the compositional nature of microbe abundance data, we used the additive log ratio transform of our relative abundance measurements in our regression models (31). In order to test the significance of each pairwise association, we performed a series of mixed-effects linear regressions between the z scores
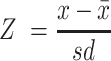 |
(1) |
of each measurement. R package lme4 (32), available through CRAN, was used to produce a mixed model for every taxon–marker pair, with the abundance of the taxon as a fixed effect and participant as a random effect. Taxa were only included in this analysis if they had a mean relative abundance of at least 0.1% across all samples. We performed FDR correction on P values derived from these mixed models, and we report the β coefficients for all models with an FDR P value ≤ 0.1.
Results
Impact of carbohydrate type on microbial composition and diversity
We obtained 330,000–545,000 16S rRNA reads from each sample after quality control filtering. Of these, Qiime2 was able to assign between 12,472 and 139,194 reads per sample to ASVs with phylum-level (or better) taxonomy annotations (median = 104,117); 8,511–128,610 of those reads per sample were annotated all the way to the genus level (median = 87,638). We found 11 distinct phyla and 73 distinct genera with a minimum relative abundance of at least 0.01% in at least 1 sample. The 2 most dominant phyla, Bacteroidetes and Firmicutes, represented 93.4% of all estimated abundance (Figure 1A). Bacteroides and Prevotella were the 2 most abundant genera, collectively representing 37.8% of all estimated abundance (Figure 1B).
FIGURE 1.

Relative abundance of phyla (A) and the 20 most abundant genera (B), based on 16S rDNA sequencing. Data are depicted as estimated percentage relative abundance grouped by study participant (n = 11), designated S01–S15, and ordered by carbohydrate type, with simple carbohydrate diet labeled “S,” refined carbohydrate diet labeled “R,” and unrefined carbohydrate diet labeled “U.” Phyla and genera were assigned using Qiime2 and based on the Greengenes database. rDNA, ribosomal DNA
The abundance of 2 genera was significantly altered by carbohydrate type
Roseburia had a higher abundance after consumption of the unrefined carbohydrate diet (11.3%) compared with the simple carbohydrate diet (5.1%) (FDR: P = 0.04; Figure 2A), but due to larger variance, it was not significantly different from the refined carbohydrate diet (5.3%). Anaerostipes had a higher abundance after the simple (0.35%) and unrefined carbohydrate diet (0.27%) compared with the refined carbohydrate diet (0.12%) (Figure 2B). However, only the difference in abundance between the simple and refined carbohydrate diets was significant (FDR: P = 0.04). Abundance of all microbial taxa examined by carbohydrate type and participants is presented in the supplementary data link.
FIGURE 2.

Relative abundance of the genera Roseburia (A) and Anaerostipes (B) in response to dietary carbohydrate type at the end of the study (n = 11 for each diet). Data are depicted as box-and-whisker plots that display the IQR (box) and extremities (whiskers) and percentage relative abundance. Differential abundance between dietary carbohydrate type was estimated using a negative binomial model including adjustment for participant. Statistical significance was determined using a false discovery rate P value ≤ 0.1.
We calculated the Shannon index of α-diversity H′ within each sample (range: 3.33–6.63; Figure 3A) and found no significant differences in α-diversity between diets (P = 0.37). However, we observed a broad range of stability in α-diversity scores; the value of H′ for several participants remained within a 0.5 window for all 3 diets, whereas that for other participants increased or decreased by as much as 1.8 between diets (Figure 3B).
FIGURE 3.

α-Diversity, within samples grouped by diet (n = 11 for each diet) (A) and each study participant (n = 3 for each participant) (B). α-Diversity was calculated as the Shannon index, calculated with a rarefaction depth of 80,000 sequencing reads. Data are depicted as box-and-whisker plots that display the IQR (box) and extremities (whiskers) of α-diversity. (C) β-Diversity between samples, depicted in a heatmap, was calculated as the weighted UniFrac distance between each pair of samples. (D) The top 2 principal coordinate axes (PC1 and PC2) based on a principal coordinate analysis of the weighted UniFrac distance matrix depicting the bacterial community by participant and carbohydrate type (n = 33). Each participant is represented by a different color, and the diets are represented as a circle for simple carbohydrate, triangle for refined carbohydrate, and square for unrefined carbohydrate. The RCE of each principal coordinate axis indicates the relative portion of total variance within the data set is captured by that axis. Statistical significance was determined using a P value ≤ 0.05. RCE, relative corrected eigenvalue.
We calculated β-diversity metrics among samples using weighted UniFrac distances, which indicate that most of the participants’ microbiomes remained relatively stable across diets and show no evidence of structural ecological shifts in response to carbohydrate type (P = 0.81; Figure 3C). Principal coordinate analysis (Figure 3D, Supplemental Figures 2 and 3) indicated that samples largely clustered by participants rather than distinguishing among dietary carbohydrate type.
