

Abstract
The neurovascular unit, consisting of neurons, astrocytes, and vascular cells, has become the focus of much discussion in the last two decades and emerging literature now suggests an association between neurovascular dysfunction and neurological disorders. In this review, we synthesize the known and suspected contributions of astrocytes to neurovascular dysfunction in disease. Throughout the brain, astrocytes are centrally positioned to dynamically mediate interactions between neurons and the cerebral vasculature, and play key roles in blood-brain barrier maintenance and neurovascular coupling. It is increasingly apparent that the changes in astrocytes in response to a variety of insults to brain tissue – commonly referred to as “reactive astrogliosis” – are not just an epiphenomenon restricted to morphological alterations, but comprise functional changes in astrocytes that also contribute to the phenotype of neurological diseases with both beneficial and detrimental effects. In the context of the neurovascular unit, astrocyte dysfunction accompanies, and may contribute to, blood-brain barrier impairment and neurovascular dysregulation, highlighting the need to determine the exact nature of the relationship between astrocyte dysfunction and neurovascular impairments. Targeting astrocytes may represent a new strategy in combinatorial therapeutics for preventing the mismatch of energy supply and demand that often accompanies neurological disorders.
Keywords: astrocytes, neurovascular unit, blood-brain barrier, neurovascular coupling, astrogliosis, neurodegeneration
1. Introduction
Astrocytes play a pivotal role in the generation, maturation, and regulation of the neurovascular unit (NVU). Astrocytes are also extremely well-tuned to their microenvironment and respond to any disruptions therein, as occurs in disease and injury, with morphological and functional changes referred to as reactive astrogliosis. The question then arises: does reactive astrogliosis functionally alter the NVU in disease? A summary review of the literature suggests a close relationship between astrogliosis, changes in blood-brain barrier (BBB) permeability, and neurovascular dysfunction in several disorders. Indeed, astrogliosis is often prominent around blood vessels, and numerous disorders of the central nervous system (CNS) that exhibit astrogliosis – e.g. traumatic brain injury (TBI), ischemic stroke, Alzheimer’s disease (AD) and related dementias, subarachnoid hemorrhage (SAH), etc. – are also accompanied by neurovascular dysregulation. Such dysregulation can include impaired autoregulation, loss of resting tone, neurovascular uncoupling and/or BBB dysfunction: all processes that are regulated, to some extent, by astrocytes. From a purely economic viewpoint, when there is a decrease in blood flow to the brain, particularly under pathological stress when cells need even more energy to repair and recover, not all neurons and/or glial cells are likely to survive, or survive undamaged. Further, the extremely limited neurogenic capacity of the adult brain would then ensure that such degeneration will have a lasting impact on brain function. In other words, impairment of neurovascular regulation could cause neurodegeneration by producing a state of malnourishment of the brain (Fig. 1). This is the basic premise of the ‘vascular’ hypothesis of AD (Zlokovic, 2002), but might such dysregulation also contribute to a broader range of neuropathologies? Here, we attempt to summarize evidence gleaned from animal and human studies to put forth the hypothesis that reactive astrogliosis may be the causative factor producing neurovascular dysfunction, which then exacerbates or perhaps, in some instances, even initiates neurodegeneration. This understudied topic has remarkable potential to help us understand the basic biology of neurological disorders and suggest new therapeutic interventions in the treatment and care of such conditions.
Figure 1. Pathological implications of an imbalance in energy supply and demand.
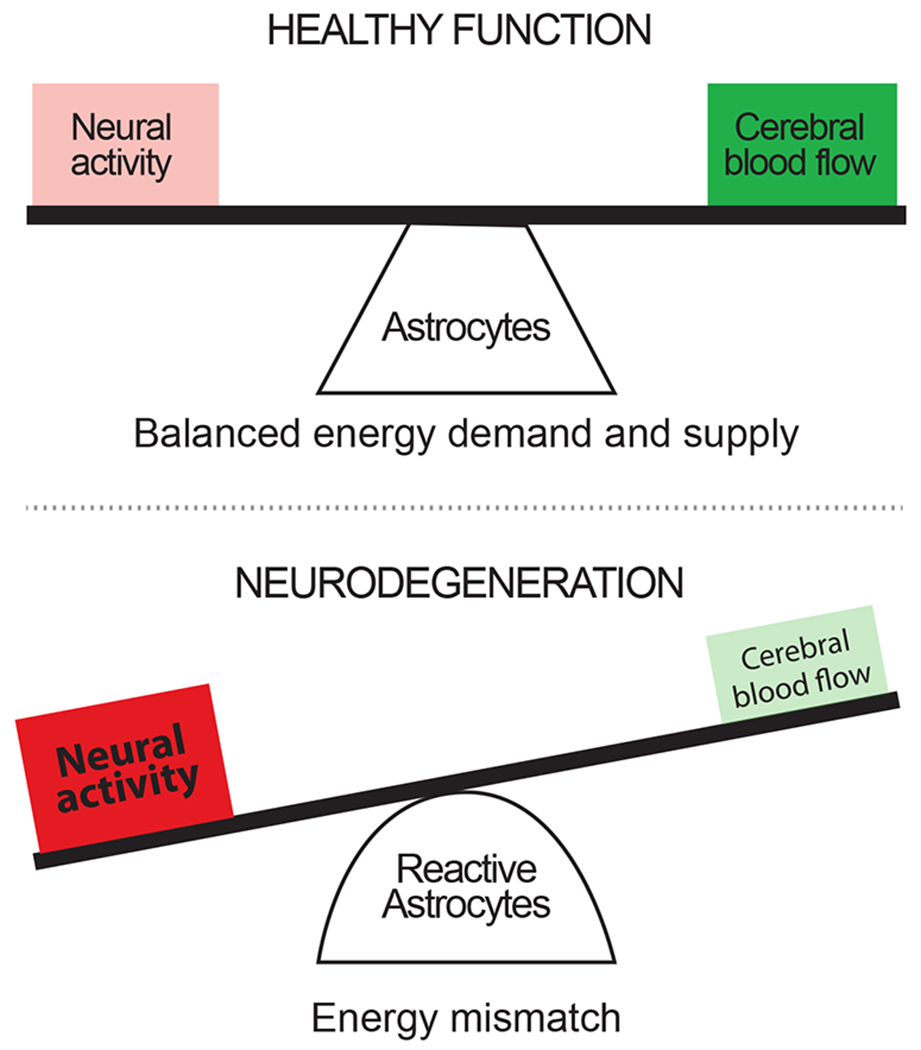
Under healthy physiological conditions, the energy demands of an active brain are supplied by nutrients delivered by the cerebral vasculature. The flow of blood and nutrients within this vasculature is fine-tuned by astrocytes via resting tone regulation and neurovascular coupling pathways. Under pathological conditions, neurons and glial cells still require energy and this requirement may even be increased to allow the cells to cope with, and recover from, the pathological environment. However, during pathology, cerebral blood flow is often reduced and neurovascular coupling is disrupted, which we hypothesize is due to altered signaling from reactive astrocytes. The resulting energy mismatch leads to the gradual development of a hypoxic condition, which is expected to contribute to progressive neuronal and astrocyte degeneration over time.
1.1. Astrocytes
Astrocytes were first identified in the mid-nineteenth century as a population of non-neuronal cells in the CNS that lacked axons and dendrites but bore short, highly ramified processes. They were initially identified as neuroglia by the pathologist Andriezen (Andriezen, 1893), but later named astrocytes – the star cells – by Cajal (Cajal, 1995). Historically, astrocytes were divided into two subtypes: protoplasmic astrocytes, which are found in the gray matter and possess a dense cloud of fine processes that fill the interstitial space in the neuropil, and fibrous astrocytes, which are found in the white matter and possess fewer but thicker, longer processes. This basic nomenclature maintains a stronghold in the glial literature even today, despite accumulating evidence suggesting there are numerous subpopulations with striking heterogeneity, both between and within brain regions (Bayraktar et al., 2015; Farmer and Murai, 2017). Most of what we know about astrocyte functions today are based on investigations of grey matter protoplasmic astrocytes, and our understanding of white matter astrocytes, especially as it pertains to vascular regulation, is relatively limited. White matter astrocytes contact axons at the nodes of Ranvier (Butt et al., 1994), express excitatory neurotransmitter transporters and may function to protect both oligodendroglia and axons from excitotoxicity (Baltan et al., 2011). They also facilitate myelination in the developing and mature brain via secretion of enzymes and other soluble factors, ion buffering, and metabolic substrate delivery (Lundgaard et al., 2014). Therefore, although the hypotheses forwarded in this review are largely based on findings from protoplasmic astrocytes of the grey matter, they are likely also relevant to gliovascular structure and function in white matter.
In the grey matter, the fine processes of astrocytes permeate the neuropil to contact neuronal cell bodies, dendrites, synapses, nodes of Ranvier, ependymal cells, and the pia mater. A single astrocyte can interact with several neurons and >100,000 synapses (Ventura and Harris, 1999; Bushong et al., 2004), forming the morphological basis of the tripartite synapse. At the other end, the endfoot processes of astrocytes almost completely encapsulate the parenchymal microvasculature (Simard et al., 2003; Mathiisen et al., 2010). Astrocytes express several ion channels, transporters and receptors, which contribute to their critical roles in modulating synaptic activity via potassium buffering, pH regulation, neurotransmitter uptake and gliotransmitter release, and general maintenance of neuronal homeostasis (Verkhratsky and Nedergaard, 2018). Mature astrocytes form a highly organized, gap junctionally-coupled network that tiles the brain, with each astrocyte occupying a unique spatial domain (Bushong et al., 2002). This spatially segregated yet intercellularly connected astrocyte syncytium allows astrocytes to discriminate and integrate neuronal signals, earning them recognition as partners in information processing (Araque et al., 2014). This syncytium may also enable astrocytes to supply energy substrates to neurons (Pellerin and Magistretti, 1994) and oligodendrocytes (Niu et al., 2016), although this is debated (DiNuzzo et al., 2010; Diaz-Garcia et al., 2017; Dienel, 2017). Central to the argument presented in this review, astrocytes are also key players in cerebral blood flow (CBF) regulation. Based on their morphological characteristics, the visionary Cajal first hypothesized that astrocytes might control vascular diameter, and much evidence now supports their integral role in maintaining resting vascular tone as well as regulating NVC (Zonta et al., 2003; Mulligan and Macvicar, 2004; Metea and Newman, 2006; Takano et al., 2006; Kim et al., 2015; Rosenegger et al., 2015; Mishra et al., 2016).
1.2. Cerebral Blood Flow
The enhanced cognitive power with which the brain endows mammals and especially humans comes at a high price – both the size (Aiello, 1995) and metabolic rate (Karbowski, 2007) of the mammalian brain have evolved to be larger than that predicted by body size alone. Indeed, increased cerebral metabolic rate is considered one of the main factors driving higher cognitive development. This increase in metabolic activity results in a correspondingly large energy demand (Howarth et al., 2012), which is supplied by the extensive cerebrovascular network originating largely from the internal carotid arteries. Evolutionary changes in skull structure show that the carotid foramen, through which the internal carotid artery passes into the skull, is enlarged in the human skull compared to other hominids and may have been the initial evolutionary step that permitted the human brain to attain its current complexity (Seymour et al., 2016), stressing the importance of cerebral blood supply for higher cognitive function.
CBF is regulated via several mechanisms. Autoregulation ensures that the brain receives a relatively constant supply of blood and nutrients despite changes in systemic blood pressure (Tzeng and Ainslie, 2014; Filosa et al., 2016). Neurogenic signals from the basal forebrain and brain stem nuclei on penetrating arterioles as well as from peripheral trigeminal ganglionic innervation of pial blood vessels also contribute to the tone of cerebral vasculature and, therefore, CBF (Hamel, 2006; Cauli and Hamel, 2010). However, as the brain lacks energy stores and is reliant upon blood glucose-dependent aerobic respiration (Holmes and Holmes, 1926), CBF is also regulated locally in response to increased neuronal activity via NVC, thereby giving rise to functional hyperemia. NVC is regulated by signaling between neurons, astrocytes, and vascular cells (endothelial cells, vascular smooth muscle cells, and pericytes), which collectively make up the NVU (Iadecola, 2017; McConnell et al., 2017) (Fig. 2). The NVU is thus the epicenter of several tightly controlled, dynamic intercellular interactions orchestrated to maintain optimal global and local CBF (Filosa et al., 2016; Mishra, 2016). It is also the structural basis of the BBB, which isolates the brain from most circulating factors, antigens and, under healthy conditions, immune cells (Daneman and Barres, 2005; Iadecola, 2017).
Figure 2. Organization of the neurovascular unit.
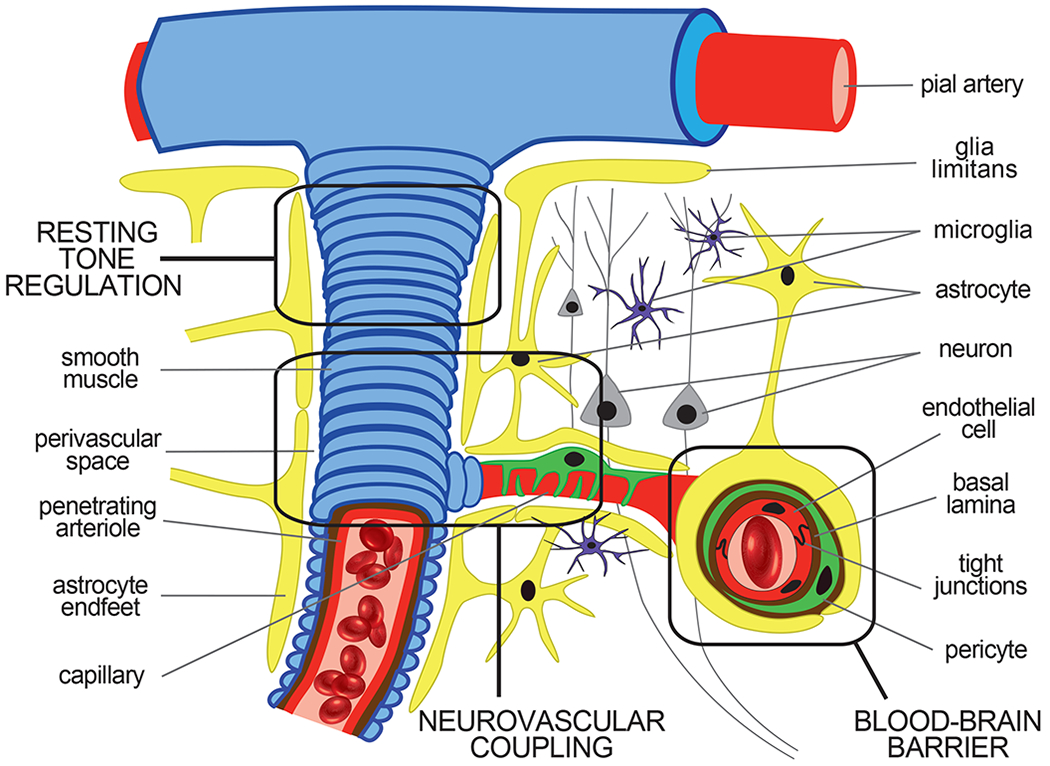
Pial arteries give rise to penetrating arterioles that dive into the brain parenchyma where they branch into smaller arterioles and capillaries. These arterioles receive intrinsic and extrinsic innervation perivascularly to facilitate resting tone regulation via their smooth muscle cell layer. The capillary microvessels lose this smooth muscle cell layer completely and are instead ensconced by astrocyte endfeet and contractile pericytes, which contact the capillary basement membrane directly. These capillary microvessels are further distinguished by their endothelial cells, which are held together by tight junction proteins that form the innermost component of the blood-brain barrier. The endothelial cells, pericytes, and astrocytes, along with the neurons and microglia within a particular vascular domain, together comprise the neurovascular unit. These neuronal and vascular cellular components interact with each other to ensure precise spatiotemporal delivery of blood oxygen and nutrients to metabolically active regions of the brain via neurovascular coupling. Astrocytes are particularly important for this interaction, as they are centrally located between the metabolically active neurons and the cerebral vessels that are poised to respond to such activity.
1.3. Astrocytes as Mediators of Neurovascular Coupling
Historically, NVC was believed to be a feedback system produced by metabolic demand: active neurons produce an energy deficit that triggers an increase in blood supply. When neuronal activity returns to normal, the energy deficit is no longer present and so blood supply returns to resting levels (Koehler et al., 2006; Attwell et al., 2010). Evidence from the past couple of decades, however, suggests that NVC is instead a feed-forward mechanism (Vaucher et al., 1997; Koehler et al., 2006; Attwell et al., 2010): neuronal activity stimulates a complex intercellular signaling cascade, resulting in the production and release of several vasoactive compounds that produce vascular dilation and an increase in blood flow. This feed-forward signaling is triggered regardless of local oxygen level (Lindauer et al., 2010). An emerging hypothesis suggests that a combination of both of these scenarios may be at play: the feed-forward system may regulate the rapid phase of the neurovascular response, whereas the feedback signaling may contribute to the slower, tonic phase (Baslow, 2017).
The role of astrocytes in conveying neurovascular signals to parenchymal arterioles has been extensively studied and reviewed (Howarth, 2014; Mishra, 2016; Iadecola, 2017). Astrocytes respond to neurotransmitters released during synaptic activity, such as glutamate (Alkayed et al., 1997; Zonta et al., 2003; Takano et al., 2006; Gordon et al., 2008) and ATP (Wells et al., 2015; Mishra et al., 2016), with an increase in their intracellular Ca2+ concentration (Mishra et al., 2016). This increased Ca2+ prompts astrocytes to synthesize and release an assortment of vasoactive substances that act on vascular smooth muscle cells to elicit vessel dilation and increase blood flow. Such vasoactive substances include ions such as K+ (Filosa et al., 2006), metabolites of arachidonic acid such as prostaglandin E2 (PGE2) (Zonta et al., 2003; Takano et al., 2006; Gordon et al., 2008; Mishra et al., 2011; Hall et al., 2014) and epoxyeicosatrienoic acids (EETs) (Alkayed et al., 1997; Metea and Newman, 2006), metabolic by-products such as lactate (Gordon et al., 2008), and products of ATP breakdown such as adenosine (Xu and Pelligrino, 2007; Vetri et al., 2011). Astrocyte-derived arachidonic acid can also be metabolized downstream in vascular smooth muscle cells to produce 20-hydroxyeicosatetraenoic acid (20-HETE) to induce vessel constriction (Mulligan and Macvicar, 2004; Metea and Newman, 2006; Mishra et al., 2011; Hall et al., 2014). Interestingly, astrocytic Ca2+-dependent NVC is bidirectional – moderate increases in Ca2+ produce dilations, while large increases produce constrictions (Girouard et al., 2010). It must also be noted that NVC can also be triggered by direct signaling from neurons (Attwell et al., 2010; Cauli and Hamel, 2010) and is not dependent on any single signaling mechanism; rather, it appears to be product of several parallel and redundant pathways (Liu et al., 2012; Hosford and Gourine, 2019), underscoring the significance of NVC in maintaining healthy brain function. Further, not all factors involved in this parallel signaling have yet been identified: simultaneous blockade of almost all the above-described pathways is not enough to completely inhibit NVC.
