

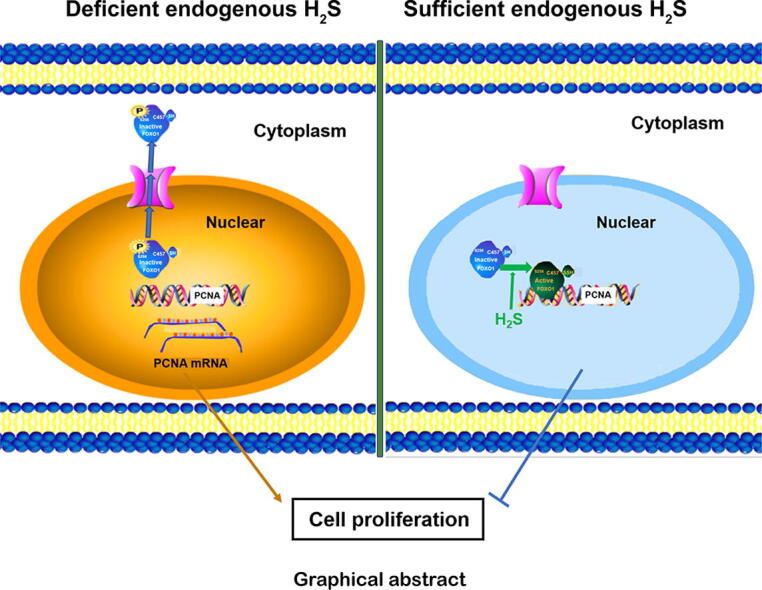
Graphical abstract
Schematic diagram of the mechanism by which H2S inhibits VSMC proliferation targeting on the FOXO1. H2S persulfidates FOXO1 at Cys457, which reduces FOXO1 phosphorylation at Ser256, prevents the FOXO1 nucleus exclusion to the cytoplasm, and further protecting against ET-1-induced VSMC proliferation.
Keywords: FOXO1, Persulfidation, Hydrogen sulfide, Vascular smooth muscle cell, Proliferation
Highlights
-
•
FOXO1 is involved in the inhibitory effect of H2S on vascular smooth muscle cell proliferation.
-
•
H2S inhibits vascular smooth muscle cell proliferation by maintaining FOXO1 activity.
-
•
H2S preserves FOXO1 activity by persulfidation.
-
•
H2S persulfidates FOXO1 at Cys457 and subsequently prevents FOXO1 phosphorylation at Ser256.
-
•
The results provide new ideas for therapeutic strategies for anti-vascular remodeling.
Abstract
Introduction
The proliferation of vascular smooth muscle cells (VSMCs) is an important physiological and pathological basis for many cardiovascular diseases. Endogenous hydrogen sulfide (H2S), the third gasotransmitter, is found to preserve vascular structure by inhibiting VSMC proliferation. However, the mechanism by which H2S suppresses VSMC proliferation has not been fully clear.
Objectives
This study aimed to explore whether H2S persulfidates the transcription factor FOXO1 to inhibit VSMC proliferation.
Methods
After the proliferation of VSMC A7r5 cells was induced by endothelin-1 (ET-1), FOXO1 phosphorylation and proliferating cell nuclear antigen (PCNA) expression were detected by Western blotting, the degree of FOXO1 nuclear exclusion and PCNA fluorescent signals in the nucleus were detected by immunofluorescence, and the persulfidation of FOXO1 was measured through a biotin switch assay.
Results
The results showed that ET-1 stimulation increased cell proliferation, FOXO1 phosphorylation and FOXO1 nuclear exclusion to the cytoplasm in the cells. However, pretreatment with NaHS, an H2S donor, successfully abolished the ET-1-induced increases in the VSMC proliferation, FOXO1 phosphorylation, and FOXO1 nuclear exclusion to the cytoplasm. Mechanistically, H2S persulfidated the FOXO1 protein in A7r5 and 293T cells, and the thiol reductant DTT reversed this effect. Furthermore, the C457S mutation of FOXO1 abolished the H2S-induced persulfidation of FOXO1 in the cells and the subsequent inhibitory effects on FOXO1 phosphorylation at Ser256, FOXO1 nuclear exclusion to the cytoplasm and cell proliferation.
Conclusion
Thus, our findings demonstrated that H2S might inhibit VSMC proliferation by persulfidating FOXO1 at Cys457 and subsequently preventing FOXO1 phosphorylation at Ser256.