Impact of carbohydrate type on microbial metatranscriptomics
We obtained between 15,041,570 and 45,692,257 rRNA-depleted RNA-seq reads per sample after quality control filtering. Total reads (769,040,612) across all samples were assembled into 5,124,369 unique bacterial transcripts and subsequently clustered into 504,115 bacterial gene families, which we used as a reference library for sequence alignment. As a minimum threshold for further analysis, we considered only assembled transcript clusters to which at least 100 reads aligned across all samples, yielding 79,467 assembled transcript clusters with such coverage. After filtering out human RNA and rRNA, each sample had between 358,314 and 4,963,992 reads align to the assembled transcript clusters.
We performed differential expression on the 79,467 assembled transcript families between each pair of diets. Following FDR correction, 9 transcript families were found to be significantly differentially expressed by carbohydrate type (all FDR: P ≤ 0.10: Figure 4A–E). BLASTn searches on these 9 transcripts revealed that 4 of them have no known homologs in the National Center for Biotechnology Information's nucleotide database (33). Of the remaining 5 genes, 3 (heat shock protein 60/chaperonin GroEl, Clp protease, and flavin adenine dinucleotide–containing oxidoreductase) had higher expression after consumption of the simple compared with the refined carbohydrate diet (all FDR: P ≤ 0.1). Notably, these 3 genes also tended to have higher expression with the simple carbohydrate diet compared with the unrefined carbohydrate diet, but this was not statistically significant (Figure 4A–C). Expression of heat shock protein 20/α-crystalin was higher after consumption of the unrefined carbohydrate diet relative to both refined and simple carbohydrate diets. However, only the difference between the unrefined and simple diets was statistically significant (FDR: P = 0.03). Last, expression of CsbD-like protein was higher after consumption of the unrefined compared with the refined carbohydrate diet (FDR: P = 0.08).
FIGURE 4.

Differentially expressed bacterial gene families: (A) heat shock protein 60/chaperon groEl, (B) heat shock protein 20/α-crystalin, (C) Clp protease, (D) CsbD-like protein, and (E) FAD-containing oxidoreductase. Data are depicted as box-and-whisker plots that display the IQR (box) and extremities (whiskers) and bacterial gene family expression (read counts per million). Differential expression between dietary carbohydrate type was estimated using a negative binomial model including adjustment for participant (n = 11 for each diet). Statistical significance was determined using a false discovery rate P value ≤ 0.1. Asterisk (*) indicates a significant difference between diets. Points over the individual box plots indicate participant data above the third quartile.
Impact of carbohydrate type on fecal concentrations of SBAs and SCFAs
Table 1 displays concentrations of CMRFs, as previously reported (7), and fecal concentrations of microbial-derived metabolites. Fecal concentrations of SBAs were significantly lower at the end of the unrefined carbohydrate diet phase compared with the simple carbohydrate diet phase, with intermediate concentrations at the end of the refined carbohydrate diet (both P ≤ 0.05; Table 1). Fecal concentrations of lithocholic acid were 50% lower after the unrefined compared with the simple carbohydrate diet (2.4 compared with 4.8 µmol/g; P ≤ 0.01), and deoxycholic acid concentrations were 64% lower (2.0 compared with 5.6 µmol/g; P = 0.03). No significant differences were observed in fecal concentrations of SCFAs among the 3 carbohydrate diets (Table 1).
TABLE 1.
Adjusted least-squares means of microbial-derived lipid metabolites and serum cardiometabolic risk factors at the end of each diet phase1
| Variables | Simple carbohydrate | Refined carbohydrate | Unrefined carbohydrate | P value2 |
|---|---|---|---|---|
| Fecal microbial metabolites | ||||
| Bile acids3 , μmol/g fecal weight | ||||