Astrocytes have been shown to play an essential role in NVC at the microvascular capillary level (Biesecker et al., 2016; Mishra et al., 2016). In the cortex, ATP released by active neurons causes a Ca2+ influx in astrocytes via the ionotropic P2X1 channels, which stimulates arachidonic acid production and its downstream metabolism to vasodilatory PGE2. PGE2 then acts on EP4 receptors on pericytes, the contractile cells on capillaries, thereby dilating capillaries (Mishra et al., 2016). In contrast to previous findings, these and other studies also reported that interfering with astrocyte Ca2+ is not sufficient to block arteriolar NVC, calling the importance of Ca2+-dependent astrocyte mechanisms in arteriole regulation into question (Nizar et al., 2013; Takata et al., 2013; Bonder and McCarthy, 2014; Jego et al., 2014; Biesecker et al., 2016; Mishra et al., 2016). Some of these studies only evaluated the role of astrocytic Ca2+ signals within the soma or inositol trisphosphate (IP3) dependent release of Ca2+ from internal stores in mediating vascular responses, and thus may not be representative of the role that IP3-independent astrocyte Ca2+ signals (Srinivasan et al., 2015), particularly in process microdomains or vascular endfeet, which are shown to play a role in NVC (Otsu et al., 2015). In a recent report (Mishra et al., 2016), we showed that chelating fast astrocyte Ca2+ does not change arteriole NVC but significantly reduces capillary NVC. It has also been proposed that astrocytes may only regulate arteriole NVC under conditions of high or persistent neuronal activity (Institoris et al., 2015). Taken together, these data support the notion that astrocytes regulate NVC at the microvascular capillary level and, under some circumstances, also at the arteriole level (Iadecola, 2017).
1.4. Astrocytes as Modulators of Cerebral Blood Flow
Recent evidence highlights the role of the local NVU and astrocytes in maintaining resting vascular tone (Filosa et al., 2016) via the release of both constrictive and dilatory factors. Though pressure-evoked vascular constriction of parenchymal arterioles is initiated by myogenic factors, it is now demonstrably maintained by astrocyte-dependent signaling (Kim et al., 2015). Specifically, an increase in intravascular pressure activates mechanosensitive, Ca2+-permeable TRPV4 channels on astrocytes, resulting in a rise in astrocyte Ca2+. This in turn causes astrocytes to release purinergic signals that are necessary to maintain the pressure-evoked constriction (Kim et al., 2015). Similarly, the resting vascular tone of retinal arterioles is dependent on purinergic signaling from Muller glial cells (Kur and Newman, 2014), the retinal approximate of astrocytes. Another study reported that Ca2+-dependent cyclooxygenase metabolites released from astrocytes dilates cortical arterioles at rest, and that arresting astrocyte Ca2+ results in arteriole constriction (Rosenegger et al., 2015). Further, cortical vasodilation induced by basal forebrain stimulation, a response previously thought to be entirely neurogenic, was recently shown to be, in part, dependent on astrocytic release of epoxyeicosanoids (Lecrux et al., 2012). A healthy balance between such dilatory and constrictive signals is likely important in regulating vascular tone and maintaining resting blood flow at physiologically desirable levels.
1.5. Astrocytes and the Blood-Brain Barrier
The microvasculature in most organ systems is fenestrated, lacks endothelial tight junctions and is readily permeable to substances in the blood. The brain vasculature, in contrast, is endowed with the BBB, which regulates permeability across the vascular wall in order to maintain homeostasis of the CNS microenvironment and, under healthy conditions, to protect the brain from the peripheral immune system. Tight junctions form a physical barrier at the BBB: they exist between overlapping processes of endothelial cells and prevent substances in the blood from leaking into the CNS extracellular space (Reese and Karnovsky, 1967; Coomber and Stewart, 1985). Thus, passage of substances from the blood to the brain requires either a transporter-mediated or a transcytosis-dependent pathway.
Intricate crosstalk between NVU components, including astrocytes, pericytes and endothelial cells, generates and maintains the BBB. During development, pericytes help establish the BBB by inhibiting the expression of endothelial genes that enhance vascular permeability (Daneman et al., 2010) and inducing polarization of the astrocyte endfeet abutting the vasculature (Armulik et al., 2010). The BBB is often impaired in neurologic disease, suggesting that continued maintenance is required during adult life to preserve normal barrier function (Daneman and Barres, 2005; Abbott et al., 2006). Astrocytes play a key role in this maintenance by secreting trophic factors such as transforming growth factor β1 (TGFβ1), glial-derived neurotrophic factor (GDNF), fibroblast growth factor (FGF) and angiopoietin-1 that preserve the BBB phenotype in endothelial cells (Abbott et al., 2006; Daneman and Prat, 2015).
2. Reactive Astrogliosis and Astrocyte Dysfunction
Astrocytes respond to CNS disease and injury via an overt process known as reactive astrogliosis. Astrogliosis encompasses a host of morphological, transcriptomic, epigenetic, and proliferative changes in astrocytes (Anderson et al., 2014) and often occurs in a graded manner (Sofroniew, 2009). Mild to moderate astrogliosis (e.g. mild trauma, diffuse immune activation), comprises a small and reversible increase in astrocytic expression of cytoskeletal intermediate filament (IF) proteins such as glial fibrillary acidic protein (GFAP) and vimentin (Brenner, 2014) (Fig. 3A–C) together with cellular hypertrophy. Severe astrogliosis (e.g. major infection, chronic neurodegeneration) produces an irreversible, lasting change in the cytoarchitecture and functional properties of astrocytes. In extreme cases, this leads to the formation of a glial scar wherein astrocytes proliferate and form dense intertwined webs to fill in and wall off the empty spaces left by dead or dying cells (Fig. 3D), thereby protecting normal CNS tissue by suppressing the spread of infectious agents, inflammatory cells and the cytokines and metabolites, and cell death. Chronic glial scarring from this kind of astrogliosis was, until recently, thought to detrimentally and physically prevent axon regrowth and thus functional recovery in spinal cord injury (Davies et al., 1999). This prevailing dogma is challenged by a recent study demonstrating that molecular signals expressed by the glial scar tissue supports axon growth and, indeed, allows nerve fibers to regrow across spinal lesions (Anderson et al., 2016). Importantly, the exact cellular changes underlying astrogliosis are likely context-dependent (Zamanian et al., 2012), defined by the specific disease and/or injury (Sofroniew, 2014; Liddelow and Barres, 2017), and perhaps even the stage of the disease. Notably, the increase in IF proteins and astrogliosis are not only induced by injury, but also in aging (Sabbatini et al., 1999; Clarke et al., 2018). An emerging area of research centers on the question of whether senescence or degenerative changes that affect astrocytes, rather than neurons, promote aging and age-related disease in humans.
Figure 3. Conventional immunohistochemical markers demonstrate astrogliosis grading in tissue from epilepsy patients.
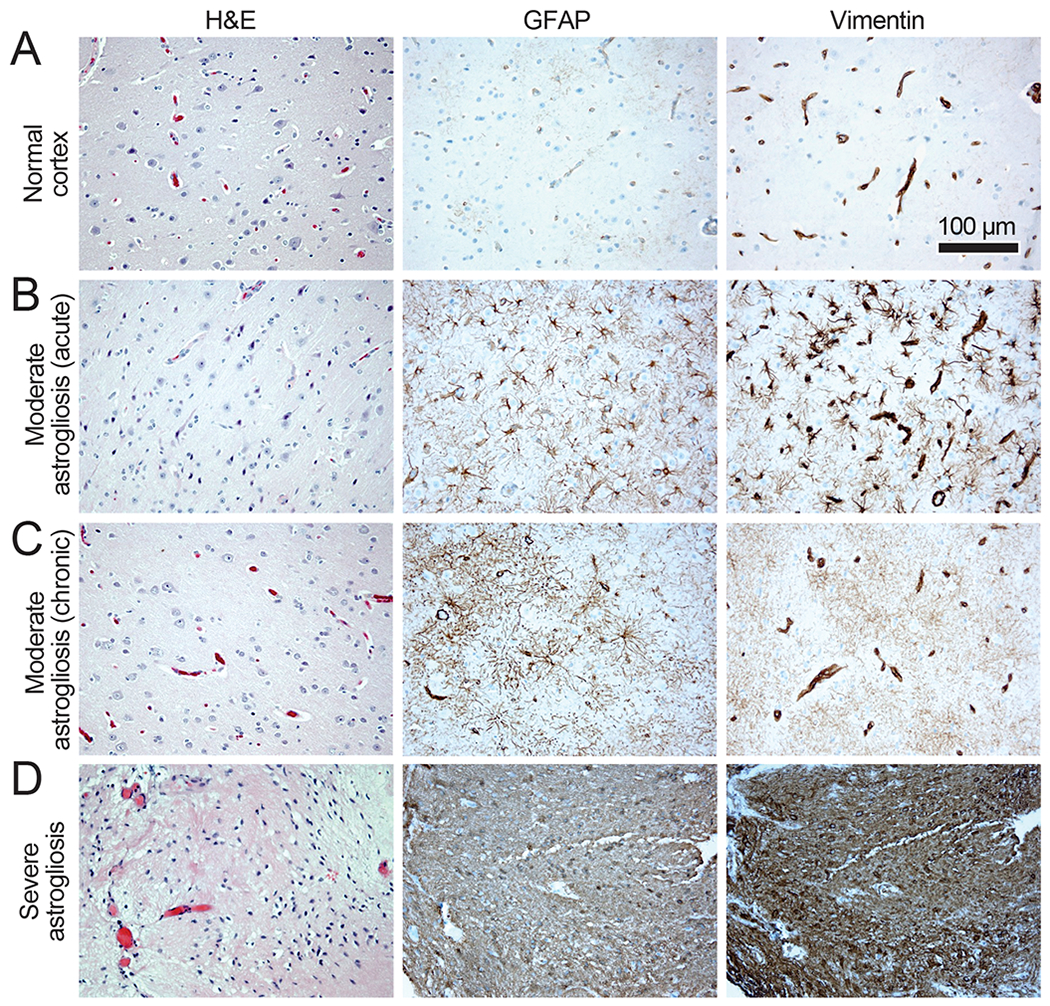
Cytoskeletal changes in reactive astrocytes as manifested in intermediate filament expression. All subjects were patients with epilepsy who underwent temporal lobectomy for resection of seizure focus. Images of hematoxylin- and eosin-stained sections are depicted in the first column, and corresponding brain regions immunostained for the intermediate filament proteins GFAP and vimentin are shown in the second and third columns, respectively. A: A relatively normal-appearing cortex. GFAP is minimally expressed and vimentin expression is restricted to vascular endothelial and smooth muscle cells. B: A subacute, moderate phase of relatively moderate astrogliosis with increased astrocyte GFAP in proximal cell processes and a similar pattern of more striking vimentin expression. C: A moderate astrogliosis that is more long-standing, with GFAP and vimentin expression distributed widely throughout the dense cloud of astrocyte fine processes. With moderate gliosis, morphologic changes detected by histologic assessment of hematoxylin- and eosin-stained sections are subtle. Neuronal loss may be inconspicuous. Vimentin expression is still more pronounced in the vascular cells than astrocytes. D: A severe chronic gliosis associated with a cortical penetrating injury that was the cause of epilepsy. Hematoxylin- and eosin-staining shows complete loss of neurons and dense eosinophilic astrocyte cytoplasm. Astrocyte GFAP and vimentin are highly expressed to the degree that vascular structures are present but obscured on vimentin immunohistochemistry; this tissue appears to have completely converted to a glial scar.
Transcriptomic analysis of reactive astrocytes has highlighted the striking heterogeneity of these cells and led to the idea of ‘good’ verses ‘bad’ astrogliosis (Liddelow and Barres, 2017). Insults that mimic infection, such as injections of the bacterial toxin lipopolysaccharide (LPS) in vivo or treatment with inflammatory cytokines ex vivo, induce expression of cytokines that are likely to aggravate pathology, while insults that induce ischemia induce the expression of proteins involved in tissue repair and neuroprotection (Zamanian et al., 2012). These indications are partly borne out by mechanistic studies: astrocytes responding to LPS stimulation actively phagocytose neurons and negatively impact recovery (Liddelow et al., 2017), while astrocytes responding to ischemia (Liu et al., 2014; Becerra-Calixto and Cardona-Gomez, 2017; Tachibana et al., 2017), spinal cord injury (Faulkner et al., 2004; Anderson et al., 2016) or TBI (Shinozaki et al., 2017) can enhance recovery. This is not entirely unexpected as astrocytes are very adaptable cells that continuously tune in to their environment and respond to it accordingly to establish and maintain homeostasis. In this sense, reactive astrogliosis is a response that aims to re-establish homeostasis after injury and attenuate damage to the nervous tissue. However, similar to peripheral inflammatory responses, astrogliosis may itself become injurious under specific conditions and exacerbate damage (Barres, 2008; Anderson et al., 2014). It is also likely that a spatial and temporal heterogeneity of astrocyte reactivity exists relative to the site of injury. Future research should focus on identifying and categorizing the disease-ameliorating and disease-exacerbating aspects of astrocyte reactivity. Results from such studies may facilitate therapeutic fine-tuning of astrogliosis to skew the process towards neuroprotection and functional recovery.
Features that characterize astrogliosis include decreased expression of inwardly rectifying K+ (Kir) channels (Ji et al., 2012), the glutamate transporters GLT-1 and GLAST (Piao et al., 2015; Hubbard et al., 2016), the purinergic receptor P2Y1 (Shinozaki et al., 2017) and adenosine kinase (Gouder et al., 2004; Aronica et al., 2013). Loss of perivascular localization of the water transporter AQP4 (Eid et al., 2005; Alvestad et al., 2013) and an increase in the expression or activation of metabotropic glutamate receptors (Aronica et al., 2000; Anneser et al., 2004; Zamanian et al., 2012; Rusnakova et al., 2013) have also been reported. Such changes can produce glutamate and potassium imbalances in the neuropil, leading to excitotoxicity or epileptiform activity. However, they do not always occur concurrently and, instead, are remarkably context-dependent. It is also important to note that overt astrogliosis, as defined by upregulation of IF proteins and/or hypertrophy, is not obligatory for astrocytes to dysfunction and disrupt neuronal function. For example, in Huntington’s disease, dysregulation of astrocytic Kir channels and Ca2+ signaling occur in the absence of IF protein upregulation (Jiang et al., 2016), and in diabetic retinopathy, glial-dependent neurovascular uncoupling can occur before gliosis is pronounced (Mishra and Newman, 2010). Another example is presented by multiple sulfatase deficiency, a lysosomal storage disorder caused by mutations in the sulfatase modifying factor 1 gene – transgenic mice with this mutation only in astrocytes display no overt astrogliosis, despite exhibiting degeneration of cortical neurons and anxiety behavior (Di Malta et al., 2012). These findings stress the need for a more nuanced sub-classification of astrocyte dysfunction states. For the purposes of this review, we use the terms ‘astrogliosis’ and ‘reactive astrocytes’ to encompass both morphologically overt astrogliosis and astrocyte dysfunction.
3. Astrocyte-mediated neuropathologies
Defects that primarily affect astrocytes are relatively rare but highlight some important aspects of the roles of astrocytes in health and disease. These tend to fall into two categories: young-onset inherited disease due to mutations in genes that are expressed in astrocytes and contribute to important astrocyte functions, and older-onset disease with morphologic or biochemical features that implicate astrocytes more than other cells as contributors to disease etiology.
Of the inherited diseases, Alexander disease is a clear example, caused by mutations in the GFAP gene. It features not only morphological changes and functional losses in astrocytes, but prominent secondary effects on other glial cells (e.g. microglial activation and oligodendrocyte loss that produce noticeable demyelination), BBB disruption, and variable neuronal loss (Mignot et al., 2004; Sosunov et al., 2018). Another example is hereditary spastic paraplegia, in which mutations in EAAT2 (GLT-1), the astrocyte glutamate transporter, produce loss of upper motor neurons (Meyer et al., 1998; Parodi et al., 2017). Amyotrophic lateral sclerosis is yet another case, where SOD1 mutations are thought to produce motor neuron degeneration through a non-cell autonomous mechanism that involves astrocyte stimulation of neuronal excitability (Papadeas et al., 2011; Fritz et al., 2013; Hayashi et al., 2016).
Some diseases produce neuronal or axonal loss through an acquired immune-mediated attack that targets astrocytes. Immune-mediated attacks on astrocyte AQP4 was recently shown to be the basis of most cases of neuromyelitis optica (Jarius and Wildemann, 2010), with astrocyte dysfunction leading to NVU disruption, oligodendroglial death, and demyelination (Ratelade and Verkman, 2012). Another similar autoimmune condition, GFAP astrocytopathy, was described in 2016, whereby patients’ immune systems generate antibodies against GFAP that is associated with subventricular and perivascular damage (Fang et al., 2016), ultimately resulting in encephalopathy, inflammatory myelitis, tremor, and ataxia. The primary astrocyte insult appears to begin around the pial, ventricular, or perivascular regions, perhaps because these are regions where the peripheral immune system can first interact with brain tissue. The pathophysiology underlying this condition is not fully understood but it is suggested that antigen-specific cytotoxic T-cells (rather than the anti-GFAP antibody itself) may play a role in some cases, or that an immune attack triggered by occult neoplasm expressing astrocyte antigens may be responsible in others (Zekeridou et al., 2018). It could also be downstream of other astrocyte dysfunctions, a question currently under investigation (Zekeridou et al., 2018).
Several late-onset conditions associated with astrocyte dysfunction have also recently gained scientific attention. Age-related neuropathologies associated with protein aggregation within astrocytes, especially hyperphosphorylated tau, have been described in frontotemporal lobar degeneration (FTLD), Pick’s disease, progressive supranuclear palsy, cortico-basal degeneration, and aging-related tau astrogliopathy (ARTAG) (Komori, 1999; Kovacs et al., 2017). Astrocytic tauopathy tends to be the dominant pathologic feature in some tau mutation-associated FTLD as well as the pathologic lesion of ARTAG (Kovacs et al., 2017). All of these diseases feature prominent neuronal loss and/or demyelination. Key roles for astrocytes have been proposed in additional diseases that feature neuronal loss, including epilepsy, Huntington’s disease, and Parkinson’s disease, and often involve blood-brain barrier dysfunction and altered astrocyte metabolism (Weissberg et al., 2015; Booth et al., 2017; Khakh et al., 2017; Boison and Steinhauser, 2018; Skotte et al., 2018; Yan et al., 2018).
