

Abstract
Acquired drug resistance in epidermal growth factor receptor (EGFR) mutant non-small cell lung cancer is a persistent challenge in cancer therapy. Previous studies of trisubstituted imidazole inhibitors lead to the serendipitous discovery of inhibitors that target the drug resistant EGFR(L858R/T790M/C797S) mutant with nanomolar potencies in a reversible binding mechanism. To dissect the molecular basis for their activity, we determined the binding modes of several trisubstituted imidazole inhibitors in complex with the EGFR kinase domain with X-ray crystallography. These structures reveal that the imidazole core acts as an H-bond acceptor for the catalytic lysine (K745) in the “αC-helix out” inactive state. Selective N-methylation of the H-bond accepting nitrogen ablates inhibitor potency, confirming the role of the K745 H-bond in potent, non-covalent inhibition of C797S variant. Insights from these studies offer new strategies for developing next generation inhibitors targeting EGFR in non-small cell lung cancer.
Graphical Abstract
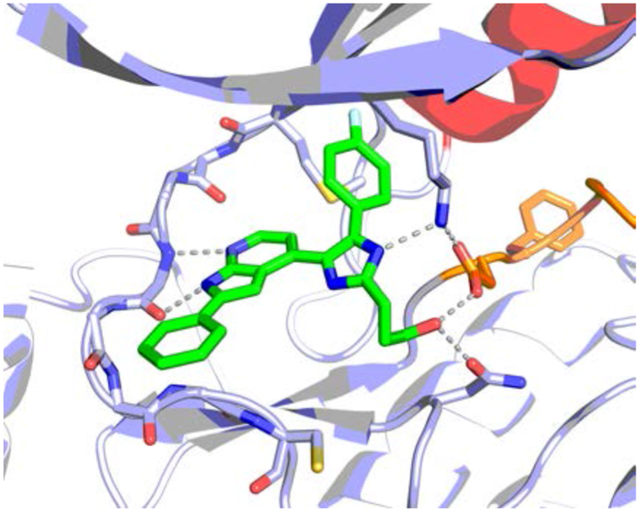
Introduction
Activating mutations within the kinase domain of the epidermal growth factor receptor (EGFR), including L858R and in-frame exon-19 deletions (del), drive tumor carcinogenesis in non-small cell lung cancer (NSCLC) and are prognostic indicators for the clinical effectiveness for treatment with tyrosine kinase inhibitors (TKIs), such as gefitinib and erlotinib.1–2 Although patients initially respond well to TKIs, drug resistance occurs due to the acquisition of the T790M “gatekeeper” mutation rendering TKIs ineffective due to increased ATP affinity within the kinase domain.3–4 Development of a novel irreversible binding anilinopyrimidine inhibitor (WZ4002), containing a C797S-targeting acrylamide Michael acceptor, exhibited exquisite potency against T790M mutant while minimally inhibited WT EGFR.5 This tool compound provided proof of principle for mutant-selective irreversible inhibition of T790M-mutant EGFR, and subsequently the third generation EGFR TKI osimertinib was approved for treatment of NCSLC patients harboring T790M positive tumors6–7 and more recently as a front-line therapy in untreated EGFR mutant NSCLC.8 However, in the context of T790M-positive NSCLC, patients acquire resistance to osimertinib through several mechanisms, including around 30–40% of patients acquiring an additional C797S mutation that renders third-generation TKIs incapable of forming their potency-enabling covalent bond.9–10 Therefore, efforts to develop inhibitors targeting both EGFR(T790M) and EGFR(C797S) mutations represent a critical unmet need in the treatment of NSCLC.
To overcome resistance to third-generation inhibitors via the acquisition of the C797S mutation, various drug discovery efforts have been dedicated to discovery of novel scaffolds that selectively target this mutation.11 While screening for highly effective p38 MAP kinase inhibitors, trisubstituted imidazole compounds were discovered to exhibit off-target inhibition of EGFR.12 Medicinal chemistry optimization and molecular docking approaches produced a series of reversible and irreversible trisubstituted imidazole-based compounds that effectively inhibited mutant EGFR (Figure 1, 1–5).13 Surprisingly, these molecules exhibited low nanomolar potency against EGFR(L858R/T790M/C797S) irrespective of whether they contained the covalent warhead.14–15 Subsequently, several examples of reversible ATP-competitive inhibitors targeting EGFR(del/T790/C797S) and/or EGFR(L858R/T790M/C797S) variants have been described, including Brigatinib,16 2-aryl-4-aminoquinazolines,17 pyrimidopyrimidinones,18–19 pyrrolopyrimidines,20 9-heterocyclyl-purines,21 and rationally designed macrocycles based on aminobenzimidazoles.22 Despite these successes in developing trisubstituted imidazole EGFR(T790M/C797S) targeting reversible inhibitors, the structural basis for their potency for this drug resistant oncogenic EGFR variant remains unclear.
