

Abstract
Clinical studies have suggested that the renin-angiotensin system (RAS) may be a promising therapeutic target in treating diabetic retinopathy (DR). While AT1 receptor blockade decreased the incidence of DR in the DIRECT trial, it did not reduce the DR progression. Lack of understanding of the molecular mechanism of retinal microvascular damage induced by RAS is a critical barrier to the use of RAS blockade in preventing or treating DR. The purpose of this study is to investigate the interaction between soluble epoxide hydrolase (sEH) and the AT1 receptor in Angiotensin II (Ang II)- and diabetes-induced retinal microvascular damage. We demonstrate that Ang II increases retinal sEH levels, which is blunted by an AT1 blocker; administration of 11,12-epoxyeicosatrienoic acid (EET) exacerbates intravitreal Ang II-induced retinal albumin leakage; while sEH knockout (KO) and blockade reduce Ang II-induced retinal vascular remodeling, sEH KO causes retinal vascular leakage in Ang II-sEH KO mice; and sEH KO potentiates diabetes-induced retinal damage via promoting retinal vascular endothelial growth factor (VEGF) but reducing expression of tight junction proteins (ZO-1 and occludin). Our studies hold the promise of providing a new strategy, the use of combined EETs blockade with AT1 blocker, to prevent or reduce DR.
Keywords: Diabetic retinopathy, Cytochrome P-450, EETs, Angiotensin II, RAS
1. Introduction
Diabetic retinopathy (DR) is a common complication of diabetes that affects 7.7 million working-age adults in the U.S. It is predicted that approximately one-third of the total adult population in the U.S. will have diabetes by the year 2050. Increasing numbers of diabetic patients can lead to a significant increase in the number of patients with DR because nearly all patients with type 1 diabetes and 60 % of those with type 2 diabetes develop DR [1]. Thus, DR is a serious neurovascular complication of diabetic patients and is the leading cause of blindness in people of working age. Because of the pronounced socioeconomic burden resulting from these patients’ lack of productivity, high treatment costs, and diminished quality of life, it underscores the urgent need for an effective new therapy for DR.
There are several treatment strategies for DR, including laser photocoagulation, anti-vascular endothelial growth factor (VEGF), and corticosteroid therapy. Laser photocoagulation is a procedure to use the heat generated from a laser to seal or terminate abnormal and leaking blood vessels in proliferative DR (PDR)[2]. There are two approaches of laser photocoagulation, including focal and scatter photocoagulation. The focal approach is a treatment to identify the leaky retinal blood vessels in PDR patients and use a limited number of laser burns to seal them off. A scatter approach is a treatment to use hundreds of laser burns on the retina of PDR patients to halt the growth of blood vessels. Although laser photocoagulation is an effective strategy to restrict proliferative retinopathy changes in PDR patients, it causes damage to the neural retina [3]. Anti-VEGF therapy is a treatment to inhibit the angiogenesis of PDR patients by blocking the action of VEGF. VEGF is a growth factor that initiates the growth of retinal blood vessels in PDR patients. Despite the fact that anti-VEGF therapy provides substantial visual improvement in PDR patients, this strategy comes with adverse side effects, such as endophthalmitis and intraocular inflammation [3]. Since corticosteroids remain the mainstay of treatment for inflammatory diseases after their first clinical use, intravitreal administration of corticosteroids is a good approach to prevent progression to proliferative DR in diabetic patients [4]. However, some patients are resistant to this therapy and have significant adverse effects, including cataract and elevated intraocular pressure [4]. Thus, currently available treatment options for DR, including laser photocoagulation, anti-VEGF, and corticosteroid therapy, have improved care, but owing to side effects, they are not sufficient in eliminating blindness [5], and there is a crucial need for a new therapy to treat DR.
A robust body of literature has well-established the role of the renin-angiotensin system (RAS) in DR [6–13]. In RAS, prorenin is activated to form renin [6–10], which converts angiotensinogen to Ang I [6,7,14]. Ang I is then hydrolyzed by angiotensin-converting enzyme (ACE) to produce Ang II [6,7]. Ang II is a major component of RAS, and the AT1 receptor is the primary receptor to mediate retinal Ang II signaling and function [6–13]. The retina contains major components of the RAS, including ACE, renin, and Ang II receptors, and Ang II is an angiogenic growth factor in experimental research [8]. Several clinical studies have focused on determining the effects of RAS blockade in the development of DR because activation of RAS contributes to retinal microvascular damage [6–10]. Accordingly, in the EUCLID trial (530 participants), after two years of treatment, Lisinopril, an ACE inhibitor, reduced the progression of DR by 50 % and progress to proliferative DR by 80 % [15]. These positive results led to the implementation of the DIRECT trial, the largest clinical trial in retinopathy [6,16]. Participants were divided into two groups: the DIRECT-Prevent group (n > 1400), which tested whether the AT1 blockade can prevent the onset of DR and the DIRECT-Protect group (n > 1900), which examined whether AT1 blockade can reduce DR progression [16]. While treatment with candesartan, an AT1 blocker, decreased the incidence of DR, AT1 blockade did not reduce the progression of established DR [16]. This may explain why the FDA still does not approve the use of RAS blockade to treat DR [17].
Long-term efforts by several investigators have shed invaluable insight into the role of lipoxygenase-derived [18–21] and cyclooxygenase-derived eicosanoids [1,22,23] in DR. CYP enzymes constitute a major metabolic pathway for arachidonic acid (AA). In the presence of NADPH and oxygen, AA is epoxidated by the CYP enzyme system in four epoxyeicosatrienoic acids (EETs), 5, 6-EET, 8, 9-EET, 11, 12-EET, and 14, 15-EET. EETs, which are synthesized by CYP2C and CYP2J enzymes [24,25], are generated predominantly in the blood micro-vessels, and soluble epoxide hydrolase (sEH) converts EETs to inactive dihydroxyeicosatrienoic acids (DHETs) [25–30]. It is well known that sEH blockade or sEH KO increases EETs levels [30–36]. Studies have established that sEH blockade has protective effects in renal disorders and cardiovascular diseases [37,38], yet, the role of sEH in retinal damage and DR remains unclear. The present study was designed to determine the interaction between sEH and the AT1 receptor in Ang II-induced retinal microvascular damage because Ang II is the central effector molecule of RAS. We then assessed the impact of sEH KO on diabetes-induced retinal microvascular damage. This study provides valuable information regarding the interaction between sEH/EETs and RAS in Ang II-induced and diabetes-induced retinal microvascular damage.
2. Materials and methods
2.1. Animal preparation and experimental design
sEH KO (B6.129X-Ephx2tm1gonz/J) mice and male C57BL/6 J mice were obtained from Jackson Laboratory (Bar Harbor, ME) as described previously [39]. We obtained tail snips from litters at weaning (about three weeks of age). The DNA from tail snips was used to identify murine genotype by PCR. Routine genotyping of sEH (+/+), sEH (+/−), and sEH (−/−) mice was done using the following primers: F1, 5′-CTTGGCAGGGTTTCTAGTCCTTAG-3′; R1, 5′-CACGCTGGCATTTTAACACCAG-3′; F2, 5′-CGCTTCCTCGTGCTTTACGGTATC-3′; and R2, GTCAAGGTCGAACGCGGCTACAC-3′. Primer F1/R1 predicts a 510-base pair amplicon for the sEH (+/+) allele. For the sEH (−/−) allele, primer F2/R2 predicts a 160-base pair product of a neomycin-resistance sequence as described previously [39]. These primers were designed by Lasergene 7 software (DNAstar, Madison, WI). The genotypic analysis was examined using 3% Nusieve GTG agarose gel (Lonza, Rockland, ME). All mice were maintained on a 12:12-h light-dark cycle and were housed five mice to a cage. All animal protocols were approved by the Augusta University Institutional Animal Care and Use Committee. Also, these protocols were in accord with the requirements stated in the National Institute of Health Guide for the Care and Use of Laboratory Animals (NIH Publications No. 8023, revised 1978) and following ARVO Statement for the Use of Animals in Ophthalmic and Vision Research.
To determine the role of sEH blockade in Ang II-induced retinal microvascular damage, we treated 8-week-old male C57BL/6 J mice with Ang II (3 mg/kg/day, s.c.; Alzet) + trans-4-[4-(3-adamantan-1-ylureido)-cyclohexyloxy]-benzoic acid (t-AUCB; Cayman) (40 mg/L in drinking water), Ang II, or vehicle (a hydroxypropyl-β-cyclodextran solution) as described previously [40] for 4 weeks. We administered t-AUCB after mice recovered from Ang II surgery. For the Ang II infusion procedure, mice were anesthetized with 2% isoflurane, and an ALZET osmotic pump (Durect Corp., Cupertino, CA, USA) was subcutaneously implanted into each mouse. t-AUCB is a highly selective sEH inhibitor [39,41], and the Ang II infusion is a well-established model for hypertension [42,43]. To determine the role of sEH KO in Ang II-induced retinal microvascular damage, we divided 8-week-old male sEH (−/−) and sEH (+/+) mice into: sEH (−/−) + Ang II (3 mg/kg/day, s.c.; Alzet), sEH (+/+) + Ang II, and sEH (+/+) + vehicle for four weeks. At four weeks post-Ang II, systolic arterial pressure was measured by a tail-cuff method using CODA® non-invasive blood pressure system (Kent Scientific). To determine the role of the AT1 receptor on retinal, heart, and renal sEH expression in Ang II-infusion model, we treated 8-week-old male C57BL/6 J mice with Ang II (3 mg/kg/day, s.c.; Alzet) + telmisartan (2.5 mg/kg/day; Cayman), Ang II, or vehicle for four weeks. We treated mice with 11, 12-EET (15 μg/kg/day, s.c.; Alzet; Cayman), 1415-EET (15 μg/kg/day, s.c.; Alzet; Cayman), or vehicle. We used this protocol as described previously [31,35]. We used 11,12-EET and 14,15-EET because they are the major products of CYP epoxygenases [31,44–46]. On day 3 after treatment, we intravitreally injected these mice with Ang II (5 μg/1 μL). Intravitreal injection was performed as previously described [47]. Briefly, Ang II (Sigma-Aldrich, Louis, MO) was dissolved in water, and a working solution of 10x was prepared to achieve 1x vitreal concentration by administrating 1 μL of this working solution into the mouse eye, assuming the volume of the mouse vitreous is ~10 μL [48].
To determine the role of sEH KO in diabetes-induced retinal microvascular damage, diabetes was induced in male sEH (+/+) and sEH (−/−) mice by injection of streptozotocin (STZ) as previously described [49]. Weekly blood glucose levels of mice were determined after overnight fasting. The blood glucose levels were determined by a One Touch blood glucose monitoring system, and blood glucose exceeded 300 mg/dL was used as an index of diabetes. We will use males only because females have been shown to have little or no response to STZ-induced diabetes [50–52] and females may have reduced reproducibility in becoming diabetes with STZ induction [50]. After different treatments, we perfused the mice with phosphate-buffered saline (PBS) and collected retinas to measure albumin leakage (permeability) by Western blot [19]. We also determined VEGF levels in the serum and retinal samples isolated from sEH (+/+), sEH (+/+) + STZ, and sEH (−/−) + STZ mice by ELISA (Mouse VEGF ELISA, R & D Systems, SMMV00).
2.2. Measurement of fluorescein angiography
The anesthetized mice were placed on the imaging platform of the Phoenix Micron III retinal imaging microscope, and Genteal gel was applied liberally to keep the eye moist during imaging. Mice were administered 10–20 μL 10 % fluorescein sodium and rapid acquisition of fluorescent images was ensued for 5 min, as previously described [20].
