

Abstract

Capacity retention in lithium metal batteries needs to be improved if they are to be commercially viable, the low cycling stability and Li corrosion during storage of lithium metal batteries being even more problematic when there is no excess lithium in the cell. Herein, we develop in situ NMR metrology to study “anode-free” lithium metal batteries where lithium is plated directly onto a bare copper current collector from a LiFePO4 cathode. The methodology allows inactive or “dead lithium” formation during plating and stripping of lithium in a full-cell lithium metal battery to be tracked: dead lithium and SEI formation can be quantified by NMR and their relative rates of formation are here compared in carbonate and ether-electrolytes. Little-to-no dead Li was observed when FEC is used as an additive. The bulk magnetic susceptibility effects arising from the paramagnetic lithium metal were used to distinguish between different surface coverages of lithium deposits. The amount of lithium metal was monitored during rest periods, and lithium metal dissolution (corrosion) was observed in all electrolytes, even during the periods when the battery is not in use, i.e., when no current is flowing, demonstrating that dissolution of lithium remains a critical issue for lithium metal batteries. The high rate of corrosion is attributed to SEI formation on both lithium metal and copper (and Cu+, Cu2+ reduction). Strategies to mitigate the corrosion are explored, the work demonstrating that both polymer coatings and the modification of the copper surface chemistry help to stabilize the lithium metal surface.
Introduction
Lithium (Li) metal is considered a promising future anode material for next-generation batteries because of its highest theoretical specific capacity of all lithium-ion anodes (3860 mAh/g, calculated based on the lithiated anode material) and low negative potential (−3.04 V vs the standard hydrogen electrode, SHE).1 Cycling of lithium metal batteries (LMBs) entails deposition of lithium metal during charging and dissolution (stripping) during discharging. The greatest obstacles to the commercialization of LMBs are safety issues associated with dendrite growth of electrodeposited Li, as well as their low capacity retention and short cycle life.2
The low capacity retention of LMBs is often overlooked because an excess amount of Li metal is typically used in research-scale cells, which leads to an artificially enhanced cycling efficiency.3−5 However, for practical, commercially viable cells, it is important to limit the amount of excess Li in order to make use of the high specific capacity of Li metal anodes. Practical LMBs will need to have the so-called negative-to-positive (N:P) ratio as close to 1:1 as possible, that is, an amount of Li metal close to that needed to fully lithiate the positive cathode material.1,3 Realistic LMB designs thus either limit the amount of excess Li, e.g., by using thin Li foils,3 or they operate in an “anode-free” battery design where the Li metal anode is replaced with a bare Cu current collector.4,6 The latter design has the obvious practical advantage that it is easier to assemble as it does not require Li metal handling. Both these battery designs tend to have a fast capacity fade, which is directly associated with the irreversible loss of active Li in the cell. This has been ascribed to both the formation of the solid electrolyte interphase (SEI) that forms both spontaneously on the Li metal and during plating and the formation of inactive Li metal typically known as “dead Li”.7,8 Dead Li corresponds to Li that no longer has an electronic contact with the current collector.8−11
Improvements in the capacity retention of LMBs have been attributed to both decreasing the extent of SEI formation and to the formation of more dense Li deposits, the latter decreasing the dead Li and SEI formation.4,12−14 Dead Li formation is thought to be caused by faster stripping of Li at sites with relatively low impedance, e.g., on fresh Li deposits with relatively thin SEI or where the SEI has ruptured.15,16 Thus, electrolytes that result in fast SEI formation kinetics and ensure full and homogeneous SEI coverage on the Li metal surface, leading to more uniform plating, should stabilize these capacity losses in LMBs.17−19
Methods to observe dead Li include scanning electron microscopy (SEM)3,9 and in situ optical microscopy.8,11,20 Quantitative methods have been recently developed; using quantitative titration gas chromatography, Fang et al. determined, for a range of electrolytes and additives, that, contrary to common belief, capacity losses in LMBs are mainly due to the dead Li formation and not SEI formation.19 A recent study performed in situ nuclear magnetic resonance (NMR) on Li metal deposition on Cu and quantified the dead Li formation in a Cu–Li cell ex situ after disassembling the cell.21 A nondestructive in situ methodology that allows the dead Li formation to be quantified during the operation of the battery is preferable. Our previous work on in situ NMR in Li metal cells allowed direct quantification of the microstructures formed during plating.22,23 The methodology uses the intensity of the pristine 7Li metal peak before passing any current (with a known surface area) to calibrate the NMR intensity and allows the NMR intensity to be converted into mass of deposited Li.22 We recently demonstrated, using in situ NMR, that the Li metal deposition in a Cu-LiFePO4 (LFP) cell and the formation of dead Li can be monitored during constant current plating and stripping.24 We also used the rate of exchange between 6Li metal and 7Li+ in a natural abundance (92.5% 7Li) electrolyte to quantify Li exchange between the solid and liquid phases, quantifying both SEI formation and the (effective) exchange current on Li metal strips.23 Here, we develop and apply a quantitative in situ NMR metrology to determine the origin of lithium losses in the Cu−LFP full cell during operation.
In an anode-free battery, the Li deposits and Cu metal are in intimate contact with each other and the electrolyte, potentially creating the conditions of a short-circuited galvanic cell. Two recent studies have drawn attention to this phenomenon,25,26 the first attributing the enhanced rates of corrosion to electrolyte reduction to form the SEI on Cu, reduction on copper being promoted by the poorer (less protective/passivating) SEI formed on the Cu surface.25 A galvanic current can also result from the reduction of copper oxides, via the various reduction reactions widely studied on Cu.27−31 Li metal deposition on Cu is analogous to the formation of a sacrificial coating on a surface, where the Li metal serves to inhibit Cu corrosion: while desirable for the Cu, this phenomenon potentially results in enhanced rates of corrosion for Li.27
Li corrosion is defined here as the removal of Li, via (i) the chemical formation of the SEI directly on the Li metal surface, which is accompanied by Li oxidation and dissolution of Li+ ions, and (ii) the galvanic corrosion of Li by a Li-oxidation reaction that is coupled with the reduction of copper oxides and/or reduction of the electrolyte on the Cu electrode surface. It is crucial to understand how to mitigate the dissolution of Li metal by forming a protective SEI on the Li deposits, but it is equally important to passivate the Cu surface,25,26 both limiting the corrosion current. The morphology of the Li deposits will have an influence on the galvanostatic corrosion mechanism, with both a smoother morphology and a greater surface coverage of Li (the latter minimizing areas where Cu is in contact with the electrolyte) expected to result in lower corrosion current. We note that the terms dissolution and corrosion are used interchangeably here, and in prior literature,25,26 because both processes involve the loss of Li metal; however, strictly the process is not a straightforward dissolution as it first involves Li metal oxidation and subsequent Li+ ion dissolution.
One strategy is to replace the natural SEI with an artificial SEI, to help ensure chemical passivation of the electrodes and to mitigate side reactions with the electrolyte.32 Polymer-coatings are an attractive option as they are easily scalable and have been shown to enhance the cycle life of LMBs by promoting homogeneous plating and stripping.1,33,34 Here we chose to study three common polymers (PEO, PMMA, and PVDF) with different chemical and mechanical properties,35 as a case study for screening different artificial SEIs. PEO is a common solid polymer electrolyte and PVDF and PMMA are used as part of gel polymer electrolytes.35−39 Furthermore, PVDF is commercially used as a binder in composite battery electrodes.40
In this work we develop a 7Li in situ NMR metrology to study the corrosion and cycling behavior of Li metal in an anode-free LMB cell assembled with a Cu current collector. LFP was chosen as a stable cathode material with a flat voltage profile at 3.5 V vs Li/Li+,41 but in principle, any lithiated cathode material can be used. Based on quantitative NMR and Coulombic efficiency (CE) measurements, the amount of dead Li and SEI formation is quantified and compared in three different electrolytes, 1 M LiPF6 in EC/DMC (LP30), with a fluoroethylene carbonate additive (LP30 + FEC), and 1 M LiTFSI in DOL/DME + 2 wt % LiNO3 (DOL/DME). The FEC additive has been shown to be beneficial for plating and stripping Li, resulting in higher CE in Li–Cu cells and better capacity retention in Li−NMC cells.42,43 The DOL/DME electrolyte is widely used in Li–S battery research,44 and the Li deposits have been shown to have a characteristic round-shape morphology.45−47 In addition, we use the bulk magnetic susceptibility (BMS) effects of Li metal and LFP, performing a careful analysis of the 7Li NMR shift, to provide insight into the surface coverage and the Li deposit morphology.48,49 The Li metal dissolution that occurs during rest periods was monitored by in situ NMR, the results revealing that the total corrosion of Li (both the chemical SEI formation and galvanic corrosion) remains a major concern for rechargeable LMBs and is expected to be especially important for batteries with a limited amount of Li present in the cell. The effect of Cu surface treatments, electrolytes, and polymer-coatings on the Cu current collector (PEO, PVDF, and PMMA) is then studied as an approach to mitigate Li corrosion.
Materials and Methods
Materials
LiFePO4 (LFP) positive electrode contained 90% active material and 5 wt % SuperP-Li (Timcal) and 5 wt % PVDF (Arkema, HSV 900) was casted with N-methyl-2-pyrrolidone (NMP, Sigma-Aldrich, 99.5%, anhydrous) on an aluminum foil, initially dried at 80 °C and calendared. Hydrochloric acid (Honeywell Fluka) was diluted to 1.1 M HCl in deionized water. The Cu foil was soaked in 1.1 M HCl (aq) for 10 min for a surface oxide removal, followed by a rinse with ethanol. The Cu foil was then quickly transferred into a glovebox antechamber, where it was dried under vacuum before transferring into an argon glovebox for storage. For the experiment using AcH-treated Cu, the Cu foil was soaked in concentrated acetic acid (Fischer Chemical, lab reagent grade) for 10 min and dried with N2 gas before being transferred into a vacuum oven at 100 °C where it was dried for 24 h. This procedure ensures a homogeneous passivation layer on the Cu surface.24,50 Poly(ethylene oxide) (PEO, Mw = 600,000, Sigma-Aldrich), poly(vinylidene fluoride) (PVDF, Kynar HSV 900), and poly(methyl methacrylate) (PMMA, Mw = 75,000, Polysciences) were used as received. The PEO and PMMA were dissolved in 0.02 g/mL acetonitrile (Sigma-Aldrich) and the PVDF dissolved in 0.02 g/mL NMP. All polymers were spin-coated on an untreated Cu foil (15 mm diameter) at 4000 rpm for 30 s. After the spin-coating, the polymer-coated Cu foil was allowed to dry at room temperature overnight. Then, the PEO-coated foil was dried at 50 °C overnight, while the PVDF and PMMA coatings were vacuum-dried at 100 °C overnight.
