

Abstract
Cholinesterase (ChE) and monoamine oxidase (MAO) inhibitors have been attracted as candidate treatments for Alzheimer's disease (AD). Fifteen khellactone-type coumarins from the roots of Peucedanum japonicum Thunberg were tested for acetylcholinesterase (AChE), butyrylcholinesterase (BChE), and MAO inhibitory activities. Compound 3′-angeloyl-4′-(2-methylbutyryl)khellactone (PJ13) most potently inhibited AChE (IC50 = 9.28 µM), followed by 3′-isovaleryl-4′-(2-methylbutyroyl)khellactone (PJ15) (IC50 = 10.0 μM). Compound senecioyl-4′-angeloyl-khellactone (PJ5) most potently inhibited BChE (IC50 = 7.22 μM) and had the highest selectivity index (> 5.54), followed by 3′-senecioyl-4′-(2-methylbutyryl)khellactone (PJ10) and 3′,4′-disenecioylkhellactone (PJ4) (IC50 = 10.2 and 10.7 μM, respectively). Compounds PJ13, PJ15, and PJ5 showed reversible and mixed-types of inhibition with Ki values of 5.98, 10.4 (for AChE), and 4.16 µM (for BChE), respectively. However, all 15 compounds weakly inhibited MAO-A and MAO-B. Molecular docking simulation revealed that PJ13 had a higher binding affinity (− 9.3 kcal/mol) with AChE than PJ15 (− 7.8 kcal/mol) or PJ5 (− 5.4 kcal/mol), due to the formation of a hydrogen bond with Tyr121 (distance: 2.52 Å). On the other hand, the binding affinity of PJ5 (− 10.0 kcal/mol) with BChE was higher than for PJ13 (− 7.7 kcal/mol) or PJ15 (− 8.1 kcal/mol), due to the formation of a hydrogen bond with Ser198 (distance: 2.05 Å). These results suggest that PJ13 and PJ5 are potential reversible selective inhibitors of AChE and BChE, respectively, for the treatment of AD.
Subject terms: Biochemistry, Biological techniques, Chemical biology, Drug discovery
Introduction
Acetylcholinesterase (AChE) is a member of α/β hydrolase protein superfamily and breaks down an acetylcholine (ACh) into acetate and choline1. Alzheimer's disease (AD) is an age-associated memory/cognitive disorder, and its mechanism has not been determined, and no curative therapy has been developed2. Since cholinergic deficiency is present in AD, the relation between AChE and AD has been extensively studied2,3. AChE inhibitors (AChEIs) inhibit the hydrolysis of ACh (a neurotransmitter in the central nervous system), and as a result, increase ACh levels and ACh half-lives in autonomic ganglia and neuromuscular junctions, which are rich in ACh receptors4. AChEIs may be reversible or irreversible5,6. Commercially available AChEIs include piperidine-based (e.g., donepezil, Aricept)7, carbamate-based (rivastigmine, Exelon)8, phenanthrene-based (galantamine, Reminyl)9, and other inhibitors. The common potential side effects of AChEIs are diarrhea, headache, insomnia, nausea, and vomiting10. Butyrylcholinesterase (BChE) is mainly expressed in glial cells and white matter in the human brain, and as its name indicated, it breaks down butyrylcholine (BCh). BChE levels are significantly elevated in AD11, and in BChE knockout AD mice, a reported reduction in fibrin Aβ plaque by up to 70% suggests that BChE inhibition has therapeutic value12. Furthermore, AChE and BChE are known to be related to AD and to act independently of each other, which may lead to the diagnosis of disease and the development of potential drug targets13.
Recently dual- or multi-targeting inhibitors of acetylcholinesterase (AChE) and monoamine oxidase (MAO) have attracted research attention as candidate treatments for AD14–19. MAO catalyzes the oxidation of monoamines20, and has two isoforms (MAO-A and MAO-B). MAO was discovered almost a century ago and has been the subject of many structural, pharmacological, and biochemical studies on neurotransmitters21. MAO inhibitors (MAOIs) are currently used to treat depression22 and Parkinson's disease23, and several studies have concluded that MAOIs reduce Aβ plaque24–26, and thus, MAOIs are considered possible future treatments for AD27.
Peucedanum japonicum Thunberg is a herb found on the cliffs of islands in Korea, Japan, and the Philippines, and has traditionally been used to treat coughs, cramps, pain, rheumatism, asthma, and angina28,29. Furthermore, it has been shown to have anti-diabetic and anti-obesity30,31, anti-nociceptive32, anti-osteoporotic33, and anti-allergic lung inflammatory effects34. In traditional medicine, P. japonicum Thunberg is also believed to prevent stroke and vascular disease. On the other hand, an extract of P. japonicum Thunberg (KH020) has been reported to reduce Y-maze alternation behavior, and suggested to have therapeutic value for the prevention and treatment of vascular dementia35. From P. japonicum Thunberg, several compounds such as rutin, 3-O-caffeoylquinic acid, 4-O-caffeoylquinic acid, 5-O-caffeoylquinic acid, cnidioside A, praeroside II, praeroside III, apterin, esculin, (R)-peucedanol, and (R)-peucedanol 7-O-ß-d-glucopyranoside were identified36. In addition, a P. japonicum Thunberg extract was fractionated and found to contain a norisoprenoid glucoside, (3S)-O-ß-d-glucopyranosyl-6-[3-oxo-(2S)-butenylidenyl]-1,1,5-trimethylcyclohexan-(5R)-ol, and two phenylpropanoid glucosides, namely, 3-(2-O-ß-d-glucopyranosyl-4-hydroxyphenyl)-propanoic acid and methyl 3-(2-O-ß-d-glucopyranosyl-4-hydroxyphenyl) propanoate37. In another study, 80% EtOH was found to also contain peucedanol 7-O-β-d-glucopyranoside and myo-inositol38. Recently, khellactone coumarins were isolated from subfractions of P. japonicum roots by recycling HPLC, and reported to reduce NO levels in LPS-stimulated RAW264.7 cells and to inhibit anti-inflammatory response39.
However, little information is available about the anticholinergic actions of khellactone coumarins. Accordingly, we investigated the inhibitory effects of khellactone coumarins from P. japonicum Thunberg on AChE, BChE, and MAOs. In addition, we investigated the bindings and kinetics of the potent inhibitors senecioyl-4′-angeloyl-khellactone (PJ5), 3′-angeloyl-4′-(2-methylbutyryl)khellactone (PJ13), and 3′-isovaleryl-4′-(2-methylbutyroyl)khellactone (PJ15), and performed molecular docking simulations of these three compounds with AChE and BChE.