The distinction between astrogliosis and astrocytopathy in disease is noteworthy: astrogliosis is defined as the active response of astrocytes to an injury (neuronal death, ischemia or TBI etc.) and involves aberrant loss or gain of astrocyte function, while astrocytopathy is defined as astrocyte degeneration characterized by swollen cell bodies, retracted short processes, and loss of fine processes and endfeet (Pekny et al., 2016; Kim et al., 2017). Particularly in conditions where astrocytopathy is observed, the disease is likely to be secondary to unhealthy astrocytes. Is astrocytopathy a separate independent phenomenon, or is it the end-product of prolonged astrogliosis and functional disruption? We do not know. However, evidence provided above supports the concept of astrocytopathy or ‘astrodegeneration’ as another important contributor to the pathogenesis of disease, including those that have been considered primarily or exclusively neuronal in nature and should be considered a possible etiological factor in neurodegenerative diseases.
4. Astrogliosis and neurovascular unit impairment in neurodegenerative disorders
Many CNS diseases feature NVU abnormalities. Some are readily attributable to NVU abnormalities, such as stroke and vasculitis. In many others – including AD, TBI, chronic traumatic encephalopathy (CTE), etc. – NVU abnormalities are rampant, but the reason for these abnormalities and their contribution to disease etiology is unclear (reviewed in (McConnell et al., 2017) and (Liebner et al., 2018). Given the central positioning of astrocytes between neurons and vascular cells within the NVU, it is plausible that they aberrantly influence the NVU, thus disrupting the BBB and NVC, and contributing to neurological disorders. Here we focus our attention on three disorders - AD, TBI, and stroke – and discuss the evidence implying a possible causal relationship between astrogliosis and NVU dysfunction. Although TBI and stroke are acute-onset conditions, they have chronic impacts on neurological health, including increased incidence and rate of cognitive decline (Savva et al., 2010; Levine et al., 2015; Ozen et al., 2015). Further, accumulating evidence that AD patients have a history of evident or silent infarcts further stresses the interaction of such acute injuries with chronic conditions (Vermeer et al., 2003; Yang et al., 2015). These chronic consequences of acute injuries may partially originate from impairments in astrocyte-vascular signaling. We also briefly discuss the association between astrogliosis and vascular dysfunction in white matter diseases.
4.1. Alzheimer’s disease and related dementias
AD is the leading neurodegenerative cause of cognitive decline and dementia. AD diagnosis is based on neurological evaluation and neuroimaging data, and is confirmed port-mortem by histological presence of extracellular plaques of amyloid β protein (Aβ) and intracellular neurofibrillary tangles of hyperphosphorylated tau protein within neurons. In many cases, Aβ deposition also occurs around the vasculature in the form of cerebral amyloid angiopathy (CAA). However, aside from a few mutations responsible for familial cases, the actual cause of AD remains unknown. While much of the past work on AD has predominantly focused on neurogenic mechanisms and neuroprotection (Kuruva and Reddy, 2017), epidemiological data suggest that vascular pathology is also a key contributing factor. Although the vascular hypothesis of AD and dementia was proposed over two decades ago (de la Torre and Mussivand, 1993; de la Torre, 1997; Farkas and Luiten, 2001; Zlokovic, 2002), only recently has it become the focus of renewed attention (Kalaria, 2010; Snyder et al., 2015; Montagne et al., 2016). Indeed, the National Institute of Neurological Disorders and Stroke has focused a new research framework for investigations specifically into vascular contributions to cognitive impairment and dementia (Corriveau et al., 2016).
BBB dysfunction and cerebral hypoperfusion both occur early in AD (Kelleher and Soiza, 2013; Iturria-Medina et al., 2016), and decreased tissue oxygenation is observed even in patients with mild cognitive impairment (Tarumi et al., 2014), suggesting disruption of CBF. Indeed, impairment of NVC occurs in AD patients (Rosengarten et al., 2009; Nicolakakis and Hamel, 2011; Kotliar et al., 2017), with regions of amyloid deposition being characterized by large reductions in CBF (Mattsson et al., 2014). A recent analysis of the patient data deposited in the multicenter AD Neuroimaging Initiative showed that cerebrovascular dysregulation was not only the strongest and earliest detectable event during the development of cognitive impairment, but was correlated with disease progression even before amyloid β (Aβ) accumulation and functional/metabolic changes (Iturria-Medina et al., 2016). Animal models of AD are also characterized by impaired NVC (Rancillac et al., 2012; Kimbrough et al., 2015; Joo et al., 2017; Tarantini et al., 2017; Gutierrez-Jimenez et al., 2018), lower resting CBF (Niwa et al., 2002) and lower oxygen extraction fraction (Gutierrez-Jimenez et al., 2018), often preceding plaque deposition (Niwa et al., 2002). In an animal model of CAA, cerebral hypoperfusion accelerated the rate of CAA burden and the appearance of cortical microinfarcts (Okamoto et al., 2012). CAA can interrupt NVC signals (Kimbrough et al., 2015), likely producing local hypoxia, which can, in turn, induce Aβ generation (Zhang et al., 2007). Thus, a vicious feedback loop is initiated wherein Aβ and hypoxia exacerbate each other. Consistent with this, a study of demented patients experiencing hypoperfusion due to a unilateral carotid artery stenosis were found to have a much higher Aβ load preferentially in the stenosed hemisphere (Huang et al., 2012). These findings extend strong support for the hypothesis that neurovascular dysfunction may be causative in AD rather than purely correlative.
What, if any, is the role of astrocytes in this dysfunction? Reactive astrogliosis is a common and widespread histologic feature of AD (Rodriguez-Arellano et al., 2015; Taipa et al., 2017). In both patients and animals models, reactive astrocytes associate strongly with senile plaques (Mandybur and Chuirazzi, 1990; Cullen, 1997; Rodriguez-Arellano et al., 2015) where they play an important role in Aβ degradation and clearance (Wyss-Coray et al., 2003). These astrocytes exhibit not only local, but global, network-wide increases in Ca2+ signaling (Kuchibhotla et al., 2009) and clear presynaptic dystrophic neurites by engulfing and degrading them (Gomez-Arboledas et al., 2018). Thus, generally speaking, astrocytes appear to take on a phagocytic role in AD, conferring neuroprotection. However, there is also ample evidence for detrimental roles of astrocytes; they exhibit decreased expression and activity of glutamine synthetase (Olabarria et al., 2011), overexpress inducible nitric oxide synthase (Akama and Van Eldik, 2000), and can inhibit neuronal function by synthesizing and releasing GABA (Jo et al., 2014). At the NVU, astrocytes exhibit several changes that may alter their ability to maintain the BBB and regulate NVC. Reactive astrocytes surrounding Aβ plaques exhibit Ca2+ hyperactivity driven by metabotropic purinergic signaling (Delekate et al., 2014) and, in models of CAA, the vascular coverage by astrocyte endfeet is reduced with concurrent downregulation of K+ and AQP4 channels (Wilcock et al., 2009). As Ca2+-dependent K+ release from astrocytes is one of the established pathways regulating NVC, dysfunction of these signals in reactive astrocytes could have an immediate and strong effect on local blood flow. Further, dysfunction of the glymphatic clearance system, which relies of astrocytic AQP4 channels, was also recently suggested to increase Aβ accumulation in an AD model (Peng et al., 2016). Further research needs to directly address whether reactive astrogliosis, via these or other mechanisms (AA metabolites, adenosine, lactate etc.), attenuates NVC or alters perivascular Aβ clearance in AD.
Astrocyte dysfunction has been demonstrated to directly induce AD-like vascular and amyloid pathology in one interesting case. In mice overexpressing astrocyte-specific TGFβ1, astrogliosis develops at a young age and is followed by a reduction in CBF, an increase in perivascular Aβ accumulation (even in the absence of an Aβ-overexpressing mutant genotype), and finally cerebral hemorrhage – similar to the progression of AD pathology in human disease (Wyss-Coray et al., 2000a; Wyss-Coray et al., 2000b; Gaertner et al., 2005). TGFβ receptor-dependent signaling produces astrogliosis in response to blood-borne proteins that leak into the brain in the event of a BBB failure (Heinemann et al., 2012). It is therefore possible to imagine a scenario whereby an early disruption of the BBB (perhaps induced by peripheral inflammation (Cattaneo et al., 2017)) and induction of astrogliosis initiates a molecular and cellular domino effect that, under repeated stress or in the right genetic background, dysregulates the NVU and overwhelms Aβ clearance systems, thus leading to AD pathology. Indeed, loss of BBB integrity is common in animal models of AD (Montagne et al., 2017), and blood constituents such as albumin, fibrinogen and immunoglobulins accumulate in aging human brains (Goodall et al., 2017), particularly in astrocytes. Even more compellingly, TGFβ1 levels are increased in the brains (Wyss-Coray et al., 1997) and cerebrospinal fluid of AD patients (Zetterberg et al., 2004). These findings lend credence to the general hypothesis that an initial BBB failure event leads to astrogliosis (and also microglial activation (Heppner et al., 2015; Sarlus and Heneka, 2017), not discussed in this review), which induces neurovascular dysregulation and hypoperfusion, which then increase microinfarcts and Aβ formation. Upon prolonged and repeated insults, or within the right genetic background, such astrocyte-directed NVU dysregulation could lead to increased Aβ deposition, reduced Aβ clearance and, ultimately, increased neuronal injury to result in the typical “sporadic” cases of AD. Now that we are beginning to understand the connections between astrocytes, neurovascular regulation, and BBB maintenance in physiology and their impairment in AD pathology, it is time to revise our strategy to one that aims to preserve not only neurons but also astrocytes, other glial cells and vascular cells: indeed, the whole community must survive for cognitive function to be saved (Barres, 2008).
4.2. Traumatic Brain Injury
TBI occurs when the brain sustains physical trauma due to impact, penetration, or rapid movement (vibrations) within the skull. Because TBI can result from multiple injury paradigms, and affect all genders and ages, the exact pathophysiology underlying the disorder is unique to each patient in both region and extent. Age and injury severity can both strongly influence pathological outcomes (Reuler and Gardner, 1987). In general, pathophysiological changes are characterized as either primary (occurring immediately as a result of the trauma) or secondary (occurring later as a result of neuroinflammation and altered cellular signaling). Among these changes, disequilibrated ionic flux and glucose metabolism are neurochemical hallmarks of the disorder. The latter causes what is referred to as a post-TBI energy crisis: vulnerable brain cells need a relatively increased glucose supply to repair and recover, but a pathological decrease in glucose uptake into the brain due to decreased CBF (Golding et al., 1999; Selwyn et al., 2013), impaired glucose transport (Cornford et al., 1996) and/or metabolic processing (Bartnik et al., 2005) results in an energy mismatch between the energy demands and supply. Further, hypoperfusion and reduced autoregulation are observed in both TBI patients (Soustiel and Sviri, 2007; Newsome et al., 2012; Hinzman et al., 2014; Sours et al., 2015) and animal models (Yuan et al., 1988) with attendant astrogliosis (Burda et al., 2015).
TBI can initiate a host of secondary injury cascades that lead to widespread dysfunction of the NVU, neurovascular uncoupling, and changes in microvascular ultrastructure (Logsdon et al., 2015; Kenney et al., 2016; Toth et al., 2016). Specifically, trauma can result in vascular endothelial cell injury, leading to vasospasm, vasoconstriction, micro-thrombosis and oxidative stress, all of which exacerbate the inflammatory response (Lenzlinger et al., 2002; Veenith et al., 2016). TBI is also strongly associated with induction of cortical spreading depolarizations (CSD), which produce severe alterations in resting and reactive CBF, including NVC (Hartings et al., 2009; Lauritzen et al., 2011; Hinzman et al., 2014) (see section 4.3 on Stroke for further discussion of CSD and cerebrovascular dysregulation). Mechanical vascular injury can also directly result in ischemia and, in some cases, hemorrhage, allowing iron and blood components to be released into the parenchyma where they produce neuronal toxicity (Wagner et al., 2003) and may also produce neurovascular uncoupling and vasospasm (Balbi et al., 2017). Microvascular injury is also a feature of CTE, which has become the topic of intense study in recent years (Kenney et al., 2016). A NINDS/NIBIB consensus group recently defined CTE to be neuropathologically identified by tau accumulation in neurons and astrocytes specifically in perivascular locations (McKee et al., 2016). The relationship between this perivascular location of tau accumulation and vascular dysfunction is particularly interesting and future studies are required to address whether perivascular astrogliosis produces or hastens the later cognitive manifestations of both TBI and CTE.
Both immediate and long-term loss of BBB properties have been reported following experimental TBI (Baskaya et al., 1997). BBB loss causes edema and increased expression of matrix metalloproteinase 9 (Suehiro et al., 2004), which results in a positive feedback cycle inducing further damage to blood vessels (Underly et al., 2017). Post-mortem studies in humans show that BBB loss persists for decades after a traumatic incident (Hay et al., 2015). Given that TBI can mechanically impair the BBB and that blood-borne substances like albumin, thrombin and fibrinogen can activate astrogliosis via TGFβ receptors (Schachtrup et al., 2010; Heinemann et al., 2012; Piao et al., 2017), this chronic loss of BBB may contribute to lasting astrocyte dysfunction and alter the neuronal environment. There is contrasting evidence for the downstream effects of astrogliosis and astrocyte dysfunction – while some studies find that astrogliosis exacerbates tissue loss and behavioral deficits after TBI (Chen et al., 2017; Menzel et al., 2017), others have found that astrogliosis enhances neuroprotection (Shinozaki et al., 2017) and helps stabilize the NVU and normalizes CBF after TBI (Villapol et al., 2014). Such divergent findings suggest that the consequences of astrogliosis after TBI are multifaceted and complex (Burda et al., 2015). How does the long-lasting failure of the BBB affect the evolution of astrogliosis over time? And how do these two events together impact the NVU, blood flow regulation and neuronal survival? These are questions that have yet to be answered (Fig. 4).
Figure 4. Interrelationship between astrogliosis and cerebrovascular dysfunction.
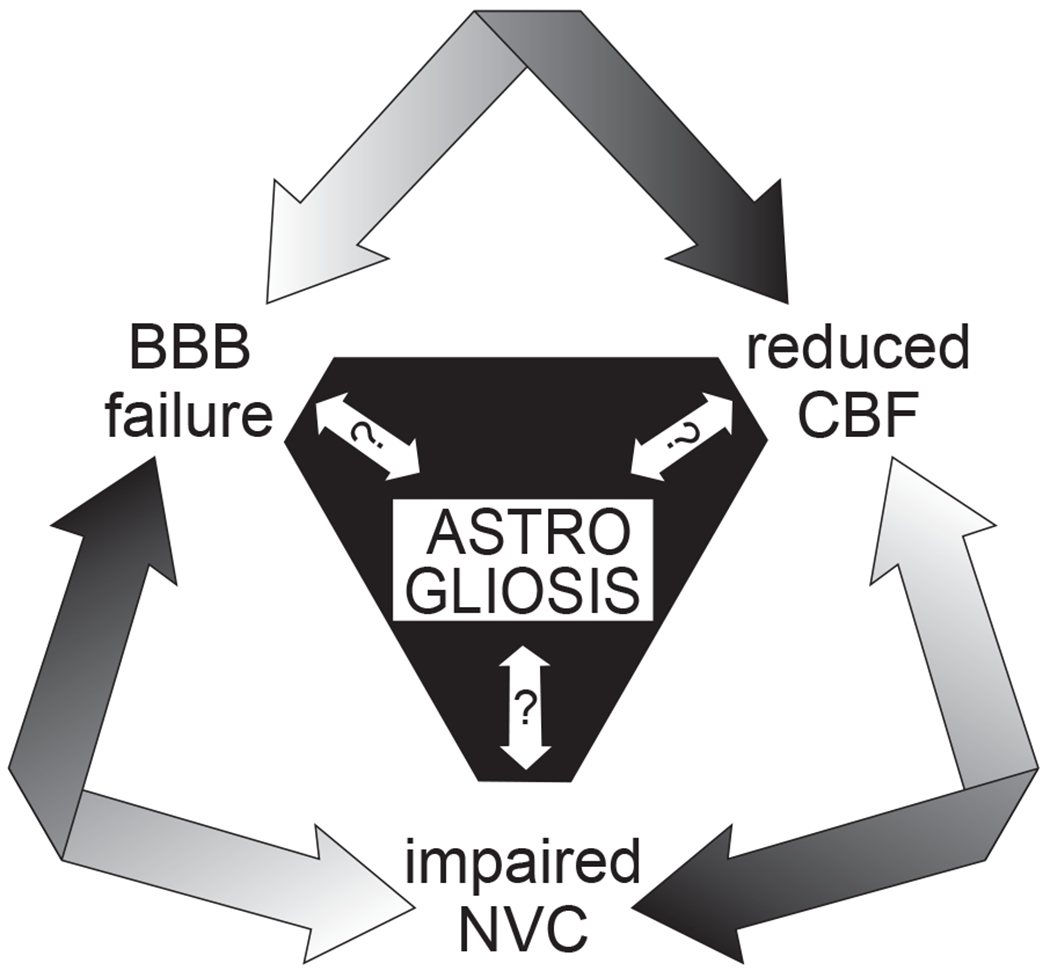
Do reactive astrocytes contribute to pathological cerebrovascular dysfunction in disease? With their central positioning between neurons and cerebral vessels and their multifaceted roles in homeostatic blood-brain barrier (BBB) maintenance, cerebral blood flow (CBF) regulation, and neurovascular coupling (NVC), astrocytes have the potential to adversely affect all of these processes when they are reactive and in a morphologically and functionally altered state. Although astrogliosis almost certainly contributes to and exacerbates the dysfunction of these processes, it is unclear whether it precedes them or results from them.
4.3. Stroke
Stroke is a common presentation of cerebrovascular disease in the brain. Ischemic stroke results from a thrombus or embolus blocking an artery supplying the brain, causing an immediate infarct in that artery’s territory. In contrast, hemorrhagic stroke results from a ruptured vascular defect such as an aneurysm, often in the sub-arachnoid space, and has a very high rate of mortality; among surviving patients, delayed cerebral ischemia (DCI) is the leading cause of poor outcome (Xiao et al., 2017). Focal neurological deficits due to the insult are an initial physiological hallmark of stroke and vary based on the severity, duration and location of the injury. Pathological and cellular changes also develop in the surrounding brain regions, where neuronal, glial, and cerebrovascular injury all co-occur. Following ischemic stroke, the most pronounced among these is the glial response: phagocytic microglia within and around the infarct engulf debris from dying cells, proliferating reactive astrocytes generate a protective glial scar around the perimeter of the infarct, and astrocytes in the non-ischemic regions beyond the infarct perimeter become reactive in a spatial gradient.