2.3. Western blot analysis
Retinal lysates were subjected to Western blot analysis. Identical amounts of protein samples were separated by NuPAGE 4 %–12 % Bis-Tris gel (Invitrogen, Carlsbad, CA) at 125 V for three h. We have previously described the detailed procedures for transfer, blocking, and washing the samples [39]. The membranes were incubated with antibody against Albumin (Abcam, ab106582), CYP2C (Detroit R & D, P2C23DR), (CYP2J (Santa Cruz, sc-67,275), GADPH (Abcam, ab8245), Occludin (Thermo Fisher, 711,500), sEH (Santa Cruz, Sc-166,961), or ZO-1 (Abcam, ab96587). The membranes were incubated with secondary antibody for Albumin, GADPH, Occludin, sEH, or ZO-1. We developed the immunoblots using an ECL detection kit (GE Healthcare, Little Chalfont, Buckinghamshire, UK).
2.4. Immunofluorescence
We isolated retinal samples from mice given different treatments. We precooled a specimen cup containing 2-methyl butane for 45 min in a styrofoam cooler containing dry ice and ethanol. After embedding retinal samples in a specimen mold containing OCT compound (Miles Scientific, Naperville), we placed the specimen mold in the specimen cup for 2 min. We then kept the samples frozen at −80°C until cutting. The samples were cut on a cryostat at a thickness of 10 μm and thawed onto glass slides. Sections were treated with proteinase K for 10 min and washed twice in PBS followed by blocking with 10 % normal goat serum and then incubated with CYP2C or sEH antibody overnight in a humidified container at 4 °C. The next day the sections were incubated in Texas Red labeled anti-mouse antibody. Sections were also incubated with Isolectin-IB4-Alexa Fluro 488 (a marker of the retinal blood vessels), and images were obtained with confocal microscopy (LSM 510; Carl Zeiss).
2.5. Statistical analysis
All values are expressed as means ± SE. All data were analyzed by GraphPad Instat Software (LaJolla, CA). Differences among three or more groups were analyzed using a one-way ANOVA, followed by Tukey’s post-test. Student’s t-test was used for the comparisons of two groups. For all comparisons, P < 0.05 was considered statistically significant.
3. Results
3.1. Expression of CYP2C and sEH in mouse retinas
Previous studies have established that EETs are CYP2C- and CYP2J-derived eicosanoids [25,26]. However, what major CYP enzymes are responsible for their synthesis in retinas is unclear. We isolated retinal samples from wild type mouse eyes and then determined the expression of CYP2C, CYP2J, and sEH by Western blot analysis in comparison with renal tissues as a positive control. Intriguingly, while Fig. 1A showed the absence of CYP2J expression, it showed the presence of CYP2C enzymes as the major epoxygenases in the retina. Fig. 1A also showed that sEH is expressed in the retina as well. Likewise, using Isolectin-IB4 as a marker of the retinal blood vessels, we found that both CYP2C and sEH are expressed in the retinal blood vessels (Fig. 1B–C).
Fig. 1.
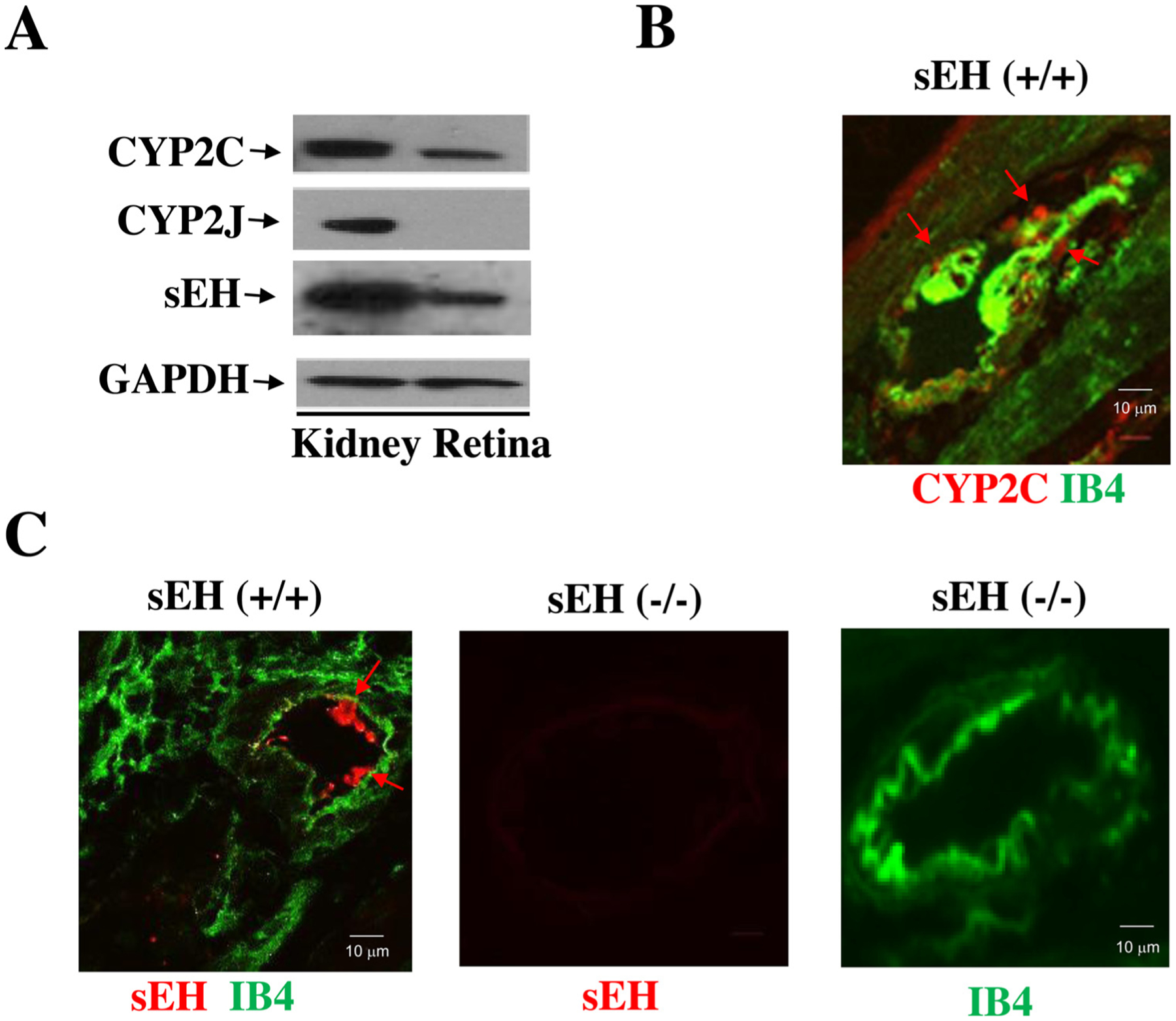
(A) The major CYP epoxygenases in the mouse retina are CYP2C rather than CYP2J isoforms. (B) CYP2C (red; red arrow) and Isolectin B4 (IB4, green; a marker of the retinal blood vessel) expression in the retina of sEH (+/+) mice. (C) sEH (red) and IB4 (green) in the retina of sEH (+/+) mice. Retinal samples from sEH (−/−) mice were used to demonstrate the specificity of the sEH antibody.
3.2. Effects of sEH blocker and AT1 blocker on retinal sEH expression in the Ang II infusion model
We investigated whether sEH blockade affects Ang II-induced retinal microvascular damage. To achieve this, we treated male mice with Ang II + t-AUCB, Ang II, or vehicle for four weeks. We used t-AUCB because t-AUCB is a highly selective sEH inhibitor (Ki = 8 nM to mouse sEH [53]) with excellent bioavailability, and we have used it successfully in our previous studies [39,41].
As shown in Fig. 2A, at 4 weeks post-Ang II, Ang II raised systolic blood pressure (BP), which was attenuated by t-AUCB, suggesting that sEH blockade has an antihypertensive effect. Notably, sEH blockade enhanced Ang II-induced retinal albumin leakage (an index of blood-retinal barrier (BRB) injury) (Fig. 2C). Remarkably, Ang II augmented retinal sEH levels by twofold as compared with control, whereas t-AUCB treatment had no significant effect on retinal sEH expression (Fig. 2B). We next determined whether Ang II induces retinal sEH levels through the AT1 receptor because it is the primary receptor to mediate retinal Ang II signaling and function [46–49]. Strikingly, at 4 weeks post-Ang II, Ang II augmented retinal sEH expression, which was reversed by the AT1 blockade (Fig. 3A). Likewise, telmisartan attenuated Ang II-induced sEH expression in the kidney and heart (Fig. 3A and B). Collectively, these data demonstrate that Ang II augments retinal sEH levels through the AT1 receptor.
Fig. 2.
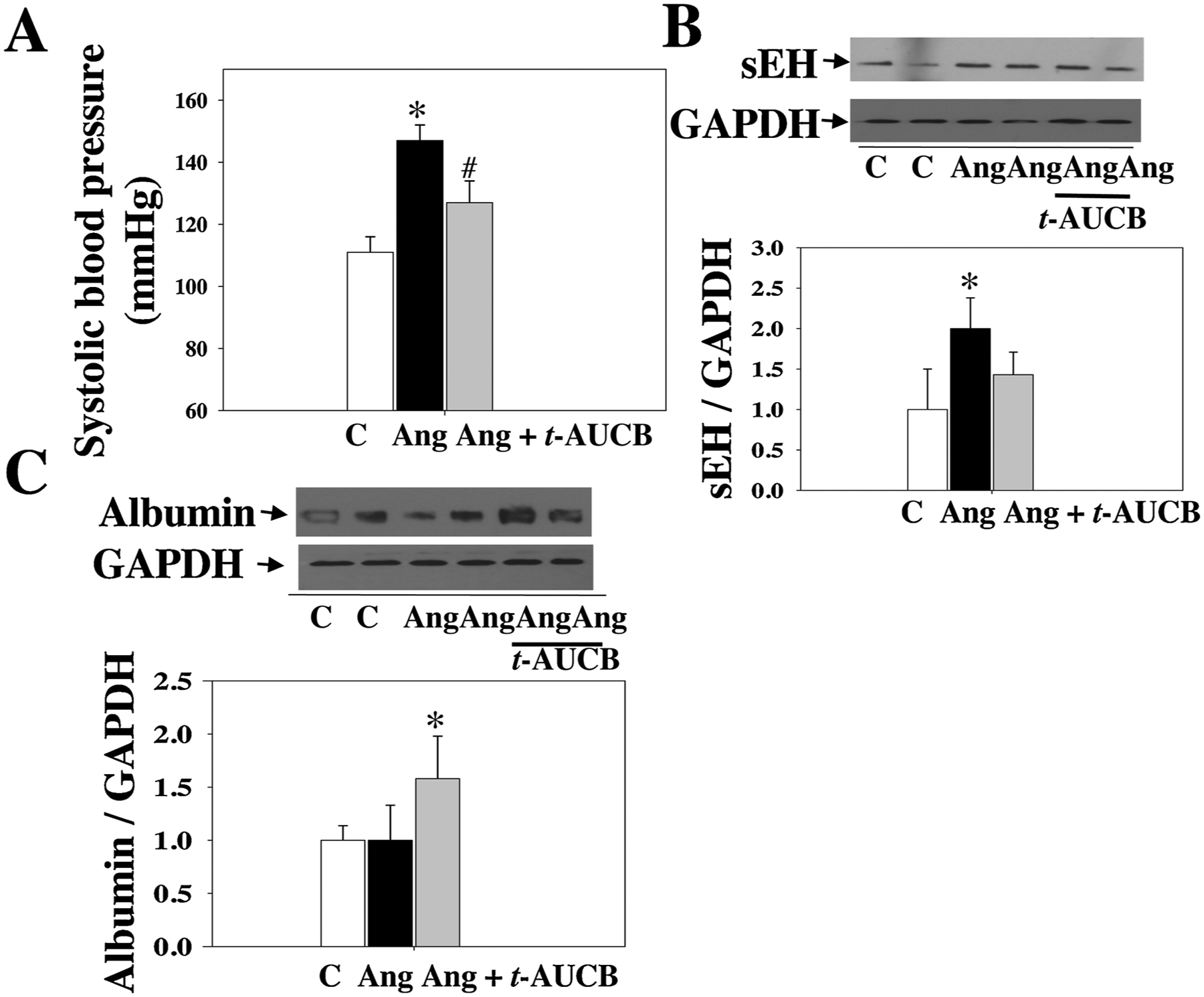
(A) Systolic blood pressure of mice treated with Ang II + t-AUCB, Ang II, and vehicle. We treated these mice given different treatments for four weeks. (B) Ang II up-regulated retinal sEH levels. (C) sEH blockade significantly increased retinal albumin leakage in the Ang II + t-AUCB group. n = 8. *P < 0.05 vs. vehicle. C = vehicle and Ang = Ang II.