| Lithocholic acid | 4.8 (3.5, 6.1)a | 3.5 (2.1, 4.8)a,b | 2.4 (1.1, 3.7)b | 0.005 |
| Deoxycholic acid | 5.6 (3.6, 7.6)a | 3.8 (1.8, 5.9)a,b | 2.0 (0.03, 4.1)b | 0.03 |
| SCFAs, µmol/g fecal weight | ||||
| Acetate | 12.7 (9.8, 15.6) | 10.8 (7.9, 13.7) | 10.4 (7.5, 13.3) | 0.81 |
| Propionate | 4.5 (3.4, 5.6) | 3.8 (2.7, 4.9) | 3.7 (2.6, 4.8) | 0.74 |
| Butyrate | 4.1 (2.9, 5.3) | 3.5 (2.29, 4.7) | 3.2 (2.0, 4.4) | 0.95 |
| Isobutyrate | 1.1 (0.7, 1.4) | 0.9 (0.5, 1.3) | 0.7 (0.3, 1.1) | 0.43 |
| 2-Methylbutyrate | 0.8 (0.5, 1.1) | 0.7 (0.4, 1.0) | 0.5 (0.2, 0.8) | 0.53 |
| Valerate | 0.9 (0.7, 1.1) | 0.8 (0.6, 1.0) | 0.8 (0.6, 1.0) | 0.89 |
| Isovalerate | 0.5 (0.3, 0.7) | 0.4 (0.2, 0.6) | 0.3 (0.1, 0.5) | 0.49 |
| 3-Methylvalerate | 0.03 (0.02, 0.04) | 0.03 (0.01, 0.04) | 0.02 (0.01, 0.04) | 0.80 |
| Isocaproate | 0.04 (0.02, 0.06) | 0.03 (0.01, 0.05) | 0.03 (0.01, 0.05) | 0.26 |
| Caproate | 0.3 (0.1, 0.5) | 0.3 (0.2, 0.5) | 0.3 (0.1, 0.5) | 0.25 |
| Serum cardiometabolic risk factors | ||||
| Glycemic | ||||
| Glucose, mmol/L | 5.3 (5.0, 5.6) | 5.2 (5.0, 5.5) | 5.1 (4.8, 5.4) | 0.52 |
| Insulin, mU/L | 12.4 (9.3, 16.5) | 12.0 (9.0, 16.0) | 10.7 (8.0, 14.3) | 0.36 |
| Glycated hemoglobin, % | 5.7 (4.9, 6.6) | 5.8 (5.0, 6.7) | 5.7 (4.9, 5.7) | 0.65 |
| HOMA-IR | 2.9 (2.1, 4.0) | 2.8 (2.0, 3.9) | 2.4 (1.7, 3.4) | 0.36 |
| Blood lipids, mmol/L | ||||
| Total cholesterol | 5.2 (3.8, 7.2)b | 5.5 (4.0, 7.6)a | 5.2 (3.8, 7.1)b | <0.01 |
| LDL cholesterol | 3.2 (2.2, 4.5)b | 3.4 (2.4, 4.8)a | 3.1 (2.2, 4.4)b | <0.01 |
| Non-HDL cholesterol | 4.0 (2.8, 5.7)b | 4.3 (3.0, 6.1)a | 4.0 (2.8, 5.6)b | <0.01 |
| HDL cholesterol | 1.2 (1.0, 1.5) | 1.2 (1.0, 1.5) | 1.2 (1.0, 1.5) | 0.32 |
| VLDL cholesterol | 0.8 (0.5, 1.3) | 0.8 (0.5, 1.4) | 0.8 (0.5, 1.3) | 0.17 |
| Total cholesterol: HDL cholesterol | 4.3 (3.6, 5.1)b | 4.5 (3.8, 5.4)a | 4.3 (3.6, 5.2)b | <0.01 |
| LDL cholesterol: HDL cholesterol | 2.6 (2.1, 3.2)b | 2.8 (2.2, 3.4)a | 2.6 (2.1, 3.2)b | 0.01 |
| Triglyceride | 1.7 (1.0, 2.9) | 1.9 (1.1, 3.1) | 1.7 (1.0, 2.9) | 0.19 |
| Inflammatory markers | ||||
| hcCRP, mg/L | 1.9 (0.6, 4.4) | 2.0 (0.6, 4.6) | 2.1 (0.7, 4.7) | 0.84 |
| IL-6, pg/mL | 0.6 (0.4, 0.7) | 0.6 (0.4, 0.8) | 0.6 (0.4, 0.8) | 0.77 |
Values are least-squares means and 95% CIs from the repeated-measures ANOVA model, with the main effect of diet and covariates (phase, sequence, age, BMI, and sex) and random effect of participant. Bile acids and SCFAs were log-transformed to normalize their distributions. When a diet effect was significant at P ≤ 0.05, multiple comparisons were carried out with the Tukey–Kramer method. Least-squares means with different letters were significantly different from each other. hsCRP, high-sensitivity C-reactive protein.
P values for repeated-measures ANOVA.
Bile acid analyses were completed on a sample of n = 10.
Associations of microbial composition with microbial-derived metabolites and cardiometabolic risk factors
All associations of microbial composition with microbial-derived metabolites and CMRFs are displayed in the supplementary data link. We observed 15 phylum-level and 1 genus-level (Acidaminococcus) associations with serum concentrations of CMRFs and fecal concentrations of microbial-derived metabolites that remained significant after FDR correction (all FDR: P ≤ 0.1; Table 2). The phylum Verrucomicrobia had a negative association with fecal concentrations of deoxycholic acid. There was a positive association between the phylum Lentisphaerae and fecal concentrations of lithocholic acid. The phyla Lentisphaerae and Cyanobacteria both had positive associations with individual SCFA concentrations. A negative association was observed between the phylum Actinobacteria and serum concentrations of the proinflammatory cytokine IL-6, and the genus Acidaminococcus was positively associated with serum concentrations of hsCRP. No significant associations were observed among the other CMRFs (blood lipids and glycemic markers) and microbial taxon abundance (see the supplementary data link).
TABLE 2.
