

Abstract
Nonalcoholic hepatitis (NASH) is the progressive inflammatory form of nonalcoholic fatty liver disease. Although, the mechanisms of hepatic inflammation in NASH remain incompletely understood, emerging literature implicate the proinflammatory environment created by toxic lipid-induced hepatocyte injury, termed lipotoxicity. Interestingly, numerous NASH promoting kinases in hepatocytes, immune cells, and adipocytes are activated by the lipotoxic insult associated with obesity. In the current review, we discuss recent advances in NASH promoting kinases as disease mediators and therapeutic targets. The focus of the review is mainly on the mitogen activated protein kinases including mixed lineage kinase 3 (MLK3), apoptosis signal-regulating kinase 1 (ASK1), c-Jun N-terminal kinase (JNK) and p38 MAPK; the ER stress kinases protein kinase RNA-like ER kinase (PERK) and inositol-requiring protein-1α (IRE-1α); as well as the Rho-associated protein kinase 1 (ROCK1). We also discuss various pharmacological agents targeting these stress kinases in NASH that are under different phases of development.
Keywords: Mitogen activated protein kinases, Rho-associated protein kinase, Endoplasmic reticulum stress, Nonalcoholic Steatohepatitis
INTRODUCTION
Nonalcoholic fatty liver disease (NAFLD) characterized by hepatic steatosis, in the absence of alcohol overuse, is the leading cause of liver diseases worldwide, with an estimated global prevalence rate of about 24%.1 A subset of patients with NAFLD (up to 30%) develop nonalcoholic steatohepatitis (NASH) characterized by hepatocellular injury, hepatic inflammation, and liver fibrosis.2 Current concepts suggest that excess circulating free fatty acids (FFAs) and accumulation of lipid intermediates in hepatocytes cause hepatocellular lipotoxicity, leading to cellular stress, dysfunction and eventually cell death.3 Lipotoxicity-induced hepatocyte cell death appears to be mediated, in part, by the apoptotic machinery activated by the death receptors and intracellular stress pathways, and potentiated by the engagement of the mitogen activated protein kinases (MAPK)4 and the endoplasmic reticulum (ER) stress response.5 This pro-apoptotic machinery may culminate in cell death, or sublethal insult promoting liver injury and inflammation. Moreover, NASH patients are at risk of end stage liver disease, mainly secondary to the unrelenting sterile inflammatory response triggered by hepatocyte lipotoxicity.5 Hence, identifying molecular mediators in hepatocyte lipotoxicity and liver inflammation in NASH is of critical biomedical importance. In this review, we discuss the concept that toxic lipids initiate signaling processes, and activate multiple stress kinases in the hepatocyte resulting in both lethal and sublethal injury that eventually culminates in a sterile inflammatory response.6 We will also provide a brief overview about the role of different stress kinases in myeloid cells, and adipocyte in NASH. We will highlight established and potential NASH therapeutic targets in the stress kinases family. Given the broad nature of the subject, we elected to discuss recent conceptual advances in the field.
MITOGEN ACTIVATED PROTEIN KINASES (MAPK)
In mammals, three major groups of MAPK have been identified. Each activated by a protein kinase signaling cascade in response to a wide variety of stress signals and mediates signal transduction cascades involved in cell growth, differentiation, and apoptosis. MAPK signaling cascades are phosphorylated on regulatory tyrosine and threonine residues and activated through a relay module, in which MAP kinase kinase kinase (MAP3K) activates MAPK kinase (MAP2K), which in turn activates MAPK.7 The three major subgroups are classified based on sequence similarity, differential activation by agonists, and substrate specificity, and are named according to their executing downstream MAPK, such as the extracellular signal-regulated kinase (ERK), the p38 kinase, and the c-Jun N-terminal kinase (JNK) families. Both JNK1/2 and p38 α/β are activated by the MAP3Ks apoptosis signal-regulating kinase 1 (ASK1),8 as well as mixed lineage kinase 3 (MLK3) during obesity and will be a focus of the current review.9–11 On the other hand ERK1/2 are preferentially activated by growth factors rather than stressors associated with obesity, and were recently reviewed elsewhere.8 Conversely, the MAPKs are inactivated by direct dephosphorylation of their threonine and tyrosine residues by a group of dual specificity protein tyrosine phosphatases (DUSPs) called MAPK phosphatases (MKPs), which play a crucial role in regulating the magnitude and temporal kinetics of MAPK activity.12
Apoptosis signal-regulating kinase 1 (ASK1)
Due to its pleiotropic functions and ubiquitous expression, the activity of ASK1 is tightly regulated by molecules that induce ASK1 conformational changes facilitating the homodimerization or post-translational modifications, such as phosphorylation, ubiquitylation and deubiquitylation (Figure 1).13 During lipotoxicity ASK1 is activated through homodimerization and subsequent autophosphorylation14. ASK1 forms a high-molecular-mass complex through its carboxy‑terminal coiled-coil (CCC) domain. The thioredoxin (Trx) binding site is located near the amino‑terminal coiled-coil (NCC) domain and keeps ASK1 inactive. Under lipotoxic stress, Trx dissociates from ASK1 and the adaptor protein TNF receptor-associated factor (TRAF) is recruited and induces homo-oligomerization and autophosphorylation of ASK1, with subsequent activation of JNK, and p38. TRAF1 expression was increased in patients with NAFLD, and TRAF1 deficiency was hepatoprotective in a mouse model of NASH.15 Nonetheless other TRAFs including TRAF 2 and TRAF6 are known to induce ASK1 activation.16 A recent study reported that TRAF6 promotes lysine 6-linked polyubiquitination and subsequent activation of ASK1. ASK1 activation triggers the release of proinflammatory and profibrotic factors including CXCL10 from hepatocytes which, in turn, activate hepatic stellate cells and induce hepatic fibrosis.17 Likewise interruption of ASK1 N-terminus-mediated dimerization by CASP8 and FADD-like apoptosis regulator (CFLAR) also known as CFLIP (Cellular FLICE (FADD-like IL-1β converting enzymes)-like inhibitory protein),14 blocks ASK1 downstream signaling, and attenuates the progression of steatohepatitis and metabolic disorders in both mice and nonhuman primates fed an obesity-induced diet (OID).
Figure 1. ASK-interacting protein 1 (ASK1) activation during lipotoxicity:
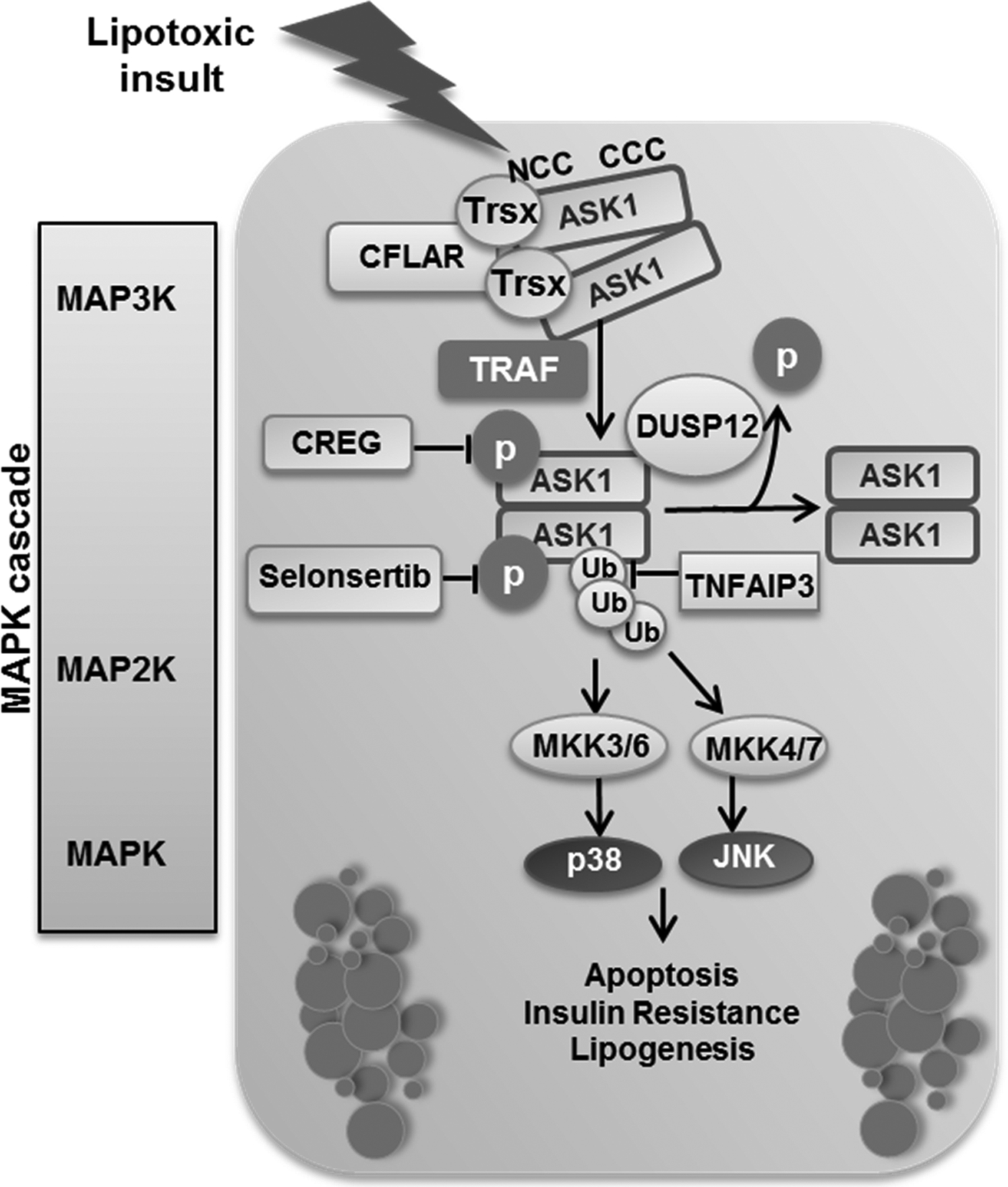
ASK1 forms a high-molecular-mass complex through its carboxy‑terminal coiled-coil (CCC) domain. The thioredoxin (Trx) binding site is located near the amino‑terminal coiled-coil (NCC) domain and keeps ASK1 inactive. Under conditions of stress, Trx dissociates from ASK1 and tumor necrosis factor (TNF) receptor-associated factor (TRAF) molecules are recruited and induce oligomerization of ASK1, resulting in its autophosphorylation and activation. CASP8 and FADD-like apoptosis regulator (CFLAR) target the N‑terminal-mediated dimerization of ASK1 and blocks its activation. Dual specificity phosphatase (DUSP) 12 dephosphorylates ASK1, which leads to its inactivation. Selonsertib binds to the catalytic kinase domain of ASK1 in an ATP-competitive manner and inhibits its activity. Cellular repressor of E1A-stimulated genes (CREG) interacts directly with ASK1 and inhibits its phosphorylation. The deubiquitinase TNF alpha-induced protein 3 (TNFAP3) deubiquitinates and suppresses ASK1. ASK1 activation triggers a mitogen activated protein kinase (MAPK) signaling cascade leading to c-Jun N‑terminal kinase (JNK) and p38 activation, which results in insulin resistance, lipogenesis, and apoptosis. MKK, mitogen-activated protein (MAP) kinase kinase; P, phosphate group; Ub, ubiquitin.
