

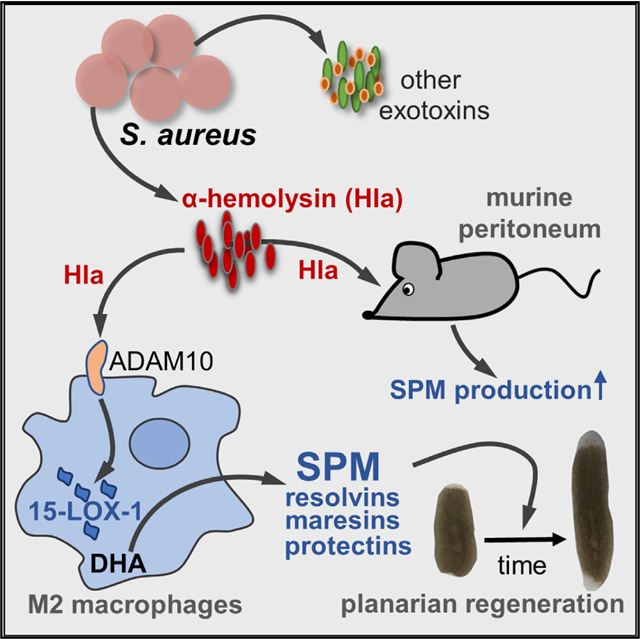
SUMMARY
Underlying mechanisms of how infectious inflammation is resolved by the host are incompletely understood. One hallmark of inflammation resolution is the activation of specialized pro-resolving mediators (SPMs) that enhance bacterial clearance and promote tissue repair. Here, we reveal α-hemolysin (Hla) from Staphylococcus aureus as a potent elicitor of SPM biosynthesis in human M2-like macrophages and in the mouse peritoneum through selective activation of host 15-lipoxygenase-1 (15-LOX-1). S. aureus-induced SPM formation in M2 is abolished upon Hla depletion or 15-LOX-1 knockdown. Isolated Hla elicits SPM formation in M2 that is reverted by inhibition of the Hla receptor ADAM10. Lipid mediators derived from Hla-treated M2 accelerate planarian tissue regeneration. Hla but not zymosan provokes substantial SPM formation in the mouse peritoneum, devoid of leukocyte infiltration and pro-inflammatory cytokine secretion. Besides harming the host, Hla may also exert beneficial functions by stimulating SPM production to promote the resolution of infectious inflammation.
In Brief
Jordan et al. reveal that α-hemolysin from Staphylococcus aureus stimulates specialized pro-resolving mediator (SPM) formation through activation of 15-lipoxygenase-1 in human macrophages involving ADAM10. The host may exploit α-hemolysin as an SPM inducer to better cope with S. aureus infections and to promote inflammation resolution and tissue regeneration.
Graphical Abstract
INTRODUCTION
Upon bacterial infection, the host reacts with an inflammatory response to eliminate the invading bacteria, followed by active defense mechanisms to limit and resolve inflammation and facilitate repair of damaged tissue (Chiang et al., 2012; Serhan, 2014). The inflammatory response is initiated and maintained by host-derived pro-inflammatory lipid mediators (LMs) that encompass prostaglandins (PGs) and leukotrienes (LTs), produced from arachidonic acid (AA, C20:4, ω−6) by cyclooxygenases (COXs) and 5-lipoxygenase (LOX), respectively (Funk, 2001; Figure 1A). In contrast, the specialized pro-resolving mediators (SPMs) are distinct LMs, formed from ω−3 fatty acids such as eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA) by different LOX pathways (Figure 1A), which actively resolve inflammation enabling the return to homeostasis (Serhan, 2014; Serhan and Levy, 2018). These SPMs are grouped into lipoxin (LX), resolvin (Rv), protectin (PD), and maresin (MaR) families (Serhan et al., 2015), which are endogenous relievers of infectious inflammation that enhance containment and lower antibiotic requirements for bacterial clearance (Chiang et al., 2012). Along these lines, unresolved inflammation and persistent infection in sepsis might be due to failure in the biosynthesis of appropriate amounts of endogenous SPMs (Dalli et al., 2015a; Spite et al., 2009). Given the current antibiotic crisis, there is a need to better understand the molecular mechanisms of how bacterial infections contribute to active resolution of inflammation through the induction and biosynthesis of SPMs (Chiang et al., 2012).
Figure 1. S. aureus Evokes Differential LM Pathways in Human M1 and M2 Macrophages.
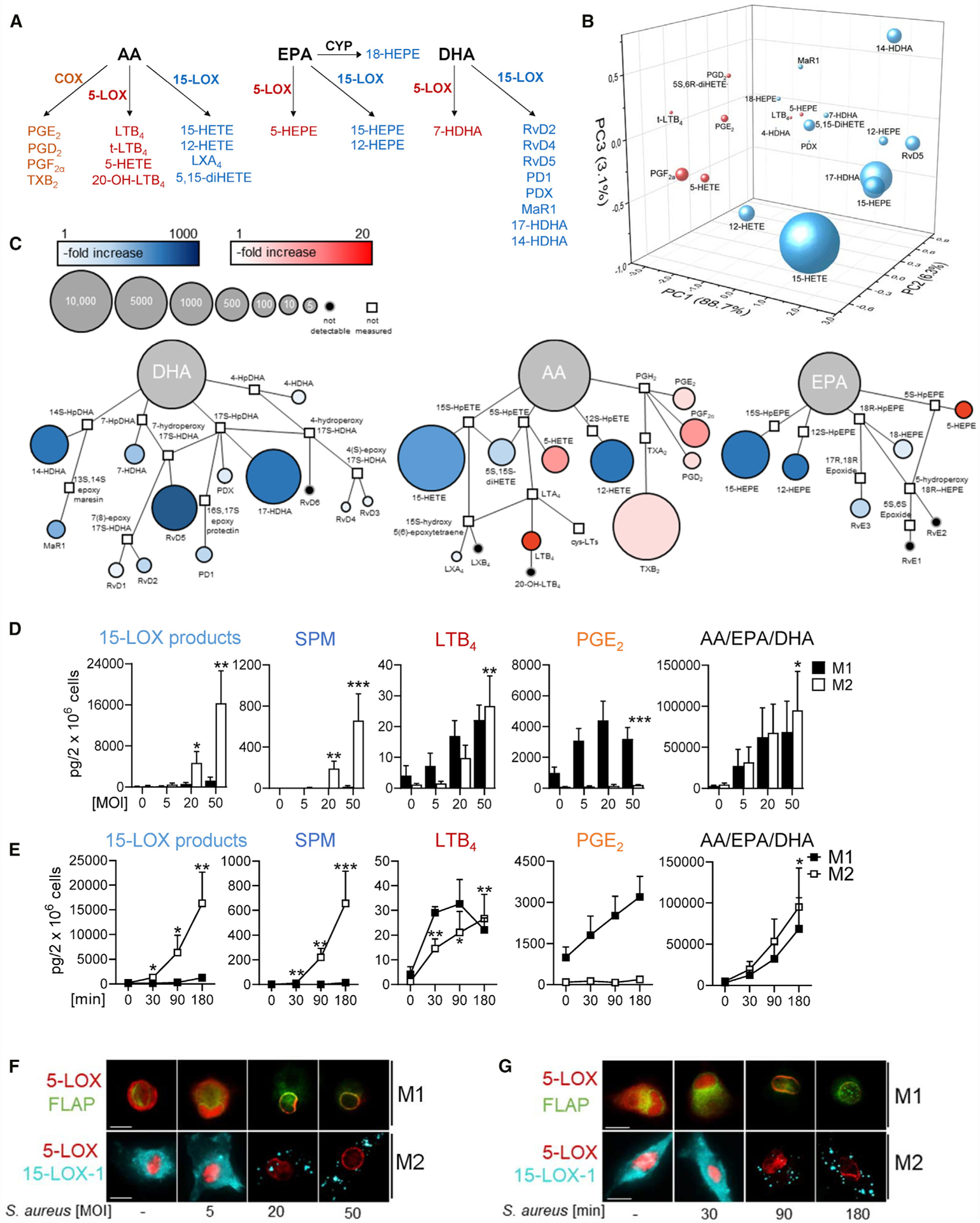
(A) Schematic representation of LM-biosynthetic pathways involving COX, 5-LOX, or 15-LOX-1.
(B and C) Three-dimensional score plot (B) and quantitative pathway network analysis (C) of the LM profile of M2 after exposure to S. aureus for 180 min compared to untreated control. Node size represents the mean values in pg, and intensity of color denotes the fold increase for each LM; n = 4.
(D and E) MOI-dependent (D) and time-dependent activation (E) of M1 (black) and M2 (white) with S. aureus for LM formation. M1 and M2 (2 × 106 cells/mL PBS plus 1 mM CaCl2) were incubated for 180 min with S. aureus at 37°C at the indicated MOI (ratio of S. aureus: macrophages) (D) or at MOI = 50 for indicated times (E); mono-hydroxylated 15-LOX products (17-HDHA, 15-HEPE, and 15-HETE), SPM (RvD5, MaR1, and PD1), LTB4, PGE2, and AA/EPA/DHA are shown as pg/2 × 106 cells. Results are means + SEM, n = 4; *p < 0.05; **p < 0.01; ***p < 0.001 versus vehicle. Data were log-transformed for statistical analysis; paired one-way ANOVA with Dunnett’s multiple comparisons test.
(F and G) MOI-dependent and time-dependent subcellular redistribution of 5-LOX and 15-LOX-1 in M1 and M2. Cells were treated for 180 min with S. aureus at the indicated MOI (F) or with MOI = 50 for the indicated times (G). Then, cells were fixed, permeabilized, and incubated with antibodies against 5-LOX (red), 15-LOX-1 (cyan-blue), and FLAP (green); scale bars, 10 μm. Results shown for one single cell are representative for approximately 100 individual cells analyzed in n = 4 independent experiments with separate donors, each.
See also Figure S1.
Staphylococcus aureus infections are a major cause for sepsis with a chronic disease pattern and high relapse rate despite antimicrobial treatment (Goldmann and Medina, 2018; Knox et al., 2015; Tong et al., 2015). We recently showed that pathogenic Escherichia coli stimulate COX and LOX pathways in human macrophages to produce differential LM profiles in a phenotype-dependent manner (Werz et al., 2018). Thus, inflammation-promoting LTs and PGs are predominantly generated by pro-inflammatory M1-like macrophages upon exposure to pathogenic bacteria, whereas SPM biosynthesis involving 15-LOX-1 is characteristic for anti-inflammatory M2-like macrophages (Werner et al., 2019; Werz et al., 2018). Regardless of the macrophage phenotype and LOX isoform, release of substrate fatty acids (i.e., AA, DHA, and EPA), elevation of intracellular Ca2+ levels ([Ca2+]i), and subcellular redistribution of the LOXs from soluble to membranous compartments are prerequisites for LM production induced by bacteria (Werz et al., 2018) as well as by other stimuli (Gijón et al., 2000; Ivanov et al., 2015; Rådmark et al., 2015). However, the underlying factors and mechanisms that originate from pathogenic bacteria to induce SPM generation in macrophages are unknown.
The pathogenicity of S. aureus relies on several virulence factors including secreted pore-forming toxins (PFTs) that inhibit neutrophil chemotaxis or lyse neutrophils, block complement activation, and neutralize antimicrobial defensive peptides (Blake et al., 2018; Foster, 2005; Otto, 2014b; von Hoven et al., 2019). PFTs play key roles in damaging host cell membranes, and they induce multiple cellular host responses including macrophage recruitment (DuMont and Torres, 2014; Foster, 2005). Here, we show that PFTs secreted from S. aureus evoke LM biosynthesis in human M2 macrophages as well as in murine peritoneum in vivo. We reveal α-hemolysin (Hla), a small β-PFT (von Hoven et al., 2019), as a potent and selective activator of 15-LOX-1 in M2 macrophages leading to substantial formation of SPM. Our results suggest that Hla may affect the combat between S. aureus and the host in different ways: despite direct deleterious effects of Hla on host cells by cytotoxic lysis and pro-inflammatory mediator and cytokine secretion, the host may also take advantage of this PFT as an elicitor of SPM formation to promote inflammation resolution.
RESULTS
S. aureus Evokes Differential LM Profiles in Human M1 and M2 Macrophages
Targeted LM metabololipidomics using ultra-performance liquid chromatography-tandem mass spectrometry (UPLC-MS-MS) revealed differential LM profiles in human M1 and M2 macrophages upon a 3-h exposure to S. aureus (LS1 strain) at a multiplicity of infection (MOI) = 50. The pro-inflammatory COX-derived PGs were mainly produced by M1, whereas the pro-resolving 15-LOX-derived SPM and their precursors were formed by M2 (Figures 1A, 1B, and S1A). Thus, M1 exposed to S. aureus produced only <20 pg SPMs (i.e., RvD5, PD1 and MaR1), whereas abundant PGs (>3,500 pg) were formed. In contrast, M2 biosynthesized substantial amounts (>700 pg) of SPMs (i.e., RvD2, RvD5, PD1, PDX, and MaR1), their precursors and pathway markers (i.e., 7-HDHA, 14-HDHA, 17-HDHA, and 18-HEPE), as well as other 15-LOX-derived LMs (i.e., 15-HEPE, 15-HETE, and 5,15-diHETE), but relatively minor levels of PGs. Notably, both M1 and M2 released abundant LM substrates (i.e., AA, DHA, and EPA) without marked differences between phenotypes (Figure S1A). Quantitative pathway network analysis in M2 supports the activation of selective LM pathways by S. aureus, in which mainly SPM and 15-LOX-products are formed (Figure 1C).
We studied if activation of the different LM pathways requires distinct thresholds of bacterial challenge and if their activation follows distinct kinetics. The amounts of LM substrates increased upon exposure to S. aureus with incremental MOI in both M1 and M2 (Figure 1D). LTB4 was elevated in M1 and M2 starting at MOI = 20; albeit, relatively low levels (<30 pg) were formed even at the highest MOI. PGE2 peaked already at low MOI but only in M1 (Figure 1D). In contrast, induction of 15-LOX-1-mediated LM production, especially the SPM in M2, required higher MOI but was much more striking (approximately 500-fold increase) than that for 5-LOX or COX products (Figure 1D). LTB4 was rapidly generated within 30 min, but formation of 15-LOX-derived products and SPM was delayed starting at 90 min (Figure 1E). MOI- and time-dependent LOX-mediated LM formation correlated with the subcellular redistribution of the respective LOXs, i.e., translocation of 5-LOX to the nuclear membrane and 15-LOX-1 accumulation at yet unknown cellular (membranous) structures within the cytosol (Figures 1F and 1G), prerequisites for cellular LOX activity (Werz et al., 2018). The requirement of different MOI thresholds for the generation of COX-/5-LOX-derived LM versus 15-LOX-derived SPM with various kinetics suggests that distinct stimuli/mechanisms activate these LM pathways.