Fig. 3.
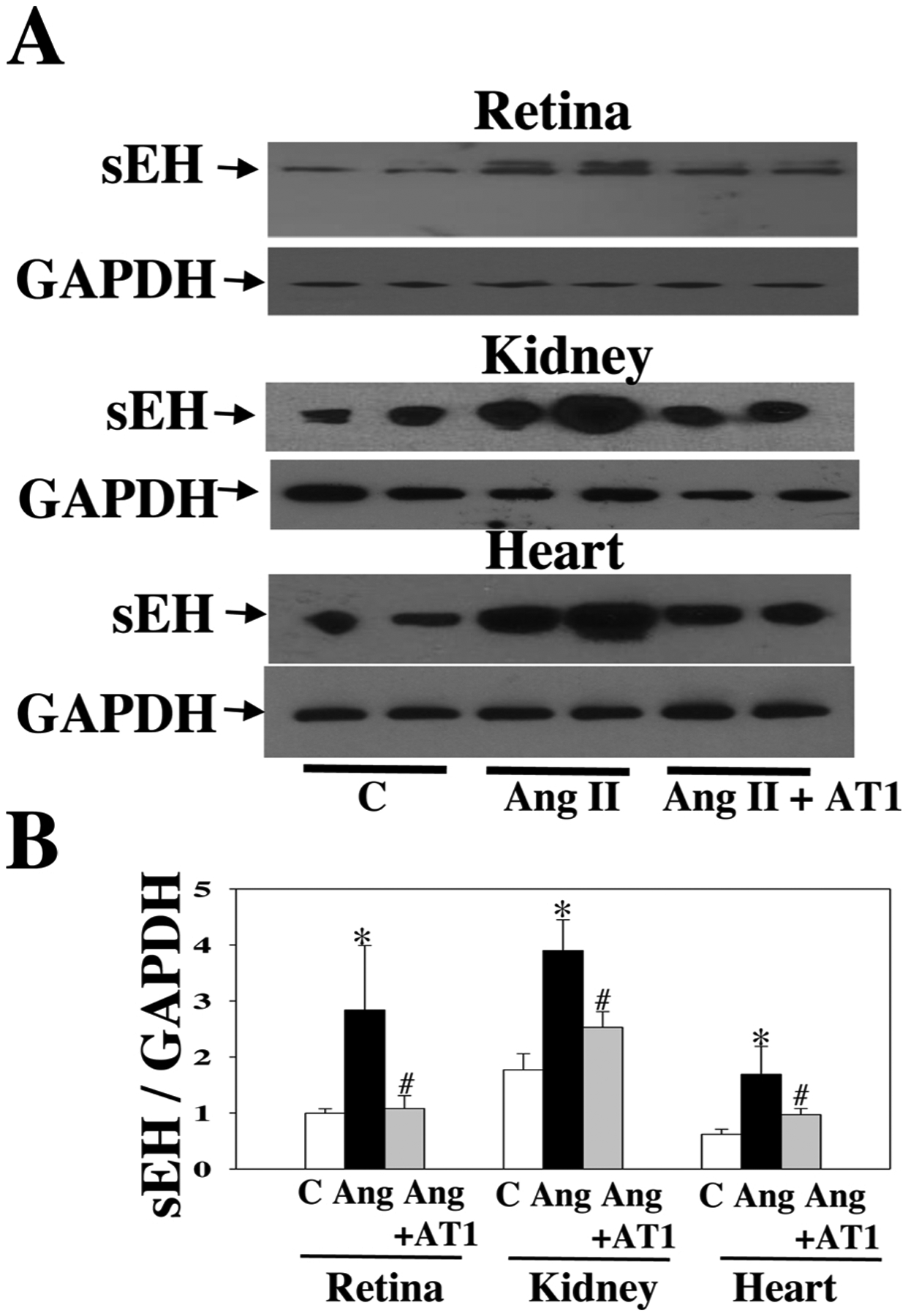
Representative Western blot of sEH in the retina, kidney, and heart of mice treated with Ang II + telmisartan (an AT1 blocker, 2.5 mg/kg/day), Ang II, or a vehicle. We treated mice given different treatments for four weeks (A). AT1 blocker attenuated the up-regulation of sEH expression by Ang II in the retina, kidney, and heart (B). n = 6. *P < 0.05 vs. vehicle; #P < 0.05 vs. Ang. C = vehicle and Ang = Ang II.
3.3. Effects of sEH KO in Ang II-induced retinal microvascular damage
Given that sEH blockade potentiates Ang II-induced retinal damage, we asked whether sEH KO affects Ang II-induced retinal permeability. We used global sEH KO mice, which accumulate EETs [31,35], to evaluate the consequence of elevated EETs on the retinal damage. First, we confirmed the absence of sEH protein in retinas of sEH KO mice (Fig. 4A). Second, we divided sEH (−/−) and sEH (+/+) mice into: sEH (−/−) + Ang II, sEH (+/+) + Ang II, and sEH (+/+) + vehicle. Similar to sEH blockade, at 4 weeks post-Ang II, sEH KO reduced BP of Ang II-treated mice (Fig. 4B). Strikingly, sEH KO resulted in a three-fold increase in retinal albumin content as compared with control mice at 4 weeks post-Ang II (Fig. 4C), suggesting that sEH KO leads to retinal hyperpermeability in Ang II-treated mice.
Fig. 4.
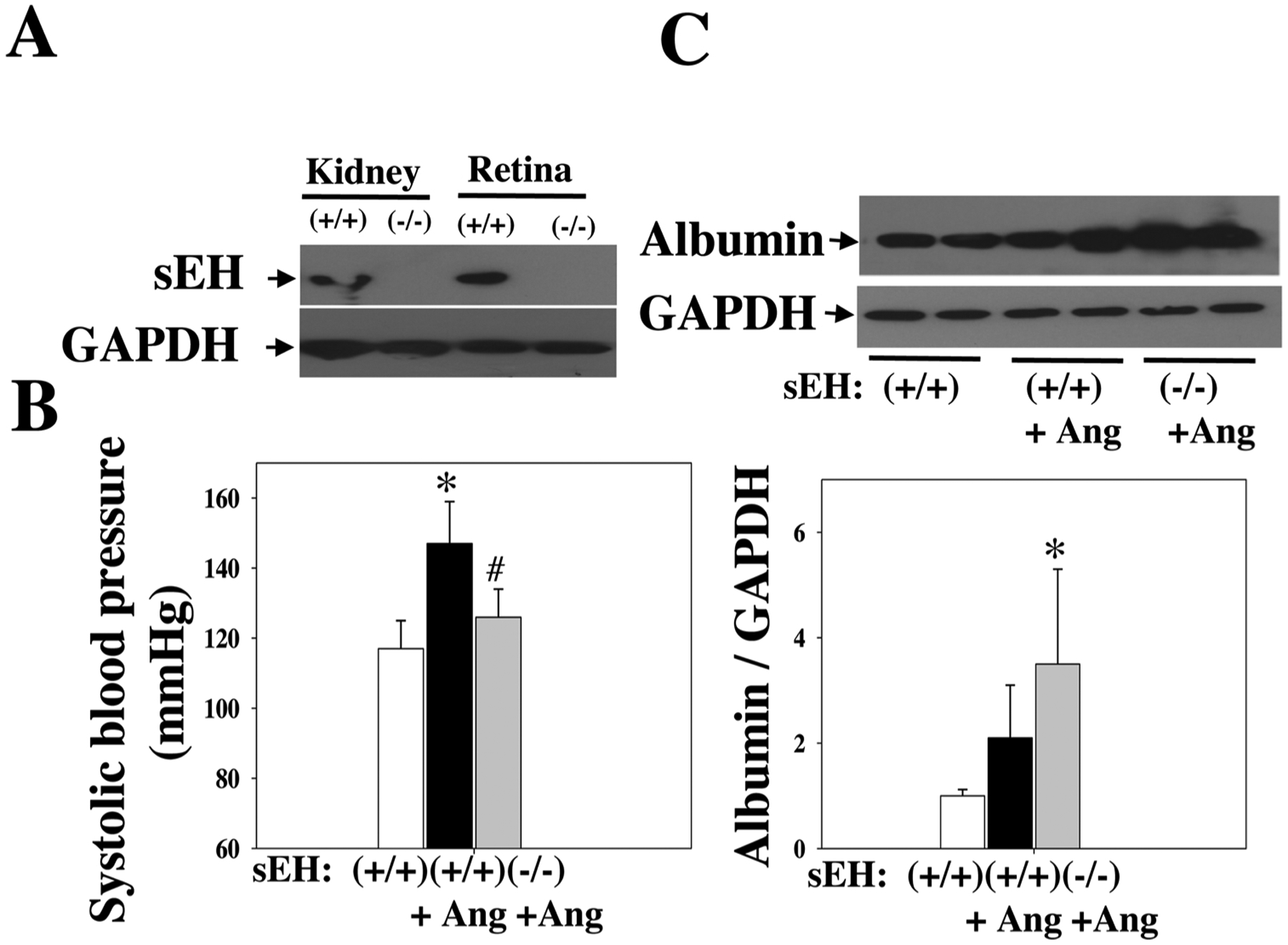
(A) There is an absence of sEH expression in the kidney and retina of sEH (−/−) mice. (B) Systolic blood pressure of sEH (−/−) + Ang II, sEH (+/+) + Ang II, and sEH (+/+) + vehicle mice. We treated these mice given different treatments for four weeks. (C) sEH KO increased retinal albumin leakage in the resulted in sEH (−/−) + Ang II group. n = 8. *P < 0.05 vs. vehicle. #P < 0.05 vs. Ang II. Ang = Ang II.
We tested the hypothesis that sEH blockade and KO attenuate Ang II-induced retinal vascular remodeling. The rationale for this hypothesis is that it is well established that rising EETs levels by sEH blockade stimulates vasodilation [54,55], decreases blood pressure [56,57], and reduces vascular remodeling [58]. The signs of retinal vascular remodeling, including right-angle crossing and increased vascular tortuosity, were found in the Ang II group at 4 weeks post-Ang II. The tortuosity and right-angle crossing were much less in the Ang II-t-AUCB group or Ang II-sEH-KO group. Remarkably, there was an increase in retinal hyperfluorescence in the Ang II-sEH-KO group at 4 weeks post-Ang II, suggesting retinal vascular leakage (Fig. 5D). These results (Fig. 5A–D) indicate that while both sEH blockade and KO attenuate Ang II-induced vascular remodeling, sEH KO causes retinal vascular leakage in the Ang II-sEH-KO group.
Fig. 5.