The current standard of care for ischemic stroke is removal of the clot, achieved chemically by administering tissue plasminogen activator or mechanically by thrombectomy. Despite recanalization of the culprit artery, a state of microvascular hypoperfusion often continues to exist – the so-called ‘no-reflow’ phenomenon. Indeed, the level of capillary perfusion is more predictive of functional recovery in patients (Al-Ali et al., 2016) than recanalization of the blocked artery alone (Cho et al., 2015). This microvascular block was believed to be the result of platelet or leukocyte adhesion in small vessels (Abumiya et al., 2000), but evidence now suggests that active capillary constriction (Leffler et al., 1989; Hauck et al., 2004) due to pericyte contraction (Hall et al., 2014) also plays a significant role. Capillary hypoperfusion produced by pericyte contraction is also a feature of DCI following SAH (Johshita et al., 1990; Li et al., 2016). As astrocytes play a central role in regulating capillary level blood flow (Biesecker et al., 2016; Mishra et al., 2016), the possibility that astrocyte dysfunction is contributing to this hypoperfusion warrants further investigation.
Several studies also suggest a significant loss of NVC in stroke patients in the asymptomatic, non-ischemic brain tissue surrounding the infarct, either unilaterally within the stroke hemisphere (Krainik et al., 2005; Salinet et al., 2015) or bilaterally in both hemispheres (Lin et al., 2011; Salinet et al., 2018). This loss of local blood flow regulation, which likely results in hypoperfusion, is proposed to underlie the increased rate and incidence of dementia in stroke patients (Savva et al., 2010; Levine et al., 2015). However, mechanistic studies investigating the cause of NVC loss are still lacking (El Amki and Wegener, 2017). An even worse case occurs following SAH, where NVC is inverted - neuronal activity gives rise to vascular constriction and CBF reduction both in vitro (Koide et al., 2013; Pappas et al., 2015) and in vivo (Balbi et al., 2017). This extreme and evidently detrimental response likely contributes to the development of DCI after SAH.
Ischemia and injury can also trigger a continuum of CSDs, a phenomenon that is characterized by failure of neuronal ion homeostasis, mass depolarization of neurons and astrocytes, excitotoxicity and edema (Lauritzen et al., 2011; Dreier et al., 2018). After a stroke, CSDs in the ischemic region are often terminal and contribute to the neuronal damage and death in the infarct core. In the peri-infarct region, CSDs are shorter in duration and spread across the cortex at the rate of approximately 2-5 mm (Lauritzen et al., 2011). Importantly, CSDs are associated with large changes in CBF. In otherwise healthy tissue, CSDs evoke a large hyperemic response (Dreier, 2011) that does not cause tissue dysfunction or damage (Nedergaard and Hansen, 1988). However, in pathological contexts such as after ischemic stroke, SAH or TBI, CSDs induce an inverse neurovascular coupling response, resulting in vasoconstriction and decreased CBF (Dreier et al., 1998; Dreier, 2011; Hinzman et al., 2014). CSD can further induce a lack of vascular reactivity and loss of evoked NVC (Lauritzen et al., 2011). The combined effect of low CBF, lack of cerebrovascular reactivity and neurovascular uncoupling, in the face of the increased energy metabolism during pathological CSD, produces a condition of cortical spreading ischemia and results in widespread neuron death (Dreier et al., 2000). This is one of the rare cases where inverse neurovascular coupling has been directly associated with tissue damage (Dreier et al., 2018).
A breakdown of the BBB occurs in patients with ischemic stroke (Merali et al., 2017; Villringer et al., 2017) and SAH (Lampl et al., 2005), as well as in animal models (Cipolla et al., 2004; Pan et al., 2017). This breakdown results from the loss of tight junction proteins such as occludin (Pan et al., 2017) and an increase in pinocytic transcytosis across the endothelium (Cipolla et al., 2004). Loss of BBB precedes the reduction in CBF (Tamaki et al., 1984) and predicts long-term functional outcome in patients (Jiang et al., 2018), and thus, appears to be an early event after stroke.
Reactive astrogliosis is also prominent after stroke (Hayakawa et al., 2012; Abeysinghe et al., 2016; Becerra-Calixto and Cardona-Gomez, 2017; Sims and Yew, 2017). The transcriptomic profile of astrocytes after ischemic injury shows that they take on a beneficial anti-inflammatory phenotype (Zamanian et al., 2012; Rusnakova et al., 2013), which may function to stabilize and resolve the injury. Astrogliosis is correlated with CNS remodeling and motor recovery (Liu et al., 2014), and with enhanced survival of neuronal and vascular cells following early reperfusion (Tachibana et al., 2017). However, not all aspects of the astrocyte response are positive: morphologically, swelling of astrocyte endfeet can compress brain microvessels and thereby decreases microvascular perfusion (Ito et al., 2011). As cerebral vessels lose BBB properties after stroke, this vessel compression may be an astrocytic response to limit the extravasation of blood-borne solutes into the brain or a response to blood-borne solutes that have already extravasated (Xiang et al., 2016). Further, although astrogliosis and BBB breakdown are concurrently observed even in brain regions far from the site of ischemia (Garbuzova-Davis et al., 2013), the temporal relationship between them is still an open question. Does astrogliosis cause BBB breakdown, or is BBB breakdown (via leakage of blood proteins such as albumin, thrombin and fibrinogen) producing astrogliosis? It is likely that these factors feed into each other bidirectionally, hence exacerbating the damage caused by ischemia.
Post-stroke astrogliosis is particularly noticeable in astrocyte endfeet around cerebral vessels (Fig. 5), indicating that CBF reduction and NVC impairment after stroke may be due to astrocyte dysfunction. After SAH, astrocytes display large amplitude Ca2+ oscillations, which result in K+ efflux from their endfeet onto the vasculature, raising the extracellular K+ concentration high enough to constrict vessels and resulting in NVC inversion (Koide et al., 2012). Similarly large astrocyte Ca2+ signals are observed immediately following ischemic stroke (Ding et al., 2009; Rakers and Petzold, 2017) with a negative impact on neuronal recovery. Large astrocytic Ca2+ waves also occur during CSD (Chuquet et al., 2007), which are common after stroke, and both the vasoconstriction and NVC inversion observed following CSD depend on astrocyte Ca2+ (Chuquet et al., 2007; Major et al., 2017) and, to some extent, the vasoconstrictive arachidonic acid metabolite 20-HETE (Fordsmann et al., 2013). In addition, a recent study reported that the decrease in CBF associated with ischemic stroke was mitigated when astrogliosis was attenuated (Begum et al., 2018). These findings suggest a causal role for astrogliosis in blood flow impairment after stroke; however, more work is required before this relationship is sufficiently experimentally corroborated.
Figure 5. Stroke-induced astrogliosis is prominent at astrocyte endfeet in rat cortex.
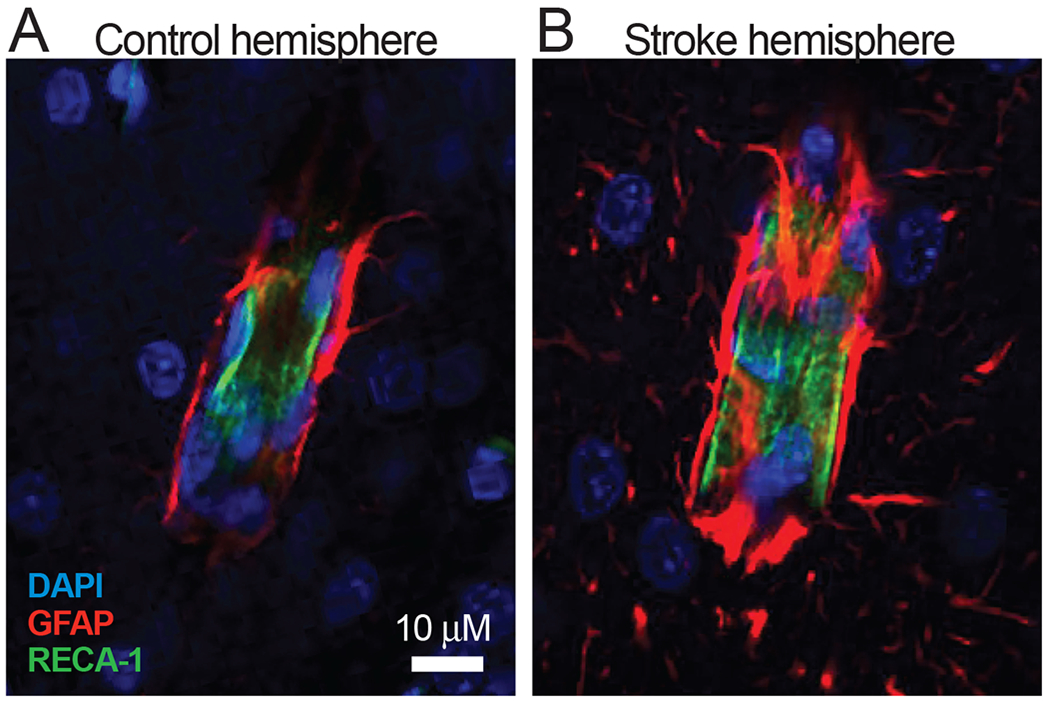
A: GFAP immunolabeled astrocyte endfeet (red) terminating on a cortical vessel in the control hemisphere of a rat that underwent transient experimental stroke. B: Astrocyte endfeet terminating on a vessel in the peri-infarct intact cortical tissue of the stroke hemisphere. Note the increased expression of GFAP and hypertrophy of the endfeet processes, suggesting astrogliosis. DAPI is shown in blue and the rat endothelial cell marker (RECA-1) is shown in green. Images were obtained on a Zeiss LSM 780 (Objective Plan-Apochromat 63x/1.40 Oil) 3 days after middle cerebral artery occlusion in 6 week-old Long Evans rat.
Although it is certain that early astrogliosis is an adaptive, neuroprotective response following ischemia (Zamanian et al., 2012), could it evolve past a threshold beyond which, perhaps depending on the extent and duration of injury, it becomes a detrimental process? Or perhaps the vascular effects of astrogliosis are maladaptive ‘side effects’. Resolving the temporal dynamics and the nature of the relationship between BBB failure, astrogliosis and vascular regulation following stroke might provide insights into this evolution and suggest new targeted therapies aimed at modulating astrogliosis and/or restoring cerebral microvascular flow in a temporally fine-tuned manner to protect the stroke penumbra.
5. Possible Roles of Astrogliosis in White Matter Disorders
With advancing age, an increasing burden of cognitive impairment appears to be related to cerebral small vessel disease, the later stages of which are characterized by demyelination and axonal loss observed as regions of white matter hyperintensities (WMH) on T2-weighted MRI images (Hase et al., 2017). WMH are observed in 90-95% of people over the age of 60 (de Leeuw et al., 2001) and are also prominent in younger patients with depression (Thomas et al., 2002) and post-stroke dementia (Chen et al., 2016). Despite their high incidence in the general population, very little is known about their underlying cause. Could astrogliosis or astrocytopathy of white matter astrocytes and the resultant cerebrovascular insufficiency be producing these WMH? Post mortem analyses show that regions of WMH often display enlarged perivascular spaces, lacunar infarcts, and changes in the NVU compartments. In particular, drastic reductions in astrocyte numbers (Zambenedetti et al., 2002) as well as structural changes and loss of perivascular endfeet are observed (Chen et al., 2016). Further, astrocytes in regions of WMH undergo clasmatodendrosis, a phenomenon that looks very much like an explosive astrocyte degeneration (Hulse et al., 2001). Whether, and how, astrogliosis, astrocytopathy and/or astrodegeneration contribute to white matter disease is not easily answered by clinical studies or a single-time point post-mortem analysis of human tissue. Therefore, there is an urgent need develop reliable animal models of WMH and other white matter pathologies to address these questions.
6. Conclusions
Essentially every approach to therapy for brain diseases due to any cause is focused on prevention or reversal of pathologic alterations in neurons – witness the common use of the term “neuro-protection” to summarize these therapeutic strategies as a whole. Certainly, there is likely to be utility in this approach for many diseases. However, despite years of dedicated effort aimed at these approaches, successes have been limited for most diseases and thoroughly disappointing in others. With this in mind, we consider whether it might be helpful to modify the current paradigm for understanding brain disease to incorporate approaches that ask whether susceptibility to major brain diseases might not lie entirely or even primarily in neurons, but rather in astrocytes, the quintessential “neuroprotectant” cells, with effects on neurons as a secondary consequence (Fig. 6). If a castle is under siege, the survival of the royal family is best assured if the fort is strengthened, the front-line soldiers are well-armed, and the food and water supply are protected. Ultimately, the development of strategies for glio-protection and vasculo-protection may prove the most effective ways to achieve neuroprotection by harnessing the ability of astrocytes to support neuronal function and by providing the energy supply to fuel recovery and function. We anticipate that future research exploring these possibilities will open the door to new combinatorial therapeutics that have a renewed chance of success in human patients.
Figure 6. A proposed model of astrogliosis progression and pathological manifestation.
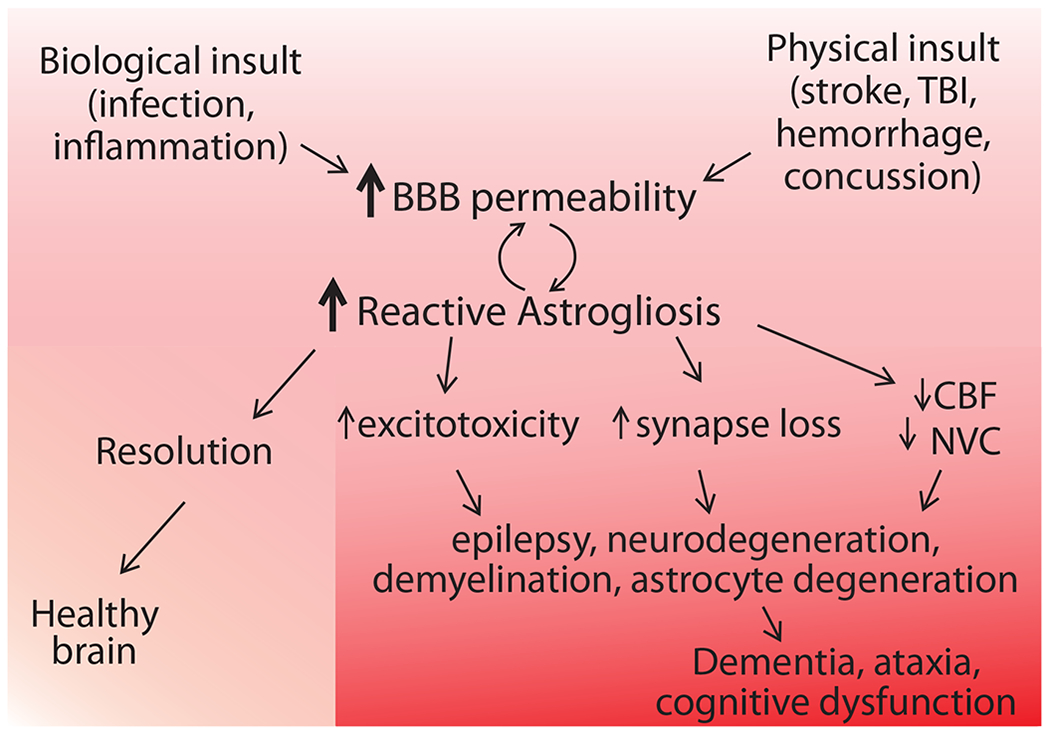
Biological insults such as peripheral infection or systemic inflammation and physical insults such as concussion, TBI, and stroke may result in BBB failure, allowing (normally blood-restricted) substances such as albumin and thrombin to leak into the brain. The presence of these blood-borne proteins in the neuronal microenvironment is detected by astrocytes and reactive astrogliosis ensues. This response is likely initiated in order to contain the damage and re-establish homeostasis. When the attempt is successful, the insult is resolved and healthy physiology prevails. Occasionally, this gliotic response might be too exaggerated and surpasses an as-yet unidentified threshold when it becomes damaging instead, resulting in excitotoxicity (lack of neurotransmitter uptake and/or K+ buffering), synapse loss, and reduction of cerebral blood flow (CBF) and neurovascular coupling (NVC). Interactions between these neurotoxic end-effects may also occur; for example, a decrease in CBF and NVC might precede and cause synapse loss. Together, these effects of astrogliosis synergize to trigger degeneration of neurons and glia, ultimately precipitating the symptoms of many neurological disorders, including dementia, cognitive disorder, and ataxia.
Acknowledgments
Funding: This work was supported by a National Institutes of Health (NIH) Ruth L. Kirschstein National Research Service Award T32 [T32HL094294] to H.M.; a Western Light Talent Training Fellowship to Z.L.; National NIH Grants [R01AG056712 and P30AG008017] to R.W.; a Collins Medical Trust grant to A.M.; and NIH [P30NS061800] (PI: Aicher), which supports the OHSU Advanced Light Microscopy Core.
Abbreviations:
- 20-HETE
20-hydroxyeicosatetraenoic acid
- AD
Alzheimer’s disease
- ARTAG
aging-related tau astrogliopathy
- BBB
blood-brain barrier
- CBF
cerebral blood flow
- CSD
cortical spreading depolarization
- CTE
chronic traumatic encephalopathy
- DCI
delayed cerebral ischemia
- EETs
epoxyeicosatrienoic acids
- FGF
fibroblast growth factor
- FTLD
frontotemporal lobar degeneration
- GDNF
glial-derived neurotrophic factor
- GFAP
fibrillary acidic protein
- IF
intermediate filament
- LPS
lipopolysaccharide
- NVC
neurovascular coupling
- NVU
neurovascular unit
- PGE2
prostaglandin E2
- SAH
subarachnoid hemorrhage
- TBI
traumatic brain injury
- TGFβ1
transforming growth factor β1
Footnotes
Conflict of Interest
None.
