

Abstract
Introduction
Intrahepatic cholangiocarcinoma (ICC) is a rare hepatobiliary cancer characterized by a poor prognosis and a limited response to conventional therapies. Currently chemotherapy is the only therapeutic option for patients with Stage IV ICC. Due to the poor response rate, there is an urgent need to identify novel molecular targets to develop novel effective therapies. Precision oncology tests utilizing targeted next-generation sequencing (NGS) platforms have rapidly entered into clinical practice. Profiling the genome and transcriptome of cancer to identify potentially targetable oncogenic pathways may guide the clinical care of the patient.
Case presentation
We present a 56-year-old male patient affected with metastatic ICC, whose cancer underwent several precision oncology tests by different NGS platforms. A novel BAP1 mutation (splice site c.581-17_585del22) and a RAD21 amplification were identified by a commercial available platform on a metastatic lesion. No germline BAP1 mutations were identified. Several lines of evidences indicate that PARP inhibitor administration might be an effective treatment in presence of BAP1 and/or RAD21 alterations since both BAP1 and RAD21 are involved in the DNA repair pathway, BAP1 interacts with BRCA1 and BRCA1-mediated DNA repair pathway alterations enhance the sensitivity to PARP inhibitor administration. In this case, after failing conventional therapies, patient was treated with PARP inhibitor olaparib. The patient had a partial response according to RECIST criteria with an overall survival of 37.2 months from the time of diagnosis of his ICC. Following 11.0 months on olaparib treatment, sustained stable disease control is ongoing. The patient is still being treated with olaparib and no significant toxicity has been reported.
Conclusion
These findings have clinical relevance since we have shown PARP inhibitor as a potential treatment for ICC patients harboring BAP1 deletion and RAD21 amplification. We have also highlighted the utility of NGS platforms to identify targetable mutations within a cancer.
Keywords: BAP1, precision oncology, cholangio carcinoma, Poly ADP ribose polymerase (PARP) inhibitor, RAD21, olaparib
Introduction
Cholangiocarcinoma (CCA) is historically classified by location into intrahepatic, perihilar (or Klatskintumor) and distal cancers. Intrahepatic cholangiocarcinoma (ICC) is the second most common primary intrahepatic tumor, with an estimated incidence of 1.6 per 100,000/year in the United States (1). Unfortunately, ICC carries an extremely poor prognosis with an overall 5-year survival of 5–15% (1). For patients with early stage ICC, surgical resection of the cancer and removal of local lymph nodes remains the only curative option (2). However, even with a complete resection, most patients succumb to both loco-regional and distant metastases (3). Unfortunately, most patients present with advanced disease. Palliative chemotherapy is of limited efficacy (4), highlighting the urgent need for novel effective therapies.
Different cancers express different oncogenic alterations which drive tumor progression. Several lines of evidences demonstrate that some of these alterations can be effectively targeted by tailored targeted agents, improving the overall survival of treated patients (5). These results have increased the use of precision oncology tests by targeted next-generation sequencing (NGS) platforms into clinical practice, to inform clinicians in making appropriate therapeutic decisions (6). Unselected ICC patients have been often included in “basket” trials (7), most of which have unfortunately failed to demonstrate a clinical benefit (7). As a result, there is a high interest to identifying oncogenic alterations in ICC to design potentially effective strategies in biomarker-enriched populations.
NGS of ICC has already allowed identification of molecular alterations which are involved in ICC carcinogenesis such as those in KRAS, BRAF, IDH1, IDH2, EGFR, FGFR2, ROS1, ARID1A, PBRM1, BRCA1, and BAP1 (8–16). FGFR kinase inhibitors have demonstrated anti-tumor activity in ICC patients harboring activating FGFR2 gene fusions (17–19). However, no effective therapeutic strategies have currently changed the standard of care of ICC patients harboring different types of alterations.
Here, we describe the case of a chemorefractory patient with ICC harboring BAP1 mutation and RAD21 amplification. The patient was successfully treated with the PARP inhibitor olaparib.
Case Presentation
In March 2017, a 56-year-old Caucasian male was admitted to San Giovanni di Dio and Ruggi D’Aragona University Hospital for mild abdominal pain and nausea. The patient’s past medical history included i) Hodgkin’s lymphoma of the spleen in 1987, treated with splenectomy and radiotherapy; ii) myocardial infarction in 2006, treated with coronary angioplasty; and iii) myocardial infarction in 2012, treated with multiple coronary artery bypass grafting. He was also a former-smoker. Patient did not present with any ICC risk factors including biliary lithiasis, alcoholic liver disease, chronic hepatitis B or C infections, or primary sclerosing cholangitis. His family history was negative for any inherited-familial cancers. Abdominal ultrasound and computed tomography (CT) scan revealed a 10 cm intrahepatic lesion in the left lobe of the liver, as well as stable right basal lung thickening ( Figure 1A ). The latter was already described in a previous chest CT scan. Ultrasound guided biopsy of the liver mass demonstrated ICC (CK7+, CK19+, HepPar1-, AFP-). In April 2017, the patient underwent a left hepatectomy and sub-total gastrectomy and cholecystectomy. Histological examination demonstrated a Stage II ICC with vascular invasion [TNM staging, American Joint Committee on Cancer (AJCC) 8th edition]. Post operatively he was seen by the multidisciplinary team. Genomic analysis of NRAS, KRAS and BRAF V600 by polymerase chain reaction (PCR) sequencing, as well as immunohistochemical (IHC) staining for detection of HER2 amplification were performed on ICC tumor tissue. Both analyses did not show any type of alteration ( Supplementary Table 1 ). Further genomic testing of EGFR was performed by sanger sequencing, but no alterations were found in exons 18, 19, 20, and 21 ( Supplementary Table 1 ). In October 2017, a whole body CT scan demonstrated a 2.0 cm local recurrence in segment V of the liver ( Figure 1B ). Patient received a percutaneous thermal ablation (PTA) of the lesion. In February 2018, a whole body CT scan demonstrated a new 3.6 cm local recurrence in segment V of the liver, close to the previously treated lesion ( Figure 1C ) for which patient received a new PTA. In May 2018, a whole body CT scan demonstrated a new local recurrence in segment V of liver and multiple lesions in segment VII and VIII ( Figure 1D ). He then started a chemotherapeutic regimen with cisplatin (25 mg/m2) followed by gemcitabine (1,000 mg/m2), each administered on days 1 and 8 every 3 weeks. Due to his poor prognosis, patient requested additional testing of the ICC specimen. An IHC analysis of ROS1 rearrangements and NTRK fusions did not demonstrate any alterations ( Supplementary Table 2 ). A Short Tandem Repeat (STR) analysis by PCR of BAT25, BAT26, D2S123, D5S346, D17S250, NR-21, and MONO-27 showed a Microsatellite Stable (MSS) tumor profile. Lastly an IHC analysis of MSH2, MSH6, PMS2, and MLH1 demonstrated no alterations of the mismatch repair system ( Supplementary Table 2 ). Following six cycles of cisplatin and gemcitabine, in September 2018, a whole-body CT scan demonstrated a stable disease (according to RECIST criteria v 1.1). The patient received an additional PTA of the lesions in segments V, VII, and VIII of the liver. In February 2019, the CT scan demonstrated progression of disease (PD) (according to RECIST criteria v 1.1) due to the development of multiple small lesions localized at the hepatic dome and around the area of previous PTA, long with a large bone metastasis to the 12th vertebral body and a left upper lobe pulmonary nodule ( Figure 2A ). Based on the availability of additional formalin fixed tumor tissue obtained from a novel tumor biopsy, three different NGS platform studies were requested by the patient: Oncomine Comprehensive Assay (implemented at Istituto Tumori Milano, Milan, Italy) ( Table 1 ), Oncofocus test [Oncologica® UK ltd (Cambridge, UK)] ( Table 2 ) and Foundation One CDx [Foundation Medicine (Cambridge, MA)] ( Table 3 ). Both the Oncomine Comprehensive Assay and the Oncofocus test did not detect any alterations of analyzed genes. In contrast the Foundation One CDx demonstrated the presence of a deletion in BAP1 (splice site c.581-17_585del22) and amplification of RAD21. Analysis of BAP1 by sanger sequencing on primary ICC tumor tissue confirmed the presence of BAP1 (splice site 581-17_585del22) alteration ( Figure 3 ). In contrast no alterations were identified in BAP1 from nucleic acids extracted from buffy coat ( Figure 3 ). Because of the involvement of RAD21 in the DNA repair pathway, the interaction of BAP1 with BRCA1 and the enhanced sensitivity to PARP inhibitor administration in presence of alterations in the BRCA1-mediated DNA repair pathway, it was decided first to treat the patient with FOLFIRI every 2 weeks [irinotecan 180 mg/m2, folinic acid 400 mg/m2, 5-fluorouracil (5-FU) 400 mg/m2 intravenous infusion bolus, then 5-FU 2400 mg/m2 intravenous infusion over 46 h] and then to start a PARP inhibitor. FOLFIRI is a conventional second-line chemotherapy regimen for ICC. In addition, irinotecan is a DNA-damaging agent. Following six cycles of FOLFIRI, in June 2019, a whole-body CT scan demonstrated PD ( Figure 2B ). A third-line therapy of off-label use with the PARP inhibitor olaparib at 800 mg/die and palliative radiotherapy (10 Gy) on the vertebral lesion was begun. In September 2019, a whole-body CT scan demonstrated a partial response (PR) ( Figure 2C ). The latter was confirmed on successive restaging scans in November 2019 ( Figure 2D ) and February 2020 ( Figure 2E ). Following 11 cycles of olaparib, the progression free survival has been 11.0 months. Currently, the patient has an overall survival of 37.2 months from the time of diagnosis of his ICC and has continued treatment with olaparib. He is in good health conditions and no treatment-related adverse events have been reported.