Cell assembly and handling of air sensitive materials was done in an argon glovebox (MBraun, O2, H2O < 1 ppm). The electrolytes used were the following: 1 M LiPF6 in ethylene carbonate and dimethyl carbonate (EC:DMC 1:1 volume ratio, Sigma-Aldrich, battery grade), termed LP30 in this study. LP30+FEC was prepared by mixing LP30 with fluoroethylene carbonate additive in 10:1 volume ratio (FEC, Sigma-Aldrich, 99%). The electrolyte referred to as DOL/DME was prepared using 1 M lithium bis(trifluoromethanesulphonyl)imide (LiTFSI, Acros Organic, 99%) in 1,3-dioxolane (Sigma-Aldrich, anhydrous, 99.8%) and 1,2-dimethoxyethane (Sigma-Aldrich, anhydrous, 99.5%) (DOL/DME in 1:1 volume ratio) with 2 wt % lithium nitrate (LiNO3, Alfa Aesar, anhydrous, 99%). The LiTFSI and LiNO3 salts were dried for 20 h at 100 °C under vacuum before use. A capsule cell (NMR Service) made out of PEEK (polyether ether ketone) was used for all in situ NMR experiments and has been described before.51 Working electrodes consisted of either a bare Cu current collector or polymer-coated Cu foil. The amount of electrolyte added to each cell was 75 μL for the carbonate electrolytes and 80–100 μL for the DOL/DME electrolyte. Glass fiber (Whatman GF/A) separators were used after being dried in vacuum at 100 °C. Cells with polymer-coated Cu were rested for 2 h after assembly before any current was applied.
Electrochemistry
Galvanostatic cycling was performed using current density of 0.5 mA/cm2 for an areal capacity of 1 mAh/cm2 on the Cu current collector, unless otherwise stated. At the end of each charge/discharge step, the cell was rested for 10 min to make sure there that around 3–5 NMR experiments were measured in order to get an accurate value of the integrated Li metal intensity. A cutoff capacity of 1 mAh/cm2 was used for plating and stripping and a cutoff voltage of 2.8 V during stripping. Note that the LFP cathode is not fully delithiated, and the areal capacity of the LFP cathode is roughly 2.3 mAh/cm2. The first cycle for plating and stripping on PVDF-coated Cu was performed using 0.1 mA/cm2 current density, and the subsequent cycles were all performed at 0.5 mA/cm2. The lower current density of 0.1 mA/cm2 for PVDF was originally chosen as a precycling step to limit the influence of ionic resistance at the PVDF-polymer/Cu interface. All in situ experiments of Li corrosion used 0.5 mA/cm2 unless otherwise stated. For the experiments with longer SEI formation periods on Cu, a current density of 0.02 mA/cm2 was used before the Li deposition at 0.5 mA/cm2. The Coulombic efficiency was calculated as follows:
| 1 |
In Situ Nuclear Magnetic Resonance
The in situ NMR experiments were conducted on a Bruker Avance 300 MHz spectrometer (the Larmor frequency for 7Li being 116.6 MHz) using a solenoidal Ag-coated Cu coil. The spectra were recorded using an in situ automatic-tuning-and-matching probe (ATM VT X in situ WB NMR probe, NMR Service) that allows for an automatic recalibration of the NMR rf-circuit during an in situ electrochemistry experiment. The retuning of the rf-circuit becomes essential in order to quantify the NMR signals when the sample conditions are changing during the electrochemistry.52 The probe has highly shielded wire connections to the electrochemistry with low-pass filters (5 MHz) attached to the probe, minimizing the interferences between the NMR- and the electrochemistry-circuit, as described in a previous publication.52 Overall, the in situ setup allows for highly reproducible NMR measurements. Single-pulse experiments were used to collect the NMR data, with a recycle delay of 1 s (>5 × T1) and 128 transients recorded. This resulted in an experimental time of about 2.5 min. The shift of 7Li was referenced to 1 M LiCl in water at 0 ppm. The spectra were processed in the Bruker Topspin software, using the automatic phase and baseline correction. Further data processing was done in R. The total intensity of the Li metal peak was integrated over the 7Li shift range of 310–220 ppm and normalized to the intensity measured at the end of plating in the first cycle.
Results
Quantifying Dead Li Formation on Cu
In situ NMR was performed to study Li deposition and stripping in a Cu−LFP full cell. Figure 1a shows the 7Li in situ NMR spectra of the Cu−LFP cell before cycling. The resonances in the 7Li diamagnetic region at around 0 ppm correspond to the electrolyte (and the SEI after its formation).53 The LFP cathode resonance is extremely broad, spreading over thousands of ppm, with a range of hyperfine and BMS shifts that depend on the LFP particles’ aspect ratio, packing density of the film, and orientation in the magnetic field.54,55 The broad resonance consequently overlaps with the diamagnetic (and Li metal) peaks, and in the current study, with an NMR spectral window range of 800 ppm and the carrier frequency centered at around 257 ppm, it will simply be seen as a contribution to the broad baseline.52 This is adjusted automatically in our data-processing via a baseline correction. Upon charging the Cu−LFP cell, Li deposition takes place on the Cu electrode and the Li metal resonance appears in the spectrum (Figure 1b). The Li metal resonates on average at around 260 ppm, the shift arising from the Knight shift, a measure of the density of states at the Fermi level (as probed by the Li 2s orbital).56 Thus, the Li metal resonance is easily distinguishable from the diamagnetic electrolyte-SEI peak.53 The Li metal peak still remains at the end of discharge (stripping, Figure 1c), indicative of the formation of electrically isolated Li deposits, termed “dead LiNMR” to denote the dead Li measured by NMR. Upon further cycling, the intensity of the Li metal peak seen at the end of stripping grows, indicating further accumulation of the dead Li in the cell (Figure 1d).
Figure 1.

Schematic of the 7Li in situ NMR technique used to study dead Li formation and the resulting 7Li NMR spectra. (a) The Cu-LiFePO4(LFP) cell before cycling and the corresponding 7Li NMR spectrum showing the resonance of the diamagnetic Li (the SEI and Li+ ions) and the absence of the Li metal peak. (b) Charging the cell results in Li deposition, as shown in the 7Li NMR spectrum of the Li metal region. (c) At the end of discharge, the Li metal signal can still be observed, which is attributed to dead Li. (d) Further cycling of the Cu−LFP cell results in an accumulation of dead Li over the next cycles (cycle 2–5), the intensity of the Li metal signal increasing at the end of stripping in each cycle.
Skin depth effects must be considered when NMR is performed on metallic samples.22,57 The rf-field used to excite the nuclear spins penetrates conductors only up to a certain depth on the order of the skin depth, which is 12 μm in this study (eq S2). The skin depth effects can be observed in a so-called nutation experiment, described in the Supporting Information, which is used here to measure the radio frequency (rf) field felt by the Li metal spins. A nutation experiment was performed after plating Li on to Cu for 1 mAh/cm2. The sinusoidal nutation curve of the Li deposits (Figure S1), typically observed for samples that do not experience skin effects, confirms that the Li deposits are fully excited and are thus less than 12 μm in thickness.58 For the remainder of the study we assume that the whole volume of the Li metal deposits are being excited.
Effect of the Electrolyte on Li Metal Cycling
Figure 2 shows one in situ NMR data set for measurements in LP30 during galvanostatic plating and stripping at a current density of 0.5 mA/cm2 and a capacity of 1 mAh/cm2 on charge (Li plating). The integrated intensity of the Li metal peak grows linearly with charge (Figure 2b). Upon discharge (Li stripping), the metal peak decreases in intensity until the cell hits the cutoff voltage and the active Li metal has been stripped off the Cu electrode. As can be seen in Figure 2b, the normalized intensity at the end of discharge is not equal to zero due to the formation of dead LiNMR.
Figure 2.

In situ7Li NMR measurement of a Cu−LFP cell cycled in LP30 electrolyte with 0.5 mA/cm2 current density and 1 mAh/cm2 capacity for each plating step. (a) The 7Li NMR spectra acquired during the plating and stripping of Li metal. The Li metal resonance appears at around 270 ppm. (b) Corresponding integrated intensity of the Li metal peak normalized to the intensity at the end of plating in the first charge and (c) voltage profile for the galvanostatic cycling.
In the next cycles, a capacity of 1 mAh/cm2 can still be passed in each plating-step as the LFP cathode has not been fully delithiated (it holds ∼2.3 mAh/cm2 areal capacity). The dead LiNMR accumulates over the first five cycles reaching approximately 40% of the Li metal deposited in the first cycle. Similarly, the intensity of Li metal at the end of plating, termed “total LiNMR” hereafter, increases in each cycle (Figure 2b). The increase in total LiNMR and dead LiNMR does not fully correlate, as seen in Figure 2b where the dead LiNMR is roughly 8% of the metal deposited in the first cycle but the increase at the end of plating in the second cycle is only 5%. This can be explained in terms of the changes in the charge wasted in parasitic reactions and the SEI formation (termed SEI capacity), which affects the amount of total LiNMR measured in each cycle.
The methodology and cycling protocol were implemented in the electrolytes LP30, LP30 + FEC, and DOL/DME, with three sets of in situ cells measured for each electrolyte. For the average total LiNMR (Figure 3a, green) in the LP30 electrolyte, we see the same trends as in Figure 2, with a roughly 20% increase over the first five cycles and the accumulation of the average dead LiNMR accounting to roughly 40% of the initial Li (Figure 3b, green). The CE is 82–85% over the first five cycles. For LP30 + FEC, almost no dead Li is detected (Figure 3c, orange), consistent with the study by Fang et al.19 The CE was noticeably lower for DOL/DME in the first cycle compared to the other electrolytes, around 75%, and correlates with greater amount of dead Li being formed. High CE in DOL/DME is typically reported in Cu–Li cells.19,47 Low CE in the DOL/DME electrolyte was however observed in Cu−LFP cells.47
Figure 3.