The small molecule ASK1 inhibitor Selonsertib (GS‑4997) binds to the catalytic kinase domain of ASK1 in an ATP-competitive manner and inhibits its activity. Although beneficial in preclinical studies, Selonsertib was not efficacious in NASH clinical trials.18,19 However, ASK1 is highly regulated positively and negatively, and the patients were not stratified by biomarkers for ASK1 activation in these trials. A protective role of dual specificity protein tyrosine phosphatases 12 (DUSP12) in murine NASH was reported.20 DUSP12 binds to ASK1 and promotes its dephosphorylation, blocking its downstream signaling cascade, leading to reduced lipogenesis and apoptosis, and improvement of high fat diet (HFD)-induced hepatic steatosis, insulin resistance and inflammation in mice.20 Cellular repressor of E1A-stimulated genes (CREG) interacts directly with ASK1 and inhibits its phosphorylation, thereby blocking the downstream MKK4/7-JNK1 signaling pathway and alleviating obesity, insulin resistance, and hepatic steatosis in mice.21 Interestingly polyubiquitination of ASK1 is related to its activation, Zhang et al. identified the deubiquitinase tumor necrosis factor alpha–induced protein 3 (TNFAIP3) as a key endogenous suppressor of ASK1 activation that interacts with and deubiquitinates ASK1 in hepatocytes. Furthermore, tnfaip3 gene delivery in the liver in both mouse and nonhuman primate models of NASH substantially blocked the disease progression.22 Interestingly a recent study in a mouse model of NASH showed that ASK1 hepatocyte depletion blunts autophagy, thereby enhancing lipid droplet accumulation and liver fibrosis. ASK1 expression in this study correlated negatively with liver fat content and NASH scores in obese humans, but positively with markers for autophagy.23 This study highlights the pleotropic function of ASK1 in NASH. Furthermore, results from phase 3 studies of selonsertib in patients with NASH and bridging fibrosis (STELLAR-3) and compensated cirrhosis (STELLAR-4)18,19 have not shown improvement in fibrosis or NASH amelioration. However, as stated above, clinical trials need to be designed employing biomarkers to better identify patients with markers of ASK1 activation.
Mixed lineage kinase (MLK)
Members of the MLK family include MLK1, MLK2, MLK3, dual leucine zipper-bearing kinase, and leucine zipper-bearing kinase. Many of these protein kinases are expressed in a limited number of tissues, however MLK3 is ubiquitously expressed.24 Previous studies have demonstrated that MLK3 is the MAP3K that mediates FFAs induced JNK activation.25,26 Mlk3 genetic deficiency in a murine nutritional model of DIO and NASH is protective against disease progression by decreasing liver steatosis, injury, inflammation and fibrosis, independent of body weight. These effects were accompanied by a reduction in the hepatic activating phosphorylation of JNK1/2.9 Furthermore, reduced JNK activation in liver and protection from HFD-induced insulin resistance and obesity was observed in compound mutant global Mlk2–/– and Mlk3–/– mice.27 Moreover, we have reported that during sublethal hepatocyte lipotoxic injury, MLK3 mediates the induction of C-X-C motif chemokine 10 (CXCL10) via an MKK3/6-p38- signal transducer and activator of transcription 1 (STAT1) signaling cascade, and the release of CXCL10-enriched extracellular vesicles (EV)s from hepatocytes by a JNK-dependent mechanism (Figure 2).10,28 The release of proinflammatory EVs by hepatocytes during lipotoxic stress is an example of sublethal injury promoting liver inflammation. CXCL10 is a potent chemotactic ligand that links hepatocyte lipotoxicity to macrophage-associated liver inflammation in NASH.29 Furthermore, we and others have demonstrated increased CXCL10 hepatic expression and serum levels in patients with NASH; this increase correlates with disease severity.28,30 Moreover, we reported that the small-molecule MLK3 inhibitor URMC099 was well tolerated, and efficacious in reversing diet-induced NASH in mice.11 Nevertheless, MLK3 remains an unexplored therapeutic target in human NASH.
Figure 2. Mixed lineage kinase 3 (MLK3) activation during hepatocyte sublethal lipotoxic injury:
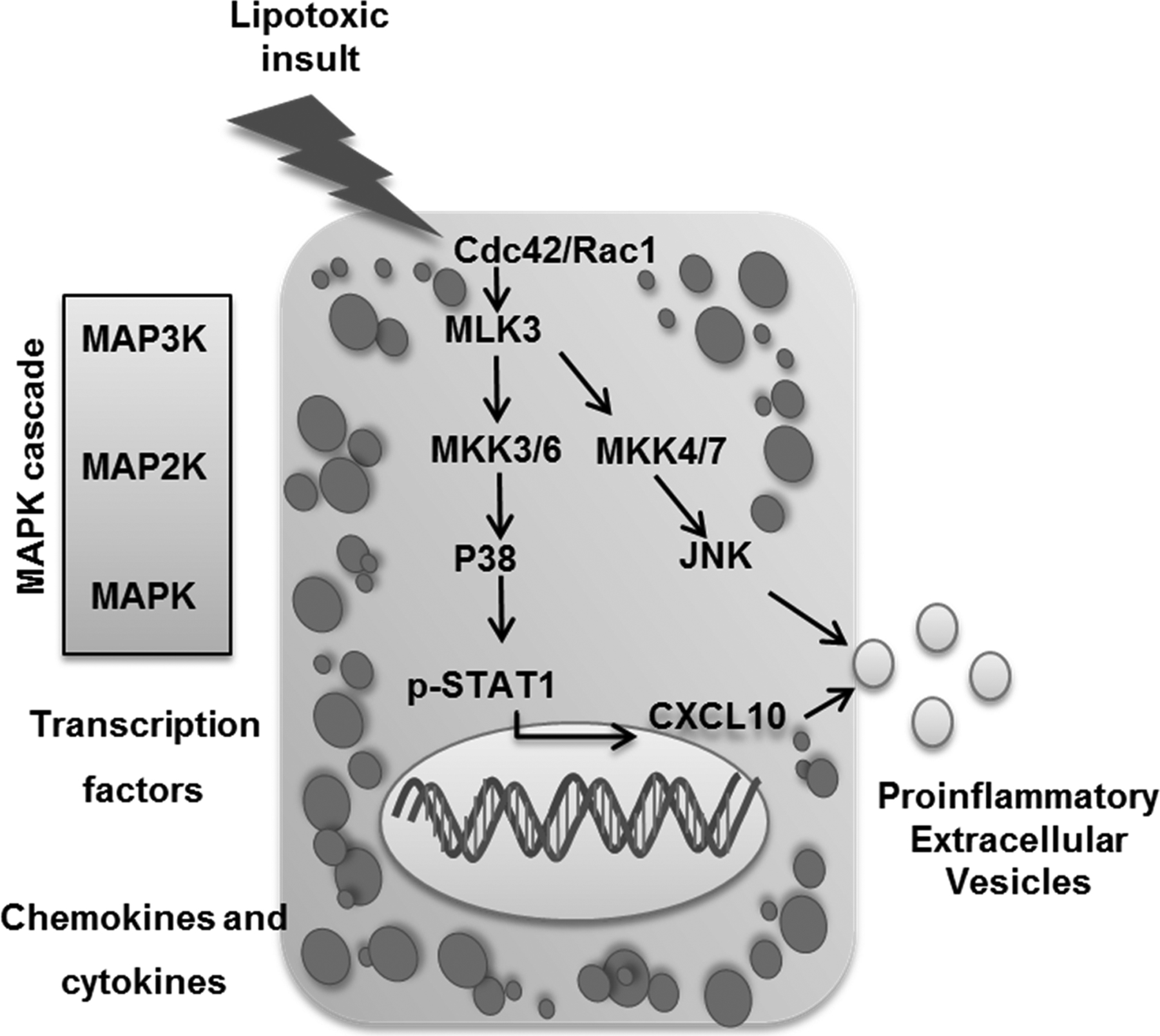
Lipotoxicity during nonalcoholic steatohepatis (NASH) activates MLK3 mainly via the small GTP-binding proteins Cdc42/Rac1, MLK3 activation triggers a downstream MAPK signaling cascade that promotes the release of C-X-C motif chemokine 10 (CXCL10)‐bearing extracellular vesicles (EV)s from hepatocytes by a mechanism that involves CXCL10 induction through a p38/ signal transducer and activator of transcription 1(STAT1)‐dependent mechanism, and CXCL10 trafficking into EVs through a JNK‐dependent mechanism.
Jun N-terminal kinase (JNK)
In the liver, JNK is a dominant effector MAPK, which phosphorylates numerous substrates including nuclear activator protein 1 (AP1) transcription factors as well as protein kinases, phosphatases, and scaffold proteins.7 Of the three jnk genes, jnk1 and jnk2 are expressed in the liver and each encodes two isoforms, 54 and 46 kiloDaltons in size. Further, in the liver JNK1 is mostly comprised of p46 and JNK2 of p54.31 JNK signaling plays significant roles in NASH by regulating cell death and metabolism in the liver.7 JNK is activated in dietary and genetic animal models of NASH,32–34 and in human NASH.35,36 JNK is activated in both macrophages and hepatocytes in NASH causing both apoptosis and liver inflammation.37,38 Interestingly, JNK1/2 in macrophages is required for tissue infiltration, proinflammatory polarization, cytokine release, and suppression of insulin sensitivity in the liver.39 During OID in mice, JNK activation in hepatocytes phosphorylates insulin receptor substrate-1 (IRS-1), suppressing insulin receptor signaling, and inducing insulin resistance.40 On the other hand, JNK1 activation in adipocytes promotes the secretion of the inflammatory cytokine interleukin 6 (IL-6). IL-6 upregulates hepatic suppressor of cytokine signaling 3 (SOCS3), which induces IRS1 degradation, and hepatic insulin resistance.41 Hence, JNK1 activation in adipose tissue promotes insulin resistance in the liver. On the other hand, mice with hepatic deficiency of JNK1 exhibit increased insulin resistance,42 whereas compound deletion of both JNK1 and JNK2 improved insulin resistance in HFD-fed mice. Interestingly, hepatic deletion of JNK2 alone recapitulates the enhanced insulin sensitivity of JNK1/JNK2 hepatic deletion,43 suggesting that JNK2 in the liver is the main player in hepatic insulin resistance, while JNK1 is the main isoform activated downstream of ASK1 with a dominant role in mediating the HFD-induced steatohepatitis.14,21
Moreover, during hepatocyte lipotoxicity JNK1 is the only isoform to phosphorylate c-Jun, a member of AP-1 transcription factor complex, which induces expression of the Bcl-2 homology 3 (BH3)-only protein p53-upregulated modulator of apoptosis (PUMA), and subsequently activates the mitochondrial pathway of apoptosis.36 Sustained JNK1/2 activation appears to be mediated by an activation loop where JNK-mediated phosphorylation of its outer mitochondrial membrane docking protein and substrate, Sab (SH3 homology associated BTK binding protein), leads to oxidative stress, mitochondrial outer membrane polarization, and apoptosis.44
JNK activation in HFD-mice mediates cross-talk between different metabolically active cells by repressing the nuclear hormone receptor peroxisome proliferator-activated receptor alpha (PPARα), and its target gene fibroblast growth factor 21 (Fgf21), through increased expression of the PPARα corepressor nuclear receptor corepressor 1 (NcoR1).43 FGF21 is a hepatokine which possesses potent regulatory effects on lipid metabolism, and adipose tissue insulin sensitivity.45 Feeding stimulates JNK activation concomitant with downregulation of FGF21, and ablation of hepatic JNK abrogates this nutritional responsiveness. Hence, it is proposed that hepatic JNK mediates the fasting and feeding cues of circulating FGF21 expression.8 However, the protective effects of hepatic JNK deficiency in suppressing the metabolic syndrome in HFD-fed mice were not observed in mice with hepatocyte-specific FGF21 deficiency, including reduced insulin resistance.46 Interestingly, treatment with subcutaneously administered PEGylated fibroblast growth factor 21 analogue (pegbelfermin) for 16 weeks was well tolerated and significantly reduced hepatic fat fraction in a randomized, double-blind, placebo-controlled, phase 2a trial in patients with non-alcoholic steatohepatitis.47
Thus, JNK activation plays a key role in the metabolic syndrome and lipotoxicity associated with NASH (Figure 3), although direct inhibition of JNK is unfavorable given the potential interference with physiological JNK signaling. Targeting the JNK activation loop is a potential therapeutic strategy in NASH, by blocking JNK binding to Sab, either by the herbal chemical, Anctin H, membrane permeable blocking peptide, or antisense oligonucleotide that target hepatic Sab.7,48 Interestingly an ongoing phase 2, randomized, double-blind, placebo-controlled, clinical trial is evaluating the efficacy and safety of a second generation JNK inhibitor CC-90001 in subjects with NASH and stage 3 or stage 4 liver fibrosis (NCT04048876-clinical trial.gov).