Materials and methods
Compounds
Fifteen khellactone-type compounds were isolated from P. japonicum Thunberg (voucher specimen: PBC-484), and the structures were determined, as described previously39. Briefly, the dried roots of P. japonicum (5.0 kg) were extracted with 80% ethanol (EtOH) at room temperature three times to obtain 1.62 kg of solid extract. The 80% EtOH extract was further partitioned between n-hexane (114.2 g) and H2O (1.50 kg), and the n-hexane extract so obtained was subjected to preparative reverse phase chromatography (Xbridge Prep C18, 5 μm, Waters Corporation, Milford, MA, USA) using methanol (MeOH) and H2O (0–52.0 min, 66–88% MeOH; 52.0–53.0 min, 88–100% MeOH; 53.0–60.0 min, 100% MeOH). The fractions (Frs. 1–8) were collected and concentrated on a rotary evaporator under reduced pressure. Purification was conducted by recycling preparative HPLC. The yield of the khellactone-type coumarins obtained was ~ 1.5% from 80% EtOH extract determined by using ultra-performance liquid chromatography (UPLC) charged with photodiode array (PDA). Chemical structures of the compounds were identified by 1H NMR, 13C NMR, CD spectrum, UV spectrum, MS/MS, and HR-ESI–MS data (Supplementary Information 1) and their purities were determined by HPLC. The structures are shown in Fig. 1.
Figure 1.

Chemical structures of khellactone coumarin derivatives from Peucedanum japonicum Thunberg39. PJ1, Isosamidin; PJ2, Pteryxin; PJ3, hyuganin; PJ4, 3′,4′-disenecioylkhellactone; PJ5, senecioyl-4′-angeloyl-khellactone; PJ6, calipteryxin; PJ7, anomalin; PJ8, 3′-senecioyl-4′-isovalerylkhellactone; PJ9, 3′-isovaleryl-4′-senecioylkhellactone; PJ10, 3′-senecioyl-4′-(2-methylbutyryl)khellactone; PJ11, 3′-isovaleryl-4′-angeloylkhellactone; PJ12, 3′-isovaleryl-4′-angeloylkhellactone; PJ13, 3′-angeloyl-4′-(2-methylbutyryl)khellactone; PJ14, 3′,4′-diisovalerylkhellactone; PJ15, 3′-isovaleryl-4′-(2-methylbutyroyl)khellactone.
Chemicals and enzymes
AChE (Type VI-S; from Electrophorus electricus), recombinant human MAO-A, MAO-B, and BChE (from equine serum), acetylthiocholine iodide (ATCI), kynuramine, benzylamine, S-butyrylthiocholine iodide (BTCI), 5,5′-dithiobis (2-nitrobenzoic acid) (DTNB), tacrine, donepezil, toloxatone, and lazabemide were purchased from Sigma-Aldrich (St. Louis, MO, USA). Clorgyline and pargyline (irreversible reference inhibitors of MAO-A and MAO-B, respectively) were from BioAssay Systems (Hayward, CA, USA)40.
Enzyme assays
AChE assays were performed as described by Ellman et al.41 with slight modifications42. In brief, assays were performed using ~ 0.2 U/mL of AChE in the presence of 0.5 mM DTNB and 0.5 mM ACTI in 0.5 mL reaction mixtures, and continuously monitored for 10 min at 412 nm. DTNB was used for color development, caused by reaction between it and thiocholine (a product of AChE). For inhibitory assays, compounds were preincubated with AChE for 15 min prior to ATCI and DTNB addition. BChE activity was assayed using the same method, but BTCI was used instead of ATCI. MAO-A activity was continuously assayed using kynuramine (a substrate) at 316 nm for 20 min, and MAO-B activity was assayed using benzylamine at 250 nm for 30 min, as described previously40.
Inhibitory activities and enzyme kinetics
Inhibitions of the activities of AChE, BChE, MAO-A, and MAO-B by the 15 compounds were investigated at an inhibitor concentration of 10 µM. IC50 values were also determined. The reference reversible inhibitors of AChE and BChE, MAO-A, and MAO-B used were tacrine (or donepezil), toloxatone and lazabemide, respectively, and the reference irreversible inhibitors of MAO-A and MAO-B used were clorgyline and pargyline, respectively. Kinetic parameters, inhibition types, and Ki values of PJ5 (for BChE), PJ13 and PJ15 (for AChE) were determined as the methods previously described43. Enzyme kinetics were investigated at five different substrate concentrations, that is, at 0, ~ 1/2 × IC50, IC50, and 2 × IC50 for each inhibitor. The inhibition types and Ki values were determined using Lineweaver–Burk Plots and secondary plots.
Analysis of inhibitor reversibilities
Inhibitor reversibilities were examined using the dialysis method44, using with AChE or BChE, rather than MAO enzymes. In brief, the experiment was performed by preincubating an inhibitor at ~ 2 × IC50 concentration with AChE or BChE for 30 min in 0.1 M sodium phosphate buffer (pH 7.2). Dialysis was conducted for 6 h with stirring and two buffer changes. Residual activities before (AU) and after (AD) dialysis were compared to those of non-treated controls, and reversibility types were determined by comparing AD and AU values.
Docking simulations of PJ5, PJ13, and PJ15 with AChE or BChE
To simulate the dockings of PJ5, PJ13, and PJ15 with AChE or BChE, we used Autodock Vina45, which has an automated docking facility. To define enzyme pockets, we used predefined active sites obtained from complexes between AChE and 3-[(1S)-1-(dimethylamino)ethyl]phenol (PDB ID: 1GQS) or donepezil (PDB ID: 6O4W), BChE and butyl-[(2 ~ {S})-1-(2-cycloheptylethylamino)-3-(1~{H}-indol-3-yl)-1-oxidanylidene-propan-2-yl]azanium (PDB ID: 6QAA), MAO-A and 7-methoxy-1-methyl-9H-β-carboline complex (PDB ID: 2Z5X), and MAO-B and pioglitazone complex (PDB ID: 4A79). To prepare PJ5, PJ13, and PJ15 for docking simulation, ChemOffice program (http://www.cambridgesoft.com) was used to create the 2D structures of PJ5, PJ13, and PJ15, to convert them into 3D structures, and to perform energy minimizations. Docking simulations of the enzymes with PJ5, PJ13, and PJ15 were performed using Chimera46. Based on the results of docking simulations, we checked for possible hydrogen bonding using bonding relaxation constraints of 0.4 Å and 20.0° using Chimera47.
Analysis of pharmacokinetic properties using in silico method
Drug-like properties of the lead compounds of PJ5, PJ13, and PJ15 were analyzed using a web tool of SwissADME at http://www.swissadme.ch/48.
Results
Analysis of inhibitory activities
The structures and purities of the 15 compounds isolated from the P. japonicum Thunberg, were determined by 1D and 2D NMR spectra, UPLC-QTOF-MS analysis, and electronic circular dichroism spectra39. All were tested for AChE and BChE inhibitory activities at a concentration of 10 µM. PJ13 and PJ15 resulted in AChE residual activity of < 50% (Table 1). PJ13 most potently inhibited AChE with an IC50 value of 9.28 µM, followed by PJ15 and PJ7 (IC50 = 10.0 and 17.9 µM, respectively). The other 12 compounds had IC50 values of ≥ 20 µM. In addition, four compounds resulted in BChE residual activity of < 50% (Table 1). PJ5 most potently inhibited BChE with an IC50 value of 7.22 µM, followed by PJ10 PJ4, and PJ9 (IC50 = 10.16, 10.66, 12.5 µM, respectively) (Table 1). The other 11 compounds had IC50 values of ≥ 40 µM. PJ5 had the highest selectivity index of > 5.54. To examine the multi-targeting abilities of the compounds, we evaluated their inhibitory effects on MAO-A or MAO-B, which are auxiliary targets in AD. However, all compounds only weakly inhibited MAO-A or MAO-B with residual activities of > 63.1% at 10 µM (Table 1).
