

Graphical abstract
Keywords: Hydrogen sulfide, Blood vessels, Hypertension, Atherosclerosis, Pulmonary hypertension
Abstract
Background
Hydrogen sulfide (H2S) is considered to be the third gasotransmitter after carbon monoxide (CO) and nitric oxide (NO). It plays an important role in the regulation of vascular homeostasis. Vascular remodeling have has proved to be related to the impaired H2S generation.
Aim of Review
This study aimed to summarize and discuss current data about the function of H2S in vascular physiology and pathophysiology as well as the underlying mechanisms.
Key Scientific Concepts of Review
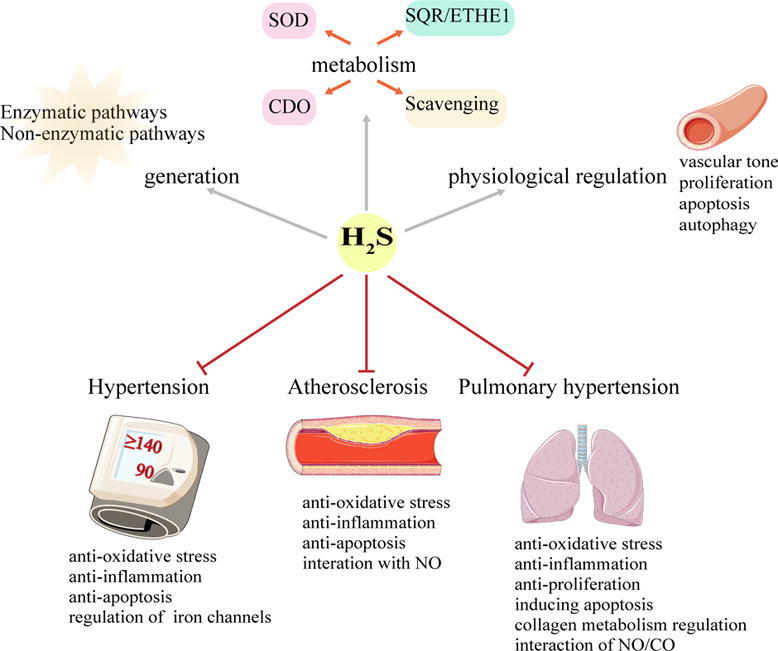
Endogenous hydrogen sulfide (H2S) as a third gasotransmitter is primarily generated by the enzymatic pathways and regulated by several metabolic pathways. H2S as a physiologic vascular regulator, inhibits proliferation, regulates its apoptosis and autophagy of vascular cells and controls the vascular tone. Accumulating evidence shows that the downregulation of H2S pathway is involved in the pathogenesis of a variety of vascular diseases, such as hypertension, atherosclerosis and pulmonary hypertension. Alternatively, H2S supplementation may greatly help to prevent the progression of the vascular diseases by regulating vascular tone, inhibiting vascular inflammation, protecting against oxidative stress and proliferation, and modulating vascular cell apoptosis, which has been verified in animal and cell experiments and even in the clinical investigation. Besides, H2S system and angiotensin-converting enzyme (ACE) inhibitors play a vital role in alleviating ischemic heart disease and left ventricular dysfunction. Notably, sulfhydryl-containing ACEI inhibitor zofenopril is superior to other ACE inhibitors due to its capability of H2S releasing, in addition to ACE inhibition. The design and application of novel H2S donors have significant clinical implications in the treatment of vascular-related diseases. However, further research regarding the role of H2S in vascular physiology and pathophysiology is required.
Introduction
Hydrogen sulfide (H2S) was discovered to be the third gasotransmitter after nitric oxide (NO) and carbon monoxide (CO). This novel gaseous molecule has been proved to be widely involved in the regulation of various systems in human body [1]. Moreover, H2S has attracted great attention in regulating the structure and function of blood vessels. Many researchers have shown that H2S exerts vital effects on vascular cellular processes, such as inflammation, apoptosis, cell cycle, cytoprotection, and mitochondrial metabolic function and biogenesis [2].
In the vasculature, H2S modulates vascular tension, suppresses the proliferation, and exerts a bidirectional effect on apoptosis and autophagy of vascular smooth muscle cells (VSMCs). Furthermore, the development of many vascular remodeling-associated diseases, including hypertension, atherosclerosis and pulmonary hypertension has been proved to be related to the impaired H2S generation. In addition, H2S and the use of zofenopril, one of the ACE inhibitors that can promote the release of H2S, in cardiovascular diseases are also gradually being valued. Therefore, the understanding how H2S is endogenously generated, as well as the regulation of blood vessels by H2S under physiological and pathological conditions, may elucidate the pathogenesis of vascular diseases and uncover new promising targets for the prevention and treatment of vascular diseases.
Endogenous H2S generation and metabolism
The generation of endogenous H2S is mostly catalyzed by enzymes, while only a small part is produced by non-enzymatic pathways [3], [4]. The enzymes that catalyze H2S production mainly include cystathionine-β-synthase (CBS), cystathionine-γ-lyase (CSE), 3-mercaptopyruvate sulfur transferase (3-MST) and cysteine aminotransferase (CAT) [5], [6]. CBS and CSE are the primary enzymes involved in H2S production [7] that catalyze the substrate L- cysteine with tissue specificity. CBS is abundant in the brain, liver, and kidney, with small amount of expression in the uterine artery, mesenteric artery, and carotid ball [8]. CSE predominantly catalyzes synthesis of H2S in the liver, ileum, portal vein, thoracic aorta and non-vasculature [2], [9]. Recently, 3-MST has been found to catalyze H2S synthesis in the central and peripheral nervous systems, vascular endothelium and other tissues [10]. It catalyzes 3-mercaptopyruvate (3-MPT), which produces H2S and pyruvate in vivo. Among these three enzymes, homocysteine is converted into cystathionine and cysteine in turn by sulfur transfer under the catalysis of CBS and CSE. Cysteine and thiols are catalyzed by CBS via β-substitution. Different from CBS, CSE catalyzes three kinds of reactions, including the α, β-cleavage of cysteine, the α, γ-cleavage of homocysteine, and the γ-substitution of homocysteine through a second mole of homocysteine. As a sulfurtransferase, 3-MST is responsible for transferring sulfur from mercaptopyruvate to an active cysteine site, and then forms MST-SSH, a persulfide intermediate. Except for thioredoxin, a variety of small molecules such as dihydrolipoic acid, homocysteine, cysteine and glutathione (GSH) release H2S by receiving the persulfide group in the presence of reductant [11]. Opposite to the enzymatic reaction, the non-enzymatic reaction of H2S generation is partial for cysteine as a substrate and is catalyzed by coordinated activities of VitB6 and iron. Non-enzymatic production of H2S occurs in the spleen, heart, lung, muscles, bone marrow and plasma, especially in RBCs [12]. The aortic H2S production rate is reported to be 5.8 ± 1.7 pmol s−1 mg protein-1[13]. In addition, various arteries demonstrate different production rates of H2S. The H2S production rate in the caudal artery, the mesenteric artery, the pulmonary artery and the thoracic aorta was 8.12 ± 0.85, 6.17 ± 0.56, 5.31 ± 0.70 and 4.06 ± 0.28 pmol s−1 mg wet tissue-1, respectively [14].
After synthesis by transsulfuration from L-cysteine, various metabolic pathways participate in the regulation of H2S concentration in the cell. Significant pathways for H2S metabolism include oxidation by sulfide quinone oxidoreductase (SQR) and persulfide dioxgenase (ETHE1) in the mitochondrion and methylation by cysteine dioxygenase (CDO) in the cytoplasm [15]. Sulfide is oxidized in the mitochondrion by SQR to generate persulfide. Persulfide is further oxidized to sulfite by ETHE1, and sulfite is finally oxidized by rhodanese or sulfite oxidase. After ubiquinone captures electrons released in the SQR reaction, the electrons are transferred to complex III in the electron transport chain [16]. In addition to the above oxidation pathway metabolism, Olson et al [17] proved that superoxide dismutase (SOD) also oxidizes H2S to produce polysulfides. Methemoglobin and molecules containing metallo or disulfides such as oxidized glutathione may also eliminate H2S [3], [18].
Physiological regulation of blood vessels by H2S
H2S on vascular tone
H2S has a bidirectional regulatory effect on vascular tone. H2S can not only relax blood vessels, but also contract blood vessels [19]. A study published in Science [20] showed that the activation of CSE by calcium-calmodulin (CaM) under physiological conditions is the main mechanism of H2S production in the vascular system. Mutant mice lacking CSE displayed lower levels of H2S, with abnormally elevated blood pressure and loss of endothelium-dependent vasodilatory function. These findings directly prove the significance of H2S for the maintenance of vascular function. Intriguingly, the vasodilation of H2S on the portal vein and the ileum was notable stronger than that on the thoracic aorta [21]. In addition, compared with H2S, hydrogen polysulfides (H2Sn) tended to contain more sulfane sulfur atoms which have a relaxing effect and ultimately lowered blood pressure [22], [23].
H2S also has vasoconstrictive effects under certain conditions. NaHS contracts VSMCs at concentrations between 5 × 10−6 M and 10−4 M [24]. A study by Ping reported similar results [25]. NaHS at concentrations ranging from 10 to 300 μM induced coronary artery constriction in rats. Therefore, the regulation of H2S on vascular tone is bidirectional.
The mechanisms underlying H2S-induced vasodilation are not fully understood. The effects of vasodilation have been attributed to iron channels that are activated by H2S according to previous studies [26]. It is suggested that H2S exerts a vasorelaxant effect via opening ATP-sensitive potassium channels (KATP channels) in VSMCs [27]. H2S mediates a new type of protein post-translational modification that is sensitive to redox, namely sulfhydration. [28]. More specifically, H2S causes sulfhydration of cysteine-43 (C43) in Kir6.1 (a subunit of KATP channel), resulting in a decrease in the capacity of Kir 6.1 binding to ATP, while the capacity of Kir 6.1 binding to PIP2 is enhanced. This event eventually causes KATP channels to open and VSMCs to relax [29]. Excepting the KATP channel, growing evidence demonstrates that calcium-activated potassium channels (Kca channels) are also activated by H2S [30], [31]. H2S increases smooth muscle Ca2+ spark activity to activate endothelial large-conductance calcium-activated potassium channels (BKCa channel) [32]. Transient receptor potential cation channel V4 (TRPV4) is also modified by H2S through sulfhydration. This is followed by the activation and the opening of TRPV4-dependent Ca2+ internal flow and the endothelial BKCa channel and results in vasodilation [33]. In addition, the SK2.3 channel which acts as an α-subunit isoform of the SKCa channel is activated by H2S through S-sulfhydration [34]. Moreover, the activation of voltage sensitive potassium channels (KV channels) and Kv7.4 voltage-gated potassium channels which are predominantly expressed in VSMCs are seen as targets for H2S action on vascular tone [35], [36]. Recent reports have also demonstrated that H2S caused S-sulfhydration of L-type Ca2+ channels, leading to a decrease in intracellular Ca2+ concentration ([Ca2+] i) [37].
Whether H2S participates in the regulation of the cyclic guanosine monophosphate (cGMP) pathway remains controversy. A compelling amount of evidence indicates that H2S exerts a vasodilative effect through the activation of endothelial nitric oxide synthase (eNOS) and the inhibition of cGMP degradation [38], [39], [40]. There are several primary mechanisms thought to participate: (1) H2S directly reacts with NO to produce nitroxyl (HNO), thereby activating the HNO– transient receptor potential ankyrin 1 (TRPA1)–calcitonin gene-related peptide (CGRP) pathway to regulate vascular tone [41]. (2) H2S inhibits the activity of phosphodiesterase 5 (PDE5) by reducing cGMP degradation and promoting cGMP signaling, followed by the activation of cGMP-dependent protein kinase (PKG) to phosphorylate the vasodilator-stimulated phosphoprotein (VASP), eventually resulting in vasodilation [42]. In addition, Sun et al. [43] believed that H2S sulfhydrated associated PDE5A dimerization to exert the vasorelaxant function. (3) H2S may alleviate oxidative stress, resulting in increased eNOS coupling by phosphorylation of eNOSS1177 [44], [45]. (4) The reaction of soluble guanylyl cyclases (sGCs) to NO can be enhanced by H2S [40], [46]. It might be related to the reduction of sGC heme Fe by H2S, so as to facilitate NO-regulated cellular signaling processes [47]. However, there is disagreement over the role of H2S. For instance, Wang [48] et al. suggested that H2S did not rely on cGMP pathway to exert vasodilation, although vasodilation was strengthened by specific sGC inhibitors (ODQ and NS-2028). Similarly, NaHS-induced relaxation was unaffected by ODQ in rat coronary arteries [49]. Taken together, the vasorelaxation of H2S varied very widely in different species and cell types. This might explain the conflicting results [46].