Figure 3. C-Jun N‑terminal kinase (JNK) activation in obesity-induced diet:
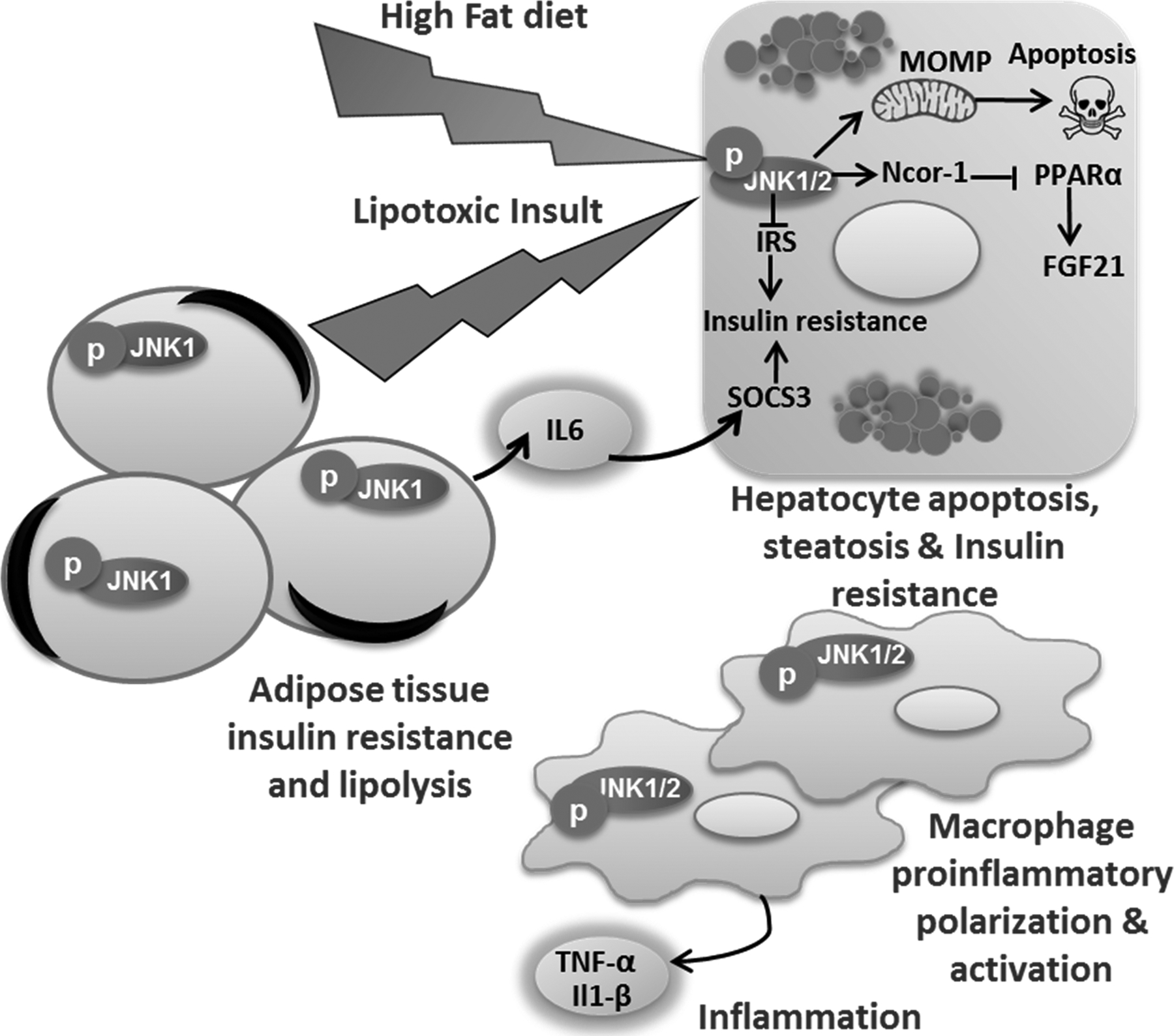
Obesity-induced diet activates JNK in hepatocytes, macrophage, and adipocytes. JNK signaling in hepatocytes inhibits insulin signaling and causes insulin resistance, JNK also activates the mitochondrial pathway of apoptosis. Sustained JNK1/2 activation up-regulates the nuclear receptor corepressor 1 (NcoR1), which, in turn, inhibits peroxisome proliferator-activated receptor alpha (PPARα) responsive gene including fibroblast growth factor 21 (FGF21). FGF21 in turn increases adipose tissue insulin sensitivity. P-JNK1 in adipocytes upregulates interleukin (IL-6) expression, IL6 in turn increases hepatocytes suppressor of cytokine signaling (SOCS) 3 which inhibits insulin signaling in hepatocytes. JNK1/2 in macrophage regulates proinflammatory polarization, hepatic infiltration, and activation.
P38 MAPK
The p38α/β MAPK isoforms are the widely expressed, extensively studied isoforms. Emerging concepts highlight the complexities of hepatic p38α/β MAPK in regulating glucose homeostasis and lipid management in physiological lean conditions and pathological obese states.8 Mice lacking hepatic p38α exhibited reduced fasting glucose level and impaired gluconeogenesis that was associated with increased AMPKα phosphorylation.49 Further evidence for a role for p38α/β MAPK in gluconeogenesis is derived from studying MAPK phosphatase-1 (MKP-1) which negatively regulates gluconeogenesis by opposing p38α/β MAPK-mediated activation of the gluconeogenic pathway.50 Interestingly p38δ was found to be elevated in the livers of obese patients with NAFLD, while mice lacking p38γ/δ in myeloid cells were resistant to diet‐induced fatty liver, hepatic steatosis and glucose intolerance. This protective effect was attributed to defective migration and hepatic infiltration of p38γ/δ‐deficient neutrophils. Neutrophil hepatic infiltration is implicated in hepatic steatosis mainly through modulating oxidative lipids metabolism and inflammation.51 Likewise p38α expression was increased in the livers of humans with NAFLD, while macrophage p38α in a mouse model of NASH promoted the progression of steatohepatitis by inducing proinflammatory cytokine secretion and polarization.52 Furthermore, in hepatocytes under lipotoxic stress p38α/β was activated through an MLK3-mediated MAPK signaling cascade resulting in STAT1 Ser727 phosphorylation, and transcriptional activation with subsequent induction of the potent chemotactic ligand CXCL10,10 which promotes macrophage trafficking to the NASH liver.29 Interestingly a recent study demonstrated that the p38 MAPK inhibitors SB203580 or BIRB796 attenuated murine NASH,52 suggesting that p38 may have a potential role as a therapeutic target in human NASH.
Interestingly, p38 activated signaling engages various targets including transcription factors and downstream protein kinases resulting in a plethora of cellular responses of opposing roles in different liver diseases.53 For example, p38 reduced liver fibrogenesis and consequent hepatocarcinogenesis through attenuating reactive oxygen species accumulation in response to thioacetamide-induced chronic liver injury.54 In this study, mice with p38α-deficient hepatocytes had enhanced reactive oxygen species (ROS) accumulation, through reduced expression of the antioxidant protein heat shock protein (HSP) 25, a mouse homolog of HSP27. Its re-expression in p38α-deficient hepatocytes prevented ROS accumulation and thioacetamide-induced fibrosis. Furthermore, p38α deficiency increased expression of SOX2, a marker for cancer stem cells and the liver oncoproteins c-Jun and Gankyrin and led to enhanced thioacetamide-induced hepatocarcinogenesis. Likewise, p38 has been reported to suppress diethylnitrosamine (DEN)-induced cancer cell proliferation by antagonizing the JNK–c-Jun pathway.55 Mice with p38α-deficient hepatocytes showed increased DEN-induced liver tumor that correlated with enhanced JNK–c-Jun activity. Furthermore, when JNK–c-Jun pathway was inactivated, proliferation of p38α-deficient hepatocytes and liver tumor cells was reduced to control levels.55
ENDOPLASMIC RETICULUM STRESS
Obesity-associated metabolic stress is associated with activation of the endoplasmic reticulum (ER) stress response.40 Early studies on glucose deprivation and protein misfolding identified the signaling pathways activated when the normal functions of the ER were impaired; these were collectively labelled the unfolded protein response (UPR).56 UPR signaling is conserved across eukaryotic cells and begins with recognition of ER stress by the three transmembrane UPR sensors, activating transcription factor 6 alpha (ATF6α), inositol-requiring enzyme 1 alpha (IRE1α), and protein kinase RNA (PKR)-like ER kinase (PERK).56,57 Canonical proteotoxic ER stress is initiated via luminal accumulation of misfolded proteins that recruit immunoglobulin binding protein/glucose regulatory protein-78 (BiP/GRP78) away from the UPR sensors.58 This allows the UPR sensors to oligomerize, and their luminal domains may directly bind misfolded proteins. Newer studies have identified direct sensing of lipid perturbations or lipotoxic stress by the transmembrane domains of the UPR sensors. With an increase in obesity-associated metabolic diseases, emerging studies have identified a role for the UPR sensors in lipid homeostasis and in the pathogenesis of NASH. The role of the UPR sensors in lipid and metabolic homeostasis has been reviewed elsewhere57,59 and here we focus on PERK and IRE1α kinases and their mechanistic role in liver steatosis, inflammation, cell death, and fibrosis in NASH (Figure 4).
