

Abstract
Objective:
Adults with Down syndrome (DS) develop Alzheimer disease (AD) pathology by their 5th decade. Compared with the general population, traditional vascular risks in adults with DS are rare, allowing examination of cerebrovascular disease in this population and insight into its role in AD without the confound of vascular risk factors. We examined in vivo magnetic resonance imaging (MRI)-based biomarkers of cerebrovascular pathology in adults with DS, and determined their cross-sectional relationship with age, beta-amyloid pathology, and mild cognitive impairment or clinical AD diagnostic status.
Methods:
Participants from the Biomarkers of Alzheimer’s Disease in Down Syndrome study (n = 138, 50 ± 7 years, 39% women) with MRI data and a subset (n = 90) with amyloid positron emission tomography (PET) were included. We derived MRI-based biomarkers of cerebrovascular pathology, including white matter hyperintensities (WMH), infarcts, cerebral microbleeds, and enlarged perivascular spaces (PVS), as well as PET-based biomarkers of amyloid burden. Participants were characterized as cognitively stable (CS), mild cognitive impairment–DS (MCI-DS), possible AD dementia, or definite AD dementia based on in-depth assessments of cognition, function, and health status.
Results:
There were detectable WMH, enlarged PVS, infarcts, and microbleeds as early as the 5th decade of life. There was a monotonic increase in WMH volume, enlarged PVS, and presence of infarcts across diagnostic groups (CS < MCI-DS < possible AD dementia < definite AD dementia). Higher amyloid burden was associated with a higher likelihood of an infarct.
Interpretation:
The findings highlight the prevalence of cerebrovascular disease in adults with DS and add to a growing body of evidence that implicates cerebrovascular disease as a core feature of AD and not simply a comorbidity.
With advances in medical insights and improvements in the quality of care, the life expectancy in Down syndrome (DS) has increased dramatically over the past 30 years.1 By the time individuals with DS are in their 5th decade, virtually all have sufficient cerebral beta-amyloid and tau pathology to meet pathological criteria for Alzheimer disease (AD) and most will have developed clinical dementia by their 60s.2–9 However, there is variability in the age of clinical onset of dementia as well as variability in symptoms and course of decline once the disease manifests.3,10–13 As a result, the impact of AD, for which no disease-modifying treatment is currently available, is an increasing public health threat to this population. An understanding of AD-related biological changes and their modifiers in adults with DS is needed to develop viable treatment and preventative strategies. Characterizing AD-related biomarker profiles with neuroimaging in DS has the additional potential to provide insight into late onset AD, an even more significant public health concern for the general population.
Although it is widely acknowledged that cerebrovascular pathology contributes to the clinical symptoms of AD, there is little agreement about its role in disease pathogenesis. Previous work established that neuroimaging markers of cerebrovascular disease, such as white matter hyperintensities (WMH), are associated with increased risk for and progression of AD14–16 and are elevated in autosomal dominant AD.17 Vascular risk factors are associated with a higher risk of clinical AD and with AD biomarkers in later life.18,19 Unlike the general population, however, individuals with DS have low prevalence of traditional vascular risk factors, such as hypertension, type 2 diabetes, and atherosclerosis.20–24 Assessment of cerebrovascular changes in DS has been restricted primarily to the analysis of cerebral amyloid angiopathy (CAA),25–28 attributed to the overproduction of amyloid. However, more comprehensive consideration of cerebrovascular changes among individuals with DS provides the unique opportunity to both characterize cerebrovascular biomarkers in vivo and study cerebrovascular disease in AD without the confound of vascular risk factors or advanced systemic arterial disease. An observed low prevalence of cerebrovascular disease in adults with DS would be consistent with their low vascular risk profiles, and would suggest that previously observed cerebrovascular abnormalities in late onset AD might reflect comorbidity. On the other hand, observing high rates of cerebrovascular disease among individuals with DS with an AD diagnosis, given their low vascular risk profiles, would provide evidence of a more central role of cerebrovascular disease in AD pathogenesis.
The current study examined structural magnetic resonance imaging (MRI) markers of cerebrovascular disease in DS, including WMH, cerebral microbleeds, infarcts, and enlarged perivascular spaces (PVS). The study also examined the emergence of cerebrovascular lesions in the context of classical AD neuropathology as measured by amyloid positron emission tomography (PET). Our overall hypothesis was that, despite the low prevalence of traditional vascular risk factors and arterial disease in DS, MRI-derived markers of cerebrovascular disease would be common, increase in an age-dependent manner, and be associated with clinical AD diagnosis, reflecting a key role in disease pathogenesis.
Patients and Methods
Participants
Participants in the current study were enrolled in the Biomarkers of Alzheimer’s Disease in Adults with Down Syndrome (ADDS; U01 AG051412) study, designed to characterize the development of AD among adults with DS clinically, genetically, and with cerebrospinal fluid, blood, and neuroimaging biomarkers. To date, the ADDS study has enrolled >200 individuals >40 years old at Columbia University/New York State Institute for Basic Research in Developmental Disabilities, Massachusetts General Hospital/Harvard Medical School (MGH), and University of California, Irvine (UC-Irvine). Participants with completed MRI scanning with sequences necessary for quantification of cerebrovascular changes (see below) and a clinical evaluation with AD diagnostic consensus (N = 138) were included in the analyses. The study was approved by the institutional review boards at participating institutions, and written informed consent was obtained from participants and/or their legal guardian or legally authorized representative. Furthermore, we received assent from every participant before every procedure.
Clinical Assessment
Participants underwent neuropsychological testing to assess cognition in domains typically affected by AD, supplemented by reviews of clinical charts and interviews with knowledgeable informants as described previously.29 The evaluations also included a standardized clinical and neurological examination. A consensus panel, including clinicians expert in the assessment and diagnosis of dementia in individuals with DS, adjudicated diagnoses following standardized procedures, which considered health history, functional and vocational abilities, performance in direct assessment, and neuropsychiatric symptoms. One of 4 AD-related diagnoses was assigned to each participant. A diagnosis of cognitively stable (CS) indicated no evidence of clinically significant cognitive decline, with performance consistent with preclinical intellectual functioning and age. A diagnosis of mild cognitive impairment–DS (MCI-DS) indicated evidence of cognitive decline over time beyond what would be expected with age but of insufficient severity to suggest dementia. A diagnosis of possible AD dementia indicated evidence of substantial decline of breadth and severity greater than indicative of MCI-DS. A diagnosis of definite AD dementia indicated clear evidence of substantial cognitive and functional decline with a high degree of confidence in the dementia rating. For any participant who was rated as having MCI-DS or possible or definite dementia, findings were reviewed to establish an etiological differential diagnosis. These were AD, AD in combination with possible other pathology, or dementia due to uncertain causes. Participants were excluded if the consensus committee determined there to be a complication or substantial concern unrelated to a neurodegenerative disorder (eg, severe sensory loss, new psychiatric diagnosis). In addition to diagnostic information related to cognitive impairment status, participants received full neurological examinations and were assessed for histories of medical illnesses. All classifications of clinical status were determined without reference to neuroimaging or other AD biomarkers.
Magnetic Resonance Imaging
Participants were scanned on a Siemens (Erlangen, Germany) Prisma 3T (Columbia University, n = 29; MGH, n = 53) or Philips (Best, the Netherlands) Achieva 3T (UC-Irvine, n = 56) MRI system. Following protocols put forth by the Alzheimer’s Disease Neuroimaging Initiative (ADNI), each participant received a high-resolution T1-weighted anatomical scan (repetition time [TR]/echo time [TE]/inversion time [TI] = 2,300/2.96/900 milliseconds, voxel size = 1 × 1 × 1mm3), a T2-weighted fluid-attenuated inversion recovery (FLAIR) scan (TR/TE/TI = 5,000/386/1,800 milliseconds, voxel size = 0.4 × 0.4 × 0.9mm3), and a T2*-weighted gradient echo (GRE) scan (TR/TE = 650/20 milliseconds, voxel size = 0.8 × 0.8 × 4mm3) or susceptibility-weighted image (SWI; TR/TE = 27/20 milliseconds, voxel size = 0.9 × 0.9 × 1.5mm3). These images were used to quantitate major classes of small- and large-vessel cerebrovascular abnormalities, including WMH, presence of infarct, severity of enlarged PVS, and presence of microbleeds using the following procedures. Examples of these cerebrovascular biomarkers are displayed in Figure 1.