Associations of microbial taxon abundance with fecal secondary bile acids, fecal SCFAs, and serum inflammatory markers1
| Variables | Taxon | β coefficient2 | SE of β | FDR P value3 |
|---|---|---|---|---|
| Bile acids, μmol/g fecal weight | ||||
| Lithocholic acid | Lentisphaerae | 0.28 | 0.17 | 0.09 |
| Deoxycholic acid | Verrucomicrobia | −0.09 | 0.19 | 0.03 |
| SCFAs, µmol/g fecal weight | ||||
| Propionate | Cyanobacteria | 0.47 | 0.15 | 0.03 |
| Isobutyrate | Cyanobacteria | 0.29 | 0.17 | 0.03 |
| Lentisphaerae | 0.56 | 0.15 | <0.001 | |
| 2-Methylbutyrate | Cyanobacteria | 0.29 | 0.17 | 0.03 |
| Lentisphaerae | 0.60 | 0.14 | <0.001 | |
| Valerate | Cyanobacteria | 0.41 | 0.16 | 0.03 |
| Lentisphaerae | 0.31 | 0.17 | 0.03 | |
| Isovalerate | Cyanobacteria | 0.25 | 0.18 | 0.08 |
| Lentisphaerae | 0.63 | 0.14 | <0.001 | |
| 3-Methylvalerate | Cyanobacteria | 0.28 | 0.10 | <0.01 |
| Lentisphaerae | 0.28 | 0.13 | <0.001 | |
| Isocaproate | Cyanobacteria | 0.58 | 0.13 | <0.01 |
| Inflammatory markers | ||||
| hcCRP, mg/L | Acidaminococcus | 0.21 | 0.15 | 0.03 |
| IL-6, pg/mL | Actinobacteria | −0.20 | 0.14 | 0.10 |
Linear mixed effects models to examine the associations of taxon abundance with concentrations of serum cardiometabolic risk indicators, fecal secondary bile acids, and fecal SCFAs. All variables were assessed as standardized z scores. Taxon abundance was modeled as the fixed effect and participant as a random effect. An FDR P value ≤0.1 was considered statistically significant. FDR, false discovery rate, hsCRP, high-sensitivity C-reactive protein.
β estimates represent the difference in the respective risk marker z score per SD unit increase in the abundance of the respective taxon.
FDR P value for linear mixed effect model.
Discussion
The current study provides novel information about the impact of dietary carbohydrate type (simple, refined, and unrefined) on the gut microbiome phylogenetic structure and functional capacity. In this secondary analysis of a clinical trial with 3 isocaloric dietary intervention phases, we document that different types of carbohydrate have modest but distinctive effects on the human gut microbiome (composition and function) and fecal SBA but not SCFA fecal concentrations. These results suggest that the favorable effects of diets enriched in unrefined carbohydrate could be mediated, in part, by the gut microbiome. Interestingly, consistent with the effects on CMRFs (7), the diet enriched in simple carbohydrate had unique effects on human gut microbiota composition and gene expression that were distinct from refined carbohydrate.
A majority of intervention studies on carbohydrate quality and gut microbial composition have compared diets enriched in whole grains to refined grains (34–39). However, simple and refined carbohydrate may have differential effects on cardiometabolic health that are mediated by the gut microbiota. Similar to the current work, some clinical trials comparing whole to refined grains have found modest changes (≥2 taxa) in gut microbial composition (34, 35), although other studies have found no significant changes (36–39). We observed that the abundance of Roseburia, a butyrate producing genera, was higher after consumption of the unrefined carbohydrate diet compared with the simple carbohydrate diet. Results were similar but nonsignificant when comparing the unrefined carbohydrate diet with the refined carbohydrate diet. A similar trend of higher abundance of Roseburia in participants fed whole grain compared with a refined grain enriched diet has been reported (35). Roseburia is a dietary fiber fermenting genus that increases in abundance in response to dietary fiber (17, 40). We observed that the abundance of the genera Anaerostipes was lowest after participants consumed the refined carbohydrate diet compared with the simple carbohydrate and unrefined carbohydrate diets, although the latter did not reach statistical significance. Anaerostipes is also a butyrate producer; thus, low abundance after consumption of the refined carbohydrate diet may contribute to the unfavorable effects of diets rich in refined carbohydrate.
Our finding that carbohydrate type had no significant impact on gut microbial α-diversity (within participants) and β-diversity (among participants) is in agreement with most (35, 38, 39) but not all prior work (34). Notably, we observed 2 distinct patterns of Shannon index shift among participants, suggesting individual differences in diet induced shifts in microbial diversity. In addition, we found that the fecal microbial composition at the end of each intervention phase clustered by participant rather than carbohydrate type. These results support prior evidence (41) suggesting that diet-induced intraindividual variation in gut microbial composition may be insufficient to overcome inherent interindividual variation and long-term dietary habits.