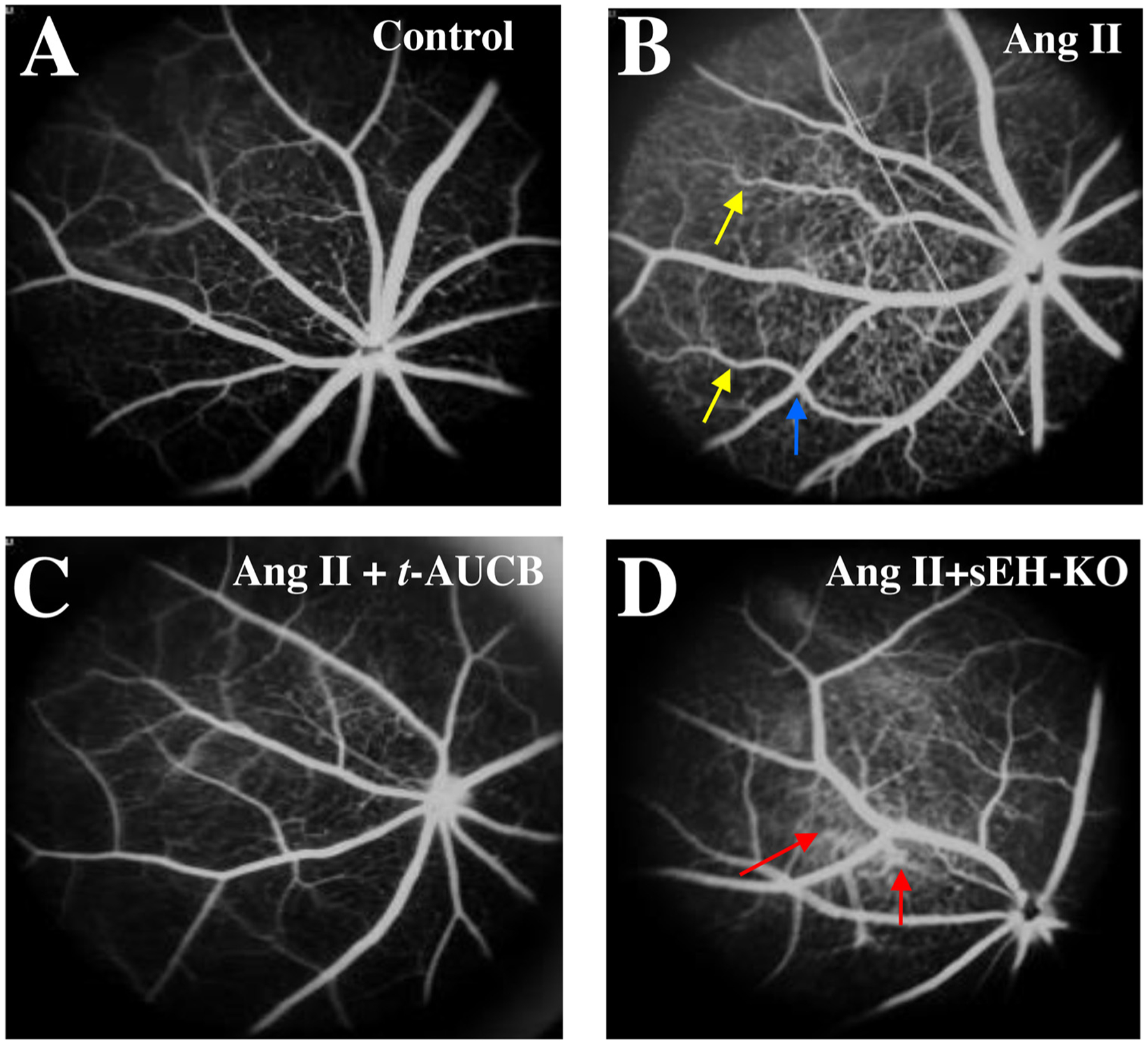
Fluorescein angiograms (FA) of the retina in (A) sEH (+/+) + vehicle, (B) sEH (+/+) + Ang II, (C) sEH (+/+) + Ang II + t-AUCB, and (D) sEH (−/−) + Ang II mice. We treated mice given different treatments for four weeks. There were some signs of hypertensive retinal vascular remodeling, such as right-angle crossing (blue arrow) and increased vascular tortuosity (yellow arrow) in the Ang II group. There was marked hyperfluorescence (red arrow) in the sEH (−/−) + Ang II group, suggesting retinal vascular leakage (D).
3.4. Effects of EETs in ang II-induced retinal microvascular damage
We reasoned that exogenous EETs might affect Ang II-induced retinal damage because sEH blockade or sEH KO increases endogenous EETs [31–36]. We observed significant retinal albumin leakage after intravitreal injection with Ang II (5 μg/1 μL). Next, we treated mice with 11, 12-EET (15 μg/kg/day, s.c.; Alzet; Cayman) + intravitreal Ang II, 1415-EET (15 μg/kg/day, s.c.; Alzet; Cayman) + intravitreal Ang II, or vehicle + intravitreal Ang II. We used this protocol as previously described [31,35]. We used 11,12-EET and 14,15-EET because they are the predominant isomers in the vasculature [31,44–46,59]. Indeed, both 11,12-EET and 14,15-EET promoted Ang II-induced retinal albumin leakage, only 11,12-EET reached significance (Fig. 6). These results establish that 11, 12-EET is more potent than 14,15-EET in potentiating Ang II-induced retinal microvascular damage. In complementary experiments, we determined whether 11,12-EET and 14,15-EET affect retinal albumin leakage in mice without Ang II treatment. We found that neither 11,12-EET nor 14,15-EET affected retinal albumin leakage in mice without Ang II treatment (Fig. 6C).
Fig. 6.
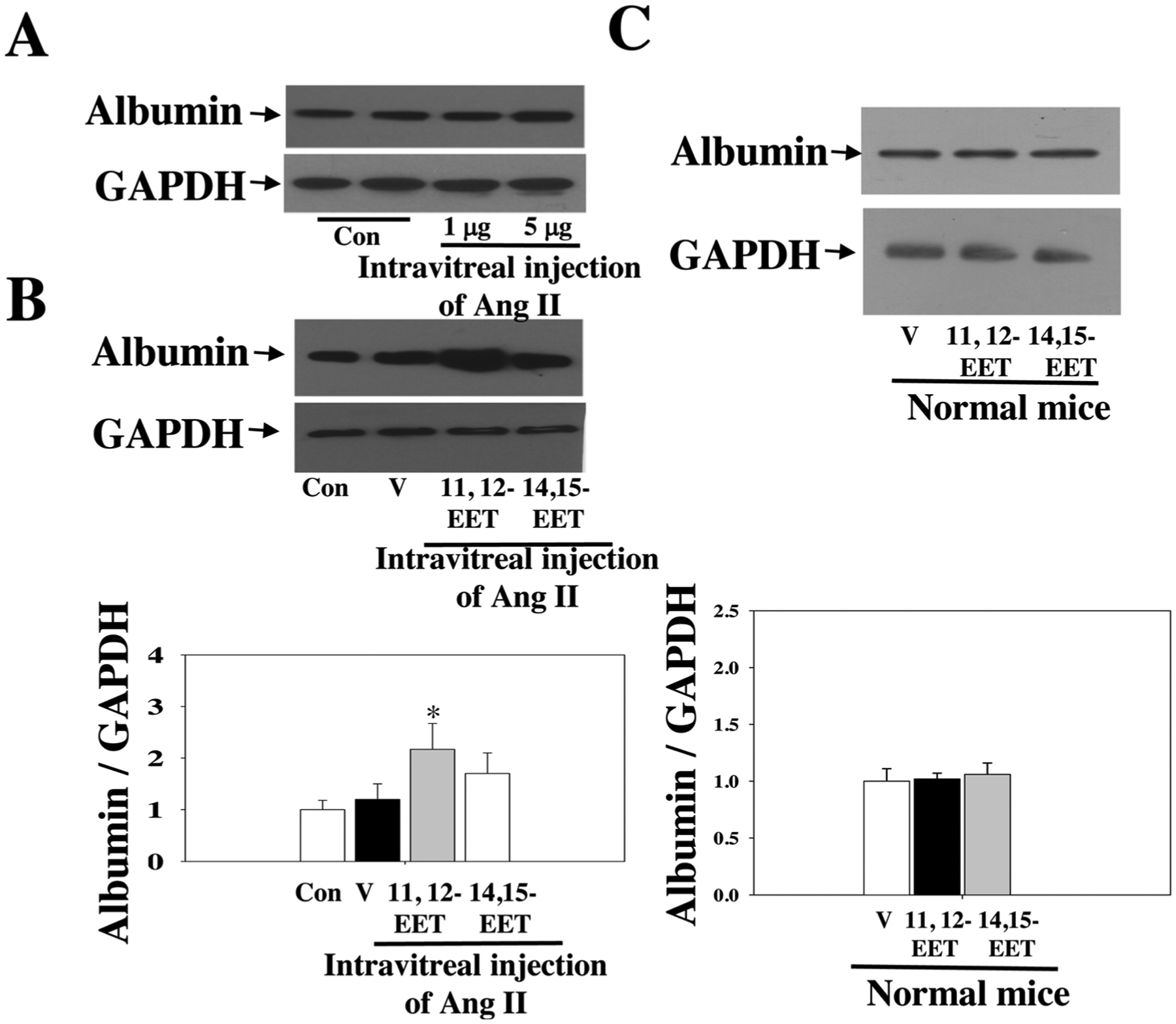
(A) Injection with 5 μg of Ang II caused significant retinal damage. (B) 1112-EET significantly promoted intravitreal Ang II-induced retinal albumin leakage. n = 6. (C) Neither 11,12-EET nor 14,15-EET affected retinal albumin leakage in mice without Ang II treatment. n = 4. * P < 0.05 vs. Ang II. V = vehicle.
3.5. Effects of sEH KO in diabetes-induced retinal microvascular damage
We tested the hypothesis that sEH KO potentiates diabetes-retinal microvascular damage. The rationale for this hypothesis is that sEH KO increases Ang II-induced retinal albumin leakage (Fig. 5). Diabetes was induced in sEH (+/+) and sEH (−/−) mice by injection of STZ as previously described [49]. Weekly blood glucose levels were measured in mice given different treatments. After administration of STZ, sEH (+/+) mice showed significant hyperglycemia during week 1 to week 6, and sEH KO did not affect hyperglycemia in STZ mice (Fig. 7A). At 11 weeks post-STZ, we isolated retinal samples from sEH (+/+) and diabetic sEH (+/+). We found that diabetes caused elevated expression of angiotensinogen and AT1 receptor, which supports the notion [60,61] that RAS is activated in the diabetic retinas. Strikingly, RAS activation is associated with a significant increase of retinal sEH levels (Fig. 7B). Moreover, sEH KO caused a detrimental effect on retinal albumin in diabetic mice (Fig. 8A), which correlated with a reduction of retinal occludin and ZO-1 expression (Fig. 8B). Furthermore, sEH KO augmented retinal and serum VEGF levels (Fig. 8C), which is consistent with previous studies [31,35,44,62] along with caused retinal vascular leakage in diabetic mice (Fig. 9C). Notably, we did not find that significant retinal vascular remodeling in either sEH-KO-STZ or sEH (+/+)-STZ group at 11 weeks post-STZ (Fig. 9C). Taken together, these findings suggest a molecular mechanism that sEH KO potentiates diabetes-induced retinal damage via promoting VEGF but reducing occludin and ZO-1 expression.
Fig. 7.