S. aureus-Released Factors Are Key to Activate the 15-LOX-1 Pathway
S. aureus possess high capacities to secrete PFTs, and we hypothesized that S. aureus releases such toxins that induce LM biosynthesis in macrophages. The addition of S. aureus-conditioned medium (SACM) from overnight cultures to M2 induced LM formation within 3 h, with a comparable LM profile as that obtained from M2 exposed to intact S. aureus (Figure 2A). Induction of LM by the SACM was concentration dependent (Figure S2A), and the temporal formation of LM was similar to the time course obtained with intact bacteria. Thus, PG and LT formation was evident 30 min upon exposure, whereas SPMs and their precursors were first produced after 90 min (Figure S2B).
Figure 2. Role of PFTs from S. aureus on the M2-Specific LM Profile.
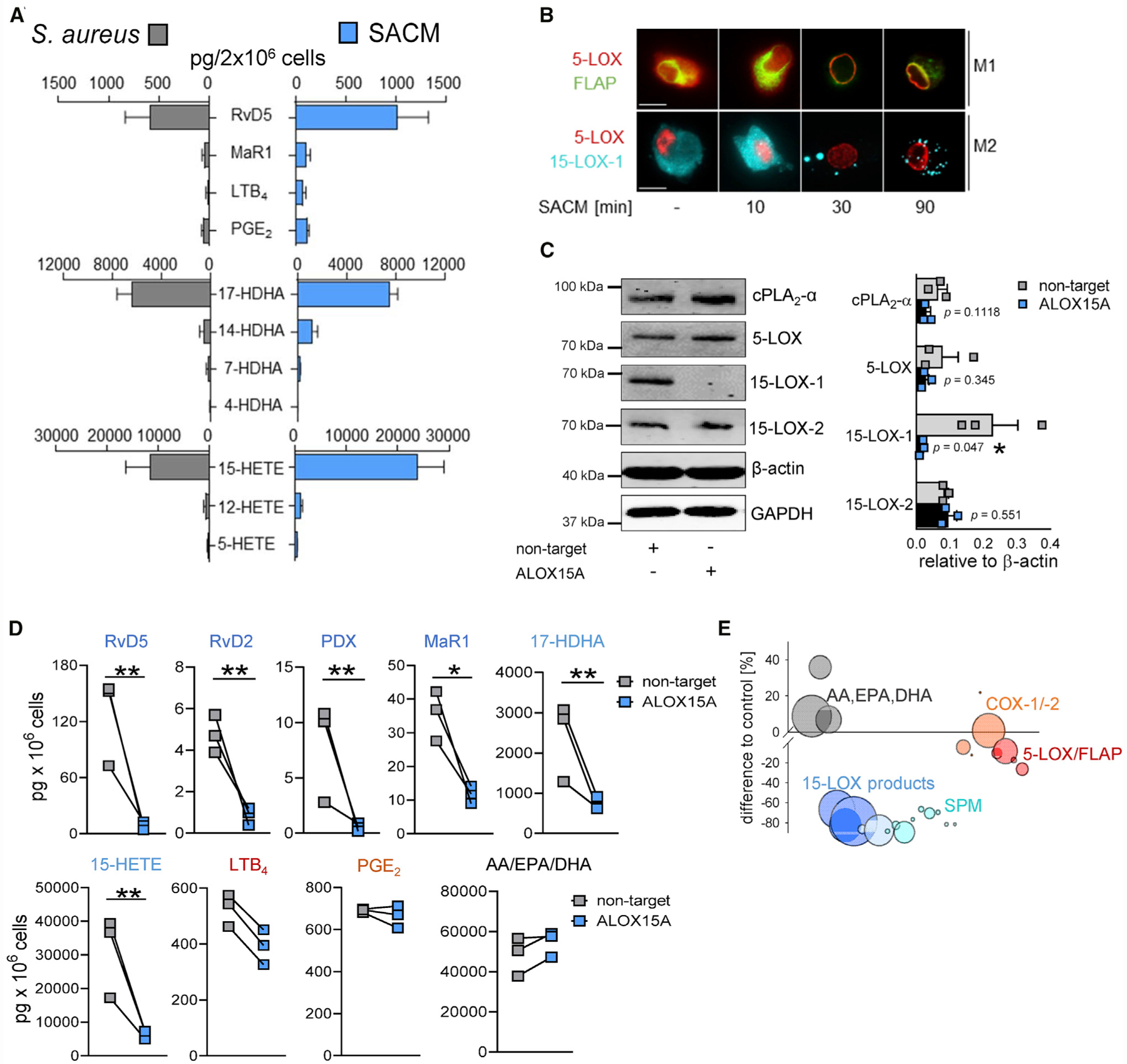
(A) Bar chart of selected LMs after exposure to intact S. aureus (MOI = 50) or S. aureus-conditioned medium (SACM; 0.5%) at 37°C for 180 min; formed LMs are shown as pg/2 × 106 cells. Results are means + SEM, n = 4 for intact S. aureus; n = 3 for SACM.
(B) Subcellular redistribution of 5-LOX and FLAP in M1 and of 5-LOX and 15-LOX-1 in M2 exposed to SACM (0.5%) for the indicated times at 37°C. Cells were fixed, permeabilized, and incubated with antibodies against 5-LOX (red), FLAP (green), and 15-LOX-1 (cyan blue); scale bars, 10 μm. Results shown for one single cell are representative for approximately 100 individual cells analyzed in n = 3 independent experiments with separate donors, each.
(C) Protein expression of non-target-siRNA- and ALOX15A-siRNA-treated M2 after 48 h of polarization by western blot, normalized to β-actin, and densitometric analysis, n = 3. Student’s unpaired t test; *p < 0.05.
(D) LM profile of non-target-siRNA- and ALOX15A-siRNA-treated M2 stimulated with 0.5% SACM for 180 min at 37°C; formed LMs are shown as pg/106 cells; results are means + SEM, n = 3 separate donors. Data were log-transformed for statistical analysis; *p < 0.05; **p < 0.01, ***p < 0.001 non-target versus ALOX15A; unpaired Student’s t test.
(E) Bubble plot of the LM profile shown in (D) (given as % of control) from ALOX15A-siRNA-treated M2; n = 3. See also Figure S2.
An analysis of LOX translocation showed that after 30 min of exposure of M1 and M2 to SACM, nucleosolic 5-LOX and cytosolic 15-LOX-1 redistributed to membranous compartments (Figure 2B), as observed with intact S. aureus. To further study the requirement of 15-LOX-1 for SACM-provoked SPM formation, we silenced the ALOX15A gene during M2 polarization using small interfering RNA (siRNA). Western blot analysis confirmed depletion of the 15-LOX-1 protein, whereas other LM-biosynthetic enzymes (i.e., 15-LOX-2, 5-LOX, and cPLA2-α) were not affected (Figures 2C and S2C). When 15-LOX-1-depleted M2s were exposed to SACM, formation of SPM, their precursors, and other 15-LOX-1-derived products were substantially impaired, whereas the levels of COX-derived PGE2 and 5-LOX-derived LTB4 and 5-HETE, as well as AA, EPA, and DHA, were largely unaffected (Figures 2D and 2E), as expected.
Depletion of α-Hemolysin or Exotoxins from S. aureus Impairs the Induction of LM in M2
S. aureus produces three major classes of exotoxins, i.e., Hla, phenol-soluble modulins (PSMs), and in some strains Panton-Valentine leukocidins (PVLs), for which the Agr/SarA system is a common regulator for their expression (Berube et al., 2014; Bronner et al., 2004; Cheung et al., 2011). We took advantage of S. aureus LS1 mutant strains deficient in Hla (Δhla) or in multiple exotoxins (Δagr/sarA) to investigate if these exotoxins are needed for provoking LM biosynthesis. The growth rates of the wild-type (WT) LS1, Δhla mutant, and Δagr/sarA mutant strains were comparable, and the generation time was only slightly prolonged for the Δagr/sarA mutant (Figures S3A and S3B). The PVLs can be excluded, as they are absent in the LS1 strain (Strobel et al., 2016). The deficiency of Hla in the LS1 Δhla mutant and Δagr/sarA mutant versus the WT LS1 strain was confirmed by PCR (Figure 3A) and western blot (Figures 3B and S3C) analysis. The SACM from the Δagr/sarA mutant failed to induce LM formation in M2, except very minor PGE2 production and release of fatty acid substrates (Figure 3C). When M2s were exposed to SACM from the Δhla mutant, SPMs and related 15-LOX-1 products were hardly formed, whereas 5-LOX and COX products, as well as DHA, EPA, and AA, were still released, albeit at lower levels than the WT strain (Figure 3C). Hla induces a rapid and massive increase in [Ca2+]i in host cells due to the formation of pores in the plasma membrane and/or activation of Ca2+-channels (Bouillot et al., 2018). In line with the impaired capacity to generate LM in M2, the SACM from the Δhla strain and the Δagr/sarA strain were less (Δhla) or not (Δagr/sarA) capable of elevating [Ca2+]i (Figure 3D), a prerequisite for 5-LOX/15-LOX-1 activation in M2 (Werz et al., 2018), and capable of inducing 5-LOX and 15-LOX-1 subcellular redistribution (Figure 3E). Moreover, depletion of Hla from SACM by using an Hla-neutralizing antibody markedly impaired the capacity to elicit formation of SPM and other 15-LOX-1 products, whereas again the release of LTB4, 5-HETE, and PGE2, as well as of DHA, EPA, and AA, was hardly impaired (Figures 3F and S3D). Of note, pro-inflammatory cytokines like tumor necrosis factor alpha (TNF-a) and interleukin-6 (IL-6) were equally released upon exposure to SACM irrespective of the presence of Hla (Δhla mutant or Hla-neutralizing antibody) or multiple exotoxins (Δagr/sarA) in M1, except for IL-1b that was not induced by the Δagr/sarA mutant (Figure S3E). Together, Hla causes activation of the 15-LOX-1 pathway and thus SPM production in M2 but is less efficient for inducing the formation of pro-inflammatory 5-LOX/COX products and cytokines.
Figure 3. Hla Deletion in S. aureus Impedes Activation of the 15-LOX Pathway.
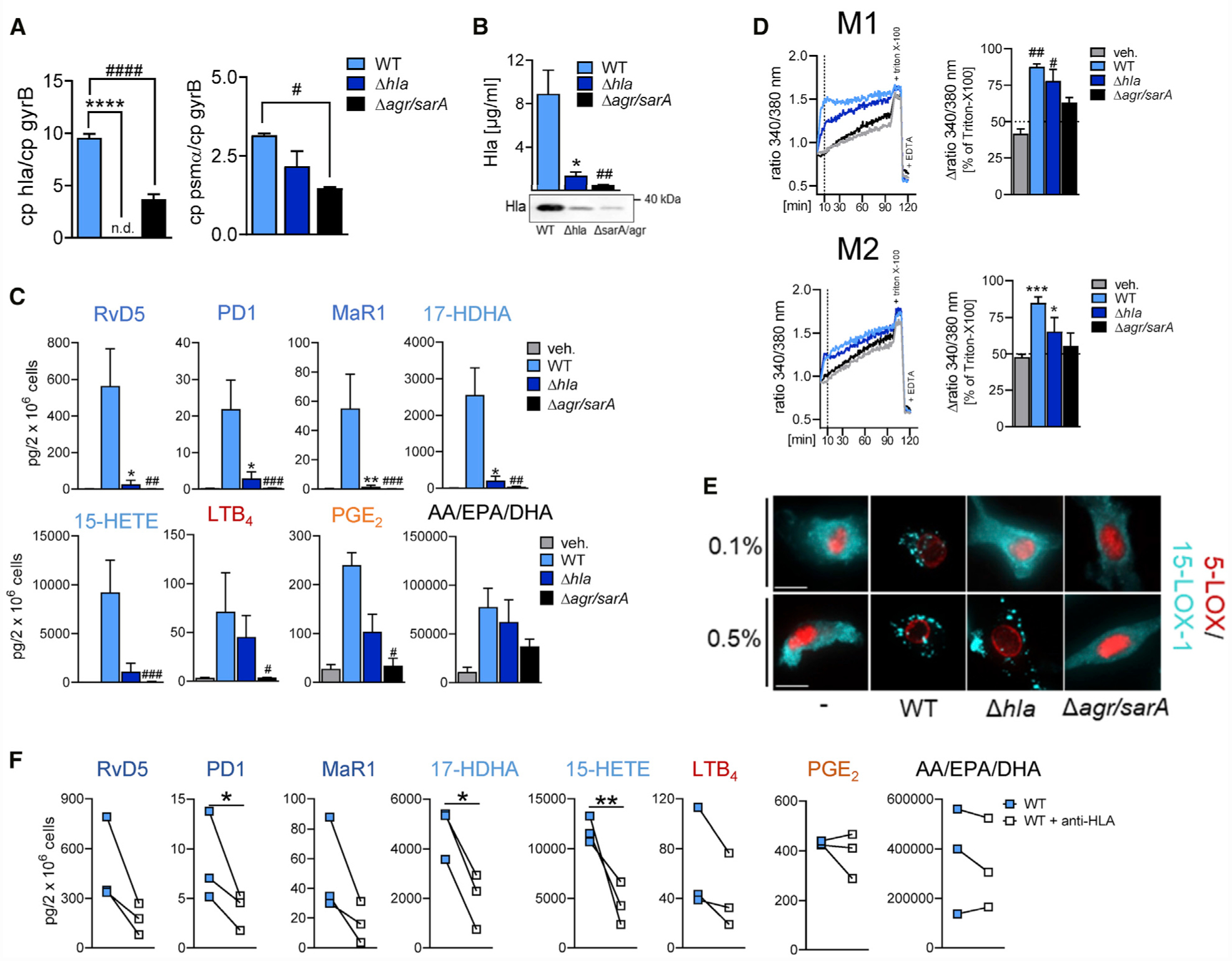
(A) Quantitative mRNA analysis of Hla and PSMα for WT, Δhla, or Δagr/sarA LS1 S. aureus strains measured by RT-PCR, normalized to the gyrB gene.
(B) Expression of Hla on the protein level from overnight cultures of the respective LS1 S. aureus strains measured by western blot and densitometric analysis thereof, n = 3; one-way ANOVA with Dunnett’s multiple comparisons test.
(C) M2s (2 × 106 cells) were incubated with 0.5% SACM of WT, Δhla, or Δagr/sarA LS1 S. aureus for 90 min at 37°C; formed LMs are shown as pg/2 × 106 cells. Results are means + SEM, n = 3; data were log-transformed for statistical analysis; unpaired one-way ANOVA with Dunnett’s multiple comparisons test. *p < 0.05, **p < 0.01 WT versus Δhla; #p < 0.05, ##p < 0.01, ###p < 0.001 WT versus Δagr/sarA.