In addition to using 16S amplicon sequencing to measure the relative abundances of organisms in microbial communities, we utilized metatranscriptomics to examine microbial genes differentially expressed by dietary carbohydrate type. Metatranscriptomics based on RNA-seq allows us to accurately measure the expression levels of living bacteria, viruses, and fungi and may capture the relative abundance of both living and dead (or quiescent) cells. The 3 annotated genes (heat shock protein 60/chaperonin GroEl, Clp protease, and flavin adenine dinucleotide–containing oxidoreductase) with higher expression after participants consumed the simple compared with the refined carbohydrate diet have been implicated in stress response and oxidative stress. Particularly, bacterial heat shock protein 60 has been implicated in immune response and inflammatory bowel disease (42, 43). The 2 genes with higher expression levels after the unrefined compared with refined (csbD-like protein) or simple (heat shock protein 20/α-crystalin) carbohydrate diets have also been implicated in bacterial stress response. However, the expression of csbD-like protein was relatively low in all groups. Hence, significant differences may be due to the higher variation in expression with the unrefined carbohydrate diet.
Favorable associations between carbohydrate quality and chronic disease could be mediated in part by the production of microbial-derived metabolites. Conjugated primary bile acids (cholic and chenodeoxycholic acids) are synthesized in the liver and secreted into the intestine with the release of bile. In the intestine, bile acids that are not reabsorbed may undergo microbial deconjugation and dehydroxylation to form SBAs, which have been considered inflammatory and carcinogenic (44). Our findings indicate that concentrations of fecal SBAs (lithocholic and deoxycholic acid) were lowest after participants consumed the unrefined carbohydrate diet. Consistent with prior work (45, 46), these data suggest a protective effect that supports previous observations of reduced risk for colon cancer by whole grain–rich and high-fiber diets (47). Despite our finding that the abundance of butyrate-producing genera (Roseburia and Anaerostipes) was altered by the dietary carbohydrate type, there was no significant effect of carbohydrate type on fecal SCFA concentrations or associations between abundance of these genera and SCFAs. Similar findings have been reported with some (34, 39) but not all studies that examined diets enriched in either whole or refined grains (35). The phylum Actinobacteria includes the genus Bifidobacterium, which has been shown to influence systemic concentrations of IL-6 and TNF-α (48). Bifidobacterium has been reported to inhibit lipopolysaccharide-induced activation of NF-κB and subsequent secretion of inflammatory cytokines (49). It has been suggested that Bifidobacterium may influence inflammation by accumulating SBAs to reduce colonic concentrations (50), consistent with our observation that abundance of the phylum Actinobacteria was inversely associated with serum concentrations of IL-6. Although the associations among other microbial taxa with CMRFs and microbial-derived metabolites are intriguing, they warrant further investigation because these taxa were present in low abundance.
The current study has several strengths. Foremost, we conducted a well-controlled dietary intervention, in which all meals were provided to the study participants. In addition, we used a crossover study design to reduce interindividual variation, which is of particular importance in studies of gut microbiota. We collected a broad array of data on microbial composition and function and microbial-derived metabolites, targeted due to prior associations with dietary carbohydrate type. Limitations include a modest sample size that was originally determined to examine the effect of carbohydrate type on CMRFs. A larger study, based on the primary outcome of changes in gut microbial composition and metabolome, is needed to confirm our findings. Although metatranscripomics provides novel insight into differentially expressed microbial genes, current limitations in gene annotation may limit the clinical interpretation of results. Last, assessment of the gut microbiome at baseline and postintervention would allow for a better inference of interindividual variation.
In summary, we observed modest differential effects of carbohydrate type on microbial composition and function and fecal concentrations of SBAs. A diet enriched in unrefined carbohydrate had favorable effects, including higher abundance of the butyrate-producing genus Roseburia and lowered SBA fecal concentrations. Although exploratory in nature, our results suggest distinct effects of simple and refined carbohydrate on gut microbial composition and function. Last, we noted that the effect of our carbohydrate intervention was insufficient to overcome a high degree of synergy within participants’ gut microbial communities. These findings provide novel information on potential mechanisms linking carbohydrate quality to human health and warrant replication in a larger study.
Supplementary Material
Acknowledgments
We acknowledge the assistance of the Mass Spectrometry and Nutrition Evaluation Laboratories at the Jean Mayer USDA Human Nutrition Research Center on Aging at Tufts University for assistance with the bile acid and SCFA analyses.
The authors’ responsibilities were as follows—NRM and AHL: designed the research; MEW, JRM, HM, JEG, JMG, AHL, and NRM: conducted the research; TF and WEJ: analyzed the data and performed the statistical analysis; MEW and TF: wrote the manuscript; NRM: has primary responsibility for final content; and all authors: contributed to critically reviewing the manuscript and read and approved the final manuscript. All authors declare no conflicts of interest.
Notes
Pilot funds were provided by the Jean Mayer USDA Human Nutrition Research Center on Aging at Tufts University and Boston Obesity Research Center to AHL; the US Department of Agriculture (agreement 58-1950-4-401) to AHL and NRM; and grant NHLBI T32-HL069772 from the National Heart, Lung, and Blood Institute/NIH to AHL. MEW is supported by grant 5T32-HL125232 from the National Heart, Lung, and Blood Institute/NIH Multidisciplinary Training Program in Cardiovascular Epidemiology. Any opinions, findings, conclusions, or recommendations expressed in this publication are those of authors and do not necessarily reflect the view of the US Department of Agriculture or the National Institutes of Health.