FIGURE 1:
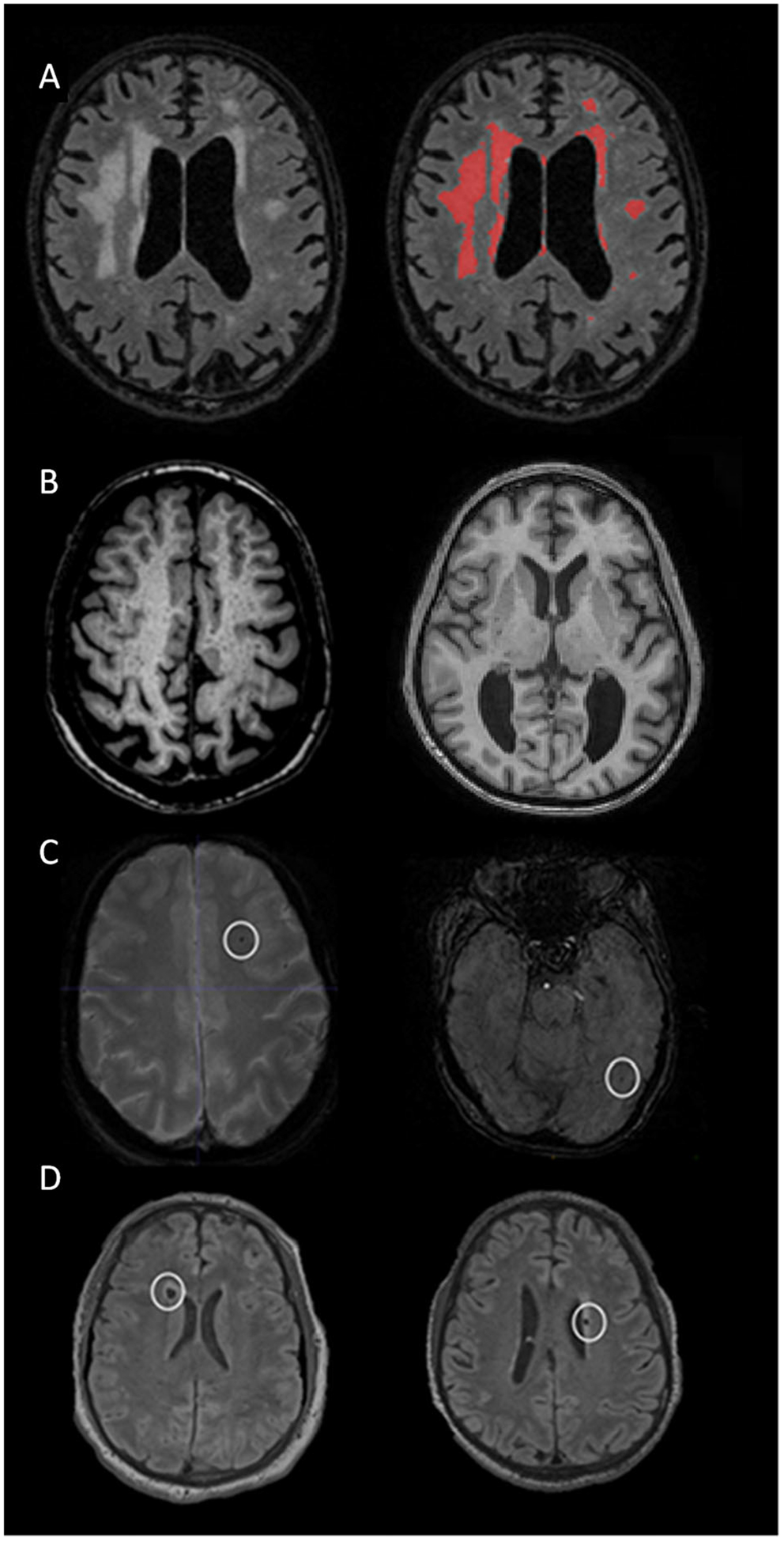
Examples of the cerebrovascular markers considered in the current study from typical study participants. (A) Distributed white matter hyperintensities displayed on T2-weighted fluid-attenuated inversion recovery (FLAIR) scan (left) labeled with in-house–developed software (right). (B) Widespread enlarged perivascular spaces throughout the white matter appreciated on T1-weighted scans in 2 participants displayed in axial (left) and coronal (right) orientations. (C) Lobar microbleeds in 2 study participants displayed on axial susceptibility-weighted images. (D) Cerebral infarcts in 2 participants displayed on axial T2-weighted FLAIR scans.
White Matter Hyperintensities
White matter hyperintensity volume was derived from T2-weighted FLAIR images. Briefly, each image was brain extracted and intensity normalized. Brain-extracted voxel intensities were high-pass filtered at the distribution mode and log-transformed for normality. A half Gaussian mixture model (eg, lower half Gaussian, higher full Gaussian) was fit to the log-transformed intensity histogram, and the voxels in the higher Gaussian distribution were labeled as hyperintense (≥5 contiguous voxels). The primary outcome was global WMH volume, but because previous work demonstrated that spatially posterior distributions of WMH (ie, parietal and occipital lobes) were related to amnestic MCI and AD,17,30 we further investigated lobar WMH volumes across diagnostic groups.
Infarct
Infarcts were assessed on FLAIR and T1-weighted images. Infarcts were rated on FLAIR images as discrete hypointense lesions >5mm with a partial or complete hyperintense ring. Lesions were confirmed on T1-weighted images as areas of hypointensity. The primary outcome was the presence of at least 1 infarct.
Microbleeds
Microbleeds were rated by visual inspection,31–33 using criteria that included a hypointense round or ovoid lesion on GRE (n = 43) or SWI (n = 51), surrounded at least halfway by parenchyma, and accompanied by a “blooming” effect. Microbleeds are not hyperintense on accompanying T1-weighted or FLAIR scans, distinguishing them from iron or calcium deposits, bone, or vessel flow voids. Because most participants had 0 or 1 detectable microbleed, the primary outcome measure was Boolean, reflecting the presence of at least 1 microbleed.
Perivascular Spaces
Enlarged PVS were visually read on T1-weighted scans as hypointensities in 13 regions and scored 0 to 2 based on characteristics on T2-weighted scans (eg, hyperintense ring), resulting in an overall score ranging from 0, indicating no enlarged PVS in any of the 13 regions, to 26, indicating the most severe enlarged PVS score in each of the 13 regions.34 This method has suitable intrarater (intraclass correlation coefficient [ICC] = 0.90) and inter-rater reliability (ICC = 0.73).34, 35
Amyloid PET
A subgroup of participants (n = 90) underwent amyloid PET with [18F]florbetapir (AV45) at each site. Individuals at Columbia University were scanned on a Siemens Biograph 64 (voxel size = 1 × 1 × 2mm3, reconstruction = OSEM3D + TOF, n = 10), those at MGH were scanned on a Siemens Biograph mMR (voxel size = 2.1 × 2.1 × 2.0mm3, reconstruction = OP-OSEM, n = 31), and those at UC-Irvine were scanned on a Siemens high-resolution research tomograph (voxel size = 1.2 × 1.2 × 1.2mm3, reconstruction = OP-OSEM3D, n = 49), with each site/scanner following the ADNI protocol (4 × 5 minute frames, 50–70 minutes postinjection).36 PET data were attenuation, radioactive decay, detector normalization, randoms, and scatter corrected. PET data were realigned and averaged, then T1-weighted MRI was coregistered to averaged PET data. Standard uptake value ratio (SUVR) was calculated using a cerebellar cortex reference region. Regions of interest (ROIs) were defined by FreeSurfer v6.037 segmentations, visually inspected for accuracy, and SUVR was averaged in a weighted composite region, encompassing anterior cingulate, frontal cortex, parietal cortex, precuneus, striatum, and temporal cortex. SUVR in the striatum alone was further investigated across diagnostic groups, because previous work showed that it was the ROI with the earliest amyloid deposition in DS.4,5 Analyses were done without and with partial volume correction (PETSurfer38).
