

Cholera is a potentially lethal disease that is endemic in much of the developing world. Vibrio cholerae, the bacterium underlying the disease, infects humans utilizing proteins encoded on horizontally acquired genetic material. Here, we provide evidence that TsrA, a Vibrionaceae-specific protein, plays a critical role in regulating these genetic elements and is essential for V. cholerae virulence in a mouse intestinal model.
KEYWORDS: Vibrio cholerae, gene regulation, H-NS, TsrA, computational biology, genetics, virulence regulation
ABSTRACT
Pathogenic strains of Vibrio cholerae require careful regulation of horizontally acquired virulence factors that are largely located on horizontally acquired genomic islands (HAIs). While TsrA, a Vibrionaceae-specific protein, is known to regulate the critical HAI virulence genes toxT and ctxA, its broader function throughout the genome is unknown. Here, we find that deletion of tsrA results in genomewide expression patterns that heavily correlate with those seen upon deletion of hns, a widely conserved bacterial protein that regulates V. cholerae virulence. This correlation is particularly strong for loci on HAIs, where all differentially expressed loci in the ΔtsrA mutant are also differentially expressed in the Δhns mutant. Correlation between TsrA and H-NS function extends to in vivo virulence phenotypes where deletion of tsrA compensates for the loss of ToxR activity in V. cholerae and promotes wild-type levels of mouse intestinal colonization. All in all, we find that TsrA broadly controls V. cholerae infectivity via repression of key HAI virulence genes and many other targets in the H-NS regulon.
IMPORTANCE Cholera is a potentially lethal disease that is endemic in much of the developing world. Vibrio cholerae, the bacterium underlying the disease, infects humans utilizing proteins encoded on horizontally acquired genetic material. Here, we provide evidence that TsrA, a Vibrionaceae-specific protein, plays a critical role in regulating these genetic elements and is essential for V. cholerae virulence in a mouse intestinal model.
INTRODUCTION
Vibrio cholerae is the causative agent of the potentially lethal disease cholera. Several factors on the progenitor genome and horizontally acquired genetic islands (HAIs) (1–4) act in concert to control V. cholerae virulence gene expression. While multiple HAIs play some role in virulence (2, 5–11), genes on V. cholerae pathogenicity island 1 (VPI-1) and the cholera toxin (CTX) prophage are most involved with the major virulence pathway, the ToxR regulon (4, 12). ToxR and the histone-like nucleoid structuring protein (H-NS) coordinate to activate or repress, respectively, the master virulence regulator ToxT and other virulence genes on VPI-1 (4–7, 13–15). Since H-NS is highly abundant and broadly controls genomic structure and expression patterns (12, 16–26), factors that modulate and fine-tune H-NS repression are common in enteric bacteria (27–32). To date, however, no such factors have emerged in V. cholerae.
TsrA is a Vibrionaceae-specific protein that is by far most common in the genomes of organisms within the Vibrio genus, as determined via BLAST-based (33) protein homology searches (data not shown). TsrA has been shown to regulate type VI secretion system (T6SS) genes (namely, Hcp) in coordination with quorum-sensing pathways and further affects both expression of toxT and the ability of V. cholerae to colonize the small intestine (34). Despite this knowledge, TsrA’s larger impacts on V. cholerae gene regulation have not been explored. Here, we elaborate on previous findings and provide more clarity regarding TsrA’s impact on gene regulation throughout the V. cholerae genome. Our transcriptomics analyses demonstrate that TsrA mimics the ability of H-NS to repress acquired genetic elements on canonical pathogenicity islands. We further show that this gene plays a critical role in controlling intestinal colonization, with deletion of tsrA completely compensating for the attenuation observed when ToxR, an essential virulence gene regulator, is also deleted in an infant mouse intestinal model. Our findings illustrate a large role in controlling V. cholerae virulence for this small protein.
RESULTS
TsrA deletion promotes expression of H-NS regulon.
Previous work showed that TsrA regulates V. cholerae gene expression of ctxA and toxT and that TsrA is structurally similar to the oligomerization domain of H-NS (34). These observations suggested TsrA might have a similar function to H-NS. To investigate this hypothesis, we compared the global transcriptome profiles of the parental C6706 V. cholerae strain to isogenic Δhns and ΔtsrA strains (see Table S1 in the supplemental material). All strains were grown exponentially at 37°C in Luria-Bertani (LB) medium, since H-NS is known to repress virulence-associated genes under this growth condition (21). As an initial control, we verified that any effects observed upon tsrA deletion are not a function of decreased H-NS protein concentration by Western blotting. Using RpoB as a loading control, we saw no difference in H-NS protein levels between an H-NS-V5 strain and a tsrA mutant derivative of said strain, as detected with anti-V5 antibody (Fig. 1A).
FIG 1.