Figure 1.
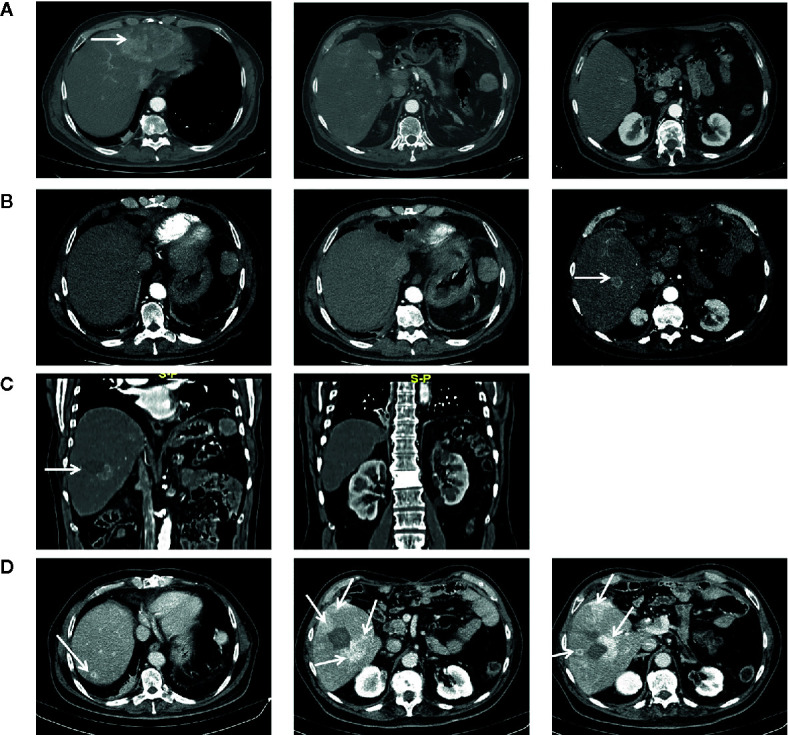
Chest CT-scan performed at diagnosis in March 2017 (A), in October 2017 following first relapse (B), in February 2018 at tumor progression following first percutaneous thermal ablation (C), in May 2018 at tumor progression following second percutaneous thermal ablation and before starting chemotherapy with cisplatin and gemcitabine (D). Arrows indicate tumor lesion.
Figure 2.

Chest CT-scan performed at diagnosis in February 2019 at tumor progression following chemotherapy with cisplatin and gemcitabine and a third percutaneous thermal ablation and before to start treatment with FOLFIRI (A), in June 2019 at tumor progression following six cycles of FOLFIRI administration and before to start treatment with olaparib (B), in September 2019 following three cycles of olaparib (C), in November 2019 following six cycles of olaparib (D), and in February 2020 following 11 cycles of olaparib (E). Arrows indicate tumor lesion.
Table 1.
Oncomine Comprehensive Assay.
| March 1st, 2019 | ||||||||
|---|---|---|---|---|---|---|---|---|
| NGS: Hot spot Cancer Panel with PGM (Personal Genome Machine) Ion Torrent technology [Thermo Fisher Scientific Life Technologies (Waltham, MA)] | ||||||||
| ABL1 | AKT1 | ALK | APC | ATM | BRAF | CDH1 | CDKN2A | CSF1R |
| CTNNB1 | EGFR | ERBB2 | ERBB4 | EZH2 | FBXW7 | FGFR1 | FGFR2 | FGFR3 |
| FLT3 | GNA11 | GNAQ | GNAS | HNF1A | HRAS | IDH1 | IDH2 | JAK2 |
| JAK3 | KDR (VEGFR2) | KIT | KRAS | MET | MLH1 | MPL | NOTCH1 | NPM1 |
| NRAS | PDGFRA | PIK3CA | PTEN | PTPN11 | RB1 | RET | SMAD4 | SMARCB1 |
| SMO | SRC | STK11 | TP53 | VHL | ||||
| Results: No hot spot mutations detected. | ||||||||
Table 2.
Oncofocus test.