(D) Analysis of [Ca2+]i in Fura-2/AM-loaded M1 and M2, incubated with 0.5% SACM of WT, Δhla, or Δagr/sarA LS1 S. aureus for up to 120 min at 37°C. Data are shown as ratio of absorbance at 340/380 nm reflecting [Ca2+]i given as % of maximum [Ca2+]i determined by cell lysis with Triton X-100; n = 3.
(E) Subcellular redistribution of 5-LOX and 15-LOX-1 in M2 stimulated with 0.1 and 0.5% SACM of WT, Δhla, or Δagr/sarA for 90 min. Cells were fixed, permeabilized, and incubated with antibodies against 5-LOX (red) and 15-LOX-1 (cyan-blue); scale bars, 10 μm. Results shown for one single cell are representative for approximately 100 individual cells analyzed in n = 3 independent experiments (separate donors), each.
(F) LM formation in M2s preincubated with 2.5 μg/mL anti-Hla antibody for 15 min and stimulated with 0.5% SACM of LS1 S. aureus for another 90 min; formed LMs are shown as pg/2 × 106 cells. Results are means + SEM, n = 3; data were log-transformed for statistical analysis; *p < 0.05, **p < 0.01 WT versus WT + anti-Hla; unpaired Student’s t test. See also Figure S3.
Hla Elicits SPM Formation and 15-LOX-1 Activation in M2
We tested if isolated Hla (1 μg/mL) could mimic the effects of SACM or of intact S. aureus to induce LM biosynthesis. In M2, the formation of SPM and other 15-LOX-derived products strongly increased upon 3-h Hla treatment (Figure 4A). In contrast, LTB4 and PGE2 formation was less elevated and the response to Hla was more prominent in M2 versus M1 (Figure 4A). Hla (0.5 or 1 μg/mL) caused subcellular redistribution of 5-LOX in M1 and 5-LOX and 15-LOX-1 in M2 (Figure S4A), and it also elevated the [Ca2+]i, although in a delayed manner compared to the Ca2+-ionophore ionomycin used for direct reference (Figure 4C). Detrimental effects of Hla (1 μg/mL) on M1 and M2 viability (3 h) were moderate (Figure S4B), similar as the cytotoxic effects of SACM (0.5%) or intact S. aureus (MOI = 50) that hardly differed, independent of the phenotype (Figure S4C).
Figure 4. Hla Elicits SPM Formation and 15-LOX-1 Activation in M2.
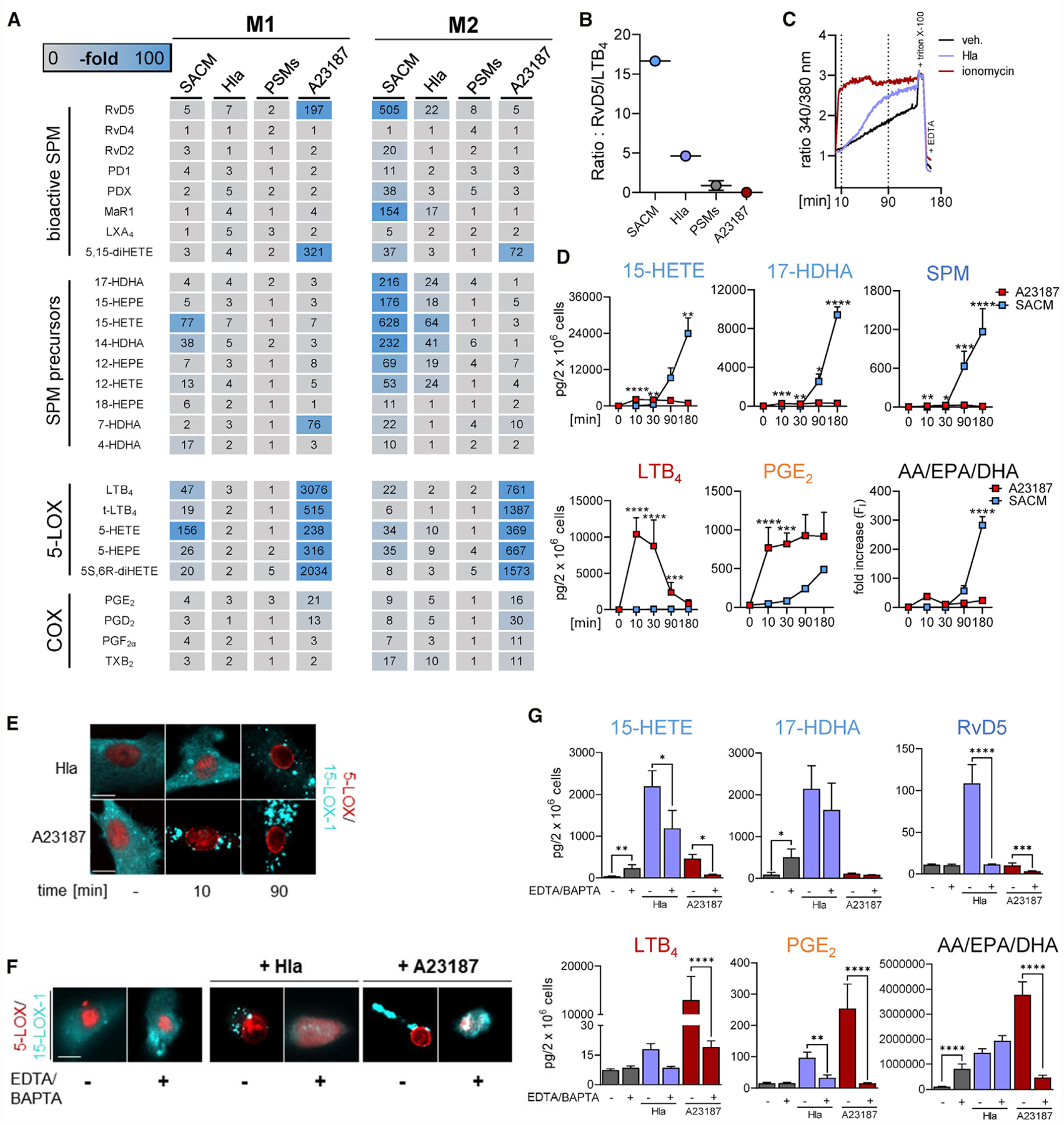
(A) LM profiles of M2s stimulated with SACM (0.5%), Hla (1 μg/mL), or PSMs (sum of 30 μg/mL PSMα3 and 10 μg/mL δ-toxin incubations) for 180 min or with A23187 (0.5 μM) for 10 min. Data are presented as fold increase to untreated control and shown as a heatmap; n = 3.
(B) Scatter dot plot of the ratio of the total amounts of RvD5 to LTB4 related to (A).
(C) Analysis of [Ca2+]i from Fura-2AM-loaded M2 treated with ionomycin (2 μM) or Hla (1 μg/mL) for up to 180 min.
(D) Time course of LM formation in M2s induced by A23187 or SACM. M2s (2 × 106 cells/mL PBS plus 1 mM CaCl2) were incubated with 0.5 μM A23187 or 0.5% SACM of LS1 S. aureus at 37°C for the indicated times. The amounts of formed 15-HETE, 17-HDHA, SPM (RvD5, MaR1 and PD1), LTB4, and PGE2 are given as pg/2 × 106 cells; AA, EPA, and DHA are presented as fold increase of activated versus resting cells. Results are means + SEM, n = 3. Data were log-transformed for statistical analysis; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 SACM versus A23187 stimulation for each indicated time point; two-way ANOVA with Sidak’s multiple comparisons test.
(E and F) Subcellular redistribution of 5-LOX and 15-LOX-1 in M2 (in PBS plus 1 mM CaCl2) stimulated with 1 μg/mL Hla or 0.5 μM A23187 for 10 and 90 min (E) and in M2s incubated in PBS containing 0.5 mM EDTA and 20 μM BAPTA/AM and stimulated with 1 μg/mL Hla for 90 min or with 0.5 μM A23187 for 10 min (F). Then, cells were fixed, permeabilized, and incubated with antibodies against 5-LOX (red) and 15-LOX-1 (cyan-blue); scale bars, 10 μm. Results shown for one single cell are representative for approximately 100 individual cells analyzed in n = 3 independent experiments (separate donors), each.
(G) Effects of Ca2+ depletion on LM formation. M2s were incubated in PBS containing 0.5 mM EDTA and 20 μM BAPTA/AM and stimulated with 1 μg/mL Hla for 90 min and with 0.5 μM A23187 for 10 min. LMs are shown as pg/2 × 106 cells. Results are means + SEM, n = 3–4; data were log-transformed for statistical analysis; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 versus Ca2+ depletion, one-way ANOVA with Tukey’s multiple comparisons test.
See also Figure S4.
Next, we tested the PFTs PSMα3 and δ-toxin (Cheung et al., 2014; Otto, 2014a) as reasonable elicitors of LM formation using also the Ca2+-ionophore A23187 as a well-known stimulus of LM formation in various leukocytes due to robust elevation of [Ca2+]I (Rådmark et al., 2015). In contrast to SACM and Hla, the PSMα3 and δ-toxin failed to evoke significant LM formation regardless of the macrophage phenotype (Figures 4A and S4D). Also, PSMα3 and δ-toxin failed to increase [Ca2+]i in these cells (Figure S4D).
The side-by-side comparison of Hla and A23187 revealed quite different and even opposing LM profiles: in M2, in which Hla strikingly induced the formation of 15-LOX-1-derived LM, A23187 essentially failed in this respect (Figure 4A). However, A23187 caused strong formation of 5-LOX- and COX-derived LM in M2, which was less pronounced for the SACM and was hardly achieved with Hla (Figure 4A). Also in M1, A23187 potently induced formation of 5-LOX products and, to a lesser extent, also PG, for which Hla was almost inactive. SPM and other 15-LOX-derived products were sparsely generated in M1 regardless of the stimulus (Figure 4A), seemingly due to the lack of 15-LOX-1 in this phenotype (Werner et al., 2019; Werz et al., 2018). A comparison of the ratios of generated RvD5 as a representative SPM versus the pro-inflammatory LTB4 in M2 indicated that Hla predominantly induces SPM, whereas with A23187, the formation of LTB4 dominates (Figure 4B).
Elevation of [Ca2+]i is considered crucial for fatty acid substrate release and LM formation upon cell activation (Gijón et al., 2000; Rådmark et al., 2015) and was connected to 5-LOX and 15-LOX-1 activation in neutrophils and macrophages upon exposure to bacteria (Romp et al., 2019; Werz et al., 2018). The increase of [Ca2+]i in M2 (or M1) was immediately apparent upon addition of A23187 (<1 min), as expected, but was markedly delayed (>30 min) upon Hla (Figures 4C and S4E). Accordingly, a maximal release of fatty acid substrates and concomitant formation of LTB4 and PGE2 was rapidly (10 min) achieved with A23187 (Figures 4D and S4F). In contrast, substrate release and generation of SPM and 15-LOX products was retarded upon SACM exposure and occurred solely after 30–90 min, with no or only marginal formation of LTB4 and PGE2 (Figures 4D and S4F). Along these lines, 15-LOX-1 rapidly redistributed within the cytosol of M2 after exposure to A23187 but was delayed after Hla treatment (Figure 4E). Removal of intra- and extracellular Ca2+ by using BAPTA/AM and EDTA abolished Hla- or A23187-induced 5-LOX and 15-LOX-1 translocation (Figure 4F) and abrogated LM formation in M2 (Figure 4G). Note that Hla-evoked fatty acid substrate release was not blocked by Ca2+ chelation. Together, these results suggest that Hla slowly induces Ca2+ mobilization, substrate release, and LOX translocation resulting in prominent 15-LOX-1 activation and SPM biosynthesis, whereas A23187 rapidly induces these processes leading to abundant LT and PG biosynthesis essentially devoid of detectable amounts of SPM or 15-LOX-derived products.
ADAM10 Mediates Hla-Induced SPM Formation and 15-LOX-1 Activation
Hla mediates many effects by ADAM10 (a disintegrin and metalloprotease 10) (von Hoven et al., 2019; Wilke and Bubeck Wardenburg, 2010) that was recently described to play a role in inflammation resolution (Zhou et al., 2019). ADAM10 is expressed in M1 and M2 with some higher tendency in M1 (p = 0.08, unpaired t test; Figure 5A). The specific ADAM10 inhibitor GI254023X strongly reduced the surface expression of ADAM10 (Figure 5B) as described before (Ezekwe et al., 2016), without cytotoxic effects in M1 and M2 (Figure S5A). Importantly, depletion of ADAM10 by GI254023X abolished the Hla-induced SPM/15-LOX-derived LM formation (Figures 5C and S5B) and LOX redistribution (Figure 5D) in M2 and reversed the cytotoxic effects of Hla in M1 and M2 (LDH assay; Figure S5C). In contrast, A23187-induced LOX translocation and LM production were unaffected by GI254023X (Figures 5C and 5D). These data suggest that Hla evokes 15-LOX-1 activation and related LM formation in M2 through its receptor ADAM10.
Figure 5. ADAM10 Mediates Hla-Induced SPM Formation and 15-LOX-1 Activation.