Validation of protein and RNA levels. (A) Western blot showing protein levels of H-NS across conditions. H-NS-V5 was identified using an anti-V5 antibody. An anti-RNA polymerase β subunit (RpoB) immunoblot served as the loading control. The image is representative of three biological replicates per condition. (B) Relative expression levels of tsrA as determined via qPCR. Expression levels normalized to 16S RNA levels under each condition. The change in expression is compared to tsrA expression in C6706 V5-hns strain containing the empty plasmid pWSK30. Bars indicate the standard errors of the mean for three biological replicates per condition. (C) Western blot showing relative protein levels of TsrA compared to H-NS. H-NS-V5 and TsrA-V5 were identified using an anti-V5 antibody. An anti-RNA polymerase β subunit (RpoB) immunoblot served as the loading control. Image is representative of three biological replicates per condition. (D) Relative expression levels of select genes, as determined via qPCR. Expression levels were normalized to the 16S RNA levels under each condition. The change in expression was normalized to relative levels in the C6706 strain. Bars indicate standard errors of the mean for three biological replicates per condition.
Aggregate log fold change data for RNA-seq experiments. Download Table S1, XLSX file, 0.3 MB (328.2KB, xlsx) .
Copyright © 2020 DuPai et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
In line with previous estimates (24), our data show that the H-NS regulon encompasses nearly 600 genes (Fig. 2A; see also Table S1 in the supplemental material). These include, as expected, genes associated with virulence and T6SS (Table 1). Although generally less extreme, RNA expression changes upon deletion of tsrA heavily mirror those observed in the Δhns mutant for a large subset of genes, especially genes on HAIs (Table 1; see also Table S1). When looking at all significantly differentially expressed loci in both strains regardless of fold change, the expression levels of HAI genes were more strongly correlated (adjusted R2 = 0.644) than their progenitor genome counterparts (adjusted R2 = 0.582) (Fig. 2B). With regard to effect size across all genes that significantly changed expression by 2-fold or more in the ΔtsrA strain versus the wild type, 181 loci (roughly 86%) exhibited similar behavior in the Δhns strain (Fig. 2A). These 181 overlapping loci include all 37 HAI genes that are differentially expressed in the ΔtsrA strain.
FIG 2.
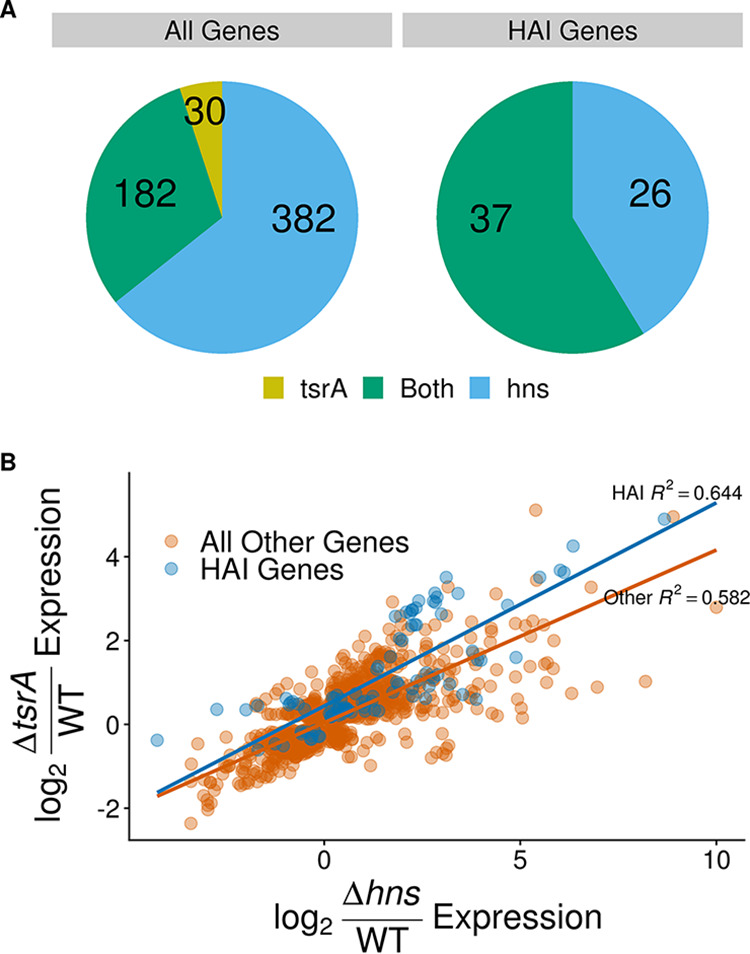
tsrA deletion impacts expression of H-NS regulon genes. (A) Distribution of differentially regulated genes in strains ΔtsrA and Δhns mutant strains. Blue and yellow represent the proportions of genes that are significantly differentially expressed in only the ΔtsrA or only the Δhns strain, respectively, whereas green indicates genes that are significantly differentially expressed in both strains. HAI genes include genes on VPI-1, VPI-2, VSP-1, VSP-2, and the CTX prophage. The included genes have a false discovery rate (FDR) adjusted P value of <0.05 and show a log2-fold increase or decrease in expression compared to the wild type of at least 1 in the indicated strain(s). (B) Summary of genes that are significantly differentially expressed in either of two mutant strains (ΔtsrA and Δhns). Each point represents a distinct gene. HAI genes (blue) include genes on VPI-1, VPI-2, VSP-1, VSP-2, and the CTX prophage, while “All Other Genes” (orange) include all other V. cholerae genes. Included genes have an FDR adjusted P value of <0.05 in both strains.