| March 2nd, 2019 | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| NGS: Oncofocus test (Oncologica® UK ltd (Cambridge, UK) | ||||||||||||||
| A2M | ABCB5 | ACACA | ACADM | ACBD5 | ACTG2 | ADAM32 | ADAMTS16 | AES | AFAP1 | AFF3 | AGAP3 | AGBL4 | AGGF1 | AGK |
| AGTRAP | AHCYL1 | AKAP12 | AKAP13 | AKAP9 | AKT1 | AKT2 | AKT3 | ALK | AP3B1 | AR | ARAF | ARHGEF2 | ARID1A | ARMC10 |
| ARMT1 | ASIC2 | ATAD2 | ATAD5 | ATF7IP | ATG7 | ATIC | ATM | ATP1B1 | ATR | ATRNL1 | ATRX | AXL | B4GALT1 | BAG4 |
| BAIAP2L1 | BAP1 | BBS9 | BCAM | BCAN | BCL2L11 | BCR | BEND5 | BICC1 | BICD2 | BIN2 | BIRC6 | BRAF | BRCA1 | BRCA2 |
| BRD3 | BRD4 | BTAF1 | BTBD1 | BTF3L4 | BTK | C11orf95 | C7orf73 | C8ORF34 | C9orf153 | CAD | CAND1 | CAPRIN1 | CAPZA2 | CARS |
| CASP7 | CBL | CCAR2 | CCDC170 | CCDC6 | CCDC88A | CCDC91 | CCND1 | CCND2 | CCND3 | CCNE1 | CCNY | CD44 | CD74 | CDC27 |
| CDK12 | CDK2 | CDK4 | CDK5RAP2 | CDK6 | CDKN1B | CDKN2A | CDKN2B | CEL | CEP85L | CEP89 | CHD9 | CHEK1 | CHEK2 | CHTOP |
| CIC | CIITA | CIT | CLCN6 | CLIP1 | CLIP2 | CLIP4 | CLTC | CNTLN | CNTRL | COL14A1 | COX5A | CPSF6 | CREB3L2 | CREB5 |
| CREBBP | CSF1R | CTNNB1 | CUL1 | CUX1 | DAB2 | DAB2IP | DCTN1 | DDR2 | DIP2C | DNAJB1 | DTD1 | DYM | DYNC1I2 | DYNC2H1 |
| EBF1 | EGFR | EIF3E | ELAVL3 | EML4 | EPHB2 | EPS15 | ERBB2 | ERBB3 | ERBB4 | ERC1 | ERCC2 | ERG | ERLIN2 | ERP44 |
| ERVK3_1 | ESR1 | ESRP1 | ETV1 | ETV4 | ETV5 | ETV6 | EZH2 | EZR | FAM114A2 | FAM131B | FAM76A | FANCA | FANCD2 | FANCI |
| FA1 | FBXO28 | FBXW7 | FCHSD1 | FGF3 | FGFR1 | FGFR19 | FGFR1OP | FGFR1OP2 | FGFR2 | FGFR3 | FGFR4 | FGR | FP1L1 | FKBP15 |
| FLT3 | FN1 | FNDC3B | FOXL2 | FOXP1 | FXR1 | FYCO1 | GABBR2 | GATA2 | GATM | GFPT1 | GHR | GIT2 | GLIS3 | GNA11 |
| GNAI1 | GNAQ | GNAS | GNS | GOLGA4 | GOLGA5 | GOLGB1 | GOPC | GRB7 | GRHL2 | GTF2I | GTF2IRD1 | GTF3C2 | H3F3A | HACL1 |
| HERPUD1 | HIP1 | HIST1H3B | HLA_A | HMGA2 | NHNF1A | HOMER1 | HOOK3 | HRAS | IDH1 | IDH2 | IGF1R | IRF2BP2 | JAK1 | JAK2 |
| JAK3 | JAKMIP1 | KANK1 | KANK2 | KCNQ5 | KCTD1 | KCTD7 | KDELR2 | KDM7A | KDR | KIAA1468 | KIAA1549 | KIAA1598 | KIF5B | KIT |
| KLC1 | KLHL7 | KNSTRN | KRAS | KTN1 | LMNA | LRIG3 | LRRFIP1 | LSM12 | LSM14A | LYN | MACF1 | MAD1L1 | MAGOH | MAP2K1 |
| MAP2K2 | MAP2K4 | MAPK1 | MAX | MBIP | MCFD2 | MDM2 | MDM4 | MED12 | MEMO1 | MET | MGEA5 | MIR143HG | MKRN1 | MLH1 |
| MPRIP | MRE11A | MRPL24 | MRPS33 | MSH2 | MSH6 | MSN | MTFHD1L | MTMR12 | MTOR | MYB | MYBL1 | MYC | MYCL | MYCN |
| MYD88 | MYH13 | MYH9 | MYO18A | MYO5A | MYRIP | MZT1 | NACC2 | NAV1 | NBN | NCOA1 | NCOA4 | NCOR2 | NDE1 | NF1 |
| NF2 | NFASC | NFIB | NFKB2 | NIN | NOL4 | NOTCH1 | NOTCH2 | NOTCH3 | NOTCH4 | NPC2 | NPM1 | NRAS | NRG1 | NSD1 |
| NTM | NTRK1 | NTRK2 | NTRK3 | NUB1 | NUDCD3 | NUP214 | NUTM1 | OFD1 | OPHN1 | OXR1 | PALB2 | PAPD7 | PAPSS1 | PARK2 |
| PAX5 | PAX8 | PCDHGA1 | PCM1 | PCNX | PDE10A | PDE4DIP | PDE7A | PDGFRA | PDGFRB | PDHX | PDP1 | PDZRN3 | PHEB | PIK3CA |
| PIK3CB | PIK3R1 | PLAG1 | PLIN3 | PMS2 | POLE | POLH | PPARG | PPFIBP1 | PPHLN1 | PPL | PPM1G | PPP2R1A | PPP4R3B | PRKACA |
| PRKACB | PRKAR1A | PRKG2 | PSMD11 | PSPH | PTCH1 | PTEN | PTPN11 | PTPN3 | PTPRK | PTPRZ1 | PWWP2A | QKI | RABEP1 | RABGAP1L |
| RAC1 | RAD18 | RAD50 | RAD51 | RAD51B | RAD51C | RAD51D | RAF1 | RANBP2 | RB1 | RBMS3 | RBPMS | RELA | RET | RHOA |
| RICTOR | RNF11 | RNF130 | RNF213 | RNF43 | ROS1 | RP2 | RSPO2 | RSPO3 | RUFY2- | SART3 | SCAF11 | SDC4 | SDCCAG3 | SEC16A |
| SEC31A | SEC61G | SETD2 | SF3B1 | SHROOM4 | SHTN1 | SLC12A7 | SLC26A4 | SLC34A2 | SLC3A2 | SLC45A3 | SLMAP | SLX4 | SMAD4 | SMARCA4 |
| SMARCB1 | SMOP | SND1 | SNHG7 | SNX19 | SOX6 | SPAG9 | SPECC1 | SPECC1L | SPOP | SPTBN1 | SQSTM1 | SRC | SRGAP3 | SSBP2 |
| STAT3 | STK11 | STK32B | STRN | STRN3 | SUGCT | TACC1 | TACC3 | TANK | TAX1BP1 | TBL1XR1 | TENM4 | TERF2 | TERT | TPM1 |
| TFG | TMEM106B | TMEM178B | TMPRSS2 | TNIP1 | TNKS2 | TOP1 | TP53 | TP53BP1 | TPM3 | TPM4 | TPR | TRAF1 | TRAK1 | TRIM24 |
| TRIM27 | TRIM33 | TRIM4 | TRIO | TRIP11 | TRMT61B | TSC1 | TSC2 | TSEN2 | TTLL7 | TXLNA | TYK2 | U2AF1 | UBE2L3 | UBN2 |
| USP10 | VAMP2 | VCL | VOPP1 | WASF2 | WDR48 | WHSC1L1 | WIPF2 | XPO1 | YAP1 | YTHDF3 | YWHAE | ZC3HAV1 | ZCCHC8 | ZEB2 |
| ZKSCAN1 | ZKSCAN5 | ZMYM2 | ZMYND8 | ZNF226 | ZNF703 | ZSCAN30 | ||||||||
| Results: - Mutations: No actionable variant detected - Copy Number Variations: No actionable variant detected - Fusion Genes: No actionable variant detected | ||||||||||||||
Table 3.
Foundation One CDx.
