

Abstract
The General AMBER Force Field (GAFF) has been broadly used by researchers all over the world to perform in silico simulations and modelings on diverse scientific topics, especially in the field of computer-aided drug design whose primary task is to accurately predict the affinity and selectivity of receptor–ligand binding. The atomic partial charges in GAFF and the second generation of GAFF (GAFF2) were originally developed with the quantum mechanics derived restrained electrostatic potential charge, but in practice, users usually adopt an efficient charge method, Austin Model 1-bond charge corrections (AM1-BCC), based on which, without expensive ab initio calculations, the atomic charges could be efficiently and conveniently obtained with the ANTECHAMBER module implemented in the AMBER software package. In this work, we developed a new set of BCC parameters specifically for GAFF2 using 442 neutral organic solutes covering diverse functional groups in aqueous solution. Compared to the original BCC parameter set, the new parameter set significantly reduced the mean unsigned error (MUE) of hydration free energies from 1.03 kcal/mol to 0.37 kcal/mol. More excitingly, this new AM1-BCC model also showed excellent performance in the solvation free energy (SFE) calculation on diverse solutes in various organic solvents across a range of different dielectric constants. In this large-scale test with totally 895 neutral organic solvent–solute systems, the new parameter set led to accurate SFE predictions with the MUE and the root-mean-square-error of 0.51 kcal/mol and 0.65 kcal/mol, respectively. This newly developed charge model, ABCG2, paved a promising path for the next generation GAFF development.
I. INTRODUCTION
In computer-aided drug design (CADD) efforts, especially at the stages of lead-identification and lead-optimization, a major task is to accurately predict the binding affinities of receptors (proteins or nucleic acids) and ligands.1–4 The prediction quality generally depends on the free energy calculation algorithm and force field (FF).5 The former includes various efficient end-point free energy methods, such as LIE and MM-PBSA/GBSA,6–12 and rigorous alchemical free energy methods, such as TI, FEP, λ-dynamics, and other advanced-sampling-based methods.13–18 The latter includes specific macromolecular force fields (FFs)19–23 and general FFs for arbitrary compounds, which may be encountered as ligands.24–27 In the past, the accuracy and transferability issues have challenged the general FF development. One thought has been moving from additive fixed-charge models to polarizable models28–33 because the latter allow the electrostatic effect across different dielectric environments (from polar to nonpolar) to be naturally handled. While polarizable FFs are currently undergoing active development, so far they have been seldom applied on simulating and calculating protein-ligand binding interactions.30–32 Due to the efficiency requirement, particularly burdened by the need of a comprehensive description of the enormous chemical space, classical pair-wise additive FFs based on fixed point charge models not only are the current main-stream but also are expected to prevail in the near and a perceivable future. With the above thought, this work explores whether additive general FFs can be further optimized to meet the accuracy and transferability requirement, for instance reliably treating a range of polar and nonpolar environments.
The general AMBER force field (GAFF)24,25 is the first general FF developed in academia, primarily to model arbitrary organic molecules. It has been widely applied on a variety of scientific topics by researchers all over the world. So far it has been cited more than 7600 times according to the databases of Web of Sciences (https://www.webofknowledge.com, accessed on June 3, 2020) and more than 9600 times according to Google Scholar Citation (https://scholar.google.com, accessed on June 3, 2020). Since 2015, the second generation of GAFF (GAFF2) has been released to the public via the AMBER program and AmberTools. Related description can be found in the footer of the released gaff2.dat file and with the completion of the remaining work on representative chemical space expansion and some parameter improvement, the development detail is soon to be submitted for publication. In a very brief summary, compared to GAFF, GAFF2 has updated the bonded parameters to reproduce molecular geometries, vibrational spectra, and potential energy surfaces from higher level quantum mechanics (QM) calculations on more model compounds, and updated the non-bonded parameters to better reproduce ab initio interaction energies and experimental neat liquid properties. Positive feedback from users (in private communications and in literature34) also revealed the encouraging performance of GAFF2 in various aspects. For example, Slochower et al.34 performed a free energy calculation study on 43 α and β-cyclodextrin (CD) host–guest pairs with different FFs: GAFF, GAFF2, and SMIRNOFF99Frosst.35 It was found that GAFF2 “has statistically significant(ly) better correlation with the experimental data” on the binding free energy and enthalpy, “excellent agreement with experiment on predicted binding entropy,” and “better model for the flexibility of the CD cavity” compared to GAFF and SMIRNOFF99Frosst, although the latter two are “arguably better than GAFF v2.1 on estimated binding free energies … based on the mean signed error relative to experiment.”
Originally, GAFF was developed based on the restrained electrostatic potential (RESP)36,37 charge model to assign atomic charges, which fits atomic charges against the electrostatic potential (ESP) from the QM calculation at the HF/6-31G* level of theory, just like the AMBER biomolecular FFs21,22 for the biomolecules. For the fact that ab initio calculations are expensive, in practice, GAFF users usually adopt the fast semiempirical method of Austin Model 1 with bond charge corrections (AM1-BCC)38,39 to generate atomic charges. AM1-BCC charges could be efficiently and conveniently obtained via the ANTECHAMBER40 module of the AMBER program tools. The basic idea of AM1-BCC is to use AM1 Mulliken charges to capture primary electronic structural features of a molecule and then apply a set of additive bond charge corrections (BCCs) upon AM1 Mulliken charges to emulate the HF/6-31G* ESP. The advantages of AM1-BCC lie in that not only the process is convenient and efficient (because ab initio calculations are not necessary) but also the produced atomic charges are less dependent on the input molecular conformation. GAFF2 was also developed based on the RESP charge model. It is reasonable to expect that many users would still prefer employing the AM1-BCC charge scheme instead of RESP. Therefore, in this work, we sought to develop an optimized AM1-BCC set for GAFF2. Specifically, we employed a solvation free energy (SFE) based strategy for this optimization. In our previous development of GAFF and GAFF2, this important physiochemical property was not directly targeted in the parameterization process but only employed for later validation, mostly due to the fact that the SFE calculation is much more computationally costly than the calculation of density or heat of vaporization. Indeed, the SFE based strategy has not been commonly adopted except for GROMOS 53A623 and 53A6OXY,41 which considered the SFEs of amino acid analogues and functional groups containing oxygen, respectively, also as the parameterization target.
SFE is a critical property in physical, chemical, and biological processes. SFE is closely related to many other important properties, such as solubility, partition coefficient, membrane permeability, and protein-ligand binding free energy in drug-discovery projects.42 Generally, the accuracy level of the SFE calculation limits the accuracy expectation for the prediction of the above properties. For example, the absolute or relative SFE calculation is an elementary step in the thermodynamic cycle used for the absolute or relative protein-ligand binding free energy calculation.15 Moreover, a ligand binding process can be viewed as a special dissolution process; both involve the conformation rearrangement of the small molecule and relaxation of the environment (solvent or protein). In certain aspect, the difference between these two processes only lies in complexity and dielectrics of the corresponding environment. Therefore, the SFE calculation is often utilized to test sampling algorithms and potential energy functions, as the basic benchmark for binding free energy prediction.5 As a popular force field, GAFF has also been extensively validated in the aspect of SFE in water [i.e., hydration free energy (HFE)].43–54 Test studies have been performed across the diverse categories of the organic molecules, on different charge models (RESP, or AM1-BCC, or the commercial Merck-Frosst version of AM1-BCC52–55), and through various free energy methods in different molecular dynamics (MD) programs.43–55
In principle, both van der Waals (VDW) parameters and atom charges can be optimized against SFEs. For instance, Nerenberg et al.55 and Jämbeck and Lyubartsev56 tried specifically tuning the VDW interactions between the solute atoms and the oxygen atom in water with the Lorentz–Berthelot combination rules kept or abandoned.56 Based on our experience and general expert understanding in the field, we prefer to focus on the charge adjustment because SFEs are more sensitive to atomic partial charges, whilst neat liquid properties are more sensitive to VDW parameters. Therefore, the VDW parameters in both GAFF and GAFF2 were mainly calibrated against pure liquid properties, such as density and heat of vaporization. In terms of charge optimization, upon the initial atomic charge assignment, three options can be taken: (1) reassigning charges based on the higher level of QM theories and basis sets, (2) applying scale factors, and (3) specifically tuning BCCs. Previous studies revealed that the atomic charges directly derived from the higher level of the QM methods can only introduce marginal improvement to the SFE calculation.57–60 A scale factor applied on the QM derived atomic charges could improve the overall performance on the SFE calculation, but different optimal scale factors may be needed for different solvents.61 In this study, we chose the strategy of optimizing the specific BCC terms in the AM1-BCC model to focus the reduction of the systematic errors of SFEs on certain functional groups, which were identified as the bottleneck of achieving accurate transfer free energy or binding free energy prediction. In this work, we (1) for the first time tested the performance of the original AM1-BCC charge model with GAFF2 on the HFEs of a large dataset of more than 400 neutral molecules, (2) developed a new set of AM1-BCC parameters, which significantly improves the accuracy of the HFE calculations in explicit TIP3P water, and (3) verified that the new AM1-BCC charge model also has an outstanding performance on the SFE calculation in various nonpolar and polar organic solvents, which have different dielectric constants. The results not only enhanced the capability of the quantitative prediction of key properties in CADD but also paved a feasible way for the development of the next generation general AMBER force field.