Apolipoprotein Genotyping
In our imaging sample of 138 participants, a subset (n = 113) have had their DNA samples genotyped for 2 APOE single nucleotide polymorphisms (SNPs; rs429358 and rs7412) with the Kompetitive allele-specific polymerase chain reaction genotyping system by LGC Genomics (Berlin, Germany). Genotype data for these 2 SNPs were used to define APOE-ε2, −ε3, and -ε4 alleles. For these analyses, individuals with at least 1 copy of the APOE-ε4 allele were classified as APOE-ε4 carriers, which has been demonstrated to be associated with a 4- to 5-fold increased risk for AD in adults with DS.39 Also, as the presence of an APOE-ε2 allele has been shown to be protective against AD in adults with DS,40 individuals with APOE-ε2/ε4 (n = 2) were excluded from all APOE analyses.
Statistical Analysis
Each biomarker had a slightly different sample size due to not completing the scan or poor scan quality due to motion or truncated acquisition (WMH, n = 124; enlarged PVS, n = 130; infarcts, n = 127; microbleeds, n = 94). Whether data were missing was unrelated to age, APOE-ε4, or diagnostic group for WMH volume, enlarged PVS, or infarct status. However, microbleed reads were more likely to be missing for older adults. Associations between demographic data, including age, sex, and APOE-ε4, and neuroimaging markers of cerebrovascular disease were tested with linear or logistic regressions, t tests, or chi-squared tests. For analyses involving age, a linear model fit better than a quadratic model. Age analyses were rerun in only CS participants to separate the effects of age and diagnosis.
General linear models were used to test a monotonic increase of WMH volume and PVS scores across diagnostic groups (CS, MCI-DS, possible AD dementia, definite AD dementia), whereas logistic regression models were used to test whether the presence of infarcts or microbleeds differed across groups. We examined the relationship among cerebrovascular and amyloid markers with a similar set of general linear and logistic regression models. Analyses involving regional WMH associations were corrected using the false discovery rate.41
We performed 2 separate post hoc analyses. First, to explore the robustness of our findings by diagnostic group, we combined possible and definite AD dementia into a single group. Second, to explore the extent to which vascular marker differences across diagnostic groups and demographic data are attributable to CAA, which we operationally defined as the presence of at least 1 microbleed,26 we reran analyses involving WMH, PVS, and infarct after adjusting for presence of cerebral microbleed. It should be noted that adjusting for presence of microbleeds reduced the sample size of other vascular biomarkers to the number of available microbleed reads. All statistical tests were performed in SPSS Statistics v26 (IBM, Armonk, NY), adjusted for site/scanner in this multisite study.
Results
Demographic characteristics across diagnostic groups for participants with MRI are presented in Table 1. Compared with CS participants, those with cognitive decline (MCI-DS, possible or definite AD dementia) were older, had a greater proportion of APOE-ε4 carriers, and had a similar sex distribution. The proportion of participants with various vascular risk factors across diagnostic groups was different for type 1 diabetes and systolic blood pressure, but not for traditional vascular risk factors such as hypertension, type 2 diabetes, and high cholesterol (see Table 1). Neither type 1 diabetes nor systolic blood pressure was associated with vascular or amyloid biomarkers, and these were not included as covariates in subsequent analyses. Acetylcholinesterase inhibitor usage was greater in those with cognitive decline (MCI-DS, possible or definite AD dementia), as expected, but statin usage was similar across diagnostic groups (see Table 1). Demographic characteristics across diagnostic groups for the subset of participants with PET (n = 90) were similar to those with MRI (n = 138) in terms of increasing age and similar proportion of men and women, but the frequency of APOE-ε4 carriers was not different by diagnostic group (CS, 21%; MCI-DS, 44%; possible AD dementia, 0%; definite AD dementia, 50%; χ2[3] = 7.1, p = 0.07).
TABLE 1.
Demographic Features across the Diagnostic Groups for the Complete Subgroup That Completed Magnetic Resonance Imaging
| Feature | Total | Cognitively Stable | MCI-DS | Possible AD Dementia | Definite AD Dementia | Test Statistic |
|---|---|---|---|---|---|---|
| n | 138 | 91 | 28 | 7 | 12 | |
| Demographic | ||||||
| Age, yr | 50 ± 7 | 49 ± 7 | 53 ± 6 | 55 ± 8 | 57 ± 7 | F(3) = 8.4, p = 4E-5a |
| Women, n (%) | 54 (39%) | 39 (43%) | 6 (21%) | 4 (57%) | 5 (42%) | χ2(3) = 5.2, p = 0.16 |
| APOE4 carrier, n (%) | 32 (28%) | 16 (22%) | 9 (41%) | 0 (0%) | 7 (64%) | χ2(3) = 12.5, p = 0.006a |
| Vascular risk | ||||||
| Hypertension, n (%) | 10 (7%) | 6 (7%) | 2 (7%) | 0 (0%) | 2 (17%) | χ2(3) = 2.2, p = 0.54 |
| Hypotension, n (%) | 18 (13%) | 12 (13%) | 5 (18%) | 1 (14%) | 0 (0%) | χ2(3) = 2.4, p = 0.50 |
| Blood pressure, systolic/diastolic, mmHg | 110 ± 14/66 ± 10 | 110 ± 13/67 ± 10 | 105 ± 14/63 ± 10 | 106 ± 16/63 ± 8 | 120 ± 15/66 ± 10 | F(3) = 3.2, p = 0.02a/F(3) = 1.2, p = 0.30 |
| Type 1 diabetes, n (%) | 2 (2%) | 0 (0%) | 2 (8%) | 0 (0%) | 0 (0%) | χ2(3) = 8.0, p = 0.05a |
| Type 2 diabetes, n (%) | 8 (6%) | 6 (7%) | 2 (8%) | 0 (0%) | 0 (0%) | χ2(3) = 1.4, p = 0.70 |
| High cholesterol, n (%) | 57 (42%) | 38 (42%) | 14 (50%) | 1 (14%) | 4 (33%) | χ2(3) = 3.3, p = 0.35 |
| Congenital heart disease, n (%) | 24 (26%) | 16 (28%) | 5 (22%) | 2 (67%) | 1 (13%) | χ2(3) = 3.6, p = 0.30 |
| Hyperthyroidism, n (%) | 4 (3%) | 3 (3%) | 0 (0%) | 1 (14%) | 0 (0%) | χ2(3) = 4.5, p = 0.22 |
| Hypothyroidism, n (%) | 85 (62%) | 58 (64%) | 19 (68%) | 2 (29%) | 6 (50%) | χ2(3) = 4.7, p = 0.20 |
| Syncope, n (%) | 24 (26%) | 16 (28%) | 6 (26%) | 0 (0%) | 2 (25%) | χ2(3) = 1.1, p = 0.77 |
| Sleep apnea, n (%) | 44 (39%) | 24 (33%) | 14 (54%) | 2 (33%) | 4 (44%) | χ2(3) = 3.8, p = 0.29 |
| Medication | ||||||
| Statins, n (%) | 44 (32%) | 28 (31%) | 13 (46%) | 0 (0%) | 3 (25%) | χ2(3) = 6.3, p = 0.10 |
| Acetylcholine esterase inhibitors, n (%) | 20 (14%) | 5 (5%) | 7 (25%) | 3 (43%) | 5 (42%) | χ2(3) = 20, p = 2E-4a |
Significant at p < 0.05.
AD = Alzheimer disease; MCI-DS = mild cognitive impairment–Down syndrome.
Descriptively, global WMH volume, enlarged PVS, infarcts, and microbleeds were present even in CS individuals (Fig 2) as young as 40, 40, 41, and 42 years old, respectively. Infarct frequency increased most from MCI-DS to possible or definite AD dementia, whereas the presence of microbleeds increased most from CS to MCI-DS (see Fig 2). However, we observed no microbleeds in those with possible AD dementia, but observed microbleeds in 43% of those with definite AD dementia. The proportion of individuals with detectable microbleeds did not differ by T2*-weighted sequence used for microbleed assessment (SWI, 14%; GRE, 16%; χ2[2] = 0.16, p = 0.93). Of the 15 individuals with microbleeds, 13 had 1 detectable microbleed, and 2 had 2. In terms of regional distribution, all microbleeds were seen in strictly lobar regions, apart from 2, which were detected in cerebellum. Overall findings related to microbleeds did not change after removal of individuals with cerebellar microbleeds or those with >1 microbleed. Assessment of global amyloid levels indicated most individuals had measurable pathophysiology, as expected for their age range (>40 years4,6). Note that the change in global amyloid SUVR due to partial volume correction increased across diagnostic groups (CS < MCI-DS, possible AD dementia, definite AD dementia; p = 0.002), suggesting that neurodegeneration begins in the MCI-DS stage along the AD continuum.