TABLE 1.
Select differentially expressed genesa
| Locus tag | Gene | ΔtsrA |
Δhns |
Normalized GC | ||
|---|---|---|---|---|---|---|
| L2FC | q value | L2FC | q value | |||
| VC0070 | tsrA | –11.71 | 9.00E–21 | –0.51 | 6.70E–01 | 0.98 |
| VC1130 | hns | –0.16 | 1.00E+00 | –11.48 | 0.00E+00 | 0.91 |
| HAI genes | ||||||
| VC0184 | 1.75 | 1.30E–190 | 3.78 | 0.00E+00 | 0.83 | |
| VC0185 | 1.04 | 4.00E–60 | 2.36 | 0.00E+00 | 0.77 | |
| VC0828 | tcpA | 2.95 | 5.90E–285 | 2.41 | 4.10E–191 | 0.91 |
| VC0829 | tcpB | 2.68 | 0.00E+00 | 2.09 | 2.70E–260 | 0.85 |
| VC0830 | tcpQ | 2.65 | 1.30E–187 | 2.18 | 1.70E–128 | 0.75 |
| VC0835 | tcpT | 3.03 | 9.50E–305 | 2.88 | 2.10E–274 | 0.76 |
| VC0836 | tcpE | 3.13 | 0.00E+00 | 2.82 | 0.00E+00 | 0.75 |
| VC0837 | tcpF | 3.51 | 0.00E+00 | 3.11 | 3.50E–248 | 0.73 |
| VC0838 | toxT | 1.32 | 1.30E–69 | 1.34 | 7.50E–73 | 0.59 |
| VC0841 | acfC | 2.35 | 1.80E–212 | 2.23 | 2.30E–191 | 0.76 |
| VC0844 | acfA | 2.63 | 2.10E–149 | 2.99 | 1.20E–192 | 0.66 |
| VC0845 | acfD | 3.12 | 0.00E+00 | 3.41 | 0.00E+00 | 0.81 |
| VC1456 | ctxB | 2.38 | 2.70E–107 | 2.31 | 7.10E–102 | 0.68 |
| VC1457 | ctxA | 2.37 | 4.10E–133 | 2.35 | 2.40E–131 | 0.81 |
| VC1806 | 1.60 | 2.90E–78 | 4.89 | 0.00E+00 | 0.91 | |
| VC1807 | 4.90 | 3.90E–65 | 8.67 | 4.90E–209 | 0.68 | |
| T6SS genes | ||||||
| VCA0105 | 1.86 | 1.90E–48 | 2.61 | 7.70E–97 | 1.17 | |
| VCA0106 | 1.93 | 3.50E–92 | 2.61 | 2.30E–170 | 0.98 | |
| VCA0107 | vipA | 2.07 | 2.00E–67 | 3.74 | 1.90E–234 | 1.02 |
| VCA0108 | vipB | 1.52 | 2.80E–50 | 3.05 | 1.20E–209 | 1.01 |
| TCA cycle | ||||||
| VC0792 | oadB | –1.70 | 2.60E–39 | –3.08 | 1.30E–119 | 1.10 |
| VC0793 | oadA | –1.38 | 2.90E–41 | –2.90 | 3.60E–169 | 1.10 |
| VC0794 | oadG-2 | –1.32 | 2.30E–28 | –3.06 | 2.90E–114 | 1.01 |
| VC0800 | –1.43 | 2.00E–42 | –2.74 | 5.90E–147 | 1.13 | |
| VC0801 | citG | –1.57 | 9.80E–42 | –2.80 | 1.20E–127 | 1.09 |
| Chitin utilization | ||||||
| VC0616 | 1.64 | 3.20E–29 | 0.50 | 1.10E–03 | 1.03 | |
| VC0617 | 2.10 | 3.40E–55 | 1.04 | 3.30E–14 | 1.02 | |
| VC0618 | 2.92 | 3.80E–62 | 1.74 | 1.40E–22 | 1.08 | |
| VC0619 | 1.86 | 1.20E–24 | 0.88 | 2.40E–06 | 1.01 | |
The indicated genes showed significant differences in expression between one or both mutant strains and a wild-type C6706 Vibrio cholerae strain. ΔtsrA L2FC = log2(ΔtsrA gene abundance/wild-type gene abundance); Δhns L2FC = log2(Δhns gene abundance/wild-type gene abundance); q value = FDR adjusted P value; normalized GC = GC content/average chromosomal GC content.
TsrA demonstrates GC and HAI independent effects on both V. cholerae chromosomes as well (Table 1). Expression of genes associated with T6SS, such as vipAB (35, 36), was increased in both mutants despite exhibiting GC content comparable to background levels. These findings agree with and expand upon previously observed links between TsrA and HCP levels (34). In addition, tricarboxylic acid (TCA) cycle enzyme genes, such as oadB and citG, are downregulated in both knockout strains. Since TCA cycle products are known to repress ToxT expression in V. cholerae (37), transcriptional regulation of these genes by TsrA provides a link between cellular response to environmental cues and regulation of virulence genes. A few metabolism-related genes also appeared to be regulated by TsrA but not H-NS, most notably loci involved in chitin utilization (VC0616-VC0619) (38–40) (Table 1). In sum, TsrA, like H-NS, functions in and regulates key pathways controlling the broader V. cholerae life cycle (19).