II. METHODS
A. Dataset preparation
The experimental data of SFEs of the neutral molecules in water (HFEs) were taken from the FreeSolv v0.52 database,54 and the experimental data of SFEs in organic solvents were obtained from the Minnesota Solvation Database v2012.62 The initial structures of solutes/solvents were taken from the mol2 files in the FreeSolv v0.52 database or from the xyz files in Minnesota Solvation Database v2012, then were imported to Schrödinger Maestro v11.263 for visual check and necessary manual modification, such as setting correct bond types for the molecule structures from the xyz files, and then all the structures were saved in the mol2 format.
B. Force fields and preparation of systems
Geometry optimization in the gas phase was performed for all mol2 structures by Gaussian 1664 at the Hartree–Fock (HF) level of theory with 6-31G* basis. The Gaussian output files were directed to ANTECHAMBER40 to generate the corresponding topology files (containing AM1-BCC atomic charges) and parameter files (including bonded terms and VDW terms) using the GAFF2 force field. The SQM module in Amber Tools was called by ANTECHAMBER to produce AM1 charges, which was further modified by the predefined BCC terms (either original ones or adjusted ones in this study) to produce the final AM1-BCC atomic charges. When parameterizing the specific BCC terms, the corresponding values in the file $AMBERHOME/dat/antechamber/BCCPARM.DAT were tuned. The organic solvent molecules were processed in the same procedure as the solute molecules. Water was treated with the TIP3P65 model.
For each solvent–solute pair system, a single solute molecule was solvated in a cubic box of pure solvent molecules with the TLEAP module in AMBER18.66 The size of the simulation box varies depending on the size of solute and the type of solvents. The rule of thumb is that the minimum solute-box distance should be larger than the short-range cutoff (10 Å here) to avoid image violations. For water solvent, the minimum distance between any solute atom and an edge of the box was set to 12 Å. For organic solvents, the initial solution box generated by TLEAP often have a vacuum space due to the size of solvent molecules and the need of deleting solvent molecules to avoid steric conflicts (Fig. S4 in supplementary material). The solution box would shrink after equilibrium runs. Therefore, the input of the initial thickness of the solvent shell provided to TLEAP was set individually for different solvent–solute pairs to ensure the side sizes of cubic boxes after equilibration were ∼39 Å, which is enough to get a solvent shell of at least 10 Å for all of the considered solutes in this study.
C. Simulation protocols
The thermodynamic integration (TI) modules of both central processing unit (CPU) and graphic processing unit (GPU) versions67–69 implemented in AMBER1866 were adopted to carry out simulations and calculate the SFEs. Periodic boundary condition and the isothermal-isobaric NPT ensemble were produced in all simulations, including both the equilibration and production phases. The temperature was kept at 298 K using Langevin dynamics with the collision frequency gamma_ln being set to 2.0 ps−1. The pressure was kept at 1.013 25 bar with Monte Carlo barostat and the pressure relaxation time being set to 2.0 ps. Because the bond constraint SHAKE algorithm is disabled for TI mutations in AMBER GPU-TI module pmemdGTI, a time step of 1 fs was used for all MD simulations. The whole solute molecule was incorporated into the soft-core region for both VDW and electrostatic interactions.70,71 The default soft-core potential implemented in AMBER18 package66 was applied. The single-step decoupling protocol71 was performed at nine discrete λ windows (0.015 92, 0.081 98, 0.193 31, 0.337 87, 0.5, 0.662 13, 0.806 69, 0.918 02, and 0.984 08). The energy information was saved every 0.5 ps for post-analysis. The Gaussian quadrature rule was adopted to integrate ∂V/∂λ (marked as “DV/DL” in AMBER output files) as described by Eqs. (1)–(3) and the weights (wi) corresponding to the λi values were 0.040 64, 0.090 32, 0.130 31, 0.156 17, 0.165 12, 0.156 17, 0.130 31, 0.090 32, and 0.040 64 according to Table 21.1 in the AMBER18 manual,66
| (1) |
| (2) |
| (3) |
where Hamiltonian V(λ) is the mixed potential of the initial state, V0, and the final state, V1.
After setting-up of the systems, initial equilibrations were conducted at λ = 0.5 with CPU-TI for 200 ps before switching to GPU-TI runs because AMBER CPU-TI has more tolerance for the changes in the size of the simulation box than AMBER GPU-TI in the NPT ensemble simulations. Five snapshots were extracted at even intervals from the last 100 ps of the CPU-TI equilibration trajectory as the starting configurations of five individual GPU-TI runs at λ = 0.5. A 2-ns simulation was conducted for each individual GPU-TI run, and the final snapshot was used as the starting configuration for the two neighboring λ windows, i.e., λ = 0.337 87 and 0.662 13, and their final snapshots of these two λ windows were used as the starting configurations of their neighboring λ windows toward two λ endpoints, respectively (Fig. S1). Similarly, for each independent run of a λ window, a 2-ns MD simulation was conducted and the beginning 0.5 ns simulation was considered as further equilibration steps, and therefore, excluded for post-analysis. The above TI protocol is shown in Fig. S1. The SFE values were separately calculated from five replicas starting with different randomly generated initial velocities (run1 to run5) at all λ windows. Then the arithmetic average was used as the final SFE for each solvent–solute pair, and the associated standard deviation was used to indicate the precision.
D. Strategy and procedure of parameterizing BCC terms
The HFEs of various solutes with single or multifold functional groups in TIP3P water were calculated with the GAFF2 force field parameters and AM1-BCC charges. The BCC terms for those functional groups with mean unsigned errors (MUEs) of >0.6 kcal/mol were adjusted, and new AM1-BCC charges were re-generated, followed by a new round of HFE calculations. The BCC terms were verified and adjusted if needed in a sequential manner according to the scheme shown in Fig. S2. In brief, the parameters for alkanes and aromatic hydrocarbons were first evaluated and adjusted if necessary, followed by alkenes, alkynes, and other chemical functional groups. The adjusted BCC parameters in earlier steps will be applied directly in the subsequent steps. After all optimizations targeting HFEs were done, the updated BCC terms were directly applied to the calculations of SFEs in various organic solvents to test their performance and transferability in different dielectric environments. The overall procedure of developing and validating the new AM1-BCC charge model is demonstrated in Fig. S3.
III. RESULTS
For most solvent–solute pairs, the standard deviation of the calculated SFE from five individual TI replicas in this study is usually <0.2 kcal/mol. Considering the uncertainty of individual experimental measurements of SFEs can be up to ∼0.6 kcal/mol,54 the precision of our TI protocol is sufficient for the parameterization process.
A. Performance of GAFF2 with the original and new AM1-BCC charge models on hydration free energies
Just as GAFF, GAFF2 has been developed with the RESP charge model for atomic partial charge assignment and the TIP3P water model for studying the molecular properties in aqueous solution, following the similar force field parameterization strategy in the AMBER biomolecule force field development in order to maximize the compatibility between the biomolecular and organic molecular force fields.21,22 The HFEs and other SFEs have not been utilized as the targets of parameterization. The overall better performance of GAFF2/RESP on the HFE in TIP3P water than that of GAFF/RESP has been validated by us, and will be demonstrated in another manuscript of GAFF2 description. In this study, we focus on the performance of the original AM1-BCC (interchangeable as the old AM1-BCC in this manuscript) charge model with the GAFF2 parameters on HFEs of a variety of organic solute molecules, and the corresponding improvement after we optimize certain BCC parameters.
As presented in Fig. 1 and Table I, the mean unsigned error (MUE) of hydration free energies for 27 linear and branched alkanes is 0.34 kcal/mol, and the MUE of nine cycloalkanes is 0.51 kcal/mol both less than the default uncertainty (0.6 kcal/mol) of experimental measurements in the FreeSolv database.54 Therefore, the BCC terms for alkanes were considered as good enough and were kept unchanged. Except for alkanes, the rest of the functional groups shown in Fig. 1 and Table I involved more or less adjustment of the related BCC parameters. 12 types of functional groups with original AM1-BCC charges show systematic errors >1.0 kcal/mol, including chlorinated aliphatic and aromatic hydrocarbons, brominated hydrocarbons, alcohols, amines, pyrazines and pyridines, nitriles, nitro hydrocarbons, nitrooxy alkanes, amides, and thioethers. After the optimization of the BCC terms, all functional groups have the MUE ≤ 0.6 kcal/mol for the benchmarked molecules. For most benchmarked functional groups shown in Table I, the new AM1-BCC charge model makes the predictive index PI72,73 and Pearson’s correlation coefficient R increase, the slope k value closer to 1.0, and the intercept b value closer to 0.0, except for several functional groups with very few data points and a narrow range of the experimental data. For the tested 442 neutral solutes in total, the MUE from the original AM1-BCC charge model is 1.03 kcal/mol, and the MUE from the new AM1-BCC charge model is significantly decreased to only 0.37 kcal/mol. The predictive index PI increases from 0.91 to 0.98. Pearson’s correlation coefficient R increases from 0.92 to 0.98. When fitting the calculated data (y) vs the experimental data (x) with function y = kx + b, the slope k value increases from 0.86 to 0.98, and the intercept b value changes from 0.27 kcal/mol to −0.06 kcal/mol. Figure 2 shows the overall performance of the original and the newly proposed AM1-BCC charges on the hydration free energies calculated with GAFF2 parameters.