FIGURE 2:
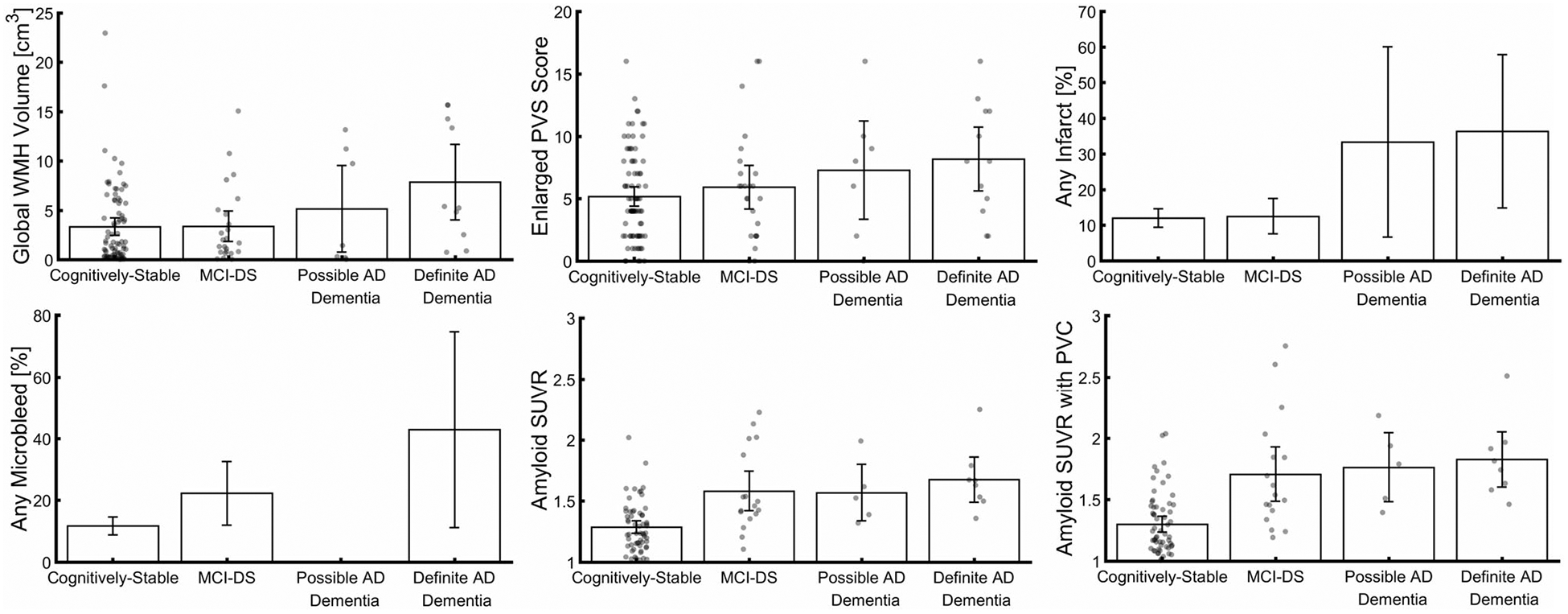
Cerebrovascular markers and global amyloid standard uptake value ratio (SUVR) across diagnostic groups. Severity and presence of cerebrovascular markers and amyloid SUVR increased across diagnostic groups. Scatter points represent individual values, bars represent mean values, and errors bars represent 95% confidence intervals. AD = Alzheimer disease; MCI-DS = mild cognitive impairment–Down syndrome; PVC = partial volume correction; PVS = perivascular space; WMH = white matter hyperintensity.
There were age-associated increases in global WMH volume (driven by frontal, parietal, and occipital lobes), enlarged PVS score, and infarcts, but not microbleeds (Tables 2 and 3). There were age-associated increases of amyloid globally (see Table 2) and in the striatum after partial volume correction (see Table 3). Restricting the analysis to the CS group, higher age was not associated with global WMH volume, enlarged PVS score, infarcts, microbleeds, or global amyloid (see Table 2). Regionally, within the CS group, higher age was associated with higher WMH volume in the frontal and occipital lobes, but not with amyloid in the striatum, regardless of partial volume correction (see Table 3). There were no sex or APOE-ε4 differences in global WMH volume, enlarged PVS score, infarcts, microbleeds, or global amyloid (see Table 2). Regionally, there was greater WMH volume in the occipital lobe in men compared to women and in the temporal lobe in APOE-ε4 carriers compared to noncarriers, but no sex or APOE-ε4 differences in striatal amyloid (see Table 3). After multiple comparisons correction, higher age was associated with higher WMH volume in the frontal, parietal, and occipital lobes in all participants, and men had greater WMH volume in occipital lobe compared to women.
TABLE 2.
Associations of Cerebrovascular and Amyloid Biomarkers with Demographic Features and Diagnostic Groups
| Feature/Group | Global White Matter Hyperintensity Volume | Enlarged Perivascular Spaces | Infarcts | Microbleeds | Global Amyloid SUVR | Global Amyloid SUVR with Partial Volume Correction |
|---|---|---|---|---|---|---|
| Age | 0.14 [0.04, 0.24] p = 0.004a |
0.17 [0.07, 0.27] p = 0.001a |
1.1 [1.0, 1.2] p = 0.02a |
1.1 [1.0, 1.2] p = 0.09 |
0.01 [0.002, 0.02] p = 0.001a |
0.02 [0.01, 0.03] p = 2E-4a |
| Age (cognitively stable only) | 0.13 [0.01, 0.25] p = 0.04a |
0.03 [−0.11, 0.17] p = 0.67 |
1.1 [0.44, 2.6] p = 0.89 |
1.1 [0.95, 1.3] p = 0.16 |
0.002 [−0.006, 0.1] p = 0.70 |
0.007 [−0.003, 0.02] p = 0.20 |
| Sex (F > M) | −1.2 [−2.6, 0.19] p = 0.11 |
−0.80 [−2.3, 0.65] p = 0.28 |
1.2 [0.45, 3.4] p = 0.69 |
1.0 [0.30, 3.4] p = 0.98 |
−0.03 [−0.15, 0.09] p = 0.59 |
−0.06 [−0.22, 0.10] p = 0.50 |
| APOE-ε4 (carrier > noncarrier) | 1.2 [−0.47, 2.9] p = 0.15 |
0.70 [−1.0, 2.4] p = 0.43 |
1.0 [0.32, 3.2] p = 0.99 |
3.3 [0.91, 12.1] p = 0.08 |
0.06 [−0.08, 0.20] p = 0.42 |
0.04 [−0.14, 0.22] p = 0.64 |
| Diagnosis | 2.8 [1.0, 4.6] p = 0.003a |
2.2 [0.42, 4.0] p = 0.02a |
1.6 [1.0, 2.5] p = 0.05a |
1.4 [0.84, 2.5] p = 0.19 |
0.26 [0.12, 0.40] p = 3E-4a |
0.36 [0.18, 0.54] p = 9E-5a |
| Diagnosis, adjusted for age | 2.3 [0.46, 4.1] p = 0.02a |
1.4 [−0.48, 3.3] p = 0.14 |
1.3 [0.82, 2.2] p = 0.25 |
1.3 [0.69, 2.3] p = 0.46 |
0.24 [0.10, 0.38] p = 0.001a |
0.33 [0.15, 0.51] p = 5E-4a |
Each row represents a separate model. Parameters represent unstandardized, mean differences, or odds ratios and their 95% confidence intervals.
Significant at p < 0.05.
F = female; M = male; SUVR = standard uptake value ratio.
TABLE 3.
