

Abstract
In recent decades, traumatic brain injury (TBI) has become one of the most important health problems worldwide and is a major cause of morbidity, mortality and economic losses. Mild traumatic brain injury (mTBI) is less considered, with clinical underestimation leading to an epidemiological underevaluation of its incidence. Many of the signs and symptoms induced by mTBI are difficult to highlight clinically, especially those related to cognitive, behavioral, or emotional impairment. The complexity of the biological mechanisms induced by mTBI in the elderly determines synchronous pathogenic actions in which the vascular, inflammatory and neurodegenerative elements are intertwined. It is difficult to highlight a major pathogenic factor, since they act simultaneously, multimodally, in a real pathogenic cascade. The identification of mTBI and cerebral vascular changes by neuroimaging techniques, transcranial Doppler (TCD) or biological markers, suggests a potential prophylactic intervention by using neuroprotective factors as early as possible. Proper prophylaxis measures with neurotrophic treatment, rebalancing the gamma-aminobutyric acid (GABA)/glutamate balance and combating the chronic inflammatory process, can become important pharmacological therapeutic targets.
Keywords: mild traumatic brain injury, Alzheimer’s disease, axonal dysfunction, brain vascular abnormality, neurodegenerative mechanism
Introduction
Alzheimer’s disease (AD) is a neurodegenerative disease that affects millions of people worldwide and begins clinically with mild cognitive impairment (MCI) that progress to severe dementia [1,2]. It is a chronic, progressive condition that decreases the quality of life of the patient, creates special problems for caregivers and represents a heavy economic burden for the health systems of each country [3]. The etiopathogenesis of the disease is still incompletely elucidated, but it is currently believed that AD is caused by complex interactions between a multitude of genetic or epigenetic changes and environmental factors [4]. In recent years, numerous studies have shown a possible involvement of cranio-cerebral trauma in the etiopathogenesis of AD [5,6].
Traumatic brain injury (TBI) has become one of the most important health problems in the world in recent decades, being a major cause of morbidity, mortality and economic losses [7]. In the US alone, in 2013, about 2.8 million people went to hospital emergency departments for TBI [8].
Some studies estimate that three out of four cases of TBI are mild traumatic brain injury (mTBI) that disrupts normal brain function for a limited time. However, traumatic forces acting on the brain, directly or indirectly, can lead to damage to several brain structures or functions, including the pituitary gland [9]. Most mTBI-induced neurological lesions can occur immediately or in the following hours and days [10].
Epidemiological data on TBI refer, in most studies, to severe forms of TBI, whose neurological and psychological consequences as well as their risks in the medium and long term, can be included in an iceberg epidemiological model. In contrast, mTBI are less considered, with clinical underestimation leading to an epidemiological under-evaluation of their incidence. It is no coincidence that this condition is also called “silent epidemic” since 1997, because many of the signs and symptoms induced are difficult to highlight clinically, especially those related to cognitive, behavioral or emotional impairment [11]. Diagnosis, but especially treatment, becomes much more difficult when “silent epidemic” is associated with “silent population”, represented by the elderly [12].
At the beginning of 2000, the incidence of mTBI in the USA was 1.5 million cases, of which 90 000 patients had long-term disabilities and 50 000 died [13]. The most common causes of mTBI are car accidents, violent or suicidal behavior, and falls in the elderly. In 2018, the incidence was 1299/100 000 inhabitants in North America and 1012/100 000 inhabitants in Europe, respectively. Worldwide, the incidence of TBI is estimated at 69 million cases [14], and the incidence of mTBI has been estimated at approximately 224/100 000 people [15]. It is assessed that of the total TBI, 75–90% of them are considered as mild [16]. This alarming increase associated with the large number of invalidities, the unfavorable prognosis as well as the very high costs of care, requires a better understanding of the neurobiological mechanisms involved in the pathogenesis and evolution of mTBI.
Originally defined as “traumatic head injury”, mTBI was difficult to classify nosographically due to reduced clinical and epidemiological data and partial understanding of the neurobiological mechanisms involved. In 2010, a consensus of experts from The Demographics and Clinical Assessment Working Group of the International and Interagency Initiative toward Common Data Elements for Research on Traumatic Brain Injury and Psychological Health defined mTBI as “an alteration in brain function, or other evidence of brain pathology, caused by an external force” [17].
Epidemiological data from a meta-analysis performed on 32 observational studies showed that TBI, both mild and severe, causes an increased risk of AD and dementia [18]. mTBI, especially repeated ones, triggers neurodegenerative processes that favor the aggregation of amyloid beta (Aβ) and tau proteins, involved in the pathogenesis of AD [19,20].
Clinical particularities of mTBI in the elderly
From a clinical point of view, consecutive mTBI brain disorders trigger a symptomatology that is characterized by: loss of consciousness for a period of time depending on the severity of the mTBI, amnesia with altered mental status and neurological disorders. The generally accepted clinical diagnostic criteria for mTBI are: loss of consciousness for up to 30 minutes, a score of 13–15 points, on the Glasgow Coma Scale (GCS) after 30 minutes, neurological changes that do not require surgery and amnesia for up to 24 hours [16, 21]. Even if only in approximately 5–20% of patients with mTBI, the clinical manifestations do not resolve naturally, it is important to diagnose correctly and early to improve the symptoms that may occur in the medium or long term. In this way, the mechanisms and risk factors that determine an unfavorable evolution of this pathology can be identified [22].
If the consequences of severe traumas are immediate or at a short distance, in mTBI neurological symptoms and cognitive decline are progressive, over a medium time (deteriorative cognitive impairment) or long (chronic post-traumatic encephalopathy). Apparently, mTBI frequency is higher in childhood and adulthood, but they onset in old age is much less recorded, due to their occurrence in the majority at home. The major causes of mTBI produced at home are domestic violence or falls in conditions of lipothymic, hypoglycemic or orthostatic hypotension in direct relation to a somatic pathology or the use of drugs for various pathologies [23,24].
The frequency of orthostatic hypotension is estimated at 10–30% of the elderly population, accompanied by decreased cognitive abilities through mechanisms of cerebral hypoperfusion and dysfunction of the sympathetic autonomic nervous system [25]. mTBI in the elderly amplifies the imbalance in the autonomic system and causes a decrease in dopaminergic transmission, mechanisms similar to those in Parkinson’s disease (PD) (post-traumatic Parkinsonism syndrome). It is known that the number of somatic comorbidities increases in elderly patients, being frequently associated: diabetes, hypertension with heart failure, heart rhythm disorders, risk of thromboembolic accidents and excessive use of anticoagulant medication. It is important to note that, in mTBI conditions in the elderly undergoing anticoagulant treatment, minor traumatic shock may be the cause of subarachnoid or subdural cerebral hemorrhage, and intraparenchymal petechial type microhemorrhages or cortical-type microbleeds [26,27].
These lesions may go unnoticed, but can be identified by computed tomography (CT) evaluation as areas of siderosis (Figure 1, A and B). Cortical superficial siderosis (cSS) accompanied by cerebral amyloid angiopathy (CAA) is a major cause of cerebral small vessel disease (CSVD). Risks of cerebral hemorrhage and cognitive impairment may be associated with transient focal neurological symptoms [28].
Figure 1.