FIG. 1.
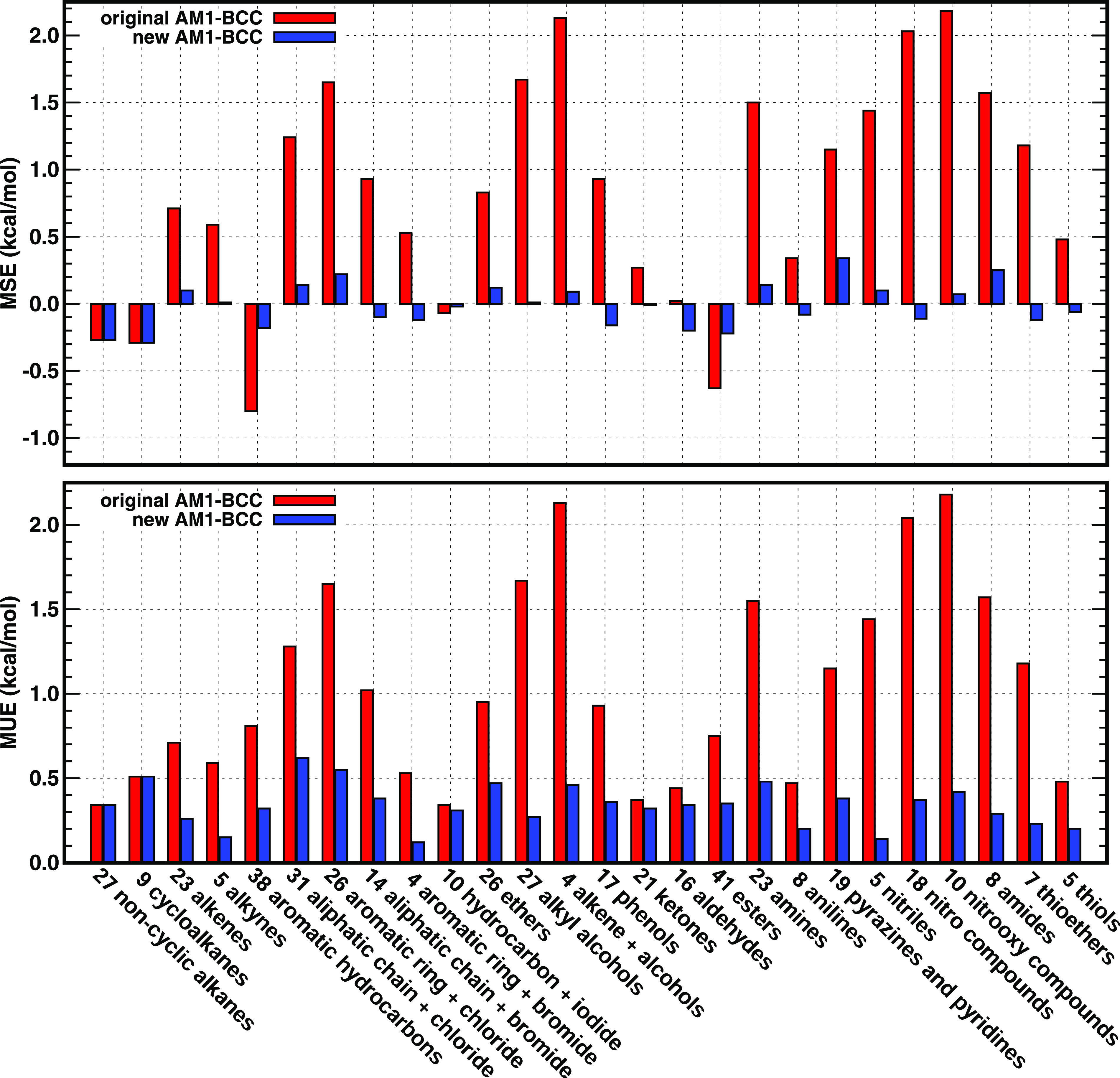
Performance of the original and the proposed new AM1-BCC charge models on the hydration free energies of 442 neutral solutes with different functional groups.
TABLE I.
The performance of the original and the proposed new AM1-BCC atomic charges in combination with the GAFF2 parameters on the hydration free energies of neutral organic solutes in TIP3P water. Expt. stands for experimental; MSE stands for mean signed error; MUE stands for mean unsigned error; PI stands for predictive index; R stands for Pearson’s correlation coefficient; k and b stand for the slope and intercept values when fitting the calculated data (y) vs experimental data (x) with function y = kx + b.
| Expt. data | Original AM1-BCC | New AM1-BCC (ABCG2) | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Data | range | MSE | MUE | b | MSE | MUE | b | |||||||
| Solutes | No. | (kcal/mol) | (kcal/mol) | (kcal/mol) | PI | R | k | (kcal/mol) | (kcal/mol) | (kcal/mol) | PI | R | k | (kcal/mol) |
| Non-cyclic alkanes | 27 | 1.33 | −0.27 | 0.34 | 0.56 | 0.61 | 0.40 | 1.27 | −0.27 | 0.34 | 0.56 | 0.61 | 0.40 | 1.27 |
| Cycloalkanes | 9 | 1.80 | −0.29 | 0.51 | 0.35 | 0.36 | 0.20 | 1.04 | −0.29 | 0.51 | 0.35 | 0.36 | 0.20 | 1.04 |
| Alkenes | 23 | 1.92 | 0.71 | 0.71 | 0.79 | 0.80 | 0.74 | 1.02 | 0.10 | 0.26 | 0.88 | 0.86 | 1.02 | 0.07 |
| Alkynes | 5 | 0.87 | 0.59 | 0.59 | 0.43 | 0.60 | 0.27 | 0.80 | 0.01 | 0.15 | 0.95 | 0.96 | 0.49 | 0.16 |
| Aromatic hydrocarbons | 38 | 4.68 | −0.80 | 0.81 | 0.96 | 0.98 | 1.36 | −0.21 | −0.18 | 0.32 | 0.97 | 0.98 | 1.20 | 0.15 |
| Aliphatic chain + chloride | 31 | 4.53 | 1.24 | 1.28 | 0.83 | 0.84 | 0.75 | 1.03 | 0.14 | 0.62 | 0.71 | 0.76 | 1.17 | 0.29 |
| Aromatic ring + chloride | 26 | 3.83 | 1.65 | 1.65 | 0.24 | 0.17 | 0.13 | −0.31 | 0.22 | 0.55 | 0.89 | 0.84 | 0.81 | −0.22 |
| Aliphatic chain + bromide | 14 | 3.17 | 0.93 | 1.02 | 0.73 | 0.83 | 0.34 | 0.57 | −0.10 | 0.38 | 0.80 | 0.88 | 0.53 | −0.36 |
| Aromatic ring + bromide | 4 | 0.99 | 0.53 | 0.53 | 0.08 | 0.46 | 0.47 | −0.47 | −0.12 | 0.12 | 1.00 | 0.99 | 1.02 | −0.08 |
| Hydrocarbon + iodide | 10 | 2.76 | −0.07 | 0.34 | 0.91 | 0.91 | 0.53 | −0.39 | −0.02 | 0.31 | 0.84 | 0.90 | 0.64 | −0.27 |
| Eithers | 26 | 5.20 | 0.83 | 0.95 | 0.92 | 0.90 | 0.97 | 0.73 | 0.12 | 0.47 | 0.95 | 0.94 | 1.01 | 0.14 |
| Alkyl alcohols | 27 | 5.66 | 1.67 | 1.67 | 0.88 | 0.95 | 0.75 | 0.51 | 0.01 | 0.27 | 0.86 | 0.95 | 0.90 | −0.45 |
| Alkene + alcohols | 4 | 0.59 | 2.13 | 2.13 | 0.10 | 0.05 | 0.09 | −5.67 | 0.09 | 0.46 | 0.21 | 0.14 | 0.23 | −2.97 |
| Phenols | 17 | 2.90 | 0.93 | 0.93 | 0.73 | 0.84 | 0.83 | −1.25 | −0.16 | 0.36 | 0.86 | 0.92 | 0.91 | 0.39 |
| Ketones | 21 | 4.35 | 0.27 | 0.37 | 0.97 | 0.94 | 0.74 | −0.68 | −0.01 | 0.32 | 0.96 | 0.93 | 0.82 | −0.65 |
| Aldehydes | 16 | 2.20 | 0.02 | 0.44 | 0.90 | 0.81 | 1.16 | 0.53 | −0.20 | 0.34 | 0.96 | 0.83 | 0.85 | −0.68 |
| Esters | 41 | 7.82 | −0.63 | 0.75 | 0.97 | 0.95 | 0.87 | −1.11 | −0.22 | 0.35 | 0.97 | 0.98 | 1.07 | 0.04 |