Regional Associations of White Matter Hyperintensity and Amyloid Biomarkers with Demographic Features and Diagnostic Groups
| Feature/Group | Frontal White Matter Hyperintensity Volume | Temporal White Matter Hyperintensity Volume | Parietal White Matter Hyperintensity Volume | Occipital White Matter Hyperintensity Volume | Striatal Amyloid SUVR | Striatal Amyloid SUVR with Partial Volume Correction |
|---|---|---|---|---|---|---|
| Age | 0.07 [0.03, 0.11] p = 0.006a |
0.01 [−6E-3, 0.03] p = 0.43 |
0.02 [4E-4, 0.04] p = 0.02a |
0.05 [0.03, 0.07] p = 0.001a |
0.01 [2E-3, 0.02] p = 0.06 |
0.01 [2E-4, 0.02] p = 0.008a |
| Age (cognitively stable only) | 0.07 [0.01, 0.13] p = 0.02a |
0.01 [−6E-3, 0.03] p = 0.43 |
0.02 [4E-4, 0.04] p = 0.14 |
0.04 [1E-3, 0.08] p = 0.04a |
−1E-3 [−0.02, 0.02] p = 0.85 |
2E-3 [−0.01, 0.01] p = 0.66 |
| Sex (F > M) | −0.31 [−1.0, 0.38] p = 0.39 |
−0.13 [−0.37, 0.11] p = 0.30 |
−0.12 [−0.41, 0.17] p = 0.41 |
−0.54 [−0.95, −0.13] p = 0.01a |
−0.06 [−0.18, 0.06] p = 0.28 |
−0.09 [−0.23, 0.05] p = 0.22 |
| APOE-ε4 (carrier > noncarrier) | 0.32 [−0.52, 1.2] p = 0.46 |
0.34 [0.07, 0.61] p = 0.02a |
0.30 [−0.05, 0.65] p = 0.09 |
0.16 [−0.33, 0.65] p = 0.54 |
0.09 [−0.05, 0.23] p = 0.17 |
0.13 [−0.03, 0.29] p = 0.09 |
| Diagnosis | 1.1 [0.24, 2.0] p = 0.01a |
0.39 [0.08, 0.70] p = 0.01a |
0.82 [0.47, 1.2] p = 2E-5a |
0.44 [−0.11, 0.99] p = 0.11 |
0.20 [0.06, 0.34] p = 0.005a |
0.23 [0.07, 0.39] p = 0.004a |
| Diagnosis, adjusted for age | 0.90 [−2E-3, 1.8] p = 0.06 |
0.40 [0.08, 0.72] p = 0.01a |
0.76 [0.39, 1.1] p = 9E-5a |
0.25 [−0.48, 0.98] p = 0.37 |
0.19 [0.05, 0.33] p = 0.007a |
0.22 [0.06, 0.38] p = 0.008a |
Each row represents a separate model. Parameters represent unstandardized, mean differences, or odds ratios and their 95% confidence intervals.
Significant at p < 0.05.
F = female; M = male; SUVR = standard uptake value ratio.
Global WMH volume increased monotonically across diagnostic groups (CS < MCI-DS < possible AD dementia < definite AD dementia), even after age adjustment (see Table 2). This effect was attributable primarily to WMH in the parietal and temporal lobes and, to a lesser extent, in the frontal lobe (see Table 3). After multiple comparisons correction, there was a monotonic increase across diagnostic groups (CS < MCI-DS < possible AD dementia < definite AD dementia) in WMH volume in the temporal and parietal lobes; the increase in parietal lobe survived age adjustment. Enlarged PVS scores and infarcts also increased across diagnostic groups, but not with age adjustment (see Table 2). Finally, microbleeds did not increase across diagnostic groups, regardless of age adjustment (see Table 2). As expected from the pathological definition of AD, global amyloid increased across diagnostic groups, even after age adjustment (see Table 2), and in the striatum alone (see Table 3). The diagnosis-related increase in amyloid was more apparent with partial volume correction (see Tables 2 and 3).
Greater enlarged PVS score was associated with greater global WMH volume, but this association did not survive age adjustment (Table 4). The presence of infarcts was associated with greater global WMH volume and greater enlarged PVS score, even after age adjustment (see Table 4). Greater global amyloid was associated with the presence of infarcts; this association was not significant with age adjustment (see Table 4).
TABLE 4.
Associations between Cerebrovascular and Amyloid Biomarkers
| Global White Matter Hyperintensity Volume | Enlarged Perivascular Spaces | Inforcts | Microbleeds | |
|---|---|---|---|---|
| Unadjusted | ||||
| Enlarged perivascular spaces | 0.25 [0.05, 0.45] p = 0.01a |
— | — | — |
| Infarcts | 2.9 [0.98, 4.8] p = 0.004a |
3.2 [1.2, 5.2] p = 0.002a |
— | — |
| Microbleeds | 1.4 [−0.56, 3.4] p = 0.18 |
0.75 [−1.6, 3.1] p = 0.53 |
1.4 [0.34, 6.1] p = 0.63 |
— |
| Global amyloid SUVR | 0.10 [−0.35, 0.55] p = 0.67 |
1.6 [−1.6, 4.9] p = 0.35 |
5.5 [0.87, 34.5] p = 0.08 |
3.7 [0.52, 26.0] p = 0.20 |
| Global amyloid SUVR with partial volume correction | 0.07 [−0.28, 0.42] p = 0.69 |
1.6 [−0.75, 4.0] p = 0.19 |
4.1 [0.99, 16.6] p = 0.05a |
2.4 [0.54, 10.7] p = 0.25 |
| Age-adjusted | ||||
| Enlarged perivascular spaces | 0.19 [−0.006, 0.39] p = 0.06 |
— | — | — |
| Infarcts | 2.4 [0.52, 4.3] p = 0.02a |
2.5 [0.56, 4.4] p = 0.01a |
— | — |
| Microbleeds | 0.84 [−1.1, 2.8] p = 0.40 |
−0.37 [−2.5, 1.8] p = 0.74 |
0.99 [0.22, 4.5] p = 0.99 |
— |
| Global amyloid SUVR | −0.11 [−0.58, 0.36] p = 0.65 |
−0.42 [−3.6, 2.7] p = 0.80 |
2.3 [0.32, 16.3] p = 0.42 |
3.0 [0.29, 31.6] p = 0.36 |
| Global amyloid SUVR with partial volume correction | −0.11 [−0.48, 0.26] p = 0.55 |
0.02 [−2.3, 2.4] p = 0.99 |
2.2 [0.40, 12.5] p = 0.32 |
2.0 [0.36, 10.9] p = 0.43 |
Each row represents a separate model. Parameters represent unstandardized, mean differences, or odds ratios and their 95% confidence intervals.
Significant at p < 0.05.
SUVR = standard uptake value ratio.
In post hoc analyses, combining possible and definite AD dementia (Supplemental Table S1), there was a monotonic increase across diagnostic groups in global WMH, WMH in all lobes, enlarged PVS score, global amyloid (with and without partial volume correction), and striatal amyloid (with and without partial volume correction). Additionally, adjusting for age, there was a monotonic increase across diagnostic groups in global WMH (and in temporal and parietal lobes), global amyloid (with and without partial volume correction), and striatal amyloid (with and without partial volume correction). Adjusting for microbleeds (Supplemental Table S2), there was a monotonic increase across diagnostic groups in global WMH (and in temporal and parietal lobes), global amyloid (with and without partial volume correction), and striatal amyloid (with and without partial volume correction). Additionally adjusting for age did not change results.
Discussion
There is much debate about the primary role of cerebrovascular changes in AD pathogenesis and progression. Because the vast majority of individuals with DS develop clinical AD but have a low prevalence of vascular risk factors, the examination of MRI markers of cerebrovascular disease in this population provides insight into AD-related cerebrovascular disease progression. Very few participants had traditional vascular risk factors, including hypertension and type 2 diabetes, but image-based biomarkers of cerebrovascular disease were prominent. Our study uniquely reports the frequency of 4 image-based cerebrovascular biomarkers across the AD continuum in older adults with DS. As with the risk for amyloidosis and AD symptomatology in DS,3 cerebrovascular markers, particularly WMH volume, increased with age; yet what was most striking was the increase across clinical AD diagnostic categories in a “dose-dependent,” monotonic manner across diagnostic groups (CS < MCI-DS < possible AD dementia < definite AD dementia). Although cerebrovascular markers were prevalent and associated with diagnosis, they were not strongly associated with amyloid pathology, particularly after adjustment for their shared association with age. We conclude that cerebrovascular disease may be a fundamental feature of AD in adults with DS that tracks with clinical progression of the disease.
Our conclusion is supported by studies in autosomal dominant AD, which like AD in adults with DS can be considered a highly penetrant genetic form of AD. We previously examined autosomal dominant AD, which included younger adults with generally low vascular risk, and found that MRI markers of cerebrovascular disease were prevalent and elevated ≤20 years before expected symptom onset.17 In late onset AD, increased image-based markers of cerebrovascular disease are well documented42 and place asymptomatic individuals at increased risk for clinical AD.43 Although it is now apparent that most individuals with clinical late onset AD have mixed brain pathology,44 the increased cerebrovascular disease is typically cited as comorbidity that contributes additively to clinical symptoms, not a component of disease pathogenesis or fundamental to disease progression. However, our findings support an alternative hypothesis that—at least in subgroups of adults—cerebrovascular changes are core features of the development of AD and not a separate comorbid pathology.