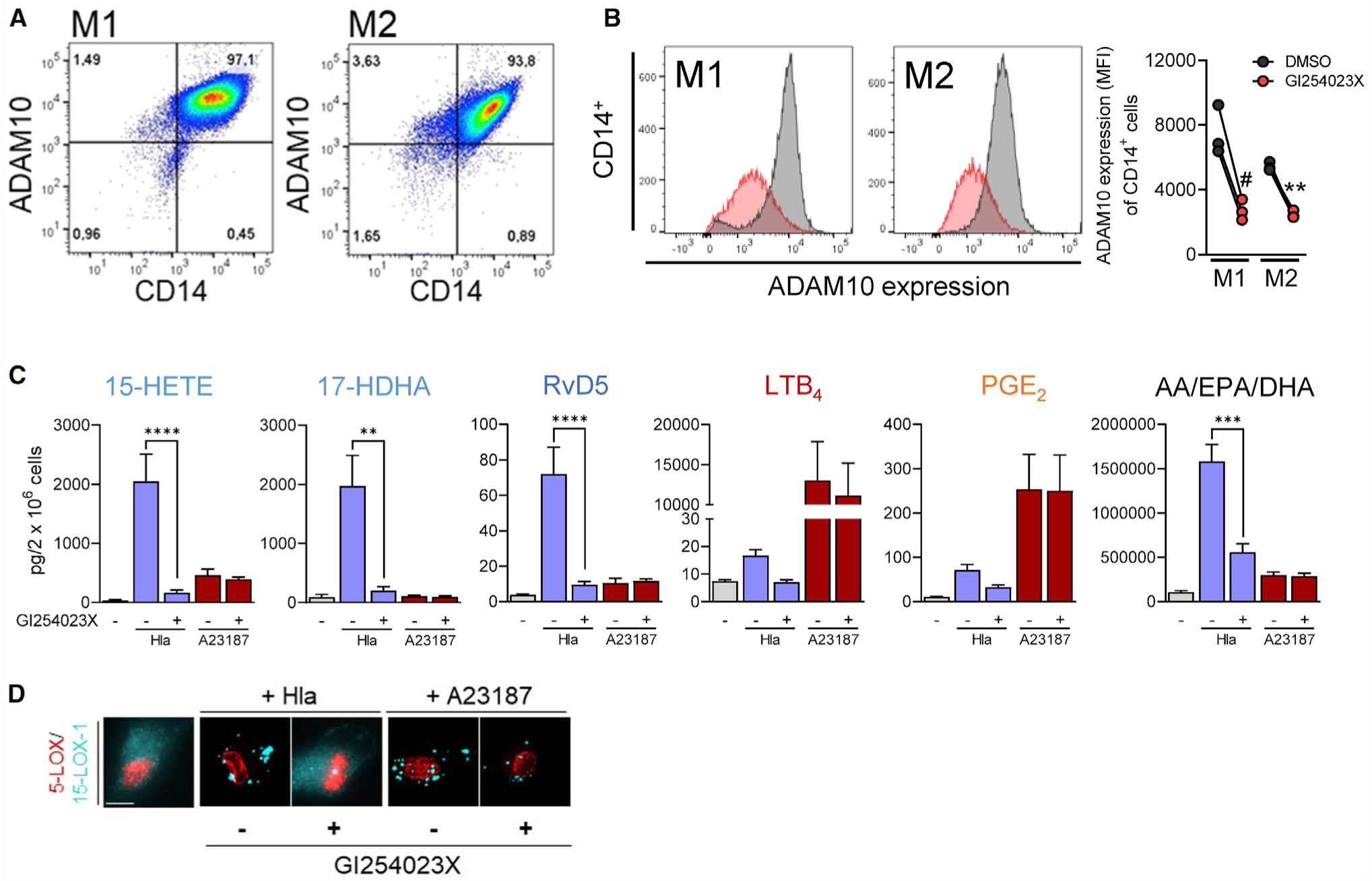
(A) Representative pseudocolor dot plots of ADAM10 expression on CD14+ M1 and M2 macrophages, measured by flow cytometry; n = 3.
(B–D) M1 or M2 macrophages (2 × 106, each) were incubated during the last 24 h of polarization with 40 mM GI254023X or vehicle (0.1% DMSO). (B) Representative histograms of ADAM10 expression on CD14+ M1 and M2 after treatment with GI254023X (red) or vehicle (gray). Data are presented as MFI (mean fluorescent intensity) in a repeated-measurement dot plot; n = 3. # p < 0.05; ** p < 0.01 GI254023X versus DMSO; unpaired Student’s t test. (C) M2s (2 × 106 cells) after treatment with GI254023X or vehicle were incubated for 180 min with Hla (1 μg/mL), 10 min with A23187 (0.5 μM), or vehicle at 37°C; 15-HETE, 17-HDHA, RvD5, PGE2, and LTB4 or AA, EPA, and DHA were analyzed by UPLC-MS-MS and shown as pg/2 × 106 cells. Results are means + SEM, n = 3–4; **p < 0.01, ***p < 0.001, ****p < 0.0001, GI254023X versus DMSO. Data were log-transformed for statistical analysis, unpaired one-way ANOVA with Tukey’s multiple comparisons test. (D) Subcellular redistribution of 5-LOX and 15-LOX-1 in M2 (in PBS plus 1 mM CaCl2) with or without pre-treatment with GI254023X. Cells were stimulated with 1 μg/mL Hla for 90 min or with 0.5 μM A23187 for 10 min, fixed, permeabilized, and incubated with antibodies against 5-LOX (red) and 15-LOX-1 (cyan-blue); scale bars, 10 μm. Results shown for one single cell are representative for approximately 100 individual cells analyzed in n = 3 independent experiments (separate donors), each.
See also Figure S5.
Hla-Induced LM Produced from M2 Promote Planarian Regeneration
Given the pivotal roles of SPM in self-limited inflammation, we tested if the LM produced by M2 upon Hla stimulation could promote tissue regeneration as a bioassay system. The established planarian tissue self-repair model was used to study the time-dependent regeneration of the head after resection. The tissue-regeneration indexes (TRIs) (Serhan et al., 2012) were calculated with TRImax (maximum tissue regeneration, 100%) at day 7. Treatment with a SPM mix (RvD2, RvD5, MaR1, PD1, and 18-HEPE) only showed a slight enhancement of regeneration at day 6, whereas LM isolates from Hla-stimulated M2 (composition see Table S1) clearly accelerated head regeneration (Figures 6A and 6B), shortening TRI50 (50% regeneration) from day 7 of vehicle-treated worms to day 5.
Figure 6. Enhancement of Planarian Regeneration by Exposure to Hla-Treated M2 Macrophage Isolates.
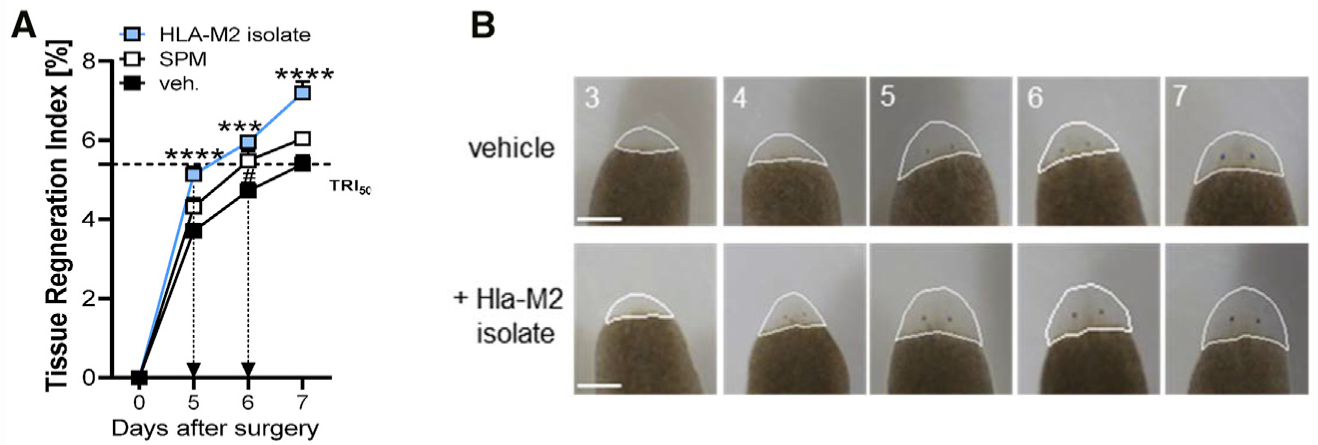
(A) Tissue regeneration index of planarians treated with Hla-M2 isolates, SPMs (200 nM RvD5, RvD2, MaR1, PD1, and 18-HEPE), or vehicle (0.5% MeOH) after the indicated days. Results are means + SEM; n = 14–15 planarians. ***p < 0.001, ****p < 0.0001 Hla-M2 isolate versus vehicle; #p < 0.05 SPMs versus vehicle; one-way ANOVA with Tukey’s multiple comparisons test.
(B) Planarians were treated with LM isolates obtained from M2s that were treated with 1 μg/mL Hla (Hla-M2 isolates) or with 0.5% MeOH (vehicle). Images shown are representative planarians from n = 15 taken at the indicated time points. The outline of the quantified area in each of the images is shown.
Hla Evokes SPM Formation in the Peritoneum of Mice In Vivo
To study how Hla modulates LM profiles in vivo, we made use of murine zymosan-induced peritonitis (Pace et al., 2017), a well-established experimental model of self-resolving inflammation relevant to human infections (Chiang et al., 2012). ADAM10 also mediates effects of Hla in mice (Inoshima et al., 2011); however, murine and human 15-LOX-1 orthologs differ in the 12-/15-lipoxygenation specificity: humans express the ortholog with dominantly 15-lipoxygenating activity and mice an ortholog with 12-lipoxygenating activity (Adel et al., 2016). Thus, we first isolated peritoneal macrophages (PMs) from mice, a major source of murine 15-LOX-1 (Dioszeghy et al., 2008), to confirm the induction of LM formation upon exposure to Hla in vitro. As observed for human M2, the levels of SPM and 12/15-LOX products were markedly increased in murine PMs upon Hla, whereas PGs and LTs were only marginally formed (Figures 7A and S6A). We then injected Hla (0.5 mg/kg) and for comparison zymosan (1 mg per mouse) into the peritoneum (intraperitoneal [i.p.]) of mice. Infiltration of leukocytes (Figure 7B) and the levels of the pro-inflammatory cytokines TNF-α, IL-6, and IL-1β (Figure 7C) in the exudates were significantly increased after 90 min upon zymosan but were unchanged upon Hla, which was evident for TNF-α and IL-6 also in the plasma (Figure S6B). Also, COX- and 5-LOX-derived LMs, especially LTB4, were markedly elevated by zymosan (Figure 7D). In contrast, mice that received Hla generated high levels of various SPMs including PD1, PDX, MaR1/2, RvD1, RvD2, RvD4, RvD5, and LXA4, as well as their bioactive precursors (Figure 7D). Importantly, zymosan failed to induce most of the SPMs, except LXA4, RvD4, and RvD5, and caused no or only minor formation of 12/15-LOX-derived products, with much lower ratio of RvD5 to LTB4 than that obtained with Hla (Figure 7E). In plasma and in spleen, i.p. injection of Hla and zymosan caused elevated levels of SPMs, 12-lipoxygenated LMs, and LTB4, but only zymosan increased TNF-α and IL-6 (Figures S6C and S6D). Together, our data suggest that zymosan and Hla may address different cell types and/or LM biosynthetic pathways and confirm potent induction of SPM formation by Hla.
Figure 7. Hla Evokes SPM Formation in the Peritoneum of Mice In Vivo.
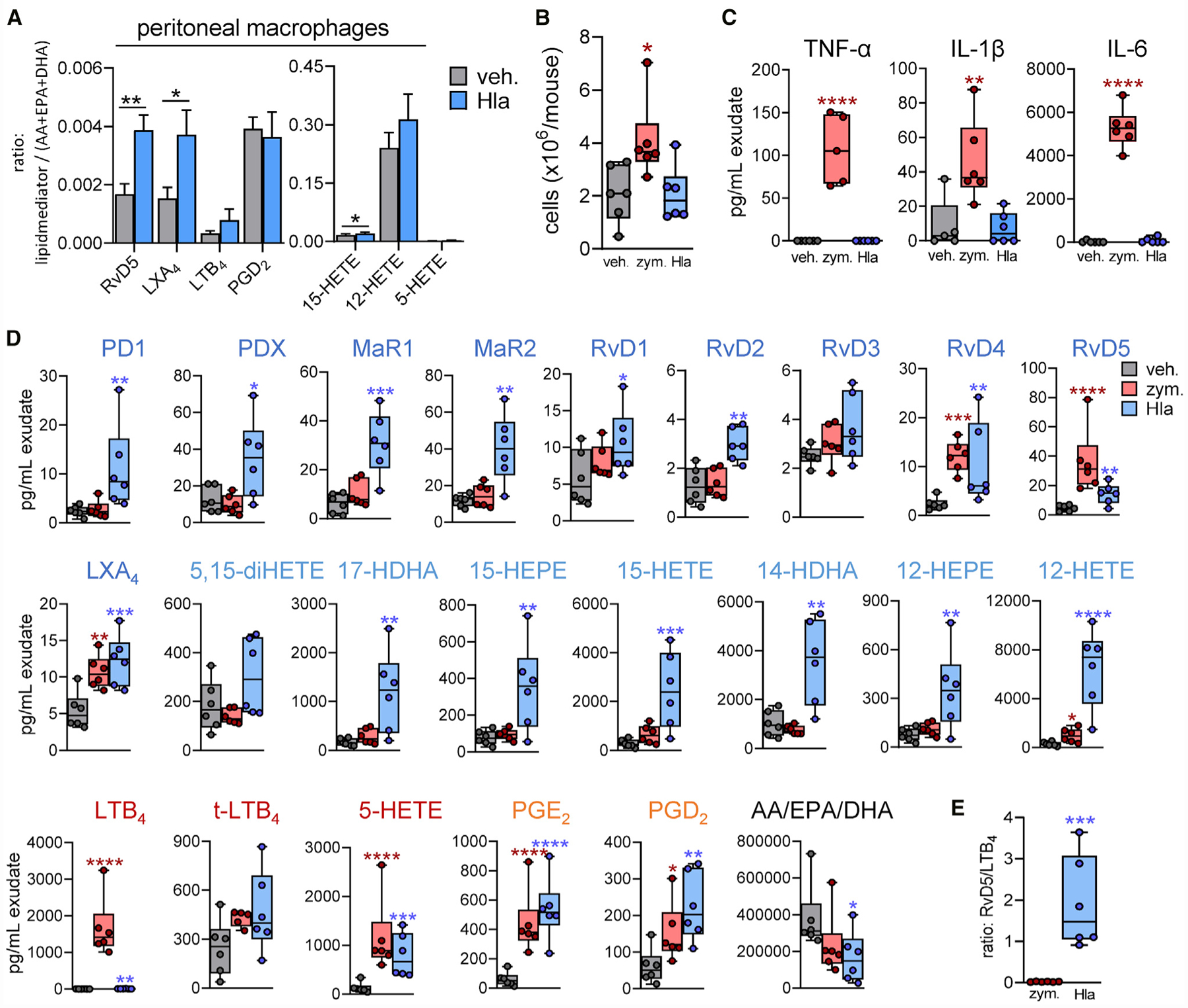
(A) Freshly isolated murine PMs were incubated for 24 h at 37°C. Then, 2 × 106 cells were treated with 1 μg/mL Hla for 90 min or vehicle at 37°C; formed LMs were analyzed by UPLC-MS-MS. Results are means + SEM of the ratio of each LM against the sum of AA, EPA plus DHA; *p < 0.05, **p < 0.01 Hla versus vehicle, paired Student’s t test.
(B–E) Male mice (n = 6 per group) received 0.5 mg/kg Hla, 1 mg zymosan, or saline buffer (0.5 mL) by i.p. injection. Analysis was performed 90 min after injection.
(B) Cell infiltration in the peritoneal exudates. Results are given as cell numbers (×106/mouse); *p < 0.05 versus vehicle; unpaired one-way ANOVA with Tukey’s multiple comparisons test. (C) Cytokine levels (pg/mL) in the exudates; n = 6; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 versus vehicle; unpaired one-way ANOVA with Tukey’s multiple comparisons test. (D) LM levels in 1 mL exudate, analyzed by UPLC-MS-MS, given in pg/mL; n = 6; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 versus vehicle; unpaired one-way ANOVA with Tukey’s multiple comparisons test. (E) Ratio of RvD5 to LTB4 from zymosan- or Hla-treated mice; each dot represents the ratio for a single mouse; n = 6; *p < 0.05, *p < 0.01, ***p < 0.001, ****p < 0.0001 Hla versus zymosan; unpaired Student’s t test. See also Figure S6.