The vasodilation of H2S was also related to the suppression of mitochondrial complexes I and III. It was shown that NaHS (100–1000 μM) suppressed mitochondrial electron transport to exert a vasodilation effect in rat mesenteric arterioles. This effect was inhibited by complex I and complex III inhibitors [30].
Accumulating evidence from H2S studies demonstrates that H2S derived from perivascular adipose tissue (PVAT) also exerts a critical effect in the regulation of vascular tension [33], [50]. PVAT exerts predominantly anti-contractile effects, which is induced by adipocyte-derived relaxing factor (ADRF) [51], [52]. Schleifenbaum et al. [53] suggested that H2S could be an ADRF to regulate vascular tone. The mechanism of H2S as ADRF could relate to activate KATP and (or) voltage-sensitive KCNQ potassium channels [54], [55]. Importantly, the findings from Kohn et al. [55] suggest that with technical progress, future studies on the vascular H2S/KCNQ pathways make it possible to relieve vascular dysfunction.
In summary, H2S-induced vasorelaxation takes place via the activation of iron channels, the interactions with NO-cGMP signaling, the inhibition of mitochondrial complexes I and III, and H2S as an ADRF. However, under certain conditions, H2S has vasoconstrictive effects which appear to involve the activation of Na+-K+-2Cl-–co-transporters and voltage-gated calcium ion channels by H2S [24]. Additionally, Ping et al. [25] suggested that the activation of the Rho kinase signaling pathway by H2S may participates in the contraction of rat coronary arteries.
Effects of H2S on proliferation and apoptosis of vascular smooth muscle cells
Accumulating evidence implicates H2S as an inhibitor of VSMC proliferation. It was shown that the VSMC proliferation rate in CSE knockout mice was dramatically increased. However, endogenous H2S significantly inhibited the proliferation of smooth muscle cell (SMC) in CSE knockout mice [56]. Similarly, NaHS, a commonly used H2S donor, dose-dependently suppressed the proliferation of VSMCs [57]. The potential mechanisms for H2S-induced proliferation are as follows: Du et al. [57] demonstrated that H2S suppressed the activity of mitogen-activated protein kinase (MAPK), which might be responsible for H2S-inhibited VSMC proliferation. Furthermore, endogenous CSE/H2S pathway can inhibit the cascade conduction of MAPK/thioredoxin interacting protein (TXNIP) signals [58], thereby protecting endothelial function. In addition, H2S dramatically inhibited the transcription and expression of Brg1 gene, reduced the recruitment of Brg1 in the promoter region of proliferating genes (pcna, ntf3 and PDGFα) and consequently inhibited the proliferation of VSMCs [59]. On the other hand, H2S not only decreased the expression of insulin-like growth factor-1 receptor (IGF-1R), but also modified IGF-1R through sulfhydration to prevent IGF-1 binding, ultimately inhibiting VSMC proliferation [60]. Recently, Wang et al. [61] demonstrated that calcium-sensing receptor (CaSR) increased endogenous generation of H2S via calcium-CaM signal pathways, ultimately inhibiting the proliferation of VSMCs. Therefore, several genes, molecules, and signaling pathways (such as MAPK/TXNIP signals, Brg1, ERK1/2, IGF-1R and CaSR) have been identified in the regulation by H2S, and contribute to the suppression of VSMC proliferation.
H2S can promote or inhibit vascular cell apoptosis. Several studies agree with the view that H2S promotes apoptosis. Studies [62], [63] have demonstrated that H2S can activate the ERK/caspase 3 pathway and promote the apoptosis of human aorta smooth muscle cell (HASMC). CSE overexpression or exogenous H2S supplementation promotes apoptosis via stimulating extracellular regulated protein kinases (ERK) 1/2, p38 MAPK, and p21 Cip/WAK-1 but suppressing cyclin D1 [56], [62]. In contrast, several studies suggest that H2S inhibits apoptosis. H2S decreased the elevated ratio of Bcl2-associated x (Bax)/B-cell lymphoma-2 (Bcl-2) and the activity of caspase-3, thus inhibiting apoptosis caused by high glucose [64]. It was also shown that NaHS suppressed apoptosis by reducing the expression of caspase-12, C/EBP homologous protein (CHOP), and glucose-regulated protein 78 (GRP78) which are related to endoplasmic reticulum stress (ERS), thus protecting vascular endothelial function [65]. Therefore, the regulation of apoptosis by H2S is bidirectional. It can promote and inhibit apoptosis under different pathological conditions.
Effect of H2S on vascular autophagy
Autophagy is essential for homeostasis in processes including cell development and differentiation, regulation of cell longevity and programmed cell death, degradation of invading pathogens, and provision of antigens to the immune system [66]. Pathogens, abnormal proteins and organelles are engulfed by autophagosomes and undergo lysosomal degradation [67], [68]. H2S is reported to either promote or inhibit autophagy depending on the different pathological process [69], [70]. NaHS was shown to activate mitophagy in rat aortic endothelial cells (RAECs) [71]. Mechanistically, NaHS facilitates Parkin recruited by PTEN induced putative kinase 1 (PINK1), and then ubiquitylates mitofusin 2 (Mfn2), leading to the upregulation of mitophagy [71]. However, several studies showed that both supplementation of H2S and the overexpression of its synthetases mitigated mitophagy [72]. H2S inhibited adenosine 5‘-monophosphate (AMP)-activated protein kinase (AMPK)/mammalian target of rapamycin (mTOR) pathway, which is closely associated with autophagy [73]. On the other hand, the ratio of microtubule-associated protein 1A/1B-light chain 3 (LC3)-II to LC3-I is commonly used as an indicator of autophagy. Expression of LC3A I/II was significantly decreased with supplementation of H2S (30 μM) [72]. NaHS could also inhibit the excessive autophagy of vascular endothelial cells by suppressing nuclear factor erythroid-2-related factor 2 (Nrf2)- reactive oxygen species (ROS) -AMPK signaling pathway [74]. Taken together, there are still different opinions of vascular autophagy regulation by H2S. A variety of pathological conditions likely contribute to the differences in the effect that have been observed.
Pathophysiological regulation of H2S on blood vessels
H2S and hypertension
Treating hypertension which is defined as ≥ 140/90 mmHg with chronically increased blood pressure remains a great challenge. Several clinical studies showed a close correlation between hypertension and reduction of H2S. The reduction of endogenous H2S synthesis and H2S-dependent vasodilation led to a microvascular dysfunction in hypertensive patients [75]. Notably, CBS, CSE and 3-MST as the H2S generating enzymes, were markedly decreased in humans with hypertension [76], suggesting that H2S generation pathway may be involved in the pathogenesis of hypertension. Similar results have also been shown in animal research. For instance, a decreased endogenous H2S content in the aorta was observed in the development and progression of spontaneously hypertensive rats (SHRs) [77]. The use of DL-propargylglycine (PPG), a CSE inhibitor, dramatically elevated the level of basal blood pressure in WKY rats and promoted vascular remodeling, demonstrating that a sufficient H2S level is necessary for the maintenance of basal blood pressure [78]. Similar to that of SHRs, it was shown that CBS/H2S pathway was down-regulated in salt-sensitive Dahl rats [79].
Extensive evidence shows that H2S exerts a crucial effect on blood pressure regulation in pathological cases. For instance, studies by Sun et al. [80] suggested that NaHS lowered tail artery pressure in SHRs. Similarly, it was shown that H2S delayed the shift from prehypertensive to hypertensive status in SHRs [81]. Notably, H2S improved endothelial function in renovascular hypertensive rats and ameliorated the damaged endothelium-dependent contraction (EDC) and endothelium-dependent relaxation (EDR) [82], [83]. Furthermore, the H2S donor alleviated hypertension, reversed aortic remodeling, and inhibited the renin–angiotensin–aldosterone (RAS) system in renal tissue of Dahl rats [79]. These experimental results demonstrate that H2S dramatically suppressed the elevation in blood pressure in two animal models.
Many scholars have discussed the protective effect of H2S on hypertension and its potential mechanisms. Previous studies [82], [83] showed that the H2S donor NaHS significantly suppressed the activation of NOD-like receptors (NLRP3), inflammasomes, and oxidative stress in SHRs. Moreover, the amelioration of excessive EDC of H2S was associated with the inhibition of the bone morphogenetic protein 4 (BMP4) and its downstream signal molecules [84]. NaHS can also protect renal artery endothelial cells and improve endothelial function through the activation of the peroxisome proliferator-activated receptor δ (PPARδ) signaling [85]. In addition to improvements in the vascular endothelium, NaHS also regulated immune function by reducing the expression of connexin 40 (Cx40)/connexin 43 (Cx43) T lymphocytes in SHRs, and reversed changes in multiple T lymphocyte subtypes in SHRs [86], which may explain the anti-inflammatory effect of H2S. Ion channels are considered as key targets for H2S depressurization. A report from Sun et al. [80] suggested that the KATP channel is activated by H2S and causes vasodilation. Furthermore, H2S may activate KATP channel by inhibiting Forkhead box O1 (FOXO1) and Forkhead box O3a (FOXO3a) phosphorylation, subsequently inducing their nuclear binding to SUR2B and Kir6.1. In addition to the regulation of the KATP channel, H2S can also activate the TRP vanilloid 1(TRPV1) ion channel through S-sulfhydration, increasing the sensitivity of carotid sinus pressure receptors in SHRs [87]. TRPA1 channels were also activated by H2S, inducing the release of CGRP and promoting vasodilation [88], [89]. On the other hand, H2S also inhibited the pathological state of SHRs by regulating the RAS system. H2S reduced the expression of RAS-related mRNA (Ren, Atp6ap2, Agt, Ace, and Agtr1a) in the kidneys of SHRs, which blocked the RAS system and exerted a vasomotor effect [81]. Finally, an underlying H2S mechanism may be related to the inhibition of collagen deposition. H2S dose-dependently inhibited MAPK activation induced by angiotensin II in SHRs and down-regulated the affinity of angiotensin II type 1 (AT1), ultimately inhibiting vascular remodeling and collagen deposition in SHRs [90]. Furthermore, reduced collagen deposition by H2S may be related to the suppression of transforming growth factor-β /Smad signaling pathway [91].
The mechanism by which H2S regulates blood pressure in high-salt Dahl rats may be as follows. Liang et al. [92] showed that H2S reduced the oxidative stress response in the paraventricular nucleus of high-salt Dahl rats, attenuated sympathetic activity, and promoted the secretion of anti-inflammatory factors, thus inhibiting the inflammatory response. H2S may also regulate blood pressure by the inactivation of epithelial sodium channels (ENaC). Reabsorption of sodium by the ENaC promotes the progress of salt-sensitive hypertension. It was shown that H2S completely blocked abnormal activation of ENaC caused by excessive H2O2. H2O2 increased sodium reabsorption by up-regulating phosphatidylinositol 3, 4, 5-trisphosphate. H2S can significantly inhibit PTEN inactivation caused by H2O2, thereby reducing oxidative stress [93].
To summarize, the mechanisms by which H2S inhibits hypertension are complicated, including the reduction of oxidative stress and inflammation, the modulation of immune function and ion channels, and the inhibition of collagen deposition and vascular remodeling.
H2S and atherosclerosis
Atherosclerosis (AS) is a chronic, complicated and progressive pathological process of large and medium-sized arteries. Several studies have shown that H2S deficiency is related to the pathogenesis of AS. For example, Gao et al. [94] suggested that H2S deficiency may predispose stable coronary artery disease (CAD) patients to vulnerable plaque rupture. As reported in many clinical studies, Wang et al. [95] found disorders of the vascular CSE/H2S pathway in apolipoprotein E (ApoE)-knockout mice. Another study from Meng et al. [96] also demonstrated that decreased endogenous H2S generation accelerated AS in CSE-knockout mice. Accumulating evidence [97], [98] has shown that endogenous H2S produced by CSE in blood vessels has an anti-AS effect. Unstable plaques generated by AS are prone to rupture and have the risk of infarction [99]. In ApoE-knockout mice, H2S stabilizes atherosclerotic plaques and suppresses lipid deposition [100], [101].
Key mechanisms for the anti-AS effect of H2S include anti-oxidative stress, anti-inflammatory effect, and regulation of ion channels [102] to protect the vascular endothelium. Intriguingly, it was reported that vascular CSE/H2S, as the target of estrogen, was involved in the mechanism by which estrogen protected against AS [103]. The detailed mechanism is as follows.
First, H2S attenuates oxidative stress to protect against AS. It induces S-sulfhydration of glutathione peroxidase 1 (GPx1) to prompt glutathione synthesis, resulting in alleviating lipid peroxidation and improving antioxidant capacities [104]. Several studies [105], [106] further found that H2S may induce Nrf2 to dissociate from kelch-like ec-associated protein 1 (Keap1) by sulfhydration of Cys151 in Keap1, enhancing nuclear translocation of Nrf2 and thereby exerting antioxidant stress and cardiovascular protection. Moreover, translocation of Nrf2 further stimulated its downstream molecules, including the NADPH quinoneoxidoreductase 1 (NQO1), thus preventing the release of inflammatory cytokines [107]. H2S was found to attenuate atherosclerotic lesions by blocking oxidative modification of low density lipoprotein (LDL) and elevating antioxidative ability [108]. A recent study shows that H2S-induced antioxidant stress is also related to its elimination of oxidized hemoglobin (Hb) and inhibition of the interaction between Hb and lipid in AS [109]. Through the regulation of above molecules, H2S exerts a critical role in prevention of collagen deposition and protection of vascular function.