Data described in the manuscript, code book, and analytic code will be made available pending review and approval of the request by e-mailing the corresponding author.
TF and MEW contributed equally to this work.
Supplemental Figures 1–3 are available from the “Supplementary data” link in the online posting of the article and from the same link in the online table of contents at https://academic.oup.com/ajcn/.
Abbreviations used: ASV, amplicon sequence variant; CMRF, cardiometabolic risk factor; CVD, cardiovascular disease; FDR, false discovery rate; hsCRP, high-sensitivity C-reactive protein; RNA-seq, RNA sequencing; rRNA, ribosomal RNA; SBA, secondary bile acid.
Contributor Information
Tyler Faits, Division of Computational Biomedicine, Department of Medicine, Boston University School of Medicine, Boston, MA, USA.
Maura E Walker, Division of Computational Biomedicine, Department of Medicine, Boston University School of Medicine, Boston, MA, USA; Section of Preventive Medicine and Epidemiology, Department of Medicine, Boston University School of Medicine, Boston, MA, USA.
Jose Rodriguez-Morato, Pompeu Fabra University, Barcelona, Spain; Cardiovascular Nutrition, Jean Mayer USDA Human Nutrition Research Center on Aging at Tufts University, Boston, MA, USA.
Huicui Meng, Cardiovascular Nutrition, Jean Mayer USDA Human Nutrition Research Center on Aging at Tufts University, Boston, MA, USA; School of Public Health (Shenzhen), Sun Yat-sen University, Guangzhou, Guangdong Province, China.
Julie E Gervis, Cardiovascular Nutrition, Jean Mayer USDA Human Nutrition Research Center on Aging at Tufts University, Boston, MA, USA.
Jean M Galluccio, Cardiovascular Nutrition, Jean Mayer USDA Human Nutrition Research Center on Aging at Tufts University, Boston, MA, USA.
Alice H Lichtenstein, Cardiovascular Nutrition, Jean Mayer USDA Human Nutrition Research Center on Aging at Tufts University, Boston, MA, USA.
W Evan Johnson, Division of Computational Biomedicine, Department of Medicine, Boston University School of Medicine, Boston, MA, USA.
Nirupa R Matthan, Cardiovascular Nutrition, Jean Mayer USDA Human Nutrition Research Center on Aging at Tufts University, Boston, MA, USA.
References
- 1.Benjamin Emelia J, Paul M, Alvaro A, Bittencourt Marcio S, Callaway Clifton W, Carson April P, Chamberlain Alanna M, Chang Alexander R, Susan C, Das Sandeep Ret al. Heart disease and stroke statistics—2019 update: a report from the American Heart Association. Circulation. 2019;139:e56–528. [DOI] [PubMed] [Google Scholar]
- 2.Zong G, Gao A, Hu FB, Sun Q. Whole grain intake and mortality from all causes, cardiovascular disease, and cancer: a meta-analysis of prospective cohort studies. Circulation. 2016;133:2370–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Reynolds A, Mann J, Cummings J, Winter N, Mete E, Morenga LT. Carbohydrate quality and human health: a series of systematic reviews and meta-analyses. Lancet North Am Ed. 2019;393:434–45. [DOI] [PubMed] [Google Scholar]
- 4.Yang Q, Zhang Z, Gregg EW, Flanders WD, Merritt R, Hu FB. Added sugar intake and cardiovascular diseases mortality among US adults. JAMA Intern Med. 2014;174:516–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Steffen LM, Jacobs DR, Stevens J, Shahar E, Carithers T, Folsom AR. Associations of whole-grain, refined-grain, and fruit and vegetable consumption with risks of all-cause mortality and incident coronary artery disease and ischemic stroke: the Atherosclerosis Risk in Communities (ARIC) study. Am J Clin Nutr. 2003;78:383–90. [DOI] [PubMed] [Google Scholar]
- 6.Williams PG. Evaluation of the evidence between consumption of refined grains and health outcomes. Nutr Rev. 2012;70:80–99. [DOI] [PubMed] [Google Scholar]
- 7.Meng H, Matthan NR, Fried SK, Berciano S, Walker ME, Galluccio JM, Lichtenstein AH. Effect of dietary carbohydrate type on serum cardiometabolic risk indicators and adipose tissue inflammatory markers. J Clin Endocrinol Metab. 2018;103(9):3430–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kirwan AM, Lenighan YM, O'Reilly ME, McGillicuddy FC, Roche HM. Nutritional modulation of metabolic inflammation. Biochem Soc Trans. 2017;45:979–85. [DOI] [PubMed] [Google Scholar]
- 9.Tighe P, Duthie G, Vaughan N, Brittenden J, Simpson WG, Duthie S, Mutch W, Wahle K, Horgan G, Thies F. Effect of increased consumption of whole-grain foods on blood pressure and other cardiovascular risk markers in healthy middle-aged persons: a randomized controlled trial. Am J Clin Nutr. 2010;92:733–40. [DOI] [PubMed] [Google Scholar]