References
- Abbott NJ, Ronnback L, Hansson E, 2006. Astrocyte-endothelial interactions at the blood-brain barrier. Nat Rev Neurosci 7, 41–53. doi: 10.1038/nrn1824 [DOI] [PubMed] [Google Scholar]
- Abeysinghe HC, Phillips EL, Chin-Cheng H, Beart PM, Roulston CL, 2016. Modulating Astrocyte Transition after Stroke to Promote Brain Rescue and Functional Recovery: Emerging Targets Include Rho Kinase. Int J Mol Sci 17, 288. doi: 10.3390/ijms17030288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abumiya T, Fitridge R, Mazur C, Copeland BR, Koziol JA, Tschopp JF, Pierschbacher MD, del Zoppo GJ, 2000. Integrin alpha(IIb)beta(3) inhibitor preserves microvascular patency in experimental acute focal cerebral ischemia. Stroke 31, 1402–1409; discussion 1409-1410 [DOI] [PubMed] [Google Scholar]
- Aiello LCWP, 1995. The Expensive-Tissue Hypothesis: The Brain and the Digestive System in Human and Primate Evolution. Current Anthropology 36 199–221 [Google Scholar]
- Akama KT, Van Eldik LJ, 2000. Beta-amyloid stimulation of inducible nitric-oxide synthase in astrocytes is interleukin-1beta- and tumor necrosis factor-alpha (TNFalpha)-dependent, and involves a TNFalpha receptor-associated factor- and NFkappaB-inducing kinase-dependent signaling mechanism. J Biol Chem 275, 7918–7924 [DOI] [PubMed] [Google Scholar]
- Al-Ali F, Berkhemer OA, Yousman WP, Elias JJ, Bender EN, Lingsma HF, van der Lugt A, Dippel DW, Roos YB, van Oostenbrugge RJ, van Zwam WH, Dillon WP, Majoie CB, 2016. The Capillary Index Score as a Marker of Viable Cerebral Tissue: Proof of Concept-The Capillary Index Score in the MR CLEAN (Multicenter Randomized Clinical Trial of Endovascular Treatment for Acute Ischemic Stroke in the Netherlands) Trial. Stroke 47, 2286–2291. doi: 10.1161/STROKEAHA.116.013513 [DOI] [PubMed] [Google Scholar]
- Alkayed NJ, Birks EK, Narayanan J, Petrie KA, Kohler-Cabot AE, Harder DR, 1997. Role of P-450 arachidonic acid epoxygenase in the response of cerebral blood flow to glutamate in rats. Stroke 28, 1066–1072 [DOI] [PubMed] [Google Scholar]
- Alvestad S, Hammer J, Hoddevik EH, Skare O, Sonnewald U, Amiry-Moghaddam M, Ottersen OP, 2013. Mislocalization of AQP4 precedes chronic seizures in the kainate model of temporal lobe epilepsy. Epilepsy Res 105, 30–41. doi: 10.1016/j.eplepsyres.2013.01.006 [DOI] [PubMed] [Google Scholar]
- Anderson MA, Ao Y, Sofroniew MV, 2014. Heterogeneity of reactive astrocytes. Neurosci.Lett 565, 23–29. doi:S0304-3940(13)01111-7 [pii]; 10.1016/j.neulet.2013.12.030 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson MA, Burda JE, Ren Y, Ao Y, O’Shea TM, Kawaguchi R, Coppola G, Khakh BS, Deming TJ, Sofroniew MV, 2016. Astrocyte scar formation aids central nervous system axon regeneration. Nature 532, 195–200. doi: 10.1038/nature17623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andriezen WL, 1893. The neuroglia elements of the brain. British Medical Journal 2, 227–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anneser JM, Chahli C, Ince PG, Borasio GD, Shaw PJ, 2004. Glial proliferation and metabotropic glutamate receptor expression in amyotrophic lateral sclerosis. J Neuropathol Exp Neurol 63, 831–840 [DOI] [PubMed] [Google Scholar]
- Araque A, Carmignoto G, Haydon PG, Oliet SH, Robitaille R, Volterra A, 2014. Gliotransmitters travel in time and space. Neuron 81, 728–739. doi:S0896-6273(14)00105-6 [pii]; 10.1016/j.neuron.2014.02.007 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armulik A, Genove G, Mae M, Nisancioglu MH, Wallgard E, Niaudet C, He L, Norlin J, Lindblom R, Strittmatter K, Johansson BR, Betsholtz C, 2010. Pericytes regulate the blood-brain barrier. Nature 468, 557–561. doi: 10.1038/nature09522 [DOI] [PubMed] [Google Scholar]
- Aronica E, Sandau US, Iyer A, Boison D, 2013. Glial adenosine kinase--a neuropathological marker of the epileptic brain. Neurochem Int 63, 688–695. doi: 10.1016/j.neuint.2013.01.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aronica E, van Vliet EA, Mayboroda OA, Troost D, da Silva FH, Gorter JA, 2000. Upregulation of metabotropic glutamate receptor subtype mGluR3 and mGluR5 in reactive astrocytes in a rat model of mesial temporal lobe epilepsy. Eur.J.Neurosci 12, 2333–2344. doi:ejn131 [pii] [DOI] [PubMed] [Google Scholar]
- Attwell D, Buchan AM, Charpak S, Lauritzen M, Macvicar BA, Newman EA, 2010. Glial and neuronal control of brain blood flow. Nature 468, 232–243. doi:nature09613 [pii]; 10.1038/nature09613 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balbi M, Koide M, Wellman GC, Plesnila N, 2017. Inversion of neurovascular coupling after subarachnoid hemorrhage in vivo. J Cereb Blood Flow Metab 37, 3625–3634. doi: 10.1177/0271678X16686595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baltan S, Murphy SP, Danilov CA, Bachleda A, Morrison RS, 2011. Histone deacetylase inhibitors preserve white matter structure and function during ischemia by conserving ATP and reducing excitotoxicity. J Neurosci 31, 3990–3999. doi: 10.1523/JNEUROSCI.5379-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barres BA, 2008. The mystery and magic of glia: a perspective on their roles in health and disease. Neuron 60, 430–440. doi:S0896-6273(08)00886-6 [pii]; 10.1016/j.neuron.2008.10.013 [doi] [DOI] [PubMed] [Google Scholar]
- Bartnik BL, Sutton RL, Fukushima M, Harris NG, Hovda DA, Lee SM, 2005. Upregulation of pentose phosphate pathway and preservation of tricarboxylic acid cycle flux after experimental brain injury. J Neurotrauma 22, 1052–1065. doi: 10.1089/neu.2005.22.1052 [DOI] [PubMed] [Google Scholar]
- Baskaya MK, Rao AM, Dogan A, Donaldson D, Dempsey RJ, 1997. The biphasic opening of the blood-brain barrier in the cortex and hippocampus after traumatic brain injury in rats. Neurosci Lett 226, 33–36 [DOI] [PubMed] [Google Scholar]
- Baslow MH, and Guilfoyle DN, 2017. The Bimodal Nature of Neurovascular Coupling: Slow Tonic and Rapid Phasic Responses are Separately Controlled by Specific Astrocyte Metabotropic and Ionotropic Glutamate Receptors. Journal of Molecular and Genetic Medicine 11. doi: 10.4172/1747-0862.1000266 [DOI] [Google Scholar]
- Bayraktar OA, Fuentealba LC, Alvarez-Buylla A, Rowitch DH, 2015. Astrocyte development and heterogeneity. Cold Spring Harb Perspect Biol 7, a020362. doi: 10.1101/cshperspect.a020362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becerra-Calixto A, Cardona-Gomez GP, 2017. The Role of Astrocytes in Neuroprotection after Brain Stroke: Potential in Cell Therapy. Front Mol Neurosci 10, 88. doi: 10.3389/fnmol.2017.00088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begum G, Song S, Wang S, Zhao H, Bhuiyan MIH, Li E, Nepomuceno R, Ye Q, Sun M, Calderon MJ, Stolz DB, St Croix C, Watkins SC, Chen Y, He R, Shull GE, Sun D, 2018. Selective knockout of astrocytic Na(+) /H(+) exchanger isoform 1 reduces astrogliosis, BBB damage, infarction, and improves neurological function after ischemic stroke. Glia 66, 126–144. doi: 10.1002/glia.23232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biesecker KR, Srienc AI, Shimoda AM, Agarwal A, Bergles DE, Kofuji P, Newman EA, 2016. Glial Cell Calcium Signaling Mediates Capillary Regulation of Blood Flow in the Retina. J Neurosci 36, 9435–9445. doi: 10.1523/JNEUROSCI.1782-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boison D, Steinhauser C, 2018. Epilepsy and astrocyte energy metabolism. Glia 66, 1235–1243. doi: 10.1002/glia.23247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonder DE, McCarthy KD, 2014. Astrocytic Gq-GPCR-linked IP3R-dependent Ca2+ signaling does not mediate neurovascular coupling in mouse visual cortex in vivo. J.Neurosci 34, 13139–13150. doi:34/39/13139 [pii]; 10.1523/JNEUROSCI.2591-14.2014 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booth HDE, Hirst WD, Wade-Martins R, 2017. The Role of Astrocyte Dysfunction in Parkinson’s Disease Pathogenesis. Trends Neurosci 40, 358–370. doi: 10.1016/j.tins.2017.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner M, 2014. Role of GFAP in CNS injuries. Neurosci Lett 565, 7–13. doi: 10.1016/j.neulet.2014.01.055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burda JE, Bernstein AM, Sofroniew MV, 2015. Astrocyte roles in traumatic brain injury. Exp.Neurol doi:S0014-4886(15)00089-8 [pii]; 10.1016/j.expneurol.2015.03.020 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bushong EA, Martone ME, Ellisman MH, 2004. Maturation of astrocyte morphology and the establishment of astrocyte domains during postnatal hippocampal development. Int J Dev Neurosci 22, 73–86. doi: 10.1016/j.ijdevneu.2003.12.008 [DOI] [PubMed] [Google Scholar]
- Bushong EA, Martone ME, Jones YZ, Ellisman MH, 2002. Protoplasmic astrocytes in CA1 stratum radiatum occupy separate anatomical domains. J Neurosci 22, 183–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butt AM, Duncan A, Berry M, 1994. Astrocyte associations with nodes of Ranvier: ultrastructural analysis of HRP-filled astrocytes in the mouse optic nerve. J Neurocytol 23, 486–499 [DOI] [PubMed] [Google Scholar]
- Cajal S.R.y., 1995. Histology of the Nervous System of Man and Vertebrates. Oxford University Press, New York. [Google Scholar]
- Cattaneo A, Cattane N, Galluzzi S, Provasi S, Lopizzo N, Festari C, Ferrari C, Guerra UP, Paghera B, Muscio C, Bianchetti A, Volta GD, Turla M, Cotelli MS, Gennuso M, Prelle A, Zanetti O, Lussignoli G, Mirabile D, Bellandi D, Gentile S, Belotti G, Villani D, Harach T, Bolmont T, Padovani A, Boccardi M, Frisoni GB, Group, I.-F., 2017. Association of brain amyloidosis with pro-inflammatory gut bacterial taxa and peripheral inflammation markers in cognitively impaired elderly. Neurobiol Aging 49, 60–68. doi: 10.1016/j.neurobiolaging.2016.08.019 [DOI] [PubMed] [Google Scholar]
- Cauli B, Hamel E, 2010. Revisiting the role of neurons in neurovascular coupling. Front Neuroenergetics 2, 9. doi: 10.3389/fnene.2010.00009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen A, Akinyemi RO, Hase Y, Firbank MJ, Ndung’u MN, Foster V, Craggs LJ, Washida K, Okamoto Y, Thomas AJ, Polvikoski TM, Allan LM, Oakley AE, O’Brien JT, Horsburgh K, Ihara M, Kalaria RN, 2016. Frontal white matter hyperintensities, clasmatodendrosis and gliovascular abnormalities in ageing and post-stroke dementia. Brain 139, 242–258. doi: 10.1093/brain/awv328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Zhong X, Smith DK, Tai W, Yang J, Zou Y, Wang LL, Sun J, Qin S, Zhang CL, 2017. Astrocyte-Specific Deletion of Sox2 Promotes Functional Recovery After Traumatic Brain Injury. Cereb Cortex, 1–16. doi: 10.1093/cercor/bhx303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho TH, Nighoghossian N, Mikkelsen IK, Derex L, Hermier M, Pedraza S, Fiehler J, Ostergaard L, Berthezene Y, Baron JC, 2015. Reperfusion within 6 hours outperforms recanalization in predicting penumbra salvage, lesion growth, final infarct, and clinical outcome. Stroke 46, 1582–1589. doi: 10.1161/STROKEAHA.114.007964 [DOI] [PubMed] [Google Scholar]
- Chuquet J, Hollender L, Nimchinsky EA, 2007. High-resolution in vivo imaging of the neurovascular unit during spreading depression. J Neurosci 27, 4036–4044. doi: 10.1523/JNEUROSCI.0721-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cipolla MJ, Crete R, Vitullo L, Rix RD, 2004. Transcellular transport as a mechanism of blood-brain barrier disruption during stroke. Front Biosci 9, 777–785 [DOI] [PubMed] [Google Scholar]
- Clarke LE, Liddelow SA, Chakraborty C, Munch AE, Heiman M, Barres BA, 2018. Normal aging induces A1-like astrocyte reactivity. Proc Natl Acad Sci U SA 115, E1896–E1905. doi: 10.1073/pnas.1800165115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coomber BL, Stewart PA, 1985. Morphometric analysis of CNS microvascular endothelium. Microvasc Res 30, 99–115 [DOI] [PubMed] [Google Scholar]
- Cornford EM, Hyman S, Cornford ME, Caron MJ, 1996. Glutl glucose transporter activity in human brain injury. J Neurotrauma 13, 523–536. doi: 10.1089/neu.1996.13.523 [DOI] [PubMed] [Google Scholar]
- Corriveau RA, Bosetti F, Emr M, Gladman JT, Koenig JI, Moy CS, Pahigiannis K, Waddy SP, Koroshetz W, 2016. The Science of Vascular Contributions to Cognitive Impairment and Dementia (VCID): A Framework for Advancing Research Priorities in the Cerebrovascular Biology of Cognitive Decline. Cell Mol Neurobiol 36, 281–288. doi: 10.1007/s10571-016-0334-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullen KM, 1997. Perivascular astrocytes within Alzheimer’s disease plaques. Neuroreport 8, 1961–1966 [DOI] [PubMed] [Google Scholar]
- Daneman R, Barres BA, 2005. The blood-brain barrier-lessons from moody flies. Cell 123, 9–12. doi: 10.1016/j.cell.2005.09.017 [DOI] [PubMed] [Google Scholar]
- Daneman R, Prat A, 2015. The blood-brain barrier. Cold Spring Harb Perspect Biol 7, a020412. doi: 10.1101/cshperspect.a020412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daneman R, Zhou L, Kebede AA, Barres BA, 2010. Pericytes are required for blood-brain barrier integrity during embryogenesis. Nature 468, 562–566. doi: 10.1038/nature09513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies SJ, Goucher DR, Doller C, Silver J, 1999. Robust regeneration of adult sensory axons in degenerating white matter of the adult rat spinal cord. J Neurosci 19, 5810–5822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Torre JC, 1997. Hemodynamic consequences of deformed microvessels in the brain in Alzheimer’s disease. Ann N Y Acad Sci 826, 75–91 [DOI] [PubMed] [Google Scholar]
- de la Torre JC, Mussivand T, 1993. Can disturbed brain microcirculation cause Alzheimer’s disease? Neurol Res 15, 146–153 [DOI] [PubMed] [Google Scholar]
- de Leeuw FE, de Groot JC, Achten E, Oudkerk M, Ramos LM, Heijboer R, Hofman A, Jolles J, van Gijn J, Breteler MM, 2001. Prevalence of cerebral white matter lesions in elderly people: a population based magnetic resonance imaging study. The Rotterdam Scan Study. J Neurol Neurosurg Psychiatry 70, 9–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delekate A, Fuchtemeier M, Schumacher T, Ulbrich C, Foddis M, Petzold GC, 2014. Metabotropic P2Y1 receptor signalling mediates astrocytic hyperactivity in vivo in an Alzheimer’s disease mouse model. Nat Commun 5, 5422. doi: 10.1038/ncomms6422 [DOI] [PubMed] [Google Scholar]
- Di Malta C, Fryer JD, Settembre C, Ballabio A, 2012. Astrocyte dysfunction triggers neurodegeneration in a lysosomal storage disorder. Proc Natl Acad Sci U SA 109, E2334–2342. doi: 10.1073/pnas.1209577109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz-Garcia CM, Mongeon R, Lahmann C, Koveal D, Zucker H, Yellen G, 2017. Neuronal Stimulation Triggers Neuronal Glycolysis and Not Lactate Uptake. Cell Metab 26, 361–374 e364. doi: 10.1016/j.cmet.2017.06.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dienel GA, 2017. Lack of appropriate stoichiometry: Strong evidence against an energetically important astrocyte-neuron lactate shuttle in brain. J Neurosci Res 95, 2103–2125. doi: 10.1002/jnr.24015 [DOI] [PubMed] [Google Scholar]
- Ding S, Wang T, Cui W, Haydon PG, 2009. Photothrombosis ischemia stimulates a sustained astrocytic Ca2+ signaling in vivo. Glia 57, 767–776. doi: 10.1002/glia.20804 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiNuzzo M, Mangia S, Maraviglia B, Giove F, 2010. Glycogenolysis in astrocytes supports blood-borne glucose channeling not glycogen-derived lactate shuttling to neurons: evidence from mathematical modeling. J Cereb Blood Flow Metab 30, 1895–1904. doi: 10.1038/jcbfm.2010.151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreier JP, 2011. The role of spreading depression, spreading depolarization and spreading ischemia in neurological disease. Nat Med 17, 439–447. doi: 10.1038/nm.2333 [DOI] [PubMed] [Google Scholar]
- Dreier JP, Ebert N, Priller J, Megow D, Lindauer U, Klee R, Reuter U, Imai Y, Einhaupl KM, Victorov I, Dirnagl U, 2000. Products of hemolysis in the subarachnoid space inducing spreading ischemia in the cortex and focal necrosis in rats: a model for delayed ischemic neurological deficits after subarachnoid hemorrhage? J Neurosurg 93, 658–666. doi: 10.3171/jns.2000.93.4.0658 [DOI] [PubMed] [Google Scholar]
- Dreier JP, Korner K, Ebert N, Gorner A, Rubin I, Back T, Lindauer U, Wolf T, Villringer A, Einhaupl KM, Lauritzen M, Dirnagl U, 1998. Nitric oxide scavenging by hemoglobin or nitric oxide synthase inhibition by N-nitro-L-arginine induces cortical spreading ischemia when K+ is increased in the subarachnoid space. J Cereb Blood Flow Metab 18, 978–990. doi: 10.1097/00004647-199809000-00007 [DOI] [PubMed] [Google Scholar]
- Dreier JP, Lemale CL, Kola V, Friedman A, Schoknecht K, 2018. Spreading depolarization is not an epiphenomenon but the principal mechanism of the cytotoxic edema in various gray matter structures of the brain during stroke. Neuropharmacology 134, 189–207. doi: 10.1016/j.neuropharm.2017.09.027 [DOI] [PubMed] [Google Scholar]