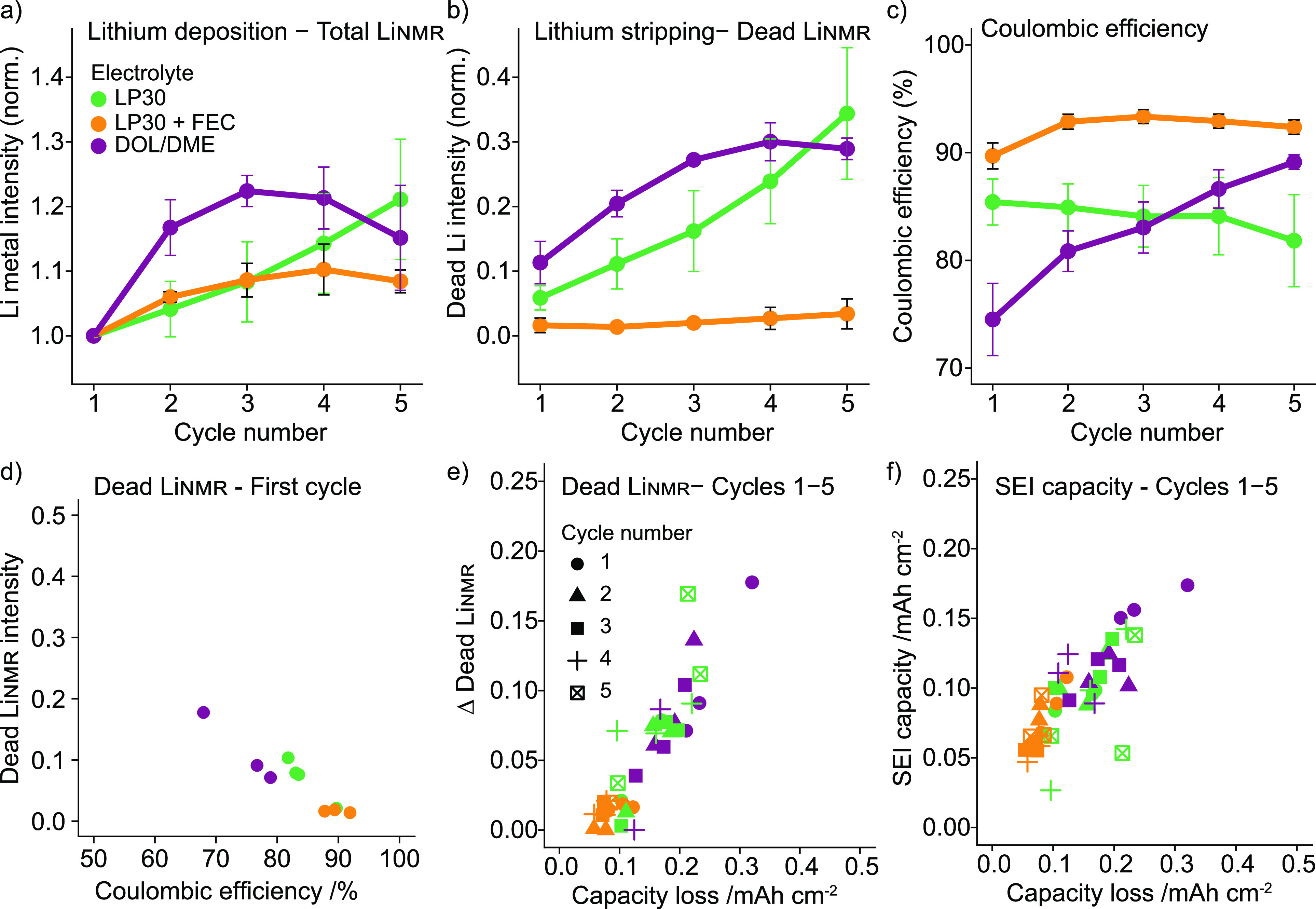
Average value of the (a) normalized total LiNMR intensity at the end of plating, (b) normalized dead LiNMR intensity at the end of stripping, and (c) electrochemically obtained CE for the first five cycles in the three electrolytes, LP30 (green), LP30 + FEC (orange), and DOL/DME (purple). The error bars represent the standard deviation of the average values obtained in three different experiments. (d) The dead LiNMR measured in the first cycle plotted against the CE showing three separate experiments for each electrolyte. (e) The difference in dead LiNMR between subsequent cycles plotted against the capacity loss (mAh/cm2) calculated from the CE. (f) The SEI capacity (mAh/cm2) calculated in each cycle against the corresponding capacity loss (mAh/cm2).
The capacity loss due to the electrochemical SEI formation (the SEI capacity, defined here as the capacity lost due to all irreversible side reactions when the current is being passed) can be estimated from the dead LiNMR measured by NMR and the capacity loss from the electrochemistry (see the Supporting Information for full derivation). Briefly, the total capacity loss measured electrochemically is defined as CL = Cplating – Cstripping, i.e., the difference between the full plating capacity and the stripping capacity. The CL results from a combination of the capacity lost due to dead Li formation and the SEI capacity, CSEI,n:
| 2 |
where the subscript n denotes the cycle number n. The dead LiNMR value measured by the NMR denoted here as χdead LiNMR is the fractional amount of dead Li measured by NMR normalized to the Li metal intensity at the end of the charge in the first cycle (which depends on both the charge passed and the charge consumed in forming the SEI). Thus, Cdead Li,n is not directly proportional to χdead LiNMR and a correction is needed to account for the capacity used to form the SEI electrochemically in the first cycle. The dead Li capacity, Cdead Li,n is instead given:
| 3 |
Cplating – CSEI,n=1 corresponding to the capacity used to deposit the Li metal that gives rise to the resonance observed at the end of the charge. Eq 2 can now be rewritten as
| 4 |
By solving for CSEI,1 in the first cycle, CSEI,n and Cdead Li,n can now be determined.
As an example of this method, the SEI capacity is now calculated for the set of data presented for LP30 in Figure 2. The CLn=1 on first cycle is 0.17 mAh/cm2 and χdead LiNMR,n=1 is 0.08 after one cycle (Figure 2b). This results in an SEI capacity of approximately 0.1 mAh/cm2 and dead Li capacity of 0.07 mAh/cm2. This indicates that in the case of LP30, the CE is influenced significantly by both the dead Li formation and the SEI formation capacity. The CE in the first cycle as well as the SEI capacity and dead Li are displayed in Table 1. A slightly higher value for the SEI capacity is observed for each electrolyte compared to the amount of dead Li. It should be noted that this calculation neglects Li corrosion, that is, the processes involving dissolution of Li due to chemical SEI formation and galvanic corrosion that do not result in a net current in the cell. We explored the effect of this assumption in the Supporting Information, taking into account the Li corrosion current as quantified below, and the effect was seen to be negligible.
Table 1. CE, SEI Formation Capacity and Dead Li Capacity in the First Cycle Calculated Using the Dead LiNMR Intensity Determined by In Situ NMR and the CE Determined from Electrochemistrya.
| electrolyte | coulombic efficiency | SEI formation capacity (mAh/cm2) | dead Li capacity (mAh/cm2) |
|---|---|---|---|
| LP30 | 85 ± 2% | 0.09 ± 0.005 | 0.06 ± 0.02 |
| LP30 + FEC | 90 ± 1% | 0.09 ± 0.01 | 0.01 ± 0.001 |
| DOL/DME | 75 ± 3% | 0.16 ± 0.01 | 0.11 ± 0.03 |
The standard deviation is that derived from the three separate in situ NMR experiments.
The difference in the dead Livalue between cycles, “Δ dead Li”, which corresponds to the dead Li formation per cycle, and the SEI capacity per cycle are shown against the corresponding capacity loss (Figure 3e,f). Both the dead Li and SEI capacity displayed a linear relationship with the capacity loss per cycle, demonstrating that both processes determine the cycling stability of LMBs. The same values (per cycle) are shown as a function of cycle number to visualize whether any stabilization occurs with cycling (Figure S2). For the carbonate electrolytes there is continuous SEI formation in each cycle, the SEI capacity remaining steady at around 0.05–0.1 mAh/cm2. For the DOL/DME however, the SEI capacity drops significantly after the first cycle before stabilizing at around 0.1 mAh/cm2 (Figure S2). Similarly, the dead Li formation in DOL/DME drops with cycling in contrast to in LP30 where it increases slightly (Figure S2).
Bulk Magnetic Susceptibility Effects
The shift of the 7Li metal peak is sensitive to orientation and morphology of the Li deposits. Chandrashekar et al. showed with 7Li MRI how the shift of Li metal can be used to distinguish between electrodeposits growing close to the Li metal (at around 260 ppm) and dendritic structure extending further away from the surface (at around 270 ppm).48 This was later confirmed in a detailed study by Chang et al., where different resonances in the in situ NMR spectra were correlated with SEM images of Li microstructure morphology formed under different stack pressures and using different separators.49
To understand the shifts observed in the in situ Cu−LFP cells, the effect of the paramagnetic LFP cathode on the 7Li metal shift is first studied in a pristine Li-LFP cell, since previous studies have shown that paramagnetic cathode electrodes can cause significant BMS effects and broadening, the shifts depending on the orientation of the electrode films with respect to the static magnetic field.55 The Li metal peak of a pristine Li–Li cell (when the electrodes are oriented perpendicular to the static, applied magnetic field) appears at 245 ppm, but when paired with the LFP cathode, the Li metal peak shifts to 260 ppm (Figure S10). In a Li–Cu cell, there is no significant shift in the 7Li metal peak (Figure S10), since Cu metal has only a small diamagnetic contribution (see the Supporting Information). Thus, the 7Li metal peak in Cu−LFP cells is expected to be shifted by approximately 15 ppm to higher frequencies from the signal typically observed in Li–Li cells.
The 7Li metal peak in the Cu−LFP cells emerges at around 275 ppm at the start of plating (Figure 4a, in LP30 electrolyte) and shifts to lower ppm values both during plating and on further cycling (as seen for the NMR spectra on fourth cycle, Figure 4b). The shift of the peak maximum was extracted (Figure 4c) and interestingly is shown to cycle with the electrochemistry, moving to lower shifts during Li plating and to higher shifts during Li stripping. During plating, the LFP cathode is delithiated (charged) and the oxidation state of iron changes from Fe2+ in LiFePO4 to Fe3+ in FePO4, which increases the susceptibility of the cathode.59 Thus, the change in shift observed for the Li metal can either be due to changes in the susceptibility of the LFP cathode that will influence the susceptibility of the whole cell or due to the dependence of the shift on the Li morphology as reported in previous studies.49,59
Figure 4.
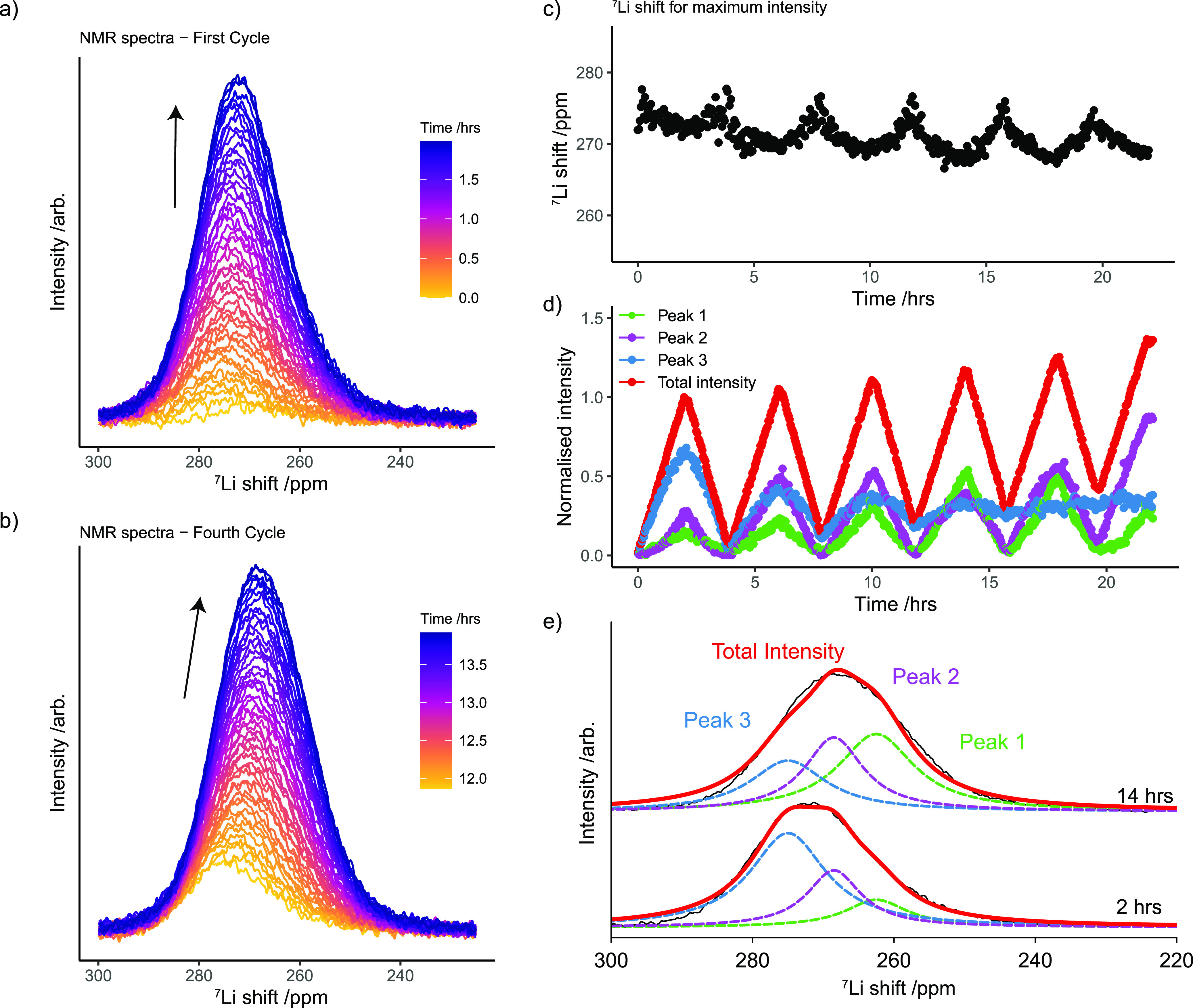
BMS effects for the Li metal peak in the LP30 electrolyte. (a) Stack plot of the Li metal spectra during charge (plating) in the first cycle (where the same metal spectra are shown versus time in Figure 2a). (b) Li metal spectra during plating in the fourth cycle. (c) Frequency of the 7Li metal shift, measured at the maximum intensity of the Li metal resonance, during cycling. (d) Deconvoluted intensities of the Li metal spectra during cycling. (e) Example of the fitted spectra at the end of charge in the first and fourth cycle.