Introduction
Vascular smooth muscle cells (VSMCs) are not only a key component of the medial layer of blood vessels but also an important metabolic and endocrine organ of the body. The excessive proliferation of VSMCs participates in the pathogenesis of a variety of vascular injury diseases, such as atherosclerosis and hypertension [1]. Therefore, the inhibition of VSMC proliferation has become a therapeutic target for many cardiovascular diseases, and thus, it is important to explore the molecular mechanisms responsible for VSMC proliferation. Endogenous hydrogen sulfide (H2S) is believed to act as the third gasotransmitter. Cystathionine-γ-lyase (CSE) is one of the key enzymes responsible for the main production of H2S in the cardiovascular system [2], [3]. H2S exerts a variety of cardiovascular protective effects, such as inhibiting the proliferation of VSMCs and collagen synthesis, mediating the relaxation of blood vessels and lowering blood pressure. [4], [5], [6]. Du et al. discovered that H2S can inhibit VSMC proliferation via the MAPK pathway [7]. Other studies have confirmed that H2S suppresses human pulmonary artery smooth muscle cell proliferation by increasing the expression of COX-2/PGI2 [8]. It has also been reported that the VSMCs isolated from CSE-knockout mice exhibit increased proliferation, which involves the cell cycle-related controllers p21Cip/WAF-1 and cyclin D, compared with those isolated from WT mice [4]. Although previous studies have attempted to explore the pathways involved in the H2S-mediated inhibition of VSMC proliferation, the mechanisms are not entirely understood. Recent studies have shown that the transcription factor forkhead box transcription factor (FOXO) is widely involved in various cell behaviors, such as the cell cycle, cell proliferation, apoptosis and responses to antioxidative stress. In mammals, the FOXO family mainly includes the FOXO1, FOXO3a, FOXO4, and FOXO6 subtypes [9], [10], [11], [12], and among these subtypes, FOXO1 can be expressed in the cardiovascular system and can inhibit cell proliferation [13]. In our previous studies, we demonstrated that H2S promotes the vasorelaxation of aortae isolated from spontaneously hypertensive rats by inhibiting the phosphorylation of FOXO1 and then stimulating the expression of the subunit KATP channel SUR2B and Kir6.1 [14]. Majumder et al. found that H2S mitigates mesangial cell apoptosis and remodeling of the matrix via the Akt/FOXO1 pathway [15]. A recent report suggested that FOXO1 downregulates cyclin B1, cyclin D1, and PCNA but upregulates cyclin p27 in pulmonary VSMCs [16]. FOXO1 also decreases the number of PCNA-positive osteoblast cells [17]. The above-mentioned studies suggest that FOXO1 is closely related to cell proliferation. Hence, we hypothesized that H2S inhibits VSMC proliferation by targeting FOXO1. Therefore, in the present study, we attempted to explore whether H2S inhibits VSMC proliferation by targeting FOXO1 and the related mechanisms.
Materials and methods
A7r5 and 293T cell culture and grouping
Rat VSMCs (A7r5 cell line) and 293T cells were purchased from National Infrastructure of Cell Line Resource, China. The cells were grown and cultured in Dulbecco’s modified Eagle’s medium (DMEM) (Gibco, USA) containing 10% FBS at 37 °C in an atmosphere with 5% CO2. The VSMCs were grouped into the control, ET-1, NaHS and ET-1 + NaHS groups. The cells in the ET-1 and ET-1 + NaHS groups were treated with 100 nmol/L ET-1 for 72 h, whereas those in the NaHS and ET-1 + NaHS groups were pretreated with 100 µmol/L NaHS for 30 min. For the mechanistic studies, the VSMCs and 293T cells were grouped into the control, NaHS and NaHS + DTT groups.
Plasmid transfection
Wild-type (WT) pENTER-FOXO1, S256A (mutation of Ser256 to alanine) pENTER-FOXO1 and C457S (mutation of Cys457 to Ser) pENTER-FOXO1 plasmids with His and Flag tags at their C termini were purchased from Vigene Biosciences (Shandong, China). When the VSMCs and 293T cells had grown to approximately 60% to 70% confluence, the above three plasmids were transfected with jetPEI™ reagent (Polyplus-transfection, France) for 24 h prior to treatment.
Cell proliferation assay
Cell proliferation was evaluated using the CCK-8 assay and by counting the cell number. 1) The CCK-8 assay is based on a water-soluble tetrazolium salt, which is reduced by dehydrogenase in the mitochondria and thereby converted to formazan dye. The latter has an absorbance at 450 nm, and the optical density is positively related to the number of viable cells. In this study, a commercial CCK-8 kit (Beyotime, China) was used according to the manufacturer’s recommended protocol. Briefly, A7r5 cells were seeded into 96-well plates and incubated overnight. After treatment, the CCK-8 working solution (10 µL) was added to the wells, and the plates were then incubated for 4 h at 37 °C. A microplate reader (Thermo, USA) was subsequently used to measure the absorbance at 450 nm. 2) Cell proliferation was also analyzed by counting the cell number. Equal numbers of A7r5 cells were plated into six-well plates and incubated overnight. After the treatments, the cells were isolated with 0.25% trypsin solution and loaded into the counting slide (Bio-Rad, USA). The slide was inserted into the TC10 automated cell counter (Bio-Rad, USA), and the cell number was then counted automatically.
Quantitative reverse transcription polymerase chain reaction
The PCNA mRNA levels were detected by real-time quantitative reverse transcription polymerase chain reaction (qRT-PCR) using a commercial qPCR kit (Promega, USA). Total RNA was extracted and reverse transcribed as previously reported [14]. The PCR amplification program was the following: 95 °C for 10 min followed by 40 cycles of 95 °C for 15 s and 60 °C for 1 min. The final results were analyzed using the formula 2−ΔΔCT with β-actin mRNA as an internal reference. The primers for PCNA were GGAGCTTGGCAATGGGAACA (forward) and GCAAAAGTTAGCTGAACTGGCT (reverse), and those for β-actin were ACCCGCGAGTACAACCTTCTT (forward) and TATCGTCATCCATGGCGAACT (reverse).
Western blotting
After treatment, the cells were lysed, and total protein was collected. The collected protein was loaded into SDS polyacrylamide gels and spotted onto nitrocellulose (NC) membranes, and nonspecific binding was blocked with 5% milk. The NC bands were separately incubated at 4 °C for 12 h with the following primary antibodies: PCNA antibody (1:1000; Anbo Biotechnology, USA), FOXO1 antibody (1:1000; Abcam, USA), p-FOXO1 antibody (1:500; CST, USA), His antibody (1:1000; ZSGB-BIO, China), Flag antibody (1:1000; ZSGB-BIO, China), GAPDH antibody (1:5000; Kangcheng, China) and β-actin antibody (1:5000; Santa Cruz, USA). The bands were then washed and incubated with the corresponding secondary antibodies (1:3000; CST, USA) for approximately 1 h. The immunoreactions were detected with a chemiluminescence detection reagent (GE Healthcare, UK).