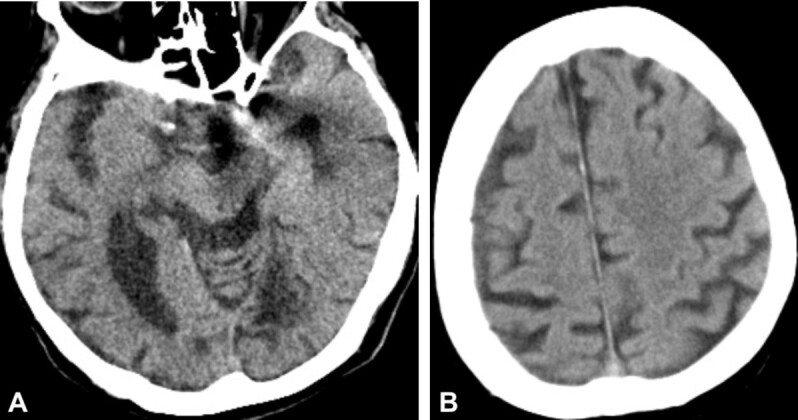
Post mTBI siderosis: (A) CT view – multiple areas of ischemic vascular injury in the occipital and left temporal lobes, with area of siderosis on the left temporal horn and microbleeds; (B) CT view – superficial cortical siderosis with microbleeds, in the right temporoparietal area. (Professor Simona Bondari collection). CT: Computed tomography
Genetic vulnerability and cognitive impairment in the elderly with mTBI
The genetic spectrum of apolipoprotein E4 (ApoE4) is involved in the genetic vulnerability of the cognitive deficit in the elderly, through a directly related action to the onset of CAA, which may precede by several years the stage of MCI in AD. Under these conditions, mTBI causes acceleration in the rate of cognitive decline, being an independent pathogenic factor of intensification for neurodegenerative mechanisms specific to AD. These mechanisms may be correlated with the disconnection of cognitive circuits and the rapid transition from the MCI stage to the medium or severe form of AD.
Thus, mTBI acts through several pathogenic mechanisms, especially against the background of the ApoE4 genetic vulnerability:
(i) Intensification of neurodegenerative processes with the onset of amyloid cascade and excessive deposit of Aβ and neurofibrils in cognitive circuits.
(ii) Impaired cerebral circulation that causes chronic cerebral ischemia.
(iii) Alteration of neurobiochemical homeostasis involved in cognitive processes: mTBI increases the activity of cholinacetyltransferase, decreases the level of acetylcholine and decreases the number and activity of gamma-aminobutyric acid (GABA) receptors in the dentate gyrus of the hippocampus. Thus, the hyperactivity of glutamate and the apoptotic mechanisms are favored, as well as the imbalance between the sympathetic and parasympathetic system with the installation of a sympathomimetic hyperactivity that favors cerebral arterial spasm.
These pathogenic mechanisms may favor rapid cognitive decline in the elderly with or without a link to the clinical evidence of MCI stage suspicion, but its rate of progression appears to be conditioned by the ApoE4 spectrum. CAA identification is the most important marker that anticipates neurodegenerative evolution [29]. This type of evolution accelerates the rate of onset for moderate or severe cognitive impairment (Figure 2).
Figure 2.
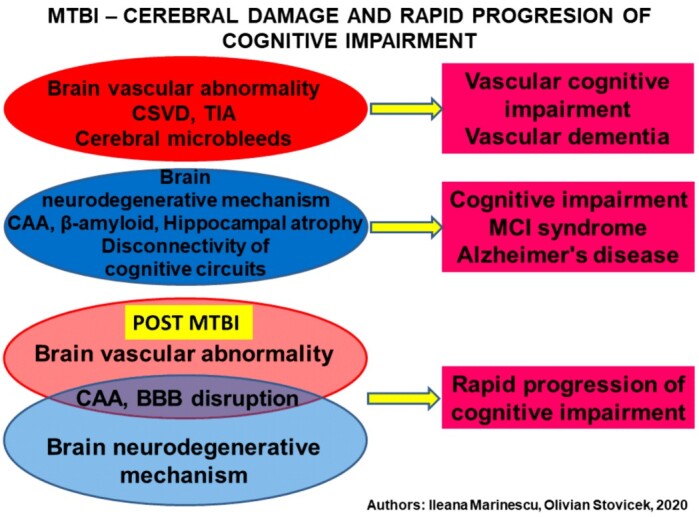
Multifactorial neurobiological mechanisms triggered by mTBI, which favors rapid cognitive impairment. MTBI: Mild traumatic brain injury; CSVD: Cerebral small vessel disease; TIA: Transient ischemic attack; CAA: Cerebral amyloid angiopathy; BBB: Blood–brain barrier; MCI: Mild cognitive impairment
Pathogenic mechanisms involved in rapid cognitive impairment
Association of the neuroinflammatory process with mTBI
Patients with mTBI adding a neuroinflammatory process have an amplified risk for a rapid AD-type neurodegenerative decline. Neuroinflammation is a proinflammatory response of glial cells that have the direct consequence of activating microglia or astrocytes. As a result of this activation process, cytokine levels increase greatly and cause macrophage activation, having as final effect the neuronal destruction. Neural damage causes dysfunction of the cognitive circuits at several levels:
(i) Neocortex, frontal and temporoparietal cortex, causing altered working memory, multisensory integration and the appearance of MCI syndrome symptoms.
(ii) Hippocampal area dominated by disconnectivity between the dentate gyrus and the cornu Ammonis (CA) 1 and CA3 regions, which can be considered as a marker of MCI amnestic syndrome.
(iii) Striatum, whose disconnectivity amplifies the dysfunction of the frontal cortex but is also accompanied by the presence of extrapyramidal symptoms.
(iv) Entorhinal and piriform cortex whose disconnectivity is clinically signaled by marked decrease in olfactory capacity.
Under these conditions, the presence of depressive disorders in the history of patients with mTBI has a high importance as risk factor. Depressive status favors pro-inflammatory processes that can be biologically objectified by high levels of C-reactive protein (CRP) and interleukin (IL) 2 and IL6. The association of proinflammatory factors with endothelial dysfunction causes major risks for somatic comorbidities and cerebral, coronary or renal microvascular damage amplified by the administration of antipsychotic drugs [30]. This therapy is used both in the conditions of depressive disorders with a suicidal component and in the case of impulsive and aggressive behavioral manifestations. This behavior may be a consequence of glutamate excitotoxicity. The proinflammatory mechanisms are involved in the imbalance between the excitatory function of glutamate and the inhibitory function of GABA neurons, in favor of hyperglutamatergia. Thus, the increase of glutamate activity determines the excitotoxicity effects for glutamate, a phenomenon that determines the synaptic disconnectivity at the level of the hippocampus, the piriform cortex and areas of cortical sensory association (Figure 3).
Figure 3.