| Amines | 23 | 6.10 | 1.50 | 1.55 | 0.82 | 0.78 | 0.90 | 1.02 | 0.14 | 0.48 | 0.96 | 0.94 | 1.09 | 0.57 |
| Anilines | 8 | 4.02 | 0.34 | 0.47 | 0.85 | 0.89 | 0.80 | −1.47 | −0.08 | 0.20 | 0.98 | 0.98 | 0.95 | −0.34 |
| Pyrazines and pyridines | 19 | 2.83 | 1.15 | 1.15 | 0.67 | 0.60 | 0.94 | 0.86 | 0.34 | 0.38 | 0.71 | 0.89 | 1.09 | 0.75 |
| Nitriles | 5 | 0.58 | 1.44 | 1.44 | 1.00 | 0.90 | 2.00 | 5.23 | 0.10 | 0.14 | 1.00 | 0.90 | 1.12 | 0.56 |
| Nitro compounds | 18 | 9.13 | 2.03 | 2.04 | 0.94 | 0.94 | 0.90 | 1.44 | −0.11 | 0.37 | 0.98 | 0.98 | 0.94 | −0.47 |
| Nitrooxy compounds | 10 | 6.52 | 2.18 | 2.18 | 0.92 | 0.95 | 0.58 | 0.45 | 0.07 | 0.42 | 0.96 | 0.97 | 0.88 | −0.41 |
| Amides | 8 | 3.19 | 1.57 | 1.57 | 0.98 | 0.98 | 0.90 | 0.63 | 0.25 | 0.29 | 0.95 | 0.95 | 0.93 | −0.46 |
| Thioethers | 7 | 0.62 | 1.18 | 1.18 | 0.42 | 0.60 | 0.62 | 0.61 | −0.12 | 0.23 | 0.34 | 0.26 | 0.37 | −1.06 |
| Thiols | 5 | 1.56 | 0.48 | 0.48 | 0.85 | 0.98 | 0.90 | 0.33 | −0.06 | 0.20 | 0.85 | 0.97 | 0.66 | −0.54 |
| Sum | 442 | 15.11 | 0.65 | 1.03 | 0.91 | 0.92 | 0.86 | 0.27 | −0.03 | 0.37 | 0.98 | 0.98 | 0.98 | −0.06 |
FIG. 2.
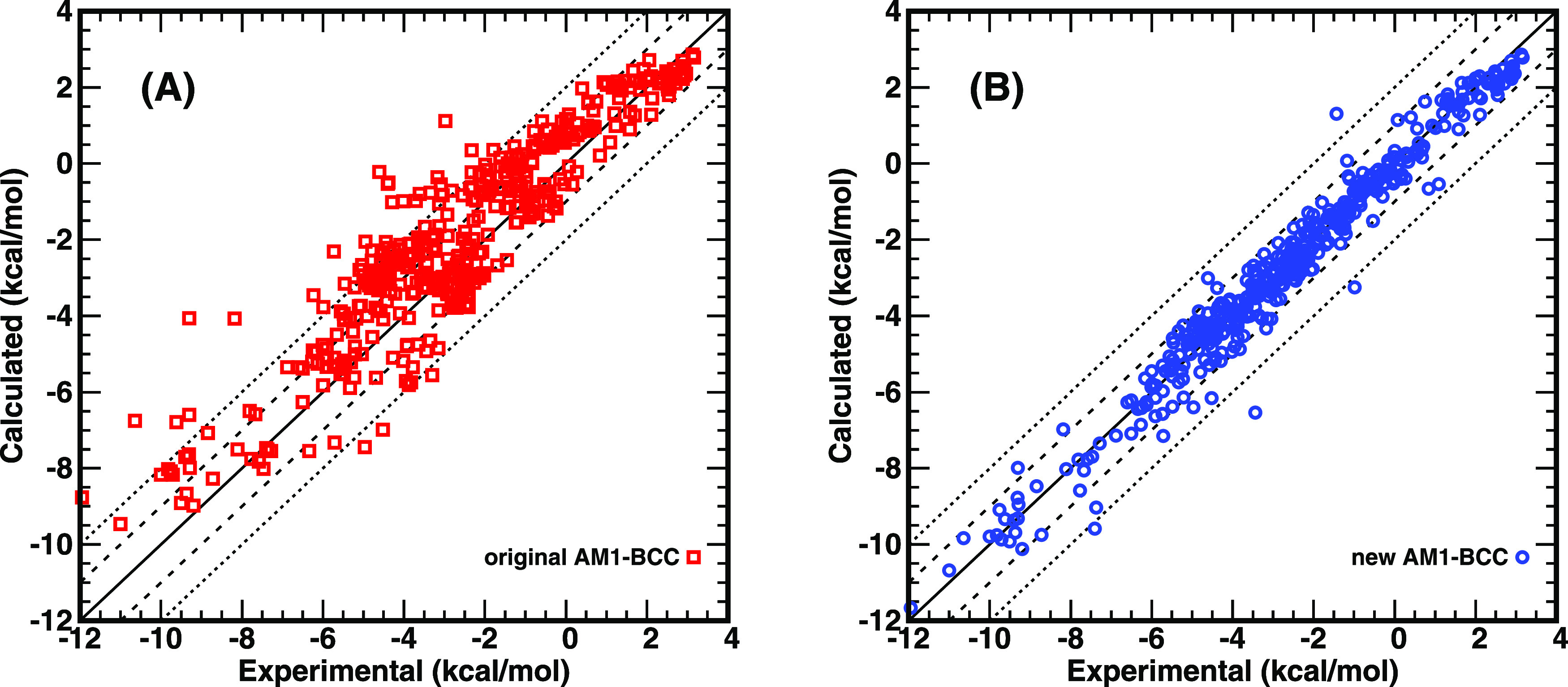
Predicted vs experimental data of hydration free energies of various organic solutes calculated with the GAFF2 parameters combined with (A) original AM1-BCC and (B) new AM1-BCC (ABCG2, an abbreviation of AM1-BCC-GAFF2) proposed in this study.
B. Performance of the two AM1-BCC models on solvation free energies in organic solvents
The updated BCC terms targeting HFEs were applied to the calculations of SFEs in various organic solvents without adjusting any BCC parameters. The calculated results compared to the experimental data of 895 neutral organic solvent–solute pairs are shown in Fig. 3. Encouragingly, the BCC terms optimized in the polar solvent TIP3P water work very well for a variety of polar and nonpolar organic solvents with different dielectric constants. Among the 895 calculated data, 66.9% have unsigned errors (UEs) <0.6 kcal/mol, 21.5% have UEs between 0.6 kcal/mol and 1.0 kcal/mol, and 12.4% of the data have UEs >1.0 kcal/mol. The MUE of all 895 neutral organic solvent–solute pairs is only 0.51 kcal/mol, and the root mean square error (RMSE) is only 0.65 kcal/mol, both close to the experimental uncertainty of SFE. The predictive index PI for all 895 pairs is 0.95, and Pearson’s correlation coefficient is 0.94 (Table II); when fitting the calculated data (y) vs the experimental data (x) of all 895 pairs with function y = kx + b, the slope k is 1.06, and the intercept b is 0.31 kcal/mol (Table II).
FIG. 3.

Calculated results vs the experimental data of solvation free energies of various organic solute compounds in different organic solvents: (a), saturated alkanes and aromatic hydrocarbons as solvents; (b), alcohols as solvents; (c), ethers, ketones, and esters as solvents; and (d), nitrogen-containing solvents.
TABLE II.