- Eid T, Lee TS, Thomas MJ, Amiry-Moghaddam M, Bjornsen LP, Spencer DD, Agre P, Ottersen OP, de Lanerolle NC, 2005. Loss of perivascular aquaporin 4 may underlie deficient water and K+ homeostasis in the human epileptogenic hippocampus. Proc Natl Acad Sci U S A 102, 1193–1198. doi: 10.1073/pnas.0409308102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Amki M, Wegener S, 2017. Improving Cerebral Blood Flow after Arterial Recanalization: A Novel Therapeutic Strategy in Stroke. Int J Mol Sci 18. doi: 10.3390/ijms18122669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang B, McKeon A, Hinson SR, Kryzer TJ, Pittock SJ, Aksamit AJ, Lennon VA, 2016. Autoimmune Glial Fibrillary Acidic Protein Astrocytopathy: A Novel Meningoencephalomyelitis. JAMA Neurol 73, 1297–1307. doi: 10.1001/jamaneurol.2016.2549 [DOI] [PubMed] [Google Scholar]
- Farkas E, Luiten PG, 2001. Cerebral microvascular pathology in aging and Alzheimer’s disease. Prog Neurobiol 64, 575–611 [DOI] [PubMed] [Google Scholar]
- Farmer WT, Murai K, 2017. Resolving Astrocyte Heterogeneity in the CNS. Front Cell Neurosci 11, 300. doi: 10.3389/fncel.2017.00300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faulkner JR, Herrmann JE, Woo MJ, Tansey KE, Doan NB, Sofroniew MV, 2004. Reactive astrocytes protect tissue and preserve function after spinal cord injury. J.Neurosci. 24, 2143–2155. doi: 10.1523/JNEUROSCI.3547-03.2004 [doi];24/9/2143 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filosa JA, Bonev AD, Straub SV, Meredith AL, Wilkerson MK, Aldrich RW, Nelson MT, 2006. Local potassium signaling couples neuronal activity to vasodilation in the brain. Nat.Neurosci. 9, 1397–1403. doi:1779 [pii]; 10.1038/nn1779 [doi] [DOI] [PubMed] [Google Scholar]
- Filosa JA, Morrison HW, Iddings JA, Du W, Kim KJ, 2016. Beyond neurovascular coupling, role of astrocytes in the regulation of vascular tone. Neuroscience 323, 96–109. doi: 10.1016/j.neuroscience.2015.03.064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fordsmann JC, Ko RW, Choi HB, Thomsen K, Witgen BM, Mathiesen C, Lonstrup M, Piilgaard H, MacVicar BA, Lauritzen M, 2013. Increased 20-HETE synthesis explains reduced cerebral blood flow but not impaired neurovascular coupling after cortical spreading depression in rat cerebral cortex. J Neurosci 33, 2562–2570. doi: 10.1523/JNEUROSCI.2308-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritz E, Izaurieta P, Weiss A, Mir FR, Rojas P, Gonzalez D, Rojas E, Brown RH Jr., Madrid R, van Zundert B, 2013. Mutant SOD1-expressing astrocytes release toxic factors that trigger motoneuron death by inducing hyperexcitability. J Neurophysiol 109, 2803–2814. doi: 10.1152/jn.00500.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaertner RF, Wyss-Coray T, Von Euw D, Lesne S, Vivien D, Lacombe P, 2005. Reduced brain tissue perfusion in TGF-beta 1 transgenic mice showing Alzheimer’s disease-like cerebrovascular abnormalities. Neurobiol Dis 19, 38–46. doi: 10.1016/j.nbd.2004.11.008 [DOI] [PubMed] [Google Scholar]
- Garbuzova-Davis S, Rodrigues MC, Hernandez-Ontiveros DG, Tajiri N, Frisina-Deyo A, Boffeli SM, Abraham JV, Pabon M, Wagner A, Ishikawa H, Shinozuka K, Haller E, Sanberg PR, Kaneko Y, Borlongan CV, 2013. Blood-brain barrier alterations provide evidence of subacute diaschisis in an ischemic stroke rat model. PLoS One 8, e63553. doi: 10.1371/journal.pone.0063553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girouard H, Bonev AD, Hannah RM, Meredith A, Aldrich RW, Nelson MT, 2010. Astrocytic endfoot Ca2+ and BK channels determine both arteriolar dilation and constriction. Proc Natl Acad Sci U SA 107, 3811–3816. doi: 10.1073/pnas.0914722107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golding EM, Robertson CS, Bryan RM Jr., 1999. The consequences of traumatic brain injury on cerebral blood flow and autoregulation: a review. Clin Exp Hypertens 21,299–332 [DOI] [PubMed] [Google Scholar]
- Gomez-Arboledas A, Davila JC, Sanchez-Mejias E, Navarro V, Nunez-Diaz C, Sanchez-Varo R, Sanchez-Mico MV, Trujillo-Estrada L, Fernandez-Valenzuela JJ, Vizuete M, Cornelia JX, Galea E, Vitorica J, Gutierrez A, 2018. Phagocytic clearance of presynaptic dystrophies by reactive astrocytes in Alzheimer’s disease. Glia 66, 637–653. doi: 10.1002/glia.23270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodall EF, Wang C, Simpson JE, Baker DJ, Drew DR, Heath PR, Saffrey MJ, Romero IA, Wharton SB, 2017. Age-associated changes in the blood-brain barrier: comparative studies in human and mouse. Neuropathol Appl Neurobiol. doi: 10.1111/nan.12408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon GR, Choi HB, Rungta RL, Ellis-Davies GC, Macvicar BA, 2008. Brain metabolism dictates the polarity of astrocyte control over arterioles. Nature 456, 745–749. doi:nature07525 [pii]; 10.1038/nature07525 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouder N, Scheurer L, Fritschy JM, Boison D, 2004. Overexpression of adenosine kinase in epileptic hippocampus contributes to epileptogenesis. J Neurosci 24, 692–701. doi: 10.1523/JNEUROSCI.4781-03.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutierrez-Jimenez E, Angleys H, Rasmussen PM, West MJ, Catalini L, Iversen NK, Jensen MS, Frische S, Ostergaard L, 2018. Disturbances in the control of capillary flow in an aged APP(swe)/PS1DeltaE9 model of Alzheimer’s disease. Neurobiol Aging 62, 82–94. doi: 10.1016/j.neurobiolaging.2017.10.006 [DOI] [PubMed] [Google Scholar]
- Hall CN, Reynell C, Gesslein B, Hamilton NB, Mishra A, Sutherland BA, O’Farrell FM, Buchan AM, Lauritzen M, Attwell D, 2014. Capillary pericytes regulate cerebral blood flow in health and disease. Nature 508, 55–60. doi:nature13165 [pii]; 10.1038/nature13165 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamel E, 2006. Perivascular nerves and the regulation of cerebrovascular tone. J.Appl.Physiol 100, 1059–1064. doi:100/3/1059 [pii]; 10.1152/japplphysiol.00954.2005 [doi] [DOI] [PubMed] [Google Scholar]
- Hartings JA, Strong AJ, Fabricius M, Manning A, Bhatia R, Dreier JP, Mazzeo AT, Tortella FC, Bullock MR, Co-Operative Study of Brain Injury, D., 2009. Spreading depolarizations and late secondary insults after traumatic brain injury. J Neurotrauma 26, 1857–1866. doi:10.1089/neu.2009-096110.1089/neu.2009-096110.1089/neu.2009.096110.1089/neu.2009.0961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hase Y, Horsburgh K, Ihara M, Kalaria RN, 2017. White Matter Degeneration in Vascular and Other Ageing-Related Dementias. J Neurochem. doi: 10.1111/jnc.14271 [DOI] [PubMed] [Google Scholar]
- Hauck EF, Apostel S, Hoffmann JF, Heimann A, Kempski O, 2004. Capillary flow and diameter changes during reperfusion after global cerebral ischemia studied by intravital video microscopy. J.Cereb.Blood Flow Metab 24, 383–391. doi:00004647-200404000-00003 [pii]; 10.1097/00004647-200404000-00003 [doi] [DOI] [PubMed] [Google Scholar]
- Hay JR, Johnson VE, Young AM, Smith DH, Stewart W, 2015. Blood-Brain Barrier Disruption Is an Early Event That May Persist for Many Years After Traumatic Brain Injury in Humans. J Neuropathol Exp Neurol 74, 1147–1157. doi: 10.1097/NEN.0000000000000261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayakawa K, Pham LD, Katusic ZS, Arai K, Lo EH, 2012. Astrocytic high-mobility group box 1 promotes endothelial progenitor cell-mediated neurovascular remodeling during stroke recovery. Proc Natl Acad Sci U S A 109, 7505–7510. doi: 10.1073/pnas.1121146109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi Y, Homma K, Ichijo H, 2016. SOD1 in neurotoxicity and its controversial roles in SOD1 mutation-negative ALS. Adv Biol Regul 60, 95–104. doi: 10.1016/j.jbior.2015.10.006 [DOI] [PubMed] [Google Scholar]
- Heinemann U, Kaufer D, Friedman A, 2012. Blood-brain barrier dysfunction, TGFbeta signaling, and astrocyte dysfunction in epilepsy. Glia 60, 1251–1257. doi: 10.1002/glia.22311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heppner FL, Ransohoff RM, Becher B, 2015. Immune attack: the role of inflammation in Alzheimer disease. Nat Rev Neurosci 16, 358–372. doi: 10.1038/nrn3880 [DOI] [PubMed] [Google Scholar]
- Hinzman JM, Andaluz N, Shutter LA, Okonkwo DO, Pahl C, Strong AJ, Dreier JR, Hartings JA, 2014. Inverse neurovascular coupling to cortical spreading depolarizations in severe brain trauma. Brain 137, 2960–2972. doi: 10.1093/brain/awu241 [DOI] [PubMed] [Google Scholar]
- Holmes EG, Holmes BE, 1926. Contributions to the Study of Brain Metabolism: Carbohydrate Metabolism Relationship of Glycogen and Lactic Acid. Biochem J 20, 1196–1203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosford PS, Gourine AV, 2019. What is the key mediator of the neurovascular coupling response? Neurosci Biobehav Rev 96, 174–181. doi: 10.1016/j.neubiorev.2018.11.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howarth C, 2014. The contribution of astrocytes to the regulation of cerebral blood flow. Front Neurosci 8, 103. doi: 10.3389/fnins.2014.00103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howarth C, Gleeson P, Attwell D, 2012. Updated energy budgets for neural computation in the neocortex and cerebellum. J Cereb Blood Flow Metab 32, 1222–1232. doi: 10.1038/jcbfm.2012.35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang KL, Lin KJ, Ho MY, Chang YJ, Chang CH, Wey SP, Hsieh CJ, Yen TC, Hsiao IT, Lee TH, 2012. Amyloid deposition after cerebral hypoperfusion: evidenced on [(18)F]AV-45 positron emission tomography. J Neurol Sci 319, 124–129. doi: 10.1016/j.jns.2012.04.014 [DOI] [PubMed] [Google Scholar]
- Hubbard JA, Szu JI, Yonan JM, Binder DK, 2016. Regulation of astrocyte glutamate transporter-1 (GLT1) and aquaporin-4 (AQP4) expression in a model of epilepsy. Exp Neurol 283, 85–96. doi: 10.1016/j.expneurol.2016.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulse RE, Winterfield J, Kunkler PE, Kraig RP, 2001. Astrocytic clasmatodendrosis in hippocampal organ culture. Glia 33, 169–179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iadecola C, 2017. The Neurovascular Unit Coming of Age: A Journey through Neurovascular Coupling in Health and Disease. Neuron 96, 17–42. doi: 10.1016/j.neuron.2017.07.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Institoris A, Rosenegger DG, Gordon GR, 2015. Arteriole dilation to synaptic activation that is sub-threshold to astrocyte endfoot Ca2+ transients. J Cereb Blood Flow Metab 35, 1411–1415. doi: 10.1038/jcbfm.2015.141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito U, Hakamata Y, Kawakami E, Oyanagi K, 2011. Temporary [corrected] cerebral ischemia results in swollen astrocytic end-feet that compress microvessels and lead to delayed [corrected] focal cortical infarction. J Cereb Blood Flow Metab 31, 328–338. doi: 10.1038/jcbfm.2010.97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iturria-Medina Y, Sotero RC, Toussaint PJ, Mateos-Perez JM, Evans AC, Alzheimer’s Disease Neuroimaging, I., 2016. Early role of vascular dysregulation on late-onset Alzheimer’s disease based on multifactorial data-driven analysis. Nat Commun 7, 11934. doi: 10.1038/ncomms11934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarius S, Wildemann B, 2010. AQP4 antibodies in neuromyelitis optica: diagnostic and pathogenetic relevance. Nat Rev Neurol 6, 383–392. doi: 10.1038/nrneurol.2010.72 [DOI] [PubMed] [Google Scholar]
- Jego P, Pacheco-Torres J, Araque A, Canals S, 2014. Functional MRI in mice lacking IP3-dependent calcium signaling in astrocytes. J Cereb Blood Flow Metab 34, 1599–1603. doi: 10.1038/jcbfm.2014.144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji M, Miao Y, Dong LD, Chen J, Mo XF, Jiang SX, Sun XH, Yang XL, Wang Z, 2012. Group I mGluR-mediated inhibition of Kir channels contributes to retinal Muller cell gliosis in a rat chronic ocular hypertension model. J Neurosci 32, 12744–12755. doi: 10.1523/JNEUROSCI.1291-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang R, Diaz-Castro B, Looger LL, Khakh BS, 2016. Dysfunctional Calcium and Glutamate Signaling in Striatal Astrocytes from Huntington’s Disease Model Mice. J Neurosci 36, 3453–3470. doi: 10.1523/JNEUROSCI.3693-15.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X, Andjelkovic AV, Zhu L, Yang T, Bennett MVL, Chen J, Keep RF, Shi Y, 2018. Blood-brain barrier dysfunction and recovery after ischemic stroke. Prog Neurobiol 163-164, 144–171. doi: 10.1016/j.pneurobio.2017.10.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo S, Yarishkin O, Hwang YJ, Chun YE, Park M, Woo DH, Bae JY, Kim T, Lee J, Chun H, Park HJ, Lee DY, Hong J, Kim HY, Oh SJ, Park SJ, Lee H, Yoon BE, Kim Y, Jeong Y, Shim I, Bae YC, Cho J, Kowall NW, Ryu H, Hwang E, Kim D, Lee CJ, 2014. GABA from reactive astrocytes impairs memory in mouse models of Alzheimer’s disease. Nat Med 20, 886–896. doi: 10.1038/nm.3639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johshita H, Kassell NF, Sasaki T, Ogawa H, 1990. Impaired capillary perfusion and brain edema following experimental subarachnoid hemorrhage: a morphometric study. J Neurosurg 73, 410–417. doi: 10.3171/jns.1990.73.3.0410 [DOI] [PubMed] [Google Scholar]
- Joo IL, Lai AY, Bazzigaluppi P, Koletar MM, Dorr A, Brown ME, Thomason LA, Sled JG, McLaurin J, Stefanovic B, 2017. Early neurovascular dysfunction in a transgenic rat model of Alzheimer’s disease. Sci Rep 7, 46427. doi: 10.1038/srep46427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalaria RN, 2010. Vascular basis for brain degeneration: faltering controls and risk factors for dementia. Nutr Rev 68 Suppl 2, S74–87. doi: 10.1111/j.1753-4887.2010.00352.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karbowski J, 2007. Global and regional brain metabolic scaling and its functional consequences. BMC Biol 5, 18. doi: 10.1186/1741-7007-5-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelleher RJ, Soiza RL, 2013. Evidence of endothelial dysfunction in the development of Alzheimer’s disease: Is Alzheimer’s a vascular disorder? Am J Cardiovasc Dis 3, 197–226 [PMC free article] [PubMed] [Google Scholar]
- Kenney K, Amyot E, Haber M, Pronger A, Bogoslovsky T, Moore C, Diaz-Arrastia R, 2016. Cerebral Vascular Injury in Traumatic Brain Injury. Exp Neurol 275 Pt 3, 353–366. doi: 10.1016/j.expneurol.2015.05.019 [DOI] [PubMed] [Google Scholar]
- Khakh BS, Beaumont V, Cachope R, Munoz-Sanjuan I, Goldman SA, Grantyn R, 2017. Unravelling and Exploiting Astrocyte Dysfunction in Huntington’s Disease. Trends Neurosci 40, 422–437. doi: 10.1016/j.tins.2017.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KJ, Iddings JA, Stern JE, Blanco VM, Croom D, Kirov SA, Filosa JA, 2015. Astrocyte contributions to flow/pressure-evoked parenchymal arteriole vasoconstriction. J Neurosci 35, 8245–8257. doi: 10.1523/JNEUROSCI.4486-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim R, Healey KL, Sepulveda-Orengo MT, Reissner KJ, 2017. Astroglial correlates of neuropsychiatric disease: From astrocytopathy to astrogliosis. Prog Neuropsychopharmacol Biol Psychiatry. doi: 10.1016/j.pnpbp.2017.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimbrough IF, Robel S, Roberson ED, Sontheimer H, 2015. Vascular amyloidosis impairs the gliovascular unit in a mouse model of Alzheimer’s disease. Brain 138, 3716–3733. doi: 10.1093/brain/awv327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koehler RC, Gebremedhin D, Harder DR, 2006. Role of astrocytes in cerebrovascular regulation. J Appl Physiol (1985) 100, 307–317. doi: 10.1152/japplphysiol.00938.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koide M, Bonev AD, Nelson MT, Wellman GC, 2012. Inversion of neurovascular coupling by subarachnoid blood depends on large-conductance Ca2+-activated K+ (BK) channels. Proc.Natl.Acad.Sci.U.S.A 109, E1387–E1395. doi:1121359109 [pii]; 10.1073/pnas.1121359109 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koide M, Bonev AD, Nelson MT, Wellman GC, 2013. Subarachnoid blood converts neurally evoked vasodilation to vasoconstriction in rat brain cortex. Acta Neurochir.Suppl 115, 167–171. doi: 10.1007/978-3-7091-1192-5_32 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komori T, 1999. Tau-positive glial inclusions in progressive supranuclear palsy, corticobasal degeneration and Pick’s disease. Brain Pathol 9, 663–679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotliar K, Hauser C, Ortner M, Muggenthaler C, Diehl-Schmid J, Angermann S, Hapfelmeier A, Schmaderer C, Grimmer T, 2017. Altered neurovascular coupling as measured by optical imaging: a biomarker for Alzheimer’s disease. Sci Rep 7, 12906. doi: 10.1038/s41598-017-13349-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovacs GG, Lee VM, Trojanowski JQ, 2017. Protein astrogliopathies in human neurodegenerative diseases and aging. Brain Pathol 27, 675–690. doi: 10.1111/bpa.12536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krainik A, Hund-Georgiadis M, Zysset S, von Cramon DY, 2005. Regional impairment of cerebrovascular reactivity and BOLD signal in adults after stroke. Stroke 36, 1146–1152. doi: 10.1161/01.STR.0000166178.40973.a7 [DOI] [PubMed] [Google Scholar]