To gain more insight into this phenomenon, the spectra were fitted using the least-squares method with three overlapping peaks (using pseudo-Voigt curves, Figure 4e); peaks at 260.5 ppm (peak 1), 268.5 ppm (peak 2), and 272.5 ppm (peak 3) were used, and their position was allowed to vary ±2.5 ppm from the set values. In order to explore the sensitivity of the deconvolution method, a fit with two components (two peaks) was attempted but found that in later cycles at least three peaks were essential to obtain a reasonable fit. The normalized intensity of the deconvoluted peaks shows how the Li metal resonance is mainly captured with peak 3 in the first cycle (at the highest Li metal shift, Figure 4d, in blue). Upon further cycling, peak 2 and peak 1 become more dominant, indicating that the main resonance is moving toward lower shifts. Of note, the dead LiNMR at the end of stripping and the initial Li deposits resonate at shifts of around 272–275 ppm at the same position as peak 3 (Figure 4d).
Taking into account the additional shift observed for a pristine Li metal strip assembled with an LFP electrode (15 ppm higher than in Cu–Li cell), a measured shift at 275 ppm can be corrected for the LFP BMS effect; this results in a shift of approximately 260 ppm, which is consistent for the Knight shift of Li metal60 and explains the generally higher Li shifts seen in this system as compared to those seen in Li–Li symmetric systems.22
The 7Li shift in LP30 + FEC and DOL/DME shows a different behavior to the LP30 with the Li metal signal appearing at lower values after plating (Figure S13 and Figure S14). For both electrolytes, peak 1, at the lowest shift, is more pronounced in the deconvolution during cycling of the cell. This difference between electrolytes indicates that the Li metal shift is not dictated wholly by the state of charge of the LFP cathode (which is similar for all the cells) but that the nature of the Li deposits, that is the morphology and surface coverage, must influence the shift.
Susceptibility calculations were then performed to explore the shifts observed during plating in more detail (described in detail in the Supporting Information). The surface coverage of Li microstructures (randomly placed voxels on the electrode surface) was varied between 2.5 and 95% on both Li metal and Cu foil (Figure S12). The overall trend is that a higher surface coverage of Li leads to a lower 7Li NMR shift due to BMS effects, the deposits starting to resemble more bulk Li metal (Figure S12b). On the basis of these susceptibility calculations, the lower shift seen for Li metal plated in DOL/DME and LP30 + FEC versus in LP30 only is, therefore, ascribed to the more homogeneous coverage of Li deposits that are formed in these electrolytes during plating and on further cycling.
The trends in metal shift observed for the different electrolytes is summarized in Figure S15, showing the intensity of the fitted peaks at the end of plating and the end of stripping. The peak at the lowest shift becomes more apparent in all electrolytes with cycling, suggesting the Li coverage on Cu is becoming greater. The intensity of peak 1 is greatest in the DOL/DME electrolyte (Figure S15a, green), which is consistent with ether-based electrolytes having more complete surface coverages compared to the LiPF6–carbonate electrolytes.16 Of note, the high shift of the dead Li peak, close to 275 ppm, indicates that it is affected by the LFP BMS effect and not by the BMS effects due to Li metal. This is consistent with the removal of smoothly deposited Li and the formation of randomly oriented and disconnected Li deposits in a diamagnetic (SEI) matrix.
Polymer-Coated Cu
To extend the in situ NMR method further, three common polymers (PEO, PMMA, and PVDF) that have been heavily studied in batteries were investigated. The polymers were cast onto Cu current collectors, the cell was assembled, and then left to rest for two hours before any current was passed.
The in situ NMR data of the polymer-coated Cu electrodes cycled in LP30 and the CE calculated from the electrochemistry are presented in Figure 5. The PEO- and PVDF- coatings show considerably lower first cycle CE of about 65% (Figure 5c) compared to 85% on bare Cu and 83% with PMMA-coated Cu. Interestingly, lower dead Li formation was detected (Figure 5b), which indicates increased capacity losses due to SEI formation when using these coatings. The cycling efficiency for both the PEO- and PVDF-coated Cu increases, however, after the first cycle indicating that surface reactions have occurred to form a more stable SEI/coating on the Cu. The PMMA presents a similar CE to that of a bare Cu in LP30, but less dead Li formation is seen. The SEI capacity and dead Li values are summarized for the first cycle in Figure S3. In all cases, less dead Li formation but higher SEI capacity is measured on the polymer-coated compared to bare Cu, indicating more side reactions in the polymer systems (Figure S3). Although we have not explored the effect of polymer swelling and/or interaction with the electrolyte, and we did not optimize polymer coating thickness,33,35 the results demonstrate the power of the quantitative NMR technique to deconvolute the contributions to the capacity losses, which cannot be determined from the electrochemistry alone. The NMR spectra of the Li metal on polymer-coated Cu was deconvoluted into three peaks using the same method as described above. The Li metal peak for PMMA and PEO-coated Cu appears at a higher shift than that seen for the PVDF system, as seen via the greater contribution of Peak 3, indicating less homogeneous deposition on cycling (Figure S16). The lower shift for the PVDF-coated Cu is likely due to the lower current used in the first cycle resulting in smoother deposition and is further discussed in the Supporting Information.
Figure 5.
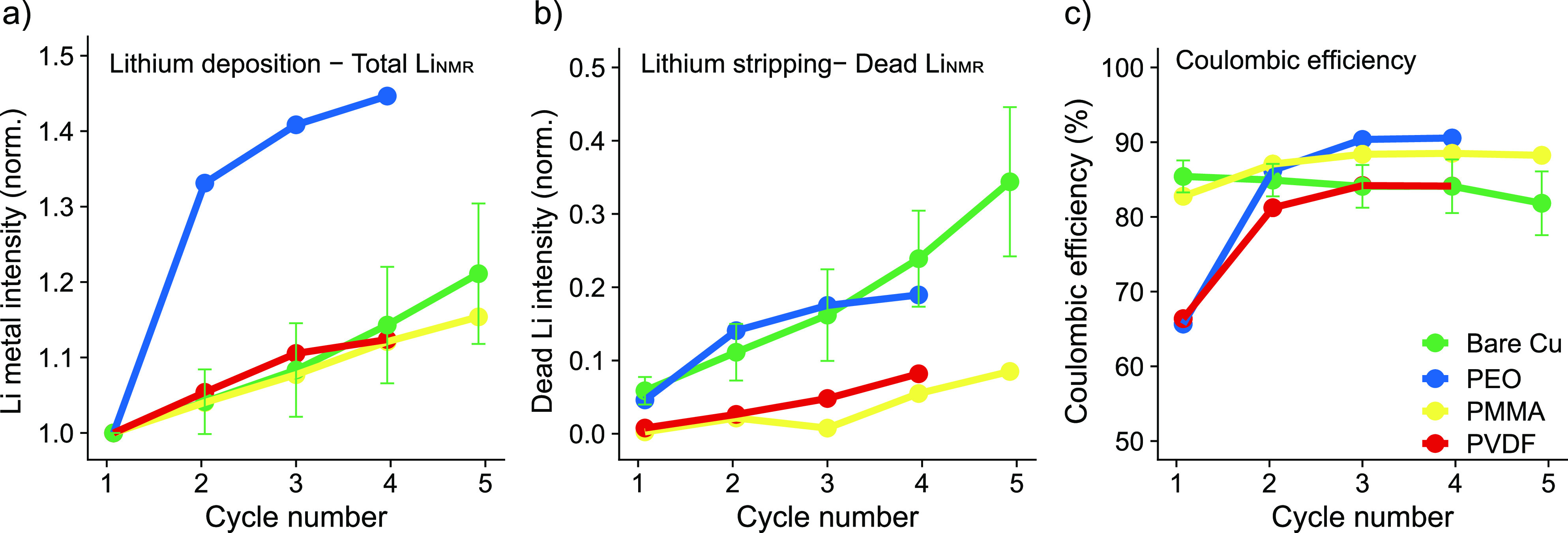
Li metal intensity for the 7Li in situ NMR measurements when cycling Cu−LFP cells in LP30 with polymer-coated Cu. (a) Normalized total LiNMR intensity at the end of plating, (b) normalized dead LiNMR intensity at the end of stripping, and (c) the CE for PEO- (blue), PMMA- (yellow), and PVDF-coated (red) Cu electrodes. Measurements for bare Cu (in green) are shown here to aid comparison.
Dissolution of Li Metal by In Situ NMR
In practice, batteries are not constantly in use, and it is important to understand the processes that occur during the periods when no current is passed. To investigate what occurs during the rest period in LMBs, Li metal was again deposited using a 0.5 mA/cm2 current density and 1 mAh/cm2 plating capacity and the evolution of the Li metal signal recorded during the open circuit voltage (OCV) using in situ NMR. Note that this experiment tracks capacity loss in a charged battery, which is particularly critical in anode-free batteries. The intensity of the Li metal grows for the first two hours during plating (Figure 6b), then a decrease in intensity is seen during the OCV period, indicating loss of Li metal. The dissolution (or corrosion) of Li metal can be due to both the chemical SEI formation on Li and the galvanic corrosion between Li and Cu that are in direct contact with the electrolyte (see the schematic, Figure 6a).25,26
Figure 6.