Secondly, H2S attenuates inflammation to protect against AS. Inactivation of nuclear factor kappa-B (NF-κB) caused by H2S reduces the expression of inflammatory factor intercellular cell adhesion molecule-1 (ICAM-1), which may be an important reason for H2S to maintain the stability of AS plaques [95]. Moreover, Du et al. [110] found that H2S modified cysteine 38 in p65 via sulfhydration, which was responsible for NF-κB inactivation. Recent studies also showed that the anti-inflammatory effect of H2S might suppress TXNIP, an activator of NLRP3, which inhibited excessive production of interleukin 18 (IL‐18) and interleukin 1β (IL‐1β) [111]. Additionally, H2S was identified as an agonist of histone deacetylase Sirtuin-1 (SIRT-1). H2S directly induced deacetylation of SIRT-1 and its target proteins (P53, P65, and sterol response element-binding protein), alleviating inflammation in the endothelium and macrophages, inhibiting macrophage cholesterol uptake in ApoE knockout mice, and eventually reducing the formation of AS plaques [112]. Furthermore, it is worth noting that the activation of matrix metalloproteinases (MMPs) was involved in AS. As a member of MMPs, MMP9 is considered to be a critical factor causing instability of AS plaques [113]. Studies have found that H2S reduced MMP9 activity by inhibiting activator protein 1 (AP-1) nuclear translocation, thus alleviating the inflammatory reaction of AS [100].
Thirdly, the interactions between NO and H2S may also be one of the anti-AS mechanisms. Specifically, H2S upregulates the expression of inducible nitric oxide synthase (iNOS) protein and promotes NO production. [114].
Fourthly, H2S has an anti-apoptotic effect. Studies showed that H2S increased the stability of plaques in ApoE knockout mice by inhibiting caspase-3/9 activity and lipoprotein receptor-1 (Lox-1) [100].
Additionally, there are other mechanisms that mediate the anti-AS effect of H2S. H2S donors can reduce the level of adrenomedullin (ADM) and increase the level of atrial natriuretic peptide (ANP) in AS rats, thus antagonizing the formation of AS [115]. Mani et al. [96] proposed that H2S plays an anti-AS effect, which may inhibit intimal proliferation and adhesion molecule expression. Recently, a study also showed that NaHS notably activated angiotensin converting enzyme 2 (ACE2)-related pathways, so as to promote the transformation from pro-atherosclerosis to anti- atherosclerosis [116].
In conclusion, H2S retarded the development of AS by a variety of molecular mechanisms that include anti-oxidative stress, anti-inflammation, anti-apoptosis, and interactions with NO.
H2S and pulmonary hypertension
Abnormal vascular remodeling and increased pulmonary artery pressure that results in right ventricular (RV) hypertrophy and heart failure are characteristic pathological features of pulmonary hypertension (PH). PH consists of hypoxic pulmonary hypertension (HPH) and PH caused by high pulmonary blood flow and so on. Acute or chronic hypoxic stimulation leads to the progression of HPH, which is typically characterized by PH and increased pulmonary vascular resistance. It was shown that both the expression of CSE and its activity were inhibited in lung tissues during HPH [117]. In another model of PH, endogenous H2S pathway was also downregulated in rat PH models caused by high pulmonary blood flow [118]. In addition, Feng et al. [119] suggested that the contents of H2S in lung tissues and serum of rats in the monocrotaline (MCT)-induced PH group were obviously inhibited, and CSE expression was dramatically co-downregulated.
However, a clinical study demonstrated that H2S at 500 μM induced an average dilation of 42.3% from the pre-constricted tension in dissected human arterial rings. In addition, H2S at 500 μM also induced an average reduction of 17.73% in pulmonary artery pressure [120]. This effect was also seen in animal models. For instance, H2S donors reduced pulmonary artery pressure and alleviated structural remodeling of pulmonary vessels during HPH [117]. In addition, exogenous H2S restored H2S contents in plasma, alleviating pulmonary artery remodeling caused by HPH.
The mechanisms by which H2S protects against PH include but are not restricted to anti-inflammation [121], anti-endoplasmic reticulum stress (ERS) [122], induction of apoptosis [123], anti-proliferation [124], [125] and upregulation of the CO/HO pathway [126]. The detailed mechanisms are as follows.
First, H2S antagonizes pulmonary vascular inflammation. Inflammation exerts a central effect on the pathogenesis of PH. Previous studies [122], [127] demonstrated that H2S inhibited pro-inflammatory and oxidative stress. It was shown that H2S alleviates pulmonary artery endothelial inflammation by inhibiting NF-κB signaling pathway [127]. Moreover, H2S not only inhibits the NF-κB signaling pathway, but also alleviates ERS by inhibiting the expression of NADPH oxidase 4 (Nox4), as well as GRP78 and CHOP the ERS-related molecule markers [122], [65].
Secondly, H2S induces PASMC apoptosis. The effect of H2S on apoptosis is bidirectional, which can promote and inhibit apoptosis. However, Li et al. [123] suggested that H2S induces apoptosis through inhibiting Bcl-2 and activating Fas signaling pathway of PASMCs in PH rats.
Thirdly, H2S significantly inhibited the expression of proliferative cell nuclear antigen (PCNA) and urotensin II (U-II), which are critical molecules related to cell proliferation [128]. This anti-proliferative effect may be related to the up-regulation of cyclooxygenase-2(COX-2)/prostaglandin I2 (PGI2) signaling pathway [124], [125].
Fourthly, H2S exerts the anti-oxidative stress effect in PH model. Oxidative stress is another important cause of elevated pulmonary arterial systolic pressure in humans. H2S enhances the ratio of GSH/ oxidized glutathione (GSSG), which represents antioxidant capacity, by scavenging GSSG, thus exerting antioxidant capacity in HPH [129]. Moreover, the expression of collagen-promoting molecules connective tissue growth factor (CTGF) and MMP-13 were increased after the application of D, L-propargylglycine (PPG), whereas the expression of tissue inhibitor of metalloproteinase 1 (TIMP-1) was significantly decreased. All of the above results indicate that H2S alleviates oxidative stress injuries, thus inhibiting pulmonary vascular remodeling [130], [131].
Lastly, H2S upregulates the CO/heme oxygenase (HO-1) pathway and is regulated by NO simultaneously in PH [132], [133]. The interaction between CO and H2S potentially contributes to the pathogenesis of HPH. Zhang et al. demonstrated that H2S might modulate the pathogenesis of HPH by activating HO-1 [126]. However, the mechanisms underlying H2S through regulation of the CO/HO pathway in PASMCs remain unknown. Accumulating evidence [134] also demonstrates that defects of NO signaling possibly contribute to the progression of PH. The NO substrate, L-arginine, is known to upregulate CSE/H2S signaling in PH caused by high blood flow [135]. Therefore, H2S protects pulmonary vascular structure through the interaction with the other two gas molecules-NO and CO.
In summary, H2S attenuates PH through several mechanisms, including anti-inflammation, induction of apoptosis, anti-proliferation, anti-oxidative stress, and regulating CO and NO signaling pathways.
H2S and other cardiovascular diseases
Previous studies have confirmed that the abnormality of endogenous H2S pathway may participate in the pathogenesis of ischemic heart disease (IHD) and left ventricular dysfunction [136]. Overexpression of CSE or supplementation of H2S donors significantly improved cardiac function and structural lesions [137], [138], [139]. The following mechanisms might be involved in the protective effect of H2S on the IHD and left ventricular dysfunction: 1) suppression of oxidative stress: H2S increases the activity of antioxidant enzymes SOD, CAT and GSH in the cardiac tissues of mice with ischemia/reperfusion (IR) injury [140]. Furthermore, a 7-day treatment of H2S donor Na2S promoted the nuclear translocation of Nrf2, an important transcription factor that regulates antioxidant genes as an adaptive response to oxidative stress, in the hearts of mice with left coronary artery occlusion and reperfusion, which might contribute to the increase in the antioxidant enzymes [137]. Moreover, the upregulation of the rhythm gene Bmal1 expression was also involved in the antioxidant effects of H2S in the ischemic cardiomyocyte H9c2 cells [141]. 2) inhibition of apoptosis and autophagy: H2S reduced the proportion of apoptotic cells in the myocardium of mice with heart failure (HF) by increasing the expression of Bcl-2 and inhibiting the expression of Bax and caspase 3 [138]. In another study, H2S alleviates autophagy of myocardial ischemia in SOD1 KO mice through the inhibition of S6 kinase (S6K) phosphorylation and AMPK phosphorylation [142]. 3) regulation of macrophage-related cardiac inflammatory response: H2S promoted the infiltration of macrophages into the infarcted myocardium in both wild type and CSE-KO mice targeting on the macrophage integrin β1 and its downstream Src-FAK/Pyk2-Rac pathway [143]. Moreover, the polarization of infiltrated macrophage in the heart of mice with MI was also governed by H2S. The results showed that H2S donor NaHS promoted the number and the proportion of anti-inflammatory M2 macrophages in left ventricular tissue after MI by increasing mitochondrial biosynthesis and fatty acid oxidation [144]. 4) interaction with other bioactive molecules: In the previous studies, the interaction between H2S and NO was involved in the vascular regulation [145]. Similarly, it is reported that H2S enhanced endogenous NO generation by increasing the mRNA level of eNOS and nNOS and decreasing the mRNA level of iNOS in the heart tissues of myocardial IR rats [146]. 5) mitochondrial protection: H2S maintains mitochondrial homeostasis by restoring the balance of Bcl-2/Bax and reducing mitochondrial-dependent apoptosis in HF rats [138], and improving mitochondrial respiration and ATP synthesis in isolated cardiac mitochondria from HF mice [137]. In addition, a blocker of mitoKATP channel 5-HD completely blocked the protective effect of H2S donor on the isolated I/R rat heart, suggesting that the opening of mitoKATP channel might be involved in the regulatory effect of H2S on the cardiac mitochondria [147].
Application of sulfhydryl group-containing angiotensin-converting enzyme (ACE) inhibitor in cardiovascular diseases
Angiotensin-converting enzyme (ACE) inhibitors are widely used as therapeutic agents in the treatment of cardiovascular diseases such as hypertension, IHD and left ventricular dysfunction in experimental studies and clinical trials [148], [149], [150]. The protective mechanisms of ACE inhibitors were mainly mediated by the inhibition of angiotension II generation and bradykinin degradation. For example, the mechanisms of cardioprotection in patients treated with ACE inhibitors might include the reduction in LV preload and afterload, suppression of sympathetic stimulation, restoration the balance of myocardial oxygen supply and demand, improvement in endogenous fibrinolysis, and alleviation of diastolic dysfunction, etc [151]. Compared with other ACE inhibitors, a sulfhydryl-group-containing ACE inhibitor zofenopril has been demonstrated to have a better clinical efficacy and safety in patients with hypertension, acute myocardial infarction (AMI) or CAD, particularly in high risk patients such as diabetes mellitus, in many clinical and preclinical studies such as SMILE series studies [152], [153], [154]. Borghi et al compared the difference in the efficacy between zofenopril and other ACE inhibitors in patients with AMI. The results showed that early administration of zofenopril in the patients ≥ 1 cardiovascular risk factor had a better prognosis and less risk of cardiovascular events than the administration of lisinopril and ramipril [153]. It has been reported that the peculiar protective effects of zofenopril including the capability of scavenging ROS, preventing of endothelial dysfunction, suppressing inflammatory response, promoting of NO generation and bioactivity, and regulating of cell apoptosis might be related to its sulfhydryl groups [151]. However, Bucci et al found that H2S could be released from S-zofenoprilat, an active metabolite of S-zofenopril, in a cell-free assay and directly play a vasorelaxant effect in vitro. Also, the key H2S-producing enzyme CSE expression in the vessel and the endothelial-dependent vasodilation in SHRs treated with S-zofenopril was recovered to normal level [155]. As well as the regulation of vessel function, H2S was found to mediate the pro-angiogenic effect of zofenopril, supported by the fact that CSE inhibitor or CSE siRNA blocked the zofenopril-induced angiogenesis in vivo and in vitro [156]. In addition, CSE-dependent H2S was also involved in the anti-inflammatory effect of zofenopril in IL-1β-induced endothelial inflammation model [157]. Interestingly, an increase in the H2S and NO level in the myocardial tissue and plasma was found to be associated with the cardioprotective effect of zofenopril pretreated before I/R injury in mouse and pig I/R [158]. Therefore, although further studies are needed, the abovementioned studies suggest that the property of H2S donor/generator might contribute to the superior clinical application of sulfhydrated ACE inhibitor zofenopril compared with other ACE inhibitors, which would open a new avenue for the treatment of cardiovascular diseases.