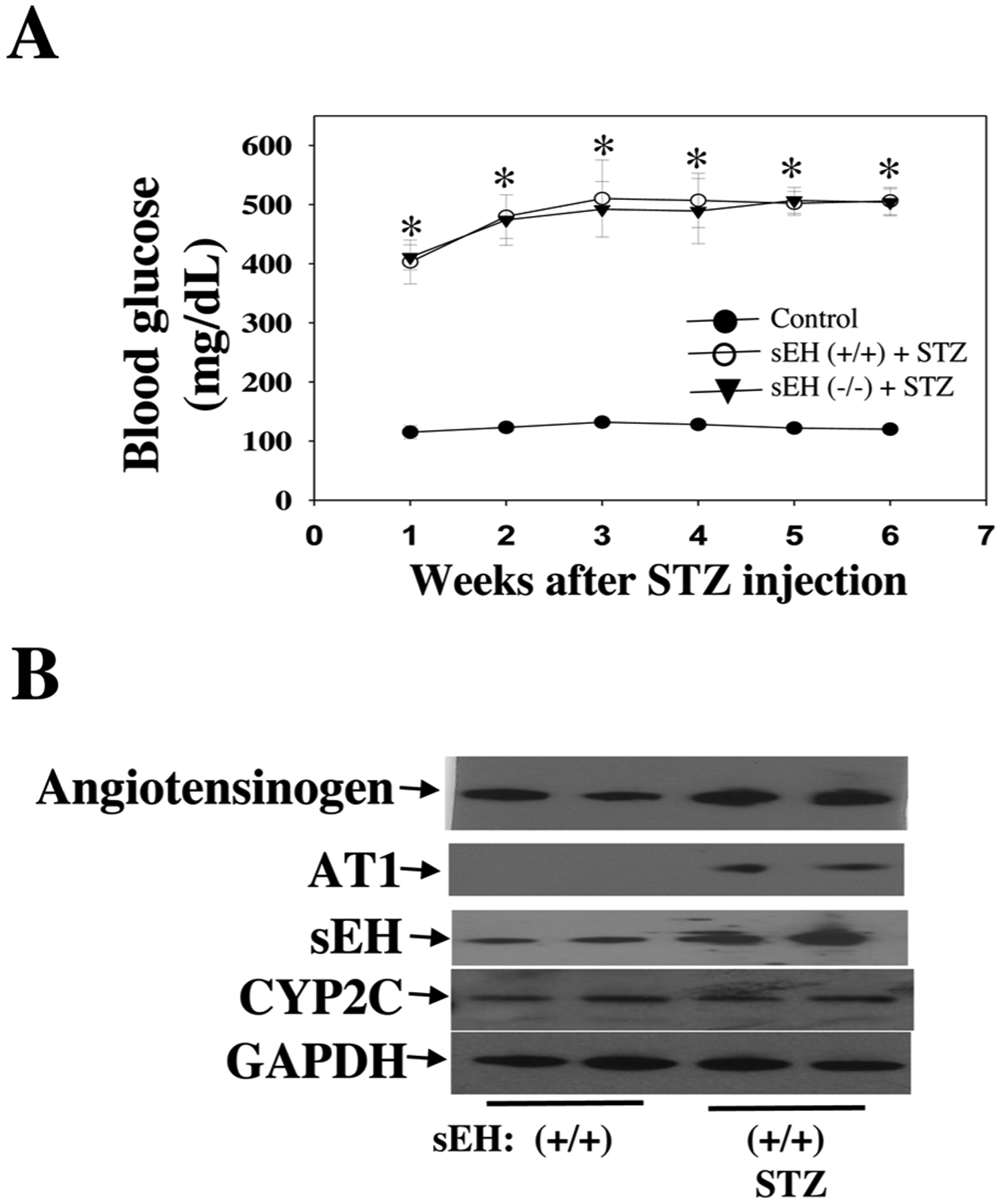
(A) Fasting blood glucose levels were determined at weekly intervals. (B) Up-regulation of angiotensinogen and AT1 (RAS activation) by diabetes at 11 weeks post-STZ is associated with increased retinal sEH expression, whereas diabetes did not affect retinal CYP2C expression. n = 6. *P < 0.05 vs. control.
Fig. 8.
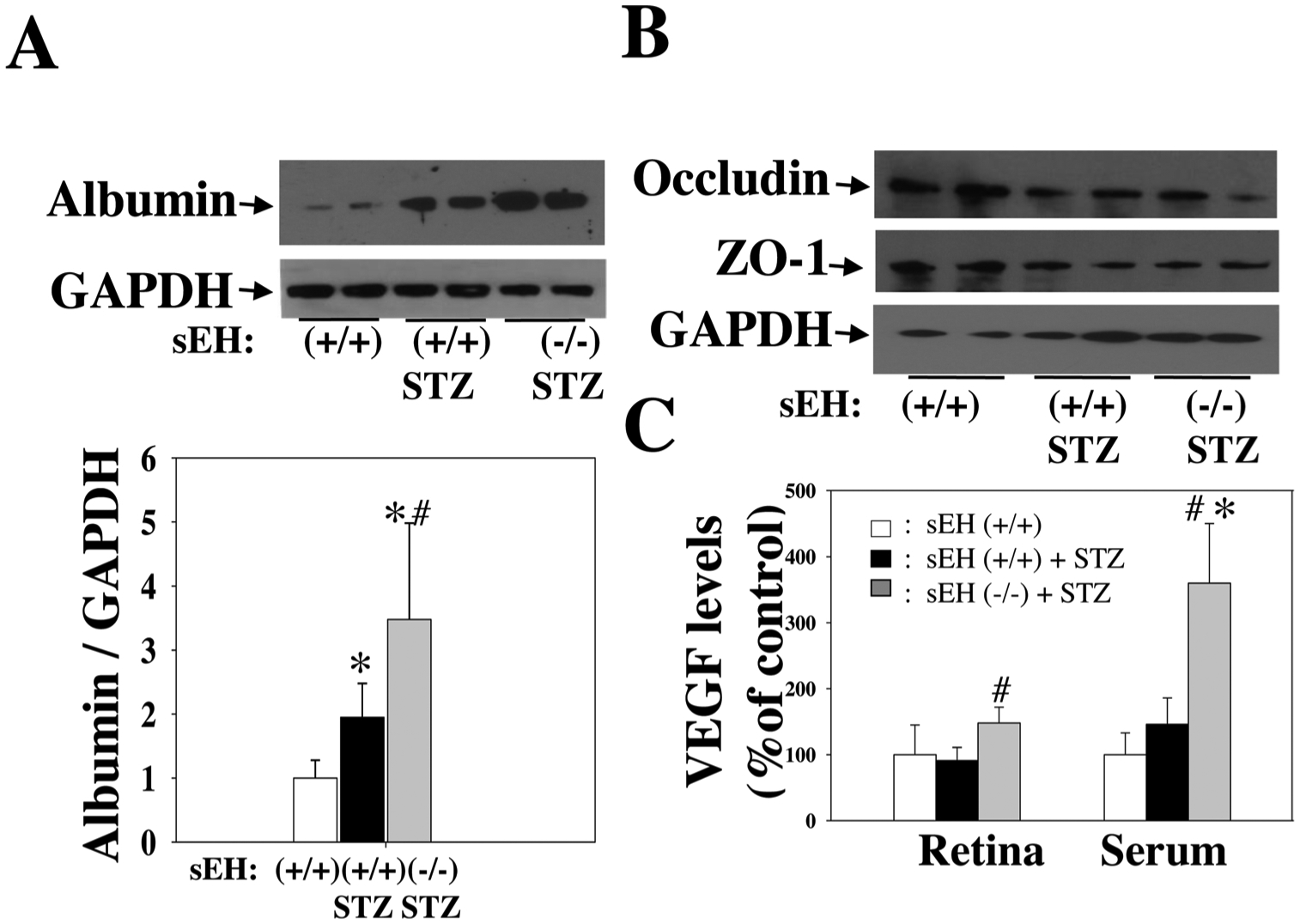
(A) sEH KO resulted in a significant increase of albumin leakage in STZ mice. (B) sEH KO caused reduced expression of occludin and ZO-1 in diabetic mice. (C) sEH KO resulted in a significant increase of retinal and serum VEGF levels in STZ mice. An ELISA kit determined VEGF levels (R& D systems). All experiments were done in diabetic mice at 11 weeks post-STZ. n = 8. *P < 0.05 vs. control; #P < 0.05 vs. sEH (+/+) + STZ.
Fig. 9.

Fluorescein angiograms (FA) of the retina in control, sEH (+/+) + STZ, and sEH (−/−) + STZ mice at 4 weeks post-STZ (A), 7 weeks post-STZ (B), and 11 weeks post-STZ (C). There was marked hyperfluorescence (red arrow) in sEH (−/−) + STZ mice at 11 weeks post-STZ, suggesting retinal vascular leakage.
4. Discussion
DR is a serious neurovascular complication of both type 1 and type 2 diabetic patients, and this disease is the leading cause of blindness in people of working age [63]. It is well established that RAS plays a central role in the pathophysiology of DR [6–13]. Ang II is the central effector molecule of RAS, and the AT1 receptor is the primary receptor to mediate the major pathogenic signaling of Ang II [64–67]. While AT1 receptor blockade (candesartan) decreased the incidence of DR in the DIRECT trial, the largest clinical trial in retinopathy, it did not reduce the progression of DR [16]. Because of these disappointing results [16], the FDA still does not approve the use of RAS blockade to treat DR [17]. Thus, a critical barrier to treat DR or reduce its progression by RAS blockade is the lack of knowledge regarding the molecular mechanism of RAS-induced retinal microvascular damage. In the present study, we focused on the interaction between the AT1 receptor and sEH in Ang II- and diabetes-induced retinal microvascular damage.
EETs biosynthesis can be carried out by several isoforms, including the CYP1A, CYP2B, CYP2C, and CYP2J families [27]. Although many CYP enzymes can epoxidize AA, it is well established that CYP2C and CYP2J are the primary enzymes responsible for EET synthesis [27,41,68]. Mouse CYP2C and CYP2J isoforms are highly expressed in different tissues, including the liver, kidneys, and brain [41]. However, the expression pattern of these CYP epoxygenases in mouse retinas is still not clear. In this study, we used CYP2C and CYP2J antibodies to determine the expression levels of these enzymes in kidneys and retinas. We found that the expression of CYP2J is absent in the retina, and CYP2C proteins are the major epoxygenases in the retina (Fig. 1). Since mouse CYP2C isoforms are highly active epoxygenases [41], these results suggest that mouse CYP2C enzymes, rather than CYP2J enzymes, are responsible for EET synthesis in retina. We also found that CYP2C and sEH are expressed in the retinal blood vessels (Fig. 1), suggesting that CYP2C/sEH are responsible for EETs production/degradation in the retinal blood vessels. These results are consistent with previous findings that a significant amount of EETs/DHETs are generated in the retinas [69,70] and retinal endothelial cells [45]. However, future investigation is needed to establish which mRNA expression of the CYP2C family is the major epoxygenase in the mouse retina.
We first determined the effects of sEH blockade on retinal sEH expression in the Ang II-infusion model because Ang II is the central effector molecule of RAS and Ang II is a well-established mouse model to cause retinal microvascular damage [71,72]. Importantly, we found that chronic Ang II infusion induces retinal sEH levels by twofold, whereas sEH blockade does not affect retinal sEH expression (Fig. 2B). These results are in agreement with the results by Imig et al. [73] that Ang II infusion causes increased expression of sEH in the kidneys. In this previous study [73], the authors hypothesized that increased renal sEH leads to increased renal EET degradation in Ang II hypertension. They also suggested that blockade of sEH could be a target for therapeutic intervention of hypertension because EETs have antihypertensive properties and play an essential role in the maintenance of renal microvascular function. Next, we tested the hypothesis that the up-regulation of retinal sEH by Ang II is mediated by the AT1 receptor because AT1 is the primary receptor to mediate retinal Ang II signaling and function [64–67]. Consistent with the results that Ang II induces heart sEH levels via the AT1 receptor [35], we found that Ang II augments retinal sEH expression, which is reversed by AT1 blockade (Fig. 3). These results provide intriguing evidence that Ang II induces retinal sEH via the AT1 receptor. However, the molecular mechanisms on how Ang II induces retinal sEH levels are still not clear. It could be due to the induction of mRNA levels of sEH in the retina because it has been shown that Ang II promotes sEH levels by the binding of c-Jun to SP-1 sites of the sEH promoter region [34], and this requires further investigation.