Table 1.
Inhibitions of AChE, BChE, MAO-A, and MAO-B by khellactone coumarins from Peucedanum japonicum Thunberg roots.
| Compounds | Residual activity at 10 µM (%) | IC50 (µM) | SIa | ||||
|---|---|---|---|---|---|---|---|
| AChE | BChE | MAO-A | MAO-B | AChE | BChE | ||
| PJ1 | 92.1 ± 1.70 | 87.2 ± 1.21 | 86.0 ± 5.47 | 85.0 ± 0.57 | > 40 | > 40 | |
| PJ2 | 73.4 ± 11.3 | 87.6 ± 7.14 | 85.4 ± 3.80 | 79.7 ± 3.47 | > 40 | > 40 | |
| PJ3 | 97.2 ± 8.30 | 78.7 ± 4.03 | 81.2 ± 3.68 | 83.7 ± 2.33 | > 40 | > 40 | |
| PJ4 | 85.4 ± 7.52 | 47.2 ± 1.95 | 100.3 ± 0.39 | 95.6 ± 4.80 | 21.3 ± 7.69 | 10.7 ± 0.060 | 1.99 |
| PJ5 | 96.0 ± 2.83 | 34.9 ± 9.64 | 84.7 ± 9.30 | 96.0 ± 0.52 | > 40 | 7.20 ± 0.79 | > 5.56 |
| PJ6 | 76.3 ± 6.46 | 84.3 ± 8.52 | 86.1 ± 1.93 | 63.1 ± 2.75 | 25.6 ± 4.50 | > 40 | < 0.64 |
| PJ7 | 75.4 ± 5.72 | 69.9 ± 6.82 | 74.0 ± 1.16 | 73.8 ± 8.23 | 17.9 ± 5.59 | > 40 | < 0.45 |
| PJ8 | 97.0 ± 1.40 | 76.8 ± 3.85 | 79.5 ± 2.05 | 68.5 ± 2.10 | 31.6 ± 4.40 | > 40 | < 0.79 |
| PJ9 | 86.9 ± 9.92 | 50.9 ± 1.11 | 80.6 ± 5.02 | 75.0 ± 8.42 | 36.1 ± 0.66 | 12.5 ± 2.82 | 2.89 |
| PJ10 | 86.4 ± 5.23 | 48.3 ± 2.78 | 92.1 ± 5.58 | 70.9 ± 2.61 | > 40 | 10.2 ± 2.25 | > 3.92 |
| PJ11 | 88.1 ± 1.35 | 86.4 ± 3.20 | 75.4 ± 0.30 | 89.1 ± 1.36 | > 40 | > 40 | |
| PJ12 | 58.3 ± 5.37 | 79.1 ± 3.32 | 78.9 ± 3.98 | 72.4 ± 3.70 | 29.0 ± 1.15 | > 40 | < 0.73 |
| PJ13 | 48.0 ± 9.40 | 75.3 ± 1.77 | 88.5 ± 5.41 | 64.6 ± 8.82 | 9.28 ± 0.094 | > 40 | < 0.23 |
| PJ14 | 51.5 ± 4.50 | 77.7 ± 0.80 | 77.0 ± 5.40 | 66.1 ± 5.23 | 28.1 ± 0.33 | > 40 | < 0.70 |
| PJ15 | 50.0 ± 2.82 | 75.0 ± 6.24 | 76.9 ± 4.20 | 73.3 ± 7.21 | 10.0 ± 0.48 | > 40 | < 0.25 |
| Toloxatone | 1.08 ± 0.025b | – | |||||
| Lazabemide | – | 0.063 ± 0.015 b | |||||
| Clorgyline | 0.007 ± 0.00070 b | – | |||||
| Pargyline | – | 0.028 ± 0.0043 b | |||||
| Tacrine | 0.27 ± 0.019 | 0.0087 ± 0.0009 | 31.0 | ||||
| Donepezil | 0.0095 ± 0.0019 | 0.18 ± 0.0038 | 0.053 | ||||
The values above are the means ± SEs of duplicate or triplicate experiments. Values for AChE and BChE were determined after preincubation of the enzymes with each compound for 15 min. a SI = IC50 of AChE/ IC50 of BChE, b IC50 value.
Reversibilities of AChE and BChE inhibitions
Inhibitory assays were carried out after preincubating AChE or BChE with inhibitors for 15 min. The reversibilities of AChE inhibitions by PJ13 and PJ15 were investigated using a dialysis-based method. Inhibitions of AChE by PJ13 and PJ15 recovered from 34.7% (AU) to 72.3% (AD) and from 32.8% to 68.7%, respectively, which were similar to those shown by tacrine (from 14.7% to 73.6%), a reversible AChE inhibitor (Fig. 2A). In addition, inhibition of BChE by PJ5 recovered from 41.2% (AU) to 86.8% (AD), which was similar to that of tacrine (from 29.9% to 100%), also a reversible BChE inhibitor (Fig. 2B). These results indicate that PJ13 and PJ15 are reversible inhibitors of AChE and PJ5 is a reversible inhibitor of BChE.
Figure 2.

Recoveries of AChE inhibitions by PJ13 and PJ15 (A) and BChE inhibition by PJ5 (B) after dialysis. Tacrine was used as the reference reversible inhibitor. The concentrations of the inhibitors used were ~ 2 × IC50: PJ13, 20 µM; PJ15, 20 µM; PJ5, 14 µM; and tacrine, 0.54 µM. For recovery experiments, preincubated enzyme mixtures were dialyzed as described in the text.
Analysis of inhibitory patterns
Modes of AChE inhibitions by PJ13 and PJ15 were investigated using Lineweaver–Burk plots. Plots of AChE inhibition by PJ13 were linear and lines intersected at a point, but not at the x- or y-axis (Fig. 3A). Secondary plots of the slopes of Lineweaver–Burk plots against inhibitor concentrations showed that the Ki value of PJ13 for AChE inhibition was 5.99 ± 0.21 µM (Fig. 3B). Plots of AChE inhibitions by PJ15 were also linear and did not intersect at the x- or y-axis (Fig. 3C), and the Ki value of PJ15 for the AChE inhibition was 10.41 ± 0.67 μM (Fig. 3D). These results show PJ13 and PJ15 acted as mixed-type inhibitors of AChE. In addition, plots of BChE inhibition by PJ5 were linear and intersected near the y-axis (Fig. 3E). Secondary plots showed the Ki value of PJ5 for BChE inhibition was 4.16 ± 0.72 µM (Fig. 3F), showing PJ5 acted as a mixed-type BChE inhibitor.
Figure 3.