Biotin switch analysis
The persulfidation of FOXO1 was detected through a biotin switch analysis developed by Zhang et al. in 2018 [18]. Briefly, total protein from the VSMCs and 293T cells was extracted in lysis buffer with protease inhibitors, and the lysates were centrifuged at 13,000 g and 4 °C for 20 min. The supernatants were then collected, and their persulfidation was then analyzed. Each supernatant (120 μL) was combined with 40 μL of 10% SDS and 0.32 µL of 10 M S-methyl methanethiosulfonate (MMTS) in a water bath at 50 °C for 20 min, and the proteins were then precipitated with acetone at −20 °C for 2 h. The precipitated protein pellets were obtained after centrifugation at 5000 g and 4 °C for 10 min. The precipitant was dissolved in 200 µL of lysis buffer containing 7 µL of EZ-linked iodoacetyl-PEG2 biotin in an incubator at 4 °C for 12 h. The biotinylated proteins were precipitated by incubating with UltraLink™ Immobilized Neutravidin™ at 4 °C for 4 h, and the persulfidated proteins were then obtained and subjected to Western blotting with 10% SDS-PAGE.
Immunofluorescence assay
The nuclear exclusion of FOXO1 and the expression of PCNA in VSMCs were detected by immunofluorescence assay as described in a previous study [19]. The VSMCs on slides were rinsed with PBS and then fixed with 4% paraformaldehyde for 15 min. The cells were incubated for 12 h at 4 °C with the following primary antibodies: FOXO1 antibody (1:100; Abcam, USA) and PCNA antibody (1:50; Sigma, USA). The fixed cells were then washed, and Alexa Fluor 488- or Alexa Fluor 594-conjugated secondary antibodies (Thermo, USA) were used to stain the corresponding primary antibodies for 1.5 h at 37 °C. The nuclei were counterstained with DAPI. Images of the cells were obtained under a confocal microscope (TCS SP5, Leica, Germany). The blue color indicates nuclei, the green or red color indicates FOXO1-positive signals, and the green color or red color indicates PCNA-positive signals.
Sequence homology analysis of FOXO1 protein
The FOXO1 protein sequences from mouse (Q9R1E0), rat (G3V7R4), human (Q12778), pig (A4L7N3), bovine (E1BPQ1), turkey (G1NRC4) and zebrafish (A3RK74) were downloaded from the UniProt database (https://www.uniprot.org/), and CLUSTALW software (https://www.genome.jp/tools-bin/clustalw) was then used to analyze the homology of the above-mentioned sequences.
Statistical analysis
SPSS 17.0 (IBM, USA) was used to analyze the experimental data. All the data are expressed as the means ± SEMs. The data from multiple groups were analyzed by one-way ANOVA. When Levene’s test showed equal variances (p > 0.05), the least significant difference (LSD) test was used for the comparisons; otherwise, the Games-Howell test was used for the post hoc comparisons. Pearson’s correlation analysis was used to identify the dependence between pairs of time and concentration series. A significant difference was indicated by a p value < 0.05.
Results
H2S inhibited FOXO1 inactivation in ET-1-stimulated VSMCs
The nuclear exclusion of the transcription factor FOXO1 to the cytoplasm after its phosphorylation results in its functional inactivation. Our results showed that the phosphorylation of FOXO1 was higher in ET-1-stimulated VSMCs than in the control cells. Furthermore, the treatment of A7r5 cells with H2S at concentrations of 50, 100 and 200 µM for 24 h significantly inhibited the ET-1-stimulated phosphorylation of FOXO1 in a dose-dependent manner, with inhibitory rates of 51%, 32% and 18% (all p < 0.05), respectively (Fig. 1a), whereas the treatment of A7r5 cells with 100 µM H2S for 6, 12 and 24 h significantly inhibited the ET-1-stimulated phosphorylation of FOXO1 at Ser256 in a time-dependent manner, with inhibitory rates of 60%, 49% and 31% (all p < 0.05), respectively (Fig. 1b). Subsequently, the degree of FOXO1 nuclear exclusion, the expression of its target gene PCNA and the level of cell proliferation were assessed in ET-1-stimulated cells treated or untreated with H2S. The results showed that in accordance with the inhibitory effect of H2S on FOXO1 phosphorylation (Fig. 1c), the ET-1 + H2S group exhibited higher levels of nuclear FOXO1 protein, lower PCNA mRNA and protein levels, reduced nuclear PCNA protein levels, and decreased proliferation than the ET-1 group (Fig. 1d–i), which suggested that H2S prevented the inactivation of FOXO1 in ET-1-treated VSMCs as well as the proliferation of these cells.
Fig. 1.