Figure 1.
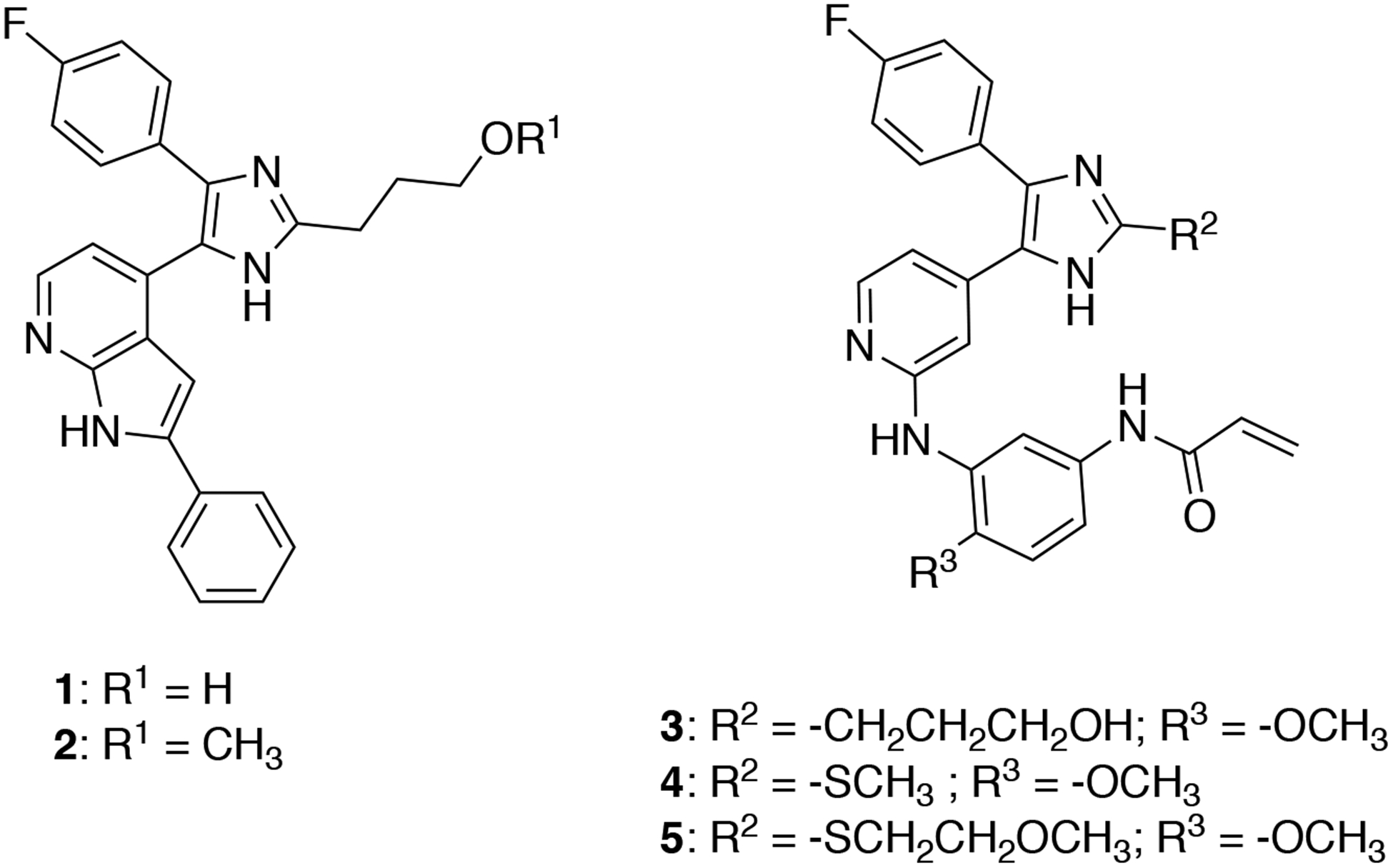
Trisubstituted imidazole inhibitor chemical structures.