| March 7nd, 2019 | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| DNA GENE LIST: ENTIRE CODING SEQUENCE FOR THE DETECTION OF BASE SUBSTITUTIONS, INSERTION/DELETIONS, AND COPY NUMBER ALTERATIONS Foundation One CDx [Foundation Medicine (Cambridge, MA)] | ||||||||||||
| ABL1 | ACVR1B | AKT1 | AKT2 | AKT3 | ALK | ALOX12B | AMER1 (FAM123B) | APC | AR | ARAF | ARFRP1 | |
| ARID1A | ASXL1 | ATM | ATR | ATRX | AURKA | AURKB | AXIN1 | AXL | BAP1 | BARD1 | BCL2 | |
| BCL2L1 | BCL2L2 | BCL6 | BCOR | BCORL1 | BRAF | BRCA1 | BRCA2 | BRD4 | BRIP1 | BTG1 | BTG2 | |
| BTK | C11orf30 (EMSY) | C17orf39 (GID34) | CALR | CARD11 | CASP8 | CBFB | CBL | CCND1 | CCND2 | CCND3 | CCNE1 | |
| CD22 | CD274 (PD-L1) | CD70 | CD79A | CD79B | CDC73 | CDH1 | CDK12 | CDK4 | CDK6 | CDK8 | CDKN1A | |
| CDKN1B | CDKN2A | CDKN2B | CDKN2C | CEBPA | CHEK1 | CHEK2 | CIC | CREBBP | CRKL | CSF1R | CSF3R | |
| CTCF | CTNNA1 | CTNNB1 | CUL3 | CUL4A | CXCR4 | CYP17A1 | DAXX | DDR1 | DDR2 | DIS3 | DNMT3A | |
| DOT1L | EED | EGFR | EP300 | EPHA3 | EPHB1 | EPHB4 | ERBB2 | ERBB3 | ERBB4 | ERCC4 | ERG | |
| ERRFI1 | ESR1 | EZH2 | FAM46C | FANCA | FANCC | FANCG | FANCL | FAS | FBXW7 | FGF10 | FGF12 | |
| FGF14 | FGF19 | FGF23 | FGF3 | FGF4 | FGF6 | FGFR1 | FGFR2 | FGFR3 | FGF4 | FH | FLCN | |
| FLT1 | FLT3 | FOXL2 | FUBP1 | GABRA6 | GATA3 | GATA4 | GATA6 | GNA11 | GNA13 | GNAQ | GNAS | |
| GRM3 | GSK3B | H3F3A | HDAC1 | HGF | HNF1A | HRAS | HSD3B1 | ID3 | IDH1 | IDH2 | IGF1R | |
| IKBKE | IKZF1 | INPP4B | IRF2 | IRF4 | IRS2 | JAK1 | JAK2 | JAK3 | JUN | KDM5A | KDM5C | |
| KDM6A | KDR | KEAP1 | KEL | KIT | KLHL6 | KMT2A (MLL) | KMT2D (MLL2) | KRAS | LTK | LYN | MAF | |
| MAP2K1 (MEK1) | MAP2K2 (MEK2) | MAP2K4 | MAP3K1 | MAP3K13 | MAPK1 | MCL1 | MDM2 | MDM4 | MED12 | MEF2B | MEN1 | |
| MEERTK | MET | MITF | MKNK1 | MLH1 | MPL | MRE11A | MSH2 | MSH3 | NBN | NF1 | NF2 | |
| NFE2L2 | NFKBIA | NKX2-1 | NOTCH1 | NOTCH2 | NOTCH3 | NPM1 | NRAS | NSD3 (WHSC1L1) | NT5C2 | NTRK1 | NTK2 | |
| NTRK3 | P2RY8 | PALB2 | PARK2 | PARP1 | PARP2 | PARP3 | PAX5 | PBRM1 | PRKAR1A | PRKCI | PTCH1 | |
| PTEN | PTPN11 | PTPRO | QKI | RAC1 | RAD21 | RAD51 | RAD51B | RAD51C | RAD51D | RAD52 | RAD54L | |
| RAF1 | RARA | RB1 | RBM10 | REL | RET | SF3B1 | SGK1 | SMAD2 | SMAD4 | SMARCA4 | SMARCB1 | |
| SMO | SNCAIP | SOCS1 | SYK | TBX3 | TEK | TET2 | TGFBR2 | TIPARP | TNFAIP3 | TNFRSF14 | TP53 | |
| TSC1 | TSC2 | TYRO3 | U2AF1 | VEGFA | VHL | WHSC1 | WT1 | XPO1 | ||||
| DNA GENE LIST: FOR THE DETECTION OF SELECT REARRANGEMENTS | ||||||||||||
| ALK | BCL2 | BCR | BRAF | BRCA1 | BRCA2 | CD74 | EGFR | ETV4 | ETV5 | ETV6 | EWSR1 | |
| EZR | FGFR1 | FGFR2 | FGFR3 | KIT | KMT2A (MLL) | MSH2 | MYB | MYC | NOTCH2 | NTRK1 | NTRK2 | |
| NUTM1 | PGFRA | RAF1 | RARA | RET | ROS1 | RSPO2 | SDC4 | SLC34A2 | TERC | TERT | TMPRSS2 | |
|
Results:
- BAP1: Splice site 581-17_585del22 - RAD21: amplification |
||||||||||||
Figure 3.

The figure shows BAP1 molecular analysis performed on tumor tissue samples and buffy coat by using sanger sequencing platform. In details, c. 581-17_585del22 mutation was found only in tumor tissue specimen (A) while nucleic acids extracted from buffy coat did not harbor this mutation (B).
Discussion and Conclusions
Novel effective therapies are urgently needed for metastatic ICC patients. The current clinical case has provided for the first-time evidence that ICC patients carrying a BAP1 deletion and RAD21 amplification might benefit from a PARP inhibitor treatment. BAP1 is a tumor suppressor gene which modulates several pathways including cell death, cell differentiation, DNA damage response and gluconeogenesis (20–28). In mediating DNA damage response, BAP1 interacts with BRCA1 (20, 21). BRCA1 plays a key role in the DNA repair mechanism as well as in cell cycle regulation (29). Germline heterozygous mutations in BAP1 cause an autosomal dominant condition known as BAP1-cancer syndrome which confers a high susceptibility to the development of several malignancies including mesothelioma, uveal melanoma, renal, cholangio and breast carcinomas (30–38). In the clinical case we have described, we identified a novel mutation in BAP1 (c.581-17_585del22). The variant was somatic and not detected in the germline. We have examined several databases (Cosmic, GenBank, ClinVar) and c.581-17_585del22 mutation was not identified. Some literature data reported a similar deletion of BAP1 with a pathogenic value (39, 40). Somatic mutations in BAP1 are reported to drive carcinogenesis in mesothelioma, lung adenocarcinoma and melanoma (30, 32, 34, 41). BAP1 mutations occur in 10–32% of ICC cases (10, 14, 30, 42–49). As a tumor suppressor gene, BAP1 seems to follow a classic two-hit model (Knudson model) in which probably the first hit involves loss of heterozygosity (LOH) induced by 3p21 deletion. The latter occurs in almost 50–75% of ICCs (36). A subsequent mutation occurring in the remaining allele might lead to impairment of protein function and/or homeostasis (36). Protein function impairment by c.581-17_585del22 is most likely to reflect a deletion in the 3’-splice site of BAP1. Previously a c.581(-5)_c.590delACTAGGGCCCTGGGG mutation has been reported causing a premature truncation of BAP1 (50). This type of alterations that disrupt the nuclear localizations signal (aminoacids 717-722) of BAP1 are predicted to be inactivating (14, 51).
As BAP1 interacts with BRCA1, several lines of evidence indicate that alterations in the BRCA-mediated DNA repair pathway confers sensitivity to PARP inhibitor administration (52). PARP inhibitors act through synthetic lethality, whereby genetic DNA repair defects are enhanced by drug-induced defects in a compensatory pathway (53). Carriers of heterozygous BRCA1/2 mutations are sensitive to PARP inhibitor treatment as they lose the wild-type allele during tumorigenesis and thereby become deficient of the homologous recombination (HR) pathway of double-strand break DNA repair by BRCA1/2-null status. Four PARP inhibitors, olaparib, rucaparib, niraparib, and talazoparib, have been approved by the U.S. Food and Drug Administration (FDA) and by the European Medicines Agency (EMA). In 2014, olaparib was approved as maintenance therapy for platinum-sensitive advanced ovarian cancer with germline mutations in BRCA1/2. In 2016, rucaparib was approved for advanced ovarian cancer with both germline and somatic BRCA1/2 mutations. In 2017 and 2018, olaparib, rucaparib, and niraparib were approved for the maintenance treatment of recurrent, epithelial ovarian, fallopian tube, or primary peritoneal cancer irrespective of the BRCA status. Last, in 2018, olaparib and talazoparib were approved for HER2-negative locally advanced or metastatic breast cancer with germline BRCA1/2 mutations. Besides in ovarian and breast cancer, PARP inhibitor efficacy has also been demonstrated in other types of cancer including prostate and pancreatic cancer, and small cell lung carcinoma, irrespective of the BRCA status (54–61). It has become clear that any form of HR deficiency in tumors that phenocopies BRCA1/2 mutations, often referred to as BRCAness, may sensitize cells to PARP inhibitors (62). Indeed mutations in DNA damage response genes such as ATM, PRKDC, ATR, RPA1, DSS1, NBN, RAD51, RAD54, CHEK1, CHEK2, FANC genes, ERCC1, POLB, FEN1, and CDK12 have shown synthetic lethality in combination with PARP inhibitors (63–67).