H2S blocked FOXO1 inactivation in ET-1-stimulated VSMCs. a. The phosphorylation of FOXO1 in ET-1-stimulated VSMCs incubated with H2S at different concentrations (50–200 μM) for 24 h. b. The phosphorylation of FOXO1 in ET-1-stimulated VSMCs incubated with 100 μM of H2S for different time (6–24 h). c. The phosphorylation of FOXO1 in VSMCs detected by Western blotting. d. FOXO1 nuclear exclusion to the cytoplasm in VSMCs determined by confocal immunofluorescence. Blue color indicates nuclei, and green color indicates FOXO1-positive signal. e. The PCNA mRNA level in VSMCs detected by qRT-PCR. f. PCNA expression in VSMCs detected by Western blotting. g. PCNA expression in VSMCs detected by confocal immunofluorescence. The blue color indicates nuclei, and the red color indicates PCNA-positive signals. h. VSMC proliferation detected by CCK-8 assay. i. VSMC proliferation evaluated by counting cell number. At least three times of replicates of experiments were conducted. ** p < 0.01, * p < 0.05 vs Con group; ## p < 0.01, # p < 0.05 vs ET-1 group. The data were shown as means ± SEMs.
Persulfidation of FOXO1 at Cys457 was required for FOXO1 activation
We subsequently aimed to identify the mechanism by which H2S inhibits FOXO1 inactivation. As shown in Fig. 2a, H2S persulfidated the FOXO1 protein in VSMCs, and this effect could be reversed by the thiol reductant DTT. The human FOXO1 protein contains seven cysteine residues: Cys23, Cys107, Cys117, Cys404, Cys457, Cys584 and Cys612. These cysteine sites are conserved among different species (Fig. 2b), and Cys457 and Cys612 show particularly high conservation. A structural analysis indicated that Cys457 of FOXO1 is located in an important functional region [20], which suggests that Cys457 is closely related to FOXO1 activity and might be persulfidated by H2S. As a result, we transfected WT and C457S-mutated FOXO1 plasmids into VSMCs and 293T cells and found that NaHS significantly persulfidated FOXO1 in the cells transfected with WT FOXO1 but could not persulfidate FOXO1 in the cells transfected with C457S FOXO1 (Fig. 2c and 2d). Furthermore, we assessed the effect of FOXO1 persulfidation on FOXO1 inactivation. The data showed that in accordance with the change in FOXO1 persulfidation, the ET-1-stimulated phosphorylation of FOXO1 at Ser256 and FOXO1 nuclear exclusion were not inhibited by H2S in the cells transfected with C457S FOXO1 (Fig. 2e and 2f). Therefore, the above-mentioned findings suggested that the persulfidation of FOXO1 by H2S at Cys457 might mediate the protective effect of H2S on FOXO1 activation.
Fig. 2.

H2S persulfidated FOXO1 at Cys457 and preserved FOXO1 activation in ET-1-treated VSMCs. a. FOXO1 persulfidation in VSMCs detected by biotin switch assay. b. Sequence homology analysis of FOXO1 proteins from different species. The sequences were obtained from the UniProt database. c. FOXO1 persulfidation in VSMCs transfected with WT or C457S-mutated FOXO1 plasmid measured by biotin switch assay. d. FOXO1 persulfidation in 293T cells transfected with WT or C457S-mutated FOXO1 plasmid measured by biotin switch assay. e. The ratios of p-FOXO1/FOXO1 in VSMCs transfected with WT or C457S-mutated FOXO1 plasmid detected by Western blotting. f. FOXO1 nuclear exclusion to the cytoplasm in VSMCs transfected with WT or C457S-mutated FOXO1 plasmid determined by confocal immunofluorescence. The blue color indicates nuclei, and the red color indicates FOXO1-positive signals. At least three times of replicates of experiments were conducted. * p < 0.05, ** p < 0.01, vs Con group; △ p < 0.05, △△p < 0.01 vs NaHS group; # p < 0.05 vs ET-1 group. The data were shown as means ± SEMs.
H2S alleviated the proliferation of ET-1-treated VSMCs by persulfidating FOXO1 at Cys457
To investigate the significance of the persulfidation of FOXO1 by H2S in the inhibition of cell proliferation, we assessed the effects of H2S on ET-1-treated VSMCs transfected with the WT or C457S-mutated FOXO1 plasmids. After ET-1 stimulation, the VSMCs transfected with the WT FOXO1 plasmids exhibited an upregulated PCNA mRNA and protein levels, an increased nuclear PCNA level and an amplified proliferation compared with the control cells (Fig. 3), whereas the pretreatment of the cells with NaHS clearly reversed the above-described effects. In contrast, in VSMCs transfected with the C457S-mutated FOXO1 plasmid, NaHS could not reverse the ET-1-induced increases in the PCNA mRNA and protein levels, nuclear PCNA levels and cell proliferation (Fig. 3). These results suggested that H2S alleviated the proliferation of ET-1-treated cells via FOXO1 persulfidation at Cys457.
Fig. 3.

H2S alleviated the proliferation of ET-1-treated VSMCs via FOXO1 persulfidation at Cys457. a. The PCNA mRNA level in VSMCs transfected with WT or C457S-mutated FOXO1 plasmid detected by qRT-PCR. b. The expression of PCNA in VSMCs transfected with WT or C457S-mutated FOXO1 plasmid determined by Western blotting. c. The expression of PCNA in VSMCs transfected with WT or C457S-mutated FOXO1 plasmid determined by confocal immunofluorescence. The blue color indicates nuclei, and the green color indicates PCNA-positive signals. d. Cell proliferation of VSMCs transfected with WT or C457S-mutated FOXO1 plasmids detected by CCK-8 assay. e. Cell proliferation of VSMCs transfected with WT or C457S-mutated FOXO1 plasmids detected by counting cell number. At least three times of replicates of experiments were conducted. ** p < 0.01, * p < 0.05 vs Con group; ## p < 0.01, # p < 0.05 vs ET-1 group; △△ p < 0.01, △ p < 0.05 vs NaHS group. The data were shown as means ± SEMs.