Figure 4. Endoplasmic reticulum stress-induced kinases:
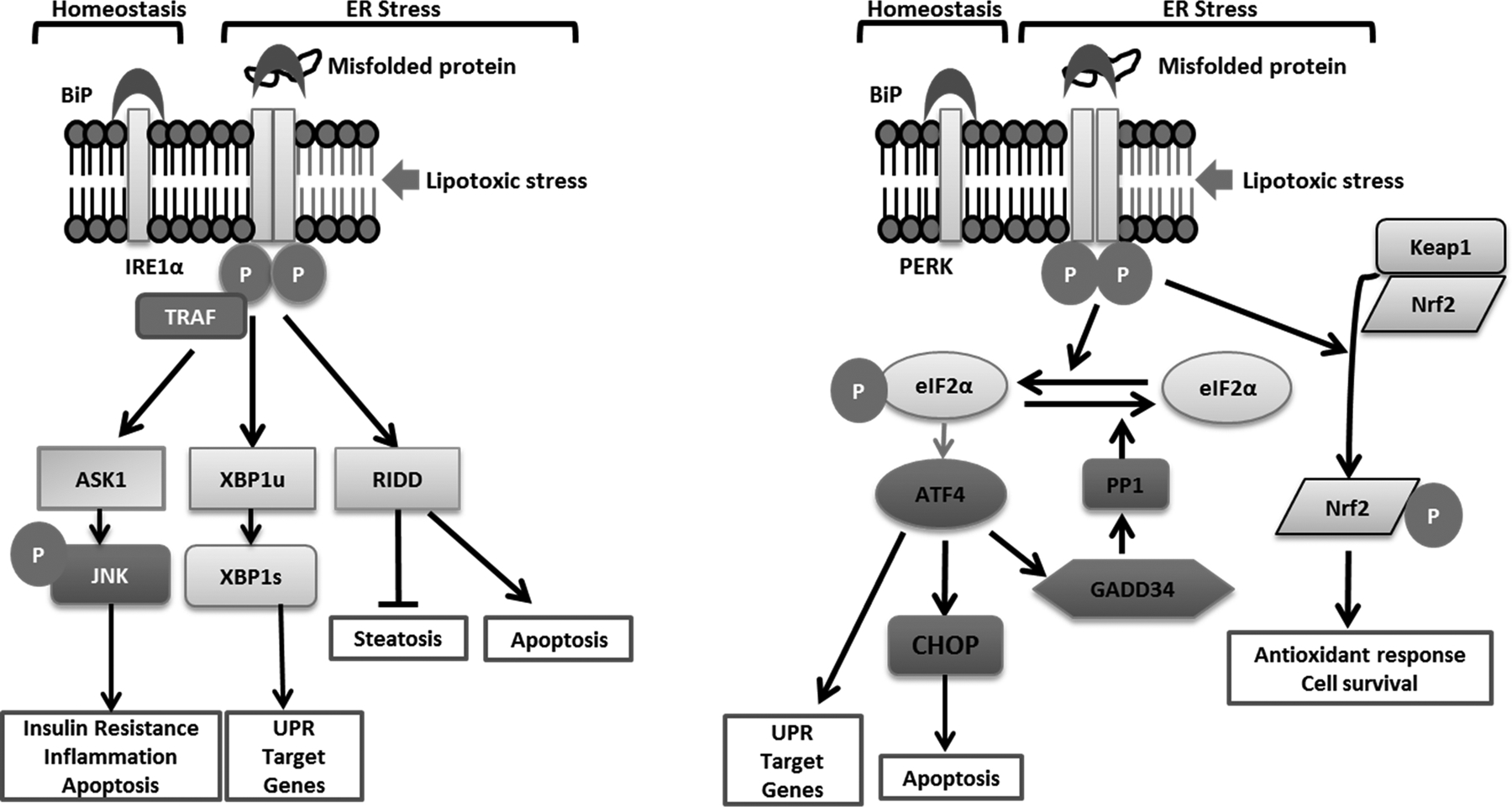
IRE1α and PERK signaling under homeostatic conditions is kept in check by inhibitory binding of their respective luminal domains to BiP. Proteotoxic ER stress due to the accumulation of misfolded proteins in the ER lumen dissociates BiP from the luminal domains allowing oligomerization-induced activation. Lipotoxic stress-induced oligomerization occurs independent of the luminal domains of IRE1α and PERK. The sensing mechanisms for proteotoxic and lipotoxic stress appear to be conserved between IRE1α and PERK. IRE1α-induced oligomerization by TRAF2 activates a kinase cascade resulting in activation of ASK1 and JNK. JNK is implicated in insulin resistance, inflammation and apoptosis. Activation of the kinase domain of IRE1α results in trans-autophosphorylation and activation of the cytosolic endoribonuclease (RNase) activity which cleaves 26 nucleotides from XBP1 mRNA to express spliced XBP1. XBP1 is a potent transcription factor upregulating a program of UPR target genes including the chaperone BiP. IRE1α also cleaves mRNAs and microRNAs in a process termed regulated IRE1α-dependent decay (RIDD). RIDD targets inhibit steatosis and promote apoptosis. PERK phosphorylates eIF2α leading to attenuation of translation with selective translation of ATF4. ATF4 induce the expression of CHOP, which sensitizes cells to ER stress-induced apoptosis. ATF4 also activates an adaptive program of stress responsive UPR target genes such as amino acid metabolism. CHOP and ATF4 together increase the expression of GADD34, which activates protein phosphatase 1 (PP1). PP1 dephosphorylates eIF2α, removing the brakes on translation. PERK also phosphorylates Nrf2 leading to its dissociation from Keap1, which leads to its nuclear translocation and activation of Nrf2 target genes including antioxidant genes which favor cell survival.
Protein kinase RNA-like ER kinase (PERK)
PERK is a type-I transmembrane protein with a cytosolic C-terminus kinase domain, a transmembrane domain and a stress-sensing luminal N-terminus.60 PERK kinase activity is activated by oligomerization and trans-autophosphorylation of a number of residues in its cytosolic domain. This leads to a conformational change, recruitment and phosphorylation of eukaryotic translation initiation factor (eIF) 2α, which leads to global and transient attenuation of protein synthesis aimed at reducing the protein folding load in the ER. PERK also phosphorylates nuclear factor (erythroid-derived)-like 2 (Nrf2) leading to its dissociation from Kelch-like ECH associating protein 1 (Keap1), translocation to the nucleus and transcriptional activation of genes containing regulatory antioxidant response elements.61 Phosphorylation of eIF2α promotes selective translation of activating transcription factor 4 (ATF4) which transcriptionally activates a subset of adaptive and stress responsive genes including amino acid metabolism and oxidative stress response.62 ATF4 also activates transcription of the proapoptotic factor C/EBP homologous protein (CHOP) which promotes cell death via expression of death receptor 5 (DR5) and the BH-3 only proteins Bim and PUMA, likely in a cell- and stimulus-specific manner given the observations that ER stress-induced cell death can occur independently of DR5 and caspase 8.63–65 PERK signaling is kept in check by growth arrest and DNA damage 34 (GADD34) which activates protein phosphatase 1 leading to dephosphorylation of eIF2α and resumption of translation.
Inositol-requiring enzyme-1α (IRE-1α)
IRE1α is a type-I transmembrane protein. Similar to PERK it has a stress-sensing luminal domain, transmembrane domain, and a cytosolic domain which contains two enzymatic activities, kinase and endoribonuclease (RNase).66 IRE1α is activated by oligomerization induced trans-autophosphorylation and RNase activation which cleaves 26 nucleotides from XBP1 mRNA to generate spliced XBP1 mRNA.67 The resultant frame shift leads to the translation of spliced XBP1 transcription factor. XBP1 target genes include genes coding for ER quality control proteins such as BiP and those involved in ER-associated degradation. Persistent activation of IRE1α leads to degradation of mRNAs and microRNAs in a process termed regulated IRE1α-dependent decay (RIDD).68 IRE1α activation also intersects with stress kinase activation via TRAF-mediated activation of JNK.69
ER stress-induced kinase signaling and steatosis
The PERK pathway regulates hepatic steatosis. Enforced expression of GADD34 in HFD-fed mice led to a reduction in liver lipogenic transcription factors PPARγ, C/EBPα and C/EBPβ.70 This was associated with improved steatosis and insulin tolerance in these mice. In cultured hepatocytes eIF2α phosphorylation led to increased translation of C/EBP suggesting that while halting global protein synthesis, eIF2α phosphorylation promotes lipogenesis. In keeping with these data, ATF4 signaling also promotes hepatic steatosis. Lack of ATF4 is associated with a reduction in steroyl-CoA desaturase 1 expression, and protection from hepatic steatosis and hypertriglyceridemia.71
More is known about the intersections between IRE1α signaling and hepatic steatosis. ER stress mediates obesity-induced insulin resistance. This was demonstrated to occur via JNK-mediated inhibitory phosphorylation of IRS-1 in the liver of ob/ob and HFD-fed mice.40 Lack of XBP1 signaling worsened ER stress due to a reduction in the compensatory capacity of the ER. This in turn enhanced JNK phosphorylation and insulin resistance. Post-translational modification of IRE1α has been reported in chronic metabolic stress-induced inflammation. S-nitrosylation of IRE1α resulted in a decline in the RNase activity without compromising its kinase activity.72 This was associated with an increase in hepatic steatosis due to lack of degradation of inhibitory microRNAs, mir-200 and miR-34.73 On the other hand, hyperactivation of IRE1α led to RIDD of lipogenic mRNAs.74 IRE1α also plays a role in very-low-density lipoprotein assembly, though the contribution of this pathway to obesity-associated dyslipidemia has not been explored.75 Thus, IRE1α signaling outputs limit hepatic steatosis. These signaling outputs depend on the RNase activity of IRE1α, which in turn is activated following kinase-mediated auto-transphosphorylation. Inhibiting the RNase activity can potentially result in hyper-phosphorylated IRE1α; on the other hand, targeting the kinase domain of IRE1α effectively inhibits the RNase activity.76 This has led to the development of small molecule IRE1α kinase-inhibiting RNase attenuators (KIRAs). Hepatic steatosis may be an inadvertent side effect that could be avoided by tissue-specific targeting of KIRAs as they are developed for therapeutic use.