We next determined the impact of sEH inhibition and sEH KO on Ang II-induced hypertension and retinal microvascular damage. We found that both sEH blockade (Fig. 2A) and sEH KO (Fig. 4B) attenuated Ang II-induced hypertension in male mice. Importantly, it should be noted that an interesting study [74] has demonstrated that there are sex differences in the control of blood pressure of sEH KO mice, whereby female mice (female-WT and female-sEHKO) had significantly lower blood pressure than their male (male-WT and male-sEHKO) counterparts. Surprisingly, despite the fact that sEH blockade reduces Ang II-induced hypertension (Fig. 2A) and attenuates Ang II-induced retinal vascular remodeling (Fig. 5), we found that its blockade (t-AUCB) enhances Ang II-induced retinal albumin leakage, but its blockade does not cause retinal hyperpermeability in Ang II-treated mice (Fig. 5C). Moreover, similar to the effects of sEH blockade on blood pressure and retinal vascular remodeling, we showed that sEH KO not only promotes Ang II-induced retinal albumin leakage but also leads to retinal hyperpermeability in Ang II-treated mice (Fig. 5D). The reasons that sEH KO rather than t-AUCB causes detrimental effects in retinal microvessels (Fig. 5) are still not clear. It could be due to that while sEH KO deletes the functions of both epoxide hydrolase (the C-terminal domain) and phosphatase (the N-terminal domain) activity, t-AUCB only affects epoxide hydrolase activity [75]. It would be interested to determine the effects of sEH phosphatase blocker [76] on retinal damage in the Ang II model in the future studies. These results suggest the notion that EETs, the upstream product of sEH, have detrimental effects in the retina. Indeed, we showed that exogenous 11, 12-EET potentiates Ang II-induced retinal damage (Fig. 6), and these results support this notion. Thus, our results support the hypothesis that AT1 blockade inhibits retinal sEH leading to the accumulation of EETs, which have detrimental effects in the retina, and rising EETs might attenuate the protective effects of AT1 blockers on retinal microvascular damage. Our hypothesis is based on previous studies [25–30] that sEH converts EETs to DHETs, which is biologically inactive metabolites. Nevertheless, we cannot rule out the possibility that DHETs might affect retinal microvascular function, which requires further investigation in the future studies.
To determine the interaction between RAS and EETs/sEH in diabetes-induced retinal microvascular damage, we addressed the following questions. Does RAS activation increase retinal sEH levels? If yes, what is the impact of sEH KO in diabetes-induced retinal microvascular damage? In addressing these questions, we found a significant elevation of the expression of angiotensinogen and AT1 receptor in diabetic retinas, which is in agreement with the concept that RAS is activated in the diabetic retinas [55,56]. Importantly, activation of RAS is associated with increased expression of retinal sEH levels without affecting retinal CYP2C expression levels (Fig. 7B), which is consistent with a previous report [69] that a time-dependent increased expression of retinal sEH levels in Akita mice, a mouse model of DR. Moreover, in our earlier study [19], we determined the lipidomic profiles (LC/MS/MS) of various polyunsaturated fatty acids (linoleic acid (LA), AA, eicosapentaenoic acid, and docosahexaenoic acid) by 12/15-LO, COX, CYP, and sEH in STZ-induced diabetic and nondiabetic mice. At six months post-STZ, a clustered heat map of circulating bioactive lipid metabolites of these mice was generated. Intriguingly, of the 107 bioactive lipids screened, only a few lipids were significantly increased in diabetic mice. Notably, we found that the levels of 5,6-DHET, 11,12-DHET, and,14,15-DHET are significantly elevated in diabetic mice [19], which supports our results that retinal sEH levels (Fig. 7B) are elevated in STZ-induced diabetic mice. We then determined the impact of sEH KO on diabetes-induced retinal microvascular damage, and we found that sEH KO causes not only a detrimental effect on retinal albumin leakage but also retinal vascular leakage in diabetic mice (Fig. 9) at 11 weeks post-STZ. These results demonstrated that RAS activation induces sEH levels in diabetic retinas, and sEH KO potentiates diabetes-induced retinal microvascular damage.
Although the present study provided new information that sEH KO exacerbates retinal microvascular damage, the exact mechanisms whereby it causes diabetes-induced retinal damage are still not clear. One possibility could be due to the effects of EETs on VEGF levels. Accumulating evidence has demonstrated that EETs promote VEGF levels in vascular endothelial cells. For example, Suzuki et al. [77] showed that the addition of 11,12-EET increases VEGF expression in human umbilical artery endothelial cells (HUAECs) under hypoxia and its effects on VEGF expression in HUAECs are blocked by sulfaphenazole (an inhibitor of CYP2C). Similarly, Cheranov et al. [78] reported that 14,15-EET induces VEGF levels in human dermal microvascular endothelial cells (HDMVECs), and the effects of 14,15-EET on VEGF expression in HDMVECs have been attributed to STAT-3 [78]. Interestingly, we found that sEH KO induces serum and retinal VEGF levels (Fig. 8C), which can contribute to the detrimental effects of diabetes-induced retinal damage. It is well established that VEGF increased retinal vascular permeability via reduction of expression of tight junction proteins, ZO-1 and occludin, and with their redistribution within the retinal vascular endothelium [79,80]. We found that sEH KO causes reduced expression of retinal occludin and ZO-1 in diabetic retinas (Fig. 8B), which supports the notion that sEH KO exacerbates diabetes-induced retinal microvascular damage via promoting VEGF but reducing expression of tight junction proteins.
To the best of our knowledge, the present study is the first to show that Ang II increases retinal sEH through the AT1, and sEH KO exacerbates Ang II- and diabetes-induced retinal damage. This is significant because it has been demonstrated that EETs, lipid mediators produced by the retinal microvasculature, have detrimental effects in the retina. For example, Michaelis et al. [45] showed that retinal endothelial cells express CYP2C protein in culture and generate significant levels of EETs. Also, this previous study [45] showed that CYP2C-derived EETs produced in retinal endothelial cells promote angiogenesis, especially under hypoxic conditions. Similarly, Capozzi et al. [44] showed that CYP-derived 11,12-EET exhibits a proangiogenic biological function in the retina following stimulation by hypoxia, and they [44] suggested that EETs blockade may provide a rational therapy against retinal neovascularization. Studies have also shown [81] that EETs promote retinal neovascularization in oxygen-induced retinopathy (OIR), and CYP2C blockade provides the protective effects on pathological retinal neovascularization in OIR [82]. Thus, the results of the present study support the hypothesis that during diabetes, RAS activation augments retinal levels of sEH, via AT1, which degrades EETs (pro-permeability and pro-angiogenic factors) to compensate for RAS-induced retinal microvascular damage. Although this hypothesis is attractive, further investigation is needed to determine whether this hypothesis can be applied to experimental and clinical studies.
5. Conclusions
We have made the novel findings that Ang II induced retinal sEH levels via the AT1 receptor. To determine the effects of the up-regulation of retinal sEH by Ang II, we investigated whether sEH blockade and sEH KO affect Ang II-induced retinal damage. We found both sEH blockade and sEH KO promote Ang II-induced retinal albumin leakage. Likewise, sEH KO exacerbates diabetes-induced retinal microvascular damage, which supports the notion that EETs, lipid mediators produced by the retinal microvasculature [83], have detrimental effects in the retina. While we propose that EETs, the substrates of sEH, have detrimental effects in the retina, an interesting study by Fleming and colleagues [69] has demonstrated that the accumulation of 19,20-dihydroxydocosapentaenoic acid (19, 20-DHDP) and overexpression of sEH in the retinal Müller glial cells causes retinopathy. While these findings support the importance of sEH and 19, 20-DHDP in the Müller glial cells, Fleming and colleagues [45,84] have also demonstrated that rising EETs in the retinal endothelial cells causes retinopathy; and these previous studies [45,84] support the hypothesis of our current research. Therefore, this study provides a novel strategy that EETs blockade combined with AT1 blocker is an appropriate approach to prevent or reduce the progression of DR.
Acknowledgments
The following grants support this study: AHA Grant-in-Aid grant (AHASE00144) to M.-H Wang; (NIH R01EY023315) to M. Al-Shabrawey; AHA (18CDA34080403) to A. Ibrahim (ASI); and (NIH R01 EY029751-01 - NEI00072) to A. Tawfik.