Conclusions
H2S participates in the physiological and pathological regulation of vasculature. The mechanisms underlying H2S-induced vasodilation are complex. H2S induced vasorelaxation predominantly by activating iron channels, interacting with NO-cGMP signaling, inhibiting mitochondrial complex I and III, and acting as an ADRF. In addition, H2S inhibits the proliferation of VSMCs in association with MAPK/ TXNIP, Brg1, ERK1/2, IGF-1R and CaSR signals. The regulation of H2S on vascular cell apoptosis and autophagy is bidirectional. It can either promote or inhibit autophagy and apoptosis depending on the different pathological process (see Fig. 1, Fig. 2, Fig. 3, Fig. 4 and Table 1, Table 2, Table 3).
Fig. 1.

Generation and metabolism of endogenous H2S.
Fig. 2.

Regulation of H2S on hypertension. → means stimulating effect, whereas ⊥means inhibiting effect. P means phosphorylation.
Fig. 3.

Regulation of H2S on atherosclerosis. → means stimulating effect, whereas ⊥ means inhibiting effect. –SSH means S- sulfhydrylation. Ace means acetylation. oxLDL, oxidized low-density lipoprotein; Ang II, angiotensin II; Ang (1–7), angiotensin (1–7).
Fig. 4.

Regulation of H2S on pulmonary hypertension. → means stimulating effect, whereas ⊥ means inhibiting effect. \ means scavenging.
Table 1.
H2S has a bidirectional regulation effect on vascular tone.
| Action | Mechanisms | Models | H2S gas/donor application (concentration) | Refs. |
|---|---|---|---|---|
| Relaxation | Activation of KATP channel | Mesenteric artery VSMCs of rats | NaHS (100–300 μM) | [27], [29] |
| Activation of KCa channel | Rat cerebral arteries | NaHS (10 and 100 μM) | [31] | |
| Activation of Ca2+ spark activity | Rat mesenteric small arteries | NaHS (10 μM) | [32] | |
| Activation of TRPV4 channel | Rat mesenteric small arteries | NaHS (1–1000 μM) | [33] | |
| Activation of BK channels | Rat mesenteric small arteries | NaHS (1–1000 μM) | [33] | |
| Activation of IKCa and SKCa channels | Mouse mesenteric arteries and aortas | NaHS (≥100 μM) | [34] | |
| Activation of Kv7 channels | Rat mesenteric small arteries | NaHS (100–3000 μM) | [30] | |
| Activation of Kv7.4 channels (subtype of Kv7) | Rat aortic rings | NaHS (1000 μM) | [35] | |
| Activation of KCNQ-type Kv channels | Rat and mouse aortas | NaHS (10–3000 μM) | [55] | |
| [37] | ||||
| Activation of HNO-TRPA1-CGRP pathway | Rat mesenteric arteries | Na2S (10 μM) | [41] | |
| Activation of cGMP-PKG-VASP pathway | Mouse aortic rings | NaHS (30 μM) | [42] | |
| Inhibition of sGC heme Fe | Mouse thoracic aorta | Na2S (50 µM) | [47] | |
| Constriction | Activation of Na+-K+-2Cl-–co-transporters and voltage-gated calcium ion channels | Rat thoracic aortas | NaHS (5–100 μM) | [24] |
| Activation of Ca2+ influx | Rat coronary arteries | NaHS (10–300 μM) | [25] |
Table 2.
Effects of H2S on proliferation and apoptosis of vascular smooth muscle cells.
| Action | Mechanisms | Cells/Models | H2S gas/donor application (concentration) | Refs. |
|---|---|---|---|---|
| Anti-proliferation | Inhibition of Brg1 transcription and expression by reducing the recruitment of Brg1 to the Pcna, Ntf3 and Pdgfα promoter regions | VSMCs | NaHS (1000 μM) | [59] |
| Anti-proliferation | Inhibition of the MAPK pathway | VSMC isolated from rat thoracic aortas | NaHS (50–500 μM) | [57] |
| Anti-proliferation | Inhibition of the MAPK/TXNIP cascade | HUVECs/CSE-KO mice | NaHS (56 µM/kg/d) | [58] |
| Anti-proliferation | Inhibition of the expression of IGF-1R and the binding of IGF-1 with IGF-1R via S-sulfhydration | SMCs isolated from mouse mesenteric arteries | NaHS (10–100 μM) | [60] |
| Inducing apoptosis/Anti-proliferation | Increasing ERK1/2, p21Cip/WAF-1, and decreasing cyclin D1 in SMCs-KO mice. Inhibition of proliferation-related genes CRL, HB-EGF and IB1 in CSE KO mice. |
SMCs-KO mice/CSE-KO mice/HASMCs | H2S (100 μM) | [56], [62] |
| Inducing apoptosis | Activation of MAPKs and caspase-3 | HASMCs | H2S (50–100 μM) | [63] |
| Inhibiting apoptosis | Activation of SOD activity Inhibition of ROS generation and MDA levels |
HUVECs | NaHS (50 μM) | [64] |
| Inhibiting apoptosis | Inhibition of caspase-12, CHOP, GRP78 | PAECs | NaHS (56 µM/kg/d) | [65] |
Table 3.
Effect of H2S on vascular autophagy.
| Action | Mechanisms | Cells/Models | H2S gas/donor application (concentration) | Refs. |
|---|---|---|---|---|
| Promoting mitophagy | Activation of Parkin recruited by PINK1 and then ubiquitination of Mfn2 | RAECs | NaHS (100 μM) | [71] |
| Inhibiting mitophagy | Phosphorylation of Akt and dephosphorylation of FoxO3a | MAECs | NaHS (30 μM) | [72] |
| Inhibiting autophagy | Dephosphorylation of AMPK and phosphorylation of mTOR | VSMCs isolated from rat thoracic aorta | NaHS (100 μM) | [73] |
| Inhibiting autophagy | Dephosphorylation of AMPK and activation of Nrf2 | RAECs/db/db mice | NaHS (100 μM) | [74] |
Recent experimental data provide evidence that H2S can prevent vascular-related diseases, such as hypertension, atherosclerosis and PH. The underlying mechanisms may include the regulation of vascular tone, anti-inflammation, anti-oxidative stress, the inhibition of VSMC proliferation, and the modulation of VSMC apoptosis. Regulating H2S level provides a novel therapeutic method against these vascular diseases. In addition, the application of H2S system and ACE inhibitors in the treatment of cardiovascular diseases has gradually been paid attention. Notably, the effectiveness of zofenopril in clinical trials is significantly better than other ACE inhibitors due to its capability of H2S releasing. Therefore, H2S has important clinical implications. Further understanding of its protective role in cardiovascular system is needed.
Future studies should investigate the interaction amongst H2S and other gaseous signaling molecules including NO and sulfur dioxide (SO2). There remain many opportunities to explore its role in atherosclerosis, PH and hypertension. Of note, drugs targeting H2S producing enzymes (CBS, CSE and 3-MST) merits further clinical research.
Conflict of Interest
The authors declare no conflict of interest.
Compliance with Ethics Requirements
This article does not contain any studies with human or animal subjects.
Acknowledgements
This research was funded by National Natural Science Foundation of China (81770422 to YH, 81670395 to HJ, 81921001 to WK, and 81622004 to HJ), Beijing Natural Science Foundation (7182168 to YH, 7191012 to HJ, and 7171010 to JD), Open Foundation from Beijing Key Laboratory of Hypertension Research to YH, and Beijing Hospitals Authority Youth Programme (QML20170302 to HZ).
Biographies

Boyang Lv has been studying for a doctor's degree in pediatrics at Peking University since 2019. She now focuses on the regulation of sulfur-containing gaseous signal molecules for cardiovascular diseases.

Selena Chen is a master’s student in the Division of Biological Sciences at the University of California, San Diego, in the laboratory of Dr. Bryan Sun. Her research interests focus on understanding the genetic regulators of skin development and molecular pathways are disrupted during skin diseases. She was awarded her Bachelor of Science Degree in Biochemistry and Cellular Biology at the University of California, San Diego in 2018.

Chaoshu Tang is a professor at the Department of Physiology and Pathophysiology, Peking University Health Science Centre, China. He chaired a National “973” Key Basic Research Project. He has been engaged in scientific research and teaching of cardiovascular physiology and pathological mechanisms. Professor Tang has won the second prize of National Science and Technology Progress (1998); the first prize of Ministry (1991, 1997, 2003 and 2005); the second and third prize and many other awards.

Hongfang Jin is a professor of Pediatric Cardiology, Peking University First Hospital, China. She majors in endogenous hydrogen sulfide research. She is an Excellent Young Scholars of National Natural Science Foundation (NSFC) and an Excellent Young Scholar in National Youth Top-notch Talent Support Program of China. Professor Jin is the president of Basic Research Committee of Chinese Pediatric Cardiology Society; the vice-president of Pediatric Committee of Cardiovascular Society of Chinese Medical Doctor Association.

Junbao Du is a professor at the Department of Pediatric Cardiology, Peking University First Hospital, China. He majors in endogenous hydrogen sulfide research. Dr. Du is a Changjiang Scholar of China, Distinguished Young Scholars of NSFC and outstanding Young-middle Aged Expert Approved by Ministry of Health of China. He serves as a PI of Gasotransmitters and Cardiovascular Diseases Laboratory, Key Lab of Cardiovascular Sciences, the Ministry of Education, P. R. China. He is also the president of Pediatric Cardiology Committee of Cardiovascular Physicians Society of Chinese Medical Doctors Association and a committee member of Asian Pacific Pediatric Cardiology Society.

Yaqian Huang is an associate professor in the Department of Pediatrics at Peking University First Hospital, China. She had obtained her PhD degree in physiology from Peking University in 2013. Since then, she has been working at Peking University First Hospital. She is currently the vice president of Youth Committee of Chinese Pediatric Cardiology Society, Chinese Medical Association. Her research interests focus on sulfur-containing gaseous signal molecules.
Footnotes
Peer review under responsibility of Cairo University.
Contributor Information
Hongfang Jin, Email: jinhongfang51@126.com.
Junbao Du, Email: junbaodu1@126.com.
Yaqian Huang, Email: yaqianhuang@126.com.