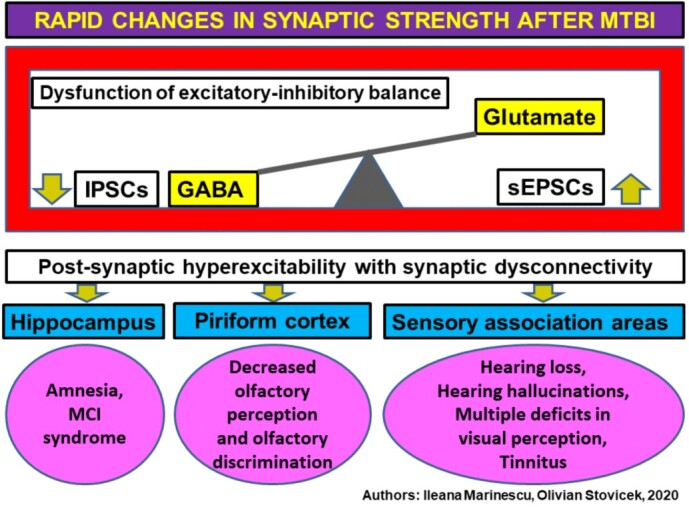
Post mTBI synaptic changes. MTBI: Mild traumatic brain injury; GABA: Gamma-aminobutyric acid; IPSCs: Inhibitory postsynaptic currents; sEPSCs: Spontaneous excitatory postsynaptic currents; MCI: Mild cognitive impairment
The relationship between inflammatory processes and dopamine
This relationship is bidirectional, the decrease of dopamine being accentuated by the increase of cytokines with the significant impairment of the dopamine level in the emergency areas represented by ventral tegmental area, substantia nigra and basal ganglia [31]. Dopamine deficiency causes depressive symptoms associated with anhedonia, psychomotor inhibition, suggesting disconnectivity between the frontal cortex and striatum. Dopamine deficiency depression can be considered a form resistant to antidepressant therapy [32]. Therapeutic resistance to antidepressant medication determines a therapeutic strategy in combination with antipsychotics or other psychotropic substances that can cause incomplete remissions predominantly symptomatic, while maintaining the proinflammatory biological background and endothelial dysfunction. Thus, a vicious circle of negative evolution of depression is generated.
In this way, dopamine deficiency is a central neurobiochemical mechanism that is installed post mTBI in the elderly, following depolarization of membranes in ventral tegmental area and substantia nigra, disrupting the release of dopamine through a synaptic mechanism of long-term depression [33]. Post mTBI dopamine deficiency influences cognitive decline by affecting the hippocampus and disconnecting corticostriatal circuits.
Therapeutic resistance of depression in the elderly patient can be an important warning sign that suggests the potential for the development of AD, having as main underlayer the dopamine deficiency and the promotion of vascular hypoperfusion by CSVD. The extrapyramidal phenomena induced by antipsychotic drugs previously used in mTBI are another important clinical marker of neurodegenerative risk. In both situations, there is a common neurobiological background characterized by chronic neuroinflammatory process, endothelial dysfunction and immune deficiency. mTBI triggers an acceleration of the disturbance of all neurobiological processes, which allows the aggression of neurodegenerative factors.
Vascular factors and disconnectivity of cognitive circuits
From a clinical point of view, widening of the periventricular spaces in the basal ganglia may associate an asymmetric extrapyramidal symptomatology (post-traumatic Parkinsonism syndrome). These lesions can be considered a potential neuroimaging marker that suggests the presence of CSVD and the disconnectivity of the basal ganglia structures with the cognitive structures represented by the striato-thalamic and striato-cortical circuits (Figure 4, A and B).
Figure 4.

Post mTBI SVD, MRI view: (A) Virchow–Robin perivascular spaces dilated in the basal ganglia, especially on the left side; (B) Infracentimetric dilatation of perivascular spaces, along the anterior commissure. (Professor Simona Bondari collection). mTBI: Mild traumatic brain injury; SVD: Small vessel disease; MRI: Magnetic resonance imaging
The thalamic area is one of the most important stations interconnecting cognitive pathways regardless of their area of emergence:
(i) Meynert basal nucleus – central acetylcholinergic structure, acetylcholine deficiency being the second central neurobiochemical mechanism in the pathogenesis of post mTBI rapid cognitive impairment.
(ii) Striatum and basal ganglia – dopaminergic structure that interconnects the emergence areas with the mesolimbic region and the cortical structures.
(iii) Hippocampal area – structure of acetylcholinergic, dopaminergic, glutamatergic and GABA-ergic interference. Thalamic lesion is a neuroimaging marker of rapid progression of cognitive impairment in the elderly with mTBI (Figure 5, A and B).
Figure 5.

Post mTBI SVD, successive CT view: (A and B) Right thalamic lesion in the lentiform nucleus and lesions of the perivascular dilation type on the left side. (Professor Simona Bondari collection). mTBI: Mild traumatic brain injury; SVD: Small vessel disease; CT: Computed tomography
Given that a depressive prodromal phase precedes many cases of mTBI in the elderly, there is a particular brain vulnerability to neuroinflammatory mechanisms favored by the imbalance between the GABA and glutamate system, incomplete remissions of depression, with onset before mTBI, and dopamine deficiency amplified by traumatic injury [33].
Neurodegenerative mechanisms in mTBI
Post mTBI pathology in AD is dominated by increased deposits of neurodegenerative elements in synaptic connections but also an increase in their production in the extracellular space. Their intersynaptic deposition is precipitated by general proinflammatory activity specific to the presence of depressive states or other associated somatic conditions present in old age [6]. Under these conditions, the transformation of Aβ precursor proteins (presenilin 1 and 2) into Aβ is activated, which is predominantly deposited in the areas of maximum vulnerability represented by temporoparietal cortex, corpus callosum and hippocampus. Excessive Aβ deposition, accompanied by excess neurofibrils and tau proteins, is significantly increased following an Aβ42 oligomerization process. The level of Aβ oligomers in cerebrospinal fluid, determined within a maximum of 72 hours from TBI, can be a marker of neurological evolution through direct involvement in neuroinflammatory and neurodegenerative processes [34].
Neurofibrils are those that cause an alteration in the functionality of axonal microtubules, with significant alteration of transport at this level, initially producing a diffuse axonal dysfunction whose progression causes axonal degeneration. If the diffuse axonal dysfunction has a reversible nature, the degeneration is irreversible.
The progression of cognitive deficit is precipitated by the rate of Aβ deposition in the cognitive circuits, but especially in the cerebral arterioles causing CAA. Rapid progression is favored by mTBI, because in the conditions of an inflammatory process and a persistent endothelial dysfunction, the global cerebral arterial perfusion decreases significantly, establishing the state of chronic cerebral ischemia (Figure 6).
Figure 6.
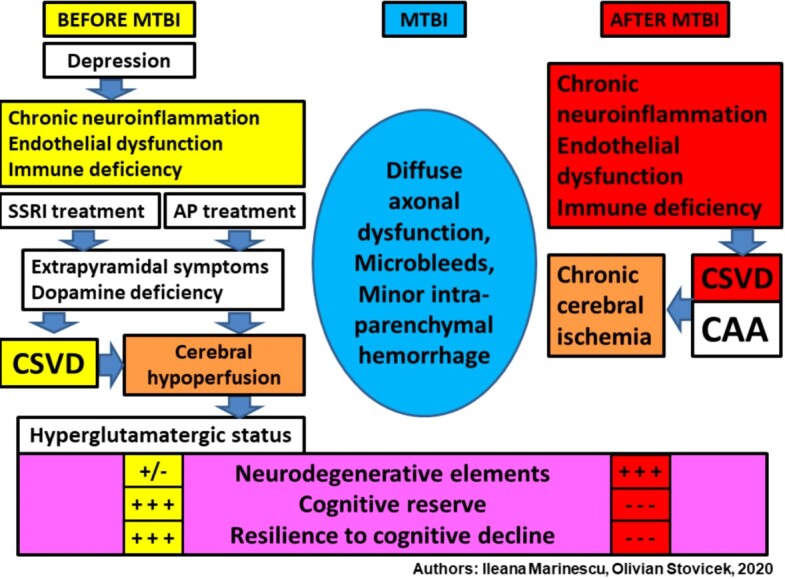
Multisystemic mechanisms involved in the onset of neurodegenerative elements, post mTBI in the elderly. MTBI: Mild traumatic brain injury; SSRI: Selective serotonin reuptake inhibitor; AP: Antipsychotic; CSVD: Cerebral small vessel disease; CAA: Cerebral amyloid angiopathy
These factors influence the evolution of clinical forms, which can have a long or rapid transition from the MCI stage to AD. Accelerated dysfunctional progression is also favored by the existence in the personal history of depression, previously mTBI. Depression through multi-systemic mechanisms is a risk factor for AD onset and rapid progression of cognitive impairment [35].