A decrease in the phosphorylation of FOXO1 at Ser256 was essential for the inhibition of VSMC proliferation via H2S-induced FOXO1 persulfidation
To confirm the role of FOXO1 phosphorylation at Ser256 in the inhibition of VSMC proliferation by H2S-induced FOXO1 persulfidation, we constructed S256A-mutated FOXO1 plasmids and transfected them into VSMCs. The results showed that H2S induced similar FOXO1 persulfidation in VSMCs transfected with WT and S256A-mutated FOXO1 plasmids (Fig. 4a). However, H2S exerted less effects on the phosphorylation of FOXO1 at Ser256 and the nuclear exclusion of FOXO1 in ET-1-treated VSMCs transfected with S256A-mutated FOXO1 plasmids than in those transfected with WT FOXO1 plasmids (Fig. 4b and c). In accordance with the uncontrolled inactivation of FOXO1, H2S treatment did not affect the PCNA mRNA and protein levels, the nuclear expression of PCNA and the proliferation of ET-1-treated A7r5 cells transfected with S256A-mutated FOXO1 plasmids (Fig. 4d–g). The above results suggested that FOXO1 dephosphorylation at Ser256 following FOXO1 persulfidation might be associated with the inhibition of VSMC proliferation by H2S-induced FOXO1 persulfidation.
Fig. 4.

The decrease in the phosphorylation of FOXO1 at Ser256 was essential for the inhibition of VSMC proliferation by H2S-induced FOXO1 persulfidation. a. FOXO1 persulfidation in VSMCs transfected with WT or S256A-mutated FOXO1 plasmids detected by biotin switch assay. b. The ratios of p-FOXO1/FOXO1 in VSMCs transfected with WT or S256A-mutated FOXO1 plasmids detected by Western blotting. c. FOXO1 location in VSMCs transfected with WT or S256A-mutated FOXO1 plasmids determined by confocal immunofluorescence. The blue color indicates nuclei, and the green color indicates FOXO1-positive signals. d. The PCNA mRNA level in VSMCs transfected with WT or S256A-mutated FOXO1 plasmids detected by qRT-PCR. e. The expression of PCNA in VSMCs transfected with WT or S256A-mutated FOXO1 plasmid determined by Western blotting. f. The expression of PCNA in VSMCs transfected with WT or S256A-mutated FOXO1 plasmid determined by confocal immunofluorescence. The blue color indicates nuclei, and the red color indicates PCNA-positive signals. g. Cell proliferation of VSMCs transfected with WT or S256A-mutated FOXO1 plasmid detected by CCK-8 assay. At least three times of replicates of experiments were conducted. * p < 0.05, ** p < 0.01 vs Con group; ##p < 0.01 vs ET-1 group; △△ p < 0.01, △ p < 0.05 vs NaHS group. The data were shown as means ± SEMs.
Discussion
It is well known that out-of-control VSMC proliferation is a main pathological change in vascular injury diseases. In the present study, we first showed that NaHS persulfidated FOXO1 at Cys457 to decrease FOXO1 phosphorylation at Ser256 and prevented FOXO1 nuclear exclusion to the cytoplasm, which further protected against the ET-1-induced proliferation of VSMCs. Therefore, our findings demonstrated a novel posttranslational modification mechanism by which H2S inhibited cell proliferation by targeting FOXO1, as shown in the graphical abstract.
As the third gasotransmitter following nitric oxide and carbon monoxide, H2S is thought to be mainly produced through the metabolic pathway of sulfur-containing amino acids and acts as an endogenous protective system in cardiovascular homeostasis [21]. The inhibition of VSMC proliferation is one of the important cardiovascular effects of H2S [22], [23]. For example, Sun et al. demonstrated that H2S suppresses mitochondrial fragmentation by downregulating Drp1 expression and inhibits the proliferation of pulmonary VSMCs [22]. Wu et al. discovered that H2S attenuates endoplasmic reticulum stress by reducing NOX-4 expression and activity and thereby inhibits smooth muscle cell proliferation [23]. However, the mechanisms by which H2S inhibits cell proliferation are not fully understood.
FOXO1 acts as a nuclear transcription factor that binds to the specific promoter region of proliferation-related target genes, such as cyclin D1/2, p27 and p21, to regulate cell proliferation [24], [25]. In the absence of survival or growth factors, FOXO1 remains in the cell nucleus, and the activation of its transcription plays a role in inhibiting cell proliferation. In the presence of growth factors, the phosphorylation of FOXO1 is increased, and FOXO1 is translocated to the cytoplasm and deactivated to stimulate cell proliferation [26]. Ye et al. revealed that the FOXO1 pathway is involved in the H2S-mediated inhibition of diabetic cardiomyopathy [27]. H2S also reportedly plays a role in promoting the binding of FOXO1 to the promoters of the KATP subunits SUR2B and Kir6.1 in VSMCs [14]. Hence, we hypothesized that H2S likely inhibited the proliferation of VSMCs through the FOXO1 pathway. As expected, the data obtained in the present study showed that the H2S donor NaHS suppressed the excessive VSMC proliferation induced by the ET-1 targeting of FOXO1, as demonstrated by the findings that NaHS successfully abolished the ET-1-induced increase in FOXO1 phosphorylation at Ser256 in a concentration- and time-dependent manner and that NaHS prevented the ET-1-stimulated nuclear exclusion of FOXO1 to the cytoplasm, the mRNA and protein expression of its downstream gene PCNA, and VSMC proliferation.