References
- 1.Yang G., Wang R. H2S and Blood Vessels: An Overview. Handb Exp Pharmacol. 2015;230:85–110. doi: 10.1007/978-3-319-18144-8_4. [DOI] [PubMed] [Google Scholar]
- 2.Rose P., Moore PK., Zhu YZ. H2S Biosynthesis and Catabolism: New Insights From Molecular Studies. Cell Mol Life Sci. 2017;74(8):1391–1412. doi: 10.1007/s00018-016-2406-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pan LL., Qin M., Liu XH., Zhu YZ. The Role of Hydrogen Sulfide on Cardiovascular Homeostasis: An Overview with Update on Immunomodulation. Front Pharmacol. 2017;8:686. doi: 10.3389/fphar.2017.00686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cao X., Ding L., Xie ZZ., Yang Y., Whiteman M., Moore PK. A Review of Hydrogen Sulfide Synthesis, Metabolism, and Measurement: Is Modulation of Hydrogen Sulfide a Novel Therapeutic for Cancer? Antioxid Redox Signal. 2019;31(1):1–38. doi: 10.1089/ars.2017.7058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Szabo C., Papapetropoulos A. International Union of Basic and Clinical Pharmacology. CII: Pharmacological Modulation of H2S Levels: H2S Donors and H2S Biosynthesis Inhibitors. Pharmacol Rev. 2017;69(4):497–564. doi: 10.1124/pr.117.014050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Olas B. Hydrogen Sulfide in Signaling Pathways. Clin Chim Acta. 2015;439:212–218. doi: 10.1016/j.cca.2014.10.037. [DOI] [PubMed] [Google Scholar]
- 7.Leffler CW., Parfenova H., Basuroy S., Jaggar JH., Umstot ES., Fedinec AL. Hydrogen Sulfide and Cerebral Microvascular Tone in Newborn Pigs. Am J Physiol Heart Circ Physiol. 2011;300(2):H440–H447. doi: 10.1152/ajpheart.00722.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Saha S., Chakraborty PK., Xiong X., Dwivedi SK., Mustafi SB., Leigh NR. Cystathionine beta-Synthase Regulates Endothelial Function via Protein S-sulfhydration. FASEB J. 2016;30(1):441–456. doi: 10.1096/fj.15-278648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hosoki RMN., Kimura H. The Possible Role of Hydrogen Sulfide as an Endogenous Smooth Muscle Relaxant in Synergy with Nitric Oxide. Biochem Biophys Res Commun. 1997;237:527–531. doi: 10.1006/bbrc.1997.6878. [DOI] [PubMed] [Google Scholar]
- 10.Coletta C., Modis K., Szczesny B., Brunyanszki A., Olah G., Rios EC. Regulation of Vascular Tone, Angiogenesis and Cellular Bioenergetics by the 3-mercaptopyruvate Sulfurtransferase/H2S Pathway: Functional Impairment by Hyperglycemia and Restoration by DL-alpha-Lipoic Acid. Mol Med. 2015;21:1–14. doi: 10.2119/molmed.2015.00035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Banerjee R., Chiku T., Kabil O., Libiad M., Motl N., Yadav PK. Assay Methods for H2S Biogenesis and Catabolism Enzymes. Methods Enzymol. 2015;554:189–200. doi: 10.1016/bs.mie.2014.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang J., Minkler P., Grove D., Wang R., Willard B., Dweik R. Non-enzymatic Hydrogen Sulfide Production from Cysteine in Blood Is Catalyzed by Iron and Vitamin B6. Commun Biol. 2019;2:194. doi: 10.1038/s42003-019-0431-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Doeller JE., Isbell TS., Benavides G., Koenitzer J., Patel H., Patel RP. Polarographic Measurement of Hydrogen Sulfide Production and Consumption by Mammalian Tissues. Anal Biochem. 2005;341(1):40–51. doi: 10.1016/j.ab.2005.03.024. [DOI] [PubMed] [Google Scholar]
- 14.Hui Y., Du J., Tang C., Bin G., Jiang H. Changes in Arterial Hydrogen Sulfide (H2S) Content During Septic Shock and Endotoxin Shock in Rats. J Infect. 2003;47(2):155–160. doi: 10.1016/s0163-4453(03)00043-4. [DOI] [PubMed] [Google Scholar]
- 15.Olson KR. H2S and Polysulfide Metabolism: Conventional and Unconventional Pathways. Biochem Pharmacol. 2018;149:77–90. doi: 10.1016/j.bcp.2017.12.010. [DOI] [PubMed] [Google Scholar]
- 16.Kabil O., Banerjee R. Enzymology of H2S Biogenesis, Decay and Signaling. Antioxid Redox Signal. 2014;20(5):770–782. doi: 10.1089/ars.2013.5339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Olson KR., Gao Y., Arif F., Arora K., Patel S., DeLeon ER. Metabolism of Hydrogen Sulfide (H2S) and Production of Reactive Sulfur Species (RSS) by Superoxide Dismutase. Redox Biol. 2018;15:74–85. doi: 10.1016/j.redox.2017.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Elsey DJ., Fowkes RC., Baxter GF. Regulation of Cardiovascular Cell Function by Hydrogen Sulfide (H(2)S) Cell Biochem Funct. 2010;28(2):95–106. doi: 10.1002/cbf.1618. [DOI] [PubMed] [Google Scholar]
- 19.Caprnda M., Qaradakhi T., Hart JL., Kobyliak N., Opatrilova R., Kruzliak P. H2S Causes Contraction and Relaxation of Major Arteries of the Rabbit. Biomed Pharmacother. 2017;89:56–60. doi: 10.1016/j.biopha.2017.01.057. [DOI] [PubMed] [Google Scholar]
- 20.Yang G., Wu L., Jiang B., Yang W., Qi J., Cao K. H2S as a Physiologic Vasorelaxant: Hypertension in Mice with Deletion of Cystathionine gamma-lyase. Science. 2008;322(5901):587–590. doi: 10.1126/science.1162667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kimura H. Hydrogen Sulfide and Polysulfides as Signaling Molecules. Proc Jpn Acad Ser B Phys Biol Sci. 2015;91(4):131–159. doi: 10.2183/pjab.91.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kimura Y., Toyofuku Y., Koike S., Shibuya N., Nagahara N., Lefer D. Identification of H2S3 and H2S Produced by 3-mercaptopyruvate Sulfurtransferase in the Brain. Sci Rep. 2015;5:14774. doi: 10.1038/srep14774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stubbert D., Prysyazhna O., Rudyk O., Scotcher J., Burgoyne JR., Eaton P. Protein Kinase G Lalpha Oxidation Paradoxically Underlies Blood Pressure Lowering by the Reductant Hydrogen Sulfide. Hypertension. 2014;64(6):1344–1351. doi: 10.1161/HYPERTENSIONAHA.114.04281. [DOI] [PubMed] [Google Scholar]
- 24.Orlov SN., Gusakova SV., Smaglii LV., Koltsova SV., Sidorenko SV. Vasoconstriction Triggered by Hydrogen Sulfide: Evidence for Na(+), K(+),2Cl(-)cotransport and L-type Ca(2+) Channel-Mediated Pathway. Biochem Biophys Rep. 2017;12:220–227. doi: 10.1016/j.bbrep.2017.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ping NN., Li S., Mi YN., Cao L., Cao YX. Hydrogen Sulphide Induces Vasoconstriction of Rat Coronary Artery via Activation of Ca(2+) Influx. Acta Physiol (Oxf). 2015;214(1):88–96. doi: 10.1111/apha.12475. [DOI] [PubMed] [Google Scholar]
- 26.Cheng Y., Ndisang JF., Tang G., Cao K., Wang R. Hydrogen Sulfide-induced Relaxation of Resistance Mesenteric Artery Beds of Rats. Am J Physiol Heart Circ Physiol. 2004;287(5):H2316–H2323. doi: 10.1152/ajpheart.00331.2004. [DOI] [PubMed] [Google Scholar]
- 27.Tang G., Wu L., Liang W., Wang R. Direct Stimulation of K(ATP) channels by Exogenous and Endogenous Hydrogen Sulfide in Vascular Smooth Muscle Cells. Mol Pharmacol. 2005;68(6):1757–1764. doi: 10.1124/mol.105.017467. [DOI] [PubMed] [Google Scholar]
- 28.Paul BD., Snyder SH. Protein Sulfhydration. Methods Enzymol. 2015;555:79–90. doi: 10.1016/bs.mie.2014.11.021. [DOI] [PubMed] [Google Scholar]
- 29.Mustafa AK., Sikka G., Gazi SK., Steppan J., Jung SM., Bhunia AK. Hydrogen Sulfide as Endothelium-derived Hyperpolarizing Factor Sulfhydrates Potassium Channels. Circ Res. 2011;109(11):1259–1268. doi: 10.1161/CIRCRESAHA.111.240242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hedegaard ER., Gouliaev A., Winther AK., Arcanjo DD., Aalling M., Renaltan NS. Involvement of Potassium Channels and Calcium-Independent Mechanisms in Hydrogen Sulfide-Induced Relaxation of Rat Mesenteric Small Arteries. J Pharmacol Exp Ther. 2016;356(1):53–63. doi: 10.1124/jpet.115.227017. [DOI] [PubMed] [Google Scholar]
- 31.Wang M., Hu Y., Fan Y., Guo Y., Chen F., Chen S. Involvement of Hydrogen Sulfide in Endothelium-Derived Relaxing Factor-Mediated Responses in Rat Cerebral Arteries. J Vasc Res. 2016;53(3–4):172–185. doi: 10.1159/000448712. [DOI] [PubMed] [Google Scholar]
- 32.Jackson-Weaver O., Osmond JM., Naik JS., Gonzalez Bosc LV., Walker BR., Kanagy NL. Intermittent Hypoxia in Rats Reduces Activation of Ca2+ Sparks in Mesenteric Arteries. Am J Physiol Heart Circ Physiol. 2015;309(11):H1915–H1922. doi: 10.1152/ajpheart.00179.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Naik JS., Osmond JM., Walker BR., Kanagy NL. Hydrogen Sulfide-Induced Vasodilation Mediated by Endothelial TRPV4 Channels. Am J Physiol Heart Circ Physiol. 2016;311(6):H1437–H1444. doi: 10.1152/ajpheart.00465.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tang G., Yang G., Jiang B., Ju Y., Wu L., Wang R. H(2)S Is An Endothelium-Derived Hyperpolarizing Factor. Antioxid Redox Signal. 2013;19(14):1634–1646. doi: 10.1089/ars.2012.4805. [DOI] [PubMed] [Google Scholar]
- 35.Martelli A., Testai L., Breschi MC., Lawson K., McKay NG., Miceli F. Vasorelaxation by Hydrogen Sulphide Involves Activation of Kv7 Potassium Channels. Pharmacol Res. 2013;70(1):27–34. doi: 10.1016/j.phrs.2012.12.005. [DOI] [PubMed] [Google Scholar]
- 36.Kohn C., Dubrovska G., Huang Y., Gollasch M. Hydrogen Sulfide: Potent Regulator of Vascular Tone and Stimulator of Angiogenesis. Int J Biomed Sci. 2012;8(2):81–86. [PMC free article] [PubMed] [Google Scholar]
- 37.Dai L., Qian Y., Zhou J., Zhu C., Jin L., Li S. Hydrogen Sulfide Inhibited L-Type Calcium Channels (CaV1.2) via Up-Regulation of the Channel Sulfhydration in Vascular Smooth Muscle Cells. Eur J Pharmacol. 2019;858 doi: 10.1016/j.ejphar.2019.172455. [DOI] [PubMed] [Google Scholar]
- 38.Yao Q., Jin H., Du J. Effect of Hydrogen Sulfide on Vasorelaxation and Content of Guanosine-Cyclic Phosphate in Vascular Tissue of Rats. Journal of Applied Clinical Pediatrics. 2015;30(10):776–778. [Google Scholar]
- 39.Beltowski J., Jamroz-Wisniewska A. Hydrogen Sulfide and Endothelium-Dependent Vasorelaxation. Molecules. 2014;19(12):21183–21199. doi: 10.3390/molecules191221183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cao X., Wu Z., Xiong S., Cao L., Sethi G., Bian JS. The Role of Hydrogen Sulfide in Cyclic Nucleotide Signaling. Biochem Pharmacol. 2018;149:20–28. doi: 10.1016/j.bcp.2017.11.011. [DOI] [PubMed] [Google Scholar]