Other pathogenic mechanisms of post mTBI rapid cognitive impairment
The clinical medical practice identifies other pathogenic mechanisms increasing the vulnerability for the rapid cognitive deficit after mTBI, by potentiating the progression of neurodegenerative elements. These mechanisms are represented by acute or chronic cerebral hypoxia (chronic hypoxic encephalopathy), cerebral vascular perfusion deficits following severe cardiac dysfunction or dysmetabolic disorders (hypoglycemia).
Histopathological (HP) evidence
In contrast to the high incidence of mTBI in old age, their clinical importance is underestimated, being considered “benign”, in contrast to severe TBI. This is determined by the lack of acute elements to indicate severity, generally being domestic accidents that do not have skin damage to the scalp and are not accompanied by loss of consciousness. There are also no positive neurological signs or those that suggest intracranial hypertension syndrome after 24 hours (nausea, vomiting, headache, balance disorders, bradycardia, neurological signs of foci or significant changes in the fundus). Neuroimaging assessments also do not suggest relevant structural or lesional changes. In order to support the clinical importance of mTBI and its consequences in the brain that takes place in a multisystemic pathogenic context, we present the evidence objectified by HP research on animals.
Translational HP research on animal models
HP studies on Wistar rats were able to highlight changes in the hippocampus (Figure 7, A–C) and in the frontal cortex (Figure 8, A and B), secondary to minimal traumatic cerebral injury. These data confirm the theoretical disconnection model following structural changes in the frontal cortex and hippocampal area. These changes cause impaired cognitive circuits, which can lead to accelerated cognitive impairment in the elderly who have had an mTBI event. Changes in the frontal cortex highlight neuronal depopulation and changes in vascular structures with intravascular and perivascular hyalinization.
Figure 7.

Post repeated mTBI hippocampal lesions, at Wistar rats sacrificed on day 21: (A) Hippocampal neuronal loss, with vacuolar disorganization; (B) Neural losses in CA1 and CA3 regions, with the tendency of focal organization, vacuolar degeneration; (C) Massive neuronal loss throughout the hippocampus, with neuronal death, marked hyaline and vacuolar degeneration. HE staining: (A–C) ×100. mTBI: Mild traumatic brain injury; CA: Cornu Ammonis; HE: Hematoxylin–Eosin
Figure 8.

Post repeated mTBI lesions of the frontal cortex, at Wistar rats sacrificed on day 21: (A) Normal frontal cortex; (B) Significant neuronal loss in the superficial layers of the cerebral cortex, nuclear pyknosis, cytoplasmic condensation and moderate perineural edema. HE staining: (A and B) ×100. mTBI: Mild traumatic brain injury; HE: Hematoxylin–Eosin
Fundamental animal research confirms the potential of mTBI to induce important brain structural changes in the hippocampus and frontal cortex, lesions that may explain the acceleration of post mTBI cognitive impairment.
Evaluation of HP changes in postmortem studies. Vascular-type mechanisms
Studies in recent decades have shown that TBIs cause, depending on their severity, a multitude of cerebrovascular lesions that include meningeal and intraparenchymal hemorrhage, cerebral edema, changes in cerebral blood flow (CBF) and morphological disturbances of the blood–brain barrier (BBB) [36,37].
Our studies allowed us to find that there is a wide range of HP changes that can explain cognitive degeneration in patients with mTBI. Thus, the most frequent changes were vascular, objectified by the appearance of intraparenchymal microhemorrhages at a time interval after an mTBI (Figure 9A), sometimes associated with hemorrhages through the Virchow–Robin perivascular space, the blood diffusing at relatively long distances through perivascular sheath, which is made of lax tissue (Figure 9B).
Figure 9.

Vascular histopathological postmortem changes in patients with mTBI: (A) Perivascular hemorrhage in the Virchow–Robin space; (B) Diffuse intraparenchymal hemorrhages with a tendency to expand in the interneuronal spaces; (C) Marked cerebral ischemia, with the appearance of red neurons (specific to cerebral ischemia), associated with pyknosis and nuclear tachychromia, neuronal necrosis and perineuronal edema. HE staining: (A) ×100; (B and C) ×200. mTBI: Mild traumatic brain injury; HE: Hematoxylin–Eosin
Currently, most studies agree that TBI produces in a first phase (acute phase) physiological and morphological changes of BBB by shear forces caused by trauma resulting in damage to endothelial cells and the basement membrane of the capillary endothelium or rupture of the podocyte extensions of astrocytes. The consequence of this first phase is a disruption of CBF, rupture of intercellular junctions between endothelial cells, increased vascular permeability, rupture of the vascular wall with perivascular or intraparenchymal hemorrhage and global dysfunction of areas of the brain because of altered neurovascular units [38]. In addition, in the elderly, meningocerebral vessels have a modified structure compared to young people, and the presence of comorbidities, such as hypertension, atherosclerosis, diabetes or ischemic heart disease, increase the risk of severe brain damage at traumas with relatively low intensity [39].
After this first phase, which is due to the mechanical energy of the damaged agent, a second phase occurs in the brain generated by metabolic disorders that occur because of perturbation of CBF and activation of immune system cells [40]. Cerebral ischemia secondary to the disturbance of CBF manifested by the appearance of red neurons and neuronal death (Figure 9C) is added to all these phenomena is added. This type of deterioration of neural structures supports the rapid progression of vascular pathogenesis of post mTBI cognitive deficit.
According to the multisystemic model which we support, a second way of cognitive dysfunction occurrence with rapid progression, post mTBI, is the consequent activation of astrocytes, microglia and macrophages. Activated astrocytes and the formation of cell extensions (podocytes) cause frequent lesions on contact with the vascular wall, following the agglutination of podocytes’ fragments (Figure 10A). In this way, it becomes possible to extravasate the endovascular content. On this vulnerable ground, entry gates are made for Aβ under AD conditions, which allow the transformation of an endothelial angiopathy into an amyloid angiopathy. A major role of the inflammatory process, including cytokine activations, is supported by intravascular and perivascular hyalinization changes. Microglial activation seems to be the first moment of cellular pathology, in which macrocytes surround the vascular walls, an aspect that announces the aggressive invasion of macrophages (Figure 10B).
Figure 10.

Immunohistochemical aspects of brain parenchyma changes in patients with mTBI: (A) Activated astrocytes, characterized by increased cell volume and the emission of multiple cell extensions of various sizes (reactive gliosis); (B) Area of the brain parenchyma with a spongy appearance, resulting from neuronal damage, with numerous macrophage-type cells (activated microglia and macrophages). Anti-GFAP antibody immunostaining: (A) ×100. Anti-CD68 antibody immunostaining: (B) ×200. mTBI: Mild traumatic brain injury; GFAP: Glial fibrillary acidic protein; CD68: Cluster of differentiation 68
A third pathogenic model suggested by HP evidence for the rapid progression of cognitive deficit is based on the action caused by chronic ischemia and microvascular destruction following activation of microglia and macrophage cells. Thus, the pathogenic binomial composed of neurodegenerative and vascular elements in AD is confirmed under conditions of synergistic multisystemic mechanisms in the pathogenesis of rapid cognitive impairment.