The performance of new AM1-BCC charges with GAFF2 parameters on the solvation free energies of neutral organic solvent–neutral organic solute pairs. Expt. stands for experimental; MSE stands for mean signed error; MUE stands for mean unsigned error; RMSE stands for root mean square error; PI stands for predictive index; R stands for Pearson’s correlation coefficient; k and b stand for the slope and intercept values, respectively, when fitting the calculated data (y) vs experimental data (x) with function y = kx + b.
| Data | Expt. data | MSE | MUE | RMSE | b | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Solvents | Solutes | No. | range (kcal/mol) | (kcal/mol) | (kcal/mol) | (kcal/mol) | PI | R | k | (kcal/mol) |
| CH | CH | 54 | 5.41 | −0.26 | 0.46 | 0.57 | 0.93 | 0.93 | 1.09 | 0.16 |
| CH | CHO | 295 | 8.44 | −0.01 | 0.50 | 0.62 | 0.93 | 0.93 | 1.12 | 0.58 |
| CH | CHN | 83 | 5.77 | 0.71 | 0.81 | 0.96 | 0.95 | 0.95 | 1.11 | 1.21 |
| CH | CHON | 20 | 7.12 | 0.31 | 0.41 | 0.58 | 0.98 | 0.97 | 0.97 | 0.15 |
| Alkanes | CHS | 5 | 2.54 | 0.17 | 0.51 | 0.54 | 0.89 | 0.95 | 1.36 | 1.59 |
| Alkanes | CHCl | 14 | 2.95 | −0.13 | 0.26 | 0.29 | 0.99 | 0.97 | 0.91 | −0.56 |
| Alkanes | CHF, CHI, CHBr | 13 | 4.07 | −0.23 | 0.31 | 0.46 | 0.91 | 0.94 | 0.91 | −0.69 |
| Ethers | CH | 16 | 3.04 | −0.21 | 0.35 | 0.42 | 0.89 | 0.93 | 1.12 | 0.45 |
| Ethers | CHO | 39 | 11.03 | −0.23 | 0.46 | 0.58 | 0.98 | 0.97 | 0.98 | −0.38 |
| Ethers | CHN | 23 | 4.35 | 0.03 | 0.44 | 0.53 | 0.96 | 0.94 | 1.11 | 0.51 |
| Ethers | CHON | 10 | 6.93 | −0.45 | 0.61 | 0.73 | 1.00 | 0.96 | 0.90 | −1.00 |
| Alcohols | CH | 54 | 7.48 | −0.16 | 0.43 | 0.54 | 0.93 | 0.95 | 1.07 | 0.08 |
| Alcohols | CHO | 96 | 10.30 | −0.08 | 0.59 | 0.73 | 0.97 | 0.95 | 1.03 | 0.10 |
| Alcohols | CHN | 55 | 5.28 | 0.45 | 0.59 | 0.75 | 0.91 | 0.89 | 1.04 | 0.68 |
| Alcohols | CHON | 16 | 8.26 | −0.10 | 0.22 | 0.31 | 1.00 | 0.99 | 0.96 | −0.34 |
| Ketones | CH | 5 | 3.21 | −0.26 | 0.46 | 0.50 | 0.67 | 0.94 | 1.06 | 0.03 |
| Ketones | CHO | 9 | 5.26 | −0.59 | 0.60 | 0.69 | 0.98 | 0.98 | 1.00 | −0.61 |
| Ketones | CHN, CHON | 6 | 4.68 | −0.11 | 0.32 | 0.37 | 1.00 | 0.99 | 0.79 | −1.11 |
| Esters | CH | 3 | 2.87 | −0.23 | 0.34 | 0.38 | 0.89 | 0.98 | 1.03 | −0.08 |
| Esters | CHO | 26 | 6.24 | −0.42 | 0.45 | 0.53 | 0.98 | 0.98 | 0.99 | −0.48 |
| Esters | CHN, CHON | 6 | 2.75 | 0.12 | 0.23 | 0.30 | 0.98 | 0.97 | 0.93 | −0.32 |
| CHN | CH | 8 | 2.14 | −0.12 | 0.43 | 0.50 | 0.65 | 0.66 | 0.67 | −1.71 |
| CHN | CHO | 22 | 5.23 | −0.26 | 0.55 | 0.63 | 0.97 | 0.92 | 0.85 | −1.07 |
| CHN | CHN, CHON | 9 | 3.98 | −0.21 | 0.59 | 0.66 | 0.85 | 0.87 | 0.58 | −2.55 |
| CHON | CHON | 8 | 3.14 | −0.31 | 0.32 | 0.44 | 0.99 | 0.99 | 1.20 | 0.94 |
| Summary | 895 | 12.87 | 0.01 | 0.51 | 0.65 | 0.95 | 0.94 | 1.06 | 0.31 |
IV. DISCUSSION
Although the first generation of GAFF was developed based on the RESP charge model rather than the AM1-BCC charge model, and the SFE was not utilized in the parameter calibration, the studies had shown that in terms of the SFE calculation accuracy, the combination of GAFF and AM1-BCC was competitive in comparison with other FFs. For instance, an average unsigned error (AVE) of 1.03 kcal/mol to the reference experimental values was obtained for the nitrogen-containing polar functional groups with GAFF/AM1-BCC,46,52 while the OPLS2005 parameters and charges led to errors greater than 1.3 kcal/mol, as revealed in the studies by Shivakumar et al.43,46 In SFE calculation related to function groups like branched alkanes, cycloalkanes, alkynes, some polar groups, and most of the halogenated molecules, Shivakumar et al. found that GAFF/AM1-BCC was superior to most force fields and on par with OPLS2005.43,44 To OPLS2005 based SFE, amides were the biggest outlier with an AUE of 2.4 kcal/mol, while GAFF led to an AUE of 1.6 kcal/mol. Notably, based on the above, Shivakumar et al. specifically incorporated the SFEs of 153 molecules as the training set to adjust the CM1A-BCC charge model for their new OPLS2.0 force field.44
As aforementioned, GAFF2/RESP has achieved better performance over GAFF/RESP on various physicochemical properties including SFE; the details on GAFF2/RESP will be described in another manuscript. The current study was motivated by our observation, as shown in the RESULTS section, that when GAFF2 was directly combined with the original AM1-BCC charge model, it could lead to a systematic error greater than 1.0 kcal/mol for the HFE calculation related to a range of functional groups, especially certain polar groups. Such a systematic error could be the major obstacle limiting the accuracy of calculating important properties, such as solubility, transfer/partition free energy, membrane permeability, and most importantly protein–ligand binding free energy. Based on the hypothesis that further BCC term optimization may provide a feasible way to greatly improve the performance of GAFF2/AM1-BCC charge model, we employed the HFE as the guiding target, conducted BCC reparameterization, and successfully obtained a new set of BCC parameters. As shown, this new ABCG2 charge model greatly reduced the MUE of HFE prediction for a set of 442 neutral molecules from 1.03 kcal/mol to 0.37 kcal/mol, which is even less than the default uncertainty of individual experimental measurement, 0.6 kcal/mol, in the FreeSolv database v0.52. Moreover, the new ABCG2 charge model also performed excellently for the SFE calculation in various polar and nonpolar organic solvents, demonstrating a very good transferability among different dielectric environments. The MUE of SFE prediction for 895 neutral organic solvent–solute systems is as small as 0.51 kcal/mol, which is still less than the aforementioned uncertainty of individual experimental measurement, 0.6 kcal/mol. Such a significant improvement is expected to drastically boost the accuracy for the future protein–ligand binding free energy calculation, which is a primary task in CADD. In summary, this new ABCG2 charge model offers not only efficiency and convenience but also the accuracy and transferability to future in silico drug development effort in either high-throughput screening or alchemical free energy prediction.
Certainly, charge model optimization based on SFE alone does not guarantee universal accuracy for all the properties or suitability of molecular dynamics simulation on all the systems because the FF parameters, including atomic charges, VDW, and other parameters are more or less correlated. They should be adjusted together in an iterative manner to get a physically faithful and reliable FF. Here, we propose an immediate feasible application of this new AM1-BCC charge model: using GAFF2/ABCG2 to re-evaluate properties, such as SFE and binding free energy, employing simulation trajectories generated based on GAFF/RESP, GAFF/old-AM1-BCC, GAFF2/RESP, or other combinations of FF and charge model. For the purpose of binding free energy prediction, we have achieved BCC parameter optimization for carboxylic and ammonium ions, which are frequently encountered in both proteins and ligands. Specifically, the MUE of hydration free energy prediction on 15 ammonium ions was reduced from 2.85 kcal/mol to 0.89 kcal/mol, and the HFE MUE of three carboxylic ions was reduced from 5.30 kcal/mol to 0.43 kcal/mol. The BCC parameter optimization work on other organic ions are currently in progress, and the corresponding new BCC parameters (together with the updated BCCs in this study) will be released to the public via the future release of the AMBER program. In addition, we would also like to test this new AM1-BCC charge model with other water models, such as the explicit OPC water model,74 since it has been recommended for the latest AMBER ff19SB protein force field.75 It is worth pointing out that such an improved AM1-BCC charge model with good performance on SFE prediction, as demonstrated in this study, also paves a feasible path to the development of the next generation of GAFF. With the astonishing progress in the computing power, including GPU computing in the recent years, it is time to incorporate the computationally intensive SFE as one of the primary targets in FF parameterization. This is also the future plan for us as a development team of the AMBER family.