The increase in WMH with age is well documented in the general population (95–96% of those aged >60 years45,46 and 100% of those aged >65 years47 in the general population). In these adults with DS, 100% had detectable WMH (range = 0.01–23.0cm3). When considering regional distribution of WMH, we found that the monotonic increase across diagnostic groups, above and beyond age, was most prominent in the parietal lobe, consistent with previous work in autosomal dominant AD and in late onset AD.16,43 When combining possible and definite AD dementia groups, this diagnosis-associated increase was observed more posterior, in the occipital lobe, suggesting that this effect was not observed initially because occipital WMH may develop early and may not continue to increase with increasing AD severity; this additional finding is consistent with occipital WMH developing earlier compared to parietal WMH in autosomal dominant AD.17 The prevailing model regards WMH formation as the result of ischemic damage in small vessels secondary to chronic hypoperfusion.48 It is unclear why posterior WMH are most specifically associated with AD risk, but it is interesting to note that AD-associated markers of metabolism, blood flow, and neurodegeneration converge in posterior brain regions.49 Cerebral amyloid angiopathy also has a propensity for posterior brain distribution,50 but after statistical adjustment for presence of cerebral microbleeds, a biomarker for CAA, our observations of WMH increases across diagnostic groups remained, suggesting that posterior increases in WMH are not simply a reflection of CAA. It is possible that chronic hypoperfusion in posterior regions, particularly in the watershed zones that comprise the parietal lobes, induces ischemic damage and promotes tau-mediated neurodegeneration.51 Some have argued that the posterior distribution of WMH in AD is secondary to tau-related pathology in a Wallerian-like pattern of axonal degeneration and not to ischemic injury.52 This pathway may also contribute to overall WMH burden, but the overwhelming evidence implicates ischemia in WMH pathogenesis.48 Furthermore, WMH predict accumulating tau pathology and not vice versa53; white matter fibers that directly connect cortical regions (so-called “U-fibers”), which would ostensibly be affected by proximal cortical tau pathology and neurodegeneration, are generally spared from WMH in aging and AD,54 and WMH formation typically precedes neurodegenerative changes related to AD and predicts future progression.43
Large-vessel ischemic damage may manifest as cavitated infarcts often attributable to arterial occlusion secondary to local arterial disease or embolism. We observed infarcts in CS participants ≥40 years old and that those with infarcts had greater WMH volumes and greater enlarged PVS scores, similar to associations in the general population.55,56 The increase in infarcts across diagnostic groups was not independent of age. Cardiovascular defects can potentially cause infarcts in individuals with DS, but there were no differences in congenital heart disease across the diagnostic groups.
There is interest in the role of PVS in the clearance of amyloid and other toxins in the aging brain.57 The increase of enlarged PVS score with age in the young, CS participants mirrored that in the older, general population (mean age = 68 years56). Qualitatively, the increase of enlarged PVS we observed in these participants with DS is similar to what we have seen in older adult (mean age = 71 years) community-based cohorts without DS.34 The increase with diagnosis, which is observed in the general population,56,58 was not reliable in adults with DS after age adjustment, and we did not observe a relationship with amyloid burden. Given that individuals with DS overproduce amyloid throughout their lifespan, enlarged PVS may be due to longstanding chronic exposure to amyloid that may not be related linearly to amyloid pathology at any given point in time.
Cerebral microbleeds are hemorrhagic lesions that are tightly associated with soluble forms of beta-amyloid pathology and CAA in the context of aging and late onset AD.25,59 We found convergent results with previous imaging studies of CAA in symptomatic individuals with DS (43%; 38.5% in Carmona-Iragui et al25), such that microbleeds increased across diagnosis similar to that in autosomal dominant AD (25–66%)60 and in late onset AD (20–30%).25,61 We sought to determine the extent to which these vascular findings in adults with DS across the AD continuum may simply be driven by CAA. The effect sizes and confidence intervals without and with adjustment for microbleeds were not largely different. We cannot preclude the possibility that the observed findings were mediated primarily by CAA, as a consequence of lifelong amyloid precursor protein overproduction. Previous work, for example, showed that in patients with CAA there were enlarged PVS in the centrum semiovale62 and lobar infarcts,63 whereas in patients with hypertension, there were enlarged PVS in the basal ganglia64 and deep infarcts.63 We used our ratings in subcortical white matter, which are less susceptible to blood hemodynamics (ie, hypertension) compared to those in basal ganglia,34 to capture PVS enlargement in the centrum semiovale and to parallel previous work.62 We did not observe spatial distributions of cerebrovascular disease biomarkers that would solely implicate CAA. Descriptively, in older adults with DS, enlarged PVS were observed both in the centrum semiovale (5.5 ± 4.0; 34% of the maximum severity rating of 16) and basal ganglia (2.9 ± 1.9; 36% of the maximum severity rating of 8) and infarcts were observed in the cortical lobes (15/132 = 11.4%) and deep, subcortical structures (6/132 = 4.5%). Potential sampling issues (ie, the possible AD dementia group was the smallest, none were APOE-ε4 carriers, and none had any microbleeds) and/or the use of 2 sequences with differing sensitivities and slice thicknesses for microbleed detection may have masked an increase with age or diagnosis. We explored the possibility of direct associations among vascular risk factors and cerebrovascular biomarkers and found no associations. In the relative absence of traditional vascular risks in individuals with DS, it will be critical to determine what factors are mediating the increased burden of WMH, enlarged PVS, and infarcts. It is possible that neuroinflammatory processes, which are elevated in DS,65 and/or genetic factors coded on chromosome 2166–68 or even beyond chromosome 21 (eg, gene interactions), may initiate or mediate arterial disease, independent of both amyloid pathology and vascular risk exposures, but in manner that is fundamental to AD. In this conceptualization, the severity and impact of cerebrovascular disease might be amplified in the presence of vascular risk factors, as is common among older adults in the general population, but vascular risk factors would not be necessary for there to be a cerebrovascular component of AD pathogenesis.
Men and APOE-ε4 carriers had greater regional WMH volume, suggesting a potential mechanism through which men69 and APOE-ε4 carriers39 with DS have an earlier onset of AD, but there was a lack of sex or APOE-ε4 effect on other cerebrovascular or amyloid biomarkers. This analysis did not consider important causes of sex-related differences compared to previous studies in AD in adults with DS that consider menopause stage and hormonal balance.69–72 Effect sizes suggest that APOE-ε4 carriers are 3.3 times more likely to have a microbleed, but not more likely to have an infarct. An autopsy study demonstrated that APOE-ε4 carriers had higher cerebral amyloid angiopathy but were not different from noncarriers in cortical microinfarcts.73 The directionality of the APOE-ε4 effect on global amyloid aligns with previous research that indicates a 4- to 5-fold increase in risk for AD in adults with DS,39,69,74 but could be masked (eg, no APOE-ε4 carriers in the possible AD dementia group).
In studies of AD in adults with DS, age is a large confound for AD diagnosis and for amyloid accumulation, which begins at birth and continues throughout life because of the triplication of chromosome 21. Here we observe image-based cerebrovascular biomarkers in adults with DS in their 40s. The age effect in adults with DS requires careful consideration, and can be best disentangled through large-scale longitudinal studies such as the ongoing study from which these data were collected. We attempted to investigate age-related increases that are distinct from AD diagnosis-related differences by restricting analyses to CS individuals only, but the relatively small sample and restricted age range may have had an impact on statistical testing. Age effects should be assessed in larger studies that include individuals with DS across the adult lifespan. We speculate that there is an inflection point, perhaps around age 40 years, where cerebrovascular disease begins to emerge in adults with DS. Nonetheless, it is important to note that we observed reliable differences in markers of cerebrovascular disease across diagnostic groups that were not simply attributable to age differences between the groups. Future studies should continue to investigate the role of cerebrovascular disease in AD in adults with DS, potentially as a core feature of AD and not necessarily as simply a comorbidity.