We subsequently examined the possible mechanisms through which H2S abolished FOXO1 inactivation and subsequent VSMC proliferation. Persulfidation, a posttranslational modification in which cysteinyl thiols (‐SH) in protein cysteine residues are converted into persulfide groups (‐SSH), is regarded as a main molecular pattern responsible for the biological function of H2S. It is well known that FOXO1 phosphorylation catalyzed by protein kinases is the essential step in FOXO1 inactivation, including the nuclear export of FOXO1 and the inhibition of its transcription factor activity [28]. Previous studies found that H2S-induced persulfidation blocks the phosphorylation of the same protein [29], [30], [31]. For example, previous studies have indicated that H2S persulfidates IKKβ at Cys179 to inhibit the phosphorylation of IKKβ and thereby control pulmonary artery endothelial cell inflammation [29]. Zhi et al. revealed that H2S persulfidates p66Shc at Cys59, which results in the prevention of the phosphorylation of p66Shc and the inhibition of the production of mitochondrial reactive oxygen species [30]. Wu et al. demonstrated that H2S promotes the persulfidation of CaMKII at Cys6 and then inhibits its phosphorylation [31]. In our study, a sequence homology analysis of the FOXO1 sequences from different species (mouse (Q9R1E0), rat (G3V7R4), human (Q12778), pig (A4L7N3), bovine (E1BPQ1), turkey (G1NRC4) and zebrafish (A3RK74)) demonstrated that the FOXO1 protein contains three to seven cysteines, which suggests the existence of a molecular basis for FOXO1 persulfidation. We thus used a specific probe of persulfidation to detect the potential persulfidation of FOXO1 by H2S. Interestingly, we found that NaHS persulfidated the FOXO1 protein in VSMCs and that DTT could reverse this effect, which suggested that H2S likely acts on a thiol group to persulfidate FOXO1.
To determine the exact site persulfidated by H2S, we first performed a sequence homology analysis of FOXO1 proteins from seven species and revealed that the cysteine residues were conserved in FOXO1 proteins from different species and that Cys457 and Cys612 were highly conserved among the seven species. Cys457 in FOXO1 is reportedly located in the transcriptional activation domain (aa 421–655) of FOXO1[20] and in the functional region (aa 283–563) of FOXO1, which interacts with Nemo-like kinase (NLK), as described in the UniProt database. Previous studies have reported that NLK activates FOXO1 phosphorylation, FOXO1 nuclear exclusion to the cytoplasm and inhibits FOXO1 transcriptional activity [32], which suggests that Cys457 is crucial for FOXO1 activity. Therefore, Cys457 in FOXO1 was selected as a preferred molecular target to determine the site persulfidated by H2S. We then constructed a mutated FOXO1 plasmid containing the site-directed C457S mutation and WT FOXO1 plasmids and transfected these into VSMCs and 293T cells separately. The results obtained with ET-1-treated cells transfected with the WT FOXO1 plasmid showed that NaHS persulfidated FOXO1, inhibited its phosphorylation at Ser256 and its nuclear exclusion to the cytoplasm, and suppressed the proliferation of the cells. However, NaHS could no longer persulfidate C457S-mutated FOXO1 protein and failed to inhibit the above-mentioned effects in cells transfected with the C457S-mutated FOXO1 plasmid. The results obtained for 293T cells were consistent with those found for VSMCs, which confirmed that H2S persulfidated FOXO1 at Cys457 to alleviate cell proliferation.
The phosphorylation of FOXO1 at Ser256 is an important regulatory switch of FOXO1 transcriptional activity [33], [34]. Therefore, the final issue that needs to be addressed is whether the decrease in the phosphorylation of FOXO1 at Ser256 is indeed an intermediate event between H2S-induced FOXO1 persulfidation and H2S-inhibited VSMC proliferation. In the present study, ET-1-treated A7r5 cells transfected with S256A-mutated and WT FOXO1 plasmids were used to explore the above question. As expected, the S256A mutation in FOXO1 did not affect the H2S-induced persulfidation of FOXO1 but abolished the H2S-induced dephosphorylation of FOXO1, the nuclear retainment of FOXO1, and the inhibition of PCNA expression and VSMC proliferation in ET-1-stimulated A7r5 cells, which suggested that a decrease in the phosphorylation of FOXO1 at Ser256 might be involved in the transduction of FOXO1 persulfidation-mediated cell signals induced by H2S to the inhibitory effects of H2S on VSMC proliferation.
Conclusions
In conclusion, this study revealed a new mechanism by which H2S inhibits the proliferation of VSMCs. As demonstrated in this study, H2S suppresses the phosphorylation of FOXO1 at Ser256 by persulfidating FOXO1 at Cys457, and this effect subsequently preserves the nuclear location and activation of FOXO1 and inhibits VSMC proliferation. The present study not only deepens our understanding of the significance of the H2S-mediated regulation of cardiovascular homeostasis but also provides new ideas and targets for anti-vascular remodeling treatment strategies for vascular injury diseases, such as hypertension and atherosclerosis.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
The present study was supported by the National Natural Science Foundation of China (81622004, 81670395 and 81921001), Beijing Natural Science Foundation (7171010) and CAMS Innovation Fund for Medical Sciences (2019-I2M-5-047).
Footnotes
Peer review under responsibility of Cairo University.
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jare.2020.06.023.
Contributor Information
Junbao Du, Email: junbaodu1@126.com.