The multifactorial mechanisms involved in the rapid progression of post mTBI cognitive impairment in the elderly with AD
Under mTBI conditions, imbalances between excitatory and inhibitory systems cause damage to the electrical synapse, producing disconnectivity in the hippocampus, piriform cortex and associative areas of multisensory integration. These imbalances may be more intense in people with various comorbidities, especially in women [41]. Damage to the hippocampal area is associated with amnesia and MCI syndrome, common symptoms present in the prodromal phase of AD. The disconnectivity of the piriform cortex causes a decrease in olfactory perception and the ability to discriminate between several olfactory stimuli. Olfactory dysfunction, correlated with olfactory bulb atrophy, has a major clinical value because it can be considered a risk factor for rapid cognitive impairment in AD [42], but also in PD [43]. Disconnections in the sensory areas of intercortical association can cause multiple disturbances of visual function, starting with decreased visual acuity and going to distorted perception of images. Subsequently, through the rapid progression of cognitive decline, the neuroimaging examination can show a widening of the calcarine sulcus and a partial atrophy of the occipital lobe, wearing the appearance of a particular form of AD, Benson’s syndrome [44]. With an incidence of 5% of cases of AD, Benson’s syndrome is characterized by progressive visual disorders, caused by neurodegenerative changes in the temporal, occipital and parietal cortex, without direct ocular pathology [45].
Alteration of the auditory integration areas causes either hearing loss with quasi-normal audiometry, or hyperexcitability with tinnitus or hallucinogenic symptomatology, the patient having partial pathological awareness of these symptoms. Hallucinogenic phenomena are often interpreted as psychotic states for which antipsychotic therapies are prescribed that cause a blockage of dopamine D2 receptors in the striatum. Thus, the striato-frontal disconnectivity is accentuated, with the aggravation of the cognitive impairment and the appearance of the extrapyramidal symptomatology that suggests a transition to Lewy body dementia or a form of AD with drug-induced Parkinsonism symptoms.
In conclusion, minor mTBI can have as a clinical marker the decrease in olfactory quality, up to anosmia or, when there is a vascular component, olfactory agnosia.
There are relevant data on the decrease of the volume of the olfactory bulb with age, the decrease being marked in the periods of progression of the MCI stage in AD. Specific to the stage with olfactory dysfunction of MCI that can anticipate the rapid transition to AD is the association of olfactory deficit with the inability to discriminate between several olfactory stimuli. The same characteristics of olfactory dysfunction are found in schizophrenia, raising the issue of dopamine involvement, a role currently highlighted in AD. In this context, the question can be raised whether the olfactory and low damage and the atrophy of the olfactory bulb can be predictive markers for the unfavorable evolution of schizophrenia and for the involvement of neurodegenerative factors in the pathogenesis of schizophrenia.
Neurodegenerative pathology occurring after an mTBI in AD in the elderly is dominated by increased deposits of neurodegenerative elements on a global background of oxidative stress and proinflammatory activation. Aβ aggression occurs through the formation and deposition of Aβ plaques in the cortical areas of maximum vulnerability, involved in cognitive circuits (hippocampus, cingulate gyrus, corpus callosum, cerebellum and temporoparietal, frontal or occipital cortex). Excessive production of neurofibrils causes a major dysfunction of the microtubules, with significant alteration of axonal transport (diffuse axonopathy – diffuse axonal dysfunction), which can lead to axonal degeneration.
The sedimentation of Aβ is dependent on the CBF, increasing at the same time with the lowering of the blood circulation speed (velocity). From our perspective, the Aβ deposition is depending on the match between the cerebral vascular structural anomalies, including the Willis polygon, and the blood flow speed. Under these circumstances, the Aβ plaques can agglutinate randomly in strategic or non-strategic areas, from the cognitive circuits’ point of view. Taking into consideration that Aβ is a “scar” factor of vascular or brain tissue lesions, the Aβ deposits are more intense in the areas where CSVD dependent vascular modifications took place. Pending the recognition of the vulnerability of elder people for oxidative stress, mTBI may trigger neurodenegerative pathology and accelerate the destructive speed for disconnection of the cognitive circuits through multifactorial mechanisms (Figure 11).
Figure 11.
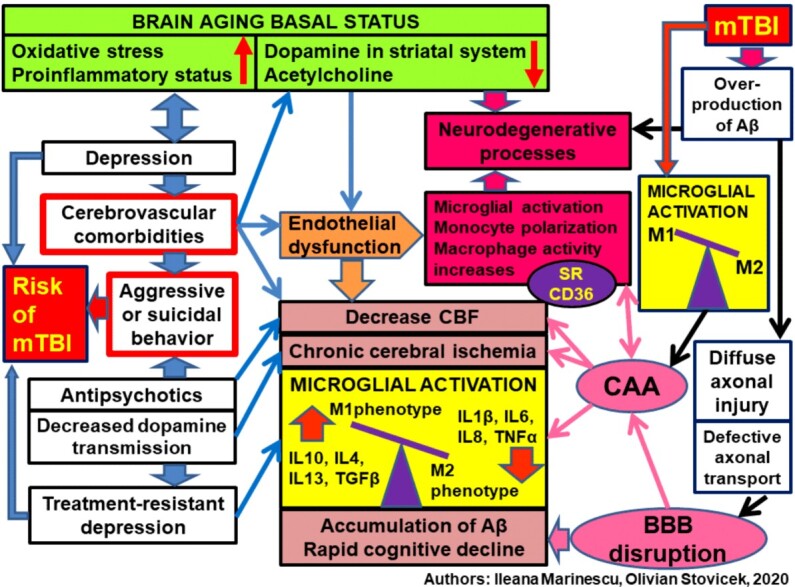
Multifactorial mechanisms and risk factors for rapid cognitive impairment post mTBI, in Alzheimer’s disease in the elderly. MTBI: Mild traumatic brain injury; SVD: Small vessel disease; CAA: Cerebral amyloid angiopathy. CBF: Cerebral blood flow; BBB: Blood–brain barrier; Aβ: Amyloid beta; SR CD36: Scavenger receptor cluster of differentiation 36; IL: Interleukin; TNFα: Tumor necrosis factor alpha; TGFβ: Transforming growth factor beta
The modification of brain flow velocity can be identified through transcranial Doppler (TCD) examination to highlight a decrease in the cerebral vasoreactivity that can worsen in the presence of ApoE4 genetic specter. This leads to an increased risk of AD in general population, without the existence of mTBI. TCD dynamic monitoring to highlight a progressive decreasing tendency in the cerebral vasoreactivity, after mTBI, may represent an indicator for cognitive deterioration. In the presence of a MCI syndrome, this indicator anticipates the rapid evolution of cognitive decline and progression to more advanced stages of AD [46]. This indicator also allows for the evaluation of the efficacy of pharmacological neuroprotective treatment in dementia. This type of medications improves the cognitive deterioration by preserving the efficiency of the cerebral vascular mechanisms [47].