- 41.Eberhardt M., Dux M., Namer B., Miljkovic J., Cordasic N., Will C. H2S and NO Cooperatively Regulate Vascular Tone by Activating a Neuroendocrine HNO-TRPA1-CGRP Signalling Pathway. Nat Commun. 2014;5:4381. doi: 10.1038/ncomms5381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Coletta C., Papapetropoulos A., Erdelyi K., Olah G., Modis K., Panopoulos P. Hydrogen Sulfide and Nitric Oxide are Mutually Dependent in the Regulation of Angiogenesis and Endothelium-Dependent Vasorelaxation. Proc Natl Acad Sci U S A. 2012;109(23):9161–9166. doi: 10.1073/pnas.1202916109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sun Y., Huang Y., Yu W., Chen S., Yao Q., Zhang C. Sulfhydration-Associated Phosphodiesterase 5A Dimerization Mediates Vasorelaxant Effect of Hydrogen Sulfide. Oncotarget. 2017;8(19):31888–31900. doi: 10.18632/oncotarget.16649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.King AL., Polhemus DJ., Bhushan S., Otsuka H., Kondo K., Nicholson CK. Hydrogen Sulfide Cytoprotective Signaling is Endothelial Nitric Oxide Synthase-Nitric Oxide Dependent. Proc Natl Acad Sci U S A. 2014;111(8):3182–3187. doi: 10.1073/pnas.1321871111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lo Faro ML., Fox B., Whatmore JL., Winyard PG., Whiteman M. Hydrogen Sulfide and Nitric Oxide Interactions in Inflammation. Nitric Oxide. 2014;41:38–47. doi: 10.1016/j.niox.2014.05.014. [DOI] [PubMed] [Google Scholar]
- 46.Bibli SI., Yang G., Zhou Z., Wang R., Topouzis S., Papapetropoulos A. Role of cGMP in Hydrogen Sulfide Signaling. Nitric Oxide. 2015;46:7–13. doi: 10.1016/j.niox.2014.12.004. [DOI] [PubMed] [Google Scholar]
- 47.Zhou Z., Martin E., Sharina I., Esposito I., Szabo C., Bucci M. Regulation of Soluble Guanylyl Cyclase Redox State by Hydrogen Sulfide. Pharmacol Res. 2016;111:556–562. doi: 10.1016/j.phrs.2016.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhao W., Wang R. H(2)S-Induced Vasorelaxation and Underlying Cellular and Molecular Mechanisms. Am J Physiol Heart Circ Physiol. 2002;283(2):H474–H480. doi: 10.1152/ajpheart.00013.2002. [DOI] [PubMed] [Google Scholar]
- 49.Cheang WS., Wong WT., Shen B., Lau CW., Tian XY., Tsang SY. 4-Aminopyridine-Sensitive K+ Channels Contributes to NaHS-Induced Membrane Hyperpolarization and Relaxation in the Rat Coronary Artery. Vascul Pharmacol. 2010;53(3–4):94–98. doi: 10.1016/j.vph.2010.04.004. [DOI] [PubMed] [Google Scholar]
- 50.Lian X., Gollasch M. A Clinical Perspective: Contribution of Dysfunctional Perivascular Adipose Tissue (PVAT) to Cardiovascular Risk. Curr Hypertens Rep. 2016;18(11):82. doi: 10.1007/s11906-016-0692-z. [DOI] [PubMed] [Google Scholar]
- 51.Cacanyiova S., Majzunova M., Golas S., Berenyiova A. The Role of Perivascular Adipose Tissue and Endogenous Hydrogen Sulfide in Vasoactive Responses of Isolated Mesenteric Arteries in Normotensive and Spontaneously Hypertensive Rats. J Physiol Pharmacol. 2019;70(2) doi: 10.26402/jpp.2019.2.13. [DOI] [PubMed] [Google Scholar]
- 52.Fang L., Zhao J., Chen Y., Ma T., Xu G., Tang C. Hydrogen Sulfide Derived from Periadventitial Adipose Tissue Is A Vasodilator. J Hypertens. 2009;27(11):2174–2185. doi: 10.1097/HJH.0b013e328330a900. [DOI] [PubMed] [Google Scholar]
- 53.Schleifenbaum J., Kohn C., Voblova N., Dubrovska G., Zavarirskaya O., Gloe T. Systemic Peripheral Artery Relaxation by KCNQ Channel Openers and Hydrogen Sulfide. J Hypertens. 2010;28(9):1875–1882. doi: 10.1097/HJH.0b013e32833c20d5. [DOI] [PubMed] [Google Scholar]
- 54.Beltowski J. Endogenous Hydrogen Sulfide in Perivascular Adipose Tissue: Role in the Regulation of Vascular Tone in Physiology and Pathology. Can J Physiol Pharmacol. 2013;91(11):889–898. doi: 10.1139/cjpp-2013-0001. [DOI] [PubMed] [Google Scholar]
- 55.Kohn C., Schleifenbaum J., Szijarto IA., Marko L., Dubrovska G., Huang Y. Differential Effects of Cystathionine-gamma-lyase-Dependent Vasodilatory H2S in Periadventitial Vasoregulation of Rat and Mouse Aortas. PLoS ONE. 2012;7(8) doi: 10.1371/journal.pone.0041951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yang G., Wu L., Bryan S., Khaper N., Mani S., Wang R. Cystathionine Gamma-Lyase Deficiency and Overproliferation of Smooth Muscle Cells. Cardiovasc Res. 2010;86(3):487–495. doi: 10.1093/cvr/cvp420. [DOI] [PubMed] [Google Scholar]
- 57.Du J., Hui Y., Cheung Y., Bin G., Jiang H., Chen X. The Possible Role of Hydrogen Sulfide as A Smooth Muscle Cell Proliferation Inhibitor in Rat Cultured Cells. Heart Vessels. 2004;19(2):75–80. doi: 10.1007/s00380-003-0743-7. [DOI] [PubMed] [Google Scholar]
- 58.Tian D., Dong J., Jin S., Teng X., Wu Y. Endogenous Hydrogen Sulfide-Mediated MAPK Inhibition Preserves Endothelial Function through TXNIP Signaling. Free Radic Biol Med. 2017;110:291–299. doi: 10.1016/j.freeradbiomed.2017.06.016. [DOI] [PubMed] [Google Scholar]
- 59.Li L., Liu D., Bu D., Chen S., Wu J., Tang C. Brg1-Dependent Epigenetic Control of Vascular Smooth Muscle Cell Proliferation by Hydrogen Sulfide. Biochim Biophys Acta. 2013;1833(6):1347–1355. doi: 10.1016/j.bbamcr.2013.03.002. [DOI] [PubMed] [Google Scholar]
- 60.Shuang T., Fu M., Yang G., Wu L., Wang R. The Interaction of IGF-1/IGF-1R and Hydrogen Sulfide on the Proliferation of Mouse Primary Vascular Smooth Muscle Cells. Biochem Pharmacol. 2018;149:143–152. doi: 10.1016/j.bcp.2017.12.009. [DOI] [PubMed] [Google Scholar]
- 61.Wang Y., Wang X., Liang X., Wu J., Dong S., Li H. Inhibition of Hydrogen Sulfide on the Proliferation of Vascular Smooth Muscle Cells Involved in the Modulation of Calcium Sensing Receptor in High Homocysteine. Exp Cell Res. 2016;347(1):184–191. doi: 10.1016/j.yexcr.2016.08.004. [DOI] [PubMed] [Google Scholar]
- 62.Yang G., Wu L., Wang R. Pro-Apoptotic Effect of Endogenous H2S on Human Aorta Smooth Muscle Cells. FASEB J. 2006;20(3):553–555. doi: 10.1096/fj.05-4712fje. [DOI] [PubMed] [Google Scholar]
- 63.Yang G., Sun X., Wang R. Hydrogen Sulfide-Induced Apoptosis of Human Aorta Smooth Muscle Cells via the Activation of Mitogen-Activated Protein Kinases and Caspase-3. FASEB J. 2004;18(14):1782–1784. doi: 10.1096/fj.04-2279fje. [DOI] [PubMed] [Google Scholar]
- 64.Guan Q., Zhang Y., Yu C., Liu Y., Gao L., Zhao J. Hydrogen Sulfide Protects Against High-Glucose-Induced Apoptosis in Endothelial Cells. J Cardiovasc Pharmacol. 2012;59(2):188–193. doi: 10.1097/FJC.0b013e31823b4915. [DOI] [PubMed] [Google Scholar]
- 65.Ding HB., Liu KX., Huang JF., Wu DW., Chen JY., Chen QS. Protective Effect of Exogenous Hydrogen Sulfide on Pulmonary Artery Endothelial Cells by Suppressing Endoplasmic Reticulum Stress in A Rat Model of Chronic Obstructive Pulmonary Disease. Biomed Pharmacother. 2018;105:734–741. doi: 10.1016/j.biopha.2018.05.131. [DOI] [PubMed] [Google Scholar]
- 66.Levine B., Klionsky DJ. Development by Self-Digestion: Molecular Mechanisms and Biological Functions of Autophagy. Dev Cell. 2004;6(4):463–477. doi: 10.1016/s1534-5807(04)00099-1. [DOI] [PubMed] [Google Scholar]
- 67.Levy JMM., Towers CG., Thorburn A. Targeting Autophagy in Cancer. Nat Rev Cancer. 2017;17(9):528–542. doi: 10.1038/nrc.2017.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wu D., Zhong P., Wang J., Wang H. Exogenous Hydrogen Sulfide Mitigates LPS + ATP-Induced Inflammation by Inhibiting NLRP3 Inflammasome Activation and Promoting Autophagy in L02 Cells. Mol Cell Biochem. 2019;457(1–2):145–156. doi: 10.1007/s11010-019-03519-6. [DOI] [PubMed] [Google Scholar]
- 69.Wu D., Wang H., Teng T., Duan S., Ji A., Li Y. Hydrogen Sulfide and Autophagy: A Double Edged Sword. Pharmacol Res. 2018;131:120–127. doi: 10.1016/j.phrs.2018.03.002. [DOI] [PubMed] [Google Scholar]
- 70.Zhang QY., Jin HF., Chen S., Chen QH., Tang CS., Du JB. Hydrogen Sulfide Regulating Myocardial Structure and Function by Targeting Cardiomyocyte Autophagy. Chin Med J (Engl). 2018;131(7):839–844. doi: 10.4103/0366-6999.228249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Liu N., Wu J., Zhang L., Gao Z., Sun Y., Yu M. Hydrogen Sulphide Modulating Mitochondrial Morphology to Promote Mitophagy in Endothelial Cells under High-glucose and High-palmitate. J Cell Mol Med. 2017;21(12):3190–3203. doi: 10.1111/jcmm.13223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sen U., Sathnur PB., Kundu S., Givvimani S., Coley DM., Mishra PK. Increased Endogenous H2S Generation by CBS, CSE, and 3MST Gene Therapy Improves Ex Vivo Renovascular Relaxation in Hyperhomocysteinemia. Am J Physiol Cell Physiol. 2012;303(1):C41–C51. doi: 10.1152/ajpcell.00398.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Qiu X., Liu K., Xiao L., Jin S., Dong J., Teng X. Alpha-Lipoic Acid Regulates the Autophagy of Vascular Smooth Muscle Cells in Diabetes by Elevating Hydrogen Sulfide Level. Biochim Biophys Acta Mol Basis Dis. 2018;1864(11):3723–3738. doi: 10.1016/j.bbadis.2018.09.005. [DOI] [PubMed] [Google Scholar]
- 74.Liu J., Wu J., Sun A., Sun Y., Yu X., Liu N. Hydrogen Sulfide Decreases High Glucose/Palmitate-Induced Autophagy in Endothelial Cells by the Nrf2-ROS-AMPK Signaling Pathway. Cell Biosci. 2016;6:33. doi: 10.1186/s13578-016-0099-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Greaney JL., Kutz JL., Shank SW., Jandu S., Santhanam L., Alexander LM. Impaired Hydrogen Sulfide-Mediated Vasodilation Contributes to Microvascular Endothelial Dysfunction in Hypertensive Adults. Hypertension. 2017;69(5):902–909. doi: 10.1161/HYPERTENSIONAHA.116.08964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ma A., Nd V. Downregulation of the Renal and Hepatic Hydrogen Sulfide (H2S)-Producing Enzymes and Capacity in Chronic Kidney Disease. Nephrol Dial Transplant. 2012;27(2):498–504. doi: 10.1093/ndt/gfr560. [DOI] [PubMed] [Google Scholar]
- 77.Du J., Yan H., Tang C. Endogenous H2S Is Involved in the Development of Spontaneous Hypertension. Beijing Da Xue Xue Bao Yi Xue Ban. 2003;35(1):102. [PubMed] [Google Scholar]
- 78.Yan H., Du J., Tang C. The Possible Role of Hydrogen Sulfide on the Pathogenesis of Spontaneous Hypertension in Rats. Biochem Biophys Res Commun. 2004;313(1):22–27. doi: 10.1016/j.bbrc.2003.11.081. [DOI] [PubMed] [Google Scholar]
- 79.Huang P., Chen S., Wang Y., Liu J., Yao Q., Huang Y. Down-regulated CBS/H2S Pathway Is Involved in High-Salt-Induced Hypertension in Dahl Rats. Nitric Oxide. 2015;46:192–203. doi: 10.1016/j.niox.2015.01.004. [DOI] [PubMed] [Google Scholar]
- 80.Sun Y., Huang Y., Zhang R., Chen Q., Chen J., Zong Y. Hydrogen Sulfide Upregulates KATP channel Expression in Vascular Smooth Muscle Cells of Spontaneously Hypertensive Rats. J Mol Med (Berl). 2015;93(4):439–455. doi: 10.1007/s00109-014-1227-1. [DOI] [PubMed] [Google Scholar]
- 81.Tain YL., Hsu CN., Lu PC. Early Short-Term Treatment with Exogenous Hydrogen Sulfide Postpones the Transition from Prehypertension to Hypertension in Spontaneously Hypertensive Rat. Clin Exp Hypertens. 2018;40(1):58–64. doi: 10.1080/10641963.2017.1313847. [DOI] [PubMed] [Google Scholar]
- 82.Xue H., Zhou S., Xiao L., Guo Q., Liu S., Wu Y. Hydrogen Sulfide Improves the Endothelial Dysfunction in Renovascular Hypertensive Rats. Physiol Res. 2015;64(5):663–672. doi: 10.33549/physiolres.932848. [DOI] [PubMed] [Google Scholar]
- 83.Li J., Teng X., Jin S., Dong J., Guo Q., Tian D. Hydrogen Sulfide Improves Endothelial Dysfunction by Inhibiting the Vicious Cycle of NLRP3 Inflammasome and Oxidative Stress in Spontaneously Hypertensive Rats. J Hypertens. 2019;37(8):1633–1643. doi: 10.1097/HJH.0000000000002101. [DOI] [PubMed] [Google Scholar]