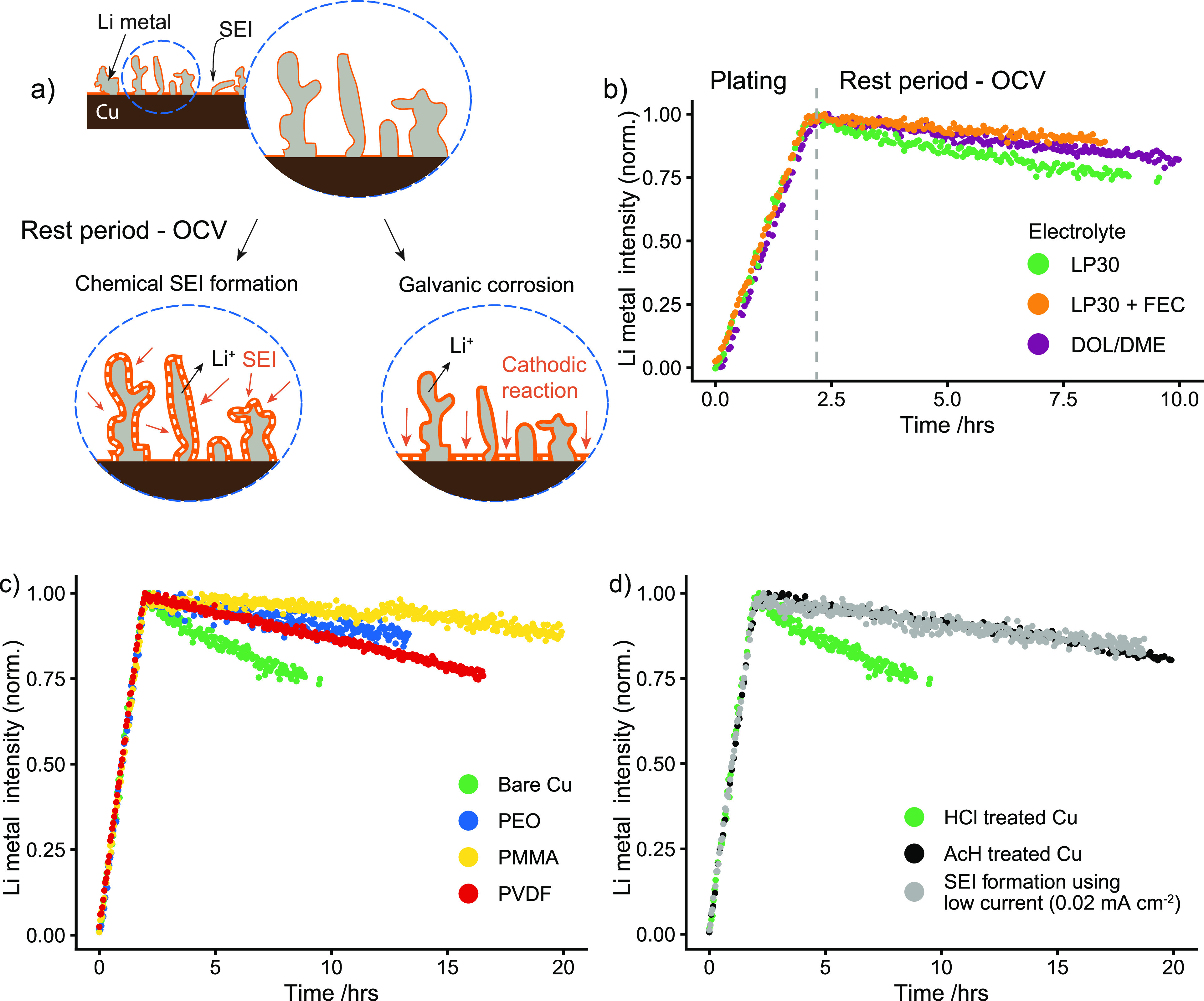
In situ NMR experiments of Li metal dissolution during the OCV period. (a) Schematic representation of the processes that lead to Li metal corrosion: the chemical formation of the SEI on Li results in the spontaneous reduction of the electrolyte and oxidation of the Li metal. Galvanic corrosion results in the dissolution of Li metal (Li oxidation) and a cathodic reaction on the Cu electrode. (b) Integrated intensity of the Li metal signal during the NMR experiment. The intensity increases during deposition for the first 2 h of the experiment (corresponding to 1 mAh/cm2) of charge and decreases constantly during the rest period at OCV for the three electrolytes, LP30, LP30 + FEC, and DOL/DME. Plating and resting experiments for (c) different polymer coatings: PEO-, PMMA-, and PVDF-coated Cu current collector and (d) different Cu treatments in LP30 electrolyte: Cu treated with HCl acid (green) and Cu treated with glacial acetic acid (AcH, black). The gray curve shows the effect of a slow SEI formation step before deposition on HCl-treated Cu at 0.02 mA/cm2 followed by a 12 h voltage hold at 3.2 V before deposition at 0.5 mA/cm2 (gray). The green curve in (b, c, and d) is for the same experiment, performed on bare Cu treated in HCl-acid and in LP30 electrolyte. Each experiment was performed twice, and the rate of dissolution was found to be highly reproducible (Figure S6 and Figure S7).
Figure 6b shows that different electrolytes have a significant impact on the rate of Li dissolution. The electrolyte effect on corrosion observed here is influenced by the Li morphology and bare Cu areas and a “protective coating effect” due to formation of a stable SEI. Among the three electrolytes studied here, the fastest dissolution of Li metal is seen in the LP30 electrolyte and the greatest stability is in LP30 + FEC (Figure 6a). The stabilization due to FEC is likely due to the interplay of greater surface coverages (as indicated by the lower 7Li metal shift seen after plating in LP30 + FEC, Figure S15) as well as the difference in the SEI formed with FEC both on Li and Cu.30,61
The same set of experiments were performed on the polymer-coated Cu current collectors in LP30 electrolyte (Figure 6c), demonstrating that all the coatings have a stabilizing effect on the Li dissolution. This is likely due to the passivating effect of the coating, passivating both the Li deposits and the Cu current collector. Interestingly, the PMMA-coating has the greatest stabilizing effect and correlates with the higher CE observed in Figure 5.
To investigate the effect of the Cu surface on corrosion, the same experiments were performed on a Cu current collector pretreated in concentrated acetic acid (denoted AcH, the Cu used in all other experiments was pretreated in the acid HCl). The surface treatments on Cu current collectors and the corresponding SEI have been studied in detail in our previous work24 where using X-ray photoelectron spectroscopy (XPS) it was shown that the surface of the AcH-treated Cu (prior to assembly in a battery cell) was better passivated with high concentrations of Cu(OH)2/CuO on the surface, whereas the HCl-treated Cu showed the Auger Cu metal peaks, indicating either a thinner or a more heterogeneous surface oxide layer on the Cu electrode with no Cu(OH)2 being observed.24
Figure 6d (black) shows that the corrosion of Li metal is slowed down using AcH-treated Cu, demonstrating the importance of surface passivation on Cu. In addition and following the approach of Lin et al.,25 the corrosion was monitored on an (HCl-pretreated) Cu electrode, following an initial low-current step of 0.02 mA/cm2 prior to Li deposition, the electrolyte reduction products precipitating on the Cu surface resulting in an SEI that grows in thickness during polarization.24,30 This was followed by Li deposition at 0.5 mA/cm2 (Figure 6d, gray). As shown previously by electrochemical measurements, the long SEI formation step on Cu slows down the Li metal dissolution under OCV consistent with passivation of the Cu electrode surface.25 This stabilization is likely a combined effect of the reduction of the copper oxides/hydroxides on the Cu surface, minimizing possible galvanostatic reduction reactions involving Cu2+ and Cu+, which will be driven to an extent by the differences in Cu2(1)+/Cu and the Li+/Li couples, and the formation of a more stable SEI layer, reducing the SEI reduction rate.
A linear fit was used to extract the slope of the dissolution curve during the OCV period, χslope in s–1 (data in Figure 6), as shown in the bar chart Figure S8. The corrosion current can be estimated from the slope by using the SEI capacity determined above. The full plating capacity is Cplating = 1 mAh/cm2 or Cplating = 3.6 C/cm2. The corrosion current density, icorr, becomes
| 5 |
The corrosion current for each electrolyte was calculated using the mean of two separate experiments (Figure S6), with the values listed in Table 2. The values assuming 100% current efficiency for Li deposition (that is ignoring any electrochemical SEI formation) are listed in Table S1. The highest corrosion current was measured in LP30 on bare Cu, of around 30 μA/cm2.
Table 2. Mean of the Slope (for Two Sets of Experiments) Obtained from a Linear Fit to the Decreasing Intensity of the 7Li NMR Metal Signal During the OCV Period (Figures S6 and S7) and the Calculated Corrosion Current icorra.
| electrolyte/polymer coating | χ/s–1 | icorr/μA cm–2 |
|---|---|---|
| LP30 | –8.7 | 29 |
| LP30–sixth cycle | –9.6 | 31 |
| LP30 – Cu passivation/SEI formation | –1.7 | 5.6 |
| LP30 + FEC | –1.7 | 5.5 |
| DOL/DME | –5.8 | 18 |
| PEO-coating | –2.1 | 5.3 |
| PMMA-coating | –1.7 | 5.2 |
| PVDF-coating | –3.9 | 10 |
For experiments using the polymer-coated Cu, LP30 electrolyte was used in all cases.
The long-term corrosion behavior of the Li electrodeposits was probed in LP30 and LP30 + FEC by acquiring ex situ NMR spectra over a 50–80 h period at different time intervals, storing the cells in an inert argon glovebox in between measurements (Figure S4). Both electrolytes show continuous Li corrosion throughout the measurement that does not seem to slow down notably with time. The Li metal deposits in LP30 have completely disappeared after 50 h, whereas there is still Li metal left in LP30 + FEC after ∼80 h, albeit only a small amount. The dissolution of the Li metal was also probed after the sixth deposition cycle in LP30 (Figure S4, black). Interestingly, the corrosion rate does not decrease after further cycling, as indicated by the similar slope of the two dissolution curves.
Discussion
The in situ NMR technique allows the capacity losses due to SEI formation and dead Li to be quantified during cycling. Importantly, it demonstrates that both the SEI formation and dead Li formation contribute significantly to the cycling stability of LMBs. In all cases, electrochemical SEI formation was shown to contribute more to the capacity losses in the anode-free battery compared to dead Li formation. This is in contrast to the results reported by Fang et al., where the authors concluded that the dead Li is the main reason behind capacity losses in LMBs in the first cycle as well as in subsequent cycles.19 The studies are not entirely comparable as Fang et al. performed the measurements in Cu–Li coin cells.19 Having Li foil in the cell has been shown to boost the cycling performance and the CE compared to anode-free cells.5 The Li foil not only supplies the battery with enough reservoir of Li+ ions but also critically may act as a scavenger for any impurities or electrolyte degradation products in the electrolyte, which likely influences the amount of SEI that forms on the Li deposits on Cu.