BAP1 is a HR DNA repair component and its loss sensitizes cancer cells to DNA repair defects (28). Currently, further investigations are needed to establish the real efficacy of PARP inhibitor on BAP1 mutated cancer cells. Some studies on various types of BAP1 mutated cancer cell lines demonstrated the potential efficacy of PARP inhibitors (68–70). A synergistic effect of PARP inhibitor and gemcitabine is described in BAP1 deficient cholangiocarcinoma cell lines (71). As a result, PARP inhibitors are currently under investigation alone or in combination with other therapies in cancer patients harboring a BAP1 mutant tumor including ICC (ClinicalTrials.gov Identifier: NCT03207347, NCT03786796, NCT03531840, and NCT03375307).
In the current clinical case, we have shown that PARP inhibitor administration can be potentially effective in BAP1 mutated ICC. Chemotherapeutic agents, such as platinum compounds which induce double-strand DNA breaks, are usually utilized prior to PARP inhibition in order to enhance DNA damage and induce PARP inhibition-mediated cell death (72). In addition PARP inhibitors are currently administered after obtaining a disease control with platinum compounds (73, 74). In the present clinical case, the PARP inhibitor olaparib was effective in controlling tumor progression, even though the patient did not benefit from FOLFIRI administration, a combination of 5-FU and topoisomerase I inhibitor irinotecan. Irinotecan exerts its anticancer effects through induction of single- and double-strand DNA breaks. 5-FU is an antimetabolite drug that exerts its anticancer effects through inhibition of DNA synthesis by inhibition of thymidylate synthase and incorporation of its metabolites into RNA and DNA. One could speculate that efficacy to PARP inhibitor was not enhanced by FOLFIRI administration, but rather by the previous administration of cisplatin. Additional studies are needed to define the timing and schedule of DNA damaging agents for PARP inhibitor enhancement in BAP1 deficient tumors.
In addition to BAP1 mutations, many other molecular alterations have been described in ICC such as KRAS, BRAF, IDH1, IDH2, EGFR, FGFR2, ROS1, ARID1A, PBRM1, and BRCA1 (8–16). These types of alterations are frequently mutually exclusive (8–16). In the current clinical case, BAP1 mutation is not associated with KRAS, BRAF, IDH1, IDH2, EGFR, FGFR2, ROS1, ARID1A, PBRM1, and BRCA1 alterations but with a RAD21 amplification. Further studies are needed to validate this type of association. RAD21 is a gene involved in the repair of DNA double-strand breaks, as well as in chromatid cohesion during mitosis (75, 76). Amplification of RAD21 is described in approximately 1.23% of cases reported in the AACR Project Genomics Evidence Neoplasia Information Exchange (AACR Project GENIE), including invasive breast carcinoma, prostate adenocarcinoma, lung adenocarcinoma and colon adenocarcinoma having the greatest prevalence (77). However, no prior data exists regarding RAD21 amplification in ICC. Whether RAD21 amplification might enhance the activity of a PARP inhibitor in BAP1 mutant ICC should be further investigated.
Both BAP1 and RAD21 alterations were detected by utilizing NGS analysis. Patient’s tumor tissue underwent analysis by several precision oncology testing methods to identify potentially oncogenic alterations. However, most of the tests performed did not detect any alterations. By comparing the results from the two most extensive tumor genomic profiles BAP1 was analyzed in both: the Foudation One CDx and Oncofocus test. However only the Foudation One CDx test was able to detect BAP1 and RAD21 alterations. These findings are likely to reflect the different methods utilized to detect potentially oncogenic alterations, the regions of the genes included in the analysis, the potential tumor heterogeneity especially with a low allele frequency of the variants and the percentage of tumor cells in the sample tested. Since there is no targeted regions for BAP1 it is unlikely that different NGS platforms only test selected exons. In our case the novel mutation c.581-17_585del22 of BAP1 was localized on exon 8 of BAP1, at the boundary of intron 7. Most of the NGS platforms include 20-25bp in the vicinity of exons. However the Oncofocus® Test did not detect the c.581-17_585del22 alteration of BAP1 alteration most likely because this region of the gene was not included in the analysis. In contrast, the Foundation One CDx platform included in the analysis the full exonic region of BAP1 besides including also RAD21 in the analysis. Foundation One CDx report contains information only about the genomic findings without allele frequency values. As limit of detection range at non-homopolymer context (insertion up to 42 bp and deletion up to 276 bp) is 6–10%, we can assume that the BAP1 c.581-17_585del mutated allele was present with a higher variant fraction in the metastatic tumor tissue analyzed. In addition, direct sequencing has a reported limit of detection of approximately 20% mutant alleles. In our case BAP1 sanger sequencing on primary ICC tumor tissue showed the unbalanced presence of the mutated allele, even if it is not possible to have a quantitative value, as with NGS or digital PCR, we can hypothesize an allele frequency close to the limit of detection. Therefore, we can assume that BAP1 c.581-17_585del mutated allele occurred with a high allele frequency, early in ICC oncogenesis.
In conclusion, genomic characterization of ICC tumors by NGS analysis can identify potential targetable oncogenic alterations in ICC, providing the possibility to improve patient survival. Specifically, BAP1 deletion and RAD21 amplification were identified and effectively targeted by PARP inhibitor administration. These results warrant further studies to define the role of PARP inhibitor in ICC harboring BAP1 and RAD21 alterations.
Data Availability Statement
The original contributions presented in the study are included in the article/ Supplementary Material . Further inquiries can be directed to the corresponding author.
Ethics Statement
Written informed consent was obtained from the patient for publication of this case report and any accompanying images.
Author Contributions
Conception and design: FSa, SP, and UM. Acquisition of data: LL, VT, and FSa. Analysis and interpretation of data: FSa, AF, VC, FSc, and UM. Writing, review, and/or revision of the manuscript: FSa, LL, and CF. Administrative, technical, or material support (i.e., reporting or organizing data, constructing databases): LL, and VT. Study supervision: SP. Other (contributed clinical and pathological material; discussed results and implications of findings): SP, GT, and CF. All authors contributed to the article and approved the submitted version.
Funding
The work was supported by Ministero dell’Università e della Ricerca (Progetti di Rilevante Interesse Nazionale (PRIN), 2017, CODICE 2017PHRC8X_003) (to SP).
Conflict of Interest
UM reports personal fees (as speaker bureau or advisor) from Boehringer Ingelheim, AstraZeneca, Roche, MSD, Amgen and Merck, unrelated to the current work.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgements
The authors wish to gratefully acknowledge the patient for allowing us to publish his clinical case.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2020.567289/full#supplementary-material
Abbreviations
AFP, alpha fetoprotein; AJCC, American Joint Committee on Cancer; ARID1A, AT-rich interactive domain-containing protein 1A; ATM, ataxia-telangiectasia mutated; ATR, ataxia telangiectasia and Rad3-related protein; BAP1, BRCA1 associated protein 1; BRAF, v-raf murine sarcoma viral oncogene homolog B1; BRCA1, Breast cancer type 1 susceptibility protein; CCA, Cholangiocarcinoma; CDK12, Cyclin Dependent Kinase 12; CHEK1-CHEK2, Checkpoint kinase 1-2; CK7, Cytokeratin 7; CK19, Cytokeratin 19; CT, computed tomography; EGFR, Epidermal Growth Factor Receptor; EMA, European Medicines Agency; ERCC1, excision repair cross-complementation group 1; FANC, Fanconi anaemia complementation group; FDA, Food and Drug Administration; FEN1, Flap endonuclease 1; FFPE, formalin fixed paraffin embedded; FGFR2, Fibroblast Growth Factor Receptor 2; 5-FU, 5-fluorouracil; HepPar1, Hepatocyte Paraffin 1; HER2, Human epidermal growth factor receptor 2; HR, homologous recombination; ICC, Intrahepatic cholangiocarcinoma; IDH1/IDH2, Isocitrate Dehydrogenase 1/2; IHC, immunohistochemical; KRAS, Kirsten ras oncogene homolog; LOH, loss of heterozygosity; MSS, Microsatellite Stable; NBN, Nibrin; NGS, next-generation sequencing; NRAS, neuroblastoma; RAS, viral oncogene homolog; RPA1, Replication protein A 70 kDa DNA-binding subunit; NTRK, neurotrophic receptor tyrosine kinase 1; PARP, poly ADP ribose polymerase; PCR, polymerase chain reaction; PR, partial response; PBRM1, Polybromo 1; PCR, polymerase chain reaction; PD, progression of disease; POLB, DNA Polymerase Beta; PRKDC, Protein Kinase; DNA-Activated, Catalytic Subunit; PTA, percutaneous thermal ablation; RAD51-RAD54, radiation-repair genes 51–54; RECIST, Response evaluation criteria in solid tumors; ROS1, ROS proto-oncogene 1receptor tyrosine kinase; STR, Short Tandem Repeat.