ER stress-induced kinase signaling and inflammation
The data on the role of IRE1α in liver inflammation in steatohepatitis are not parsimonious. Wang et al. reported an increase in hepatic inflammation in hepatocyte-specific IRE1α knockout mice challenged with a HFD for 20 weeks suggesting a protective role for IRE1α.73 Mice with hyperactivation of IRE1α due to hepatocyte XBP1 deletion demonstrate reduced steatosis, albeit an increase in liver injury.77 This may partly be mediated by RIDD of hepatoprotective mRNAs. Deletion of Bax Inhibitor 1 (BI-1), a negative regulator of IRE1α, worsened hepatocyte cell death and liver inflammation, also associated with hyperactivation of IRE1α, persistent splicing of XBP1, and activation of the NLRP3 inflammasome.78,79 Recent studies have also implicated IRE1α in the release of proinflammatory extracellular vesicles from steatotic hepatocytes.80 These data would suggest that the magnitude, timing, and persistence of IRE1α activation determine whether the signaling outputs favor or counter inflammation in NASH. The role of PERK in liver inflammation remains to be explored.
ER stress-induced kinase signaling and cell death decisions
The outcome of ER stress is either restoration of cellular homeostasis or cell death. ER stress-induced cell death is mediated by the transcription factor CHOP partially via the proapoptotic proteins DR5 and Bim.81 Though CHOP overexpression is insufficient to induce cell death by itself, ER stress-induced cell death is decreased in the absence of CHOP. However, HFD fed CHOP−/− demonstrated greater liver injury, inflammation and hepatocyte apoptosis reportedly secondary to a reduction in macrophage apoptosis in CHOP−/− mice demonstrating the importance of cellular context in determining ER stress outcomes.81 CHOP−/− mice challenged with a methionine- and choline-deficient diet demonstrated some protection against steatohepatitis,82 also highlighting the differences in stimulus-specific ER stress outcomes. Emerging studies implicate engagements of DR5 independent of its ligand, as a terminal protein-folding checkpoint during unresolved ER stress before committing to a terminal apoptotic fate.83 This is in keeping with our observations of attenuated NASH in mice deficient in TRAIL receptor, the mouse equivalent of DR5.84
Thus obesity-associated metabolic abnormalities activate two ER kinases. PERK activation may protects against steatosis, though promotes apoptosis via transcriptional upregulation of CHOP. IRE1α protects against steatosis but may promote liver injury and inflammation via the release of extracellular vesicles and RIDD.
RHO-ASSOCIATED PROTEIN KINASES
Rho-associated protein kinase (ROCK) 1 and ROCK2 belong to the AGC kinase subfamily of serine/threonine kinases which includes, among others, cAMP-dependent protein kinase 1 (PKA) or protein kinase C (PKC).85 As the name suggests, ROCKs were originally identified as downstream effectors of the small GTPase RhoA. ROCK kinases are important regulators of actin cytoskeleton and are involved in diverse cellular functions including cytoskeleton organization, cell adhesion and motility, proliferation and apoptosis, and smooth muscle cell contraction. Although ROCK1 and ROCK2 have highly related functional domains and significant amino acid identity, they possess only partially overlapping functions. On the other hand, they can be regulated both by common means as well as mechanisms unique to ROCK1 or ROCK2. Various signals activate ROCKs by releasing the protein from the autoinhibitory conformation, including Rho protein binding to Rho-binding domain and proteolytic removal of the C terminus of ROCK1 by caspase 3 or ROCK2 by granzyme B.86
Emerging data demonstrate that ROCK1 plays a role in regulating glucose metabolism, insulin signaling, and energy metabolism, all of which theoretically may be involved in the pathogenesis of NASH. Whole body ROCK1−/− mice are viable with no detectable anatomic abnormalities.87 However, global ROCK1 deficiency causes systemic insulin resistance by impairing insulin signaling in the skeletal muscle.87 On the other hand, adipose-specific deletion of ROCK1 improved insulin sensitivity in mice with obesity induced by HFD feeding, without affecting adiposity.88 In genetically obese mice and mice fed HFD, hepatic ROCK1 deficiency reduced steatosis through suppression of de novo lipogenesis, which appeared to be independent of body weight.89 Conversely, hepatic overexpression of constitutively active ROCK1 was sufficient to promote adiposity, insulin resistance, and steatosis in mice fed HFD. At a cellular level, hepatocyte ROCK1 was demonstrated to be activated by endocannabinoids 2-AG and AEA via endocannabinoid receptor. Activated ROCK1 then inhibits 5’ AMP-activated protein kinase (AMPK), resulting in stimulation of the sterol regulatory element-binding transcription factor 1 (SREBP1c)-mediated lipogenic pathways.20 Altogether, this suggests that hepatic ROCK1, through stimulation of de novo lipogenesis, is a major link between HFD-induced obesity, insulin resistance, and fatty liver disease.
ROCK1 has also been implicated in mediating hepatocyte lipotoxic signaling. In particular, toxic lipids, such as lysophosphatidylcholine, induce proteolytic activation of ROCK1, which in turn mediates EV release.90 Genetic deletion of ROCK1 or pharmacologic inhibition of ROCK1/2 significantly decreased lipotoxicity-induced EV release from hepatocytes in vitro.90 Since lipotoxic hepatocyte EVs have a proinflammatory effect on monocytes and macrophages,91,92 it is yet to be determine if hepatocyte ROCK1 may promote liver inflammation through lipotoxic EV release.
Therapeutic inhibition of ROCK activity
Pharmacologic ROCK inhibition has emerged as an attractive therapeutic approach to reverse several pathologies involving inflammation and fibrosis.93,94 There are several commercially available inhibitors of ROCK kinase activity, which are mainly dual ROCK1/2 inhibitors. Among dual ROCK1/2 inhibitors, fasudil (or HA-1077) is approved for use in Japan and China for the treatment of cerebral vasospasm.93 According to post-marketing trials, fasudil is considered to be a safe drug with no serious side effects.94 In addition, more potent and selective ROCK1/2 inhibitors are currently being developed and tested in clinical trials for various indications.95
ROCK pharmacologic inhibition has also been tested in animal models of NASH. In a murine model of NASH induced by western diet, fasudil reduced liver injury, macrophage-associated inflammation and fibrosis, without significant effect on liver steatosis.90 Interestingly, fasudil significantly decreased levels of circulating EVs that are markedly increased by western diet feeding. In a rat model of NASH, concomitant administration of ROCK kinase inhibitor Y-27632 with the choline-deficient, L-amino acid–defined diet prevented development of liver steatosis and fibrosis;96 unfortunately, the authors did not assess liver inflammation. In another rat model based on high-fat diet feeding combined with streptozotocin-induced diabetes, fasudil markedly decreased markers of hepatic stellate cell activation and liver fibrosis.97
This quite remarkable therapeutic effect of ROCK inhibition on NASH is likely due to a combination of multiple effects on various cell types involved in the disease pathogenesis (Figure 5). We can speculate that ROCK inhibition in hepatocytes may prevent the lipotoxicity-induced release of pathogenic EVs, which, in turn, may be associated with attenuated liver inflammation. In vitro studies have demonstrated that ROCK1 inhibition decreases numbers of EVs released during hepatocyte lipotoxicity.90 EVs released by lipotoxic hepatocytes were shown to be enriched in proinflammatory molecules, such as CXCL10 and damage-associated molecular patterns, which have the potential to promote the recruitment of monocytes and activation of monocyte-derived macrophages. Moreover, we have recently demonstrated that lipotoxic hepatocyte-derived EVs are enriched with adhesion molecules namely integrin β1 which enhances monocytes adhesion to liver sinusoidal endothelial cells, a key step in the inflammatory process in NASH.92 Whether ROCK1 directly regulates EV protein content along with vesicle release is currently unknown. ROCK inhibition may also have a direct effect on monocytes and macrophages, especially in regards to their recruitment and proinflammatory activation. For example, bone marrow-derived ROCK1-deficient macrophages displayed impaired chemotaxis toward monocyte chemotactic protein 1 and in a mouse model of atherosclerosis, ROCK1 deficiency in macrophages decreases their accumulation in the atherosclerotic lesion.98 Another study showed that ROCK inhibitors block the generation of inflammatory cytokines, such as interleukin-6 and TNF, in lipopolysaccharide-stimulated monocytes.99 Another way by which fasudil can attenuate NASH-associated inflammation is via its effect on liver endothelial cells. ROCK inhibition has been demonstrated to decrease chemokine production and expression of adhesion molecules in endothelial cells in various disease models.100,101 As mentioned above, ROCK inhibitors have also been demonstrated to decrease liver fibrosis in preclinical NASH models. The decrease in fibrogenesis upon ROCK inhibition may be due to mitigated inflammation and, perhaps, also due to a direct effect on hepatic stellate cells, the major liver cell type responsible for matrix deposition during hepatic injury. Indeed, ROCK1 inhibitors have been shown to inhibit several processes in hepatic stellate cells such as profibrogenic activation, contraction and migration.102–104
Figure 5. Rho-associated protein kinase (ROCK):
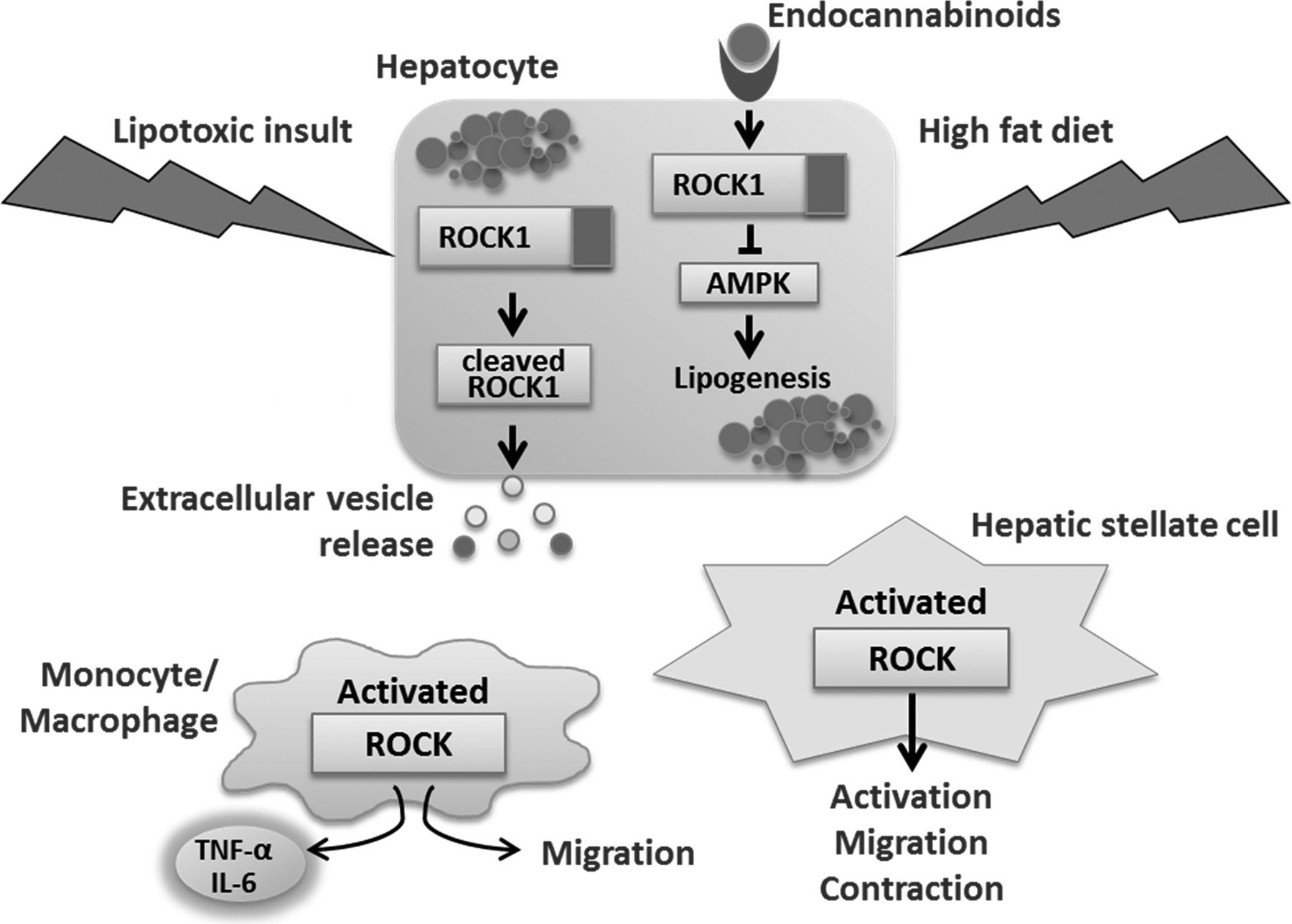
ROCK1 and ROCK2 may play role in several processes involved in NASH pathogenesis. In hepatocytes, lipotoxicity leads to cleavage-mediate ROCK1 kinase activation, which, in turn, promotes release of extracellular vesicles. Endocannabinoids increased upon high-fat diet stimulate endocannabinoid receptor on hepatocytes, leading to ROCK1 activation and subsequent inhibition of 5’ AMP-activated protein kinase (AMPK), which results in stimulation of the sterol regulatory element-binding transcription factor 1 (SREBP1c)-mediated lipogenic pathway. ROCK isoforms in myeloid cells may be involved in cytokine production and cell migration, as treatment with ROCK1/2 inhibitors prevents these processes. In hepatic stellate cells, ROCK activation has been reported during cell activation, migration and contraction.