Yaqian Huang, Email: yaqianhuang@126.com.
Hongfang Jin, Email: jinhongfang51@126.com.
Appendix A. Supplementary material
The following are the Supplementary data to this article:
Supplementary figure 1.

Supplementary figure 2.

Supplementary figure 3.

Supplementary figure 4.

References
- 1.Gibbons G.H., Dzau V.J. The emerging concept of vascular remodeling. N Engl J Med. 1994;330:1431–1438. doi: 10.1056/NEJM199405193302008. [DOI] [PubMed] [Google Scholar]
- 2.Tang C., Li X., Du J. Hydrogen sulfide as a new endogenous gaseous transmitter in the cardiovascular system. Curr Vasc Pharmacol. 2006;4:17–22. doi: 10.2174/157016106775203144. [DOI] [PubMed] [Google Scholar]
- 3.Jha S., Calvert J.W., Duranski M.R., Ramachandran A., Lefer D.J. Hydrogen sulfide attenuates hepatic ischemia-reperfusion injury: role of antioxidant and antiapoptotic signaling. Am J Physiol Heart Circ Physiol. 2008;295:H801–H806. doi: 10.1152/ajpheart.00377.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yang G., Wu L., Bryan S., Khaper N., Mani S., Wang R. Cystathionine gamma-lyase deficiency and overproliferation of smooth muscle cells. Cardiovasc Res. 2010;86:487–495. doi: 10.1093/cvr/cvp420. [DOI] [PubMed] [Google Scholar]
- 5.Liu M., Li Y., Liang B., Li Z., Jiang Z., Chu C. Hydrogen sulfide attenuates myocardial fibrosis in diabetic rats through the JAK/STAT signaling pathway. Int J Mol Med. 2018;41:1867–1876. doi: 10.3892/ijmm.2018.3419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yang G., Wu L., Jiang B., Yang W., Qi J., Cao K. H2S as a physiologic vasorelaxant: hypertension in mice with deletion of cystathionine gamma-lyase. Science. 2008;322:587–590. doi: 10.1126/science.1162667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Du J., Hui Y., Cheung Y., Bin G., Jiang H., Chen X. The possible role of hydrogen sulfide as a smooth muscle cell proliferation inhibitor in rat cultured cells. Heart Vessels. 2004;19:75–80. doi: 10.1007/s00380-003-0743-7. [DOI] [PubMed] [Google Scholar]
- 8.Li Y., Liu G., Cai D., Pan B., Lin Y., Li X. H2S inhibition of chemical hypoxia-induced proliferation of HPASMCs is mediated by the upregulation of COX-2/PGI2. Int J Mol Med. 2014;33:359–366. doi: 10.3892/ijmm.2013.1579. [DOI] [PubMed] [Google Scholar]
- 9.Tran H., Brunet A., Grenier J.M., Datta S.R., Fornace A.J., Jr, DiStefano P.S. DNA repair pathway stimulated by the forkhead transcription factor FOXO3a through the Gadd45 protein. Science. 2002;296:530–534. doi: 10.1126/science.1068712. [DOI] [PubMed] [Google Scholar]
- 10.Seoane J., Le H.V., Shen L., Anderson S.A., Massagué J. Integration of Smad and forkhead pathways in the control of neuroepithelial and glioblastoma cell proliferation. Cell. 2004;117:211–223. doi: 10.1016/s0092-8674(04)00298-3. [DOI] [PubMed] [Google Scholar]
- 11.Dijkers P.F., Medema R.H., Lammers J.W., Koenderman L., Coffer P.J. Expression of the pro-apoptotic Bcl-2 family member Bim is regulated by the forkhead transcription factor FKHR-L1. Curr Biol. 2000;10:1201–1204. doi: 10.1016/s0960-9822(00)00728-4. [DOI] [PubMed] [Google Scholar]
- 12.Nemoto S., Finkel T. Redox regulation of forkhead proteins through a p66shc-dependent signaling pathway. Science. 2002;295:2450–2452. doi: 10.1126/science.1069004. [DOI] [PubMed] [Google Scholar]
- 13.Wilhelm K., Happel K., Eelen G., Schoors S., Oellerich M.F., Lim R. FOXO1 couples metabolic activity and growth state in the vascular endothelium. Nature. 2016;529:216–220. doi: 10.1038/nature16498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sun Y., Huang Y., Zhang R., Chen Q., Chen J., Zong Y. Hydrogen sulfide upregulates KATP channel expression in vascular smooth muscle cells of spontaneously hypertensive rats. J Mol Med (Berl) 2015;93:439–455. doi: 10.1007/s00109-014-1227-1. [DOI] [PubMed] [Google Scholar]
- 15.Majumder S., Ren L., Pushpakumar S., Sen U. Hydrogen sulphide mitigates homocysteine-induced apoptosis and matrix remodelling in mesangial cells through Akt/FOXO1 signalling cascade. Cell Signal. 2019;61:66–77. doi: 10.1016/j.cellsig.2019.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Savai R., Al-Tamari H.M., Sedding D., Kojonazarov B., Muecke C., Teske R. Pro-proliferative and inflammatory signaling converge on FoxO1 transcription factor in pulmonary hypertension. Nat Med. 2014;20:1289–1300. doi: 10.1038/nm.3695. [DOI] [PubMed] [Google Scholar]