In LP30 + FEC, a small amount of dead Li was observed on the first cycle (0.01 mA/cm2), and on cycle five, the total dead Li capacity amounted to 0.04 mAh/cm2, although the CE remained 90–93% over the first five cycles demonstrating that the SEI needs reforming and continues to grow every cycle. This is consistent for all three electrolytes as shown in Figure S2 where the SEI formation capacity for the carbonate electrolytes remains around 0.05–0.1 mAh/cm2. This is also consistent with the corrosion rate seen on the sixth cycle in LP30 (Figure S4) where a stabilization after five cycles of plating and stripping was not observed. This is probably due to the volume changes associated with the complete stripping of Li metal in an anode-free battery, damaging the SEI in each cycle. But it also indicates that the film formed on Cu is still poorly passivating, even after 6 cycles. By contrast, the corrosion current drops by a factor of 5 (Table 2) when a passivating film is formed electrochemically on Cu prior to deposition. In this case, a larger contribution to the corrosion current is presumably coming from Li corrosion due to chemical SEI formation (Figure 6a). Further studies are needed to explore this phenomenon, additives such as FEC clearly affecting both the galvanic and chemical SEI formation. This may also indicate that the full stripping of the Li is too extreme in LMBs and a gentler approach where a reservoir of Li is left on the anode after fully discharging the cell may help stabilize the SEI in later cycles.62
In contrast to the carbonate electrolytes, the CE of the DOL/DME increased in the first five cycles with a concurrent drop in dead Li formation and SEI formation (Figure S2). In addition, the Li deposits in DOL/DME appear at a lower shift (Figure S14) indicating formation of a more homogeneous coverage of Li on the Cu electrode. The CE on the PEO- and PVDF-coated Cu was low in the first cycle compared to that on bare Cu (Figure 5) without an increase in the observed dead Li concentration, indicating increased side reactions during plating. This may partly be due to less homogeneous deposition, which causes enhanced SEI formation. This is indicated in the 7Li metal shift analysis, where the shift to lower frequencies with cycle number seen on bare Cu is not seen for the polymer-coatings (compare Figure 4 and Figure S16). This may indicate that instead of an increased Li metal coverage on further cycling, the polymer coating instead breaks/fractures with continued cycling, which leads to even less homogeneous plating. Assegie et al. demonstrated the importance of optimizing the PEO-film on a Cu current collector, where high CE was only achieved with a uniform and defect-free PEO coating.34 Further experiments are required to explore how different current rates and optimization of the polymer film affect the homogeneity of deposition in these systems.
The quantitative in situ NMR allows the corrosion current of Li metal on Cu to be estimated. The corrosion current density (with respect to the Cu electrode area) of Li electrodeposits on Cu was measured as 29 μA/cm2 in LP30 and 5.5 μA/cm2 in LP30 + FEC. The lower corrosion current measured for LP30 + FEC is likely due to both a denser Li morphology and differences in the SEI that forms. Our previous work, quantifying the rate of SEI formation, showed a noticeably faster SEI formation in LP30 + FEC at the OCV, that led to faster stabilization of the SEI on Li metal.23 This will lead to more effective passivation and slower Li corrosion at later times. In addition, the chemical composition and the nanostructure of the SEI on both Li metal and Cu has been shown to be different in LP30 + FEC.30,42,43,61
The rate of galvanic corrosion measured here should be contrasted with the values measured by zero resistance ammetry (ZRA).26 Kolesnikov et al. found the corrosion rate to be highly dynamic with the initial corrosion current >160 μA/cm2 that decreased rapidly to <1 μA/cm2 after 10 h of measurements, reaching a steady-state value of 0.2 μA/cm2 after 100 h in the LiPF6–carbonate electrolyte. They again attribute this decrease in the corrosion current to the formation of a passive film on the Cu, which decreases the reduction reactions at the Cu surface.26 The passivation of the Cu is consistent with the reduced corrosion rate when using a slow SEI formation step on Cu prior to Li deposition, which indicates that if the Cu electrode is properly stabilized the Li corrosion can be mitigated (Figure 6c). The results are also consistent with the data for LP30, where the measurements of Li corrosion immediately after the current is switched off showed an enhanced rate of corrosion (initially around 50 μA/cm2, Figure S9) that settled to a steadier value after two hours. However, no complete stabilization was observed here over longer time-periods in the carbonate electrolytes (Figure S4). Finally, Kolesnikov et al. describe the corrosion effect in terms of the differences in the standard electrode potentials of two metals, here Li and Cu. Strictly this is only true if the Li corrosion is coupled with either Cu+ or Cu2+ reduction to Cu metal; this process can be mitigated, for example, by applying a current above the 0 V (vs Li+/Li) to reduce any oxidized Cu species on the Cu surface24 as was performed for example in Figure 6d. Instead it is more appropriate to view galvanic corrosion as arising from a difference in the rates of SEI formation on Li and Cu, which will arise from both a difference in the passivating nature of the SEI on the Cu as discussed in ref (25) and also simply due to differences in surface area of the two metals. A factor that is also important in galvanic corrosion is the transport of Li+ ions in the SEI and the electrolyte, as Li dissolution at the Li metal deposits releases a Li+ ion, which is then charge balanced by Li+-ion transport through the SEI on the Li metal, the electrolyte, and through the passivating layer on Cu, and which combines with an anion to precipitate a (Li+ electrolyte–) salt (such as LiF, Li2CO3, or an organic, e.g., lithium diethylcarbonate) on the Cu surface. Therefore, both electronic contact (of Li and Cu) and ionic pathways are required for galvanic corrosion. The shorter path for ionic transport favors, for example, corrosion near the Li/Cu interface, a mechanism which itself could lead to dead Li formation (as seen by Kolesnikov et al.).26 This does suggest, however, that the formation of a better SEI on Li will play a role in mitigating galvanic corrosion.
Conclusions
We have demonstrated the application of in situ7Li NMR to study the plating and stripping and the corrosion of Li metal on Cu current collector. The in situ NMR method is shown to be a valuable technique for deconvoluting the multiple capacity losses that occur in lithium metal batteries, which will enable further studies on different electrolytes as well as on the compatibility of protective coatings and artificial SEIs for Li deposition.
Almost no dead Li is observed in LP30 + FEC although the CE is < 100% (∼92%), indicating that capacity losses are mainly due to the SEI formation. Similarly, for LP30 and DOL/DME electrolytes, the dead Li accumulates continuously for the first five cycles but cannot account for the entire capacity loss, and again, the SEI formation is a significant part of the Coulombic losses. The bulk magnetic susceptibility shifts caused by Li metal results in lower 7Li shift of the deposits formed in DOL/DME and LP30 + FEC, indicating higher surface coverage of Li as suggested by the susceptibility calculations. In contrast, the 7Li metal shift in LP30 on both bare Cu and polymer-coated Cu is at a higher metal shift, indicating less dense coverage of the deposits. Of note, the dead Li deposits show essentially no BMS effect due to the Li metal, consistent with their random orientations on the electrode; their shift is dominated by the BMS effect arising from the LFP electrode (and the Li Knight shift).
The evolution of Li metal was monitored under OCV conditions by in situ7Li NMR and revealed that the Li metal corrosion rate is highly sensitive to the type of electrolyte and the surface of the Cu current collector. The rate of corrosion is high and remains a critical issue for lithium metal batteries to be a viable option for energy storage technologies. Strategies to protect the Li metal deposits need to be studied systematically and with quantitative techniques such as those presented here. Polymer coatings were shown to protect the Li metal deposits from dissolution and may be studied further with respect to reactivity, conductivity, and coating homogeneity and thickness.33 Furthermore, it is crucial to perform more controlled study of the Cu films, to correlate Cu surface chemistries and the passivating films that form with the rates of galvanic corrosion. We note, however, that simple pretreatments of the Cu metal to remove surface oxides and to reduce Cu2+/Cu+ species electrochemically and form an SEI on Cu before Li deposition were shown to reduce the corrosion rate noticeably.
The in situ NMR technique can be used to study corrosion in, for example, Li–S batteries where corrosion remains a critical issue due to the dissolution of polysulfides.63 Strategies that aim to mitigate Li corrosion and will be interesting to study include artificial SEI layers such as Al2O363 and metal coatings64−67 and inactive additives such as hydrocarbons that have been shown to decrease the corrosion rate and lead to more homogeneous Li plating and lower Li+ solvation energies.68,69 Furthermore, this technique could be used to study corrosion in Na metal batteries using 23Na in situ NMR spectroscopy.
Acknowledgments
A.B.G. acknowledges the support from the Royal Society (RP/R1/180147) and EPSRC-EP/M009521/1. C.V.A. acknowledges financial support from the TomKat Center Postdoctoral Fellowship in Sustainable Energy at Stanford and a Visiting Fellowship from Corpus Christi College at the University of Cambridge. S.M. thanks the Blavatnik Cambridge Fellowships. C.P.G. thanks the EU/ERC for an Advanced Fellowship. A.B.G. thanks the NanoDTC Cambridge for travel funding. A.B.G. thanks Dr. Andrew Ilott for sharing the code for the bulk magnetic susceptibility calculations and for useful discussions. A.B.G. thanks Dr. Katharina Märker for useful discussions and careful reading of the manuscript. C.P.G. thanks Profs. Alison Davenport and Mary Ryan for insightful discussions.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.0c10258.
Details on the calculation of skin depth effects and the SEI capacity; additional NMR spectra for the different electrolytes and details on bulk magnetic susceptibly effects and simulations; and additional NMR data on the Li corrosion during the open circuit voltage and the electrochemical cycling data (PDF)
Author Present Address
§ Pritzker School of Molecular Engineering, University of Chicago, Chicago, IL 60637, USA.