References
- [1].Zheng L, Howell SJ, Hatala DA, Huang K, Kern TS, Salicylate-based antiinflammatory drugs inhibit the early lesion of diabetic retinopathy, Diabetes 56 (2) (2007) 337–345. [DOI] [PubMed] [Google Scholar]
- [2].Blindbaek SL, Peto T, Grauslund J, How do we evaluate the role of focal/grid photocoagulation in the treatment of diabetic macular edema? Acta Ophthalmol. (Copenh) 97 (4) (2019) 339–346. [DOI] [PubMed] [Google Scholar]
- [3].Phipps JA, Jobling AI, Greferath U, Fletcher EL, Vessey KA, Alternative pathways in the development of diabetic retinopathy: the renin-angiotensin and kallikrein-kinin systems, Clin. Exp. Optom 95 (3) (2012) 282–289. [DOI] [PubMed] [Google Scholar]
- [4].Whitcup SM, Cidlowski JA, Csaky KG, Ambati J, Pharmacology of corticosteroids for diabetic macular edema, Invest. Ophthalmol. Vis. Sci 59 (1) (2018) 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Shah AR, Gardner TW, Diabetic retinopathy: research to clinical practice, Clin. Diabetes Endocrinol 3 (2017) 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Fouda AY, Artham S, El-Remessy AB, Fagan SC, Renin-angiotensin system as a potential therapeutic target in stroke and retinopathy: experimental and clinical evidence, Clin. Sci 130 (4) (2016) 221–238. [DOI] [PubMed] [Google Scholar]
- [7].Bender SB, McGraw AP, Jaffe IZ, Sowers JR, Mineralocorticoid receptor-mediated vascular insulin resistance: an early contributor to diabetes-related vascular disease? Diabetes 62 (2) (2013) 313–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Phipps JA, Clermont AC, Sinha S, Chilcote TJ, Bursell SE, Feener EP, Plasma kallikrein mediates angiotensin II type 1 receptor-stimulated retinal vascular permeability, Hypertension 53 (2) (2009) 175–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Wright AD, Dodson PM, Diabetic retinopathy and blockade of the renin-angiotensin system: new data from the DIRECT study programme, Eye Lond. (Lond) 24(1) (2010) 1–6. [DOI] [PubMed] [Google Scholar]
- [10].Marin Garcia PJ, Marin-Castano ME, Angiotensin II-related hypertension and eye diseases, World J. Cardiol 6 (9) (2014) 968–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Duan Y, Beli E, Li Calzi S, Quigley JL, Miller RC, Moldovan L, Feng D, Salazar TE, Hazra S, Al-Sabah J, Chalam KV, Le Phuong Trinh T, Meroueh M, Markel TA, Murray MC, Vyas RJ, Boulton ME, Parsons-Wingerter P, Oudit GY, Obukhov AG, Grant MB, Loss of angiotensin-converting enzyme 2 exacerbates diabetic retinopathy by promoting bone marrow dysfunction, Stem Cells (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Dominguez JM 2nd., Hu P, Caballero S, Moldovan L, Verma A, Oudit GY, Li Q, Grant MB, Adeno-associated virus overexpression of angiotensin-converting Enzyme-2 reverses diabetic retinopathy in type 1 diabetes in mice, Am. J. Pathol 186 (6) (2016) 1688–1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Verma A, Shan Z, Lei B, Yuan L, Liu X, Nakagawa T, Grant MB, Lewin AS, Hauswirth WW, Raizada MK, Li Q, ACE2 and Ang-(1–7) confer protection against development of diabetic retinopathy, Mol. Ther 20 (1) (2012) 28–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Sun Z, Cade R, Zhang Z, Alouidor J, Van H, Angiotensinogen gene knockout delays and attenuates cold-induced hypertension, Hypertension 41 (2) (2003) 322–327. [DOI] [PubMed] [Google Scholar]
- [15].Chaturvedi N, Sjolie AK, Stephenson JM, Abrahamian H, Keipes M, Castellarin A, Rogulja-Pepeonik Z, Fuller JH, Effect of lisinopril on progression of retinopathy in normotensive people with type 1 diabetes. The EUCLID Study Group. EURODIAB Controlled Trial of Lisinopril in Insulin-Dependent Diabetes Mellitus, Lancet 351 (9095) (1998) 28–31. [DOI] [PubMed] [Google Scholar]
- [16].Chaturvedi N, Porta M, Klein R, Orchard T, Fuller J, Parving HH, Bilous R, Sjolie AK, Effect of candesartan on prevention (DIRECT-Prevent 1) and progression (DIRECT-Protect 1) of retinopathy in type 1 diabetes: randomised, placebo-controlled trials, Lancet 372 (9647) (2008) 1394–1402. [DOI] [PubMed] [Google Scholar]
- [17].Bolinger MT, Antonetti DA, Moving past Anti-VEGF: novel therapies for treating diabetic retinopathy, Int. J. Mol. Sci 17 (9) (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Elmasry K, Ibrahim AS, Saleh H, Elsherbiny N, Elshafey S, Hussein KA, Al-Shabrawey M, Role of endoplasmic reticulum stress in 12/15-lipoxygenase-induced retinal microvascular dysfunction in a mouse model of diabetic retinopathy, Diabetologia 61 (5) (2018) 1220–1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Ibrahim AS, Saleh H, El-Shafey M, Hussein KA, El-Masry K, Baban B, Sheibani N, Wang MH, Tawfik A, Al-Shabrawey M, Targeting of 12/15-Lipoxygenase in retinal endothelial cells, but not in monocytes/macrophages, attenuates high glucose-induced retinal leukostasis, Biochim. Biophys. Acta 1862 (6) (2017) 636–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Ibrahim AS, Elshafey S, Sellak H, Hussein KA, El-Sherbiny M, Abdelsaid M, Rizk N, Beasley S, Tawfik AM, Smith SB, Al-Shabrawey M, A lipidomic screen of hyperglycemia-treated HRECs links 12/15-Lipoxygenase to microvascular dysfunction during diabetic retinopathy via NADPH oxidase, J. Lipid Res 56 (3) (2015) 599–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Gubitosi-Klug RA, Talahalli R, Du Y, Nadler JL, Kern TS, 5-Lipoxygenase, but not 12/15-lipoxygenase, contributes to degeneration of retinal capillaries in a mouse model of diabetic retinopathy, Diabetes 57 (5) (2008) 1387–1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Kern TS, Miller CM, Du Y, Zheng L, Mohr S, Ball SL, Kim M, Jamison JA, Bingaman DP, Topical administration of nepafenac inhibits diabetes-induced retinal microvascular disease and underlying abnormalities of retinal metabolism and physiology, Diabetes 56 (2) (2007) 373–379. [DOI] [PubMed] [Google Scholar]
- [23].Kern TS, Contributions of inflammatory processes to the development of the early stages of diabetic retinopathy, Exp. Diabetes Res (2007) (2007) 95103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Imig JD, Simpkins AN, Renic M, Harder DR, Cytochrome P450 eicosanoids and cerebral vascular function, Expert Rev. Mol. Med 13 (2011) e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Fang X, Hu S, Watanabe T, Weintraub NL, Snyder GD, Yao J, Liu Y, Shyy JY, Hammock BD, Spector AA, Activation of peroxisome proliferator-activated receptor alpha by substituted urea-derived soluble epoxide hydrolase inhibitors, J. Pharmacol. Exp. Ther 314 (1) (2005) 260–270. [DOI] [PubMed] [Google Scholar]
- [26].Imig JD, Epoxyeicosatrienoic acids, 20-hydroxyeicosatetraenoic acid, and renal microvascular function, Prostaglandins Other Lipid Mediat. 104–105 (2013) 2–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Huang H, Al-Shabrawey M, Wang MH, Cyclooxygenase- and cytochrome P450-derived eicosanoids in stroke, Prostaglandins Other Lipid Mediat. 122 (2016) 45–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Iliff JJ, Jia J, Nelson J, Goyagi T, Klaus J, Alkayed NJ, Epoxyeicosanoid signaling in CNS function and disease, Prostaglandins Other Lipid Mediat. 91 (3–4) (2010) 68–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Iliff JJ, Wang R, Zeldin DC, Alkayed NJ, Epoxyeicosanoids as mediators of neurogenic vasodilation in cerebral vessels, Am. J. Physiol. Heart Circ. Physiol 296(5) (2009) H1352–H1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Liu Y, Zhang Y, Schmelzer K, Lee TS, Fang X, Zhu Y, Spector AA, Gill S, Morisseau C, Hammock BD, Shyy JY, The antiinflammatory effect of laminar flow: the role of PPARgamma, epoxyeicosatrienoic acids, and soluble epoxide hydrolase, Proc. Natl. Acad. Sci. U. S. A 102 (46) (2005) 16747–16752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Panigrahy D, Edin ML, Lee CR, Huang S, Bielenberg DR, Butterfield CE, Barnes CM, Mammoto A, Mammoto T, Luria A, Benny O, Chaponis DM, Dudley AC, Greene ER, Vergilio JA, Pietramaggiori G, Scherer-Pietramaggiori SS, Short SM, Seth M, Lih FB, Tomer KB, Yang J, Schwendener RA, Hammock BD, Falck JR, Manthati VL, Ingber DE, Kaipainen A, D’Amore PA, Kieran MW, Zeldin DC, Epoxyeicosanoids stimulate multiorgan metastasis and tumor dormancy escape in mice, J. Clin. Invest 122 (1) (2012) 178–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Sinal CJ, Miyata M, Tohkin M, Nagata K, Bend JR, Gonzalez FJ, Targeted disruption of soluble epoxide hydrolase reveals a role in blood pressure regulation, J. Biol. Chem 275 (51) (2000) 40504–40510. [DOI] [PubMed] [Google Scholar]
- [33].Qin J, Sun D, Jiang H, Kandhi S, Froogh G, Hwang SH, Hammock BD, Wolin MS, Thompson CI, Hintze TH, Huang A, Inhibition of soluble epoxide hydrolase increases coronary perfusion in mice, Physiol. Rep 3 (6) (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Ai D, Pang W, Li N, Xu M, Jones PD, Yang J, Zhang Y, Chiamvimonvat N, Shyy JY, Hammock BD, Zhu Y, Soluble epoxide hydrolase plays an essential role in angiotensin II-induced cardiac hypertrophy, Proc. Natl. Acad. Sci. U. S. A 106 (2) (2009) 564–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Panigrahy D, Kalish BT, Huang S, Bielenberg DR, Le HD, Yang J, Edin ML, Lee CR, Benny O, Mudge DK, Butterfield CE, Mammoto A, Mammoto T, Inceoglu B, Jenkins RL, Simpson MA, Akino T, Lih FB, Tomer KB, Ingber DE, Hammock BD, Falck JR, Manthati VL, Kaipainen A, D’Amore PA, Puder M, Zeldin DC, Kieran MW, Epoxyeicosanoids promote organ and tissue regeneration, Proc. Natl. Acad. Sci. U. S. A 110 (33) (2013) 13528–13533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Xu DY, Davis BB, Wang ZH, Zhao SP, Wasti B, Liu ZL, Li N, Morisseau C, Chiamvimonvat N, Hammock BD, A potent soluble epoxide hydrolase inhibitor, t-AUCB, acts through PPARgamma to modulate the function of endothelial progenitor cells from patients with acute myocardial infarction, Int. J. Cardiol 167 (4) (2013) 1298–1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Imig JD, Epoxide hydrolase and epoxygenase metabolites as therapeutic targets for renal diseases, Am. J. Physiol. Renal Physiol 289 (3) (2005) F496–F503. [DOI] [PubMed] [Google Scholar]
- [38].Imig JD, Eicosanoid regulation of the renal vasculature, Am. J. Physiol. Renal Physiol 279 (6) (2000) F965–F981. [DOI] [PubMed] [Google Scholar]
- [39].Luo HH, Chang Y. Zhou, Zhang S, Hwang SH, Morisseau C, Wang CY, Inscho EW, Hammock BD, Wang MH, Inhibition or deletion of soluble epoxide hydrolase prevents hyperglycemia, promotes insulin secretion, and reduces islet apoptosis, J. Pharmacol. Exp. Ther 334 (2) (2010) 430–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Huang H, Morisseau C, Wang J, Yang T, Falck JR, Hammock BD, Wang MH, Increasing or stabilizing renal epoxyeicosatrienoic acid production attenuates abnormal renal function and hypertension in obese rats, Am. J. Physiol. Renal Physiol 293 (1) (2007) F342–F349. [DOI] [PubMed] [Google Scholar]
- [41].Chen L, Fan C, Zhang Y, Bakri M, Dong H, Morisseau C, Maddipati KR, Luo p., Wang CY, Hammock BD, Wang MH, Beneficial effects of inhibition of soluble epoxide hydrolase on glucose homeostasis and islet damage in a streptozotocin-induced diabetic mouse model, Prostaglandins Other Lipid Mediat. 104–105 (2013) 42–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Ojeda NB, Intapad S, Royals TP, Black JT, Dasinger JH, Tull FL, Alexander BT, Hypersensitivity to acute ANG II in female growth-restricted off-spring is exacerbated by ovariectomy, Am. J. Physiol. Regul. Integr. Comp. Physiol 301 (4) (2011) R1199–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Bakri M, Yi Y, Chen LD, Aisa HA, Wang MH, Alkaloids of Nitraria sibirica Pall. decrease hypertension and albuminuria in angiotensin II-salt hypertension, Chin. J. Nat. Med 12 (4) (2014) 266–272. [DOI] [PubMed] [Google Scholar]