In this study, we structurally characterize the binding modes of reversible and irreversible binding trisubstituted imidazole EGFR inhibitors with X-ray crystallography to understand their targeting of EGFR drug resistant mutant C797S. Crystal structures of inhibitors bound to both inactive and active EGFR kinases reveal that the imidazole moiety functions as a H-bond acceptor for the e-ammonium group of the catalytic lysine (K745) in the “αC-helix out” inactive state and blocking this interaction via the synthesis of selective imidazole N-methylation abolishes potency for EGFR(L858R/T790M/C797S). This H-bonding interaction with the EGFR kinase, which is not possible with WZ4002 or osimertinib but is observed in examples of other C797S inhibitors, is key for enabling strong, reversible binding rendering the trisubstituted imidazole molecules as effective inhibitors.
Results
We determined crystal structures of reversible (1–2) and irreversible binding (3–5) imidazole-based inhibitors (Figure 1) soaked into crystals of EGFR(T790M/V948R) (Figure 2, S1–2, and Table S1).13–15 The inclusion of the V948R variant enables crystallization of EGFR in the inactive state by precluding the formation of the activating asymmetric dimer. The structural hallmark of the inactive EGFR kinase is the outward arrangement of the αC-helix (“αC-helix out”), which is rotated inward due in the active “αC-helix in” asymmetric kinase dimer.23 Expectedly, reversible imidazole inhibitors (1 and 2) bind to the ATP pocket anchored by H-bonds to kinase hinge via the pyrrolopyridine (Figure 2A–B). C797 at the periphery of the ATP binding site is distant from the inhibitor (~ 5.0 Å) indicating that the binding modes of 1 and 2 is likely agnostic with respect to the C797S mutation. The imidazole group extends away from the hinge toward K745, the catalytic lysine, and the fluorophenyl group extends into the hydrophobic pocket positioned between the T790M gatekeeper mutation and K745. The imidazole N3 forms an H-bond with K745, which is also H-bonded to D855 of the DFG motif. The flexible functional groups extending from the imidazole C2 adopt subtly different conformations in the four copies of the EGFR kinase domain in the asymmetric unit of the crystal structures of 1 and 2. As representative examples, the propanol group of 1 engages in H-bonding with D855 and N842, while the propylether moiety of 2 is incapable of such interactions and forms an intramolecular H-bond to the imidazole N1. Generally, this experimentally determined binding mode of the inhibitor is in good agreement with the previously reported poses generated with in silico docking approaches.13–15 Additionally, in both cases electron density difference maps indicate the presence of an ion bound underneath the EGFR kinase P-loop