DISCUSSION
Inflammation is a hallmark of S. aureus-mediated disorders, including sepsis and skin and urinary tract infections (Seyyed Mousavi et al., 2017; Tong et al., 2015). S. aureus uses a large arsenal of virulence factors that play important roles in pathogen invasion but also in host immunity (Bronner et al., 2004; Foster, 2005; Goldmann and Medina, 2018; Otto, 2014b; Tuchscherr et al., 2011; von Hoven et al., 2019). However, the molecular and cellular mechanisms contributing to host defense during S. aureus infection, in particular the role of LM in this process, are largely unknown (Dioszeghy et al., 2008; Pezato et al., 2012). Specific pro-inflammatory and pro-resolving LMs are temporally and differentially regulated during infections in mice exposed to E. coli or S. aureus (Chiang et al., 2012). These pathogens also evoked formation of distinct LMs in human macrophage phenotypes (Werz et al., 2018) and in neutrophils (Romp et al., 2019). Here, we uncovered that Hla as an S. aureus-derived virulence factor potently and selectively activates 15-LOX-1 in human M2 culminating in substantial SPM biosynthesis, without marked induction of PGs and LTs. SPMs orchestrate the resolution of infectious inflammation and induce cellular pathways that enable bacterial clearance, tissue repair, and appropriate return to homeostasis of the host (Serhan, 2014; Serhan and Levy, 2018). Therefore, in the course of S. aureus infections, the host might benefit from Hla by boosting SPM biosynthesis and thus promoting inflammation resolution. Recently, host beneficial effects were shown for a PFT-like protein complex from frog that promoted tissue repair and wound healing in mice (Gao et al., 2019). Alternatively, S. aureus may use Hla-induced SPMs to promote skewing of macrophages to M2-like responses, allowing the persistence of S. aureus, e.g., in the nasal passages, thereby worsening chronic rhinosinusitis (Krysko et al., 2011), and establishment of a niche within, i.e., biofilms where it may avoid killing by the host.
LM isolates from Hla-stimulated M2 accelerated head regeneration of planarians in vivo, which was even superior over a defined mixture of SPMs. Possibly, additional SPMs (including cysteinyl-SPM; Dalli et al., 2015b) or SPM precursors (e.g., 17-HDHA) potentially contained in the LM isolates may act synergistically by other receptors to promote head regeneration. The i.p. Hla administration to mice robustly elevated SPMs and 12/15-LOX products but hardly pro-inflammatory LTs, TNF-α, IL-1β, and IL-6 and neutrophil recruitment. The latter effects were in sharp contrast to zymosan, a typical inflammatory stimulus (Rao et al., 1994), which nevertheless, induces self-resolving peritonitis with SPM formation at later stages (Schwab et al., 2007; Yamada et al., 2011). Although resident PMs are likely a major source for LM production upon Hla challenge in mouse peritoneum, infiltrating neutrophils or monocytes as well as epithelial cells may also contribute.
Because S. aureus-induced LM formation was mimicked by SACM, we concluded that secreted factors from S. aureus are operative as stimuli. Notably, among all LM produced in M2, the SPMs and their precursors were most prominently upregulated due to robust activation of 15-LOX-1 as a known SPM-biosynthetic key enzyme (Dalli and Serhan, 2012; Werner et al., 2019; Werz et al., 2018). In fact, knockdown of ALOX15A abolished SACM-induced SPM formation. Activation of 15-LOX-1 and its oxidized products, free or bound to phospholipids of membranes, plays an essential role in controlling self-tolerance and the resolution of inflammation (Kim et al., 2018; Uderhardt and Krönke, 2012; Werz et al., 2018; Yamada et al., 2011). For example, in mice, 15-LOX-1 deficiency increased infiltration of pro-inflammatory macrophages and upregulated pro-inflammatory signaling in dermal adipose tissue in the dorsal skin along with a severe loss of RvD2 (Kim et al., 2018). Although S. aureus secretes many molecules, in particular PFTs (Otto, 2014b) that may be responsible, several lines of evidence point to Hla as a key candidate for eliciting the production of 15-LOX-1-derived LMs in M2: (1) the LS1 Δhla mutant failed to induce 15-LOX-1-derived LMs and 15-LOX translocation versus the WT LS1 strain, (2) inclusion of Hla-neutralizing antibodies abrogated SACM-induced 15-LOX product formation, and (3) isolated Hla selectively evoked formation of 15-LOX-1-derived LM and induced 15-LOX translocation. However, the SACM also evoked COX- and 5-LOX-derived LMs, which was hardly apparent for isolated Hla and was still evident after Hla depletion of SACM, suggesting a role for other PFTs in this respect. In fact, in human neutrophils that generate abundant LTs upon exposure to S. aureus, PSMs elicited LT biosynthesis involving the GPCR ALX/FPR2, while Hla was essentially inactive (Romp et al., 2019). In M1 or M2, however, PSMs neither induced LT formation nor 15-LOX-1 activation or SPM production. Although PSMs, released in the invasive early phase of S. aureus infections causing neutrophil activation (Kretschmer et al., 2010), represent S. aureus-derived PFTs responsible for pro-inflammatory LT formation in neutrophils, Hla causes the generation of anti-inflammatory SPMs, particularly in M2.
The cytolytic effects of Hla due to perforation of the host plasma membrane are predominantly associated with inflammatory processes, e.g., cytokine release and apoptosis of innate immune cells (Berube and Bubeck Wardenburg, 2013; von Hoven et al., 2019). Also, Hla can induce formation of pro-inflammatory eicosanoids (e.g., TXA2) in lung and heart of rodents (Chang et al., 1992; Grandel et al., 2009; Sibelius et al., 2000). The immune system, however, has evolved distinct pathways to sense such membrane damage for inducing tissue repair and regeneration (DuMont and Torres, 2014). In this respect, Hla-induced SPM formation may support termination of the inflammatory process in S. aureus infections along with bacterial clearance and return to homeostasis. In our in vitro approach, inflammation-initiating 5-LOX and COX products were rapidly generated in M1 by S. aureus at low MOI, which may reflect the early phase of a S. aureus infection, for which low numbers of bacteria initiate the inflammatory response by interactions with pro-inflammatory M1 (Atri et al., 2018; Dalli and Serhan, 2016). In contrast, SPM formation was delayed, required higher MOI, and occurred in M2, which are conditions relevant at later stages of infectious inflammation (Chiang et al., 2012; Dalli and Serhan, 2016). Such a temporal switch from pro-inflammatory to pro-resolving LMs is decisive for inflammation termination (Levy et al., 2001; Serhan and Levy, 2018) and may be promoted by Hla.
The ability of Hla to elicit marked SPM formation in human M2 or in murine peritoneum is unique. In general, stimuli that evoke cellular formation of LOX products and related SPMs must accomplish the supply of free fatty acid substrates and LOX translocation to appropriate membranous compartments (Kuhn et al., 1999; Rådmark et al., 2015). Both processes are achieved in macrophages by Ca2+-elevating agents, such as ionophores, GPCR ligands (e.g., fMLP, C5a, LTB4, or PAF), thapsigargin, ATP, or zymosan, that indeed caused formation of 5-LOX products, but in contrast to Hla, only marginally evoked SPM production (Gijón et al., 2000; Norris et al., 2014; Werz, 2002; Werz et al., 2018). Hla typically increases [Ca2+]i in many cell types with consequences on cellular functions (Bouillot et al., 2018), including AA release (Fink et al., 1989; Grimminger et al., 1997). Also in M2, isolated Hla or SACM led to elevation of [Ca2+]i, which was not the case for PSMα3 and δ-toxin that failed to evoke LM formation, and Ca2+-depletion studies confirmed the necessity of Ca2+ for Hla-evoked LM biosynthesis. This raises the intriguing question of why only Hla but not A23187 or other Ca2+-mobilizing agents induce the 15-LOX product and SPM formation. Note that both Hla and A23187 gave comparable strong fatty acid substrate release and 15-LOX translocation, and A23187 markedly increased 5-LOX- and COX-derived LMs in M1 and M2, as observed before (Werz et al., 2018). A23187 robustly increases [Ca2+]i within seconds and is the most effective stimulus for 5-LOX product formation in leukocytes (Werz, 2002). This is in sharp contrast to Hla, activating almost exclusively the 15-LOX pathway in M2. For cellular activation of 15-LOX, (lipid-)hydroperoxides are required but other mechanisms of enzyme activation are not known (Ivanov et al., 2015). Apparently, the distinct LOX activation features of A23187 and Hla are related to kinetic differences in elevating [Ca2+]i in M2, immediately rising in response to A23187 but delayed upon Hla exposure. In fact, although A23187-induced fatty acid release and LOX translocation, along with 5-LOX and COX product formation, are rapid and transient, Hla-induced responses are slow and sustained and may govern 15-LOX to produce LMs in a preferably delayed manner. Conceivably, distinct requirements for hydroperoxide-mediated activation or turnover-dependent suicide inactivation of the LOXs at different subcellular locales may account for these differences: rapid and massive substrate and hydroperoxide supply at the nuclear membrane upon A23187 (Gijón et al., 2000) may accelerate 5-LOX suicidal inactivation, whereas moderate Hla-mediated substrate/hydroperoxide release for 15-LOX at the yet undefined membranous locale may accomplish sustained enzyme activity.
Hla, a 33.2 kDa monomer, binds to the host cell membrane and assembles into a heptameric complex that perforates the plasma membrane to accomplish massive Ca2+ influx (von Hoven et al., 2019). Specific host cell binding of Hla is achieved by its proteinaceous receptor ADAM10 (Wilke and Bubeck Wardenburg, 2010). This metalloprotease is expressed on the surface of many host cells, including monocytes/macrophages and neutrophils (Kieseier et al., 2003; Pruessmeyer et al., 2014), and its activation by Hla might be caused by precedent Ca2+ influx (von Hoven et al., 2019). Our data reveal a crucial and selective role of ADAM10 in Hla-induced 15-LOX-1 activation and SPM formation, based on the complete abrogation of 15-LOX-1 translocation and LM formation by the ADAM10 inhibitor GI254023X. Note that upon stimulation of M2 with A23187, the ADAM10 inhibitor failed in this respect, even though the ionophore was shown before to cause activation of ADAM10 due to Ca2+ influx (Horiuchi et al., 2007). Again, these findings underline the uniqueness of Hla-induced 15-LOX-1 activation in M2 culminating in SPM formation. Interestingly, ADAM10 and ADAM17 were recently implied in the acceleration of inflammation resolution regulated by fibrinogen-like protein 2 (Zhou et al., 2019).
In conclusion, we identified Hla as S. aureus-derived PFT with the unique property of eliciting substantial SPM formation in human and murine macrophages through selective activation of 15-LOX-1-initiated pathways. Our findings suggest that the host may exploit this PFT as an elicitor of SPM formation to better cope with S. aureus infections by accelerating bacterial killing and clearance and tissue repair and regeneration by SPMs to eventually return to homeostasis.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Oliver Werz (oliver.werz@uni-jena.de).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
This study did not generate/analyze datasets/code.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
Male CD-1 mice (33–39 g, 8–9 weeks, Charles River Laboratories, Calco, Italy) were used, and housed in a controlledenvironment (21± 2°C) and provided with standard rodent chow and water. Animals were allowed to acclimate for four days prior to experiments and were subjected to 12 h light/12 h dark schedule. Mice were randomly assigned for the experiments, which were conducted during the light phase. The experimental procedures were approved by the Italian Ministry according to International and National law and policies (EU Directive 2010/63/ EU and Italian DL 26/2014 for animal experiments, ARRIVE guidelines and the Basel declaration including the 3R concept).
Resident PM were obtained by lavage of the peritoneal cavity of mice with 7 mL of cold Dulbecco’s modified Eagle’s medium (DMEM) with heparin (5 U/mL). PM were then centrifuged at 500 × g at 4°C for 5 min and incubated for 24 h at 37°C. Then, 2 × 106 cells were treated for 90 min at 37°C as indicated. All samples were then centrifuged at 500 × g at 4°C for 5 min. Supernatants were collected and frozen at −80°C. Formed LM were isolated by SPE and analyzed by UPLC-MS-MS.
Bacterial strains
The following bacterial strains were used:
| Strain name | PVL | ||
|---|---|---|---|
| in manuscript | Strain | Description | (PCR) |
| WT | LS1 | septic arthritis isolate | - |
| Δagr/sarA | HOM175 | LS1 derivative carrying the agr::tetM and sar::Tn917LTV1 | - |
| mutations of RN6911 and ALC136, respectively; EmR, TcR | |||
| Δhla | LS1Δhla | LS1 Δhla, ErmR, obtained by phage transduction from DU1090 | - |
For experiments with intact S. aureus, bacteria were grown overnight at 37°C in brain heart infusion (BHI) medium while shaking, diluted to OD600nm of 0.05 and grown for another 3 h (log-phase). Bacteria were washed in PBS and resuspended in PBS without CaCl2. SACM was prepared by growing the bacteria for 17 h in BHI medium, pelleted for 10 min at 3350 × g and sterile-filtered through a Millex-GP filter unit (0.22 mm; Millipore) before used at the indicated percentage.
For analysis of the growth rate and generation time, S. aureus WT LS1 and their respective mutants were grown overnight in 15 mL of BHI with shaking (160 rpm) at 37°C. The following day, 1 mL of BHI were inoculated with each strain in order to obtain the starting optical density 0.05 (578 nm) and 200 μl were added to 4 wells in a clear 96-well flat bottom microtiter plate. The growth of each strain was monitored spectrophotometrically (578 nm) every 30 min during 24 h in a microplate reader (Tecan Trading AG, Männedorf, Swiss). The growth rate (μ, growth speed) and the generation time (g) for each strain used in this study were calculated according the followings formulas.