- Kuchibhotla KV, Lattarulo CR, Hyman BT, Bacskai BJ, 2009. Synchronous hyperactivity and intercellular calcium waves in astrocytes in Alzheimer mice. Science 323, 1211–1215. doi: 10.1126/science.1169096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kur J, Newman EA, 2014. Purinergic control of vascular tone in the retina. J Physiol 592, 491–504. doi: 10.1113/jphysiol.2013.267294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuruva CS, Reddy PH, 2017. Amyloid beta modulators and neuroprotection in Alzheimer’s disease: a critical appraisal. Drug Discov Today 22, 223–233. doi: 10.1016/j.drudis.2016.10.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lampl Y, Shmuilovich O, Lockman J, Sadeh M, Lorberboym M, 2005. Prognostic significance of blood brain barrier permeability in acute hemorrhagic stroke. Cerebrovasc Dis 20, 433–437. doi: 10.1159/000088981 [DOI] [PubMed] [Google Scholar]
- Lauritzen M, Dreier JP, Fabricius M, Hartings JA, Graf R, Strong AJ, 2011. Clinical relevance of cortical spreading depression in neurological disorders: migraine, malignant stroke, subarachnoid and intracranial hemorrhage, and traumatic brain injury. J Cereb Blood Flow Metab 31, 17–35. doi: 10.1038/jcbfm.2010.191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecrux C, Kocharyan A, Sandoe CH, Tong XK, Hamel E, 2012. Pyramidal cells and cytochrome P450 epoxygenase products in the neurovascular coupling response to basal forebrain cholinergic input. J Cereb Blood Flow Metab 32, 896–906. doi: 10.1038/jcbfm.2012.4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leffler CW, Beasley DG, Busija DW, 1989. Cerebral ischemia alters cerebral microvascular reactivity in newborn pigs. Am.J.Physiol 257, H266–H271 [DOI] [PubMed] [Google Scholar]
- Lenzlinger PM, Marx A, Trentz O, Kossmann T, Morganti-Kossmann MC, 2002. Prolonged intrathecal release of soluble Fas following severe traumatic brain injury in humans. J Neuroimmunol 122, 167–174 [DOI] [PubMed] [Google Scholar]
- Levine DA, Galecki AT, Langa KM, Unverzagt FW, Kabeto MU, Giordani B, Wadley VG, 2015. Trajectory of Cognitive Decline After Incident Stroke. JAMA 314, 41–51. doi: 10.1001/jama.2015.6968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Chen Y, Li B, Luo C, Zuo S, Liu X, Zhang JH, Ruan H, Feng H, 2016. Hemoglobin induced NO/cGMP suppression Deteriorate Microcirculation via Pericyte Phenotype Transformation after Subarachnoid Hemorrhage in Rats. Sci Rep 6, 22070. doi: 10.1038/srep22070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liddelow SA, Barres BA, 2017. Reactive Astrocytes: Production, Function, and Therapeutic Potential. Immunity 46, 957–967. doi: 10.1016/j.immuni.2017.06.006 [DOI] [PubMed] [Google Scholar]
- Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, Bennett ML, Munch AE, Chung WS, Peterson TC, Wilton DK, Frouin A, Napier BA, Panicker N , Kumar M, Buckwalter MS, Rowitch DH, Dawson VL, Dawson TM, Stevens B, Barres BA, 2017. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541, 481–487. doi: 10.1038/nature21029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebner S, Dijkhuizen RM, Reiss Y, Plate KH, Agalliu D, Constantin G, 2018. Functional morphology of the blood-brain barrier in health and disease. Acta Neuropathol 135, 311–336. doi: 10.1007/s00401-018-1815-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin WH, Hao Q, Rosengarten B, Leung WH, Wong KS, 2011. Impaired neurovascular coupling in ischaemic stroke patients with large or small vessel disease. Eur J Neurol 18, 731–736. doi: 10.1111/j.1468-1331.2010.03262.x [DOI] [PubMed] [Google Scholar]
- Lindauer U, Leithner C, Kaasch H, Rohrer B, Foddis M, Fuchtemeier M, Offenhauser N, Steinbrink J, Royl G, Kohl-Bareis M, Dirnagl U, 2010. Neurovascular coupling in rat brain operates independent of hemoglobin deoxygenation. J Cereb Blood Flow Metab 30, 757–768. doi: 10.1038/jcbfm.2009.259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Li C, Falck JR, Harder DR, Koehler RC, 2012. Relative contribution of cyclooxygenases, epoxyeicosatrienoic acids, and pH to the cerebral blood flow response to vibrissal stimulation. Am J Physiol Heart Circ Physiol 302, H1075–1085. doi: 10.1152/ajpheart.00794.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Li Y, Cui Y, Roberts C, Lu M, Wilhelmsson U, Pekny M, Chopp M, 2014. Beneficial effects of gfap/vimentin reactive astrocytes for axonal remodeling and motor behavioral recovery in mice after stroke. Glia 62, 2022–2033. doi: 10.1002/glia.22723 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logsdon AE, Lucke-Wold BP, Turner RC, Huber JD, Rosen CL, Simpkins JW, 2015. Role of Microvascular Disruption in Brain Damage from Traumatic Brain Injury. Compr Physiol 5, 1147–1160. doi: 10.1002/cphy.c140057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundgaard I, Osorio MJ, Kress BT, Sanggaard S, Nedergaard M, 2014. White matter astrocytes in health and disease. Neuroscience 276, 161–173. doi: 10.1016/j.neuroscience.2013.10.050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Major S, Petzold GC, Reiffurth C, Windmuller O, Foddis M, Lindauer U, Kang EJ, Dreier JR, 2017. A role of the sodium pump in spreading ischemia in rats. J Cereb Blood Flow Metab 37, 1687–1705. doi: 10.1177/0271678X16639059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandybur TI, Chuirazzi CC, 1990. Astrocytes and the plaques of Alzheimer’s disease. Neurology 40, 635–639 [DOI] [PubMed] [Google Scholar]
- Mathiisen TM, Lehre KR, Danbolt NC, Ottersen OP, 2010. The perivascular astroglial sheath provides a complete covering of the brain microvessels: an electron microscopic 3D reconstruction. Glia 58, 1094–1103. doi: 10.1002/glia.20990 [DOI] [PubMed] [Google Scholar]
- Mattsson N, Tosun D, Insel PS, Simonson A, Jack CR Jr., Beckett LA, Donohue M, Jagust W, Schuff N, Weiner MW, Alzheimer’s Disease Neuroimaging, I., 2014. Association of brain amyloid-beta with cerebral perfusion and structure in Alzheimer’s disease and mild cognitive impairment. Brain 137, 1550–1561. doi: 10.1093/brain/awu043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McConnell HL, Kersch CN, Woltjer RL, Neuwelt EA, 2017. The Translational Significance of the Neurovascular Unit. J Biol Chem 292, 762–770. doi: 10.1074/jbc.R116.760215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKee AC, Cairns NJ, Dickson DW, Folkerth RD, Keene CD, Litvan I, Perl DP, Stein TD, Vonsattel JP, Stewart W, Tripodis Y, Crary JF, Bieniek KF, Dams-O’Connor K, Alvarez VE, Gordon WA, group TC, 2016. The first NINDS/NIBIB consensus meeting to define neuropathological criteria for the diagnosis of chronic traumatic encephalopathy. Acta Neuropathol 131, 75–86. doi: 10.1007/s00401-015-1515-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menzel L, Kleber L, Friedrich C, Hummel R, Dangel L, Winter J, Schmitz K, Tegeder I, Schafer MK, 2017. Progranulin protects against exaggerated axonal injury and astrogliosis following traumatic brain injury. Glia 65, 278–292. doi: 10.1002/glia.23091 [DOI] [PubMed] [Google Scholar]
- Merali Z, Huang K, Mikulis D, Silver E, Kassner A, 2017. Evolution of blood-brain-barrier permeability after acute ischemic stroke. PLoS One 12, e0171558. doi: 10.1371/journal.pone.0171558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metea MR, Newman EA, 2006. Glial cells dilate and constrict blood vessels: a mechanism of neurovascular coupling. J.Neurosci. 26, 2862–2870. doi:26/11/2862 [pii]; 10.1523/JNEUROSCI.4048-05.2006 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer T, Munch C, Volkel H, Booms R, Ludolph AC, 1998. The EAAT2 (GLT-1) gene in motor neuron disease: absence of mutations in amyotrophic lateral sclerosis and a point mutation in patients with hereditary spastic paraplegia. J Neurol Neurosurg Psychiatry 65, 594–596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mignot C, Boespflug-Tanguy O, Gelot A, Dautigny A, Pham-Dinh D, Rodriguez D, 2004. Alexander disease: putative mechanisms of an astrocytic encephalopathy. Cell Mol Life Sci 61, 369–385. doi: 10.1007/s00018-003-3143-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra A, 2016. Binaural blood flow control by astrocytes: listening to synapses and the vasculature. J Physiol. doi: 10.1113/JP270979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra A, Hamid A, Newman EA, 2011. Oxygen modulation of neurovascular coupling in the retina. Proc.Natl.Acad.Sci.U.S.A 108, 17827–17831. doi:1110533108 [pii]; 10.1073/pnas.1110533108 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra A, Newman EA, 2010. Inhibition of inducible nitric oxide synthase reverses the loss of functional hyperemia in diabetic retinopathy. Glia 58, 1996–2004. doi: 10.1002/glia.21068 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra A, Reynolds JR, Chen Y, Gourine AV, Rusakov DA, Attwell D, 2016. Astrocytes mediate neurovascular signaling to capillary pericytes but not to arterioles. Nat Neurosci. doi: 10.1038/nn.4428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montagne A, Nation DA, Pa J, Sweeney MD, Toga AW, Zlokovic BV, 2016. Brain imaging of neurovascular dysfunction in Alzheimer’s disease. Acta Neuropathol 131, 687–707. doi: 10.1007/s00401-016-1570-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montagne A, Zhao Z, Zlokovic BV, 2017. Alzheimer’s disease: A matter of blood-brain barrier dysfunction? J Exp Med 214, 3151–3169. doi: 10.1084/jem.20171406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulligan SJ, Macvicar BA, 2004. Calcium transients in astrocyte endfeet cause cerebrovascular constrictions. Nature 431, 195–199. doi: 10.1038/nature02827 [doi];nature02827 [pii] [DOI] [PubMed] [Google Scholar]
- Nedergaard M, Hansen AJ, 1988. Spreading depression is not associated with neuronal injury in the normal brain. Brain Res 449, 395–398 [DOI] [PubMed] [Google Scholar]
- Newsome MR, Scheibel RS, Chu Z, Hunter JV, Li X, Wilde EA, Lu H, Wang ZJ, Lin X, Steinberg JL, Vasquez AC, Cook L, Levin HS, 2012. The relationship of resting cerebral blood flow and brain activation during a social cognition task in adolescents with chronic moderate to severe traumatic brain injury: a preliminary investigation. Int J Dev Neurosci 30, 255–266. doi: 10.1016/j.ijdevneu.2011.10.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolakakis N, Hamel E, 2011. Neurovascular function in Alzheimer’s disease patients and experimental models. J Cereb Blood Flow Metab 31, 1354–1370. doi: 10.1038/jcbfm.2011.43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu J, Li T, Yi C, Huang N, Koulakoff A, Weng C, Li C, Zhao CJ, Giaume C, Xiao L, 2016. Connexin-based channels contribute to metabolic pathways in the oligodendroglial lineage. J Cell Sci 129, 1902–1914. doi: 10.1242/jcs.178731 [DOI] [PubMed] [Google Scholar]
- Niwa K, Kazama K, Younkin SG, Carlson GA, ladecola C, 2002. Alterations in cerebral blood flow and glucose utilization in mice overexpressing the amyloid precursor protein. Neurobiol Dis 9, 61–68. doi: 10.1006/nbdi.2001.0460 [DOI] [PubMed] [Google Scholar]
- Nizar K, Uhlirova H, Tian R, Saisan PA, Cheng Q, Reznichenko L, Weldy KL, Steed TC, Sridhar VB, MacDonald CL, Cui J, Gratiy SL, Sakadzic S, Boas DA, Beka TI, Einevoll GT, Chen J, Masliah E, Dale AM, Silva GA, Devor A, 2013. In vivo stimulus-induced vasodilation occurs without IP3 receptor activation and may precede astrocytic calcium increase. J. Neurosci. 33, 8411–8422. doi:33/19/8411 [pii]; 10.1523/JNEUROSCI.3285-12.2013 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto Y, Yamamoto T, Kalaria RN, Senzaki H, Maki T, Hase Y, Kitamura A, Washida K , Yamada M, Ito H, Tomimoto H, Takahashi R, Ihara M, 2012. Cerebral hypoperfusion accelerates cerebral amyloid angiopathy and promotes cortical microinfarcts. Acta Neuropathol 123, 381–394. doi: 10.1007/s00401-011-0925-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olabarria M, Noristani HN, Verkhratsky A, Rodriguez JJ, 2011. Age-dependent decrease in glutamine synthetase expression in the hippocampal astroglia of the triple transgenic Alzheimer’s disease mouse model: mechanism for deficient glutamatergic transmission? Mol Neurodegener 6, 55. doi: 10.1186/1750-1326-6-55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otsu Y, Couchman K, Lyons DG, Collot M, Agarwal A, Mallet JM, Pfrieger FW, Bergles DE, Charpak S, 2015. Calcium dynamics in astrocyte processes during neurovascular coupling. Nat Neurosci 18, 210–218. doi: 10.1038/nn.3906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozen LJ, Fernandes MA, Clark AJ, Roy EA, 2015. Evidence of cognitive decline in older adults after remote traumatic brain injury: an exploratory study. Neuropsychol Dev Cogn B Aging Neuropsychol Cogn 22, 517–533. doi: 10.1080/13825585.2014.993584 [DOI] [PubMed] [Google Scholar]
- Pan R, Yu K, Weatherwax T, Zheng H, Liu W, Liu KJ, 2017. Blood Occludin Level as a Potential Biomarker for Early Blood Brain Barrier Damage Following Ischemic Stroke. Sci Rep 7, 40331. doi: 10.1038/srep40331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadeas ST, Kraig SE, O’Banion C, Lepore AC, Maragakis NJ, 2011. Astrocytes carrying the superoxide dismutase 1 (SOD1G93A) mutation induce wild-type motor neuron degeneration in vivo. Proc Natl Acad Sci U S A 108, 17803–17808. doi: 10.1073/pnas.1103141108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pappas AC, Koide M, Wellman GC, 2015. Astrocyte Ca2+ Signaling Drives Inversion of Neurovascular Coupling after Subarachnoid Hemorrhage. J Neurosci 35, 13375–13384. doi: 10.1523/JNEUROSCI.1551-15.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parodi L, Fenu S, Stevanin G, Durr A, 2017. Hereditary spastic paraplegia: More than an upper motor neuron disease. Rev Neurol (Paris) 173, 352–360. doi: 10.1016/j.neurol.2017.03.034 [DOI] [PubMed] [Google Scholar]
- Pekny M, Pekna M, Messing A, Steinhauser C, Lee JM, Parpura V, Hoi EM, Sofroniew MV, Verkhratsky A, 2016. Astrocytes: a central element in neurological diseases. Acta Neuropathol 131, 323–345. doi: 10.1007/s00401-015-1513-1 [DOI] [PubMed] [Google Scholar]
- Pellerin L, Magistretti PJ, 1994. Glutamate uptake into astrocytes stimulates aerobic glycolysis: a mechanism coupling neuronal activity to glucose utilization. Proc Natl Acad Sci U S A 91, 10625–10629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng W, Achariyar TM, Li B, Liao Y, Mestre H, Hitomi E, Regan S, Kasper T, Peng S, Ding E, Benveniste H, Nedergaard M, Deane R, 2016. Suppression of glymphatic fluid transport in a mouse model of Alzheimer’s disease. Neurobiol Dis 93, 215–225. doi: 10.1016/j.nbd.2016.05.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piao C, Ralay Ranaivo H, Rusie A, Wadhwani N, Koh S, Wainwright MS, 2015. Thrombin decreases expression of the glutamate transporter GLAST and inhibits glutamate uptake in primary cortical astrocytes via the Rho kinase pathway. Exp Neurol 273, 288–300. doi: 10.1016/j.expneurol.2015.09.009 [DOI] [PubMed] [Google Scholar]
- Piao CS, Holloway AL, Hong-Routson S, Wainwright MS, 2017. Depression following traumatic brain injury in mice is associated with down-regulation of hippocampal astrocyte glutamate transporters by thrombin. J Cereb Blood Flow Metab, 271678X17742792. doi: 10.1177/0271678X17742792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakers C, Petzold GC, 2017. Astrocytic calcium release mediates peri-infarct depolarizations in a rodent stroke model. J Clin Invest 127, 511–516. doi: 10.1172/JCI89354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rancillac A, Geoffroy H, Rossier J, 2012. Impaired neurovascular coupling in the APPxPSI mouse model of Alzheimer’s disease. Curr Alzheimer Res 9, 1221–1230 [DOI] [PubMed] [Google Scholar]
- Ratelade J, Verkman AS, 2012. Neuromyelitis optica: aquaporin-4 based pathogenesis mechanisms and new therapies. Int J Biochem Cell Biol 44, 1519–1530. doi: 10.1016/j.biocel.2012.06.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reese TS, Karnovsky MJ, 1967. Fine structural localization of a blood-brain barrier to exogenous peroxidase. J Cell Biol 34, 207–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reuler JB, Gardner EM, 1987. An inner-city clinic and house call experience for medical students. J Med Educ 62, 183–185 [DOI] [PubMed] [Google Scholar]
- Rodriguez-Arellano JJ, Parpura V, Zorec R, Verkhratsky A, 2015. Astrocytes in physiological aging and Alzheimer’s disease. Neuroscience. doi:S0306-4522(15)00031-7 [pii]; 10.1016/j.neuroscience.2015.01.007 [doi] [DOI] [PubMed] [Google Scholar]