This work establishes the presence of MRI cerebrovascular disease markers in adults with DS and their relationship to AD-related clinical states. It will be critical to follow these observations with an in-depth examination of the potential mechanistic mediators in the relationship between cerebrovascular disease and AD clinical diagnoses in this population, the interaction of these cerebrovascular markers with other AD-related biomarkers, and the emergence of cerebrovascular changes across the entire adult lifespan in both cross-sectional and longitudinal contexts. Most experimental therapeutic approaches for late onset AD have focused on the precipitating role of amyloid pathology, but these approaches have generally been unsuccessful in clinical trials.75 Consideration of cerebrovascular pathways that are potentially involved in pathogenesis and symptom expression point to important new therapeutic avenues for AD prevention and treatment in adults with DS and in the general population.
Supplementary Material
Acknowledgment
This publication was supported by the NIH National Center for Advancing Translational Sciences (UL1TR001873). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. This work was also supported by the NIH National Institute on Aging (NIA; 1U01AG051412 and K99AG065506). Samples from the National Centralized Repository for Alzheimer Disease and Related Dementias, which receives government support under a cooperative agreement grant (U24 AG21886) awarded by the NIA, were used in this study.
We thank contributors who collected samples used in this study; patients and their families, whose help and participation made this work possible; and the ADDS staff as well as MRI and PET technologists, who also made this work possible.
Footnotes
Potential Conflicts of Interest
Nothing to report.
Data Availability
Data from the ADDS study are available upon request from the Image Data Archive at the Laboratory of Neuro Imaging (LONI).
References
- 1.Presson AP, Partyka G, Jensen KM, et al. Current estimate of Down syndrome population prevalence in the United States. J Pediatr 2013;163:1163–1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Head E, Lott IT. Down syndrome and beta-amyloid deposition. Curr Opin Neurol 2004;17:95–100. [DOI] [PubMed] [Google Scholar]
- 3.Lott IT, Head E. Dementia in Down syndrome: unique insights for Alzheimer disease research. Nat Rev Neurol 2019;15:135–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lao PJ, Betthauser TJ, Hillmer AT, et al. The effects of normal aging on amyloid-β deposition in nondemented adults with Down syndrome as imaged by carbon 11–labeled Pittsburgh compound B. Alzheimers Dement 2016;12:380–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lao PJ, Handen BL, Betthauser TJ, et al. Longitudinal changes in amyloid positron emission tomography and volumetric magnetic resonance imaging in the nondemented Down syndrome population. Alzheimers Dement (Amst) 2017;9:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Annus T, Wilson LR, Hong YT, et al. The pattern of amyloid accumulation in the brains of adults with Down syndrome. Alzheimers Dement 2016;12:538–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sabbagh MN, Fleisher A, Chen K, et al. Positron emission tomography and neuropathologic estimates of fibrillar amyloid-β in a patient with Down syndrome and Alzheimer disease. Arch Neurol 2011;68:1461–1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Neale N, Padilla C, Fonseca LM, et al. Neuroimaging and other modalities to assess Alzheimer’s disease in Down syndrome. Neuroimage Clin 2018;17:263–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tatebe H, Kasai T, Ohmichi T, et al. Quantification of plasma phosphorylated tau to use as a biomarker for brain Alzheimer pathology: pilot case-control studies including patients with Alzheimer’s disease and Down syndrome. Mol Neurodegener 2017;12:63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wiseman FK, Al-Janabi T, Hardy J, et al. A genetic cause of Alzheimer disease: mechanistic insights from Down syndrome. Nat Rev Neurosci 2015;16:564–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Karmiloff-Smith A, Al-Janabi T, D’Souza H, et al. The importance of understanding individual differences in Down syndrome. F1000Res 2016;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lautarescu BA, Holland AJ, Zaman SH. The early presentation of dementia in people with Down syndrome: a systematic review of longitudinal studies. Neuropsychol Rev 2017;27:31–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lai F, Williams RS. A prospective study of Alzheimer disease in Down syndrome. Arch Neurol 1989;46:849–853. [DOI] [PubMed] [Google Scholar]
- 14.Li J-Q, Tan L, Wang H-F, et al. Risk factors for predicting progression from mild cognitive impairment to Alzheimer’s disease: a systematic review and meta-analysis of cohort studies. J Neurol Neurosurg Psychiatry 2016;87:476–484. [DOI] [PubMed] [Google Scholar]
- 15.Brickman AM. Contemplating Alzheimer’s disease and the contribution of white matter hyperintensities. Curr Neurol Neurosci Rep 2013;13:415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yoshita M, Fletcher E, Harvey D, et al. Extent and distribution of white matter hyperintensities in normal aging, MCI, and AD. Neurology 2006;67:2192–2198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee S, Viqar F, Zimmerman ME, et al. White matter hyperintensities are a core feature of Alzheimer’s disease: evidence from the dominantly inherited Alzheimer network. Ann Neurol 2016;79:929–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gottesman RF, Schneider AL, Zhou Y, et al. Association between midlife vascular risk factors and estimated brain amyloid deposition. JAMA 2017;317:1443–1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Walker KA, Sharrett AR, Wu A, et al. Association of midlife to late-life blood pressure patterns with incident dementia. JAMA 2019;322:535–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kerins G, Petrovic K, Bruder M, Gruman C. Medical conditions and medication use in adults with Down syndrome: a descriptive analysis. Downs Syndr Res Pract 2008;12:141–147. [DOI] [PubMed] [Google Scholar]
- 21.Haveman M, Maaskant MA, Sturmans F. Older Dutch residents of institutions, with and without Down syndrome: comparisons of mortality and morbidity trends and motor/social functioning. Aust N Z J Dev Disabil 1989;15:241–255. [Google Scholar]
- 22.Murdoch JC, Rodger JC, Rao SS, et al. Down’s syndrome: an atheroma-free model? Br Med J 1977;2:226–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pucci F, Machado G, Solera E, et al. Blood pressure levels and body mass index in Brazilian adults with Down syndrome. Sao Paulo Med J 2016;134:330–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kapell D, Nightingale B, Rodriguez A, et al. Prevalence of chronic medical conditions in adults with mental retardation: comparison with the general population. Ment Retard 1998;36:269–279. [DOI] [PubMed] [Google Scholar]
- 25.Carmona-Iragui M, Balasa M, Benejam B, et al. Cerebral amyloid angiopathy in Down syndrome and sporadic and autosomal-dominant Alzheimer’s disease. Alzheimers Dement 2017;13:1251–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Head E, Phelan MJ, Doran E, et al. Cerebrovascular pathology in Down syndrome and Alzheimer disease. Acta Neuropathol Commun 2017;5:93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Head E, Powell DK, Schmitt FA. Metabolic and vascular imaging biomarkers in Down syndrome provide unique insights into brain aging and Alzheimer disease pathogenesis. Front Aging Neurosci 2018;10:191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Helman AM, Siever M, McCarty KL, et al. Microbleeds and cerebral amyloid angiopathy in the brains of people with Down syndrome with Alzheimer’s disease. J Alzheimers Dis 2019;67:103–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Krinsky-McHale SJ, Zigman WB, Lee JH, et al. Promising outcome measures of early Alzheimer’s dementia in adults with Down syndrome. Alzheimers Dement (Amst) 2020;12:e12044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brickman AM, Provenzano FA, Richards C, et al. Parietal lobe white matter hyperintensity volume, not hippocampal atrophy, predicts incident Alzheimer’s disease in a community-based cohort. Arch Neurol 2012;69:1621–1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Greenberg SM, Vernooij MW, Cordonnier C, et al. Cerebral microbleeds: a guide to detection and interpretation. Lancet Neurol 2009;8:165–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wiegman AF, Meier IB, Schupf N, et al. Cerebral microbleeds in a multiethnic elderly community: demographic and clinical correlates. J Neurol Sci 2014;345:125–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Meier IB, Gu Y, Guzaman VA, et al. Lobar microbleeds are associated with a decline in executive functioning in older adults. Cerebrovasc Dis 2014;38:377–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gutierrez J, Elkind MSV, Dong C, et al. Brain perivascular spaces as biomarkers of vascular risk: results from the Northern Manhattan Study. Am J Neuroradiol 2017;38:862–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gutierrez J, Rundek T, Ekind MS, et al. Perivascular spaces are associated with atherosclerosis: an insight from the Northern Manhattan Study. AJNR Am J Neuroradiol 2013;34:1711–1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Landau SM, Breault C, Joshi AD, et al. Amyloid-beta imaging with Pittsburgh compound B and florbetapir: comparing radiotracers and quantification methods. J Nucl Med 2013;54:70–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Desikan RS, Ségonne F, Fischl B, et al. An automated labeling system for subdividing the human cerebral cortex on MRI scans into gyral based regions of interest. Neuroimage 2006;31:968–980. [DOI] [PubMed] [Google Scholar]