V. CONCLUSION
GAFF2 has been released to the public through the AMBER program and the AMBER Tools since 2015, and has been continuingly enriched and improved. Currently, GAFF2 is in the final packing stage and the detailed description about its development is soon to be submitted for publication. In this study, we focused on the GAFF2 performance when combined with the AM1-BCC charge model in terms of the solvation free energy calculation. We found that TI calculations with GAFF2/original AM1-BCC on the HFE of 442 neutral organic solutes in TIP3P water produced a MUE of 1.03 kcal/mol. So, we optimized the BCC parameters to achieve a new set of AM1-BCC parameters, which significantly reduced the MUE of HFEs of the aforementioned 442 neutral organic solutes to only 0.37 kcal/mol. We further found that this new set of AM1-BCC charge model also led to a low MUE of 0.51 kcal/mol for SFEs of 895 pairs of neutral organic solutes in a variety of neutral organic solvents. This result demonstrated that the newly derived AM1-BCC model (ABCG2) has the capability of treating different dielectric environments. Such encouraging dielectric transferability ensures the suitability of the new force field model for quantitatively predicting the important properties in CADD such as solvation, transferring, and binding free energies. Moreover, the excellent results achieved in this study point out a feasible way of developing next generation GAFF, i.e., including the computationally intensive SFE as one of the primary parameterization targets. The new BCC parameters optimized in this study will be released to the public in the near future via a new version of the AMBER program.
SUPPLEMENTARY MATERIAL
See the supplementary material for the protocol of initial equilibration and later λ expanding in the TI calculation (Fig. S1), the detailed scheme of adjusting bond charge correction (BCC) parameters for various chemical functional groups, sequentially (Fig. S2), the overall flow chart of conducting BCC parameterization using hydration free energies and validation using solvation free energies of a large number of solutes in various organic solvents (Fig. S3), the rule and steps of setting initial box for the organic solvent–solute systems (Fig. S4), and the adjusted BCC terms with their original and new values.
DATA AVAILABILITY
The new BCC parameters of the ABCG2 charge model along with the atom type definition file will be released to the public via the future release of the AMBER program.
ACKNOWLEDGMENTS
The authors gratefully acknowledge the funding support from the National Institutes of Health (NIH) and National Science Foundation (NSF) to J.W. (Grant Nos. NIH R01GM079383, NIH P30DA035778, and NSF 1955260), to W.Y. (Grant No. NIH R01GM111886), and to T.-S.L. (Grant No. NIH R01GM107485). The authors also thank the Center for Research Computing (CRC) at the University of Pittsburgh for the computing resources.
Note: This paper is part of the JCP Special Topic on Classical Molecular Dynamics (MD) Simulations: Codes, Algorithms, Force Fields, and Applications.
Contributor Information
Wei Yang, Email: .
Tai-Sung Lee, Email: .
Junmei Wang, Email: .
REFERENCES
- 1.Gilson M. K. and Zhou H.-X., “Calculation of protein-ligand binding affinities,” Annu. Rev. Biophys. Biomol. Struct. 36, 21 (2007). 10.1146/annurev.biophys.36.040306.132550 [DOI] [PubMed] [Google Scholar]
- 2.Deng Y. and Roux B., “Computations of standard binding free energies with molecular dynamics simulations,” J. Phys. Chem. B 113, 2234 (2009). 10.1021/jp807701h [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jorgensen W. L., “The many roles of computation in drug discovery,” Science 303, 1813 (2004). 10.1126/science.1096361 [DOI] [PubMed] [Google Scholar]
- 4.Jorgensen W. L., “Efficient drug lead discovery and optimization,” Acc. Chem. Res. 42, 724 (2009). 10.1021/ar800236t [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yang W., “Advanced sampling for molecular simulation is coming of age,” J. Comput. Chem. 37, 549 (2016). 10.1002/jcc.24299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hansson T., Marelius J., and Åqvist J., “Ligand binding affinity prediction by linear interaction energy methods,” J. Comput. -Aided Mol. Des. 12, 27 (1998). 10.1023/a:1007930623000 [DOI] [PubMed] [Google Scholar]
- 7.Zhou R., Friesner R. A., Ghosh A., Rizzo R. C., Jorgensen W. L., and Levy R. M., “New linear interaction method for binding affinity calculations using a continuum solvent model,” J. Phys. Chem. B 105, 10388 (2001). 10.1021/jp011480z [DOI] [Google Scholar]
- 8.He X., Man V. H., Ji B., Xie X.-Q., and Wang J., “Calculate protein–ligand binding affinities with the extended linear interaction energy method: Application on the Cathepsin S set in the D3R grand challenge 3,” J. Comput. - Aided Mol. Des. 33, 105 (2019). 10.1007/s10822-018-0162-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Massova I. and Kollman P. A., “Combined molecular mechanical and continuum solvent approach (MM-PBSA/GBSA) to predict ligand binding,” Perspect. Drug Discovery Des. 18, 113 (2000). 10.1023/a:1008763014207 [DOI] [Google Scholar]
- 10.Genheden S. and Ryde U., “The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities,” Expert Opin. Drug Discovery 10, 449 (2015). 10.1517/17460441.2015.1032936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang C., Greene D. A., Xiao L., Qi R., and Luo R., “Recent developments and applications of the MMPBSA method,” Front. Mol. Biosci. 4, 87 (2018). 10.3389/fmolb.2017.00087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang E., Sun H., Wang J., Wang Z., Liu H., Zhang J. Z. H., and Hou T., “End-point binding free energy calculation with MM/PBSA and MM/GBSA: Strategies and applications in drug design,” Chem. Rev. 119, 9478 (2019). 10.1021/acs.chemrev.9b00055 [DOI] [PubMed] [Google Scholar]
- 13.Chodera J. D., Mobley D. L., Shirts M. R., Dixon R. W., Branson K., and Pande V. S., “Alchemical free energy methods for drug discovery: Progress and challenges,” Curr. Opin. Struct. Biol. 21, 150–160 (2011). 10.1016/j.sbi.2011.01.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang L., Wu Y., Deng Y., Kim B., Pierce L., Krilov G., Lupyan D., Robinson S., Dahlgren M. K., Greenwood J., Romero D. L., Masse C., Knight J. L., Steinbrecher T., Beuming T., Damm W., Harder E., Sherman W., Brewer M., Wester R., Murcko M., Frye L., Farid R., Lin T., Mobley D. L., Jorgensen W. L., Berne B. J., Friesner R. A., and Abel R., “Accurate and reliable prediction of relative ligand binding potency in prospective drug discovery by way of a modern free-energy calculation protocol and force field,” J. Am. Chem. Soc. 137, 2695 (2015). 10.1021/ja512751q [DOI] [PubMed] [Google Scholar]
- 15.Knight J. L. and Brooks C. L., “λ-Dynamics free energy simulation methods,” J. Comput. Chem. 30, 1692 (2009). 10.1002/jcc.21295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zheng L., Chen M., and Yang W., “Random walk in orthogonal space to achieve efficient free-energy simulation of complex systems,” Proc. Natl. Acad. Sci. U. S. A. 105, 20227 (2008). 10.1073/pnas.0810631106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gallicchio E. and Levy R. M., “Advances in all atom sampling methods for modeling protein–ligand binding affinities,” Curr. Opin. Struct. Biol. 21, 161 (2011). 10.1016/j.sbi.2011.01.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hansen N. and van Gunsteren W. F., “Practical aspects of free-energy calculations: A review,” J. Chem. Theory Comput. 10, 2632 (2014). 10.1021/ct500161f [DOI] [PubMed] [Google Scholar]
- 19.MacKerell A. D., Bashford D., Bellott M., Dunbrack R. L., Evanseck J. D., Field M. J., Fischer S., Gao J., Guo H., Ha S., Joseph-McCarthy D., Kuchnir L., Kuczera K., Lau F. T. K., Mattos C., Michnick S., Ngo T., Nguyen D. T., Prodhom B., Reiher W. E., Roux B., Schlenkrich M., Smith J. C., Stote R., Straub J., Watanabe M., Wiórkiewicz-Kuczera J., Yin D., and Karplus M., “All-atom empirical potential for molecular modeling and dynamics studies of proteins,” J. Phys. Chem. B 102(18), 3586 (1998). 10.1021/jp973084f [DOI] [PubMed] [Google Scholar]
- 20.Jorgensen W. L., Maxwell D. S., and Tirado-Rives J., “Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids,” J. Am. Chem. Soc. 118, 11225 (1996). 10.1021/ja9621760 [DOI] [Google Scholar]