TsrA plays a critical role in mouse intestinal colonization.
Deletion of tsrA has been shown to increase V. cholerae colonization in an infant mouse model and affect expression of a few genes dually regulated by H-NS and ToxR (34). We previously showed the importance of ToxR in V. cholerae host colonization could be abrogated by deleting H-NS (12). Given the ability of TsrA to regulate virulence gene expression and its parallel effects with H-NS, we hypothesized that TsrA may likewise play a critical role in host colonization. We used an infant mouse model of intestinal colonization to test this hypothesis. As found previously (34), deletion of tsrA leads to a modest hypercolonization of the infant mouse with the ΔtsrA mutant out-colonizing a wild-type C6706 strain by ∼4-fold (Fig. 3). Remarkably, we show that deletion of tsrA completely negates V. cholerae’s dependence on ToxR to colonize the infant mouse intestine (Fig. 3A). The near wild-type infection levels of the ΔtsrA ΔtoxRS strain are in stark contrast to the drastically reduced infectivity seen in the ΔtoxRS single mutant, providing a clear testament to the potency of TsrA-mediated virulence repression. This phenotype was complemented by exogenous expression of tsrA in the ΔtsrA ΔtoxRS mutant, which showed an extreme colonization defect in line with the ΔtoxRS single mutant (Fig. 3B). These results implicate TsrA as a high level regulator of critical V. cholerae virulence systems.
FIG 3.
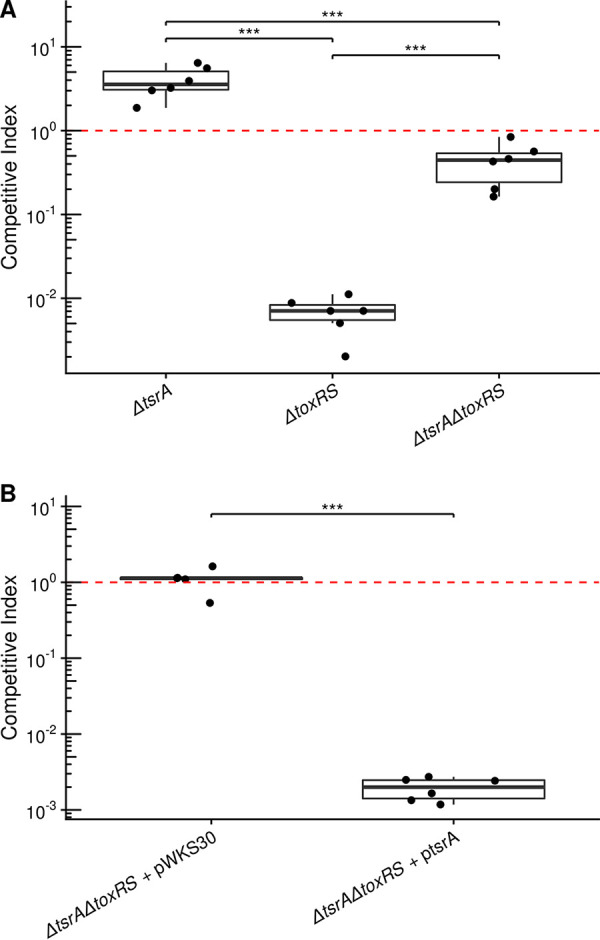
Deletion of tsrA promotes mouse intestinal colonization. Competitive indices of V. cholerae mutants compared to the wild type. (A and B) The indicated strains were competed with wild-type (C6706 with or without pWKS30) V. cholerae in an infant mouse infection model. The fold change of the mutant versus the wild-type strain is shown. The red line indicates perfect competition with the wild type (i.e., a competitive index of 1). Points indicate individual animals, and boxplots indicate the median, first and third quartiles, and range (***, P < 0.001).
DISCUSSION
Our results support previous TsrA research and suggest an expanded role for this protein in fine-tuning expression of the complex virulence cascade of V. cholerae. As a testament to TsrA’s importance, deletion of tsrA wholly overcomes the infant mouse intestinal colonization defects seen in a ΔtoxRS strain. We further show that TsrA stands as a potent coregulator of HAI genes and other portions of the H-NS regulon most responsible for virulence. TsrA’s regulatory activities mirror and supplement those of H-NS.
Since virulent strains of V. cholerae rely on ToxR and its regulon to facilitate intestinal colonization (4, 41), it is little surprise that H-NS and TsrA regulons overlap so heavily at sites, such as VPI-1 and the CTX prophage, that are also controlled by ToxR (21, 23). TsrA-mediated repression at these loci likely explains how a ΔtsrA ΔtoxRS strain maintains wild-type levels of intestinal colonization, a phenotype previously observed in a Δhns ΔtoxRS mutant (12).
Since TsrA lacks a DNA binding domain but has some weak homology to the H-NS oligomerization domain (34), it may act through interactions with H-NS that affect the latter’s ability to influence gene expression, as HHA is known to do in Escherichia coli (28, 42, 43). Unfortunately, we were unable to purify TsrA after multiple attempts to confirm an interaction with H-NS in vitro. It is clear that future genetic and biochemical studies will be needed to fully determine how TsrA functions and influences H-NS activity.