μ = growth rate N = final population t = final time g = generation time N0 = initial population t0 = initial time
Human cells
Monocytes were isolated from leukocyte concentrates obtained from freshly withdrawn peripheral blood of healthy male and female adult human donors (18–65 years) which were provided by the Institute of Transfusion Medicine at the University Hospital Jena, Germany. The experimental protocol was approved by the ethical committee of the University Hospital Jena. All methods were performed in accordance with the relevant guidelines and regulations. Peripheral blood mononuclear cells (PBMC) were separated using dextran sedimentation, followed by centrifugation on lymphocyte separation medium (Histopaque®−1077, Sigma-Aldrich). PBMC were seeded in RPMI 1640 (Sigma-Aldrich) containing 10% (v/v) heat inactivated fetal calf serum (FCS), 100 U/mL penicillin, and 100 μg/mL streptomycin in cell culture flasks (Greiner Bio-one, Frickenhausen, Germany) for 1.5 h at 37°C and 5% CO2 for adherence of monocytes. For differentiation of monocytes to macrophages and polarization toward M1 and M2, published criteria were used (Murray et al., 2014). M1 were generated by incubating monocytes with 20 ng/ml GM-CSF (Peprotech, Hamburg, Germany) for 6 days in RPMI 1640 supplemented with 10% fetal calf serum, 2 mmol/L glutamine (Biochrom/Merck, Berlin, Germany), and penicillin-streptomycin (Biochrom/Merck), followed by 100 ng/ml LPS and 20 ng/ml INF-γ (Peprotech) treatment for another 48 h. M2 were incubated with 20 ng/ml M-CSF (Peprotech) for 6 days of differentiation plus 20 ng/ml IL-4 (Peprotech) for additional 48 h of polarization.
METHOD DETAILS
Bacterial gene expression
To determine gene expression in the early stationary phase, overnight bacterial cultures were diluted to an OD578nm of 0.05 and incubated for 5 h at 37°C with rotation (160 rpm). Then 1 mL of bacterial suspension was mixed with RNAprotect Bacteria Reagent (-QIAGEN), centrifuged, and stored at −20°C. For isolation of RNA, the pellet was mixed with RNApro Solution (MP Biomedicals, Eschwege, Germany). The mixture was transferred to a Lysing Matrix B tube (MP Biomedical) and homogenized with a FastPrep® (MP Biomedicals) homogenizer. After subsequent centrifugation, the supernatant was used for RNA isolation with the peqGOLD Total RNA Kit (VWR). DNA was digested with TURBO DNase (Thermo Fisher Scientific, Darmstadt, Germany). The concentration of the RNA was determined by spectrophotometric analysis with a NanoDrop (Thermo Fisher Scientific, Darmstadt, Germany) before reverse transcription to complementary DNA (cDNA) (qScript cDNA SuperMix, Quantabio, Beverly, MA, USA). The cDNA was analyzed with QuantiNova SYBR Green PCR Kit (QIAGEN, Hilden, Germany) in a Rotor-Gene Q (QIAGEN) thermocycler. The reaction mixtures were incubated for 15 min at 95°C, followed by 40 cycles of 15 s at 95°C, 30 s at 55°C, and 30 s at 72°C. Fold-changes in expression were calculated by the Pfaffl equation (Pfaffl, 2001).
The following primers were used:
| Gene | Primer description | Primer sequence | Product length |
|---|---|---|---|
| hla | Forward | 5′-caactgataaaaaagtaggctggaaagtgat-3′ | 201 nt |
| Reverse | 5′-ctggtgaaaaccctgaagataatagag-3′ | ||
| psmα | Forward | 5′-gccattcacatggaattcgt-3′ | 151 nt |
| Reverse | 5′-caatagccatcgttttgtcct-3′ | ||
| gyrB(*) | Forward | 5′-aattgaagcaggctatgtgt-3′ | 138 nt |
| Reverse | 5′-atagaccattttggtgttgg-3′ |
housekeeping reference gene
Incubations of macrophages and LM metabololipidomics
Polarized macrophages (2 × 106/mL) were incubated in PBS containing 1 mM CaCl2. Isolated compounds, aliquots of S. aureus (LS1 or mutants) at the indicated MOIs or SACM were added at 37°C. After the indicated incubation periods, the supernatants were transferred to 2 mL of ice-cold methanol containing 10 mL of deuterium-labeled internal standards (200 nM d8–5S-HETE, d4-LTB4, d5-LXA4, d5-RvD2, d4-PGE2 and 10 μM d8-AA; Cayman Chemical/Biomol GmbH, Hamburg, Germany) to facilitate quantification and sample recovery. Sample preparation was conducted by adapting published criteria (Werz et al., 2018). In brief, samples were kept at −20°C for 60 min to allow protein precipitation. After centrifugation (1200 g, 4°C, 10 min) 8 mL acidified H2O was added (final pH = 3.5) and samples were subjected to solid phase extraction. Solid phase cartridges (Sep-Pak® Vac 6cc 500 mg/ 6 mL C18; Waters, Milford, MA) were equilibrated with 6 mL methanol and 2 mL H2O before samples were loaded onto columns. After washing with 6 mL H2O and additional 6 mL hexane, LM were eluted with 6 mL methyl formate. Finally, the samples were brought to dryness using an evaporation system (TurboVap LV, Biotage, Uppsala, Sweden) and resuspended in 100 μL methanol-water (50/50, v/v) for UPLC-MS-MS automated injections. LM profiling was analyzed with an Acquity UPLC system (Waters, Milford, MA, USA) and a QTRAP 5500 Mass Spectrometer (ABSciex, Darmstadt, Germany) equipped with a Turbo V Source and electrospray ionization. LM were eluted using an ACQUITY UPLC® BEH C18 column (1.7 μm, 2.1 × 100 mm; Waters, Eschborn, Germany) at 50°C with a flow rate of 0.3 ml/min and a mobile phase consisting of methanol-water-acetic acid of 42:58:0.01 (v/v/v) that was ramped to 86:14:0.01 (v/v/v) over 12.5 min and then to 98:2:0.01 (v/v/v) for 3 min (Werner et al., 2019). The QTrap 5500 was operated in negative ionization mode using scheduled multiple reaction monitoring (MRM) coupled with information-dependent acquisition. The scheduled MRM window was 60 s, optimized LM parameters were adopted (Colas et al., 2014), and the curtain gas pressure was set to 35 psi. The retention time and at least six diagnostic ions for each LM were confirmed by means of an external standard (Cayman Chemical/Biomol GmbH, Hamburg, Germany). Quantification was achieved by calibration curves for each LM. Linear calibration curves were obtained for each LM and gave r2 values of 0.998 or higher (for fatty acids 0.95 or higher). Additionally, the limit of detection for each targeted LM was determined (Werner et al., 2019).
Principal component analysis
PCA was performed using OriginPro 2019 software following mean centering and unit variance scaling of LM amounts. PCA serves as an unbiased, multivariate projection designed to identify the systematic variation in a data matrix (the overall bioactive LM profile of each sample) with lower dimensional plane using score plots and loading plots. The score plot shows the systematic clusters among the observations (closer plots presenting higher similarity in the data matrix).
SDS-PAGE and western blot
Cell lysates of macrophages corresponding to 2 × 106 cells as well as supernatants of bacterial overnight cultures were separated on 10% polyacrylamide gels (5-LOX, 15-LOX-1, 15-LOX-2, cPLA2-α, and Hla), and blotted onto nitrocellulose membranes (Amersham Protran Supported 0.45 μm nitrocellulose, GE Healthcare, Freiburg, Germany). Incremental amounts of isolated Hla (0.01 – 1 μg) were used to assess the Hla concentrations in bacterial supernatants. The membranes were incubated with the following primary antibodies: polyclonal rabbit anti-cPLA2-α, 1:200 (2832, Cell Signaling, Danvers, MA); rabbit polyclonal anti 5-LOX, 1:1000 (by Genscript, Piscataway to a peptide with the C-terminal 12 amino acids of 5-LOX: CSPDRIPNSVAI); mouse monoclonal anti-15-LOX-1, 1:500 (ab119774, Abcam, Cambridge, UK); rabbit polyclonal anti-15-LOX-2, 1:500 (ab23691, Abcam); mouse monoclonal anti-α-hemolysin (ab190467, Abcam, Cambridge, UK), rabbit polyclonal anti-β-actin, 1:1000 (4967S, Cell Signaling), and rabbit monoclonal anti-GAPDH, 1:1000 (D16H11, Cell Signaling). Immunoreactive bands were stained with IRDye 800CW Goat anti-Mouse IgG (H+L), 1:10,000 (926–32210, LI-COR Biosciences, Lincoln, NE), IRDye 800CW Goat anti-Rabbit IgG (H+L), 1:15,000 (926 32211, LI-COR Biosciences) and/or IRDye 680LT Goat anti-Mouse IgG (H+L), 1:40,000 (926–68020, LI-COR Biosciences), and visualized by an Odyssey infrared imager (LI-COR Biosciences, Lincoln, NE). Data from densitometric analysis were background corrected.
Immunofluorescence microscopy
Macrophages (0.8 × 106 cells) were seeded onto glass coverslips in a 12-well plate and cultured for 48 h. S. aureus, SACM, Hla, A23187 or vehicle (PBS) were added and incubations were kept at 37°C for the indicated times. Cells were then fixed using 4% paraformaldehyde solution. Acetone (3 min, 4°C) and 0.1% Triton X-100 (10 min, RT) were used for permeabilization before blocking with normal goat serum (10%, 50062Z, Thermo Fisher Scientific). Samples were incubated with mouse monoclonal anti-5-LOX antibody, 1:100 (610694, BD Biosciences), rabbit anti-5-LOX antibody, 1:100 (1550 AK6, provided by Dr. Olof Radmark, Karolinska Institutet, Stockholm, Sweden), rabbit polyclonal anti-FLAP antibody, 5 μg/ml (ab85227, Abcam, Cambridge, UK Abcam), or mouse monoclonal anti-15-LOX-1 antibody, 1:100 (ab119774, Abcam, Cambridge, UK) at 4°C overnight. 5-LOX, 15-LOX-1 and FLAP were stained with the fluorophore-labeled secondary antibodies; Alexa Fluor 488 goat anti-rabbit IgG (H+L), 1:500 (A11034, Thermo Fisher Scientific) and Alexa Fluor 555 goat anti-mouse IgG (H+L); 1:500 (A21424, Thermo Fisher Scientific). Nuclear DNA was stained with ProLong Gold Antifade Mountant with DAPI (15395816, Thermo Fisher Scientific). Samples were analyzed by a Zeiss Axiovert 200M microscope, and a Plan Neofluar × 40/1.30 Oil (DIC III) objective (Carl Zeiss, Jena, Germany). An AxioCam MR camera (Carl Zeiss) was used for image acquisition.
Flow cytometry
Fluorescent staining for flow cytometric analysis of M1 and M2 macrophages for 48 h was performed in FACS buffer (PBS with 0.5% BSA, 2 mM EDTA and 0.1% sodium azide). Non-specific antibody binding was blocked by using mouse serum (10 min at 4°C) prior to antibody staining. Then, macrophages were stained with fluorochrome-labeled antibody mixtures (4°C for 20 min). The following antibodies were used: FITC anti-human CD14 (2 μg/test, clone M5E2, BD Bioscience, San Jose, CA), APC anti-human CD156c/ADAM-10 (clone REA309, Miltenyi Biotec, Bergisch Gladbach, Germany). To determine viability upon Hla treatment, the macrophages were detached after treatment with PBS plus 0.5% BSA, 5 mM EDTA and 0.4% lidocaine for 20 min, and resuspended in 300 μL FACS buffer containing propidium iodide staining solution (40 ng/test, Biolegend, San Diego, CA). To assess macrophage viability upon treatment with intact S. aureus or SACM, the cells were detached with PBS plus 0.5% BSA, and 5 mM EDTA, stained with Zombie Aqua Fixable Viability Kit (Biolegend) for 10 min at 4°C and then fixed with 4% paraformaldehyde for 20 min at room temperature. Upon staining, macrophages were analyzed using BD LSR Fortessa (BD Bioscience). Data were analyzed using FlowJo X Software (BD Bioscience).
LDH release
To evaluate the effect of the ADAM10 inhibitor GI254023X on Hla-induced cytolysis of macrophages, lactate dehydrogenase (LDH) release was assessed by CytoTox 96®Non-Radioactive Cytotoxicity assay (Promega GmbH, Mannheim, Germany) according to manufacturer instructions. Briefly, 2 × 106 macrophages/mL in PBS plus 1 mM CaCl2 were incubated with GI254023X or vehicle (DMSO) for 24 h and then with 1 μg/mL Hla or vehicle (negative control) or lysis solution (positive control) for 180 min at 37°C. After centrifugation at 400 g (5 min, 4°C), the supernatants were collected and diluted to appropriate LDH concentrations, and the absorbance was measured by NOVOstar microplate reader (BMG Labtechnologies GmbH, Offenburg, Germany) at 490 nm.
Ca2+ imaging
Macrophages were pre-stained with Fura-2/AM (1 μM) for 30 min at 37°C in the dark. Cells were resuspended in Krebs-HEPES buffer containing 0.1% BSA at a density of 1.25 × 106/mL and 1 mM CaCl2 was added. 200 μL of the cell suspension was transferred into a 96-well plate. After 10 min, 2 mM ionomycin, SACM (0.5%), 1 μg/mL Hla, 30 μg/mL PSMα3, 10 μg/mL δ-toxin or vehicle (PBS) were added. The signal was monitored in a thermally (37°C) controlled NOVOstar microplate reader (BMG Labtechnologies GmbH); emission at 510 nm, excitation at 340 nm (Ca2+-bound Fura-2) and 380 nm (free Fura-2). After cell lysis with Triton X-100, the maximal fluorescence signals were monitored ( = 100%) and after chelating Ca2+ with 20 mM EDTA, the minimal fluorescence signals ( = 0%) were recorded.
Cytokine release
M1 and M2 macrophages (2 × 106 cells) were treated with SACM or vehicle (BHI) for 180 min and the supernatant was collected. The exudates of the mice were collected as described before. For measurement of the cytokine levels, supernatants were centrifuged (2000 × g, 4°C, 10 min). The levels of IL-6, IL-1β and TNF-α were analyzed by in-house–made ELISA kits (R&D Systems, Bio-Techne).
ALOX15A gene knockdown in human M2 macrophages
Cells (1.5 3 106) were subjected to electroporation using Neonâ Transfection System 100 mL Kit (Thermo Fisher Scientific). To maximize the efficiency, the Neon 24 optimization protocol was applied according to the manufacturer’s instruction which varied in pulse voltage, pulse width and the number of pulses. Macrophages, differentiated with M-CSF for 6 days, were harvested using PBS plus 1 mM EDTA and the cell pellet was resuspended in 1 mL of PBS. 20 mM of human non-targeting siRNA (Thermo Fisher Scientific; D-001810-01-05) and 20 mM of ALOX15 Trilencer-27 Human siRNA (OriGene, SR300171) was resuspended in the Resuspension Buffer T (Thermo Fisher Scientific) and added to the cells before electroporation. The electroporated cells were transferred immediately to a 12-well containing 1 mL of the corresponding growth medium plus 20 ng/ml IL-4 (Peprotech) and then incubated for another 48 h in a 5% CO2 incubator.