- 84.Xiao L., Dong JH., Jin S., Xue HM., Guo Q., Teng X. Hydrogen Sulfide Improves Endothelial Dysfunction via Downregulating BMP4/COX-2 Pathway in Rats with Hypertension. Oxid Med Cell Longev. 2016;2016:8128957. doi: 10.1155/2016/8128957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Xiao L., Dong JH., Teng X., Jin S., Xue HM., Liu SY. Hydrogen Sulfide Improves Endothelial Dysfunction in Hypertension by Activating Peroxisome Proliferator-Activated Receptor Delta/Endothelial Nitric Oxide Synthase Signaling. J Hypertens. 2018;36(3):651–665. doi: 10.1097/HJH.0000000000001605. [DOI] [PubMed] [Google Scholar]
- 86.Ni X., Zhang L., Peng M., Shen TW., Yu XS., Shan LY. Hydrogen Sulfide Attenuates Hypertensive Inflammation via Regulating Connexin Expression in Spontaneously Hypertensive Rats. Med Sci Monit. 2018;24:1205–1218. doi: 10.12659/MSM.908761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Yu W., Liao Y., Huang Y., Chen SY., Sun Y., Sun C. Endogenous Hydrogen Sulfide Enhances Carotid Sinus Baroreceptor Sensitivity by Activating the Transient Receptor Potential Cation Channel Subfamily V Member 1 (TRPV1) Channel. J Am Heart Assoc. 2017;6(5) doi: 10.1161/JAHA.116.004971. pii: e004971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Pozsgai G., Hajna Z., Bagoly T., Boros M., Kemény Á., Materazzi S. The Role of Transient Receptor Potential Ankyrin 1 (TRPA1) Receptor Activation in Hydrogen-Sulphide-Induced CGRP-Release and Vasodilation. Eur J Pharmacol. 2012;689(1–3):56–64. doi: 10.1016/j.ejphar.2012.05.053. [DOI] [PubMed] [Google Scholar]
- 89.Pozsgai G., Batai IZ., Pinter E. Effects of Sulfide and Polysulfides Transmitted by Direct or Signal Transduction-Mediated Activation of TRPA1 Channels. Br J Pharmacol. 2019;176(4):628–645. doi: 10.1111/bph.14514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zhao X., Zhang LK., Zhang CY., Zeng XJ., Yan H., Jin HF. Regulatory Effect of Hydrogen Sulfide on Vascular Collagen Content in Spontaneously Hypertensive Rats. Hypertens Res. 2008;31(8):1619–1630. doi: 10.1291/hypres.31.1619. [DOI] [PubMed] [Google Scholar]
- 91.Sun L., Jin H., Sun L., Chen S., Huang Y., Liu J. Hydrogen Sulfide Alleviates Myocardial Collagen Remodeling in Association with Inhibition of TGF-beta/Smad Signaling Pathway in Spontaneously Hypertensive Rats. Mol Med. 2015;20:503–515. doi: 10.2119/molmed.2013.00096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Liang YF., Zhang DD., Yu XJ., Gao HL., Liu KL., Qi J. Hydrogen Sulfide in Paraventricular Nucleus Attenuates Blood Pressure by Regulating Oxidative Stress and Inflammatory Cytokines in High Salt-Induced Hypertension. Toxicol Lett. 2017;270:62–71. doi: 10.1016/j.toxlet.2017.02.004. [DOI] [PubMed] [Google Scholar]
- 93.Zhang J., Chen S., Liu H., Zhang B., Zhao Y., Ma K. Hydrogen Sulfide Prevents Hydrogen Peroxide-Induced Activation of Epithelial Sodium Channel through A PTEN/PI(3,4,5)P3 Dependent Pathway. PLoS ONE. 2013;8(5) doi: 10.1371/journal.pone.0064304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Gao L., Xu Z., Yin Z., Chen K., Wang C., Zhang H. Association of Hydrogen Sulfide with Alterations of Monocyte Chemokine Receptors, CCR2 and CX3CR1 in Patients with Coronary Artery Disease. Inflamm Res. 2015;64(8):627–635. doi: 10.1007/s00011-015-0844-7. [DOI] [PubMed] [Google Scholar]
- 95.Wang Y., Zhao X., Jin H., Wei H., Li W., Bu D. Role of Hydrogen Sulfide in the Development of Atherosclerotic Lesions in Apolipoprotein E Knockout Mice. Arterioscler Thromb Vasc Biol. 2009;29(2):173–179. doi: 10.1161/ATVBAHA.108.179333. [DOI] [PubMed] [Google Scholar]
- 96.Mani S., Li H., Untereiner A., Wu L., Yang G., Austin RC. Decreased Endogenous Production of Hydrogen Sulfide Accelerates Atherosclerosis. Circulation. 2013;127(25):2523–2534. doi: 10.1161/CIRCULATIONAHA.113.002208. [DOI] [PubMed] [Google Scholar]
- 97.Bibli SI., Hu J., Sigala F., Wittig I., Heidler J., Zukunft S. Cystathionine gamma Lyase Sulfhydrates the RNA Binding Protein Human Antigen R to Preserve Endothelial Cell Function and Delay Atherogenesis. Circulation. 2019;139(1):101–114. doi: 10.1161/CIRCULATIONAHA.118.034757. [DOI] [PubMed] [Google Scholar]
- 98.Wang ZJ., Wu J., Guo W., Zhu YZ. Atherosclerosis and the Hydrogen Sulfide Signaling Pathway-Therapeutic Approaches to Disease Prevention. Cell Physiol Biochem. 2017;42(3):859–875. doi: 10.1159/000478628. [DOI] [PubMed] [Google Scholar]
- 99.van den Born JC., Mencke R., Conroy S., Zeebregts CJ., van Goor H., Hillebrands JL. Cystathionine gamma-lyase Is Expressed in Human Atherosclerotic Plaque Microvessels and Is Involved in Micro-Angiogenesis. Sci Rep. 2016;6:34608. doi: 10.1038/srep34608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Xiong Q., Wang Z., Yu Y., Wen Y., Suguro R., Mao Y. Hydrogen Sulfide Stabilizes Atherosclerotic Plaques in Apolipoprotein E Knockout Mice. Pharmacol Res. 2019 doi: 10.1016/j.phrs.2019.04.006. [DOI] [PubMed] [Google Scholar]
- 101.Liu Z., Han Y., Li L., Lu H., Meng G., Li X. The Hydrogen Sulfide Donor, GYY4137, Exhibits Anti-atherosclerotic Activity in High Fat Fed Apolipoprotein E(-/-) Mice. Br J Pharmacol. 2013;169(8):1795–1809. doi: 10.1111/bph.12246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Leucker TM., Nomura Y., Kim JH., Bhatta A., Wang V., Wecker A. Cystathionine gamma-lyase Protects Vascular Endothelium: a Role for Inhibition of Histone Deacetylase 6. Am J Physiol Heart Circ Physiol. 2017;312(4):H711–H720. doi: 10.1152/ajpheart.00724.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Fu XD, Zhou KW, Gao Q, Zheng SH, Chen HY, Li P. 17β-Estradiol Attenuates Atherosclerosis Development: The Possible Role of Hydrogen Sulfide. Int J Cardiol. 2013;167(3):1061–1063. doi: 10.1016/j.ijcard.2012.10.071. [DOI] [PubMed] [Google Scholar]
- 104.Cheung SH., Lau JYW. Hydrogen Sulfide Mediates Athero-Protection against Oxidative Stress via S-sulfhydration. PLoS One. 2018;13(3) doi: 10.1371/journal.pone.0194176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Xie L., Gu Y., Wen M., Zhao S., Wang W., Ma Y. Hydrogen Sulfide Induces Keap1 S-sulfhydration and Suppresses Diabetes-Accelerated Atherosclerosis via Nrf2 Activation. Diabetes. 2016;65(10):3171–3184. doi: 10.2337/db16-0020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Li Z., Polhemus DJ., Lefer DJ. Evolution of Hydrogen Sulfide Therapeutics to Treat Cardiovascular Disease. Circ Res. 2018;123(5):590–600. doi: 10.1161/CIRCRESAHA.118.311134. [DOI] [PubMed] [Google Scholar]
- 107.Aghagolzadeh P., Radpour R., Bachtler M., van Goor H., Smith ER., Lister A. Hydrogen Sulfide Attenuates Calcification of Vascular Smooth Muscle Cells via KEAP1/NRF2/NQO1 Activation. Atherosclerosis. 2017;265:78–86. doi: 10.1016/j.atherosclerosis.2017.08.012. [DOI] [PubMed] [Google Scholar]
- 108.Wen YD., Wang H., Zhu YZ. Protective Effects of Hydrogen Sulfide in the Development of Atherosclerosis in Hyperlipidemic Rabbit. Nitric Oxide. 2012;27 [Google Scholar]
- 109.Potor L., Nagy P., Mehes G., Hendrik Z., Jeney V., Petho D. Hydrogen Sulfide Abrogates Hemoglobin-Lipid Interaction in Atherosclerotic Lesion. Oxid Med Cell Longev. 2018;2018:3812568. doi: 10.1155/2018/3812568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Du J., Huang Y., Yan H., Zhang Q., Zhao M., Zhu M. Hydrogen Sulfide Suppresses Oxidized Low-Density Lipoprotein (ox-LDL)-Stimulated Monocyte Chemoattractant Protein 1 Generation from Macrophages via the Nuclear Factor KappaB (NF-kappaB) Pathway. J Biol Chem. 2014;289(14):9741–9753. doi: 10.1074/jbc.M113.517995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Yue LM., Gao YM., Han BH. Evaluation on the Effect of Hydrogen Sulfide on the NLRP3 Signaling Pathway and Its Involvement in the Pathogenesis of Atherosclerosis. J Cell Biochem. 2019;120(1):481–492. doi: 10.1002/jcb.27404. [DOI] [PubMed] [Google Scholar]
- 112.Du C., Lin X., Xu W., Zheng F., Cai J., Yang J. Sulfhydrated Sirtuin-1 Increasing Its Deacetylation Activity Is an Essential Epigenetics Mechanism of Anti-Atherogenesis by Hydrogen Sulfide. Antioxid Redox Signal. 2019;30(2):184–197. doi: 10.1089/ars.2017.7195. [DOI] [PubMed] [Google Scholar]
- 113.Vacek TP., Rehman S., Neamtu D., Yu S., Givimani S., Tyagi SC. Matrix Metalloproteinases in Atherosclerosis: Role of Nitric Oxide, Hydrogen Sulfide, Homocysteine, and Polymorphisms. Vasc Health Risk Manag. 2015;11:173–183. doi: 10.2147/VHRM.S68415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Lin Y., Chen Y., Zhu N., Zhao S., Fan J., Liu E. Hydrogen Sulfide Inhibits Development of Atherosclerosis through Up-regulating Protein S-nitrosylation. Biomed Pharmacother. 2016;83:466–476. doi: 10.1016/j.biopha.2016.07.003. [DOI] [PubMed] [Google Scholar]
- 115.Li W., Du JB., Jin HF. Effects of Hydrogen Sulfide Donor on Production of Adrenomedullin and Atrial Natriuretic Peptide in Rats with Atherosclerosis. Zhongguo Dang Dai Er Ke Za Zhi. 2015;17(10):1119–1123. [PubMed] [Google Scholar]
- 116.Lin Y., Zeng H., Gao L., Gu T., Wang C., Zhang H. Hydrogen Sulfide Attenuates Atherosclerosis in a Partially Ligated Carotid Artery Mouse model via Regulating Angiotensin Converting Enzyme 2 Expression. Front Physiol. 2017;8:782. doi: 10.3389/fphys.2017.00782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Zhang CY., Du JB., Bu DF., Yan H., Tang XY., Tang CS. The Regulatory Effect of Hydrogen Sulfide on Hypoxic Pulmonary Hypertension in Rats. Biochem Biophys Res Commun. 2003;302(4):810–816. doi: 10.1016/s0006-291x(03)00256-0. [DOI] [PubMed] [Google Scholar]
- 118.Li XH., Du JB., Shi L., Li J., Tang XY., Qi JG. Down-regulation of Endogenous Hydrogen Sulfide Pathway in Pulmonary Hypertension and Pulmonary Vascular Structural Remodeling Induced by High Pulmonary Blood Flow in Rats. Circ J. 2005;69(11):1418–1424. doi: 10.1253/circj.69.1418. [DOI] [PubMed] [Google Scholar]
- 119.Feng SS., Yu W., Du SX., Du JB., Jin HF. Change of endogenous hydrogen sulfide pathway in the monocrotaline-induced pulmonary hypertensive rats. J Appl Clin Pediatr. 2016;31(19):1489–1492. [Google Scholar]
- 120.Ariyaratnam P., Loubani M., Morice AH. Hydrogen Sulphide Vasodilates Human Pulmonary Arteries: a Possible Role in Pulmonary Hypertension? Microvasc Res. 2013;90:135–137. doi: 10.1016/j.mvr.2013.09.002. [DOI] [PubMed] [Google Scholar]
- 121.Jin HF., Liang C., Liang JM., Tang CS., Du JB. Effects of Hydrogen Sulfide on Vascular Inflammation in Pulmonary Hypertension Induced by High Pulmonary Blood Flow: Experiment with Rats. Zhonghua yi xue za zhi. 2008;88(32):2235–2239. [PubMed] [Google Scholar]