References
- 1. Massarweh NN, El-Serag HB. Epidemiology of Hepatocellular Carcinoma and Intrahepatic Cholangiocarcinoma. Cancer Control (2017) 24:1–11. 10.1177/1073274817729245 1073274817729245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bridgewater J, Galle PR, Khan SA, Llovet JM, Park J-W, Patel T, et al. Guidelines for the diagnosis and management of intrahepatic cholangiocarcinoma. J Hepatol (2014) 60:1268–89. 10.1016/j.jhep.2014.01.021 [DOI] [PubMed] [Google Scholar]
- 3. Rahnemai-Azar AA, Weisbrod A, Dillhoff M, Schmidt C, Pawlik TM. Intrahepatic cholangiocarcinoma: Molecular markers for diagnosis and prognosis. Surg Oncol (2017) 26:125–37. 10.1016/j.suronc.2016.12.009 [DOI] [PubMed] [Google Scholar]
- 4. Valle J, Wasan H, Palmer DH, Cunningham D, Anthoney A, Maraveyas A, et al. Cisplatin plus gemcitabine versus gemcitabine for biliary tract cancer. N Engl J Med (2010) 362:1273–81. 10.1056/NEJMoa0908721 [DOI] [PubMed] [Google Scholar]
- 5. Fujimura T, Fujisawa Y, Kambayashi Y, Aiba S. Significance of BRAF Kinase Inhibitors for Melanoma Treatment: From Bench to Bedside. Cancers (Basel) (2019) 11:1342. 10.3390/cancers11091342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Schwartzberg L, Kim ES, Liu D, Schrag D. Precision Oncology: Who, How, What, When, and When Not? Am Soc Clin Oncol Educ Book (2017) 37:160–9. 10.1200/EDBK_174176 [DOI] [PubMed] [Google Scholar]
- 7. Valle JW, Lamarca A, Goyal L, Barriuso J, Zhu AX. New Horizons for Precision Medicine in Biliary Tract Cancers. Cancer Discov (2017) 7:943–62. 10.1158/2159-8290.CD-17-0245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Arai Y, Totoki Y, Hosoda F, Shirota T, Hama N, Nakamura H, et al. Fibroblast growth factor receptor 2 tyrosine kinase fusions define a unique molecular subtype of cholangiocarcinoma. Hepatology (2014) 59:1427–34. 10.1002/hep.26890 [DOI] [PubMed] [Google Scholar]
- 9. Gu T-L, Deng X, Huang F, Tucker M, Crosby K, Rimkunas V, et al. Survey of tyrosine kinase signaling reveals ROS kinase fusions in human cholangiocarcinoma. PloS One (2011) 6:e15640. 10.1371/journal.pone.0015640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Jiao Y, Pawlik TM, Anders RA, Selaru FM, Streppel MM, Lucas DJ, et al. Exome sequencing identifies frequent inactivating mutations in BAP1, ARID1A and PBRM1 in intrahepatic cholangiocarcinomas. Nat Genet (2013) 45:1470–3. 10.1038/ng.2813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Moeini A, Sia D, Bardeesy N, Mazzaferro V, Llovet JM. Molecular Pathogenesis and Targeted Therapies for Intrahepatic Cholangiocarcinoma. Clin Cancer Res (2016) 22:291–300. 10.1158/1078-0432.CCR-14-3296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Saha SK, Parachoniak CA, Ghanta KS, Fitamant J, Ross KN, Najem MS, et al. Mutant IDH inhibits HNF-4α to block hepatocyte differentiation and promote biliary cancer. Nature (2014) 513:110–4. 10.1038/nature13441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yoshikawa D, Ojima H, Iwasaki M, Hiraoka N, Kosuge T, Kasai S, et al. Clinicopathological and prognostic significance of EGFR, VEGF, and HER2 expression in cholangiocarcinoma. Br J Cancer (2008) 98:418–25. 10.1038/sj.bjc.6604129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chan-On W, Nairismägi M-L, Ong CK, Lim WK, Dima S, Pairojkul C, et al. Exome sequencing identifies distinct mutational patterns in liver fluke-related and non-infection-related bile duct cancers. Nat Genet (2013) 45:1474–8. 10.1038/ng.2806 [DOI] [PubMed] [Google Scholar]
- 15. Andrici J, Goeppert B, Sioson L, Clarkson A, Renner M, Stenzinger A, et al. Loss of BAP1 Expression Occurs Frequently in Intrahepatic Cholangiocarcinoma. Med (Baltimore) (2016) 95:e2491. 10.1097/MD.0000000000002491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sia D, Hoshida Y, Villanueva A, Roayaie S, Ferrer J, Tabak B, et al. Integrative molecular analysis of intrahepatic cholangiocarcinoma reveals 2 classes that have different outcomes. Gastroenterology (2013) 144:829–40. 10.1053/j.gastro.2013.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Javle M, Lowery M, Shroff RT, Weiss KH, Springfeld C, Borad MJ, et al. Phase II Study of BGJ398 in Patients With FGFR-Altered Advanced Cholangiocarcinoma. J Clin Oncol (2018) 36:276–82. 10.1200/JCO.2017.75.5009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nakanishi Y, Akiyama N, Tsukaguchi T, Fujii T, Sakata K, Sase H, et al. The fibroblast growth factor receptor genetic status as a potential predictor of the sensitivity to CH5183284/Debio 1347, a novel selective FGFR inhibitor. Mol Cancer Ther (2014) 13:2547–58. 10.1158/1535-7163.MCT-14-0248 [DOI] [PubMed] [Google Scholar]
- 19. Goyal L, Shi L, Liu LY, Fece de la Cruz F, Lennerz JK, Raghavan S, et al. TAS-120 Overcomes Resistance to ATP-Competitive FGFR Inhibitors in Patients with FGFR2 Fusion-Positive Intrahepatic Cholangiocarcinoma. Cancer Discov (2019) 9:1064–79. 10.1158/2159-8290.CD-19-0182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jensen DE, Rauscher FJ. BAP1, a candidate tumor suppressor protein that interacts with BRCA1. Ann N Y Acad Sci (1999) 886:191–4. 10.1111/j.1749-6632.1999.tb09414.x [DOI] [PubMed] [Google Scholar]
- 21. Jensen DE, Proctor M, Marquis ST, Gardner HP, Ha SI, Chodosh LA, et al. BAP1: a novel ubiquitin hydrolase which binds to the BRCA1 RING finger and enhances BRCA1-mediated cell growth suppression. Oncogene (1998) 16:1097–112. 10.1038/sj.onc.1201861 [DOI] [PubMed] [Google Scholar]
- 22. Baughman JM, Rose CM, Kolumam G, Webster JD, Wilkerson EM, Merrill AE, et al. NeuCode Proteomics Reveals Bap1 Regulation of Metabolism. Cell Rep (2016) 16:583–95. 10.1016/j.celrep.2016.05.096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bononi A, Giorgi C, Patergnani S, Larson D, Verbruggen K, Tanji M, et al. BAP1 regulates IP3R3-mediated Ca2+ flux to mitochondria suppressing cell transformation. Nature (2017) 546:549–53. 10.1038/nature22798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Eletr ZM, Wilkinson KD. An emerging model for BAP1’s role in regulating cell cycle progression. Cell Biochem Biophys (2011) 60:3–11. 10.1007/s12013-011-9184-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Machida YJ, Machida Y, Vashisht AA, Wohlschlegel JA, Dutta A. The deubiquitinating enzyme BAP1 regulates cell growth via interaction with HCF-1. J Biol Chem (2009) 284:34179–88. 10.1074/jbc.M109.046755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ruan H-B, Han X, Li M-D, Singh JP, Qian K, Azarhoush S, et al. O-GlcNAc transferase/host cell factor C1 complex regulates gluconeogenesis by modulating PGC-1α stability. Cell Metab (2012) 16:226–37. 