[3H]-Arachidonic acid labeling of macrophages and measurement of AA metabolites
Release of [3H]-AA and its transformed metabolites from macrophages was analyzed as described (Pergola et al., 2014). In brief, cells were resuspended in RPMI 1640 medium containing 10% (v/v) heat inactivated FCS, 100 U/mL penicillin, and 100 μg/mL streptomycin and incubated with 5 nM tritium-labeled [5,6,8,9,11,12,14,15-3H]AA (corresponding to 0.5 μCi/mL, specific activity 200 Ci/mmol; Biotrend Chemicals GmbH, Cologne, Germany) for 2 h at 37°C. Cells were collected, washed to remove unincorporated [3H]AA, and resuspended in PBS containing 1 mM CaCl2. The cells were stimulated with 0.5 μM A23187 or 1 μg/mL Hla for 10 or 90 min at 37°C. The reaction was stopped on ice and cells were centrifuged. Aliquots of the supernatants were combined with 2 mL Rotiszint® eco plus and assayed for radioactivity by scintillation counting (Micro Beta Trilux, Perkin Elmer, Waltham, MA).
Planarian regeneration
Planarians used in this work belong to the species Schmidtea mediterranea asexual biotype. All animals were maintained at 19°C in 1 × Montjuïc Salts (1.6 mM NaCl, 1 mM CaCl2, 1 mM MgSO4, 0.1 mM MgCl2, 0.1 mM KCl, 0.1 g NaHCO3/L) and fed with organic veal liver. Planarians (at least 14 planarians per condition) were starved for 7 days and then amputated anterior and posterior to the pharynx. Trunks were immediately placed under any of the three treatment conditions: (i) a SPM mixture consisting of 200 nM of RvD5, PD1, MaR1, RvD2 and 18-HEPE, (ii) LM isolates from M2 produced after Hla-treatment, and (iii) 0.5% methanol as vehicle in planarian water. Amounts of LM in Hla – M2 isolate were measured by UPLC-MS-MS and are given in Table S1. Planarians were kept in the treatment solution for the entire experiment and treatment solution was replaced every two days. The process of regeneration under the different treatments was monitored daily for 7 days. Photographs on live planarians were taken every 24 hours under a stereo-microscope coupled with a Leica camera MC170 HD (Leica). The images were analyzed using ImageJ software (NIH, Bethesda, MD, USA). A tissue regeneration index (TRI) (Dalli et al., 2016) was determined by dividing the area of the head blastema by the total area of the planarian body.
Peritonitis in mice
Male CD1 mice (n = 6 per experimental group) received Hla (0.5 mg/kg), zymosan (1 mg, 2 mg/mL in saline, boiled and washed; Sigma, Milan, Italy) or vehicle (0.5 mL of 0.9% saline solution) by i.p. injection according to well-recognized experimental design for studying LM formation in acute inflammation (Rossi et al., 2014). After 90 min, mice were euthanized and blood (approximately 0.7–0.9 ml) was collected by intracardiac puncture using citrate as anticoagulant. Plasma was obtained by centrifugation at 800 × g at 4°C for 10 min. Peritoneal exudates were collected using 3 mL of PBS. After 60 s of gentle manual massage, cells in the exudates were immediately counted by using a Burker’s chamber after vital trypan blue staining. 2 mL of exudates were centrifuged (18,000 × g, 5 min, 400°C) and supernatants were collected and frozen at −80°C. Spleens were harvested from mice and homogenized. LM levels in the supernatants of peritoneal exudates, plasma and spleen were assessed by UPLC-MS/MS analyses as described above. Cytokines in the peritoneal exudates were determined by ELISA as described above.
QUANTIFICATION AND STATISTICAL ANALYSIS
Results are expressed as mean ± standard error of the mean (SEM) of n observations, where n represents the number of experiments with separate donors, performed on different days, as indicated. For the in vivo studies, n represents the number of animals. Analyses of data were conducted using GraphPad Prism 8 software (San Diego, CA). Two-tailed t test was used for comparison of two groups. For multiple comparison, one-way and two-way analysis of variance (ANOVA) with Bonferroni, Dunnetts, Tukey’s or Sidak’s post hoc tests were applied as indicated. The criterion for statistical significance is p < 0.05.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER, Catalog number |
|---|---|---|
| Antibodies | ||
| Alexa Fluor 488 goat anti-rabbit IgG (H+L) | Thermo Fisher Scientific | A11034; RRID:AB_2576217 |
| Alexa Fluor 555 goat anti-mouse IgG (H+L) | Thermo Fisher Scientific | A21424; RRID:AB_141780 |
| IRDye 800CW goat anti-rabbit | Li-Cor Biotechnology | 926-32211; RRID:AB_621843 |
| IRDye 800CW goat anti-mouse | Li-Cor Biotechnology | 926-32210; RRID:AB_621842 |
| IRDye 680LT goat anti-mouse | Li-Cor Biotechnology | 926-68020; RRID:AB_10706161 |
| mouse monoclonal anti-5-LOX | BD Bioscience | 610694; RRID:AB_2226941 |
| rabbit polyclonal anti-5-LOX | Dr. Olof Radmark, Karolinska Institutet, Stockholm, Sweden | 1550 AK6 |
| mouse monoclonal anti-15-LOX-1 | Abcam | ab119774; RRID:AB_10901109 |
| rabbit polyclonal anti-15-LOX-2 | Abcam | ab23691; RRID:AB_447612 |
| rabbit polyclonal anti-FLAP | Abcam | ab85227; RRID:AB_10673941 |
| rabbit polyclonal anti-cPLA2α | Cell Signaling | 2832; RRID:AB_2164442 |
| mouse monoclonal anti-α-hemolysin | Abcam | ab190467 |
| rabbit polyclonal anti-β-actin | Cell Signaling | 4967S; RRID:AB_330288 |
| rabbit monoclonal anti-GAPDH | Cell Signaling | D16H11; RRID:AB_10622025 |
| FITC anti-human CD14 | BD Bioscience | clone M5E2; RRID:AB_395798 |
| APC anti-human CD156c/ADAM-10 | Miltenyi Biotec | clone REA309; RRID:AB_2655305 |
| Bacterial Strains | ||
| LS1 | Ahmed et al., 2001 | N/A |
| LS1 Δagr/ΔsarA (HOM175) | Tuchscherr et al., 2015 | N/A |
| LS1 Δhla | Schmitt et al., 2012 | N/A |
| Chemicals, Peptides and Recombinant Proteins | ||
| GI254023X | Sigma | SML0789 |
| Arachidonic acid | Cayman Chemicals | 90010 |
| DAPI | Thermo Fisher Scientific | 15395816 |
| Propidium iodide | Biolegend | 421301 |
| BAPTA/AM | Sigma | 196419 |
| Fura-2AM | Thermo Fisher Scientific | 15510607 |
| Dulbecco’s modified Eagle’s high glucose medium with glutamine (DMEM) | GE Healthcare Life Sciences | FG0435 |
| penicillin/streptomycin | GE Healthcare Life Sciences | P0781 |
| PSMα3 | Genosphere Biotechnology | N/A |
| α-hemolysin | Sigma | H9395 |
| δ-toxin | Genosphere Biotechnology | N/A |
| Non-immune goat serum | Invitrogen | 50-062Z |
| Fetal calf serum | Sigma | F7524 |
| Histopaque® – 1077 | Sigma | 10771 |
| GM-CSF | Peprotech | 300-23 |
| M-CSF | Peprotech | 300-25 |
| IL-4 | Peprotech | 200-04 |
| zymosan A (Saccharomyces cerevisiae) | Sigma | Z4250-1G |
| A23187 | Cayman | Cay11016-10 |
| Brain-heart-infusion broth | Sigma | 53286 |
| methyl formate | Sigma | 291056 |
| RNAprotect Bacteria Reagent | Quiagen | 76506 |
| RNApro Solution | MP Biomedicals | 6055050 |
| TURBO DNase | Thermo Fisher Scientific | AM2239 |
| d8-5S-HETE | Cayman Chemical | Cay334230 |
| d4-LTB4 | Cayman Chemical | Cay320110 |
| d5-LXA4 | Cayman Chemical | Cay10007737 |
| d5-RvD2 | Cayman Chemical | Cay11184 |
| d4-PGE2 | Cayman Chemical | Cay314010 |
| d8-AA | Cayman Chemical | Cay390010 |
| [5,6,8,9,11,12,14,15-3H]AA | Biotrend Chemicals | MT901L-250 |
| Rotiszint® eco plus | Perkin Elmer | 6013621 |
| Experimental Models: Organisms/Strains | ||
| Male CD-1 mice | Charles River Laboratories | N/A |
| Critical Commercial Assays | ||
| peqGOLD Total RNA Kit | VWR | 12-6634 |
| Human TNFα ELISA kit | R&D Systems | DY210 |
| Huamn IL-1β ELISA kit | R&D Systems | DY201 |
| Human IL-6 ELISA kit | R&D Systems | DY206 |
| Mouse TNFα ELISA kit | R&D Systems | DY410 |
| Mouse IL-1β ELISA kit | R&D Systems | DY401 |
| Mouse IL-6 ELISA kit | R&D Systems | DY406 |
| Zombie Aqua Fixable Viability Kit | Biolegend | 423102 |
| CytoTox 96®Non-Radioactive Cytotoxicity assay | Promega | G1780 |
| siRNA | ||
| ALOX15 Trilencer-27 Human siRNA | OriGene | SR300171 |
| human non-targeting siRNA | Thermo Fisher Scientific | D-001810-01-05 |
| Software and Algorithms | ||
| AxioVision Se64 Rel. 4.9 | Carl Zeiss | http://www.zeiss.de/mikroskopie/downloads.html?vaURL=www.zeiss.de/mikroskopie/downloads/axiovision-downloads.html |
| FlowJo X Software | BD Bioscience | https://www.fiowjo.com/solutions/fiowjo/downloads |
| Analyst software 1.6.3 | AB Sciex | https://sciex.com/products/software/analyst-software |
| ImageJ software | NIH, Bethesda | https://imagej.nih.gov/ij/ |
| GraphPad InStat 3 | GraphPad Software Inc | https://www.graphpad.com/scientific-software/instat/ |
| GraphPad Prism 8 | GraphPad Software Inc | https://www.graphpad.com/scientific-software/prism/ |
| Odyssey 3.0 software | LI-COR | https://www.licor.com/bio/image-studio/ |
| SigmaPlot 13.0 software | Systat Software Inc | https://systatsoftware.com/downloads/download-sigmaplot/ |
| OriginPro 2019 | OriginLab Corporation | https://www.additive-net.de/de/software/produkte/originlab/originpro |
Highlights.
S. aureus causes SPM formation in human M2 macrophages by α-hemolysin
α-hemolysin elicits SPM biosynthesis by activation of 15-lipoxygenase-1 in M2
Inhibition of ADAM10 or 15-lipoxygenase-1 knockdown reverts α-hemolysin actions
α-hemolysin elevates SPM levels in mouse peritoneum devoid of leukocyte influx
ACKNOWLEDGMENTS
This work was supported by the Deutsche Forschungsgemeinschaft, SFB 1127/2 ChemBioSys 239748522, and SFB1278/1 Polytarget 316213987 project A04. Z.R. received funding from China Scholarship Council and J.G. received a Carl-Zeiss stipend. V.A. was supported by the International Leibniz Research School for Microbial and Biomolecular Interactions. C.G.-E. was funded by the Leibniz Institute on Aging (FLI), a member of the Leibniz Association, and is financially supported by the Federal Government of Germany and the State of Thuringia. C.N.S. contributions are supported by program project P01GM095467 from the National Institutes of Health, USA. We thank S. Wendler, R. Beier, M. Schreier, and L. Peltner for expert technical assistance.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.108247.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Adel S, Karst F, González-Lafont À, Pekárová M, Saura P, Masgrau L, Lluch JM, Stehling S, Horn T, Kuhn H, and Heydeck D (2016). Evolutionary alteration of ALOX15 specificity optimizes the biosynthesis of anti-inflammatory and proresolving lipoxins. Proc. Natl. Acad. Sci. USA 113, E4266–E4275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed S, Meghji S, Williams RJ, Henderson B, Brock JH, and Nair SP (2001). Staphylococcus aureus fibronectin binding proteins are essential for internalization by osteoblasts but do not account for differences in intracellular levels of bacteria. Infect. Immun 69, 2872–2877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atri C, Guerfali FZ, and Laouini D (2018). Role of Human Macrophage Polarization in Inflammation during Infectious Diseases. Int. J. Mol. Sci 19, 1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berube BJ, and Bubeck Wardenburg J (2013). Staphylococcus aureus α-toxin: nearly a century of intrigue. Toxins (Basel) 5, 1140–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berube BJ, Sampedro GR, Otto M, and Bubeck Wardenburg J (2014). The psma locus regulates production of Staphylococcus aureus alpha-toxin during infection. Infect. Immun 82, 3350–3358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blake KJ, Baral P, Voisin T, Lubkin A, Pinho-Ribeiro FA, Adams KL, Roberson DP, Ma YC, Otto M, Woolf CJ, et al. (2018). Staphylococcus aureus produces pain through pore-forming toxins and neuronal TRPV1 that is silenced by QX-314. Nat. Commun 9, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouillot S, Reboud E, and Huber P (2018). Functional Consequences of Calcium Influx Promoted by Bacterial Pore-Forming Toxins. Toxins (Basel) 10, E387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bronner S, Monteil H, and Prévost G (2004). Regulation of virulence determinants in Staphylococcus aureus: complexity and applications. FEMS Microbiol. Rev 28, 183–200. [DOI] [PubMed] [Google Scholar]
- Chang SW, Czartolomna J, and Voelkel NF (1992). Role of eicosanoids in staphylococcal alpha-toxin-induced lung injury in the rat. Am. J. Physiol 262, L502–L510. [DOI] [PubMed] [Google Scholar]
- Cheung GY, Wang R, Khan BA, Sturdevant DE, and Otto M (2011). Role of the accessory gene regulator agr in community-associated methicillin-resistant Staphylococcus aureus pathogenesis. Infect. Immun 79, 1927–1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung GY, Joo HS, Chatterjee SS, and Otto M (2014). Phenol-soluble modulins—critical determinants of staphylococcal virulence. FEMS Microbiol. Rev 38, 698–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang N, Fredman G, Bäckhed F, Oh SF, Vickery T, Schmidt BA, and Serhan CN (2012). Infection regulates pro-resolving mediators that lower antibiotic requirements. Nature 484, 524–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colas RA, Shinohara M, Dalli J, Chiang N, and Serhan CN (2014). Identification and signature profiles for pro-resolving and inflammatory lipid mediators in human tissue. Am. J. Physiol. Cell Physiol 307, C39–C54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalli J, and Serhan CN (2012). Specific lipid mediator signatures of human phagocytes: microparticles stimulate macrophage efferocytosis and pro-resolving mediators. Blood 120, e60–e72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalli J, and Serhan C (2016). Macrophage Proresolving Mediators-the When and Where. Microbiol. Spectr 4, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalli J, Chiang N, and Serhan CN (2015a). Elucidation of novel 13-series resolvins that increase with atorvastatin and clear infections. Nat. Med 21, 1071–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalli J, Ramon S, Norris PC, Colas RA, and Serhan CN (2015b). Novel proresolving and tissue-regenerative resolvin and protectin sulfido-conjugated pathways. FASEB J. 29, 2120–2136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalli J, Sanger JM, Rodriguez AR, Chiang N, Spur BW, and Serhan CN (2016). Identification and Actions of a Novel Third Maresin Conjugate in Tissue Regeneration: MCTR3. PLoS One 11, e0149319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dioszeghy V, Rosas M, Maskrey BH, Colmont C, Topley N, Chaitidis P, Kühn H, Jones SA, Taylor PR, and O’Donnell VB (2008). 12/15-Lipoxygenase regulates the inflammatory response to bacterial products in vivo. J. Immunol 181, 6514–6524. [DOI] [PubMed] [Google Scholar]