Lineweaver–Burk plots for the inhibitions of AChE by PJ13 (A) and PJ15 (C), and of BChE by PJ5 (E), and respective secondary plots (B, D, F) of slopes against inhibitor concentration. Substrates were used at five different concentrations (0.05–1.0 mM). Experiments were carried out at three inhibitor concentrations at around their respective IC50 values. Initial reaction rates are expressed as increases in absorbance per min. Km values of AChE and BChE were 0.1 and 0.18 mM, respectively.
Molecular docking simulation
AutoDock Vina docking simulations showed that PJ5, PJ13, and PJ15 located well at the binding site of 3-[(1S)-1-(dimethylamino)ethyl]phenol complexed with AChE and at that of butyl-[(2 ~ {S})-1-(2-cycloheptylethylamino)-3-(1 ~ {H}-indol-3-yl)-1-oxidanylidene-propan-2-yl]azanium complexed with BChE. The results of the docking simulation for AChE showed that PJ13 interacted by forming a hydrogen bond with Tyr121 (distance: 2.52 Å). However, no hydrogen bond interaction was predicted for PJ5 and PJ15 (Fig. 4A–C). Docking simulation of PJ5 with BChE implied that a hydrogen bonding interaction was established with Ser198 (distance: 2.05 Å) of BChE, whereas no hydrogen bond was proposed for PJ13 and PJ15 (Fig. 4D–F). The binding affinity of PJ13 (− 9.3 kcal/mol) for AChE was higher than that of PJ15 (− 7.8 kcal/mol) or PJ5 (− 5.4 kcal/mol) (Table 2). In addition, PJ5 had higher binding affinity for BChE (− 10.0 kcal/mol) than PJ13 (− 7.7 kcal/mol) or PJ15 (− 8.1 kcal/mol). The binding affinities of PJ5, PJ13, and PJ15 with MAO-A or MAO-B were predicted to be weaker than those with AChE or BChE (Table 2). Docking simulations were provided in Supplementary Figure S17 (A–F). The binding score (− 4.8 kcal/mol) of PJ13 for MAO-B was relatively higher than those of PJ5 and PJ15 in accordance with the residual activities at 10 µM.
Figure 4.
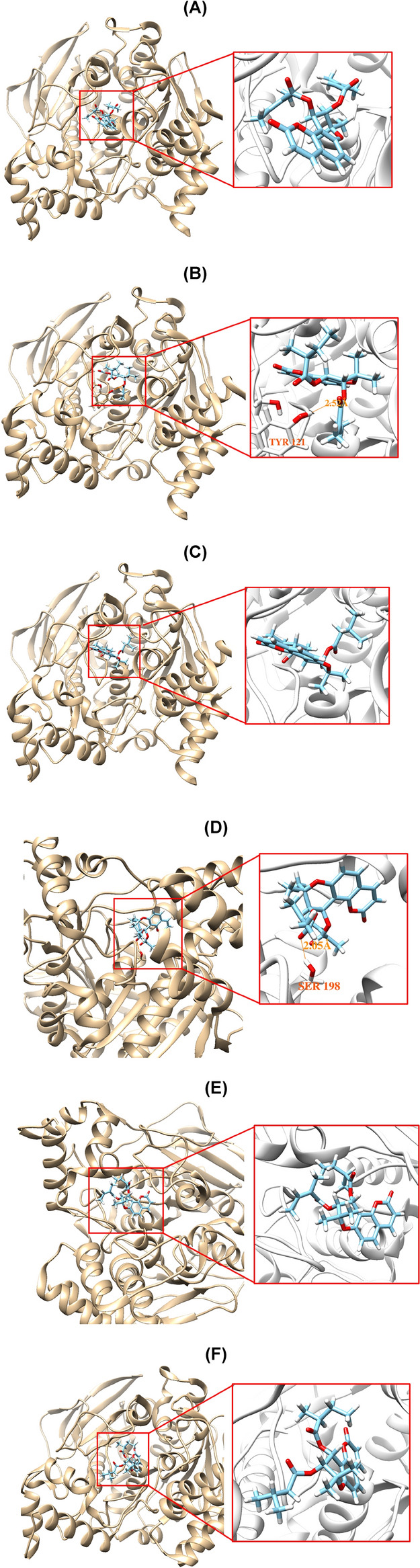
Docking simulations of PJ5, PJ13, and PJ15 with AChE (1GQS) (A–C, respectively) and with BChE (6QAA) (D–F, respectively).
Table 2.
Binding energy values of PJ5, PJ13, and PJ15 to AChE, BChE, MAO-A, and MAO-B.
| Compounds | ∆G (kcal/mol) | |||
|---|---|---|---|---|
| AChE | BChE | MAO-A | MAO-B | |
| PJ5 | − 5.4 (− 3.7) | − 10.0 (− 8.5) | 1.3 (1.7) | 0.3 (− 0.3) |
| PJ13 | − 9.3 (− 8.6) | − 7.7 (− 6.5) | − 1.2 (− 1.6) | − 4.8 (− 4.8) |
| PJ15 | − 7.8 (− 8.7) | − 8.1 (− 6.8) | − 0.6 (− 0.3) | − 3.4 (− 3.4) |
The values in parentheses were obtained from the complexed or pre-defined structures with donepezil.
When the crystal structure of AChE complexed with donepezil (PDB ID: 6O4W) and the binding pockets for BChE, MAO-A, and MAO-B defined with donepezil were used for docking simulations, the binding scores of PJ compounds were similar to the values obtained with their complexed ligands (Tables 2 and Supplementary Table S3). From docking simulations with the AChE/donepezil complex (PDB ID: 6O4W), it was predicted that PJ13 and PJ15 formed one hydrogen bond with Tyr124 (distances = 2.602 and 2.994 Å, respectively), but PJ5 did not form the bond. On the contrary, PJ5 could form a hydrogen bond with Thr120 of BChE (distance = 3.354 Å), but PJ13 and PJ15 did not form (Supplementary Fig. S18).
Pharmacokinetic properties using in silico method
From the SwissADME analysis, it was predicted that the lead compounds of PJ5, PJ13, and PJ15 had high gastrointestinal adsorption abilities and cytochrome P450 inhibitory activities for 2C19, 2C9, and 3A4, however, they did not have blood–brain barrier (BBB) permeabilities (Table 3).
Table 3.
Predicted pharmacokinetic properties of PJ5, PJ13, and PJ15.
| Compounds | GI absorption | BBB permeant | P-gp substrate | CYP1A2 inhibitor | CYP2C19 inhibitor | CYP2C9 inhibitor | CYP2D6 inhibitor | CYP3A4 inhibitor | Log Kp (Skin permeation) (cm/s) |
|---|---|---|---|---|---|---|---|---|---|
| PJ5 | High | No | No | No | Yes | Yes | No | Yes | − 5.64 |
| PJ13 | High | No | No | No | Yes | Yes | No | Yes | − 5.68 |
| PJ15 | High | No | No | No | Yes | Yes | Yes | Yes | − 5.65 |
GI, gastroinstestinal absorption; BBB, blood–brain barrier; P-gp, P-glycoprotein; CYP, cytochrome P450.