Triggering of an axonal pathology is the main pathogenic action of mTBI. The transport axonal pathology involved in AD pathogenesis can be a consequence from the diffuse axonal dysfunction induced by mTBI. The amplified oxidative stress and the microglia activation with imbalance of phenotypic programming determine a microglia response. The M1 microglia is activated which amplify the inflammatory factors and proinflammatory cytokines. The transport activity of tau proteins and Aβ at axon level is increased. In this way, the neuron tries to “clean” up the neurodegenerative elements from the circuits where they are involved. The genetic specter dominated by presenilin 1 favors the phosphorylation and diffuse axonal dysfunction. Consequently, the active anterograde and retrograde transport defects turn into reversible defects, into transport anomalies with important axonal degeneration. The anterograde transport is initially disturbed, then the integral transport, disrupted mainly by mTBI, is dependent on adenosine triphosphate (ATP) and involves especially the microtubular transport [48].
In the following stage, the macrophages are activated by polarized neurons that are blocked in a unique morphological and functional activity, thus stopping the information transfer from the cellular body to axon and dendrites. The macrophage activation is an unfavorable prognosis factor, since it is associated with the intervention of other multisystemic disruption factors. The consequences of these factors are mainly cerebral vascular modifications, with the onset of CAA and alteration in the permeability and functionality of BBB. The amyloid angiopathy in elderly people can also have a sporadic form, identified frequently in the presence of cerebral blood perfusion disorders, consecutive to CSVD. They have a major contribution in the cognitive decline of the elderly person, especially after ischemic or hemorrhagic vascular accidents [49,50].
Sporadic CAA was identified in postmortem studies in 20–40% of elderly patients without dementia and in 50–60% of those with dementia [51]. From the clinical and neuroimaging perspective, the main markers announcing the sporadic CAA component are the cerebral microbleeds, existence of a subarachnoid bleeding acute episode, cortical subarachnoid hemorrhage, identification of a transient focal neurological episode, and in neuroimaging with the presence of white matter hyperintensity (WMH), cSS, increase of periventricular spaces and microbleeds.
The cerebral hypoperfusion seems to be the main cause in the decrease of the number and functionality of pericytes, with the excessive increase in BBB permeability for Aβ. This disruption can be correlated with certain indicators that would allow early prediction of rapid cognitive decline and progression of neurodegenerative elements in AD. These cellular markers can bring a significant contribution in the prevention strategies, indicating the progression from one evolutionary stage to another. These are indicated by the decrease in platelet-derived growth factor receptor beta (PDGFRB) in precuneus, given the increase of the CSVD vascular dysfunction. The PDGFRB decrease and the fibrinogen increase indicate the CAA onset with massive inclusion of Aβ fibrinogen in the cerebral arterioles wall. This exacerbates the deficit of cerebral arterial perfusion, which is controlled through an insufficiently elucidated mechanism by scavenger receptor cluster of differentiation 36 (SR CD36) [52]. This is present in the endothelial cells, macrophages, monocytes and microglia, as well as in the periphery. Their number increases in 48 to 72 hours after mTBI, precipitating the mechanism that accelerates Aβ deposits in cerebral arterioles.
Identification of CD36 monocytes in the peripheral blood gives an early warning of the cerebral vascular modifications of atherosclerosis type [53,54].
The translational research on animal models demonstrated that CD36 signals the development of an inflammatory process consecutive to cerebral tissue lesions (intravascular and perivascular hyalinization) after an acute ischemic process. The chronic cerebral ischemia state consequent to mTBI determines neuronal and microglial lesions, subsequent to oxidative stress and excitotoxicity, through multisystemic inflammatory, vascular and neurodegenerative processes [54,55].
Thus, this receptor has become an important target in pharmacological research for the molecular treatment of AD. The genetic studies on animal model showed that by blocking CD36 receptors the AD neurodegenerative modifications are no longer produced due to the decrease in the oxidative stress and in cerebral vascular dysfunction [56].
The rapid deterioration of the cognitive status for the elderly patient is favored by the involved of several post mTBI pathogenic mechanisms, where one of the most important is the vulnerabilization in the cerebral arterioles’ wall, allowing the Aβ invasion with increase of areas affected by CAA. The multisystemic mechanisms of cognitive deficit in mTBI are impacted by the imbalance in the GABA/glutamate ratio. This imbalance is emphasized by the activation of the hypothalamic–pituitary–adrenal axis under the existence of a depressive disorder in prodromal phase of AD, of behavioral disorders and antipsychotic treatment with high capacity to block D2 dopamine receptors.
Conclusions
The severe TBI pathology benefits from a special attention, both from clinicians as well as from decision makers, being accepted as a matter of public health for the elderly persons. On the other hand, mTBIs are considered “benign”, in contrast with numerous studies that underlined their potential to determine cognitive deteriorative neuropsychiatric modifications. It is important for any clinician to identify, within MCI syndrome, the decrease in olfactory perception associated or not with auditory deficiency, visual perception deficit or tinnitus. These latter clinical modifications are correlated with the disconnection of cerebral areas for association and cognitive integration of sensory signals. The risk factor for rapid evolution of cognitive deficit in MCI stage, post mTBI, include the presence in patients history of depression with antidepressant or antipsychotic treatment with selective serotonin reuptake inhibitors (SSRI), positive markers for swelling and endothelial dysfunction that may indicated rapid deterioration of cognitive reserve and invasion of neurodegenerative elements. Also, the chronic cerebral ischemia state objectified through hypoperfusion after TCD or magnetic resonance imaging (MRI) examination represents an important neuroimaging marker associated with other markers for thalamic lesions and increase in Virchow–Robin spaces. We consider that there are sufficient clinical, biological and neuroimaging arguments to suggest the importance of such investigations in elderly patients, post mTBI, in order to identify the risk of rapid cognitive deterioration. The correct prophylaxis measures with neurotrophic agents, restoring the GABA/glutamate balance and fighting the chronic inflammatory process can become important pharmacological therapeutic targets. There are limitations in the research and multisystemic interpretations performed by our team, which we hope to overcome in future studies, with the support of colleagues involved in the fundamental and clinical research of this pathology.
Conflict of interests
The authors declare that they have no conflict of interests.