TsrA’s low relative protein abundance compared to H-NS clarifies the smaller effect size of most transcriptomic changes in the ΔtsrA strain compared to more intense changes in the Δhns strain. These data, as well as TsrA’s known role in coordinating T6SS expression in coordination with quorum-sensing pathways, generally support a model of TsrA having a more specialized function than H-NS. In this model, if H-NS is the master regulator of virulence gene expression, then TsrA is a master modulator, fine-tuning expression levels in response to some unknown environmental cues.
Our results suggest that TsrA serves an important role in V. cholerae gene regulation by controlling the expression of key virulence genes and other H-NS targets. Since V. cholerae’s survival in diverse environments depends on precise control of varied genomic elements at specific times, a factor such as TsrA that can modulate and target expression of specific genes helps potentiate V. cholerae’s impressive proclivity to adapt and thrive.
MATERIALS AND METHODS
Bacterial strains, plasmids, and media.
Strains and plasmids used in this study are listed in Table S1 in the supplemental material. Strains were grown in lysogeny broth/agar at 37°C. The antibiotics streptomycin (100 μg/ml) and carbenicillin (75 μg/ml) were used for selection as needed. X-Gal (5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside) was used at 40 μg/ml.
Plasmid and strain construction.
All cloning products were sequence verified. For in-frame deletion constructs, surrounding genomic DNA was amplified by crossover PCR and cloned into pWM91 for subsequent sacB-mediated allelic exchange (44). To add the V5 epitope tag to H-NS, hns was amplified from the genome using primers, including the epitope sequences, to add the appropriate tag to the resulting PCR product. For complementation constructs, the original genes with their native promoters were PCR amplified off chromosomal DNA and cloned into plasmid pWKS30 (45).
Western blot analysis.
Equal amounts of cells grown at 37°C in LB medium were harvested during exponential phase. Cells were pelleted, resuspended in loading buffer, and separated on a NuPAGE Bis-Tris gel (Thermo Fisher). After transfer, membranes were blotted with monoclonal anti-V5 antibody (Sigma-Aldrich) or anti-RpoB antibody (BioLegend). RpoB was blotted as a loading control. Pierce ECL Western blotting substrate (Thermo Scientific) was added before exposing the X-ray film. Experiments were carried out in at least biological triplicates.
RNA sequencing.
RNA sequencing (RNA-seq) was performed essentially as previously described (46). Total RNA was extracted from cells in exponential phase growing at 37°C in LB medium using a Direct-zol RNA MiniPrep kit with TRI-Reagent (Zymo Research). DNase treatment was carried out using a Turbo DNA-free kit (Ambion, Inc.). Ribosomal RNA was depleted using a Ribo-Zero rRNA removal kit for Gram-negative bacteria (Illumina). Sequencing libraries were then prepared for the Illumina sequencing platform. Experiments were repeated in biological triplicates.
Data analysis and visualization.
RNA-seq data were aligned to a transcriptome (47) derived from the El Tor N16961 reference genome (ASM674v1) (48). RNA abundances were quantified using Kallisto version 0.43.1 (49), and differential expression was calculated using DESeq2 version 1.18.1 (50). All other data were analyzed using R version 3.6 (51) with the Tidyverse family of packages (52). All visualizations were generated with ggplot2 version 3.2.1 (53).
Infant mouse colonization assays.
Assays were performed as previously described (12). At least five mice were tested for each mutant. P values were calculated using Tukey’s honest significant difference test following one-way analysis of variance.
Ethics approval.
The mouse experiment was reviewed and approved by the UT Austin IACUC (approval AUP-2018-00354).
Data availability.
Raw sequence reads for the RNA-seq data were uploaded to the Sequence Read Archive (SRA) (https://www.ncbi.nlm.nih.gov/sra) under accession number SRP242320. Processed RNA-seq results are provided in Table S1 in the supplemental material.
Strains and plasmids used in this study. Download Table S2, XLSX file, 0.01 MB (10.3KB, xlsx) .
Copyright © 2020 DuPai et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
ACKNOWLEDGMENTS
This research was supported by National Institutes of Health grants R21AI137546 and R01AI125337 to B.W.D. The funder had no role in the study design, data collection and interpretation, or the decision to submit the work for publication.
C.D.D. analyzed and interpreted all data and prepared all figures with guidance from C.O.W. BWD performed mouse experiments and contributed to all experimental designs. A.L.C. and A.R.C. performed all other experiments, including RNA-seq, qPCR, and Western blot analysis. C.D.D. was a major contributor in writing the manuscript. All authors read and approved the final manuscript.