- 38.Greve DN, Salat DH, Bowen SL, et al. Different partial volume correction methods lead to different conclusions: an 18F-FDG-PET study of aging. Neuroimage 2016;132:334–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schupf N, Lee JH, Kapell D, et al. Onset of dementia is associated with apolipoprotein E ε4 in Down’s syndrome. Ann Neurol 1996;40:799–801. [DOI] [PubMed] [Google Scholar]
- 40.Royston M, Mann D, Pickering-Brown S, et al. ApoE2 allele, Down’s syndrome, and dementia. Ann N Y Acad Sci 1996;777:255–259. [DOI] [PubMed] [Google Scholar]
- 41.Benjamini Y, Drai D, Elmer G, et al. Controlling the false discovery rate in behavior genetics research. Behav Brain Res 2001;125:279–284. [DOI] [PubMed] [Google Scholar]
- 42.Santos CY, Snyder PJ, Wu W-C, et al. Pathophysiologic relationship between Alzheimer’s disease, cerebrovascular disease, and cardiovascular risk: a review and synthesis. Alzheimers Dement (Amst) 2017;7:69–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brickman AM, Provenzano FA, Muraskin J, et al. Regional white matter hyperintensity volume, not hippocampal atrophy, predicts incident Alzheimer disease in the community. Arch Neurol 2012;69:1621–1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kapasi A, DeCarli C, Schneider JA. Impact of multiple pathologies on the threshold for clinically overt dementia. Acta Neuropathol 2017;134:171–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Longstreth W, Manolio TA, Arnold A, et al. Clinical correlates of white matter findings on cranial magnetic resonance imaging of 3301 elderly people: the Cardiovascular Health Study. Stroke 1996;27:1274–1282. [DOI] [PubMed] [Google Scholar]
- 46.De Leeuw F, de Groot JC, Achten E, et al. Prevalence of cerebral white matter lesions in elderly people: a population based magnetic resonance imaging study. The Rotterdam Scan Study. J Neurol Neurosurg Psychiatry 2001;70:9–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lao PJ, Vorberger RS, Narkhede A, et al. White matter regions with low microstructure in young adults spatially coincide with white matter hyperintensities in older adults. Front Aging Neurosci 2019;11:345 10.3389/fnagi.2019.00345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fernando MS, Simpson JE, Matthews F, et al. White matter lesions in an unselected cohort of the elderly: molecular pathology suggests origin from chronic hypoperfusion injury. Stroke 2006;37:1391–1398. [DOI] [PubMed] [Google Scholar]
- 49.Jack CR Jr, Bennett DA, Blennow K, et al. NIA-AA research framework: toward a biological definition of Alzheimer’s disease. Alzheimers Dement 2018;14:535–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vinters H, Gilbert J. Cerebral amyloid angiopathy: incidence and complications in the aging brain. II. The distribution of amyloid vascular changes. Stroke 1983;14:924–928. [DOI] [PubMed] [Google Scholar]
- 51.Raz L, Bhaskar K, Weaver J, et al. Hypoxia promotes tau hyper-phosphorylation with associated neuropathology in vascular dysfunction. Neurobiol Dis 2019;126:124–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.McAleese KE, Walker L, Graham S, et al. Parietal white matter lesions in Alzheimer’s disease are associated with cortical neurodegenerative pathology, but not with small vessel disease. Acta Neuropathol 2017;134:459–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tosto G, Zimmerman ME, Hamilton JL, et al. The effect of white matter hyperintensities on neurodegeneration in mild cognitive impairment. Alzheimers Dement 2015;11:1510–1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Robert H, Paul CR, Ott BR, Salloway S. Vascular dementia: cerebrovascular mechanisms and clinical management. Totowa, NJ: Humana Press, 2005. [Google Scholar]
- 55.Doubal FN, MacLullich AM, Ferguson KJ, et al. Enlarged perivascular spaces on MRI are a feature of cerebral small vessel disease. Stroke 2010;41:450–454. [DOI] [PubMed] [Google Scholar]
- 56.Potter GM, Doubal FN, Jackson CA, et al. Enlarged perivascular spaces and cerebral small vessel disease. Int J Stroke 2015;10:376–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Attems J, Yamaguchi H, Saido TC, Thal DR. Capillary CAA and perivascular Aβ-deposition: two distinct features of Alzheimer’s disease pathology. J Neurol Sci 2010;299:155–162. [DOI] [PubMed] [Google Scholar]
- 58.Chen W, Song X, Zhang Y, Alzheimer’s Disease Neuroimaging Initiative. Assessment of the Virchow-Robin spaces in Alzheimer disease, mild cognitive impairment, and normal aging, using high-field MR imaging. AJNR Am J Neuroradiol 2011;32:1490–1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Smith EE, Greenberg SM. β-Amyloid, blood vessels, and brain function. Stroke 2009;40:2601–2606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ryan NS, Bastos-Leite AJ, Rohrer JD, et al. Cerebral microbleeds in familial Alzheimer’s disease. Brain. 2011;135:e201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yamada M Predicting cerebral amyloid angiopathy-related intracerebral hemorrhages and other cerebrovascular disorders in Alzheimer’s disease. Front Neurol 2012;3:64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Charidimou A, Boulouis G, Pasi M, et al. MRI-visible perivascular spaces in cerebral amyloid angiopathy and hypertensive arteriopathy. Neurology 2017;88:1157–1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pasi M, Boulouis G, Fotiadis P, et al. Distribution of lacunes in cerebral amyloid angiopathy and hypertensive small vessel disease. Neurology 2017;88:2162–2168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Charidimou A, Jaunmuktane Z, Baron J-C, et al. White matter perivascular spaces: an MRI marker in pathology-proven cerebral amyloid angiopathy? Neurology 2014;82:57–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wilcock DM, Hurban J, Helman AM, et al. Down syndrome individuals with Alzheimer’s disease have a distinct neuroinflammatory phenotype compared to sporadic Alzheimer’s disease. Neurobiol Aging 2015;36:2468–2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Shim KS, Ferrando-Miguel R, Lubec G. Aberrant protein expression of transcription factors BACH1 and ERG, both encoded on chromosome 21, in brains of patients with Down syndrome and Alzheimer’s disease. J Neural Transm Suppl 2003;67:39–49. [DOI] [PubMed] [Google Scholar]
- 67.Zis P, Dickinson M, Shende S, et al. Oxidative stress and memory decline in adults with Down syndrome: longitudinal study. J Alzheimers Dis 2012;31:277–283. [DOI] [PubMed] [Google Scholar]
- 68.Strydom A, Dickinson MJ, Shende S, et al. Oxidative stress and cognitive ability in adults with Down syndrome. Prog Neuropsychopharmacol Biol Psychiatry 2009;33:76–80. [DOI] [PubMed] [Google Scholar]
- 69.Schupf N, Kapell D, Nightingale B, et al. Earlier onset of Alzheimer’s disease in men with Down syndrome. Neurology 1998;50:991–995. [DOI] [PubMed] [Google Scholar]
- 70.Schupf N, Winsten S, Patel B, et al. Bioavailable estradiol and age at onset of Alzheimer’s disease in postmenopausal women with Down syndrome. Neurosci Lett 2006;406:298–302. [DOI] [PubMed] [Google Scholar]
- 71.Schupf N, Lee JH, Wei M, et al. Estrogen receptor-α variants increase risk of Alzheimer’s disease in women with Down syndrome. Dement Geriatr Cogn Disord 2008;25:476–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Schupf N, Pang D, Patel BN, et al. Onset of dementia is associated with age at menopause in women with Down’s syndrome. Ann Neurol 2003;54:433–438. [DOI] [PubMed] [Google Scholar]
- 73.Yip A, McKee A, Green R, et al. APOE, vascular pathology, and the AD brain. Neurology 2005;65:259–265. [DOI] [PubMed] [Google Scholar]
- 74.Schupf N. Genetic and host factors for dementia in Down’s syndrome. Br J Psychiatry 2002;180:405–410. [DOI] [PubMed] [Google Scholar]
- 75.Makin S The amyloid hypothesis on trial. Nature 2018;559:S4–S7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.