References
- 1.Wang J, Zhang S, Wang Y, Chen L, Zhang XS. Disease-aging network reveals significant roles of aging genes in connecting genetic diseases. PLoS Comput Biol. 2009;5(9):e1000521–e1000521. doi: 10.1371/journal.pcbi.1000521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kokiko-Cochran ON, Godbout JP. The inflammatory continuum of traumatic brain injury and Alzheimer’s disease. Front Immunol. 2018;9:672–672. doi: 10.3389/fimmu.2018.00672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wyss-Coray T. Ageing, neurodegeneration and brain rejuvenation. Nature. 2016;539(7628):180–186. doi: 10.1038/nature20411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Scheltens P, Blennow K, Breteler MM, de Strooper B, Frisoni GB, Salloway S, Van der Flier WM. Alzheimer’s disease. Lancet. 2016;388(10043):505–517. doi: 10.1016/S0140-6736(15)01124-1. [DOI] [PubMed] [Google Scholar]
- 5.Fleminger S, Oliver DL, Lovestone S, Rabe-Hesketh S, Giora A. Head injury as a risk factor for Alzheimer’s disease: the evidence 10 years on; a partial replication. J Neurol Neurosurg Psychiatry. 2003;74(7):857–862. doi: 10.1136/jnnp.74.7.857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ramos-Cejudo J, Wisniewski T, Marmar C, Zetterberg H, Blennow K, de Leon MJ, Fossati S. Traumatic brain injury and Alzheimer’s disease: the cerebrovascular link. EBioMedicine. 2018;28:21–30. doi: 10.1016/j.ebiom.2018.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jiang JY, Gao GY, Feng JF, Mao Q, Chen LG, Yang XF, Liu JF, Wang YH, Qiu BH, Huang XJ. Traumatic brain injury in China. Lancet Neurol. 2019;18(3):286–295. doi: 10.1016/S1474-4422(18)30469-1. [DOI] [PubMed] [Google Scholar]
- 8.Taylor CA, Bell JM, Breiding MJ, Xu L. Traumatic brain injury-related emergency department visits, hospitalizations, and deaths - United States, 2007 and 2013. MMWR Surveill Summ. 2017;66(9):1–16. doi: 10.15585/mmwr.ss6609a1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.West TA, Sharp S. Neuroendocrine dysfunction following mild TBI: when to screen for it. J Fam Pract. 2014;63(1):11–16. [PubMed] [Google Scholar]
- 10.Werner C, Engelhard K. Pathophysiology of traumatic brain injury. Br J Anaesth. 2007;99(1):4–9. doi: 10.1093/bja/aem131. [DOI] [PubMed] [Google Scholar]
- 11.Langlois JA, Kegler SR, Butler JA, Gotsch KE, Johnson RL, Reichard AA, Webb KW, Coronado VG, Selassie AW, Thurman DJ. Traumatic brain injury-related hospital discharges. Results from a 14-state surveillance system, 1997. MMWR Surveill Summ. 2003;52(4):1–20. [PubMed] [Google Scholar]
- 12.Dams-O’Connor K, Gibbons LE, Landau A, Larson EB, Crane PK. Health problems precede traumatic brain injury in older adults. J Am Geriatr Soc. 2016;64(4):844–848. doi: 10.1111/jgs.14014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thurman DJ, Alverson C, Dunn KA, Guerrero J, Sniezek JE. Traumatic brain injury in the United States: a public health perspective. J Head Trauma Rehabil. 1999;14(6):602–615. doi: 10.1097/00001199-199912000-00009. [DOI] [PubMed] [Google Scholar]
- 14.Dewan MC, Rattani A, Gupta S, Baticulon RE, Hung YC, Punchak M, Agrawal A, Adeleye AO, Shrime MG, Rubiano AM, Rosenfeld JV, Park KB. Estimating the global incidence of traumatic brain injury. J Neurosurg. 2018;130(4):1080–1097. doi: 10.3171/2017.10.JNS17352. [DOI] [PubMed] [Google Scholar]
- 15.Nguyen R, Fiest KM, McChesney J, Kwon CS, Jette N, Frolkis AD, Atta C, Mah S, Dhaliwal H, Reid A, Pringsheim T, Dykeman J, Gallagher C. The international incidence of traumatic brain injury: a systematic review and meta-analysis. Can J Neurol Sci. 2016;43(6):774–785. doi: 10.1017/cjn.2016.290. [DOI] [PubMed] [Google Scholar]
- 16.Prince C, Bruhns ME. Evaluation and treatment of mild traumatic brain injury: the role of neuropsychology. Brain Sci. 2017;7(8):105–105. doi: 10.3390/brainsci7080105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Menon DK, Schwab K, Wright DW, Maas AI, Demographics and Clinical Assessment Working Group of the International and Interagency Initiative toward Common Data Elements for Research on Traumatic Brain Injury and Psychological Health Position statement: definition of traumatic brain injury. Arch Phys Med Rehabil. 2010;91(11):1637–1640. [Google Scholar]
- 18.Li Y, Li Y, Li X, Zhang S, Zhao J, Zhu X, Tian G. Head injury as a risk factor for dementia and Alzheimer’s disease: a systematic review and meta-analysis of 32 observational studies. PLoS One. 2017;12(1):e0169650–e0169650. doi: 10.1371/journal.pone.0169650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Edwards G, Moreno-Gonzalez I, Soto C. Amyloid-beta and tau pathology following repetitive mild traumatic brain injury. Biochem Biophys Res Commun. 2017;483(4):1137–1142. doi: 10.1016/j.bbrc.2016.07.123. [DOI] [PubMed] [Google Scholar]
- 20.Mendez MF. What is the relationship of traumatic brain injury to dementia. J Alzheimers Dis. 2017;57(3):667–681. doi: 10.3233/JAD-161002. [DOI] [PubMed] [Google Scholar]
- 21.Carroll LJ, Cassidy JD, Holm L, Kraus J, Coronado VG, WHO Collaborating Center Task Force on Mild Traumatic Brain Injury Methodological issues and research recommendations for mild traumatic brain injury: The WHO Collaborating Centre Task Force on Mild Traumatic Brain Injury. J Rehabil Med. 2004;(43 Suppl):113–125. doi: 10.1080/16501960410023877. [DOI] [PubMed] [Google Scholar]
- 22.Powell JM, Ferraro JV, Dikmen SS, Temkin NR, Bell KR. Accuracy of mild traumatic brain injury diagnosis. Arch Phys Med Rehabil. 2008;89(8):1550–1555. doi: 10.1016/j.apmr.2007.12.035. [DOI] [PubMed] [Google Scholar]
- 23.Joseph B, Khalil M, Zangbar B, Kulvatunyou N, Orouji T, Pandit V, O’Keeffe T, Tang A, Gries L, Friese RS, Rhee P, Davis JW. Prevalence of domestic violence among trauma patients. JAMA Surg. 2015;150(12):1177–1183. doi: 10.1001/jamasurg.2015.2386. [DOI] [PubMed] [Google Scholar]
- 24.Tinetti ME, Kumar C. The patient who falls: "it’s always a trade-off". JAMA. 2010;303(3):258–266. doi: 10.1001/jama.2009.2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Low PA, Tomalia VA. Orthostatic hypotension: mechanisms, causes, management. J Clin Neurol. 2015;11(3):220–226. doi: 10.3988/jcn.2015.11.3.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chauny JM, Marquis M, Bernard F, Williamson D, Albert M, Laroche M, Daoust R. Risk of delayed intracranial hemorrhage in anticoagulated patients with mild traumatic brain injury: systematic review and meta-analysis. J Emerg Med. 2016;51(5):519–528. doi: 10.1016/j.jemermed.2016.05.045. [DOI] [PubMed] [Google Scholar]
- 27.Garwood CL, Corbett TL. Use of anticoagulation in elderly patients with atrial fibrillation who are at risk for falls. Ann Pharmacother. 2008;42(4):523–532. doi: 10.1345/aph.1K498. [DOI] [PubMed] [Google Scholar]
- 28.Charidimou A, Linn J, Vernooij MW, Opherk C, Akoudad S, Baron JC, Greenberg SM, Jäger HR, Werring DJ. Cortical superficial siderosis: detection and clinical significance in cerebral amyloid angiopathy and related conditions. Brain. 2015;138(Pt 8):2126–2139. doi: 10.1093/brain/awv162. [DOI] [PubMed] [Google Scholar]