Discussion
In this study, fifteen khellactone coumarin compounds from P. japonicum were analyzed for their abilities to inhibit AChE, BChE, MAO-A, and MAO-B. Compound PJ13 (IC50 = 9.28 µM) most potently inhibited AChE, followed by PJ15 and PJ7 (10.0 and 17.9 µM, respectively), which indicated all three are highly potent natural AChE inhibitors, based on the IC50 values of < 20 µM49. The IC50 values of PJ13 and PJ15 were lower than those of the C–glucosylflavone, isovitexin-7-O-methyl ether (swertisin) (32.09 µg/mL, i.e., 71.9 µM) from Anthocleista vogelii50, the flavonoids tiliroside (23.5 µM) and quercetin (19.8 µM) from Agrimonia pilosa51, and the verbascosides decaffeoylverbascoside (16.1 µM) and acteoside (19.9 µM) from Harpagophytum procumbens52, but higher than those of sargachromanol I (SCI, 0.79 µM) from the brown alga Sargassum siliquastrum and dihydroberberine (1.18 µM) from Coptis chinensis42. Compared to other coumarin derivatives, the values of PJ13 and PJ15 were lower than those of scopoletin (52 µM) from Vaccinium oldhami Miquel53, a dihydropyranocoumarin decursinol (28 μM ) from Angelica gigas Nakai54, mansonone E (23.5 µM) from Mansonia gagei55, daphnetin (11.57 µM) from Artemisia capillaris56, and a furanocoumarin (R)-( +)-6′-hydroxy-7′-methoxybergamottin (11.2 µM) from Citrus hystrix57, and higher than those of esculetin (6.13 µM) from A. capillaris56, a dihydroxanthylectin-type coumarin 4′-hydroxy Pd–C-III (1.09 µM) from Angelica decursiva58, and a 4-phenyl coumarin mesuagenin B (0.7 µM) from Mesua elegans59.
Regarding BChE inhibition, PJ5 (IC50 = 7.22 µM) was the most potent inhibitor, followed by PJ10 and PJ4 (IC50 = 10.16 and 10.66 µM, respectively). The IC50 value of PJ5 in this study was lower than those of broussonin A (7.50 µM) from Anemarrhena asphodeloidesa42, isoacteoside (29.7 µM) from H. procumbens52, corenone B (10.9 μg/mL, i.e., 49.5 µM) from Niphogeton dissecta60, and kaempferol (62.5 µM) from Cleistocalyx operculatus61, but higher than that of 4′-hydroxy Pd–C-III (5.78 µM) from A. decursiva58. Compared to other coumarins, the IC50 value of PJ5 for BChE inhibition was lower than those of hyuganin C (38.86 µM), from Mutellina purpurea62, a coumarin pteryxin (12.96 μg/mL, i.e., 33.5 µM) from M. purpurea63, the esculetin (9.29 µM) and the daphnetin (8.66 µM)56, and it might be concluded that PJ5 is the most potent BChE inhibitor in natural coumarins reported.
These results show that PJ5 is potent and selective inhibitor of BChE, and that PJ13 and PJ15 are selective inhibitors of AChE. It might be suggested that combination of compounds effectively inhibit ChE. The possibility of dual inhibition of AChE and MAO enzymes was investigated for dual- or multi-targeting therapeutic purposes in AD15,17–19. However, in the present study, no tested khellactone coumarin showed dual inhibitory activity.
Structurally, PJ5, PJ13, and PJ15 contain a coumarin ring system, and the coumarins are known to have a variety of biological functions, which include anti-inflammatory, anticancer, antiviral, antioxidant, and antidepressant effects, and some have been shown to inhibit AChE and BChE58,64. PJ13 and PJ15 differ structurally as different substituents are bound to the 3C ester. PJ13 [(9R,10R)-8,8-dimethyl-10-((2-methylbutanoyl)oxy)-2-oxo-9,10-dihydro-2H,8H-pyrano[2,3-f]chromen-9-yl (E)-2-methylbut-2-enoate] had a substituent [(Z)-but-2-en-2-yl] with a double bond between 1 and 2C in the sec-butyl structure, whereas PJ15 [(9R,10R)-8,8-dimethyl-10-((2-methylbutanoyl)oxy)-2-oxo-9,10-dihydro-2H,8H-pyrano[2,3-f]chromen-9-yl 3-methylbutanoate] has an isobutyl group in this position. The AChE inhibitory activity of PJ15 was slightly higher than that of PJ13, which contains a (Z)-but-2-en-2-yl group. PJ4, PJ5, and PJ10 share a common 3-methylbut-2-enoate structure, and showed relatively higher BChE activities than other compounds. The higher BChE inhibitory activity of PJ9 than PJ8 appeared to be due to the different position of the double bond.
AChE or BChE inhibitors have been reported to exhibit competitive, noncompetitive, and mixed-type inhibitory patterns42,58. In the present study, potent inhibitions of AChE by PJ13 and PJ15 and of BChE by PJ5 were found to be reversible and to exhibit mixed-type inhibition, with Ki values of 5.98, 10.4, and 4.16 µM, respectively. These results suggest that PJ13, PJ15, and PJ5 bind to the allosteric site or the substrate-binding site of AChE.
Docking simulation analysis with AChE revealed that the PJ13 interacted with the phenolic hydroxyl group of Tyr121 to form a hydrogen bond, while no hydrogen-bond was predicted for PJ5 and PJ15. In addition, the oxygen of the carboxyl group of PJ5 formed a hydrogen bond with Ser198 of BChE, whereas no hydrogen bonding was suggested for PJ13 and PJ15. These results imply that the existence of the hydrogen bond in the complex has major effects on binding energies. Furthermore, the results concur with the Ki values and binding affinities of AChE or BChE for PJ5, PJ13, or PJ15.
To explain the reason PJ15 inhibits AChE more selectively than PJ5, Van der Waals (VDW) distances and interactions were examined at C16, C17, C18, and C19 (for PJ15) or C21 (for PJ5) atoms in the docked ligands, according to the difference between PJ15 and PJ5, i.e., the 2-methyl-butane and the 2-methyl-butene group, respectively (Figs. 1 and Supplementary Fig. S16). It was predicted that thirteen and five VDW interactions were formed with PJ15 and PJ5, respectively, within a distance of 4 Å (Supplementary Table S1 and S2). The VDW interactions of PJ15 could inhibit AChE more selectively than JP5.
In molecular dynamics analysis, average root mean square deviation (RMSD) values of PJ5, PJ13, and PJ15 for AChE were estimated to be 0.767, 0.684, and 0.752 Å, respectively, and those for BChE were 0.738, 0.823, 0.757 Å, respectively (Supplementary Figure S19). The results supported well the experimental data and the docking simulations in this study.
In a previous study, it was observed that PJ5, PJ13, and PJ15 were non-toxic up to 10 µg/µL (i.e., ~ 25 mM) and exhibited potent for anti-inflammatory effects at 10 µg/µL in previous study39, which suggests PJ5, PJ13, and PJ15 be considered candidates for the treatment of AD as ChE inhibitors with anti-inflammatory activities.