- 21.Hornak V., Abel R., Okur A., Strockbine B., Roitberg A., and Simmerling C., “Comparison of multiple amber force fields and development of improved protein backbone parameters,” Proteins: Struct., Funct., Bioinf. 65, 712 (2006). 10.1002/prot.21123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Maier J. A., Martinez C., Kasavajhala K., Wickstrom L., Hauser K. E., and Simmerling C., “ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB,” J. Chem. Theory Comput. 11, 3696 (2015). 10.1021/acs.jctc.5b00255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Oostenbrink C., Villa A., Mark A. E., and van Gunsteren W. F., “A biomolecular force field based on the free enthalpy of hydration and solvation: The GROMOS force-field parameter sets 53A5 and 53A6,” J. Comput. Chem. 25, 1656 (2004). 10.1002/jcc.20090 [DOI] [PubMed] [Google Scholar]
- 24.Wang J., Wolf R. M., Caldwell J. W., Kollman P. A., and Case D. A., “Development and testing of a general amber force field,” J. Comput. Chem. 25, 1157 (2004). 10.1002/jcc.20035 [DOI] [PubMed] [Google Scholar]
- 25.Wang J., Wolf R. M., Caldwell J. W., Kollman P. A., and Case D. A., “Erratum: ‘Development and testing of a general amber force field,’ Journal of Computational Chemistry (2004) 25(9) 1157–1174,” J. Comput. Chem. 26, 114 (2005). 10.1002/jcc.20145 [DOI] [PubMed] [Google Scholar]
- 26.Vanommeslaeghe K., Hatcher E., Acharya C., Kundu S., Zhong S., Shim J., Darian E., Guvench O., Lopes P., Vorobyov I., and A. D. MacKerell, Jr., “CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force field,” J. Comput. Chem. 31, 671 (2010). 10.1002/jcc.21367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yu W., He X., Vanommeslaeghe K., and A. D. MacKerell, Jr., “Extension of the CHARMM general force field to sulfonyl-containing compounds and its utility in biomolecular simulations,” J. Comput. Chem. 33, 2451 (2012). 10.1002/jcc.23067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bauer B. A. and Patel S., “Recent applications and developments of charge equilibration force fields for modeling dynamical charges in classical molecular dynamics simulations,” Theor. Chem. Acc. 131, 1153 (2012). 10.1007/s00214-012-1153-7 [DOI] [Google Scholar]
- 29.Lemkul J. A., Huang J., Roux B., and A. D. MacKerell, Jr., “An empirical polarizable force field based on the classical drude oscillator model: Development history and recent applications,” Chem. Rev. 116, 4983 (2016). 10.1021/acs.chemrev.5b00505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vanommeslaeghe K. and A. D. MacKerell, Jr., “CHARMM additive and polarizable force fields for biophysics and computer-aided drug design,” Biochim. Biophys. Acta 1850, 861 (2015). 10.1016/j.bbagen.2014.08.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Goel H., Yu W., Ustach V. D., Aytenfisu A. H., Sun D., and A. D. MacKerell, Jr., “Impact of electronic polarizability on protein-functional group interactions,” Phys. Chem. Chem. Phys. 22, 6848 (2020). 10.1039/d0cp00088d [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jing Z., Liu C., Cheng S. Y., Qi R., Walker B. D., Piquemal J.-P., and Ren P., “Polarizable force fields for biomolecular simulations: Recent advances and applications,” Annu. Rev. Biophys. 48, 371 (2019). 10.1146/annurev-biophys-070317-033349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang J., Cieplak P., Luo R., and Duan Y., “Development of polarizable Gaussian model for molecular mechanical calculations I: Atomic polarizability parameterization to reproduce ab initio anisotropy,” J. Chem. Theory Comput. 15, 1146 (2019). 10.1021/acs.jctc.8b00603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Slochower D. R., Henriksen N. M., Wang L.-P., Chodera J. D., Mobley D. L., and Gilson M. K., “Binding thermodynamics of Host–Guest systems with SMIRNOFF99Frosst 1.0.5 from the open force field initiative,” J. Chem. Theory Comput. 15, 6225 (2019). 10.1021/acs.jctc.9b00748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mobley D. L., Bannan C. C., Rizzi A., Bayly C. I., Chodera J. D., Lim V. T., Lim N. M., Beauchamp K. A., Slochower D. R., Shirts M. R., Gilson M. K., and Eastman P. K., “Escaping atom types in force fields using direct chemical perception,” J. Chem. Theory Comput. 14, 6076 (2018). 10.1021/acs.jctc.8b00640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bayly C. I., Cieplak P., Cornell W., and Kollman P. A., “A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: The RESP model,” J. Phys. Chem. 97, 10269 (1993). 10.1021/j100142a004 [DOI] [Google Scholar]
- 37.Wang J., Cieplak P., and Kollman P. A., “How well does a restrained electrostatic potential (RESP) model perform in calculating conformational energies of organic and biological molecules?,” J. Comput. Chem. 21, 1049 (2000). [DOI] [Google Scholar]
- 38.Jakalian A., Bush B. L., Jack D. B., and Bayly C. I., “Fast, efficient generation of high-quality atomic charges. AM1-BCC model: I. Method,” J. Comput. Chem. 21, 132 (2000). [DOI] [PubMed] [Google Scholar]
- 39.Jakalian A., Jack D. B., and Bayly C. I., “Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. Parameterization and validation,” J. Comput. Chem. 23, 1623 (2002). 10.1002/jcc.10128 [DOI] [PubMed] [Google Scholar]
- 40.Wang J., Wang W., Kollman P. A., and Case D. A., “Automatic atom type and bond type perception in molecular mechanical calculations,” J. Mol. Graphics Modell. 25, 247 (2006). 10.1016/j.jmgm.2005.12.005 [DOI] [PubMed] [Google Scholar]
- 41.Horta B. A. C., Fuchs P. F. J., van Gunsteren W. F., and Hünenberger P. H., “New interaction parameters for oxygen compounds in the GROMOS force field: Improved pure-liquid and solvation properties for alcohols, ethers, aldehydes, ketones, carboxylic acids, and esters,” J. Chem. Theory Comput. 7, 1016 (2011). 10.1021/ct1006407 [DOI] [PubMed] [Google Scholar]
- 42.Jämbeck J. P. M., Mocci F., Lyubartsev A. P., and Laaksonen A., “Partial atomic charges and their impact on the free energy of solvation,” J. Comput. Chem. 34, 187 (2013). 10.1002/jcc.23117 [DOI] [PubMed] [Google Scholar]
- 43.Shivakumar D., Williams J., Wu Y., Damm W., Shelley J., and Sherman W., “Prediction of absolute solvation free energies using molecular dynamics free energy perturbation and the OPLS force field,” J. Chem. Theory Comput. 6, 1509 (2010). 10.1021/ct900587b [DOI] [PubMed] [Google Scholar]
- 44.Shivakumar D., Harder E., Damm W., Friesner R. A., and Sherman W., “Improving the prediction of absolute solvation free energies using the next generation OPLS force field,” J. Chem. Theory Comput. 8, 2553 (2012). 10.1021/ct300203w [DOI] [PubMed] [Google Scholar]
- 45.Mukherjee G., Patra N., Barua P., and Jayaram B., “A fast empirical GAFF compatible partial atomic charge assignment scheme for modeling interactions of small molecules with biomolecular targets,” J. Comput. Chem. 32, 893 (2011). 10.1002/jcc.21671 [DOI] [PubMed] [Google Scholar]
- 46.Ahmed A. and Sandler S. I., “Hydration free energies of multifunctional nitroaromatic compounds,” J. Chem. Theory Comput. 9, 2774 (2013). 10.1021/ct3011002 [DOI] [PubMed] [Google Scholar]
- 47.Martins S. A., Sousa S. F., Ramos M. J., and Fernandes P. A., “Prediction of solvation free energies with thermodynamic integration using the general amber force field,” J. Chem. Theory Comput. 10, 3570 (2014). 10.1021/ct500346y [DOI] [PubMed] [Google Scholar]
- 48.Lundborg M. and Lindahl E., “Automatic GROMACS topology generation and comparisons of force fields for solvation free energy calculations,” J. Phys. Chem. B 119, 810 (2015). 10.1021/jp505332p [DOI] [PubMed] [Google Scholar]
- 49.Zhang J., Tuguldur B., and van der Spoel D., “Force field benchmark of organic liquids. II. Gibbs energy of solvation,” J. Chem. Inf. Model. 55, 1192 (2015). 10.1021/acs.jcim.5b00106 [DOI] [PubMed] [Google Scholar]
- 50.Mohamed N. A., Bradshaw R. T., and Essex J. W., “Evaluation of solvation free energies for small molecules with the AMOEBA polarizable force field,” J. Comput. Chem. 37, 2749 (2016). 10.1002/jcc.24500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mecklenfeld A. and Raabe G., “Comparison of RESP and IPolQ-mod partial charges for solvation free energy calculations of various solute/solvent pairs,” J. Chem. Theory Comput. 13, 6266 (2017). 10.1021/acs.jctc.7b00692 [DOI] [PubMed] [Google Scholar]