- 122.Wu J., Pan W., Wang C., Dong H., Xing L., Hou J. H2S Attenuates Endoplasmic Reticulum Stress in Hypoxia-Induced Pulmonary Artery Hypertension. Biosci Rep. 2019;39(7) doi: 10.1042/BSR20190304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Li W., Jin HF., Liu D., Sun JH., Jian PJ., Li XH. Hydrogen Sulfide Induces Apoptosis of Pulmonary Artery Smooth Muscle Cell in Rats with Pulmonary Hypertension Induced by High Pulmonary Blood Flow. Chin Med J (Engl). 2009;122(24):3032–3038. [PubMed] [Google Scholar]
- 124.Li Y., Liu G., Cai D., Pan B., Lin Y., Li X. H2S Inhibition of Chemical Hypoxia-Induced Proliferation of HPASMCs is Mediated by the Upregulation of COX-2/PGI2. Int J Mol Med. 2014;33(2):359–366. doi: 10.3892/ijmm.2013.1579. [DOI] [PubMed] [Google Scholar]
- 125.Yao Z., Wang C. A Novel Mechanism of Sildenafil Improving the Excessive Proliferation and H2S Production in Pulmonary Arterial Smooth Muscle Cells. J Cardiovasc Pharmacol. 2019 doi: 10.1097/FJC.0000000000000714. [DOI] [PubMed] [Google Scholar]
- 126.Zhang QY., Du JB., Zhou WJ., Yan H, Tang CS., Zhang CY. Impact of Hydrogen Sulfide on Carbon Monoxide/Heme Oxygenase Pathway in the Pathogenesis of Hypoxic Pulmonary Hypertension. Biochem Biophys Res Commun. 2004;317(1):30–37. doi: 10.1016/j.bbrc.2004.02.176. [DOI] [PubMed] [Google Scholar]
- 127.Feng S., Chen S., Yu W., Zhang D., Zhang C., Tang C. H2S Inhibits Pulmonary Arterial Endothelial Cell Inflammation in Rats with Monocrotaline-Induced Pulmonary Hypertension. Lab Invest. 2017;97(3):268–278. doi: 10.1038/labinvest.2016.129. [DOI] [PubMed] [Google Scholar]
- 128.Jin HF., Cong BL., Zhao B., Zhang CY., Liu XM., Zhou WJ. Effects of Hydrogen Sulfide on Hypoxic Pulmonary Vascular Structural Remodeling. Life Sci. 2006;78(12):1299–1309. doi: 10.1016/j.lfs.2005.07.009. [DOI] [PubMed] [Google Scholar]
- 129.Wei HL., Zhang CY., Jin HF., Tang CS., Du JB. Hydrogen Sulfide Regulates Lung Tissue-Oxidized Glutathione and Total Antioxidant Capacity in Hypoxic Pulmonary Hypertensive Rats. Acta Pharmacol Sin. 2008;29(6):670–679. doi: 10.1111/j.1745-7254.2008.00796.x. [DOI] [PubMed] [Google Scholar]
- 130.Li X., Jin H., Bin G., Wang L., Tang C., Du J. Endogenous Hydrogen Sulfide Regulates Pulmonary Artery Collagen Remodeling in Rats with High Pulmonary Blood Flow. Exp Biol Med (Maywood). 2009;234(5):504–512. doi: 10.3181/0807-RM-230. [DOI] [PubMed] [Google Scholar]
- 131.Li MX., Chen YH., Liao CC., Lin F., Bai Y., Mi WJ. Role and Mechanism of Hydrogen Sulfide in Cigarette Smoke Induced Chronic Obstructive Pulmonary Disease Related Pulmonary Vascular Remodeling in Rats. Zhonghua yi xue za zhi. 2017;97(2):137–142. doi: 10.3760/cma.j.issn.0376-2491.2017.02.012. [DOI] [PubMed] [Google Scholar]
- 132.Li X., Du J., Jin H., Tang X., Bu D., Tang C. The Regulatory Effect of Endogenous Hydrogen Sulfide on Pulmonary Vascular Structure and Gasotransmitters in Rats with High Pulmonary Blood Flow. Life Sci. 2007;81(10):841–849. doi: 10.1016/j.lfs.2007.07.021. [DOI] [PubMed] [Google Scholar]
- 133.Chen YH., Wu R., Geng B., Qi YF., Wang PP., Yao WZ. Endogenous Hydrogen Sulfide Reduces Airway Inflammation and Remodeling in a Rat Model of Asthma. Cytokine. 2009;45(2):117–123. doi: 10.1016/j.cyto.2008.11.009. [DOI] [PubMed] [Google Scholar]
- 134.Souza-Costa DC., Zerbini T., Metzger IF., Rocha JB., Gerlach RF., Tanus-Santos JE. L-arginine Attenuates Acute Pulmonary Embolism-Induced Oxidative Stress and Pulmonary Hypertension. Nitric Oxide. 2005;12(1):9–14. doi: 10.1016/j.niox.2004.10.009. [DOI] [PubMed] [Google Scholar]
- 135.Wang YF., Shi L., Du JB., Tang CS. Impact of L-arginine on Hydrogen Sulfide/Cystathionine-gamma-lyase Pathway in Rats with High Blood Flow-Induced Pulmonary Hypertension. Biochem Biophys Res Commun. 2006;345(2):851–857. doi: 10.1016/j.bbrc.2006.04.162. [DOI] [PubMed] [Google Scholar]
- 136.Papapetropoulos A., Whiteman M., Cirino G. Pharmacological Tools for Hydrogen Sulphide Research: A Brief, Introductory Guide for Beginners. Br J Pharmacol. 2015;172(6):1633–1637. doi: 10.1111/bph.12806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Calvert JW., Elston M., Nicholson CK., Gundewar S., Jha S., Elrod JW. Genetic and Pharmacologic Hydrogen Sulfide Therapy Attenuates Ischemia-Induced Heart Failure in Mice. Circulation. 2010;122(1):11–19. doi: 10.1161/CIRCULATIONAHA.109.920991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Wang X., Wang Q., Guo W., Zhu YZ. Hydrogen Sulfide Attenuates Cardiac Dysfunction in a Rat Model of Heart Failure: A Mechanism through Cardiac Mitochondrial Protection. Biosci Rep. 2011;31(2):87–98. doi: 10.1042/BSR20100003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Kar S., Shahshahan HR., Kambis TN., Yadav SK., Li Z., Lefer DJ. Hydrogen Sulfide Ameliorates Homocysteine-Induced Cardiac Remodeling and Dysfunction. Front Physiol. 2019;10:598. doi: 10.3389/fphys.2019.00598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Sun X., Wang W., Dai J., Jin S., Huang J., Guo C. A Long-Term and Slow-Releasing Hydrogen Sulfide Donor Protects against Myocardial Ischemia/Reperfusion Injury. Sci Rep. 2017;7(1):3541. doi: 10.1038/s41598-017-03941-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Hu J., Xue Y., Tang K., Fan J., Du J., Li W. The Protective Effects of Hydrogen Sulfide on the Myocardial Ischemia via Regulating Bmal1. Biomed Pharmacother. 2019;120 doi: 10.1016/j.biopha.2019.109540. [DOI] [PubMed] [Google Scholar]
- 142.Bai YD., Yang YR., Mu XP., Lin G., Wang YP., Jin S. Hydrogen Sulfide Alleviates Acute Myocardial Ischemia Injury by Modulating Autophagy and Inflammation Response under Oxidative Stress. Oxid Med Cell Longev. 2018;2018:3402809. doi: 10.1155/2018/3402809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Miao L., Xin XM., Xin H., Shen XY., Zhu YZ. Hydrogen Sulfide Recruits Macrophage Migration by Integrin β1-Src-FAK/Pyk2-Rac Pathway in Myocardial Infarction. Sci Rep. 2016;6:22363. doi: 10.1038/srep22363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Miao L., Shen X., Whiteman M., Xin H., Shen Y., Xin X. Hydrogen Sulfide Mitigates Myocardial Infarction via Promotion of Mitochondrial Biogenesis-Dependent M2 Polarization of Macrophages. Antioxid Redox Signal. 2016;25(5):268–281. doi: 10.1089/ars.2015.6577. [DOI] [PubMed] [Google Scholar]
- 145.Cirino G., Vellecco V., Bucci M. Nitric Oxide and Hydrogen Sulfide: The Gasotransmitter Paradigm of the Vascular System. Br J Pharmacol. 2017;174(22):4021–4031. doi: 10.1111/bph.13815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Jeddi S., Gheibi S., Kashfi K., Carlstrom M., Ghasemi A. Dose-dependent Effects of Long-term Administration of Hydrogen Sulfide on Myocardial Ischemia-Reperfusion Injury in Male Wistar Rats: Modulation of RKIP, NF-kappaB, and Oxidative Stress. Int J Mol Sci. 2020;21(4):1415. doi: 10.3390/ijms21041415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Testai L., Marino A., Piano I., Brancaleone V., Tomita K., Di Cesare Mannelli L. The Novel H2S-Donor 4-carboxyphenyl Isothiocyanate Promotes Cardioprotective Effects against Ischemia/Reperfusion Injury through Activation of MitoKATP Channels and Reduction of Oxidative Stress. Pharmacol Res. 2016;113(Pt A):290–299. doi: 10.1016/j.phrs.2016.09.006. [DOI] [PubMed] [Google Scholar]
- 148.Bucci M., Roviezzo F., Brancaleone V., Di Lorenzo A., Evangelista S., Gori M. ACE-inhibition Ameliorates Vascular Reactivity and Delays Diabetes Outcome in NOD Mice. Vascul Pharmacol. 2008;49(2–3):84–90. doi: 10.1016/j.vph.2008.06.002. [DOI] [PubMed] [Google Scholar]
- 149.Napoli C., Cicala C., D’Armiento FP., Roviezzo F., Somma P., de Nigris F. Beneficial Effects of ACE-inhibition with Zofenopril on Plaque Formation and Low-density Lipoprotein Oxidation in Watanabe Heritable Hyperlipidemic Rabbits. Gen Pharmacol. 1999;33(6):467–477. doi: 10.1016/s0306-3623(99)00043-9. [DOI] [PubMed] [Google Scholar]
- 150.Napoli C., Pinto A., Cirino G. Pharmacological Modulation, Preclinical Studies, and New Clinical Features of Myocardial Ischemic Preconditioning. Pharmacol Ther. 2000;88(3):311–331. doi: 10.1016/s0163-7258(00)00093-0. [DOI] [PubMed] [Google Scholar]
- 151.Borghi C., Bacchelli S., Esposti DD., Ambrosioni E. A Review of the Angiotensin-converting Enzyme Inhibitor, Zofenopril, in the Treatment of Cardiovascular Diseases. Expert Opin Pharmacother. 2004;5(9):1965–1977. doi: 10.1517/14656566.5.9.1965. [DOI] [PubMed] [Google Scholar]
- 152.Borghi C., Bacchelli S., Esposti DD., Ambrosioni E. Effects of The Early ACE Inhibition in Diabetic Nonthrombolyzed Patients with Anterior Acute Myocardial Infarction. Diabetes Care. 2003;26(6):1862–1868. doi: 10.2337/diacare.26.6.1862. [DOI] [PubMed] [Google Scholar]
- 153.Borghi C., Omboni S., Reggiardo G., Bacchelli S., Degli Esposti D., Ambrosioni E. Efficacy of Zofenopril Compared with Placebo and Other Angiotensin-converting Enzyme Inhibitors in Patients with Acute Myocardial Infarction and Previous Cardiovascular Risk Factors: A Pooled Individual Data Analysis of 4 Randomized, Double-blind, Controlled. Prospective Studies. J Cardiovasc Pharmacol. 2017;69(1):48–54. doi: 10.1097/FJC.0000000000000440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Borghi C., Omboni S., Novo S., Vinereanu D., Ambrosio G., Ambrosioni E. Zofenopril and Ramipril in Patients with Left Ventricular Systolic Dysfunction after Acute Myocardial Infarction: A Propensity Analysis of the Survival of Myocardial Infarction Long-term Evaluation (SMILE) 4 study. J Renin Angiotensin Aldosterone Syst. 2016;17(4) doi: 10.1177/1470320316656480. pii: 1470320316656480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Bucci M., Vellecco V., Cantalupo A., Brancaleone V., Zhou Z., Evangelista S. Hydrogen Sulfide Accounts for the Peripheral Vascular Effects of Zofenopril Independently of ACE Inhibition. Cardiovasc Res. 2014;102(1):138–147. doi: 10.1093/cvr/cvu026. [DOI] [PubMed] [Google Scholar]
- 156.Terzuoli E., Monti M., Vellecco V., Bucci M., Cirino G., Ziche M. Characterization of Zofenoprilat as An Inducer of Functional Angiogenesis through Increased H2S Availability. Br J Pharmacol. 2015;172(12):2961–2973. doi: 10.1111/bph.13101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Monti M., Terzuoli E., Ziche M., Morbidelli L. H2S Dependent and Independent Anti-inflammatory Activity of Zofenoprilat in Cells of the Vascular Wall. Pharmacol Res. 2016;113(Pt A):426–437. doi: 10.1016/j.phrs.2016.09.017. [DOI] [PubMed] [Google Scholar]
- 158.Donnarumma E., Ali MJ., Rushing AM., Scarborough AL., Bradley JM., Organ CL. Zofenopril Protects Against Myocardial Ischemia-Reperfusion Injury by Increasing Nitric Oxide and Hydrogen Sulfide Bioavailability. J Am Heart Assoc. 2016;5(7) doi: 10.1161/JAHA.116.003531. [DOI] [PMC free article] [PubMed] [Google Scholar]