Author Contributions
The manuscript was written with contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Zheng J.; Kim M. S.; Tu Z.; Choudhury S.; Tang T.; Archer L. A. Regulating Electrodeposition Morphology of Lithium: Towards Commercially Relevant Secondary Li Metal Batteries. Chem. Soc. Rev. 2020, 49 (9), 2701–2750. 10.1039/C9CS00883G. [DOI] [PubMed] [Google Scholar]
- Fang C.; Wang X.; Meng Y. S. Key Issues Hindering a Practical Lithium-Metal Anode. Trends Chem. 2019, 1 (2), 152–158. 10.1016/j.trechm.2019.02.015. [DOI] [Google Scholar]
- Liu J.; Bao Z.; Cui Y.; Dufek E. J.; Goodenough J. B.; Khalifah P.; Li Q.; Liaw B. Y.; Liu P.; Manthiram A.; Meng Y. S.; Subramanian V. R.; Toney M. F.; Viswanathan V. V.; Whittingham M. S.; Xiao J.; Xu W.; Yang J.; Yang X. Q.; Zhang J. G. Pathways for Practical High-Energy Long-Cycling Lithium Metal Batteries. Nat. Energy 2019, 4 (3), 180–186. 10.1038/s41560-019-0338-x. [DOI] [Google Scholar]
- Weber R.; Genovese M.; Louli A. J.; Hames S.; Martin C.; Hill I. G.; Dahn J. R. Long Cycle Life and Dendrite-Free Lithium Morphology in Anode-Free Lithium Pouch Cells Enabled by a Dual-Salt Liquid Electrolyte. Nat. Energy 2019, 4 (8), 683–689. 10.1038/s41560-019-0428-9. [DOI] [Google Scholar]
- Xiao J.; Li Q.; Bi Y.; Cai M.; Dunn B.; Glossmann T.; Liu J.; Osaka T.; Sugiura R.; Wu B.; Yang J.; Zhang J.-G.; Whittingham M. S. Understanding and Applying Coulombic Efficiency in Lithium Metal Batteries. Nat. Energy 2020, 5 (8), 561–568. 10.1038/s41560-020-0648-z. [DOI] [Google Scholar]
- Qian J.; Adams B. D.; Zheng J.; Xu W.; Henderson W. A.; Wang J.; Bowden M. E.; Xu S.; Hu J.; Zhang J. G. Anode-Free Rechargeable Lithium Metal Batteries. Adv. Funct. Mater. 2016, 26 (39), 7094–7102. 10.1002/adfm.201602353. [DOI] [Google Scholar]
- Yoshimatsu I.; Hirai T.; Yamaki J.-i. Lithium Electrode Morphology during Cycling in Lithium Cells. J. Electrochem. Soc. 1988, 135 (10), 2422. 10.1149/1.2095351. [DOI] [Google Scholar]
- Steiger J.; Kramer D.; Mönig R. Microscopic Observations of the Formation, Growth and Shrinkage of Lithium Moss during Electrodeposition and Dissolution. Electrochim. Acta 2014, 136, 529–536. 10.1016/j.electacta.2014.05.120. [DOI] [Google Scholar]
- Lu D.; Shao Y.; Lozano T.; Bennett W. D.; Graff G. L.; Polzin B.; Zhang J.; Engelhard M. H.; Saenz N. T.; Henderson W. A.; Bhattacharya P.; Liu J.; Xiao J. Failure Mechanism for Fast-Charged Lithium Metal Batteries with Liquid Electrolytes. Adv. Energy Mater. 2015, 5 (3), 1400993. 10.1002/aenm.201400993. [DOI] [Google Scholar]
- Chen K.-H.; Wood K. N.; Kazyak E.; LePage W. S.; Davis A. L.; Sanchez A. J.; Dasgupta N. P. Dead Lithium: Mass Transport Effects on Voltage, Capacity, and Failure of Lithium Metal Anodes. J. Mater. Chem. A 2017, 5 (23), 11671–11681. 10.1039/C7TA00371D. [DOI] [Google Scholar]
- Sanchez A. J.; Kazyak E.; Chen Y.; Chen K. H.; Pattison E. R.; Dasgupta N. P. Plan-View Operando Video Microscopy of Li Metal Anodes: Identifying the Coupled Relationships among Nucleation, Morphology, and Reversibility. ACS Energy Lett. 2020, 5 (3), 994–1004. 10.1021/acsenergylett.0c00215. [DOI] [Google Scholar]
- Qian J.; Henderson W. a.; Xu W.; Bhattacharya P.; Engelhard M.; Borodin O.; Zhang J.-G. High Rate and Stable Cycling of Lithium Metal Anode. Nat. Commun. 2015, 6 (1), 6362. 10.1038/ncomms7362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurng S.; Brown Z. L.; Kim J.; Lucht B. L. Effect of Electrolyte on the Nanostructure of the Solid Electrolyte Interphase (SEI) and Performance of Lithium Metal Anodes. Energy Environ. Sci. 2018, 11 (9), 2600–2608. 10.1039/C8EE00364E. [DOI] [Google Scholar]
- Cho S. J.; Yu D. E.; Pollard T. P.; Moon H.; Jang M.; Borodin O.; Lee S. Y. Nonflammable Lithium Metal Full Cells with Ultra-High Energy Density Based on Coordinated Carbonate Electrolytes. iScience 2020, 23 (2), 100844. 10.1016/j.isci.2020.100844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kushima A.; So K. P.; Su C.; Bai P.; Kuriyama N.; Maebashi T.; Fujiwara Y.; Bazant M. Z.; Li J. Liquid Cell Transmission Electron Microscopy Observation of Lithium Metal Growth and Dissolution: Root Growth, Dead Lithium and Lithium Flotsams. Nano Energy 2017, 32 (11), 271–279. 10.1016/j.nanoen.2016.12.001. [DOI] [Google Scholar]
- Wood K. N.; Kazyak E.; Chadwick A. F.; Chen K. H.; Zhang J. G.; Thornton K.; Dasgupta N. P. Dendrites and Pits: Untangling the Complex Behavior of Lithium Metal Anodes through Operando Video Microscopy. ACS Cent. Sci. 2016, 2 (11), 790–801. 10.1021/acscentsci.6b00260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X.; Zhang M.; Alvarado J.; Wang S.; Sina M.; Lu B.; Bouwer J.; Xu W.; Xiao J.; Zhang J. G.; Liu J.; Meng Y. S. New Insights on the Structure of Electrochemically Deposited Lithium Metal and Its Solid Electrolyte Interphases via Cryogenic TEM. Nano Lett. 2017, 17 (12), 7606–7612. 10.1021/acs.nanolett.7b03606. [DOI] [PubMed] [Google Scholar]
- He M.; Guo R.; Hobold G. M.; Gao H.; Gallant B. M. The Intrinsic Behavior of Lithium Fluoride in Solid Electrolyte Interphases on Lithium. Proc. Natl. Acad. Sci. U. S. A. 2020, 117 (1), 73–79. 10.1073/pnas.1911017116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang C.; Li J.; Zhang M.; Zhang Y.; Yang F.; Lee J. Z.; Lee M.-H.; Alvarado J.; Schroeder M. A.; Yang Y.; Lu B.; Williams N.; Ceja M.; Yang L.; Cai M.; Gu J.; Xu K.; Wang X.; Meng Y. S. Quantifying Inactive Lithium in Lithium Metal Batteries. Nature 2019, 572 (7770), 511–515. 10.1038/s41586-019-1481-z. [DOI] [PubMed] [Google Scholar]
- Pang Q.; Liang X.; Shyamsunder A.; Nazar L. F. An In Vivo Formed Solid Electrolyte Surface Layer Enables Stable Plating of Li Metal. Joule 2017, 1 (4), 871–886. 10.1016/j.joule.2017.11.009. [DOI] [Google Scholar]
- Hsieh Y.-C.; Leißing M.; Nowak S.; Hwang B.-J.; Winter M.; Brunklaus G. Quantification of Dead Lithium via In Situ Nuclear Magnetic Resonance Spectroscopy. Cell Reports Phys. Sci. 2020, 1, 100139. 10.1016/j.xcrp.2020.100139. [DOI] [Google Scholar]
- Bhattacharyya R.; Key B.; Chen H.; Best A. S.; Hollenkamp A. F.; Grey C. P. In Situ NMR Observation of the Formation of Metallic Lithium Microstructures in Lithium Batteries. Nat. Mater. 2010, 9 (6), 504–510. 10.1038/nmat2764. [DOI] [PubMed] [Google Scholar]
- Gunnarsdóttir A. B.; Vema S.; Menkin S.; Marbella L. E.; Grey C. P. Investigating the Effect of a Fluoroethylene Carbonate Additive on Lithium Deposition and the Solid Electrolyte Interphase in Lithium Metal Batteries Using in Situ NMR Spectroscopy. J. Mater. Chem. A 2020, 8 (30), 14975–14992. 10.1039/D0TA05652A. [DOI] [Google Scholar]
- Menkin S.; O’Keefe C. A.; Gunnarsdottir A. B.; Dey S.; Pesci F.; Shen Z.; Aguadero A.; Grey C. P.. Preprint: Towards an Understanding of the SEI Formation and Lithium Preferential Plating on Copper. ChemRxiv 2020, 10.26434/chemrxiv.12839792.v1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin D.; Liu Y.; Li Y. Y.; Li Y. Y.; Pei A.; Xie J.; Huang W.; Cui Y. Fast Galvanic Lithium Corrosion Involving a Kirkendall-Type Mechanism. Nat. Chem. 2019, 11 (4), 382–389. 10.1038/s41557-018-0203-8. [DOI] [PubMed] [Google Scholar]
- Kolesnikov A.; Kolek M.; Dohmann J. F.; Horsthemke F.; Börner M.; Bieker P.; Winter M.; Stan M. C. Galvanic Corrosion of Lithium-Powder-Based Electrodes. Adv. Energy Mater. 2020, 10, 2000017. 10.1002/aenm.202000017. [DOI] [Google Scholar]
- Gileadi E.Physical Electrochemistry: Fundamentals, Techniques and Applications; Wiley-VCH: Weinheim, 2011. [Google Scholar]
- Novák P. CuO Cathode in Lithium Cells-II. Reduction Mechanism of CuO. Electrochim. Acta 1985, 30 (12), 1687–1692. 10.1016/0013-4686(85)87015-8. [DOI] [Google Scholar]
- Klein F.; Pinedo R.; Hering P.; Polity A.; Janek J.; Adelhelm P. Reaction Mechanism and Surface Film Formation of Conversion Materials for Lithium- and Sodium-Ion Batteries: An XPS Case Study on Sputtered Copper Oxide (CuO) Thin Film Model Electrodes. J. Phys. Chem. C 2016, 120 (3), 1400–1414. 10.1021/acs.jpcc.5b10642. [DOI] [Google Scholar]
- Huang W.; Boyle D. T.; Li Y. Y.; Li Y. Y.; Pei A.; Chen H.; Cui Y. Nanostructural and Electrochemical Evolution of the Solid-Electrolyte Interphase on CuO Nanowires Revealed by Cryogenic-Electron Microscopy and Impedance Spectroscopy. ACS Nano 2019, 13 (1), 737–744. 10.1021/acsnano.8b08012. [DOI] [PubMed] [Google Scholar]
- Shu J.; Shui M.; Huang F.; Xu D.; Ren Y.; Hou L.; Cui J.; Xu J. Comparative Study on Surface Behaviors of Copper Current Collector in Electrolyte for Lithium-Ion Batteries. Electrochim. Acta 2011, 56 (8), 3006–3014. 10.1016/j.electacta.2011.01.004. [DOI] [Google Scholar]
- Yu Z.; Cui Y.; Bao Z. Design Principles of Artificial Solid Electrolyte Interphases for Lithium-Metal Anodes. Cell Reports Phys. Sci. 2020, 1 (7), 100119. 10.1016/j.xcrp.2020.100119. [DOI] [Google Scholar]