- 52.Mobley D. L., Bayly C. I., Cooper M. D., Shirts M. R., and Dill K. A., “Small molecule hydration free energies in explicit solvent: An extensive test of fixed-charge atomistic simulations,” J. Chem. Theory Comput. 5, 350 (2009). 10.1021/ct800409d [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mobley D. L., Bayly C. I., Cooper M. D., Shirts M. R., and Dill K. A., “Correction to small molecule hydration free energies in explicit solvent: An extensive test of fixed-charge atomistic simulations,” J. Chem. Theory Comput. 11, 1347 (2015). 10.1021/acs.jctc.5b00154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mobley D. L. and Guthrie J. P., “FreeSolv: A database of experimental and calculated hydration free energies, with input files,” J. Comput. - Aided Mol. Des. 28, 711 (2014). 10.1007/s10822-014-9747-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nerenberg P. S., Jo B., So C., Tripathy A., and Head-Gordon T., “Optimizing Solute–Water van der Waals interactions to reproduce solvation free energies,” J. Phys. Chem. B 116, 4524 (2012). 10.1021/jp2118373 [DOI] [PubMed] [Google Scholar]
- 56.Jämbeck J. P. M. and Lyubartsev A. P., “Update to the general amber force field for small solutes with an emphasis on free energies of hydration,” J. Phys. Chem. B 118, 3793 (2014). 10.1021/jp4111234 [DOI] [PubMed] [Google Scholar]
- 57.Mobley D. L., Liu S., Cerutti D. S., Swope W. C., and Rice J. E., “Alchemical prediction of hydration free energies for SAMPL,” J. Comput. - Aided Mol. Des. 26, 551 (2012). 10.1007/s10822-011-9528-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mobley D. L., Dumont É., Chodera J. D., and Dill K. A., “Comparison of charge models for fixed-charge force fields: Small-molecule hydration free energies in explicit solvent,” J. Phys. Chem. B 111, 2242 (2007). 10.1021/jp0667442 [DOI] [PubMed] [Google Scholar]
- 59.Mobley D. L., Dumont É., Chodera J. D., and Dill K. A., “Comparison of charge models for fixed-charge force fields: Small molecule hydration free energies in explicit solvent,” J. Phys. Chem. B 115, 1329 (2011). 10.1021/jp108173f [DOI] [PubMed] [Google Scholar]
- 60.Wallin G., Nervall M., Carlsson J., and Åqvist J., “Charges for large scale binding free energy calculations with the linear interaction energy method,” J. Chem. Theory Comput. 5, 380 (2009). [DOI] [PubMed] [Google Scholar]
- 61.Dodda L. S., Vilseck J. Z., Cutrona K. J., and Jorgensen W. L., “Evaluation of CM5 charges for nonaqueous condensed-phase modeling,” J. Chem. Theory Comput. 11, 4273 (2015). 10.1021/acs.jctc.5b00414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Marenich A. V., Kelly C. P., Thompson J. D., Hawkins G. D., Chambers C. C., Giesen D. J., Winget P., Cramer C. J., and Truhlar D. G., Minnesota Solvation Database–Version 2012, University of Minnesota, Minneapolis, 2012. [Google Scholar]
- 63.Maestro, Schrödinger, LLC, New York, NY, 2017.
- 64.Frisch M. J., Trucks G. W., Schlegel H. B., Scuseria G. E., Robb M. A., Cheeseman J. R., Scalmani G., Barone V., Petersson G. A., Nakatsuji H., Li X., Caricato M., Marenich A. V., Bloino J., Janesko B. G., Gomperts R., Mennucci B., Hratchian H. P., Ortiz J. V., Izmaylov A. F., Sonnenberg J. L., Williams-Young D., Ding F., Lipparini F., Egidi F., Goings J., Peng B., Petrone A., Henderson T., Ranasinghe D., Zakrzewski V. G., Gao J., Rega N., Zheng G., Liang W., Hada M., Ehara M., Toyota K., Fukuda R., Hasegawa J., Ishida M., Nakajima T., Honda Y., Kitao O., Nakai H., Vreven T., Throssell K., J. A. Montgomery, Jr., Peralta J. E., Ogliaro F., Bearpark M. J., Heyd J. J., Brothers E. N., Kudin K. N., Staroverov V. N., Keith T. A., Kobayashi R., Normand J., Raghavachari K., Rendell A. P., Burant J. C., Iyengar S. S., Tomasi J., Cossi M., Millam J. M., Klene M., Adamo C., Cammi R., Ochterski J. W., Martin R. L., Morokuma K., Farkas O., Foresman J. B., and Fox D. J., Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford, CT, 2016. [Google Scholar]
- 65.Jorgensen W. L., Chandrasekhar J., Madura J. D., Impey R. W., and Klein M. L., “Comparison of simple potential functions for simulating liquid water,” J. Chem. Phys. 79, 926 (1983). 10.1063/1.445869 [DOI] [Google Scholar]
- 66.Case D. A., Ben-Shalom I. Y., Brozell S. R., Cerutti D. S., Cheatham T. E. III, Cruzeiro V. W. D., Darden T. A., Duke R. E., Ghoreishi D., Gilson M. K., Gohlke H., Goetz A. W., Greene D., Harris R., Homeyer N., Izadi S., Kovalenko A., Kurtzman T., Lee T. S., LeGrand S., Li P., Lin C., Liu J., Luchko T., Luo R., Mermelstein D. J., Merz K. M., Miao Y., Monard G., Nguyen C., Nguyen H., Omelyan I., Onufriev A., Pan F., Qi R., Roe D. R., Roitberg A., Sagui C., Schott-Verdugo S., Shen J., Simmerling C. L., Smith J., Salomon-Ferrer R., Swails J., Walker R. C., Wang J., Wei H., Wolf R. M., Wu X., Xiao L., York D. M., and Kollman P. A., AMBER 2018, University of California, San Francisco, 2018. [Google Scholar]
- 67.Kaus J. W., Pierce L. T., Walker R. C., and McCammon J. A., “Improving the efficiency of free energy calculations in the amber molecular dynamics package,” J. Chem. Theory Comput. 9, 4131 (2013). 10.1021/ct400340s [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lee T.-S., Hu Y., Sherborne B., Guo Z., and York D. M., “Toward fast and accurate binding affinity prediction with pmemdGTI: An efficient implementation of GPU-accelerated thermodynamic integration,” J. Chem. Theory Comput. 13, 3077 (2017). 10.1021/acs.jctc.7b00102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lee T.-S., Cerutti D. S., Mermelstein D., Lin C., LeGrand S., Giese T. J., Roitberg A., Case D. A., Walker R. C., and York D. M., “GPU-accelerated molecular dynamics and free energy methods in Amber18: Performance enhancements and new features,” J. Chem. Inf. Model. 58, 2043 (2018). 10.1021/acs.jcim.8b00462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Steinbrecher T., Mobley D. L., and Case D. A., “Non-linear scaling schemes for Lennard-Jones interactions in free energy calculations,” J. Chem. Phys. 127, 214108 (2007). 10.1063/1.2799191 [DOI] [PubMed] [Google Scholar]
- 71.Steinbrecher T., Joung I., and Case D. A., “Soft-core potentials in thermodynamic integration: Comparing one and two-step transformations,” J. Comput. Chem. 32, 3253 (2011). 10.1002/jcc.21909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Pearlman D. A. and Charifson P. S., “Are free energy calculations useful in practice? A comparison with rapid scoring functions for the p38 map kinase protein system,” J. Med. Chem. 44, 3417 (2001). 10.1021/jm0100279 [DOI] [PubMed] [Google Scholar]
- 73.Michel J., Verdonk M. L., and Essex J. W., “Protein-Ligand binding affinity predictions by implicit solvent Simulations: A tool for lead optimization?,” J. Med. Chem. 49, 7427 (2006). 10.1021/jm061021s [DOI] [PubMed] [Google Scholar]
- 74.Izadi S., Anandakrishnan R., and Onufriev A. V., “Building water models: A different approach,” J. Phys. Chem. Lett. 5, 3863 (2014). 10.1021/jz501780a [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tian C., Kasavajhala K., Belfon K. A. A., Raguette L., Huang H., Migues A. N., Bickel J., Wang Y., Pincay J., Wu Q., and Simmerling C., “ff19SB: Amino-acid-specific protein backbone parameters trained against quantum mechanics energy surfaces in solution,” J. Chem. Theory Comput. 16, 528 (2020). 10.1021/acs.jctc.9b00591 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
See the supplementary material for the protocol of initial equilibration and later λ expanding in the TI calculation (Fig. S1), the detailed scheme of adjusting bond charge correction (BCC) parameters for various chemical functional groups, sequentially (Fig. S2), the overall flow chart of conducting BCC parameterization using hydration free energies and validation using solvation free energies of a large number of solutes in various organic solvents (Fig. S3), the rule and steps of setting initial box for the organic solvent–solute systems (Fig. S4), and the adjusted BCC terms with their original and new values.
Data Availability Statement
The new BCC parameters of the ABCG2 charge model along with the atom type definition file will be released to the public via the future release of the AMBER program.