- 17.Siqueira M.F., Flowers S., Bhattacharya R., Faibish D., Behl Y., Kotton D.N. FOXO1 modulates osteoblast differentiation. Bone. 2011;48:1043–1051. doi: 10.1016/j.bone.2011.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang D., Wang X., Tian X., Zhang L., Yang G., Tao Y. The increased endogenous sulfur dioxide acts as a compensatory mechanism for the downregulated endogenous hydrogen sulfide pathway in the endothelial cell inflammation. Front Immunol. 2018;9:882. doi: 10.3389/fimmu.2018.00882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Du J., Huang Y., Yan H., Zhang Q., Zhao M., Zhu M. Hydrogen sulfide suppresses oxidized low-density lipoprotein (ox-LDL)-stimulated monocyte chemoattractant protein 1 generation from macrophages via the nuclear factor κB (NF-κB) pathway. J Biol Chem. 2014;289:9741–9753. doi: 10.1074/jbc.M113.517995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Saline M., Badertscher L., Wolter M. Lau R, Gunnarsson A, Jacso T, et al, AMPK and AKT protein kinases hierarchically phosphorylate the N-terminus of the FOXO1 transcription factor, modulating interactions with 14–3-3 proteins. J Biol Chem. 2019;294:13106–13116. doi: 10.1074/jbc.RA119.008649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Elsey D.J., Fowkes R.C., Baxter G.F. Regulation of cardiovascular cell function by hydrogen sulfide (H(2)S) Cell Biochem Funct. 2010;28:95–106. doi: 10.1002/cbf.1618. [DOI] [PubMed] [Google Scholar]
- 22.Sun A., Wang Y., Liu J., Yu X., Sun Y., Yang F. Exogenous H2S modulates mitochondrial fusion-fission to inhibit vascular smooth muscle cell proliferation in a hyperglycemic state. Cell Biosci. 2016;6:36. doi: 10.1186/s13578-016-0102-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wu J., Pan W., Wang C., Dong H., Xing L., Hou J. H2S attenuates endoplasmic reticulum stress in hypoxia-induced pulmonary artery hypertension. Biosci Rep. 2019;39 doi: 10.1042/BSR20190304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jeong O.S., Chae Y.C., Jung H., Park S.C., Cho S.J., Kook H. Long noncoding RNA linc00598 regulates CCND2 transcription and modulates the G1 checkpoint. Sci Rep. 2016;6:32172. doi: 10.1038/srep32172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chang Y.W., Zhao Y.F., Cao Y.L., Gu X.F., Li Z.Q., Wang S.Q. Liver X receptor α inhibits osteosarcoma cell proliferation through up-regulation of FoxO1. Cell Physiol Biochem. 2013;32:180–186. doi: 10.1159/000350134. [DOI] [PubMed] [Google Scholar]
- 26.Link W. Introduction to FOXO biology. Methods Mol Biol. 2019;1890:1–9. doi: 10.1007/978-1-4939-8900-3_1. [DOI] [PubMed] [Google Scholar]
- 27.Ye P., Gu Y., Zhu Y.R., Chao Y.L., Kong X.Q., Luo J. Exogenous hydrogen sulfide attenuates the development of diabetic cardiomyopathy via the FoxO1pathway. J Cell Physiol. 2018;233:9786–9798. doi: 10.1002/jcp.26946. [DOI] [PubMed] [Google Scholar]
- 28.Zhang X., Gan L., Pan H., Guo S., He X., Olson S.T. Phosphorylation of serine 256 suppresses transactivation by FKHR (FOXO1) by multiple mechanisms: direct and indirect effects on nuclear/cytoplasmic shuttling and DNA binding. J Biol Chem. 2002;277:45276–45284. doi: 10.1074/jbc.M208063200. [DOI] [PubMed] [Google Scholar]
- 29.Zhang D., Wang X., Chen S., Chen S., Yu W., Liu X. Endogenous hydrogen sulfide sulfhydrates IKKβ at cysteine 179 to control pulmonary artery endothelial cell inflammation. Clin Sci (Lond) 2019;133:2045–2059. doi: 10.1042/CS20190514. [DOI] [PubMed] [Google Scholar]
- 30.Xie Z.Z., Shi M.M., Xie L., Wu Z.Y., Li G., Hua F. Sulfhydration of p66Shc at cysteine59 mediates the antioxidant effect of hydrogen sulfide. Antioxid Redox Signal. 2014;21:2531–2542. doi: 10.1089/ars.2013.5604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wu D., Hu Q., Tan B., Rose P., Zhu D., Zhu Y.Z. Amelioration of mitochondrial dysfunction in heart failure through S-sulfhydration of Ca2+/calmodulin-dependent protein kinase II. Redox Biol. 2018;19:250–262. doi: 10.1016/j.redox.2018.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kim S., Kim Y., Lee J., Chung J. Regulation of FOXO1 by TAK1-Nemo-like kinase pathway. J Biol Chem. 2010;285:8122–8129. doi: 10.1074/jbc.M110.101824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Biggs W.H., 3rd, Meisenhelder J., Hunter T., Cavenee W.K., Arden K.C. Protein kinase B/Akt-mediated phosphorylation promotes nuclear exclusion of the winged helix transcription factor FKHR1. Proc Natl Acad Sci U S A. 1999;96:7421–7426. doi: 10.1073/pnas.96.13.7421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gerst F., Kaiser G., Panse M., Sartorius T., Pujol A. Anita M Hennige, Protein kinase Cδ regulates nuclear export of FOXO1 through phosphorylation of the chaperone 14–3-3ζ. Diabetologia. 2015;58:2819–2831. doi: 10.1007/s00125-015-3744-z. [DOI] [PubMed] [Google Scholar]