Conclusion
Among the fifteen khellactone coumarin compounds isolated from P. japonicum Thunberg, PJ5 and PJ13 were found to potently and effectively inhibited BChE and AChE, respectively. Furthermore, these inhibitors were reversible and caused by mixed inhibition. Molecular docking simulations showed that PJ13 had the highest binding affinity for AChE at − 9.3 kcal/mol, and that PJ5 had the highest binding affinity for BChE at − 10.0 kcal/mol. These results supported the notion that PJ13 and PJ5 should be considered novel, potent, and selective inhibitors of AChE and BChE, respectively. In addition, our findings suggest that PJ5, PJ13, and PJ15 are nontoxic, reversible AChE and BChE inhibitors and candidates for the treatment of AD.
Supplementary information
Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (NRF-2019R1A2C1088967), by grants from the Korea Research Institute of Bioscience and Biotechnology Research Initiative Program, and by the R&D Program for Forest Science Technology (2017030B10-1919-BA01) provided by the Korea Forest Service (Korea Forestry Promotion Institute).
Author contributions
J.H.H., B.H.E., and J.E.P. tested biological activities of the compounds and wrote primarily the main manuscript text; H.W.R., D.-Y.K., J.-H.K., and S.-R.O. isolated and wrote the part; M.-G.K. and D.P. analyzed docking data and wrote the part; H.K. reviewed and finalized the manuscript.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Jeong Hyun Heo and Bo Hyun Eom.
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-020-78782-5.
References
- 1.Singh M, Kaur M, Kukreja H, Chugh R, Silakari O, Singh D. Acetylcholinesterase inhibitors as Alzheimer therapy: From nerve toxins to neuroprotection. Eur. J. Med. Chem. 2013;70:165–188. doi: 10.1016/j.ejmech.2013.09.050. [DOI] [PubMed] [Google Scholar]
- 2.Akıncıoğlu H, Gülçin İ. Potent acetylcholinesterase inhibitors: potential drugs for Alzheimer's disease. Mini Rev. Med. Chem. 2020;20:703–715. doi: 10.2174/1389557520666200103100521. [DOI] [PubMed] [Google Scholar]
- 3.Talesa VN. Acetylcholinesterase in Alzheimer's disease. Mech. Ageing Dev. 2001;122:1961–1969. doi: 10.1016/S0047-6374(01)00309-8. [DOI] [PubMed] [Google Scholar]
- 4.English BA, Webster AA. Acetylcholinesterase and its inhibitors. In: Robertson D, Biaggioni I, Burnstock G, Low PA, Paton JFR, editors. Primer on the Autonomic Nervous System. 3. Cambridge: Academic Press; 2012. pp. 631–633. [Google Scholar]
- 5.Colović MB, Krstić DZ, Lazarević-Pašti TD, Bondžić AM, Vasić VM. Acetylcholinesterase inhibitors: Pharmacology and toxicology. Curr. Neuropharmacol. 2013;11:315–335. doi: 10.2174/1570159X11311030006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McGleenon BM, Dynan KB, Passmore AP. Acetylcholinesterase inhibitors in Alzheimer's disease. Br. J. Clin. Pharmacol. 1999;48:471–480. doi: 10.1046/j.1365-2125.1999.00026.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kumar A. & Sharma S. Donepezil. NCBI Bookshelf. Last Update: April 21, (2020).
- 8.Onor ML, Trevisiol M, Aguglia A. Rivastigmine in the treatment of Alzheimer’s disease: an update. Clin. Interv. Aging. 2007;2:17–32. doi: 10.2147/ciia.2007.2.1.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Woodruff-Pak DS, Lander C, Geerts H. Nicotinic cholinergic modulation: Galantamine as a prototype. CNS Drug Rev. 2002;8:405–426. doi: 10.1111/j.1527-3458.2002.tb00237.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Inglis F. The tolerability and safety of cholinesterase inhibitors in the treatment of dementia. Int. J. Clin. Pract. Suppl. 2002;127:45–63. [PubMed] [Google Scholar]
- 11.Kumar A, Pintus F, Di Petrillo A, Medda R, Caria P, Matos MJ, Vina D, Pieroni E, Delogu F, Era B, Delogu GL, Fais A. Novel 2-pheynylbenzofuran derivatives as selective butyrylcholinesterase inhibitors for Alzheimer’s disease. Sci. Rep. 2018;8:4424. doi: 10.1038/s41598-018-22747-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Darvesh S. Butyrylcholinesterase as a diagnostic and therapeutic target for Alzheimer's disease. Curr. Alzheimer Res. 2016;13:1173–1177. doi: 10.2174/1567205013666160404120542. [DOI] [PubMed] [Google Scholar]
- 13.Lake F. BChE reported to be associated with plaque level in Alzheimer’s disease. Biomark Med. 2013;7:197–198. doi: 10.2217/bmm.13.37. [DOI] [PubMed] [Google Scholar]
- 14.Ramsay RR, Majekova M, Medina M, Valoti M. Key targets for multi-target ligands designed to combat neurodegeneration. Front. Neurosci. 2016;10:375. doi: 10.3389/fnins.2016.00375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Repsold BP, Malan SF, Joubert J, Oliver DW. Multi-targeted directed ligands for Alzheimer's disease: Design of novel lead coumarin conjugates. SAR QSAR Environ. Res. 2018;29:231–255. doi: 10.1080/1062936X.2018.1423641. [DOI] [PubMed] [Google Scholar]
- 16.Mathew B, Baek SC, Parambi DGT, Lee JP, Joy M, Rilda PRA, Randev RV, Nithyamol P, Vijayan V, Inasu ST, Mathew GE, Lohidakshan KK, Krishnan GK, Kim H. Selected aryl thiosemicarbazones as a new class of multi-targeted monoamine oxidase inhibitors. Med. Chem. Comm. 2018;9:1871–1881. doi: 10.1039/C8MD00399H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mathew B, Baek SC, Parambi DGT, Lee JP, Mathew GE, Jayanthi S, Vinod D, Rapheal C, Devikrishna V, Kondarath SS, Uddin MS, Kim H. Potent and highly selective dual-targeting monoamine oxidase-B inhibitors: fluorinated chalcones of morpholine versus imidazole. Arch. Pharm. 2019;352:e1800309. doi: 10.1002/ardp.201800309. [DOI] [PubMed] [Google Scholar]