Given that the frequently used ROCK inhibitors (e.g., fasudil and Y-27632) potently inhibit the kinase activity of both ROCK isoforms, it is unclear whether the abovementioned observations are due to the effects on ROCK1 or ROCK2 or both isoforms. Future research using cell type-specific mouse models along with the development of selective inhibitors for each isoform will further delineate the roles of ROCK isoforms in liver pathobiology. Such information is necessary in order to design and implement ROCK1/2 targeted therapies.
CONCLUSION AND REFLECTION
A variety of kinase signaling pathways have been implicated in promoting NASH pathogenesis. Kinases are targetable by small molecule inhibitors, kinase pocket inhibitors (reversible and irreversible), and allosteric inhibitors. Hence, identifying dominant kinase signaling pathways in NASH has potential therapeutic implications. Herein, we have examined the information relevant to these kinase pathways implicated in NASH pathogenesis to date. However, the role of kinases is in NASH pathogenesis remains a door half opened, rich for exploration.
Although targeting tyrosine kinase inhibitors is poorly tolerated in man, targeting serine/threonine kinase is better tolerated. Augmenting cytoprotective kinase pathways can also be beneficial. For example, metformin an (AMP-activated protein kinase) AMPK activator is widely used clinically for the treatment of diabetes mellitus in the context of the metabolic syndrome. Indeed, the ASK1 inhibitor was also well tolerated, albeit ineffective in unselected patients.18,19 In the emerging era of precision medicine, it will be critical to match biomarkers of kinase activation with the appropriate therapy. NASH is a heterogeneous disease and matching disease pathogenesis with targeted therapy needs to be applied to this disease. Finally, we note that at MLK3 kinase and ROCK kinase inhibitors need further examination in human disease. While MLK3 inhibitors have been and identified,11,28 developing isoform specific ROCK kinase inhibitor has remained a challenge. Nonetheless, we encourage further studies and exploration of these targetable kinases for NASH therapies.
MAIN CONCEPTS AND LEARNING POINTS
Stress kinases contribute to NASH pathogenesis through multiple mechanisms including, lethal and sublethal hepatocyte injury, inflammation, fibrogenesis, as well as insulin resistance.
ASK1, the lead MAPK therapeutic target in human NASH, was not efficacious in clinical trials likely secondary to its pleotropic functions and complex regulatory mechanisms; however, studies were not stratified for ASK1 activation.
Targeting the JNK sustained activation loop may serve as a potential therapeutic strategy in NASH, with the hope to avoid interfering with the physiological functions of JNK.
MLK3 pharmacological inhibition attenuated inflammation and fibrosis in a mouse model of NASH; however its efficacy in human NASH will need to be assessed in clinical trials.
Blocking the ER kinases PERK or IRE1α in NASH may exacerbate steatosis but have the potential to attenuate liver injury and inflammation; hence identifying a more selective and nontoxic target in this pathway will help advance the field.
Inhibition of ROCK kinase activity attenuated inflammation and fibrosis in a mouse model of NASH; further studies are needed to better understand the role of ROCK isoforms in various cell types involved in NASH pathogenesis.
KEY POINTS.
A subset of patients with nonalcoholic fatty liver disease (NAFLD) develops an inflammatory condition termed nonalcoholic steatohepatitis (NASH). NASH is characterized by hepatocellular injury, inflammation, and progressive liver fibrosis.
The mechanisms whereby hepatic inflammation occurs in NASH remain incompletely understood, but appear to be linked to the proinflammatory microenvironment created by toxic lipid-induced hepatocyte injury, termed lipotoxicity.
Numerous NASH promoting kinases are activated during obesity inducing diets, as well as by lipotoxic treatment in hepatocytes, immune cells, and adipocytes.
Among the NASH promoting kinases are the mitogen activated protein kinases (MAPKs) including mixed lineage kinase 3 (MLK3), apoptosis signal-regulating kinase 1 (ASK1), c-Jun N-terminal kinase (JNK) and p38 MAPK; the ER stress kinases protein kinase RNA-like ER kinase (PERK) and inositol-requiring protein-1α (IRE-1α); as well as the Rho-associated protein kinase 1 (ROCK1).
Multiple pharmacological agents targeting these stress kinases in NASH are under different phases of development.
Acknowledgments:
Funding support from the AASLD Foundation Bridge Award (to SHI), the NIH grant DK111397 (to SHI), Gilead Science career development award (to SHI), and the Mayo Clinic K2R pipeline (to SHI), NIH grant DK41876 (to GJG), Mayo Clinic Center for Cell Signaling in Gastroenterology (NIDDK P30DK084567) Pilot and Feasibility Award (to PH), NIH grant DK11378 (to HM) and the Mayo Foundation.
List of abbreviations:
- NAFLD
nonalcoholic fatty liver disease
- NASH
nonalcoholic steatohepatitis
- MAPK
mitogen activated protein kinases
- ROCK
Rho-associated protein kinase
- FFAs
free fatty acids
- ER
endoplasmic reticulum
- MAP3K
MAP kinase kinase kinase
- MAP2K
MAPK kinase
- ERK
extracellular signal-regulated kinase
- MLK3
mixed lineage kinase 3
- JNK
Jun N-terminal kinase
- ASK1
apoptosis signal-regulating kinase 1
- DUSP
dual specificity protein tyrosine phosphatase
- MKPs
MAPK phosphatases
- CCC
carboxy‑terminal coiled-coil
- NCC
amino‑terminal coiled-coil
- Trx
thioredoxin
- TRAF
tumor necrosis factor receptor-associated factor
- CFLAR
CASP8 and FADD-like apoptosis regulator
- OID
obesity-induced diet
- CREG
cellular repressor of E1A-stimulated genes
- TNFAIP3
tumor necrosis factor alpha–induced protein 3
- EV
extracellular vesicles
- STAT1
signal transducer and activator of transcription 1
- CXCL10
C-X-C motif chemokine 10
- HFD
high fat diet
- IRS-1
insulin receptor substrate-1
- PUMA
p53-upregulated modulator of apoptosis
- PGC1α
peroxisome proliferator-activated receptor gamma coactivator 1-alpha
- IL-6
cytokine interleukin 6
- SOCS3
suppressor of cytokine signaling 3
- PPARα
peroxisome proliferator-activated receptor alpha
- NCOR1
nuclear receptor corepressor 1
- FGF21
fibroblast growth factor 21
- ROS
reactive oxygen species
- HSP
heat shock protein
- DEN
diethylnitrosamine
- ATF6α
activating transcription factor 6 alpha
- IRE1α
inositol-requiring enzyme 1 alpha
- PERK
protein kinase RNA (PKR)-like ER kinase
- BiP/GRP78
binding protein/glucose regulatory protein-78
- UPR
unfolded protein response
- eIF2α
eukaryotic translation initiation factor 2α
- Keap1
Kelch-like ECH associating protein 1
- Nrf2
nuclear factor (erythroid-derived)-like 2
- ATF4
activating transcription factor 4
- CHOP
C/EBP homologous protein
- DR5
death receptor 5
- RNase
endoribonuclease
- GADD34
growth arrest and DNA damage 34
- RIDD
regulated IRE1α-dependent decay
- ERN1
ER to nucleus signaling 1
Footnotes
Publisher's Disclaimer: This is an Accepted Manuscript of an article published by Thieme Publishing Group in Journal Seminars in Liver Disease on 6/11/2020, available online at https://www.thieme-connect.de/products/ejournals/abstract/10.1055/s-0040-1713115
REFERENCES
- 1.Younossi ZM, Koenig AB, Abdelatif D, Fazel Y, Henry L, Wymer M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology. 2016;64(1):73–84. [DOI] [PubMed] [Google Scholar]
- 2.Machado MV, Diehl AM. Pathogenesis of Nonalcoholic Steatohepatitis. Gastroenterology. 2016;150(8):1769–1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hirsova P, Ibrabim SH, Gores GJ, Malhi H. Lipotoxic lethal and sublethal stress signaling in hepatocytes: relevance to NASH pathogenesis. J Lipid Res. 2016;57(10):1758–1770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jaeschke A, Davis RJ. Metabolic stress signaling mediated by mixed-lineage kinases. Molecular cell. 2007;27(3):498–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hirsova P, Gores GJ. Death Receptor-Mediated Cell Death and Proinflammatory Signaling in Nonalcoholic Steatohepatitis. Cellular and molecular gastroenterology and hepatology. 2015;1(1):17–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ibrahim SH, Hirsova P, Gores GJ. Non-alcoholic steatohepatitis pathogenesis: sublethal hepatocyte injury as a driver of liver inflammation. Gut. 2018;67(5):963.-+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Win S, Than TA, Zhang J, Oo C, Min RWM, Kaplowitz N. New insights into the role and mechanism of c-Jun-N-terminal kinase signaling in the pathobiology of liver diseases. Hepatology. 2018;67(5):2013–2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lawan A, Bennett AM. Mitogen-Activated Protein Kinase Regulation in Hepatic Metabolism. Trends Endocrinol Metab. 2017;28(12):868–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ibrahim SH, Gores GJ, Hirsova P, et al. Mixed lineage kinase 3 deficient mice are protected against the high fat high carbohydrate diet-induced steatohepatitis. Liver international : official journal of the International Association for the Study of the Liver. 2014;34(3):427–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tomita K, Kabashima A, Freeman BL, Bronk SF, Hirsova P, Ibrahim SH. Mixed Lineage Kinase 3 Mediates the Induction of CXCL10 by a STAT1-Dependent Mechanism During Hepatocyte Lipotoxicity. J Cell Biochem. 2017;118(10):3249–3259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tomita K, Kohli R, MacLaurin BL, et al. Mixed-lineage kinase 3 pharmacological inhibition attenuates murine nonalcoholic steatohepatitis. JCI Insight. 2017;2(15). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Caunt CJ, Keyse SM. Dual-specificity MAP kinase phosphatases (MKPs): shaping the outcome of MAP kinase signalling. FEBS J. 2013;280(2):489–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schuster S, Feldstein AE. NASH: Novel therapeutic strategies targeting ASK1 in NASH. Nat Rev Gastroenterol Hepatol. 2017;14(6):329–330. [DOI] [PubMed] [Google Scholar]