- Rosenegger DG, Tran CH, Wamsteeker Cusulin JI, Gordon GR, 2015. Tonic Local Brain Blood Flow Control by Astrocytes Independent of Phasic Neurovascular Coupling. J Neurosci 35, 13463–13474. doi: 10.1523/JNEUROSCI.1780-15.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosengarten B, Paulsen S, Burr O, Kaps M, 2009. Neurovascular coupling in Alzheimer patients: effect of acetylcholine-esterase inhibitors. Neurobiol Aging 30, 1918–1923. doi: 10.1016/j.neurobiolaging.2008.02.017 [DOI] [PubMed] [Google Scholar]
- Rusnakova V, Honsa P, Dzamba D, Stahlberg A, Kubista M, Anderova M, 2013. Heterogeneity of astrocytes: from development to injury - single cell gene expression. PLoS One 8, e69734. doi: 10.1371/journal.pone.0069734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabbatini M, Barili P, Bronzetti E, Zaccheo D, Amenta E, 1999. Age-related changes of glial fibrillary acidic protein immunoreactive astrocytes in the rat cerebellar cortex. Mech Ageing Dev 108, 165–172 [DOI] [PubMed] [Google Scholar]
- Salinet AS, Robinson TG, Panerai RB, 2015. Effects of cerebral ischemia on human neurovascular coupling, CO2 reactivity, and dynamic cerebral autoregulation. J Appl Physiol (1985) 118, 170–177. doi: 10.1152/japplphysiol.00620.2014 [DOI] [PubMed] [Google Scholar]
- Salinet AS, Silva NC, Caldas J, deAzevedo DS, de-Lima-Oliveira M, Nogueira RC, Conforto AB, Texeira MJ, Robinson TG, Panerai RB, Bor-Seng-Shu E, 2018. Impaired cerebral autoregulation and neurovascular coupling in middle cerebral artery stroke: Influence of severity? J Cereb Blood Flow Metab, 271678X18794835. doi: 10.1177/0271678X18794835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarlus H, Heneka MT, 2017. Microglia in Alzheimer’s disease. J Clin Invest 127, 3240–3249. doi: 10.1172/JCI90606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savva GM, Stephan BC, Alzheimer’s Society Vascular Dementia Systematic Review, G., 2010. Epidemiological studies of the effect of stroke on incident dementia: a systematic review. Stroke 41, e41–46. doi: 10.1161/STROKEAHA.109.559880 [DOI] [PubMed] [Google Scholar]
- Schachtrup C, Ryu JK, Helmrick MJ, Vagena E, Galanakis DK, Degen JL, Margolis RU, Akassoglou K, 2010. Fibrinogen triggers astrocyte scar formation by promoting the availability of active TGF-beta after vascular damage. J Neurosci 30, 5843–5854. doi: 10.1523/JNEUROSCI.0137-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selwyn R, Hockenbury N, Jaiswal S, Mathur S, Armstrong RC, Byrnes KR, 2013. Mild traumatic brain injury results in depressed cerebral glucose uptake: An (18)FDG PET study. J Neurotrauma 30, 1943–1953. doi: 10.1089/neu.2013.2928 [DOI] [PubMed] [Google Scholar]
- Seymour RS, Bosiocic V, Snelling EP, 2016. Fossil skulls reveal that blood flow rate to the brain increased faster than brain volume during human evolution. R Soc Open Sci 3, 160305. doi: 10.1098/rsos.160305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinozaki Y, Shibata K, Yoshida K, Shigetomi E, Gachet C, Ikenaka K, Tanaka KF, Koizumi S, 2017. Transformation of Astrocytes to a Neuroprotective Phenotype by Microglia via P2Y1 Receptor Downregulation. Cell Rep 19, 1151–1164. doi: 10.1016/j.celrep.2017.04.047 [DOI] [PubMed] [Google Scholar]
- Simard M, Arcuino G, Takano T, Liu QS, Nedergaard M, 2003. Signaling at the gliovascular interface. J.Neurosci. 23, 9254–9262. doi:23/27/9254 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sims NR, Yew WP, 2017. Reactive astrogliosis in stroke: Contributions of astrocytes to recovery of neurological function. Neurochem Int 107, 88–103. doi: 10.1016/j.neuint.2016.12.016 [DOI] [PubMed] [Google Scholar]
- Skotte NH, Andersen JV, Santos A, Aldana BI, Willert CW, Norremolle A, Waagepetersen HS, Nielsen ML, 2018. Integrative Characterization of the R6/2 Mouse Model of Huntington’s Disease Reveals Dysfunctional Astrocyte Metabolism. Cell Rep 23, 2211–2224. doi: 10.1016/j.celrep.2018.04.052 [DOI] [PubMed] [Google Scholar]
- Snyder HM, Corriveau RA, Craft S, Faber JE, Greenberg SM, Knopman D, Lamb BT, Montine TJ, Nedergaard M, Schaffer CB, Schneider JA, Wellington C, Wilcock DM, Zipfel GJ, Zlokovic B, Bain LJ, Bosetti E, Galis ZS, Koroshetz W, Carrillo MC, 2015. Vascular contributions to cognitive impairment and dementia including Alzheimer’s disease. Alzheimers Dement 11, 710–717. doi: 10.1016/j.jalz.2014.10.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sofroniew MV, 2009. Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci. 32, 638–647. doi:S0166-2236(09)00153-2 [pii]; 10.1016/j.tins.2009.08.002 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sofroniew MV, 2014. Astrogliosis. Cold Spring Harb.Perspect.Biol. doi:cshperspect.a020420 [pii]; 10.1101/cshperspect.a020420 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sosunov A, Olabarria M, Goldman JE, 2018. Alexander disease: an astrocytopathy that produces a leukodystrophy. Brain Pathol 28, 388–398. doi: 10.1111/bpa.12601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sours C, Zhuo J, Roys S, Shanmuganathan K, Gullapalli RP, 2015. Disruptions in Resting State Functional Connectivity and Cerebral Blood Flow in Mild Traumatic Brain Injury Patients. PLoS One 10, e0134019. doi: 10.1371/journal.pone.0134019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soustiel JF, Sviri GE, 2007. Monitoring of cerebral metabolism: non-ischemic impairment of oxidative metabolism following severe traumatic brain injury. Neurol Res 29, 654–660. doi: 10.1179/016164107X240017 [DOI] [PubMed] [Google Scholar]
- Srinivasan R, Huang BS, Venugopal S, Johnston AD, Chai H, Zeng H, Golshani P, Khakh BS, 2015. Ca(2+) signaling in astrocytes from Ip3r2(−/−) mice in brain slices and during startle responses in vivo. Nat.Neurosci. 18, 708–717. doi:nn.4001 [pii]; 10.1038/nn.4001 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suehiro E, Fujisawa H, Akimura T, Ishihara H, Kajiwara K, Kato S, Fujii M, Yamashita S, Maekawa T, Suzuki M, 2004. Increased matrix metalloproteinase-9 in blood in association with activation of interleukin-6 after traumatic brain injury: influence of hypothermic therapy. J Neurotrauma 21, 1706–1711. doi: 10.1089/neu.2004.21.1706 [DOI] [PubMed] [Google Scholar]
- Tachibana M, Ago T, Wakisaka Y, Kuroda J, Shijo M, Yoshikawa Y, Komori M, Nishimura A, Makihara N, Nakamura K, Kitazono T, 2017. Early Reperfusion After Brain Ischemia Has Beneficial Effects Beyond Rescuing Neurons. Stroke 48, 2222–2230. doi: 10.1161/STROKEAHA.117.016689 [DOI] [PubMed] [Google Scholar]
- Taipa R, Ferreira V, Brochado R, Robinson A, Reis I, Marques E, Mann DM, Melo-Pires M, Sousa N, 2017. Inflammatory pathology markers (activated microglia and reactive astrocytes) in early and late onset Alzheimer disease: a post mortem study. Neuropathol Appl Neurobiol. doi: 10.1111/nan.12445 [DOI] [PubMed] [Google Scholar]
- Takano T, Tian GF, Peng W, Lou N, Libionka W, Han X, Nedergaard M, 2006. Astrocyte-mediated control of cerebral blood flow. Nat.Neurosci 9, 260–267. doi:nn1623 [pii]; 10.1038/nn1623 [doi] [DOI] [PubMed] [Google Scholar]
- Takata N, Nagai T, Ozawa K, Oe Y, Mikoshiba K, Hirase H, 2013. Cerebral blood flow modulation by Basal forebrain or whisker stimulation can occur independently of large cytosolic Ca2+ signaling in astrocytes. PLoS One 8, e66525. doi: 10.1371/journal.pone.0066525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamaki K, Sadoshima S, Baumbach GL, ladecola C, Reis DJ, Heistad DD, 1984. Evidence that disruption of the blood-brain barrier precedes reduction in cerebral blood flow in hypertensive encephalopathy. Hypertension 6, 175–81 [DOI] [PubMed] [Google Scholar]
- Tarantini S, Fulop GA, Kiss T, Farkas E, Zolei-Szenasi D, Galvan V, Toth P, Csiszar A, Ungvari Z, Yabluchanskiy A, 2017. Demonstration of impaired neurovascular coupling responses in TG2576 mouse model of Alzheimer’s disease using functional laser speckle contrast imaging. Geroscience. doi: 10.1007/s11357-017-9980-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarumi T, Dunsky DI, Khan MA, Liu J, Hill C, Armstrong K, Martin-Cook K, Cullum CM, Zhang R, 2014. Dynamic cerebral autoregulation and tissue oxygenation in amnestic mild cognitive impairment. J Alzheimers Dis 41, 765–778. doi: 10.3233/JAD-132018 [DOI] [PubMed] [Google Scholar]
- Thomas AJ, Perry R, Barber R, Kalaria RN, O’Brien JT, 2002. Pathologies and pathological mechanisms for white matter hyperintensities in depression. Ann N Y Acad Sci 977, 333–339 [DOI] [PubMed] [Google Scholar]
- Toth P, Szarka N, Farkas E, Ezer E, Czeiter E, Amrein K, Ungvari Z, Hartings JA, Buki A, Koller A, 2016. Traumatic brain injury-induced autoregulatory dysfunction and spreading depression-related neurovascular uncoupling: Pathomechanisms, perspectives, and therapeutic implications. Am J Physiol Heart Circ Physiol 311, H1118–H1131. doi: 10.1152/ajpheart.00267.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzeng YC, Ainslie PN, 2014. Blood pressure regulation IX: cerebral autoregulation under blood pressure challenges. Eur JAppI Physiol 114, 545–559. doi: 10.1007/s00421-013-2667-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Underly RG, Levy M, Hartmann DA, Grant RI, Watson AN, Shih AY, 2017. Pericytes as Inducers of Rapid, Matrix Metalloproteinase-9-Dependent Capillary Damage during Ischemia. J Neurosci 37, 129–140. doi: 10.1523/JNEUROSCI.2891-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaucher E, Borredon J, Bonvento G, Seylaz J, Lacombe P, 1997. Autoradiographic evidence for flow-metabolism uncoupling during stimulation of the nucleus basalis of Meynert in the conscious rat. J Cereb Blood Flow Metab 17, 686–694. doi: 10.1097/00004647-199706000-00010 [DOI] [PubMed] [Google Scholar]
- Veenith TV, Carter EL, Geeraerts T, Grossac J, Newcombe VF, Outtrim J, Gee GS, Lupson V, Smith R, Aigbirhio FI, Fryer TD, Hong YT, Menon DK, Coles JR, 2016. Pathophysiologic Mechanisms of Cerebral Ischemia and Diffusion Hypoxia in Traumatic Brain Injury. JAMA Neurol 73, 542–550. doi: 10.1001/jamaneurol.2016.0091 [DOI] [PubMed] [Google Scholar]
- Ventura R, Harris KM, 1999. Three-dimensional relationships between hippocampal synapses and astrocytes. J.Neurosci 19, 6897–6906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verkhratsky A, Nedergaard M, 2018. Physiology of Astroglia. Physiol Rev 98, 239–389. doi: 10.1152/physrev.00042.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermeer SE, Den Heijer T, Koudstaal PJ, Oudkerk M, Hofman A, Breteler MM, Rotterdam Scan S, 2003. Incidence and risk factors of silent brain infarcts in the population-based Rotterdam Scan Study. Stroke 34, 392–396 [DOI] [PubMed] [Google Scholar]
- Vetri E, Xu H, Mao L, Paisansathan C, Pelligrino DA, 2011. ATP hydrolysis pathways and their contributions to pial arteriolar dilation in rats. Am J Physiol Heart Circ Physiol 301, H1369–1377. doi: 10.1152/ajpheart.00556.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villapol S, Byrnes KR, Symes AJ, 2014. Temporal dynamics of cerebral blood flow, cortical damage, apoptosis, astrocyte-vasculature interaction and astrogliosis in the pericontusional region after traumatic brain injury. Front Neurol 5, 82. doi: 10.3389/fneur.2014.00082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villringer K, Sanz Cuesta BE, Ostwaldt AC, Grittner U, Brunecker P, Khalil AA, Schindler K, Eisenblatter O, Audebert H, Fiebach JB, 2017. DCE-MRI blood-brain barrier assessment in acute ischemic stroke. Neurology 88, 433–440. doi: 10.1212/WNL.0000000000003566 [DOI] [PubMed] [Google Scholar]
- Wagner KR, Sharp FR, Ardizzone TD, Lu A, Clark JF, 2003. Heme and iron metabolism: role in cerebral hemorrhage. J Cereb Blood Flow Metab 23, 629–652. doi: 10.1097/01.WCB.0000073905.87928.6D [DOI] [PubMed] [Google Scholar]
- Weissberg I, Wood L, Kamintsky L, Vazquez O, Milikovsky DZ, Alexander A, Oppenheim H, Ardizzone C, Becker A, Frigerio F, Vezzani A, Buckwalter MS, Huguenard JR, Friedman A, Kaufer D, 2015. Albumin induces excitatory synaptogenesis through astrocytic TGF-beta/ALK5 signaling in a model of acquired epilepsy following blood-brain barrier dysfunction. Neurobiol Dis 78, 115–125. doi: 10.1016/j.nbd.2015.02.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells JA, Christie IN, Hosford PS, Huckstepp RT, Angelova PR, Vihko P, Cork SC, Abramov AY, Teschemacher AG, Kasparov S, Lythgoe MF, Gourine AV, 2015. A critical role for purinergic signalling in the mechanisms underlying generation of BOLD fMRI responses. J Neurosci 35, 5284–5292. doi: 10.1523/JNEUROSCI.3787-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcock DM, Vitek MP, Colton CA, 2009. Vascular amyloid alters astrocytic water and potassium channels in mouse models and humans with Alzheimer’s disease. Neuroscience 159, 1055–1069. doi: 10.1016/j.neuroscience.2009.01.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyss-Coray T, Lin C, Sanan DA, Mucke L, Masliah E, 2000a. Chronic overproduction of transforming growth factor-betal by astrocytes promotes Alzheimer’s disease-like microvascular degeneration in transgenic mice. Am J Pathol 156, 139–150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyss-Coray T, Lin C, von Euw D, Masliah E, Mucke L, Lacombe P, 2000b. Alzheimer’s disease-like cerebrovascular pathology in transforming growth factor-beta 1 transgenic mice and functional metabolic correlates. Ann N Y Acad Sci 903, 317–323 [DOI] [PubMed] [Google Scholar]
- Wyss-Coray T, Loike JD, Brionne TC, Lu E, Anankov R, Yan E, Silverstein SC, Husemann J, 2003. Adult mouse astrocytes degrade amyloid-beta in vitro and in situ. Nat Med 9, 453–457. doi: 10.1038/nm838 [DOI] [PubMed] [Google Scholar]
- Wyss-Coray T, Masliah E, Mallory M, McConlogue L, Johnson-Wood K, Lin C, Mucke L, 1997. Amyloidogenic role of cytokine TGF-beta1 in transgenic mice and in Alzheimer’s disease. Nature 389, 603–606. doi: 10.1038/39321 [DOI] [PubMed] [Google Scholar]
- Xiang J, Tang Y, Li C, Su EJ, Lawrence DA, Keep RF, 2016. Mechanisms Underlying Astrocyte Endfeet Swelling in Stroke. Acta Neurochir Suppl 121, 19–22. doi: 10.1007/978-3-319-18497-5_4 [DOI] [PubMed] [Google Scholar]
- Xiao M, Li Q, Feng H, Zhang L, Chen Y, 2017. Neural Vascular Mechanism for the Cerebral Blood Flow Autoregulation after Hemorrhagic Stroke. Neural Plast 2017, 5819514. doi: 10.1155/2017/5819514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu HL, Pelligrino DA, 2007. ATP release and hydrolysis contribute to rat pial arteriolar dilatation elicited by neuronal activation. Exp Physiol 92, 647–651. doi: 10.1113/expphysiol.2006.036863 [DOI] [PubMed] [Google Scholar]
- Yan BC, Xu R, Gao M, Wang J, Jiang D, Zhu X, Won MH, Su PQ, 2018. Changes in the Blood-Brain Barrier Function Are Associated With Hippocampal Neuron Death in a KainicAcid Mouse Model of Epilepsy. Front Neurol 9, 775. doi: 10.3389/fneur.2018.00775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Wong A, Wang Z, Liu W, Au L, Xiong Y, Chu WW, Leung EY, Chen S, Lau C, Chan AY, Lau AY, Fan F, Ip V, Soo Y, Leung T, Ho CL, Wong LK, Mok VC, 2015. Risk factors for incident dementia after stroke and transient ischemic attack. Alzheimers Dement 11, 16–23. doi: 10.1016/j.jalz.2014.01.003 [DOI] [PubMed] [Google Scholar]
- Yuan XQ, Prough DS, Smith TL, Dewitt DS, 1988. The effects of traumatic brain injury on regional cerebral blood flow in rats. J Neurotrauma 5, 289–301. doi: 10.1089/neu.1988.5.289 [DOI] [PubMed] [Google Scholar]
- Zamanian JL, Xu L, Foo LC, Nouri N, Zhou L, Giffard RG, Barres BA, 2012. Genomic analysis of reactive astrogliosis. J.Neurosci 32, 6391–6410. doi:32/18/6391 [pii]; 10.1523/JNEUROSCI.6221-11.2012 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zambenedetti R, Schmitt HP, Zatta R, 2002. Metallothionein I-II immunocytochemical reactivity in Binswanger’s encephalopathy. J Alzheimers Dis 4, 459–466 [DOI] [PubMed] [Google Scholar]
- Zekeridou A, McKeon A, Flanagan EP, 2018. A path to understanding autoimmune GFAP astrocytopathy. Eur J Neurol 25, 421–422. doi: 10.1111/ene.13527 [DOI] [PubMed] [Google Scholar]
- Zetterberg H, Andreasen N, Blennow K, 2004. Increased cerebrospinal fluid levels of transforming growth factor-betal in Alzheimer’s disease. Neurosci Lett 367, 194–196. doi: 10.1016/j.neulet.2004.06.001 [DOI] [PubMed] [Google Scholar]
- Zhang X, Zhou K, Wang R, Cui J, Lipton SA, Liao FF, Xu H, Zhang YW, 2007. Hypoxia-inducible factor 1alpha (HIF-1alpha)-mediated hypoxia increases BACE1 expression and beta-amyloid generation. J Biol Chem 282, 10873–10880. doi: 10.1074/jbc.M608856200 [DOI] [PubMed] [Google Scholar]
- Zlokovic BV, 2002. Vascular disorder in Alzheimer’s disease: role in pathogenesis of dementia and therapeutic targets. Adv Drug Deliv Rev 54, 1553–1559 [DOI] [PubMed] [Google Scholar]
- Zonta M, Angulo MC, Gobbo S, Rosengarten B, Hossmann KA, Pozzan T, Carmignoto G, 2003. Neuron-to-astrocyte signaling is central to the dynamic control of brain microcirculation Nat. Neurosci. 6, 43–50. doi: 10.1038/nn980 [doi];nn980 [pii] [DOI] [PubMed] [Google Scholar]