- 18.Reeta BSC, Lee JP, Rangarajan TM, Ayushee SRP, Singh M, Mangiatordi GF, Nicolotti O, Kim H, Mathew B. Ethyl acetohydroxamate incorporated chalcones: unveiling a novel class of chalcones for multitarget monoamine oxidase-B inhibitors against Alzheimer's disease. CNS Neurol. Disord. Drug Targets. 2019;18:643–654. doi: 10.2174/1871527318666190906101326. [DOI] [PubMed] [Google Scholar]
- 19.Kumar B, Kumar V, Prashar V, Saini S, Dwivedi AR, Bajaj B, Mehta D, Parkash J, Kumar V. Dipropargyl substituted diphenylpyrimidines as dual inhibitors of monoamine oxidase and acetylcholinesterase. Eur. J. Med. Chem. 2019;177:221–234. doi: 10.1016/j.ejmech.2019.05.039. [DOI] [PubMed] [Google Scholar]
- 20.Tipton KF, Boyce S, O'Sullivan J, Davey GP, Healy J. Monoamine oxidases: certainties and uncertainties. Curr. Med. Chem. 2004;15:1965–1982. doi: 10.2174/0929867043364810. [DOI] [PubMed] [Google Scholar]
- 21.Edmondson DE, Binda C. Monoamine oxidases. Subcell. Biochem. 2018;87:117–139. doi: 10.1007/978-981-10-7757-9_5. [DOI] [PubMed] [Google Scholar]
- 22.Cristancho MA, O’reardon JP, Thase ME. Atypical depression in the 21st century: Diagnostic and treatment issues. Psychiatric Times. 2011;28:42–47. [Google Scholar]
- 23.Dezsi L, Vecsei L. Monoamine oxidase B inhibitors in Parkinson's disease. CNS Neurol. Disord. Drug Targets. 2017;16:425–439. doi: 10.2174/1871527316666170124165222. [DOI] [PubMed] [Google Scholar]
- 24.Tran MH, Yamada K, Nabeshima T. Amyloid beta-peptide induces cholinergic dysfunction and cognitive deficits: a minireview. Peptides. 2002;23:1271–1283. doi: 10.1016/S0196-9781(02)00062-1. [DOI] [PubMed] [Google Scholar]
- 25.Tsunekawa H, Noda Y, Mouri A, Yoneda F, Nabeshima T. Synergistic effects of selegiline and donepezil on cognitive impairment induced by amyloid beta (25–35) Behav. Brain Res. 2008;190:224–232. doi: 10.1016/j.bbr.2008.03.002. [DOI] [PubMed] [Google Scholar]
- 26.Li Z, Jia K, Duan Y, Wang D, Zhou Z, Dong S. Xanomeline derivative EUK1001 attenuates Alzheimer's disease pathology in a triple transgenic mouse model. Mol. Med. Rep. 2017;16:7835–7840. doi: 10.3892/mmr.2017.7502. [DOI] [PubMed] [Google Scholar]
- 27.Cai Z. Monoamine oxidase inhibitors: Promising therapeutic agents for Alzheimer's disease (review) Mol. Med. Rep. 2014;9:1533–1541. doi: 10.3892/mmr.2014.2040. [DOI] [PubMed] [Google Scholar]
- 28.Sarkhail P. Traditional uses, phytochemistry and pharmacological properties of the genus Peucedanum: A review. J. Ethnopharmacol. 2014;156:235–270. doi: 10.1016/j.jep.2014.08.034. [DOI] [PubMed] [Google Scholar]
- 29.Nukitrangsan N, Okabe T, Toda T, Inafuku M, Iwasaki H, Oku H. Anti-obesity activity of Peucedanum japonicum Thunb extract in obese diabetic animal model C57BL/6J Ham Slc-ob/ob mice. Int. J. Life Sci. Med. Res. 2012;2:28–34. doi: 10.5963/LSMR0202004. [DOI] [Google Scholar]
- 30.Okabe T, Toda T, Nukitrangsan N, Inafuku M, Iwasaki H, Oku H. Peucedanum japonicum Thunb inhibits high-fat diet induced obesity in mice. Phytother. Res. 2011;25:870–877. doi: 10.1002/ptr.3355. [DOI] [PubMed] [Google Scholar]
- 31.Choi RY, Nam SJ, Ham JR, Lee HI, Yee ST, Kang KY, Seo KI, Lee JH, Kim MJ, Lee MK. Anti-adipogenic and anti-diabetic effects of cis-3′,4′-diisovalerylkhellactone isolated from Peucedanum japonicum Thunb leaves in vitro. Bioorg. Med. Chem. Lett. 2016;26:4655–4660. doi: 10.1016/j.bmcl.2016.08.056. [DOI] [PubMed] [Google Scholar]
- 32.Kim SH, Jong HS, Yoon MH, Oh SH, Jung KT. Antinociceptive effect of intrathecal sec-O-glucosylhamaudol on the formalin-induced pain in rats. Korean J. Pain. 2017;30:98–103. doi: 10.3344/kjp.2017.30.2.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim JM, Erkhembaatar M, Lee GS, Lee JH, Noh EM, Lee M, Song HK, Lee CH, Kwon KB, Kim MS, Lee YR. Peucedanum japonicum thunb Ethanol extract suppresses RANKL-mediated osteoclastogenesis. Exp. Ther. Med. 2017;14:410–416. doi: 10.3892/etm.2017.4480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chun JM, Lee AR, Kim HS, Lee AY, Gu GJ, Moon BC, Kwon BI. Peucedanum japonicum extract attenuates allergic airway inflammation by inhibiting Th2 cell activation and production of pro-inflammatory mediators. J. Ethnopharmacol. 2018;211:78–88. doi: 10.1016/j.jep.2017.09.006. [DOI] [PubMed] [Google Scholar]
- 35.Kim KN, Choi MJ, Lee Y, Cho SH. The protective and recovery effects of Peucedanum Japonicum Thunberg for vascular dementia. J. Orient Neuropsychiatry. 2013;24:123–130. doi: 10.7231/jon.2013.24.1.123. [DOI] [Google Scholar]
- 36.Hisamoto M, Kikuzaki H, Ohigashi H, Nakatani N. Antioxidant compounds from the leaves of Peucedanum japonicum thunb. J. Agric. Food Chem. 2003;51:5255–5261. doi: 10.1021/jf0262458. [DOI] [PubMed] [Google Scholar]
- 37.Hisamoto M, Kikuzaki H, Nakatani N. Constituents of the leaves of Peucedanum japonicum thunb. and their biological activity. J. Agric. Food Chem. 2004;52:445–450. doi: 10.1021/jf0349127. [DOI] [PubMed] [Google Scholar]
- 38.Lee SO, Choi SZ, Lee JH, Chung SH, Park SH, Kang HC, Yang EY, Cho HJ, Lee KR. Antidiabetic coumarin and cyclitol compounds from Peucedanum japonicum. Arch. Pharm. Res. 2004;27:1207–1210. doi: 10.1007/BF02975882. [DOI] [PubMed] [Google Scholar]
- 39.Won HJ, Lee SM, Kim DY, Kwon OK, Park MH, Kim JH, Ryu HW, Oh SR. Rapid securing of reference substances from Peucedanum japonicum Thunberg.z by recycling preparative high-performance liquid chromatography. J. Chromatography B. 2019;1133:121835. doi: 10.1016/j.jchromb.2019.121835. [DOI] [PubMed] [Google Scholar]
- 40.Baek SC, Park MH, Ryu HW, Lee JP, Kang MG, Park D, Park CM, Oh SR, Kim H. Rhamnocitrin isolated from Prunus padus var. seoulensis: A potent and selective reversible inhibitor of human monoamine oxidase A. Bioorg. Chem. 2019;83:317–325. doi: 10.1016/j.bioorg.2018.10.051. [DOI] [PubMed] [Google Scholar]