directly interacting with K745 in all cases and R841 in the case of 2 (Figure 2B, Figure S2–3). While we have not experimentally determined the identity of this ion, we have modeled this density as a chloride ion since it is closely associated with positive side chains and backbone amides and a water molecule was not sufficiently electron-dense to model it. We also note the high concentration of chloride present in the purification and crystallization buffer.
Figure 2.
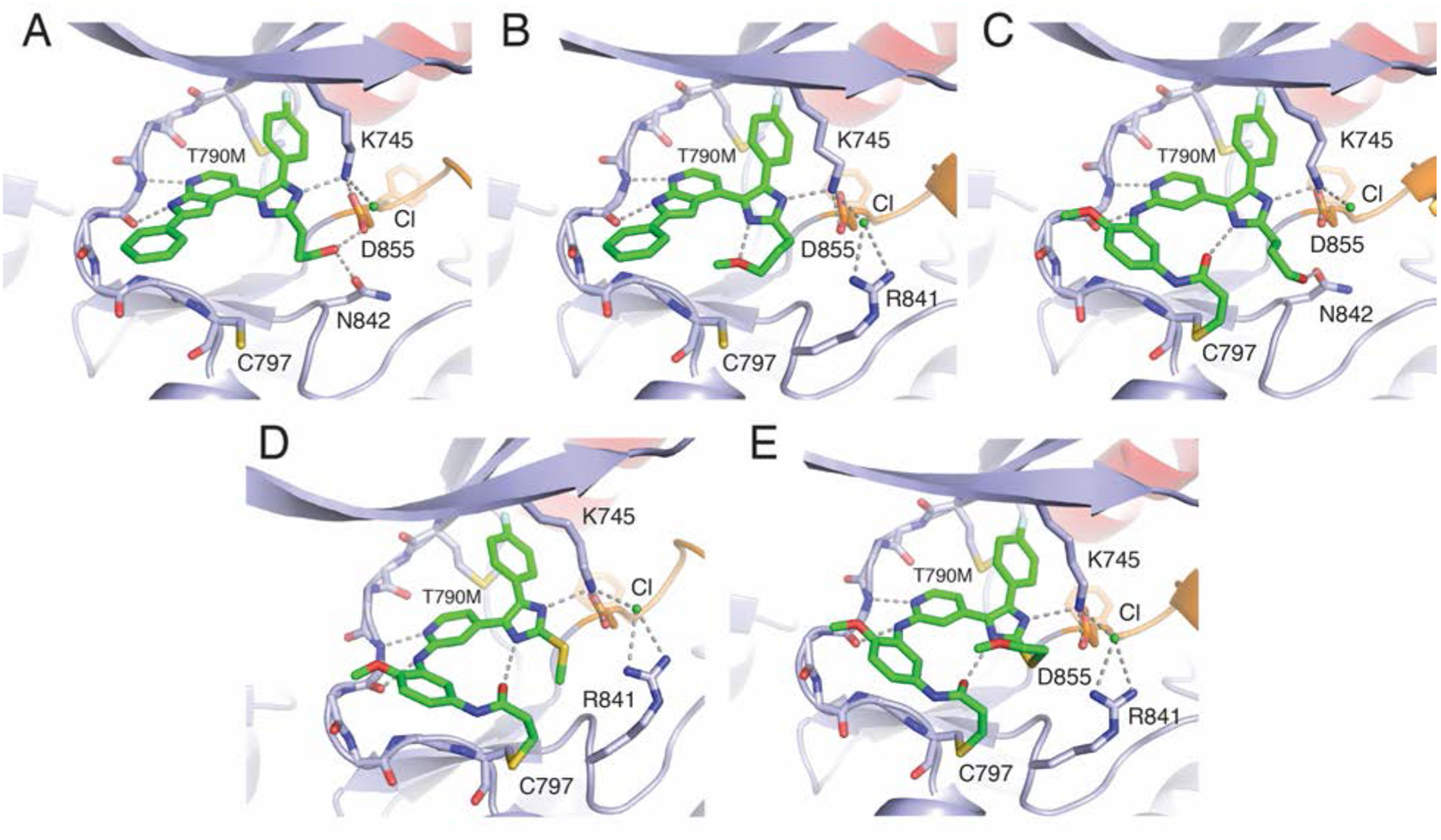
Structures and binding modes of 1 (A, PDB 6V5N), 2 (B, PDB 6V5P), 3 (C, PDB 6V6O), 4 (D, PDB 6V6K), and 5 (E, PDB 6V66) in complex with EGFR(T790M/V948R). The V948R mutation enables the kinase domain to crystallize in the inactive state.
We also determined structures of inactive “αC-helix out” EGFR(T790M/V948R) crystals soaked with irreversible compounds 3–5, which contain a 2-aminopyridine phenylacrylamide Michael acceptor to target C797S (Figure 2C–E). Indeed, crystal structure electron density maps indicate that 3–5 form their expected covalent bonds with C797 (Figure S2). The phenylacrylamide group is found to engage in intramolecular H-bonding with the imidazole N1 as predicted by molecular modeling.13 The formation of the covalent bond to C797 does not significantly alter the inhibitor binding mode in comparison to the reversibly binding compounds (1 and 2), indicating that both covalent and non-covalent imidazole inhibitors bind EGFR involving conserved H-bonds to K745.
Active “αC-helix in” kinase structures were determined though compound soaking with crystals of WT EGFR kinase domain. We were successful in obtaining crystal structures of the irreversible inhibitors (3–5), but failed to obtain structures in complex with reversibly binding compounds (1 and 2). As expected, these imidazole-based inhibitors bind to the kinase hinge in essentially the same manner observed in the inactive-state complexes (Figure 3, Figure S4, and Table S2). Due to the inward rotation of that αC-helix (Figure 3, red) in the kinase active state, the E762 carboxylate side chain is now positioned toward K745, establishing a salt bridge competing the K745 ammonium group away from the imidazole. The crystal structures of 3 and 5 show a covalent bond formed between the inhibitor and C797, while this is not the case for 4 (Figure 3). The absence of a covalent bond with 4 is unclear, but may be due to differences in compound reactivity and potency (Tables 1 and S4). A structural overlay of the “αC-helix out” inactive (green) and “αC-helix in” active (blue) kinase crystal structures in complex with 3 illustrates how K745 can toggle between functions as an H-bond donor to the imidazole and E762 carboxylate as dependent on the orientation of the kinase αC-helix (Figure 4).