- Lopez J.; Pei A.; Oh J. Y.; Wang G.-J. J. N.; Cui Y.; Bao Z. Effects of Polymer Coatings on Electrodeposited Lithium Metal. J. Am. Chem. Soc. 2018, 140 (37), 11735–11744. 10.1021/jacs.8b06047. [DOI] [PubMed] [Google Scholar]
- Assegie A. A.; Cheng J.-H.; Kuo L.-M.; Su W.-N.; Hwang B.-J. Polyethylene Oxide Film Coating Enhances Lithium Cycling Efficiency of an Anode-Free Lithium-Metal Battery. Nanoscale 2018, 10 (13), 6125–6138. 10.1039/C7NR09058G. [DOI] [PubMed] [Google Scholar]
- Manuel Stephan A. Review on Gel Polymer Electrolytes for Lithium Batteries. Eur. Polym. J. 2006, 42 (1), 21–42. 10.1016/j.eurpolymj.2005.09.017. [DOI] [Google Scholar]
- Fauteux D.; Prud’Homme J.; Harvey P. E. Electrochemical Stability and Ionic Conductivity of Some Polymer-Lix Based Electrolytes. Solid State Ionics 1988, 28–30, 923–928. 10.1016/0167-2738(88)90305-0. [DOI] [Google Scholar]
- Chiappone A.; Jeremias S.; Bongiovanni R.; Schönhoff M. NMR Study of Photo-Crosslinked Solid Polymer Electrolytes: The Influence of Monofunctional Oligoethers. J. Polym. Sci., Part B: Polym. Phys. 2013, 51 (21), 1571–1580. 10.1002/polb.23371. [DOI] [Google Scholar]
- Bohnke O.; Rousselot C.; Gillet P. A.; Truche C. Gel Electrolyte for Solid-State Electrochromic Cell. J. Electrochem. Soc. 1992, 139 (7), 1862. 10.1149/1.2069512. [DOI] [Google Scholar]
- Appetecchi G. B.; Croce F.; Scrosati B. Kinetics and Stability of the Lithium Electrode in Poly(Methylmethacrylate)-Based Gel Electrolytes. Electrochim. Acta 1995, 40 (8), 991–997. 10.1016/0013-4686(94)00345-2. [DOI] [Google Scholar]
- Obrovac M. N.; Chevrier V. L. Alloy Negative Electrodes for Li-Ion Batteries. Chem. Rev. 2014, 114 (23), 11444–11502. 10.1021/cr500207g. [DOI] [PubMed] [Google Scholar]
- Padhi A. K.; Nanjundaswamy K. S.; Masquelier C.; Okada S.; Goodenough J. B. Effect of Structure on the Fe3+/Fe2+ Redox Couple in Iron Phosphates. J. Electrochem. Soc. 1997, 144 (5), 1609. 10.1149/1.1837649. [DOI] [Google Scholar]
- Brown Z. L.; Jurng S.; Nguyen C. C.; Lucht B. L. Effect of Fluoroethylene Carbonate Electrolytes on the Nanostructure of the Solid Electrolyte Interphase and Performance of Lithium Metal Anodes. ACS Appl. Energy Mater. 2018, 1 (7), 3057–3062. 10.1021/acsaem.8b00705. [DOI] [Google Scholar]
- Zhang X. Q.; Cheng X. B.; Chen X.; Yan C.; Zhang Q. Fluoroethylene Carbonate Additives to Render Uniform Li Deposits in Lithium Metal Batteries. Adv. Funct. Mater. 2017, 27 (10), 1605989. 10.1002/adfm.201605989. [DOI] [Google Scholar]
- Hagen M.; Hanselmann D.; Ahlbrecht K.; Maça R.; Gerber D.; Tübke J. Lithium-Sulfur Cells: The Gap between the State-of-the-Art and the Requirements for High Energy Battery Cells. Adv. Energy Mater. 2015, 5 (16), 1401986. 10.1002/aenm.201401986. [DOI] [Google Scholar]
- Pei A.; Zheng G.; Shi F.; Li Y.; Cui Y. Nanoscale Nucleation and Growth of Electrodeposited Lithium Metal. Nano Lett. 2017, 17 (2), 1132–1139. 10.1021/acs.nanolett.6b04755. [DOI] [PubMed] [Google Scholar]
- Shi F.; Pei A.; Vailionis A.; Xie J.; Liu B.; Zhao J.; Gong Y.; Cui Y. Strong Texturing of Lithium Metal in Batteries. Proc. Natl. Acad. Sci. U. S. A. 2017, 114 (46), 12138–12143. 10.1073/pnas.1708224114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson V.; Kotronia A.; Lacey M.; Edström K.; Johansson P. Highly Concentrated LiTFSI-EC Electrolytes for Lithium Metal Batteries. ACS Appl. Energy Mater. 2020, 3, 200. 10.1021/acsaem.9b01203. [DOI] [Google Scholar]
- Chandrashekar S.; Trease N. M.; Chang H. J.; Du L.-S. S.; Grey C. P.; Jerschow A. 7Li MRI of Li Batteries Reveals Location of Microstructural Lithium. Nat. Mater. 2012, 11 (4), 311–315. 10.1038/nmat3246. [DOI] [PubMed] [Google Scholar]
- Chang H. J.; Trease N. M.; Ilott A. J.; Zeng D.; Du L. S.; Jerschow A.; Grey C. P. Investigating Li Microstructure Formation on Li Anodes for Lithium Batteries by in Situ 6Li/7Li NMR and SEM. J. Phys. Chem. C 2015, 119 (29), 16443–16451. 10.1021/acs.jpcc.5b03396. [DOI] [Google Scholar]
- Chavez K. L.; Hess D. W. A Novel Method of Etching Copper Oxide Using Acetic Acid. J. Electrochem. Soc. 2001, 148 (11), G640. 10.1149/1.1409400. [DOI] [Google Scholar]
- Pecher O.; Carretero-Gonzalez J.; Griffith K. J. J.; Grey C. P. P. Materials’ Methods: NMR in Battery Research. Chem. Mater. 2017, 29 (1), 213–242. 10.1021/acs.chemmater.6b03183. [DOI] [Google Scholar]
- Pecher O.; Bayley P. M.; Liu H.; Liu Z.; Trease N. M.; Grey C. P. Automatic Tuning Matching Cycler (ATMC) in Situ NMR Spectroscopy as a Novel Approach for Real-Time Investigations of Li-and Na-Ion Batteries. J. Magn. Reson. 2016, 265 (2015), 200–209. 10.1016/j.jmr.2016.02.008. [DOI] [PubMed] [Google Scholar]
- Trease N. M.; Zhou L.; Chang H. J.; Zhu B. Y.; Grey C. P. In Situ NMR of Lithium Ion Batteries: Bulk Susceptibility Effects and Practical Considerations. Solid State Nucl. Magn. Reson. 2012, 42, 62–70. 10.1016/j.ssnmr.2012.01.004. [DOI] [PubMed] [Google Scholar]
- Pigliapochi R.; O’Brien L.; Pell A. J.; Gaultois M. W.; Janssen Y.; Khalifah P. G.; Grey C. P. When Do Anisotropic Magnetic Susceptibilities Lead to Large NMR Shifts? Exploring Particle Shape Effects in the Battery Electrode Material LiFePO4. J. Am. Chem. Soc. 2019, 141 (33), 13089–13100. 10.1021/jacs.9b04674. [DOI] [PubMed] [Google Scholar]
- Zhou L.; Leskes M.; Ilott A. J.; Trease N. M.; Grey C. P. Paramagnetic Electrodes and Bulk Magnetic Susceptibility Effects in the in Situ NMR Studies of Batteries: Application to Li1.08Mn1.92O4spinels. J. Magn. Reson. 2013, 234, 44–57. 10.1016/j.jmr.2013.05.011. [DOI] [PubMed] [Google Scholar]
- Rowland T. J. Nuclear Magnetic Resonance in Metals. Prog. Mater. Sci. 1961, 9 (1), 3–91. 10.1016/0079-6425(61)90008-1. [DOI] [Google Scholar]
- Ilott A. J.; Chandrashekar S.; Klöckner A.; Chang H. J.; Trease N. M.; Grey C. P.; Greengard L.; Jerschow A. Visualizing Skin Effects in Conductors with MRI: 7Li MRI Experiments and Calculations. J. Magn. Reson. 2014, 245, 143–149. 10.1016/j.jmr.2014.06.013. [DOI] [PubMed] [Google Scholar]
- Mohammadi M.; Jerschow A. In Situ and Operando Magnetic Resonance Imaging of Electrochemical Cells: A Perspective. J. Magn. Reson. 2019, 308, 106600. 10.1016/j.jmr.2019.106600. [DOI] [PubMed] [Google Scholar]
- Kadyk T.; Eikerling M. Magnetic Susceptibility as a Direct Measure of Oxidation State in LiFePO4 Batteries and Cyclic Water Gas Shift Reactors. Phys. Chem. Chem. Phys. 2015, 17 (30), 19834–19843. 10.1039/C5CP02977E. [DOI] [PubMed] [Google Scholar]
- Gutowsky H. S.; McGarvey B. R. Nuclear Magnetic Resonance in Metals. I. Broadening of Absorption Lines by Spin-Lattice Interactions. J. Chem. Phys. 1952, 20 (9), 1472–1477. 10.1063/1.1700782. [DOI] [Google Scholar]
- Li Y.; Huang W.; Li Y.; Pei A.; Boyle D. T.; Cui Y. Correlating Structure and Function of Battery Interphases at Atomic Resolution Using Cryoelectron Microscopy. Joule 2018, 2 (10), 2167–2177. 10.1016/j.joule.2018.08.004. [DOI] [Google Scholar]
- Lin D.; Liu Y.; Liang Z.; Lee H. W.; Sun J.; Wang H.; Yan K.; Xie J.; Cui Y. Layered Reduced Graphene Oxide with Nanoscale Interlayer Gaps as a Stable Host for Lithium Metal Anodes. Nat. Nanotechnol. 2016, 11 (7), 626–632. 10.1038/nnano.2016.32. [DOI] [PubMed] [Google Scholar]
- Kozen A. C.; Lin C. F.; Pearse A. J.; Schroeder M. A.; Han X.; Hu L.; Lee S. B.; Rubloff G. W.; Noked M. Next-Generation Lithium Metal Anode Engineering via Atomic Layer Deposition. ACS Nano 2015, 9 (6), 5884–5892. 10.1021/acsnano.5b02166. [DOI] [PubMed] [Google Scholar]
- Ma L.; Kim M. S.; Archer L. A. Stable Artificial Solid Electrolyte Interphases for Lithium Batteries. Chem. Mater. 2017, 29 (10), 4181–4189. 10.1021/acs.chemmater.6b03687. [DOI] [Google Scholar]
- Liang X.; Pang Q.; Kochetkov I. R.; Sempere M. S.; Huang H.; Sun X.; Nazar L. F. A Facile Surface Chemistry Route to a Stabilized Lithium Metal Anode. Nat. Energy 2017, 2, 17119. 10.1038/nenergy.2017.119. [DOI] [Google Scholar]
- Choudhury S.; Tu Z.; Stalin S.; Vu D.; Fawole K.; Gunceler D.; Sundararaman R.; Archer L. A. Electroless Formation of Hybrid Lithium Anodes for Fast Interfacial Ion Transport. Angew. Chem., Int. Ed. 2017, 56 (42), 13070–13077. 10.1002/anie.201707754. [DOI] [PubMed] [Google Scholar]
- Tu Z.; Choudhury S.; Zachman M. J.; Wei S.; Zhang K.; Kourkoutis L. F.; Archer L. A. Fast Ion Transport at Solid-Solid Interfaces in Hybrid Battery Anodes. Nat. Energy 2018, 3 (4), 310–316. 10.1038/s41560-018-0096-1. [DOI] [Google Scholar]
- Besenhard J. O.; Gürtler J.; Komenda P.; Paxinos A. Corrosion Protection of Secondary Lithium Electrodes in Organic Electrolytes. J. Power Sources 1987, 20 (3–4), 253–258. 10.1016/0378-7753(87)80120-9. [DOI] [Google Scholar]
- Amanchukwu C. V.; Kong X.; Qin J.; Cui Y.; Bao Z. Nonpolar Alkanes Modify Lithium-Ion Solvation for Improved Lithium Deposition and Stripping. Adv. Energy Mater. 2019, 9 (41), 1902116. 10.1002/aenm.201902116. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.