10.1016/j.cmet.2012.07.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Xu J, Kadariya Y, Cheung M, Pei J, Talarchek J, Sementino E, et al. Germline Mutation of Bap1 Accelerates Development of Asbestos-Induced Malignant Mesothelioma. Cancer Res (2014) 74:4388–97. 10.1158/0008-5472.CAN-14-1328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yu H, Pak H, Hammond-Martel I, Ghram M, Rodrigue A, Daou S, et al. Tumor suppressor and deubiquitinase BAP1 promotes DNA double-strand break repair. Proc Natl Acad Sci USA (2014) 111:285–90. 10.1073/pnas.1309085110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yoshida K, Miki Y. Role of BRCA1 and BRCA2 as regulators of DNA repair, transcription, and cell cycle in response to DNA damage. Cancer Sci (2004) 95:866–71. 10.1111/j.1349-7006.2004.tb02195.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Di Nunno V, Frega G, Santoni M, Gatto L, Fiorentino M, Montironi R, et al. BAP1 in solid tumors. Future Oncol (2019) 15:2151–62. 10.2217/fon-2018-0915 [DOI] [PubMed] [Google Scholar]
- 31. Carbone M, Yang H, Pass HI, Krausz T, Testa JR, Gaudino G. BAP1 and cancer. Nat Rev Cancer (2013) 13:153–9. 10.1038/nrc3459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cheung M, Talarchek J, Schindeler K, Saraiva E, Penney LS, Ludman M, et al. Further evidence for germline BAP1 mutations predisposing to melanoma and malignant mesothelioma. Cancer Genet (2013) 206:206–10. 10.1016/j.cancergen.2013.05.018 [DOI] [PubMed] [Google Scholar]
- 33. Pilarski R, Rai K, Cebulla C, Abdel-Rahman M. BAP1 Tumor Predisposition Syndrome, in: GeneReviews®. Seattle (WA: University of Washington, Seattle; Available at: http://www.ncbi.nlm.nih.gov/books/NBK390611/ (Accessed July 28, 2020). [Google Scholar]
- 34. Abdel-Rahman MH, Pilarski R, Cebulla CM, Massengill JB, Christopher BN, Boru G, et al. Germline BAP1 mutation predisposes to uveal melanoma, lung adenocarcinoma, meningioma, and other cancers. J Med Genet (2011) 48:856–9. 10.1136/jmedgenet-2011-100156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Carbone M, Flores EG, Emi M, Johnson TA, Tsunoda T, Behner D, et al. Combined Genetic and Genealogic Studies Uncover a Large BAP1 Cancer Syndrome Kindred Tracing Back Nine Generations to a Common Ancestor from the 1700s. PloS Genet (2015) 11:e1005633. 10.1371/journal.pgen.1005633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Murali R, Wiesner T, Scolyer RA. Tumours associated with BAP1 mutations. Pathology (2013) 45:116–26. 10.1097/PAT.0b013e32835d0efb [DOI] [PubMed] [Google Scholar]
- 37. Kobrinski DA, Yang H, Kittaneh M. BAP1: role in carcinogenesis and clinical implications. Transl Lung Cancer Res (2020) 9:S60–6. 10.21037/tlcr.2019.11.24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Carbone M, Ferris LK, Baumann F, Napolitano A, Lum CA, Flores EG, et al. BAP1 cancer syndrome: malignant mesothelioma, uveal and cutaneous melanoma, and MBAITs. J Transl Med (2012) 10:179. 10.1186/1479-5876-10-179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ewens KG, Kanetsky PA, Richards-Yutz J, Purrazzella J, Shields CL, Ganguly T, et al. Chromosome 3 status combined with BAP1 and EIF1AX mutation profiles are associated with metastasis in uveal melanoma. Invest Ophthalmol Vis Sci (2014) 55:5160–7. 10.1167/iovs.14-14550 [DOI] [PubMed] [Google Scholar]
- 40. Ewens KG, Lalonde E, Richards-Yutz J, Shields CL, Ganguly A. Comparison of Germline versus Somatic BAP1 Mutations for Risk of Metastasis in Uveal Melanoma. BMC Cancer (2018) 18:1172. 10.1186/s12885-018-5079-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Masoomian B, Shields JA, Shields CL. Overview of BAP1 cancer predisposition syndrome and the relationship to uveal melanoma. J Curr Ophthalmol (2018) 30:102–9. 10.1016/j.joco.2018.02.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bogenberger JM, DeLeon TT, Arora M, Ahn DH, Borad MJ. Emerging role of precision medicine in biliary tract cancers. NPJ Precis Oncol (2018) 2:21. 10.1038/s41698-018-0064-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Simbolo M, Fassan M, Ruzzenente A, Mafficini A, Wood LD, Corbo V, et al. Multigene mutational profiling of cholangiocarcinomas identifies actionable molecular subgroups. Oncotarget (2014) 5:2839–52. 10.18632/oncotarget.1943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Fujimoto A, Furuta M, Shiraishi Y, Gotoh K, Kawakami Y, Arihiro K, et al. Whole-genome mutational landscape of liver cancers displaying biliary phenotype reveals hepatitis impact and molecular diversity. Nat Commun (2015) 6:6120. 10.1038/ncomms7120 [DOI] [PubMed] [Google Scholar]
- 45. Ross JS, Wang K, Gay L, Al-Rohil R, Rand JV, Jones DM, et al. New routes to targeted therapy of intrahepatic cholangiocarcinomas revealed by next-generation sequencing. Oncologist (2014) 19:235–42. 10.1634/theoncologist.2013-0352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Churi CR, Shroff R, Wang Y, Rashid A, Kang HC, Weatherly J, et al. Mutation Profiling in Cholangiocarcinoma: Prognostic and Therapeutic Implications. PloS One (2014) 9:e115383. 10.1371/journal.pone.0115383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zou S, Li J, Zhou H, Frech C, Jiang X, Chu JSC, et al. Mutational landscape of intrahepatic cholangiocarcinoma. Nat Commun (2014) 5:5696. 10.1038/ncomms6696 [DOI] [PubMed] [Google Scholar]
- 48. Sia D, Losic B, Moeini A, Cabellos L, Hao K, Revill K, et al. Massive parallel sequencing uncovers actionable FGFR2 – PPHLN1 fusion and ARAF mutations in intrahepatic cholangiocarcinoma. Nat Commun (2015) 6:6087. 10.1038/ncomms7087 [DOI] [PubMed] [Google Scholar]
- 49. Borad MJ, Champion MD, Egan JB, Liang WS, Fonseca R, Bryce AH, et al. Integrated genomic characterization reveals novel, therapeutically relevant drug targets in FGFR and EGFR pathways in sporadic intrahepatic cholangiocarcinoma. PloS Genet (2014) 10:e1004135. 10.1371/journal.pgen.1004135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Farshidfar F, Zheng S, Gingras M-C, Newton Y, Shih J, Robertson AG, et al. Integrative Genomic Analysis of Cholangiocarcinoma Identifies Distinct IDH-Mutant Molecular Profiles. Cell Rep (2017) 18:2780–94. 10.1016/j.celrep.2017.02.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ventii KH, Devi NS, Friedrich KL, Chernova TA, Tighiouart M, Van Meir EG, et al. BRCA1-associated protein-1 is a tumor suppressor that requires deubiquitinating activity and nuclear localization. Cancer Res (2008) 68:6953–62. 10.1158/0008-5472.CAN-08-0365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kamel D, Gray C, Walia JS, Kumar V. PARP Inhibitor Drugs in the Treatment of Breast, Ovarian, Prostate and Pancreatic Cancers: An Update of Clinical Trials. Curr Drug Targets (2018) 19:21–37. 10.2174/1389450118666170711151518 [DOI] [PubMed] [Google Scholar]