REFERENCES
- 1.Faruque SM, Mekalanos JJ. 2003. Pathogenicity islands and phages in Vibrio cholerae evolution. Trends Microbiol 11:505–510. doi: 10.1016/j.tim.2003.09.003. [DOI] [PubMed] [Google Scholar]
- 2.Rahman MH, Biswas K, Hossain MA, Sack RB, Mekalanos JJ, Faruque SM. 2008. Distribution of genes for virulence and ecological fitness among diverse Vibrio cholerae population in a cholera endemic area: tracking the evolution of pathogenic strains. DNA Cell Biol 27:347–355. doi: 10.1089/dna.2008.0737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Banerjee R, Das B, Balakrish Nair G, Basak S. 2014. Dynamics in genome evolution of Vibrio cholerae. Infect Genet Evol 23:32–41. doi: 10.1016/j.meegid.2014.01.006. [DOI] [PubMed] [Google Scholar]
- 4.Childers BM, Klose KE. 2007. Regulation of virulence in Vibrio cholerae: the ToxR regulon. Future Microbiol 2:335–344. doi: 10.2217/17460913.2.3.335. [DOI] [PubMed] [Google Scholar]
- 5.Brown RC, Taylor RK. 1995. Organization of tcp, acf, and toxT genes within a ToxT-dependent operon. Mol Microbiol 16:425–439. doi: 10.1111/j.1365-2958.1995.tb02408.x. [DOI] [PubMed] [Google Scholar]
- 6.Häse CC, Mekalanos JJ. 1998. TcpP protein is a positive regulator of virulence gene expression in Vibrio cholerae. Proc Natl Acad Sci U S A 95:730–734. doi: 10.1073/pnas.95.2.730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Higgins DE, Nazareno E, DiRita VJ. 1992. The virulence gene activator ToxT from Vibrio cholerae is a member of the AraC family of transcriptional activators. J Bacteriol 174:6974–6980. doi: 10.1128/jb.174.21.6974-6980.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Waldor MK, Mekalanos JJ. 1996. Lysogenic conversion by a filamentous phage encoding cholera toxin. Science 272:1910–1914. doi: 10.1126/science.272.5270.1910. [DOI] [PubMed] [Google Scholar]
- 9.Dziejman M, Balon E, Boyd D, Fraser CM, Heidelberg JF, Mekalanos JJ. 2002. Comparative genomic analysis of Vibrio cholerae: genes that correlate with cholera endemic and pandemic disease. Proc Natl Acad Sci U S A 99:1556–1561. doi: 10.1073/pnas.042667999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Beaber JW, Hochhut B, Waldor MK. 2002. Genomic and functional analyses of SXT, an integrating antibiotic resistance gene transfer element derived from Vibrio cholerae. J Bacteriol 184:4259–4269. doi: 10.1128/jb.184.15.4259-4269.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hochhut B, Waldor MK. 1999. Site-specific integration of the conjugal Vibrio cholerae SXT element into prfC. Mol Microbiol 32:99–110. doi: 10.1046/j.1365-2958.1999.01330.x. [DOI] [PubMed] [Google Scholar]
- 12.Kazi MI, Conrado AR, Mey AR, Payne SM, Davies BW. 2016. ToxR antagonizes H-NS regulation of horizontally acquired genes to drive host colonization. PLoS Pathog 12:e1005570. doi: 10.1371/journal.ppat.1005570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Miller VL, Taylor RK, Mekalanos JJ. 1987. Cholera toxin transcriptional activator ToxR is a transmembrane DNA binding protein. Cell 48:271–279. doi: 10.1016/0092-8674(87)90430-2. [DOI] [PubMed] [Google Scholar]
- 14.Miller VL, DiRita VJ, Mekalanos JJ. 1989. Identification of toxS, a regulatory gene whose product enhances toxR-mediated activation of the cholera toxin promoter. J Bacteriol 171:1288–1293. doi: 10.1128/jb.171.3.1288-1293.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Weber GG, Klose KE. 2011. The complexity of ToxT-dependent transcription in Vibrio cholerae. Indian J Med Res 133:201–206. [PMC free article] [PubMed] [Google Scholar]
- 16.Grainger DC. 2016. Structure and function of bacterial H-NS protein. Biochem Soc Trans 44:1561–1569. doi: 10.1042/BST20160190. [DOI] [PubMed] [Google Scholar]
- 17.Arold ST, Leonard PG, Parkinson GN, Ladbury JE. 2010. H-NS forms a superhelical protein scaffold for DNA condensation. Proc Natl Acad Sci U S A 107:15728–15732. doi: 10.1073/pnas.1006966107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yu RR, DiRita VJ. 2002. Regulation of gene expression in Vibrio cholerae by ToxT involves both antirepression and RNA polymerase stimulation. Mol Microbiol 43:119–134. doi: 10.1046/j.1365-2958.2002.02721.x. [DOI] [PubMed] [Google Scholar]
- 19.Ayala JC, Silva AJ, Benitez JA. 2017. H-NS: an overarching regulator of the Vibrio cholerae life cycle. Res Microbiol 168:16–25. doi: 10.1016/j.resmic.2016.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stoebel DM, Free A, Dorman CJ. 2008. Anti-silencing: overcoming H-NS-mediated repression of transcription in Gram-negative enteric bacteria. Microbiology (Reading) 154:2533–2545. doi: 10.1099/mic.0.2008/020693-0. [DOI] [PubMed] [Google Scholar]