- 14.Wang PX, Ji YX, Zhang XJ, et al. Targeting CASP8 and FADD-like apoptosis regulator ameliorates nonalcoholic steatohepatitis in mice and nonhuman primates. Nat Med. 2017;23(4):439–449. [DOI] [PubMed] [Google Scholar]
- 15.Xiang M, Wang PX, Wang AB, et al. Targeting hepatic TRAF1-ASK1 signaling to improve inflammation, insulin resistance, and hepatic steatosis. Journal of Hepatology. 2016;64(6):1365–1377. [DOI] [PubMed] [Google Scholar]
- 16.Nishitoh H, Saitoh M, Mochida Y, et al. ASK1 is essential for JNK/SAPK activation by TRAF2. Mol Cell. 1998;2(3):389–395. [DOI] [PubMed] [Google Scholar]
- 17.Wang Y, Wen H, Fu J, et al. Hepatocyte TNF Receptor-Associated Factor 6 Aggravates Hepatic Inflammation and Fibrosis by Promoting Lysine 6-Linked Polyubiquitination of Apoptosis Signal-Regulating Kinase 1. Hepatology. 2020;71(1):93–111. [DOI] [PubMed] [Google Scholar]
- 18.Harrison SA, Wong VWS, Okanoue T, et al. Safety and Efficacy of Selonsertib for the Treatment of Bridging Fibrosis or Compensated Cirrhosis Due to Nonalcoholic Steatohepatitis (Nash): Results of the Phase 3 Stellar Studies. Hepatology. 2019;70:45a–46a. [Google Scholar]
- 19.Loomba R, Lawitz E, Mantry PS, et al. The ASK1 Inhibitor Selonsertib in Patients with Nonalcoholic Steatohepatitis: A Randomized, Phase 2 Trial. Hepatology. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huang Z, Wu LM, Zhang JL, et al. Dual Specificity Phosphatase 12 Regulates Hepatic Lipid Metabolism Through Inhibition of the Lipogenesis and Apoptosis Signal-Regulating Kinase 1 Pathways. Hepatology. 2019;70(4):1099–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang QY, Zhao LP, Tian XX, et al. The novel intracellular protein CREG inhibits hepatic steatosis, obesity, and insulin resistance. Hepatology. 2017;66(3):834–854. [DOI] [PubMed] [Google Scholar]
- 22.Zhang P, Wang PX, Zhao LP, et al. The deubiquitinating enzyme TNFAIP3 mediates inactivation of hepatic ASK1 and ameliorates nonalcoholic steatohepatitis. Nat Med. 2018;24(1):84–94. [DOI] [PubMed] [Google Scholar]
- 23.Challa TD, Wueest S, Lucchini FC, et al. Liver ASK1 protects from non-alcoholic fatty liver disease and fibrosis. EMBO Mol Med. 2019;11(10):e10124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brancho D, Ventura JJ, Jaeschke A, Doran B, Flavell RA, Davis RJ. Role of MLK3 in the regulation of mitogen-activated protein kinase signaling cascades. Mol Cell Biol. 2005;25(9):3670–3681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jaeschke A, Davis RJ. Metabolic stress signaling mediated by mixed-lineage kinases. Molecular cell. 2007;27(3):498–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sharma M, Urano F, Jaeschke A. Cdc42 and Rac1 are major contributors to the saturated fatty acid-stimulated JNK pathway in hepatocytes. Journal of hepatology. 2012;56(1):192–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kant S, Barrett T, Vertii A, et al. Role of the mixed-lineage protein kinase pathway in the metabolic stress response to obesity. Cell Rep. 2013;4(4):681–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ibrahim SH, Hirsova P, Tomita K, et al. Mixed lineage kinase 3 mediates release of C-X-C motif ligand 10-bearing chemotactic extracellular vesicles from lipotoxic hepatocytes. Hepatology. 2016;63(3):731–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tomita K, Freeman BL, Bronk SF, et al. CXCL10-Mediates Macrophage, but not Other Innate Immune Cells-Associated Inflammation in Murine Nonalcoholic Steatohepatitis. Sci Rep. 2016;6:28786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang X, Shen JY, Man K, et al. CXCL10 plays a key role as an inflammatory mediator and a non-invasive biomarker of non-alcoholic steatohepatitis. Journal of Hepatology. 2014;61(6):1365–1375. [DOI] [PubMed] [Google Scholar]
- 31.Finch A, Davis W, Carter WG, Saklatvala J. Analysis of mitogen-activated protein kinase pathways used by interleukin 1 in tissues in vivo: activation of hepatic c-Jun N-terminal kinases 1 and 2, and mitogen-activated protein kinase kinases 4 and 7. Biochem J. 2001;353(Pt 2):275–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hirosumi J, Tuncman G, Chang L, et al. A central role for JNK in obesity and insulin resistance. Nature. 2002;420(6913):333–336. [DOI] [PubMed] [Google Scholar]
- 33.Schattenberg JM, Singh R, Wang Y, et al. JNK1 but not JNK2 promotes the development of steatohepatitis in mice. Hepatology. 2006;43(1):163–172. [DOI] [PubMed] [Google Scholar]
- 34.Singh R, Wang Y, Xiang Y, Tanaka KE, Gaarde WA, Czaja MJ. Differential effects of JNK1 and JNK2 inhibition on murine steatohepatitis and insulin resistance. Hepatology. 2009;49(1):87–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Puri P, Mirshahi F, Cheung O, et al. Activation and dysregulation of the unfolded protein response in nonalcoholic fatty liver disease. Gastroenterology. 2008;134(2):568–576. [DOI] [PubMed] [Google Scholar]
- 36.Cazanave SC, Mott JL, Elmi NA, et al. JNK1-dependent PUMA expression contributes to hepatocyte lipoapoptosis. The Journal of biological chemistry. 2009;284(39):26591–26602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kodama Y, Kisseleva T, Iwaisako K, et al. c-Jun N-terminal Kinase-1 From Hematopoietic Cells Mediates Progression From Hepatic Steatosis to Steatohepatitis and Fibrosis in Mice. Gastroenterology. 2009;137(4):1467–1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Malhi H, Bronk SF, Werneburg NW, Gores GJ. Free fatty acids induce JNK-dependent hepatocyte lipoapoptosis. J Biol Chem. 2006;281(17):12093–12101. [DOI] [PubMed] [Google Scholar]
- 39.Han MS, Jung DY, Morel C, et al. JNK expression by macrophages promotes obesity-induced insulin resistance and inflammation. Science. 2013;339(6116):218–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ozcan U, Cao Q, Yilmaz E, et al. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science. 2004;306(5695):457–461. [DOI] [PubMed] [Google Scholar]
- 41.Sabio G, Das M, Mora A, et al. A stress signaling pathway in adipose tissue regulates hepatic insulin resistance. Science. 2008;322(5907):1539–1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sabio G, Cavanagh-Kyros J, Ko HJ, et al. Prevention of steatosis by hepatic JNK1. Cell Metab. 2009;10(6):491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vernia S, Cavanagh-Kyros J, Garcia-Haro L, et al. The PPAR alpha-FGF21 Hormone Axis Contributes to Metabolic Regulation by the Hepatic JNK Signaling Pathway. Cell Metabolism. 2014;20(3):512–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Win S, Than TA, Le BH, Garcia-Ruiz C, Fernandez-Checa JC, Kaplowitz N. Sab (Sh3bp5) dependence of JNK mediated inhibition of mitochondrial respiration in palmitic acid induced hepatocyte lipotoxicity. J Hepatol. 2015;62(6):1367–1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.BonDurant LD, Ameka M, Naber MC, et al. FGF21 Regulates Metabolism Through Adipose-Dependent and -Independent Mechanisms. Cell Metab. 2017;25(4):935–944 e934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vernia S, Cavanagh-Kyros J, Barrett T, Tournier C, Davis RJ. Fibroblast Growth Factor 21 Mediates Glycemic Regulation by Hepatic JNK. Cell Rep. 2016;14(10):2273–2280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sanyal A, Charles ED, Neuschwander-Tetri BA, et al. Pegbelfermin (BMS-986036), a PEGylated fibroblast growth factor 21 analogue, in patients with non-alcoholic steatohepatitis: a randomised, double-blind, placebo-controlled, phase 2a trial. Lancet. 2019;392(10165):2705–2717. [DOI] [PubMed] [Google Scholar]
- 48.Win S, Than TA, Min RWM, Aghajan M, Kaplowitz N. c-Jun N-Terminal Kinase Mediates Mouse Liver Injury Through a Novel Sab (SH3BP5)-Dependent Pathway Leading to Inactivation of Intramitochondrial Src. Hepatology. 2016;63(6):1987–2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jing Y, Liu W, Cao H, et al. Hepatic p38alpha regulates gluconeogenesis by suppressing AMPK. J Hepatol. 2015;62(6):1319–1327. [DOI] [PubMed] [Google Scholar]
- 50.Lawan A, Zhang L, Gatzke F, et al. Hepatic mitogen-activated protein kinase phosphatase 1 selectively regulates glucose metabolism and energy homeostasis. Mol Cell Biol. 2015;35(1):26–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gonzalez-Teran B, Matesanz N, Nikolic I, et al. p38 gamma and p38 delta reprogram liver metabolism by modulating neutrophil infiltration. Embo Journal. 2016;35(5):536–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang X, Fan LN, Wu JF, et al. Macrophage p38 alpha promotes nutritional steatohepatitis through M1 polarization. Journal of Hepatology. 2019;71(1):163–174. [DOI] [PubMed] [Google Scholar]
- 53.Min L, He B, Hui L. Mitogen-activated protein kinases in hepatocellular carcinoma development. Semin Cancer Biol. 2011;21(1):10–20. [DOI] [PubMed] [Google Scholar]
- 54.Sakurai T, Kudo M, Umemura A, et al. p38alpha inhibits liver fibrogenesis and consequent hepatocarcinogenesis by curtailing accumulation of reactive oxygen species. Cancer Res. 2013;73(1):215–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hui L, Bakiri L, Mairhorfer A, et al. p38alpha suppresses normal and cancer cell proliferation by antagonizing the JNK-c-Jun pathway. Nat Genet. 2007;39(6):741–749. [DOI] [PubMed] [Google Scholar]
- 56.Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8(7):519–529. [DOI] [PubMed] [Google Scholar]
- 57.Maiers JL, Malhi H. Endoplasmic Reticulum Stress in Metabolic Liver Diseases and Hepatic Fibrosis. Semin Liver Dis. 2019;39(2):235–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bertolotti A, Zhang Y, Hendershot LM, Harding HP, Ron D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat Cell Biol. 2000;2(6):326–332. [DOI] [PubMed] [Google Scholar]