- 53. Lord CJ, Ashworth A. PARP inhibitors: Synthetic lethality in the clinic. Science (2017) 355:1152–8. 10.1126/science.aam7344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Weaver AN, Yang ES. Beyond DNA Repair: Additional Functions of PARP-1 in Cancer. Front Oncol (2013) 3:290. 10.3389/fonc.2013.00290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Sonnenblick A, de Azambuja E, Azim HA, Piccart M. An update on PARP inhibitors–moving to the adjuvant setting. Nat Rev Clin Oncol (2015) 12:27–41. 10.1038/nrclinonc.2014.163 [DOI] [PubMed] [Google Scholar]
- 56. Mirza MR, Pignata S, Ledermann JA. Latest clinical evidence and further development of PARP inhibitors in ovarian cancer. Ann Oncol (2018) 29:1366–76. 10.1093/annonc/mdy174 [DOI] [PubMed] [Google Scholar]
- 57. Franzese E, Centonze S, Diana A, Carlino F, Guerrera LP, Napoli MD, et al. PARP inhibitors in ovarian cancer. Cancer Treat Rev (2019) 73:1–9. 10.1016/j.ctrv.2018.12.002 [DOI] [PubMed] [Google Scholar]
- 58. Keung MYT, Wu Y, Vadgama JV. PARP Inhibitors as a Therapeutic Agent for Homologous Recombination Deficiency in Breast Cancers. J Clin Med (2019) 8:435. 10.3390/jcm8040435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Mateo J, Lord CJ, Serra V, Tutt A, Balmaña J, Castroviejo-Bermejo M, et al. de Bono JS. A decade of clinical development of PARP inhibitors in perspective. Ann Oncol (2019) 30:1437–47. 10.1093/annonc/mdz192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Pant S, Maitra A, Yap TA. PARP inhibition — opportunities in pancreatic cancer. Nat Rev Clin Oncol (2019) 16:595–6. 10.1038/s41571-019-0257-6 [DOI] [PubMed] [Google Scholar]
- 61. Pilié PG, Gay CM, Byers LA, O’Connor MJ, Yap TA. PARP Inhibitors: Extending Benefit Beyond BRCA-Mutant Cancers. Clin Cancer Res (2019) 25:3759–71. 10.1158/1078-0432.CCR-18-0968 [DOI] [PubMed] [Google Scholar]
- 62. Lord CJ, Ashworth A. BRCAness revisited. Nat Rev Cancer (2016) 16:110–20. 10.1038/nrc.2015.21 [DOI] [PubMed] [Google Scholar]
- 63. Bryant HE, Helleday T. Inhibition of poly (ADP-ribose) polymerase activates ATM which is required for subsequent homologous recombination repair. Nucleic Acids Res (2006) 34:1685–91. 10.1093/nar/gkl108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. McCabe N, Turner NC, Lord CJ, Kluzek K, Białkowska A, Swift S, et al. Deficiency in the Repair of DNA Damage by Homologous Recombination and Sensitivity to Poly(ADP-Ribose) Polymerase Inhibition. Cancer Res (2006) 66:8109–15. 10.1158/0008-5472.CAN-06-0140 [DOI] [PubMed] [Google Scholar]
- 65. Murai J, Huang SN, Das BB, Renaud A, Zhang Y, Doroshow JH, et al. Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors. Cancer Res (2012) 72:5588–99. 10.1158/0008-5472.CAN-12-2753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Postel-Vinay S, Bajrami I, Friboulet L, Elliott R, Fontebasso Y, Dorvault N, et al. A high-throughput screen identifies PARP1/2 inhibitors as a potential therapy for ERCC1-deficient non-small cell lung cancer. Oncogene (2013) 32:5377–87. 10.1038/onc.2013.311 [DOI] [PubMed] [Google Scholar]
- 67. Bajrami I, Frankum JR, Konde A, Miller RE, Rehman FL, Brough R, et al. Genome-wide Profiling of Genetic Synthetic Lethality Identifies CDK12 as a Novel Determinant of PARP1/2 Inhibitor Sensitivity. Cancer Res (2014) 74:287–97. 10.1158/0008-5472.CAN-13-2541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Peña-Llopis S, Vega-Rubín-de-Celis S, Liao A, Leng N, Pavía-Jiménez A, Wang S, et al. BAP1 loss defines a new class of renal cell carcinoma. Nat Genet (2012) 44:751–9. 10.1038/ng.2323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Parrotta R, Okonska A, Ronner M, Weder W, Stahel R, Penengo L. Felley-Bosco E. A Novel BRCA1-Associated Protein-1 Isoform Affects Response of Mesothelioma Cells to Drugs Impairing BRCA1-Mediated DNA Repair. J Thorac Oncol (2017) 12:1309–19. 10.1016/j.jtho.2017.03.023 [DOI] [PubMed] [Google Scholar]
- 70. Rathkey D, Khanal M, Murai J, Zhang J, Sengupta M, Jiang Q, et al. Sensitivity of Mesothelioma Cells to PARP Inhibitors Is Not Dependent on BAP1 but Is Enhanced by Temozolomide in Cells With High-Schlafen 11 and Low-O6-methylguanine-DNA Methyltransferase Expression. J Thorac Oncol (2020) 15:843–59. 10.1016/j.jtho.2020.01.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Parasramka M, Yan IK, Wang X, Nguyen P, Matsuda A, Maji S, et al. BAP1 dependent expression of long non-coding RNA NEAT-1 contributes to sensitivity to gemcitabine in cholangiocarcinoma. Mol Cancer (2017) 16:22. 10.1186/s12943-017-0587-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Mylavarapu S, Das A, Roy M. Role of BRCA Mutations in the Modulation of Response to Platinum Therapy. Front Oncol (2018) 8:16. 10.3389/fonc.2018.00016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Pennington KP, Walsh T, Harrell MI, Lee MK, Pennil CC, Rendi MH, et al. Germline and somatic mutations in homologous recombination genes predict platinum response and survival in ovarian, fallopian tube, and peritoneal carcinomas. Clin Cancer Res (2014) 20:764–75. 10.1158/1078-0432.CCR-13-2287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Wang ZC, Birkbak NJ, Culhane AC, Drapkin R, Fatima A, Tian R, et al. Profiles of Genomic Instability in High-Grade Serous Ovarian Cancer Predict Treatment Outcome. Clin Cancer Res (2012) 18:5806–15. 10.1158/1078-0432.CCR-12-0857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Peters J-M, Nishiyama T. Sister Chromatid Cohesion. Cold Spring Harb Perspect Biol (2012) 4:a011130. 10.1101/cshperspect.a011130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Hill VK, Kim J-S, Waldman T. Cohesin Mutations in Human Cancer. Biochim Biophys Acta (2016) 1866:1–11. 10.1016/j.bbcan.2016.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. AACR Project GENIE Consortium AACR Project GENIE: Powering Precision Medicine through an International Consortium. Cancer Discov (2017) 7:818–31. 10.1158/2159-8290.CD-17-0151 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The original contributions presented in the study are included in the article/ Supplementary Material . Further inquiries can be directed to the corresponding author.