- 21.Nye MB, Pfau JD, Skorupski K, Taylor RK. 2000. Vibrio cholerae H-NS silences virulence gene expression at multiple steps in the ToxR regulatory cascade. J Bacteriol 182:4295–4303. doi: 10.1128/jb.182.15.4295-4303.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nye MB, Taylor RK. 2003. Vibrio cholerae H-NS domain structure and function with respect to transcriptional repression of ToxR regulon genes reveals differences among H-NS family members. Mol Microbiol 50:427–444. doi: 10.1046/j.1365-2958.2003.03701.x. [DOI] [PubMed] [Google Scholar]
- 23.Stonehouse EA, Hulbert RR, Nye MB, Skorupski K, Taylor RK. 2011. H-NS binding and repression of the ctx promoter in Vibrio cholerae. J Bacteriol 193:979–988. doi: 10.1128/JB.01343-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang H, Ayala JC, Benitez JA, Silva AJ. 2015. RNA-seq analysis identifies new genes regulated by the histone-like nucleoid structuring protein (H-NS) affecting Vibrio cholerae virulence, stress response, and chemotaxis. PLoS One 10:e0118295. doi: 10.1371/journal.pone.0118295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gordon BRG, Li Y, Cote A, Weirauch MT, Ding P, Hughes TR, Navarre WW, Xia B, Liu J. 2011. Structural basis for recognition of AT-rich DNA by unrelated xenogeneic silencing proteins. Proc Natl Acad Sci U S A 108:10690–10695. doi: 10.1073/pnas.1102544108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ayala JC, Wang H, Silva AJ, Benitez JA. 2015. Repression by H-NS of genes required for the biosynthesis of the Vibrio cholerae biofilm matrix is modulated by the second messenger cyclic diguanylic acid. Mol Microbiol 97:630–645. doi: 10.1111/mmi.13058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Madrid C, Balsalobre C, García J, Juárez A. 2007. The novel Hha/YmoA family of nucleoid-associated proteins: use of structural mimicry to modulate the activity of the H-NS family of proteins. Mol Microbiol 63:7–14. doi: 10.1111/j.1365-2958.2006.05497.x. [DOI] [PubMed] [Google Scholar]
- 28.Ali SS, Whitney JC, Stevenson J, Robinson H, Howell PL, Navarre WW. 2013. Structural insights into the regulation of foreign genes in Salmonella by the Hha/H-NS complex. J Biol Chem 288:13356–13369. doi: 10.1074/jbc.M113.455378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nieto JM, Madrid C, Prenafeta A, Miquelay E, Balsalobre C, Carrascal M, Juárez A. 2000. Expression of the hemolysin operon in Escherichia coli is modulated by a nucleoid-protein complex that includes the proteins Hha and H-NS. Mol Gen Genet 263:349–358. doi: 10.1007/s004380051178. [DOI] [PubMed] [Google Scholar]
- 30.Olekhnovich IN, Kadner RJ. 2007. Role of nucleoid-associated proteins Hha and H-NS in expression of Salmonella enterica activators HilD, HilC, and RtsA required for cell invasion. J Bacteriol 189:6882–6890. doi: 10.1128/JB.00905-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Silphaduang U, Mascarenhas M, Karmali M, Coombes BK. 2007. Repression of intracellular virulence factors in Salmonella by the Hha and YdgT nucleoid-associated proteins. J Bacteriol 189:3669–3673. doi: 10.1128/JB.00002-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vivero A, Banos RC, Mariscotti JF, Oliveros JC, Garcia-del Portillo F, Juarez A, Madrid C. 2008. Modulation of horizontally acquired genes by the Hha-YdgT proteins in Salmonella enterica serovar Typhimurium. J Bacteriol 190:1152–1156. doi: 10.1128/JB.01206-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, Madden TL. 2009. BLAST+: architecture and applications. BMC Bioinformatics 10:421. doi: 10.1186/1471-2105-10-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zheng J, Shin OS, Cameron DE, Mekalanos JJ. 2010. Quorum sensing and a global regulator TsrA control expression of type VI secretion and virulence in Vibrio cholerae. Proc Natl Acad Sci U S A 107:21128–21133. doi: 10.1073/pnas.1014998107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Joshi A, Kostiuk B, Rogers A, Teschler J, Pukatzki S, Yildiz FH. 2017. Rules of engagement: the type VI secretion system in Vibrio cholerae. Trends Microbiol 25:267–279. doi: 10.1016/j.tim.2016.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Borgeaud S, Metzger LC, Scrignari T, Blokesch M. 2015. The type VI secretion system of Vibrio cholerae fosters horizontal gene transfer. Science 347:63–67. doi: 10.1126/science.1260064. [DOI] [PubMed] [Google Scholar]