- 10.Marckmann P, Raben A, Astrup A. Ad libitum intake of low-fat diets rich in either starchy foods or sucrose: effects on blood lipids, factor VII coagulant activity, and fibrinogen. Metab Clin Exp. 2000;49:731–5. [DOI] [PubMed] [Google Scholar]
- 11.Tang WHW, Kitai T, Hazen SL. Gut microbiota in cardiovascular health and disease. Circ Res. 2017;120:1183–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Turnbaugh PJ, Ridaura VK, Faith JJ, Rey FE, Knight R, Gordon JI. The effect of diet on the human gut microbiome: a metagenomic analysis in humanized gnotobiotic mice. Sci Transl Med. 2009;1:6ra14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kashyap PC, Marcobal A, Ursell LK, Larauche M, Duboc H, Earle KA, Sonnenburg ED, Ferreyra JA, Higginbottom SK, Million Met al. Complex interactions among diet, gastrointestinal transit, and gut microbiota in humanized mice. Gastroenterology. 2013;144:967–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Makki K, Deehan EC, Walter J, Bäckhed F. The impact of dietary fiber on gut microbiota in host health and disease. Cell Host Microbe. 2018;23:705–15. [DOI] [PubMed] [Google Scholar]
- 15.Sonnenburg ED, Sonnenburg JL. Starving our microbial self: the deleterious consequences of a diet deficient in microbiota-accessible carbohydrates. Cell Metab. 2014;20:779–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wahlström A, Sayin SI, Marschall H-U, Bäckhed F. Intestinal crosstalk between bile acids and microbiota and its impact on host metabolism. Cell Metab. 2016;24:41–50. [DOI] [PubMed] [Google Scholar]
- 17.David LA, Maurice CF, Carmody RN, Gootenberg DB, Button JE, Wolfe BE, Ling AV, Devlin AS, Varma Y, Fischbach MAet al. Diet rapidly and reproducibly alters the human gut microbiome. Nature. 2014;505:559–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Lozupone CA, Turnbaugh PJ, Fierer N, Knight R. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc Natl Acad Sci USA. 2011;108:4516–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rodríguez-Morató J, Matthan NR, Liu J, de la Torre R, Chen C-YO. Cranberries attenuate animal-based diet-induced changes in microbiota composition and functionality: a randomized crossover controlled feeding trial. J Nutr Biochem. 2018;62:76–86. [DOI] [PubMed] [Google Scholar]
- 20.Han J, Lin K, Sequeira C, Borchers CH. An isotope-labeled chemical derivatization method for the quantitation of short-chain fatty acids in human feces by liquid chromatography–tandem mass spectrometry. Anal Chim Acta. 2015;854:86–94. [DOI] [PubMed] [Google Scholar]
- 21.Bolyen E, Rideout JR, Dillon MR, Bokulich NA, Abnet CC, Al-Ghalith GA, Alexander H, Alm EJ, Arumugam M, Asnicar Fet al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat Biotechnol. 2019;37:852–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJA, Holmes SP. DADA2: High-resolution sample inference from Illumina amplicon data. Nat Methods. 2016;13:581–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K, Huber T, Dalevi D, Hu P, Andersen GL. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol. 2006;72:5069–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McMurdie PJ, Holmes S. Waste not, want not: why rarefying microbiome data is inadmissible. PLoS Comput Biol. 2014;10:e1003531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Weiss S, Xu ZZ, Peddada S, Amir A, Bittinger K, Gonzalez A, Lozupone C, Zaneveld JR, Vázquez-Baeza Y, Birmingham Aet al. Normalization and microbial differential abundance strategies depend upon data characteristics. Microbiome. 2017;5:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Benjamini Y, Drai D, Elmer G, Kafkafi N, Golani I. Controlling the false discovery rate in behavior genetics research. Behav Brain Res. 2001;125:279–84. [DOI] [PubMed] [Google Scholar]
- 28.Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng Qet al. Trinity: reconstructing a full-length transcriptome without a genome from RNA-Seq data. Nat Biotechnol. 2011;29:644–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011;12:323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Aitchison J. The statistical analysis of compositional data. J R Stat Soc Ser B Methodological. 1982;44:139–77. [Google Scholar]
- 32.Bates D, Mächler M, Bolker B, Walker S. Fitting linear mixed-effects models using lme4. J Stat Software. 2015;67:1–48. [Google Scholar]