- [44].Capozzi ME, McCollum GW, Penn JS, The role of cytochrome P450 epoxygenases in retinal angiogenesis, Invest. Ophthalmol. Vis. Sci 55 (7) (2014) 4253–4260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Michaelis UR, Xia N, Barbosa-Sicard E, Falck JR, Fleming I, Role of cytochrome P450 2C epoxygenases in hypoxia-induced cell migration and angiogenesis in retinal endothelial cells, Invest. Ophthalmol. Vis. Sci 49 (3) (2008) 1242–1247. [DOI] [PubMed] [Google Scholar]
- [46].Shahabi P, Siest G, Meyer UA, Visvikis-Siest S, Human cytochrome P450 epoxygenases: variability in expression and role in inflammation-related disorders, Pharmacol. Ther 144 (2) (2014) 134–161. [DOI] [PubMed] [Google Scholar]
- [47].Ibrahim AS, Tawfik AM, Hussein KA, Elshafey S, Markand S, Rizk N, Duh EJ, Smith SB, Al-Shabrawey M, Pigment epithelium-derived factor inhibits retinal microvascular dysfunction induced by 12/15-lipoxygenase-derived eicosanoids, Biochim. Biophys. Acta 1851 (3) (2015) 290–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Remtulla S, Hallett PE, A schematic eye for the mouse, and comparisons with the rat, Vision Res. 25 (1) (1985) 21–31. [DOI] [PubMed] [Google Scholar]
- [49].Furman BL, Streptozotocin-induced diabetic models in mice and rats, Curr. Protoc. Pharmacol 70 (47) (2015) 1–20 5. [DOI] [PubMed] [Google Scholar]
- [50].Deeds MC, Anderson JM, Armstrong AS, Gastineau DA, Hiddinga HJ, Jahangir A, Eberhardt NL, Kudva YC, Single dose streptozotocin-induced diabetes: considerations for study design in islet transplantation models, Lab Anim. (NY) 45 (3) (2011) 131–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Hayashi K, Kojima R, Ito M, Strain differences in the diabetogenic activity of streptozotocin in mice, Biol. Pharm. Bull 29 (6) (2006) 1110–1119. [DOI] [PubMed] [Google Scholar]
- [52].Le May C, Chu K, Hu M, Ortega CS, Simpson ER, Korach KS, Tsai MJ, Mauvais-Jarvis F, Estrogens protect pancreatic beta-cells from apoptosis and prevent insulin-deficient diabetes mellitus in mice, Proc Natl Acad Sci U S A 103 (24) (2006) 9232–9237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Liu JY, Tsai HJ, Hwang SH, Jones PD, Morisseau C, Hammock BD, Pharmacokinetic optimization of four soluble epoxide hydrolase inhibitors for use in a murine model of inflammation, Br. J. Pharmacol 156 (2) (2009) 284–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Imig JD, Hammock BD, Soluble epoxide hydrolase as a therapeutic target for cardiovascular diseases, Nat. Rev. Drug Discov 8 (10) (2009) 794–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Imig JD, Navar LG, Roman RJ, Reddy KK, Falck JR, Actions of epoxygenase metabolites on the preglomerular vasculature, J. Am. Soc. Nephrol 7 (11) (1996) 2364–2370. [DOI] [PubMed] [Google Scholar]
- [56].Zhao X, Yamamoto T, Newman JW, Kim IH, Watanabe T, Hammock BD, Stewart J, Pollock JS, Pollock DM, Imig JD, Soluble epoxide hydrolase inhibition protects the kidney from hypertension-induced damage, J. Am. Soc. Nephrol 15 (5) (2004) 1244–1253. [PubMed] [Google Scholar]
- [57].Zhao X, Imig JD, Kidney CYP450 enzymes: biological actions beyond drug metabolism, Curr. Drug Metab 4 (1) (2003) 73–84. [DOI] [PubMed] [Google Scholar]
- [58].Revermann M, Schloss M, Barbosa-Sicard E, Mieth A, Liebner S, Morisseau C, Geisslinger G, Schermuly RT, Fleming I, Hammock BD, Brandes RP, Soluble epoxide hydrolase deficiency attenuates neointima formation in the femoral cuff model of hyperlipidemic mice, Arterioscler. Thromb. Vasc. Biol 30 (5) (2010) 909–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Park SK, Herrnreiter A, Pfister SL, Gauthier KM, Falck BA, Falck JR, Campbell WB, GPR40 is a low-affinity epoxyeicosatrienoic acid receptor in vascular cells, J. Biol. Chem 293 (27) (2018) 10675–10691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Kurihara T, Ozawa Y, Nagai N, Shinoda K, Noda K, Imamura Y, Tsubota K, Okano H, Oike Y, Ishida S, Angiotensin II type 1 receptor signaling contributes to synaptophysin degradation and neuronal dysfunction in the diabetic retina, Diabetes 57 (8) (2008) 2191–2198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Nagai N, Izumi-Nagai K, Oike Y, Koto T, Satofuka S, Ozawa Y, Yamashiro K, Inoue M, Tsubota K, Umezawa K, Ishida S, Suppression of diabetes-induced retinal inflammation by blocking the angiotensin II type 1 receptor or its downstream nuclear factor-kappaB pathway, Invest. Ophthalmol. Vis. Sci 48 (9) (2007) 4342–4350. [DOI] [PubMed] [Google Scholar]
- [62].Sander AL, Jakob H, Sommer K, Sadler C, Fleming I, Marzi I, Frank J, Cytochrome P450-derived epoxyeicosatrienoic acids accelerate wound epithelialization and neovascularization in the hairless mouse ear wound model, Langenbecks Arch. Surg 396 (8) (2011) 1245–1253. [DOI] [PubMed] [Google Scholar]
- [63].Wilkinson-Berka JL, Rana I, Armani R, Agrotis A, Reactive oxygen species, Nox and angiotensin II in angiogenesis: implications for retinopathy, Clin. Sci 124 (10) (2013) 597–615. [DOI] [PubMed] [Google Scholar]
- [64].Sun Z, Wang X, Wood CE, Cade JR, Genetic AT1A receptor deficiency attenuates cold-induced hypertension, Am. J. Physiol. Regul. Integr. Comp. Physiol 288 (2) (2005) R433–9. [DOI] [PubMed] [Google Scholar]
- [65].Pires PW, Ko EA, Pritchard HAT, Rudokas M, Yamasaki E, Earley S, The angiotensin II receptor type 1b is the primary sensor of intraluminal pressure in cerebral artery smooth muscle cells, J. Physiol. (Paris) 595 (14) (2017) 4735–4753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Marques-Lopes J, Lynch MK, Van Kempen TA, Waters EM, Wang G, Iadecola C, Pickel VM, Milner TA, Female protection from slow-pressor effects of angiotensin II involves prevention of ROS production independent of NMDA receptor trafficking in hypothalamic neurons expressing angiotensin 1A receptors, Synapse 69 (3) (2015) 148–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Patel D, Alhawaj R, Kelly MR, Accarino JJ, Lakhkar A, Gupte SA, Sun D, Wolin MS, Potential role of mitochondrial superoxide decreasing ferrochelatase and heme in coronary artery soluble guanylate cyclase depletion by angiotensin II, Am. J. Physiol. Heart Circ. Physiol 310 (11) (2016) H1439–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Huang H, Chang HH, Xu Y, Reddy DS, Du J, Zhou Y, Dong Z, Falck JR, Wang MH, Epoxyeicosatrienoic Acid inhibition alters renal hemodynamics during pregnancy, Exp. Biol. Med. (Maywood) 231 (11) (2006) 1744–1752. [DOI] [PubMed] [Google Scholar]
- [69].Hu J, Dziumbla S, Lin J, Bibli SI, Zukunft S, de Mos J, Awwad K, Fromel T, Jungmann A, Devraj K, Cheng Z, Wang L, Fauser S, Eberhart CG, Sodhi A, Hammock BD, Liebner S, Muller OJ, Glaubitz C, Hammes HP, Popp R, Fleming I, Inhibition of soluble epoxide hydrolase prevents diabetic retinopathy, Nature 552 (7684) (2017) 248–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Shao Z, Fu Z, Stahl A, Joyal JS, Hatton C, Juan A, Hurst C, Evans L, Cui Z, Pei D, Gong Y, Xu D, Tian K, Bogardus H, Edin ML, Lih F, Sapieha P, Chen J, Panigrahy D, Hellstrom A, Zeldin DC, Smith LE, Cytochrome P450 2C8 omega3-long-chain polyunsaturated fatty acid metabolites increase mouse retinal pathologic neovascularization–brief report, Arterioscler. Thromb. Vasc. Biol 34 (3) (2014) 581–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Praddaude F, Cousins SW, Pecher C, Marin-Castano ME, Angiotensin II-induced hypertension regulates AT1 receptor subtypes and extracellular matrix turnover in mouse retinal pigment epithelium, Exp. Eye Res 89 (1) (2009) 109–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Pons M, Cousins SW, Alcazar O, Striker GE, Marin-Castano ME, Angiotensin II-induced MMP-2 activity and MMP-14 and basigin protein expression are mediated via the angiotensin II receptor type 1-mitogen-activated protein kinase 1 pathway in retinal pigment epithelium: implications for age-related macular degeneration, Am. J. Pathol 178 (6) (2011) 2665–2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Imig JD, Zhao X, Capdevila JH, Morisseau C, Hammock BD, Soluble epoxide hydrolase inhibition lowers arterial blood pressure in angiotensin II hypertension, Hypertension 39 (2 Pt 2) (2002) 690–694. [DOI] [PubMed] [Google Scholar]
- [74].Froogh G, Qin J, Kandhi S, Le Y, Jiang H, Luo M, Sun D, Huang A, Female-favorable attenuation of coronary myogenic constriction via reciprocal activations of epoxyeicosatrienoic acids and nitric oxide, Am. J. Physiol. Heart Circ. Physiol 310 (11) (2016) H1448–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Newman JW, Morisseau C, Harris TR, Hammock BD, The soluble epoxide hydrolase encoded by EPXH2 is a bifunctional enzyme with novel lipid phosphate phosphatase activity, Proc. Natl. Acad. Sci. U. S. A 100 (4) (2003) 1558–1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Hou HH, Liao YJ, Hsiao SH, Shyue SK, Lee TS, Role of phosphatase activity of soluble epoxide hydrolase in regulating simvastatin-activated endothelial nitric oxide synthase, Sci. Rep 5 (2015) 13524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Suzuki S, Oguro A, Osada-Oka M, Funae Y, Imaoka S, Epoxyeicosatrienoic acids and/or their metabolites promote hypoxic response of cells, J. Pharmacol. Sci 108(1) (2008) 79–88. [DOI] [PubMed] [Google Scholar]
- [78].Cheranov SY, Karpurapu M, Wang D, Zhang B, Venema RC, Rao GN, An essential role for SRC-activated STAT-3 in 14,15-EET-induced VEGF expression and angiogenesis, Blood 111 (12) (2008) 5581–5591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Ye EA, Liu L, Steinle JJ, miR-15a/16 inhibits TGF-beta3/VEGF signaling and increases retinal endothelial cell barrier proteins, Vision Res. 139 (2017) 23–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Ved N, Hulse RP, Bestall SM, Donaldson LF, Bainbridge JW, Bates DO, Vascular endothelial growth factor-A165b ameliorates outer-retinal barrier and vascular dysfunction in the diabetic retina, Clin. Sci 131 (12) (2017) 1225–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Gong Y, Fu Z, Liegl R, Chen J, Hellstrom A, Smith LE, omega-3 and omega-6 long-chain PUFAs and their enzymatic metabolites in neovascular eye diseases, Am. J. Clin. Nutr 106 (1) (2017) 16–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Gong Y, Fu Z, Edin ML, Liu CH, Wang Z, Shao Z, Fredrick TW, Saba NJ, Morss PC, Burnim SB, Meng SS, Lih FB, Lee KS, Moran EP, SanGiovanni JP, Hellstrom A, Hammock BD, Zeldin DC, Smith LE, Cytochrome P450 Oxidase 2C Inhibition Adds to omega-3 Long-Chain Polyunsaturated Fatty Acids Protection Against Retinal and Choroidal Neovascularization, Arterioscler. Thromb. Vasc. Biol 36 (9) (2016) 1919–1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Michaelis UR, Fisslthaler B, Barbosa-Sicard E, Falck JR, Fleming I, Busse R, Cytochrome P450 epoxygenases 2C8 and 2C9 are implicated in hypoxia-induced endothelial cell migration and angiogenesis, J. Cell. Sci 118 (Pt 23) (2005) 5489–5498. [DOI] [PubMed] [Google Scholar]
- [84].Webler AC, Michaelis UR, Popp R, Barbosa-Sicard E, Murugan A, Falck JR, Fisslthaler B, Fleming I, Epoxyeicosatrienoic acids are part of the VEGF-activated signaling cascade leading to angiogenesis, Am. J. Physiol., Cell Physiol 295 (5) (2008) C1292–301. [DOI] [PMC free article] [PubMed] [Google Scholar]