- 37.Minato Y, Fassio SR, Wolfe AJ, Häse CC. 2013. Central metabolism controls transcription of a virulence gene regulator in Vibrio cholerae. Microbiology (Reading) 159:792–802. doi: 10.1099/mic.0.064865-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Meibom KL, Li XB, Nielsen AT, Wu C-Y, Roseman S, Schoolnik GK. 2004. The Vibrio cholerae chitin utilization program. Proc Natl Acad Sci U S A 101:2524–2529. doi: 10.1073/pnas.0308707101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hunt DE, Gevers D, Vahora NM, Polz MF. 2008. Conservation of the chitin utilization pathway in the Vibrionaceae. Appl Environ Microbiol 74:44–51. doi: 10.1128/AEM.01412-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chourashi R, Das S, Dhar D, Okamoto K, Mukhopadhyay AK, Chatterjee NS. 2018. Chitin-induced T6SS in Vibrio cholerae is dependent on ChiS activation. Microbiology (Reading) 164:751–763. doi: 10.1099/mic.0.000656. [DOI] [PubMed] [Google Scholar]
- 41.Krukonis ES, Yu RR, DiRita VJ. 2000. The Vibrio cholerae ToxR/TcpP/ToxT virulence cascade: distinct roles for two membrane-localized transcriptional activators on a single promoter. Mol Microbiol 38:67–84. doi: 10.1046/j.1365-2958.2000.02111.x. [DOI] [PubMed] [Google Scholar]
- 42.Aznar S, Paytubi S, Juarez A. 2013. The Hha protein facilitates incorporation of horizontally acquired DNA in enteric bacteria. Microbiology (Reading) 159:545–554. doi: 10.1099/mic.0.062448-0. [DOI] [PubMed] [Google Scholar]
- 43.Madrid C, Garcia J, Pons M, Juarez A. 2007. Molecular evolution of the H-NS protein: interaction with Hha-like proteins is restricted to Enterobacteriaceae. J Bacteriol 189:265–268. doi: 10.1128/JB.01124-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Metcalf WW, Jiang W, Daniels LL, Kim S-K, Haldimann A, Wanner BL. 1996. Conditionally replicative and conjugative plasmids carrying lacZα for cloning, mutagenesis, and allele replacement in bacteria. Plasmid 35:1–13. doi: 10.1006/plas.1996.0001. [DOI] [PubMed] [Google Scholar]
- 45.Wang RF, Kushner SR. 1991. Construction of versatile low-copy-number vectors for cloning, sequencing and gene expression in Escherichia coli. Gene 100:195–199. doi: 10.1016/0378-1119(91)90366-J. [DOI] [PubMed] [Google Scholar]
- 46.Davies BW, Bogard RW, Young TS, Mekalanos JJ. 2012. Coordinated regulation of accessory genetic elements produces cyclic di-nucleotides for Vibrio cholerae virulence. Cell 149:358–370. doi: 10.1016/j.cell.2012.01.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Krin E, Pierlé SA, Sismeiro O, Jagla B, Dillies M-A, Varet H, Irazoki O, Campoy S, Rouy Z, Cruveiller S, Médigue C, Coppée J-Y, Mazel D. 2018. Expansion of the SOS regulon of Vibrio cholerae through extensive transcriptome analysis and experimental validation. BMC Genomics 19:373. doi: 10.1186/s12864-018-4716-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Heidelberg JF, Eisen JA, Nelson WC, Clayton RA, Gwinn ML, Dodson RJ, Haft DH, Hickey EK, Peterson JD, Umayam L, Gill SR, Nelson KE, Read TD, Tettelin H, Richardson D, Ermolaeva MD, Vamathevan J, Bass S, Qin H, Dragoi I, Sellers P, McDonald L, Utterback T, Fleishmann RD, Nierman WC, White O, Salzberg SL, Smith HO, Colwell RR, Mekalanos JJ, Venter JC, Fraser CM. 2000. DNA sequence of both chromosomes of the cholera pathogen Vibrio cholerae. Nature 406:477–483. doi: 10.1038/35020000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bray NL, Pimentel H, Melsted P, Pachter L. 2016. Near-optimal probabilistic RNA-seq quantification. Nat Biotechnol 34:525–527. doi: 10.1038/nbt.3519. [DOI] [PubMed] [Google Scholar]
- 50.Love MI, Huber W, Anders S. 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.R Core Team. 2013. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria [Google Scholar]
- 52.Wickham H, Averick M, Bryan J, Chang W, McGowan L, François R, Grolemund G, Hayes A, Henry L, Hester J, Kuhn M, Pedersen T, Miller E, Bache S, Müller K, Ooms J, Robinson D, Seidel D, Spinu V, Takahashi K, Vaughan D, Wilke C, Woo K, Yutani H. 2019. Welcome to the Tidyverse. JOSS 4:1686. doi: 10.21105/joss.01686. [DOI] [Google Scholar]
- 53.Wilkinson L. 2011. ggplot2: elegant graphics for data analysis by WICKHAM, H. Biometrics 67:678–679. doi: 10.1111/j.1541-0420.2011.01616.x. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Aggregate log fold change data for RNA-seq experiments. Download Table S1, XLSX file, 0.3 MB (328.2KB, xlsx) .
Copyright © 2020 DuPai et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Strains and plasmids used in this study. Download Table S2, XLSX file, 0.01 MB (10.3KB, xlsx) .
Copyright © 2020 DuPai et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Data Availability Statement
Raw sequence reads for the RNA-seq data were uploaded to the Sequence Read Archive (SRA) (https://www.ncbi.nlm.nih.gov/sra) under accession number SRP242320. Processed RNA-seq results are provided in Table S1 in the supplemental material.
Strains and plasmids used in this study. Download Table S2, XLSX file, 0.01 MB (10.3KB, xlsx) .
Copyright © 2020 DuPai et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.