- 41.Ellman GL, Courtney KD, Andres V, Jr, Featherstone RM. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 1961;7:88–95. doi: 10.1016/0006-2952(61)90145-9. [DOI] [PubMed] [Google Scholar]
- 42.Lee JP, Kang MG, Lee JY, Oh JM, Baek SC, Leem HH, Park D, Cho ML, Kim H. Potent inhibition of acetylcholinesterase by sargachromanol I from Sargassum siliquastrum and by selected natural compounds. Bioorg. Chem. 2019;89:103043. doi: 10.1016/j.bioorg.2019.103043. [DOI] [PubMed] [Google Scholar]
- 43.Lee HW, Ryu HW, Kang MG, Park D, Oh SR, Kim H. Potent selective monoamine oxidase B inhibition by maackiain, a pterocarpan from the roots of Sophora flavescens. Bioorg. Med. Chem. Lett. 2016;26:4714–4719. doi: 10.1016/j.bmcl.2016.08.044. [DOI] [PubMed] [Google Scholar]
- 44.Baek SC, Lee HW, Ryu HW, Kang MG, Park D, Kim SH, Cho ML, Oh SR, Kim H. Selective inhibition of monoamine oxidase A by hispidol. Bioorg. Med. Chem. Lett. 2018;28:584–588. doi: 10.1016/j.bmcl.2018.01.049. [DOI] [PubMed] [Google Scholar]
- 45.Trott O, Olson AJ. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 47.Mills JEJ, Dean PM. Three-dimensional hydrogen-bond geometry and probability information from a crystal survey. J. Comput. Aided Mol Des. 1996;10:607–622. doi: 10.1007/BF00134183. [DOI] [PubMed] [Google Scholar]
- 48.Daina A, Michielin O, Zoete V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017;7:42717. doi: 10.1038/srep42717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.dos Santos TC, Gomes TM, Pinto BAS, Camara AL, de Andrade PAM. Naturally occurring acetylcholinesterase inhibitors and their potential use for Alzheimer's disease therapy. Front. Pharmacol. 2018;9:1192. doi: 10.3389/fphar.2018.01192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ajayi OS, Aderogba MA, Obuotor EM, Majinda RRT. Acetylcholinesterase inhibitor from Anthocleista vogelii leaf extracts. J. Ethnopharmacol. 2019;231:503–506. doi: 10.1016/j.jep.2018.11.009. [DOI] [PubMed] [Google Scholar]
- 51.Jung M, Park M. Acetylcholinesterase inhibition by flavonoids from Agrimoniapilosa. Molecules. 2007;12:2130–2139. doi: 10.3390/12092130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bae YH, Cuong TD, Hung TM, Kim JA, Woo MH, Byeon JS, Choi JS, Min BS. Cholinesterase inhibitors from the roots of Harpagophytum procumbens. Arch. Pharm. Res. 2014;37:1124–1129. doi: 10.1007/s12272-013-0316-y. [DOI] [PubMed] [Google Scholar]
- 53.Lee JH, Lee KT, Yang JH, Baek NI, Kim DK. Acetylcholinesterase inhibitors from the twigs of Vaccinium oldhami Miquel. Arch. Pharm. Res. 2004;27:53–56. doi: 10.1007/BF02980046. [DOI] [PubMed] [Google Scholar]
- 54.Kang SY, Lee KY, Sung SH, Park MJ, Kim YC. Coumarins isolated from Angelica gigas inhibit acetylcholinesterase: Structure-activity relationships. J. Nat. Prod. 2001;64:683–685. doi: 10.1021/np000441w. [DOI] [PubMed] [Google Scholar]
- 55.Changwong N, Sabphon C, Ingkaninan K, Sawasdee P. Acetyl- and butyryl-cholinesterase inhibitory activities of mansorins and mansonones. Phytother. Res. 2012;26:392–396. doi: 10.1002/ptr.3576. [DOI] [PubMed] [Google Scholar]
- 56.Ali MY, Jannat S, Jung HA, Choi RJ, Roy A, Choi JS. Anti-Alzheimer's disease potential of coumarins from Angelica decursiva and Artemisia capillaris and structure-activity analysis. Asian Pac. J. Trop. Med. 2016;9:103–111. doi: 10.1016/j.apjtm.2016.01.014. [DOI] [PubMed] [Google Scholar]
- 57.Youkwan J, Sutthivaiyakit S, Sutthivaiyakit P. Citrusosides A−D and furanocoumarins with cholinesterase inhibitory activity from the fruit peels of Citrus hystrix. J. Nat. Prod. 2010;73:1879–1883. doi: 10.1021/np100531x. [DOI] [PubMed] [Google Scholar]
- 58.Ali MY, Seong SH, Jung HA, Jannat S, Choi JS. Kinetics and molecular docking of dihydroxanthyletin-type coumarins from Angelica decursiva that inhibit cholinesterase and BACE1. Arch. Pharm. Res. 2018;41:753–764. doi: 10.1007/s12272-018-1056-9. [DOI] [PubMed] [Google Scholar]
- 59.Awang K, Chan G, Litaudon M, Ismail NH, Martin MT, Gueritte F. 4-Phenylcoumarins from Mesua elegans with acetylcholinesterase inhibitory activity. Bioorg. Med. Chem. 2010;18:7873–7877. doi: 10.1016/j.bmc.2010.09.044. [DOI] [PubMed] [Google Scholar]
- 60.Calva J, Bec N, Gilardoni G, Larroque C, Cartuche L, Bicchi C, Montesinos JV. Acorenone B: AChE and BChE inhibitor as a major compound of the essential oil distilled from the Ecuadorian species Niphogeton dissecta (Benth.) J.F. Macbr. Pharmaceuticals. 2017;10:84. doi: 10.3390/ph10040084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Min BS, Cuong TD, Lee JS, Shin BS, Woo MH, Hung TM. Cholinesterase inhibitors from Cleistocalyx operculatus buds. Arch. Pharm. Res. 2010;33:1665–1670. doi: 10.1007/s12272-010-1016-5. [DOI] [PubMed] [Google Scholar]
- 62.Orhan IE, Senol Deniz FS, Traedal-Henden S, Cerón-Carrasco JP, den Haan H, Peña-García J, Pérez-Sánchez H, Emerce E, Skalicka-Wozniak K. Profiling auspicious butyrylcholinesterase inhibitory activity of two herbal molecules: hyperforin and hyuganin C. Chem. Biodivers. 2019;16:e1900017. doi: 10.1002/cbdv.201900017. [DOI] [PubMed] [Google Scholar]
- 63.Orhan IE, Senol FS, Shekfeh S, Skalicka-Wozniak K, Banoglu E. Pteryxin—A promising butyrylcholinesterase-inhibiting coumarin derivative from Mutellina purpurea. Food Chem. Toxicol. 2017;109:970–974. doi: 10.1016/j.fct.2017.03.016. [DOI] [PubMed] [Google Scholar]
- 64.Anand P, Singh B, Singh N. A review on coumarins as acetylcholinesterase inhibitors for Alzheimer’s disease. Bioorg. Med. Chem. 2012;20:1175–1180. doi: 10.1016/j.bmc.2011.12.042. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.