- DuMont AL, and Torres VJ (2014). Cell targeting by the Staphylococcus aureus pore-forming toxins: it’s not just about lipids. Trends Microbiol. 22, 21–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ezekwe EA Jr., Weng C, and Duncan JA (2016). ADAM10 Cell Surface Expression but Not Activity Is Critical for Staphylococcus aureus α-Hemolysin-Mediated Activation of the NLRP3 Inflammasome in Human Monocytes. Toxins (Basel) 8, 95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fink D, Contreras ML, Lelkes PI, and Lazarovici P (1989). Staphylococcus aureus alpha-toxin activates phospholipases and induces a Ca2+ influx in PC12 cells. Cell. Signal 1, 387–393. [DOI] [PubMed] [Google Scholar]
- Foster TJ (2005). Immune evasion by staphylococci. Nat. Rev. Microbiol 3, 948–958. [DOI] [PubMed] [Google Scholar]
- Funk CD (2001). Prostaglandins and leukotrienes: advances in eicosanoid biology. Science 294, 1871–1875. [DOI] [PubMed] [Google Scholar]
- Gao ZH, Deng CJ, Xie YY, Guo XL, Wang QQ, Liu LZ, Lee WH, Li SA, and Zhang Y (2019). Pore-forming toxin-like protein complex expressed by frog promotes tissue repair. FASEB J. 33, 782–795. [DOI] [PubMed] [Google Scholar]
- Gijón MA, Spencer DM, Siddiqi AR, Bonventre JV, and Leslie CC (2000). Cytosolic phospholipase A2 is required for macrophage arachidonic acid release by agonists that Do and Do not mobilize calcium. Novel role of mitogen-activated protein kinase pathways in cytosolic phospholipase A2 regulation. J. Biol. Chem 275, 20146–20156. [DOI] [PubMed] [Google Scholar]
- Goldmann O, and Medina E (2018). Staphylococcus aureus strategies to evade the host acquired immune response. Int. J. Med. Microbiol 308, 625–630. [DOI] [PubMed] [Google Scholar]
- Grandel U, Bennemann U, Buerke M, Hattar K, Seeger W, Grimminger F, and Sibelius U (2009). Staphylococcus aureus alpha-toxin and Escherichia coli hemolysin impair cardiac regional perfusion and contractile function by activating myocardial eicosanoid metabolism in isolated rat hearts. Crit. Care Med 37, 2025–2032. [DOI] [PubMed] [Google Scholar]
- Grimminger F, Rose F, Sibelius U, Meinhardt M, Pötzsch B, Spriestersbach R, Bhakdi S, Suttorp N, and Seeger W (1997). Human endothelial cell activation and mediator release in response to the bacterial exotoxins Escherichia coli hemolysin and staphylococcal alpha-toxin. J. Immunol 159, 1909–1916. [PubMed] [Google Scholar]
- Horiuchi K, Le Gall S, Schulte M, Yamaguchi T, Reiss K, Murphy G, Toyama Y, Hartmann D, Saftig P, and Blobel CP (2007). Substrate selectivity of epidermal growth factor-receptor ligand sheddases and their regulation by phorbol esters and calcium influx. Mol. Biol. Cell 18, 176–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoshima I, Inoshima N, Wilke GA, Powers ME, Frank KM, Wang Y, and Bubeck Wardenburg J (2011). A Staphylococcus aureus pore-forming toxin subverts the activity of ADAM10 to cause lethal infection in mice. Nat. Med 17, 1310–1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov I, Kuhn H, and Heydeck D (2015). Structural and functional biology of arachidonic acid 15-lipoxygenase-1 (ALOX15). Gene 573, 1–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kieseier BC, Pischel H, Neuen-Jacob E, Tourtellotte WW, and Hartung HP (2003). ADAM-10 and ADAM-17 in the inflamed human CNS. Glia 42, 398–405. [DOI] [PubMed] [Google Scholar]
- Kim SN, Akindehin S, Kwon HJ, Son YH, Saha A, Jung YS, Seong JK, Lim KM, Sung JH, Maddipati KR, and Lee YH (2018). Anti-inflammatory role of 15-lipoxygenase contributes to the maintenance of skin integrity in mice. Sci. Rep 8, 8856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knox J, Uhlemann AC, and Lowy FD (2015). Staphylococcus aureus infections: transmission within households and the community. Trends Microbiol. 23, 437–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kretschmer D, Gleske AK, Rautenberg M, Wang R, Köberle M, Bohn E, Schöneberg T, Rabiet MJ, Boulay F, Klebanoff SJ, et al. (2010). Human formyl peptide receptor 2 senses highly pathogenic Staphylococcus aureus. Cell Host Microbe 7, 463–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krysko O, Holtappels G, Zhang N, Kubica M, Deswarte K, Derycke L, Claeys S, Hammad H, Brusselle GG, Vandenabeele P, et al. (2011). Alternatively activated macrophages and impaired phagocytosis of S. aureus in chronic rhinosinusitis. Allergy 66, 396–403. [DOI] [PubMed] [Google Scholar]
- Kuhn H, Heydeck D, Brinckman R, and Trebus F (1999). Regulation of cellular 15-lipoxygenase activity on pretranslational, translational, and post-translational levels. Lipids 34, 273–279. [DOI] [PubMed] [Google Scholar]
- Levy BD, Clish CB, Schmidt B, Gronert K, and Serhan CN (2001). Lipid mediator class switching during acute inflammation: signals in resolution. Nat. Immunol 2, 612–619. [DOI] [PubMed] [Google Scholar]
- Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, Gordon S, Hamilton JA, Ivashkiv LB, Lawrence T, et al. (2014). Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity 41, 14–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norris PC, Gosselin D, Reichart D, Glass CK, and Dennis EA (2014). Phospholipase A2 regulates eicosanoid class switching during inflammasome activation. Proc. Natl. Acad. Sci. USA 111, 12746–12751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto M (2014a). Phenol-soluble modulins. Int. J. Med. Microbiol 304, 164–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto M (2014b). Staphylococcus aureus toxins. Curr. Opin. Microbiol 17, 32–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pace S, Pergola C, Dehm F, Rossi A, Gerstmeier J, Troisi F, Pein H, Schaible AM, Weinigel C, Rummler S, et al. (2017). Androgen-mediated sex bias impairs efficiency of leukotriene biosynthesis inhibitors in males. J. Clin. Invest 127, 3167–3176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pergola C, Gerstmeier J, Mönch B,Çalışkan B, Luderer S, Weinigel C, Barz D, Maczewsky J, Pace S, Rossi A, et al. (2014). The novel benzimidazole derivative BRP-7 inhibits leukotriene biosynthesis in vitro and in vivo by targeting 5-lipoxygenase-activating protein (FLAP). Br. J. Pharmacol 171, 3051–3064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pezato R, Świerczyńska-Krępa M, Niżankowska-Mogilnicka E, Derycke L, Bachert C, and Pérez-Novo CA (2012). Role of imbalance of eicosanoid pathways and staphylococcal superantigens in chronic rhinosinusitis. Allergy 67, 1347–1356. [DOI] [PubMed] [Google Scholar]
- Pfaffl MW (2001). A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 29, e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruessmeyer J, Hess FM, Alert H, Groth E, Pasqualon T, Schwarz N, Nyamoya S, Kollert J, van der Vorst E, Donners M, et al. (2014). Leukocytes require ADAM10 but not ADAM17 for their migration and inflammatory recruitment into the alveolar space. Blood 123, 4077–4088. [DOI] [PubMed] [Google Scholar]
- Rådmark O, Werz O, Steinhilber D, and Samuelsson B (2015). 5-Lipoxygenase, a key enzyme for leukotriene biosynthesis in health and disease. Biochim. Biophys. Acta 1851, 331–339. [DOI] [PubMed] [Google Scholar]
- Rao TS, Currie JL, Shaffer AF, and Isakson PC (1994). In vivo characterization of zymosan-induced mouse peritoneal inflammation. J. Pharmacol. Exp. Ther 269, 917–925. [PubMed] [Google Scholar]
- Romp E, Arakandy V, Fischer J, Wolz C, Siegmund A, Löffler B, Tuchscherr L, Werz O, and Garscha U (2019). Exotoxins from Staphylococcus aureus activate 5-lipoxygenase and induce leukotriene biosynthesis. Cell. Mol. Life Sci 77, 3841–3858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi A, Pergola C, Pace S, Rådmark O, Werz O, and Sautebin L (2014). In vivo sex differences in leukotriene biosynthesis in zymosan-induced peritonitis. Pharmacol. Res 87, 1–7. [DOI] [PubMed] [Google Scholar]
- Schmitt J, Joost I, Skaar EP, Herrmann M, and Bischoff M (2012). Haemin represses the haemolytic activity of Staphylococcus aureus in an Sae-dependent manner. Microbiology (Reading) 158, 2619–2631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwab JM, Chiang N, Arita M, and Serhan CN (2007). Resolvin E1 and protectin D1 activate inflammation-resolution programmes. Nature 447, 869–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serhan CN (2014). Pro-resolving lipid mediators are leads for resolution physiology. Nature 510, 92–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serhan CN, and Levy BD (2018). Resolvins in inflammation: emergence of the pro-resolving superfamily of mediators. J. Clin. Invest 128, 2657–2669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serhan CN, Dalli J, Karamnov S, Choi A, Park CK, Xu ZZ, Ji RR, Zhu M, and Petasis NA (2012). Macrophage proresolving mediator maresin 1 stimulates tissue regeneration and controls pain. FASEB J. 26, 1755–1765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serhan CN, Chiang N, and Dalli J (2015). The resolution code of acute inflammation: Novel pro-resolving lipid mediators in resolution. Semin. Immunol 27, 200–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seyyed Mousavi MN, Mehramuz B, Sadeghi J, Alizadeh N, Oskouee MA, and Kafil HS (2017). The pathogenesis of Staphylococcus aureus in autoimmune diseases. Microb. Pathog 111, 503–507. [DOI] [PubMed] [Google Scholar]
- Sibelius U, Grandel U, Buerke M, Mueller D, Kiss L, Kraemer HJ, Braun-Dullaeus R, Haberbosch W, Seeger W, and Grimminger F (2000). Staphylococcal alpha-toxin provokes coronary vasoconstriction and loss in myocardial contractility in perfused rat hearts: role of thromboxane generation. Circulation 101, 78–85. [DOI] [PubMed] [Google Scholar]
- Spite M, Norling LV, Summers L, Yang R, Cooper D, Petasis NA, Flower RJ, Perretti M, and Serhan CN (2009). Resolvin D2 is a potent regulator of leukocytes and controls microbial sepsis. Nature 461, 1287–1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strobel M, Pförtner H, Tuchscherr L, Völker U, Schmidt F, Kramko N, Schnittler HJ, Fraunholz MJ, Löffler B, Peters G, and Niemann S (2016). Post-invasion events after infection with Staphylococcus aureus are strongly dependent on both the host cell type and the infecting S. aureus strain. Clin. Microbiol. Infect 22, 799–809. [DOI] [PubMed] [Google Scholar]
- Tong SY, Davis JS, Eichenberger E, Holland TL, and Fowler VG Jr. (2015). Staphylococcus aureus infections: epidemiology, pathophysiology, clinical manifestations, and management. Clin. Microbiol. Rev 28, 603–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuchscherr L, Medina E, Hussain M, Völker W, Heitmann V, Niemann S, Holzinger D, Roth J, Proctor RA, Becker K, et al. (2011). Staphylococcus aureus phenotype switching: an effective bacterial strategy to escape host immune response and establish a chronic infection. EMBO Mol. Med 3, 129–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuchscherr L, Bischoff M, Lattar SM, Noto Llana M, Pförtner H, Niemann S, Geraci J, Van de Vyver H, Fraunholz MJ, Cheung AL, et al. (2015). Sigma Factor SigB Is Crucial to Mediate Staphylococcus aureus Adaptation during Chronic Infections. PLoS Pathog. 11, e1004870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uderhardt S, and Krönke G (2012). 12/15-lipoxygenase during the regulation of inflammation, immunity, and self-tolerance. J. Mol. Med. (Berl.) 90, 1247–1256. [DOI] [PubMed] [Google Scholar]
- von Hoven G, Qin Q, Neukirch C, Husmann M, and Hellmann N (2019). Staphylococcus aureus α-toxin: small pore, large consequences. Biol. Chem 400, 1261–1276. [DOI] [PubMed] [Google Scholar]
- Werner M, Jordan PM, Romp E, Czapka A, Rao Z, Kretzer C, Koeberle A, Garscha U, Pace S, Claesson HE, et al. (2019). Targeting biosynthetic networks of the proinflammatory and proresolving lipid metabolome. FASEB J. 33, 6140–6153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werz O (2002). 5-lipoxygenase: cellular biology and molecular pharmacology. Curr. Drug Targets Inflamm. Allergy 1, 23–44. [DOI] [PubMed] [Google Scholar]
- Werz O, Gerstmeier J, Libreros S, De la Rosa X, Werner M, Norris PC, Chiang N, and Serhan CN (2018). Human macrophages differentially produce specific resolvin or leukotriene signals that depend on bacterial pathogenicity. Nat. Commun 9, 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilke GA, and Bubeck Wardenburg J (2010). Role of a disintegrin and metalloprotease 10 in Staphylococcus aureus alpha-hemolysin-mediated cellular injury. Proc. Natl. Acad. Sci. USA 107, 13473–13478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada T, Tani Y, Nakanishi H, Taguchi R, Arita M, and Arai H (2011). Eosinophils promote resolution of acute peritonitis by producing proresolving mediators in mice. FASEB J. 25, 561–568. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Lei J, Xie Q, Wu L, Jin S, Guo B, Wang X, Yan G, Zhang Q, Zhao H, et al. (2019). Fibrinogen-like protein 2 controls sepsis catabasis by interacting with resolvin Dp5. Sci. Adv 5, eaax0629. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate/analyze datasets/code.