- 29.Smith EE. Cerebral amyloid angiopathy as a cause of neurodegeneration. J Neurochem. 2018;144(5):651–658. doi: 10.1111/jnc.14157. [DOI] [PubMed] [Google Scholar]
- 30.Udriştoiu I, Marinescu I, Pîrlog MC, Militaru F, Udriştoiu T, Marinescu D, Mutică M. The microvascular alterations in frontal cortex during treatment with antipsychotics: a post-mortem study. Rom J Morphol Embryol. 2016;57(2):501–506. [PubMed] [Google Scholar]
- 31.Miller AH, Haroon E, Raison CL, Felger JC. Cytokine targets in the brain: impact on neurotransmitters and neurocircuits. Depress Anxiety. 2013;30(4):297–306. doi: 10.1002/da.22084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Marinescu I, Vasiliu O, Vasile D. Translational approaches in treatment-resistant depression based on animal model. Rom J Morphol Embryol. 2018;59(3):955–964. [PubMed] [Google Scholar]
- 33.Chen YH, Huang EYK, Kuo TT, Miller J, Chiang YH, Hoffer BJ. Impact of traumatic brain injury on dopaminergic transmission. Cell Transplant. 2017;26(7):1156–1168. doi: 10.1177/0963689717714105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gatson JW, Warren V, Abdelfattah K, Wolf S, Hynan LS, Moore C, Diaz-Arrastia R, Minei JP, Madden C, Wigginton JG. Detection of β-amyloid oligomers as a predictor of neurological outcome after brain injury. J Neurosurg. 2013;118(6):1336–1342. doi: 10.3171/2013.2.JNS121771. [DOI] [PubMed] [Google Scholar]
- 35.Marinescu I, Enătescu VR, Ghelase ŞM, Marinescu D. Neurobiological arguments for a pathogenic multifactorial disconnective model of cognitive disorders from Alzheimer’s disease in elderly people. Rom J Morphol Embryol. 2017;58(4):1165–1173. [PubMed] [Google Scholar]
- 36.Salehi A, Zhang JH, Obenaus A. Response of the cerebral vasculature following traumatic brain injury. J Cereb Blood Flow Metab. 2017;37(7):2320–2339. doi: 10.1177/0271678X17701460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hay JR, Johnson VE, Young AMH, Smith DH, Stewart W. Blood–brain barrier disruption is an early event that may persist for many years after traumatic brain injury in humans. J Neuropathol Exp Neurol. 2015;74(12):1147–1157. doi: 10.1097/NEN.0000000000000261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Iadecola C. Untangling neurons with endothelial nitric oxide. Circ Res. 2016;119(10):1052–1054. doi: 10.1161/CIRCRESAHA.116.309927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gupta A, Iadecola C. Impaired Aβ clearance: a potential link between atherosclerosis and Alzheimer’s disease. Front Aging Neurosci. 2015;7:115–115. doi: 10.3389/fnagi.2015.00115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kawata K, Liu CY, Merkel SF, Ramirez SH, Tierney RT, Langford D. Blood biomarkers for brain injury: what are we measuring. Neurosci Biobehav Rev. 2016;68:460–473. doi: 10.1016/j.neubiorev.2016.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Preda SA, Nechita F, Comanescu MC, Albulescu DM, Tuculina MJ, Docea AD, Burada E, Vasile RC, Mitroi M. Evaluation of bone turnover and DXA markers in premature ovarian failure. Rev Chim (Bucharest) 2019;70(6):2054–2057. [Google Scholar]
- 42.Roberts RO, Christianson TJ, Kremers WK, Mielke MM, Machulda MM, Vassilaki M, Alhurani RE, Geda YE, Knopman DS, Petersen RC. Association between olfactory dysfunction and amnestic mild cognitive impairment and Alzheimer disease dementia. JAMA Neurol. 2016;73(1):93–101. doi: 10.1001/jamaneurol.2015.2952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Leverenz JB, Quinn JF, Zabetian C, Zhang J, Montine KS, Montine TJ. Cognitive impairment and dementia in patients with Parkinson disease. Curr Top Med Chem. 2009;9(10):903–912. [PMC free article] [PubMed] [Google Scholar]
- 44.Benson DF, Davis RJ, Snyder BD. Posterior cortical atrophy. Arch Neurol. 1988;45(7):789–793. doi: 10.1001/archneur.1988.00520310107024. [DOI] [PubMed] [Google Scholar]
- 45.Maia da, Millington RS, Bridge H, James-Galton M, Plant GT. Visual dysfunction in posterior cortical atrophy. Front Neurol. 2017;8:389–389. doi: 10.3389/fneur.2017.00389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wolters FJ, Zonneveld HI, Hofman A, van der Lugt A, Koudstaal PJ, Vernooij MW, Ikram MA, Heart–Brain Connection Collaborative Research Group Cerebral perfusion and the risk of dementia: a population-based study. Circulation. 2017;136(8):719–728. doi: 10.1161/CIRCULATIONAHA.117.027448. [DOI] [PubMed] [Google Scholar]
- 47.Tapu M, Bicu D, Tapu F, Stovicek O. The efficacy of cerebrolysin in vascular dementia. J Neurol Sci. 2009;283(1–2):P286–P286. [Google Scholar]
- 48.Liu XA, Rizzo V, Puthanveettil SV. Pathologies of axonal transport in neurodegenerative diseases. Transl Neurosci. 2012;3(4):355–372. doi: 10.2478/s13380-012-0044-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Viswanathan A, Greenberg SM. Cerebral amyloid angiopathy in the elderly. Ann Neurol. 2011;70(6):871–880. doi: 10.1002/ana.22516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Boulouis G, Charidimou A, Greenberg SM. Sporadic cerebral amyloid angiopathy: pathophysiology, neuroimaging features, and clinical implications. Semin Neurol. 2016;36(3):233–243. doi: 10.1055/s-0036-1581993. [DOI] [PubMed] [Google Scholar]
- 51.Charidimou A, Boulouis G, Gurol ME, Ayata C, Bacskai BJ, Frosch MP, Viswanathan A, Greenberg SM. Emerging concepts in sporadic cerebral amyloid angiopathy. Brain. 2017;140(7):1829–1850. doi: 10.1093/brain/awx047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Goncharov NV, Nadeev AD, Jenkins RO, Avdonin PV. Markers and biomarkers of endothelium: when something is rotten in the state. Oxid Med Cell Longev. 2017;2017:9759735–9759735. doi: 10.1155/2017/9759735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Marcovecchio PM, Thomas GD, Mikulski Z, Ehinger E, Mueller KAL, Blatchley A, Wu R, Miller YI, Nguyen AT, Taylor AM, McNamara CA, Ley K, Hedrick CC. Scavenger receptor CD36 directs nonclassical monocyte patrolling along the endothelium during early atherogenesis. Arterioscler Thromb Vasc Biol. 2017;37(11):2043–2052. doi: 10.1161/ATVBAHA.117.309123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Abe T, Shimamura M, Jackman K, Kurinami H, Anrather J, Zhou P, Iadecola C. Key role of CD36 in Toll-like receptor 2 signaling in cerebral ischemia. Stroke. 2010;41(5):898–904. doi: 10.1161/STROKEAHA.109.572552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Coraci IS, Husemann J, Berman JW, Hulette C, Dufour JH, Campanella GK, Luster AD, Silverstein SC, El-Khoury JB. CD36, a class B scavenger receptor, is expressed on microglia in Alzheimer’s disease brains and can mediate production of reactive oxygen species in response to beta-amyloid fibrils. Am J Pathol. 2002;160(1):101–112. doi: 10.1016/s0002-9440(10)64354-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Park L, Wang G, Zhou P, Zhou J, Pitstick R, Previti ML, Younkin L, Younkin SG, Van Nostrand WE, Cho S, Anrather J, Carlson GA, Iadecola C. Scavenger receptor CD36 is essential for the cerebrovascular oxidative stress and neurovascular dysfunction induced by amyloid-β. Proc Natl Acad Sci U S A. 2011;108(12):5063–5068. doi: 10.1073/pnas.1015413108. [DOI] [PMC free article] [PubMed] [Google Scholar]