- 59.Han J, Kaufman RJ. The role of ER stress in lipid metabolism and lipotoxicity. J Lipid Res. 2016;57(8):1329–1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Harding HP, Zhang Y, Bertolotti A, Zeng H, Ron D. Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol Cell. 2000;5(5):897–904. [DOI] [PubMed] [Google Scholar]
- 61.Cullinan SB, Zhang D, Hannink M, Arvisais E, Kaufman RJ, Diehl JA. Nrf2 is a direct PERK substrate and effector of PERK-dependent cell survival. Mol Cell Biol. 2003;23(20):7198–7209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fusakio ME, Willy JA, Wang Y, et al. Transcription factor ATF4 directs basal and stress-induced gene expression in the unfolded protein response and cholesterol metabolism in the liver. Mol Biol Cell. 2016;27(9):1536–1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lu M, Lawrence DA, Marsters S, et al. Opposing unfolded-protein-response signals converge on death receptor 5 to control apoptosis. Science. 2014;345(6192):98–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wali JA, Rondas D, McKenzie MD, et al. The proapoptotic BH3-only proteins Bim and Puma are downstream of endoplasmic reticulum and mitochondrial oxidative stress in pancreatic islets in response to glucotoxicity. Cell Death Dis. 2014;5:e1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Glab JA, Doerflinger M, Nedeva C, et al. DR5 and caspase-8 are dispensable in ER stress-induced apoptosis. Cell Death Differ. 2017;24(5):944–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Han D, Lerner AG, Vande Walle L, et al. IRE1alpha kinase activation modes control alternate endoribonuclease outputs to determine divergent cell fates. Cell. 2009;138(3):562–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tirasophon W, Lee K, Callaghan B, Welihinda A, Kaufman RJ. The endoribonuclease activity of mammalian IRE1 autoregulates its mRNA and is required for the unfolded protein response. Genes Dev. 2000;14(21):2725–2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hollien J, Weissman JS. Decay of endoplasmic reticulum-localized mRNAs during the unfolded protein response. Science. 2006;313(5783):104–107. [DOI] [PubMed] [Google Scholar]
- 69.Urano F, Wang X, Bertolotti A, et al. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science. 2000;287(5453):664–666. [DOI] [PubMed] [Google Scholar]
- 70.Oyadomari S, Harding HP, Zhang Y, Oyadomari M, Ron D. Dephosphorylation of translation initiation factor 2alpha enhances glucose tolerance and attenuates hepatosteatosis in mice. Cell Metab. 2008;7(6):520–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Li H, Meng Q, Xiao F, et al. ATF4 deficiency protects mice from high-carbohydrate-diet-induced liver steatosis. Biochem J. 2011;438(2):283–289. [DOI] [PubMed] [Google Scholar]
- 72.Yang L, Calay ES, Fan J, et al. METABOLISM. S-Nitrosylation links obesity-associated inflammation to endoplasmic reticulum dysfunction. Science. 2015;349(6247):500–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wang JM, Qiu Y, Yang Z, et al. IRE1alpha prevents hepatic steatosis by processing and promoting the degradation of select microRNAs. Sci Signal. 2018;11(530). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.So JS, Hur KY, Tarrio M, et al. Silencing of lipid metabolism genes through IRE1alpha-mediated mRNA decay lowers plasma lipids in mice. Cell Metab. 2012;16(4):487–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wang S, Chen Z, Lam V, et al. IRE1alpha-XBP1s induces PDI expression to increase MTP activity for hepatic VLDL assembly and lipid homeostasis. Cell Metab. 2012;16(4):473–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Harnoss JM, Le Thomas A, Shemorry A, et al. Disruption of IRE1alpha through its kinase domain attenuates multiple myeloma. Proc Natl Acad Sci U S A. 2019;116(33):16420–16429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Liu X, Henkel AS, LeCuyer BE, Schipma MJ, Anderson KA, Green RM. Hepatocyte X-box binding protein 1 deficiency increases liver injury in mice fed a high-fat/sugar diet. Am J Physiol Gastrointest Liver Physiol. 2015;309(12):G965–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bailly-Maitre B, Belgardt BF, Jordan SD, et al. Hepatic Bax inhibitor-1 inhibits IRE1alpha and protects from obesity-associated insulin resistance and glucose intolerance. J Biol Chem. 2010;285(9):6198–6207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lebeaupin C, Proics E, de Bieville CH, et al. ER stress induces NLRP3 inflammasome activation and hepatocyte death. Cell Death Dis. 2015;6:e1879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kakazu E, Mauer AS, Yin M, Malhi H. Hepatocytes release ceramide-enriched pro-inflammatory extracellular vesicles in an IRE1alpha-dependent manner. J Lipid Res. 2016;57(2):233–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Malhi H, Bronk SF, Werneburg NW, Gores GJ. Free fatty acids induce JNK-dependent hepatocyte lipoapoptosis. J Biol Chem. 2006;281(17):12093–12101. [DOI] [PubMed] [Google Scholar]
- 82.Toriguchi K, Hatano E, Tanabe K, et al. Attenuation of steatohepatitis, fibrosis, and carcinogenesis in mice fed a methionine-choline deficient diet by CCAAT/enhancer-binding protein homologous protein deficiency. J Gastroenterol Hepatol. 2014;29(5):1109–1118. [DOI] [PubMed] [Google Scholar]
- 83.Lam M, Marsters SA, Ashkenazi A, Walter P. Misfolded proteins bind and activate death receptor 5 to induce apoptosis during unresolved endoplasmic reticulum stress. Elife. 2020;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Idrissova L, Malhi H, Werneburg NW, et al. TRAIL receptor deletion in mice suppresses the inflammation of nutrient excess. J Hepatol. 2015;62(5):1156–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Pearce LR, Komander D, Alessi DR. The nuts and bolts of AGC protein kinases. Nat Rev Mol Cell Biol. 2010;11(1):9–22. [DOI] [PubMed] [Google Scholar]
- 86.Julian L, Olson MF. Rho-associated coiled-coil containing kinases (ROCK): structure, regulation, and functions. Small GTPases. 2014;5:e29846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lee DH, Shi J, Jeoung NH, et al. Targeted disruption of ROCK1 causes insulin resistance in vivo. J Biol Chem. 2009;284(18):11776–11780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lee SH, Huang H, Choi K, et al. ROCK1 isoform-specific deletion reveals a role for diet-induced insulin resistance. Am J Physiol Endocrinol Metab. 2014;306(3):E332–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Huang H, Lee SH, Sousa-Lima I, et al. Rho-kinase/AMPK axis regulates hepatic lipogenesis during overnutrition. J Clin Invest. 2018;128(12):5335–5350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hirsova P, Ibrahim SH, Krishnan A, et al. Lipid-induced Signaling Causes Release of Inflammatory Extracellular Vesicles from Hepatocytes. Gastroenterology. 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hirsova P, Ibrahim SH, Verma VK, et al. Extracellular vesicles in liver pathobiology: Small particles with big impact. Hepatology. 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Guo Q, Furuta K, Lucien F, et al. Integrin beta1-enriched extracellular vesicles mediate monocyte adhesion and promote liver inflammation in murine NASH. J Hepatol. 2019;71(6):1193–1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Olson MF. Applications for ROCK kinase inhibition. Curr Opin Cell Biol. 2008;20(2):242–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Jahani V, Kavousi A, Mehri S, Karimi G. Rho kinase, a potential target in the treatment of metabolic syndrome. Biomed Pharmacother. 2018;106:1024–1030. [DOI] [PubMed] [Google Scholar]
- 95.Musso G, De Michieli F, Bongiovanni D, et al. New Pharmacologic Agents That Target Inflammation and Fibrosis in Nonalcoholic Steatohepatitis-Related Kidney Disease. Clin Gastroenterol Hepatol. 2017;15(7):972–985. [DOI] [PubMed] [Google Scholar]
- 96.Kitamura K, Tada S, Nakamoto N, et al. Rho/Rho kinase is a key enzyme system involved in the angiotensin II signaling pathway of liver fibrosis and steatosis. J Gastroenterol Hepatol. 2007;22(11):2022–2033. [DOI] [PubMed] [Google Scholar]
- 97.Zhou H, Fang C, Zhang L, Deng Y, Wang M, Meng F. Fasudil hydrochloride hydrate, a Rho-kinase inhibitor, ameliorates hepatic fibrosis in rats with type 2 diabetes. Chin Med J (Engl). 2014;127(2):225–231. [PubMed] [Google Scholar]
- 98.Wang HW, Liu PY, Oyama N, et al. Deficiency of ROCK1 in bone marrow-derived cells protects against atherosclerosis in LDLR−/− mice. FASEB J. 2008;22(10):3561–3570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Thorlacius K, Slotta JE, Laschke MW, et al. Protective effect of fasudil, a Rho-kinase inhibitor, on chemokine expression, leukocyte recruitment, and hepatocellular apoptosis in septic liver injury. J Leukoc Biol. 2006;79(5):923–931. [DOI] [PubMed] [Google Scholar]
- 100.Shimada H, Rajagopalan LE. Rho-kinase mediates lysophosphatidic acid-induced IL-8 and MCP-1 production via p38 and JNK pathways in human endothelial cells. FEBS Lett. 2010;584(13):2827–2832. [DOI] [PubMed] [Google Scholar]
- 101.Rao J, Ye Z, Tang H, et al. The RhoA/ROCK Pathway Ameliorates Adhesion and Inflammatory Infiltration Induced by AGEs in Glomerular Endothelial Cells. Sci Rep. 2017;7:39727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kuroda S, Tashiro H, Igarashi Y, et al. Rho inhibitor prevents ischemia-reperfusion injury in rat steatotic liver. J Hepatol. 2012;56(1):146–152. [DOI] [PubMed] [Google Scholar]
- 103.Kawada N, Seki S, Kuroki T, Kaneda K. ROCK inhibitor Y-27632 attenuates stellate cell contraction and portal pressure increase induced by endothelin-1. Biochem Biophys Res Commun. 1999;266(2):296–300. [DOI] [PubMed] [Google Scholar]
- 104.Tangkijvanich P, Tam SP, Yee HF Jr. Wound-induced migration of rat hepatic stellate cells is modulated by endothelin-1 through rho-kinase-mediated alterations in the acto-myosin cytoskeleton. Hepatology. 2001;33(1):74–80. [DOI] [PubMed] [Google Scholar]