- 33.Sayers EW, Beck J, Brister JR, Bolton EE, Canese K, Comeau DC, Funk K, Ketter A, Kim S, Kimchi Aet al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2016;44:D7–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Martínez I, Lattimer JM, Hubach KL, Case JA, Yang J, Weber CG, Louk JA, Rose DJ, Kyureghian G, Peterson DAet al. Gut microbiome composition is linked to whole grain-induced immunological improvements. ISME J. 2013;7:269–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vanegas SM, Meydani M, Barnett JB, Goldin B, Kane A, Rasmussen H, Brown C, Vangay P, Knights D, Jonnalagadda Set al. Substituting whole grains for refined grains in a 6-wk randomized trial has a modest effect on gut microbiota and immune and inflammatory markers of healthy adults. Am J Clin Nutr. 2017;105:635–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cooper DN, Kable ME, Marco ML, De Leon A, Rust B, Baker JE, Horn W, Burnett D, Keim NL. The effects of moderate whole grain consumption on fasting glucose and lipids, gastrointestinal symptoms, and microbiota. Nutrients. 2017;9:173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ampatzoglou A, Atwal KK, Maidens CM, Williams CL, Ross AB, Thielecke F, Jonnalagadda SS, Kennedy OB, Yaqoob P. Increased whole grain consumption does not affect blood biochemistry, body composition, or gut microbiology in healthy, low-habitual whole grain consumers. J Nutr. 2015;145:215–21. [DOI] [PubMed] [Google Scholar]
- 38.Langkamp-Henken B, Nieves C, Culpepper T, Radford A, Girard S-A, Hughes C, Christman MC, Mai V, Dahl WJ, Boileau Tet al. Fecal lactic acid bacteria increased in adolescents randomized to whole-grain but not refined-grain foods, whereas inflammatory cytokine production decreased equally with both interventions. J Nutr. 2012;142:2025–32. [DOI] [PubMed] [Google Scholar]
- 39.Roager HM, Vogt JK, Kristensen M, Hansen LBS, Ibrügger S, Mærkedahl RB, Bahl MI, Lind MV, Nielsen RL, Frøkiær Het al. Whole grain-rich diet reduces body weight and systemic low-grade inflammation without inducing major changes of the gut microbiome: a randomised cross-over trial. Gut. 2019;68:83–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tomova A, Bukovsky I, Rembert E, Yonas W, Alwarith J, Barnard ND, Kahleova H. The effects of vegetarian and vegan diets on gut microbiota. Front Nutr. 2019; ;6:47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lang JM, Pan C, Cantor RM, Tang WHW, Garcia-Garcia JC, Kurtz I, Hazen SL, Bergeron N, Krauss RM, Lusis AJ. Impact of individual traits, saturated fat, and protein source on the gut microbiome. mBio. 2018;9:e01604–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ohue R, Hashimoto K, Nakamoto M, Furukawa Y, Masuda T, Kitabatake N, Tani F. Bacterial heat shock protein 60, GroEL, can induce the conversion of naïve T cells into a CD4 CD25(+) Foxp3-expressing phenotype. J Innate Immun. 2011;3:605–13. [DOI] [PubMed] [Google Scholar]
- 43.Cappello F, Mazzola M, Jurjus A, Zeenny M-N, Jurjus R, Carini F, Leone A, Bonaventura G, Tomasello G, Bucchieri Fet al. Hsp60 as a novel target in IBD management: a prospect. Front Pharmacol. 2019;; 10:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bernstein H, Bernstein C, Payne CM, Dvorakova K, Garewal H. Bile acids as carcinogens in human gastrointestinal cancers. Mutat Res Rev Mutat Res. 2005;589:47–65. [DOI] [PubMed] [Google Scholar]
- 45.Reddy BS, Sharma C, Simi B, Engle A, Laakso K, Puska P, Korpela R. Metabolic epidemiology of colon cancer: effect of dietary fiber on fecal mutagens and bile acids in healthy subjects. Cancer Res. 1987;47:644–8. [PubMed] [Google Scholar]
- 46.Srikumar TS. Effects of consumption of white bread and brown bread on the concentrations of fecal bile acids and neutral steroids and on fecal enzyme activities. Nutr Res. 2000;20:327–33. [Google Scholar]
- 47.Aune D, Chan DSM, Lau R, Vieira R, Greenwood DC, Kampman E, Norat T. Dietary fibre, whole grains, and risk of colorectal cancer: systematic review and dose-response meta-analysis of prospective studies. BMJ. 2011;343:d6617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bernini LJ, Simão ANC, Alfieri DF, Lozovoy MAB, Mari NL, de Souza CHB, Dichi I, Costa GN. Beneficial effects of Bifidobacterium lactis on lipid profile and cytokines in patients with metabolic syndrome: a randomized trial. Effects of probiotics on metabolic syndrome. Nutrition. 2016;32:716–9. [DOI] [PubMed] [Google Scholar]
- 49.Riedel CU, Foata F, Philippe D, Adolfsson O, Eikmanns BJ, Blum S. Anti-inflammatory effects of bifidobacteria by inhibition of LPS-induced NF-κB activation. World J Gastroenterol. 2006;12:3729–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sánchez B. Bile acid–microbiota crosstalk in gastrointestinal inflammation and carcinogenesis: a role for bifidobacteria and lactobacilli? Nat Rev Gastroenterol Hepatol. 2018;15:205. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.