Figure 3.
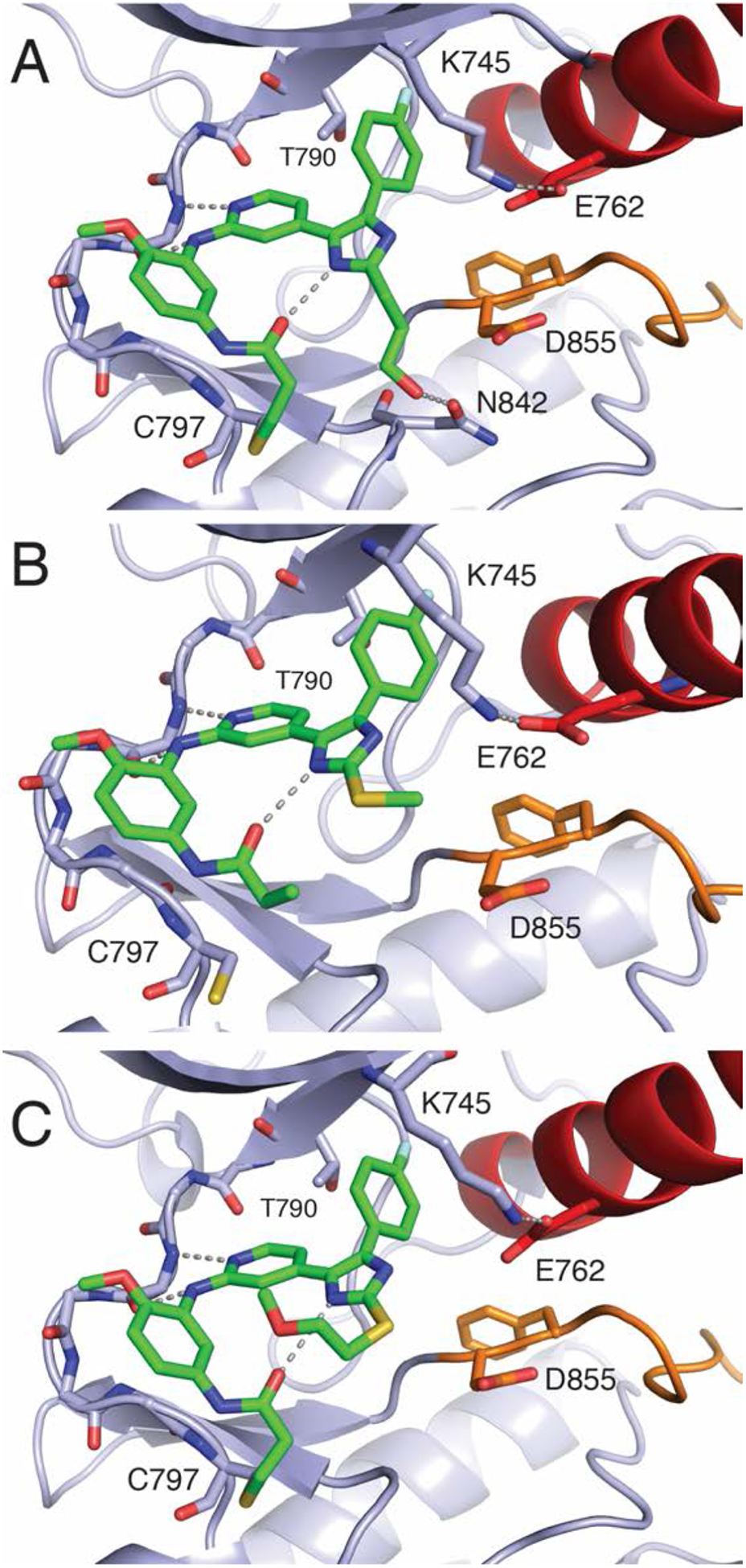
Structures and binding modes of 3 (A, PDB 6VH4), 4 (B, PDB 6VHN), and 5 (C, PDB 6VHP) bound to WT EGFR kinase domain in the active state.
Table 1.
Impact of N-methylation of 4on biochemical IC50.
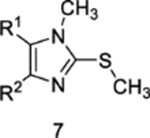
| IC50 (nM) | 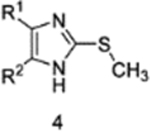 |
 |
 |
|---|---|---|---|
| L858R/T790M | < 1 | 111 | 1 |
| L858R/T790M/C797S | 90 | > 5000 | > 5000 |
Figure 4.
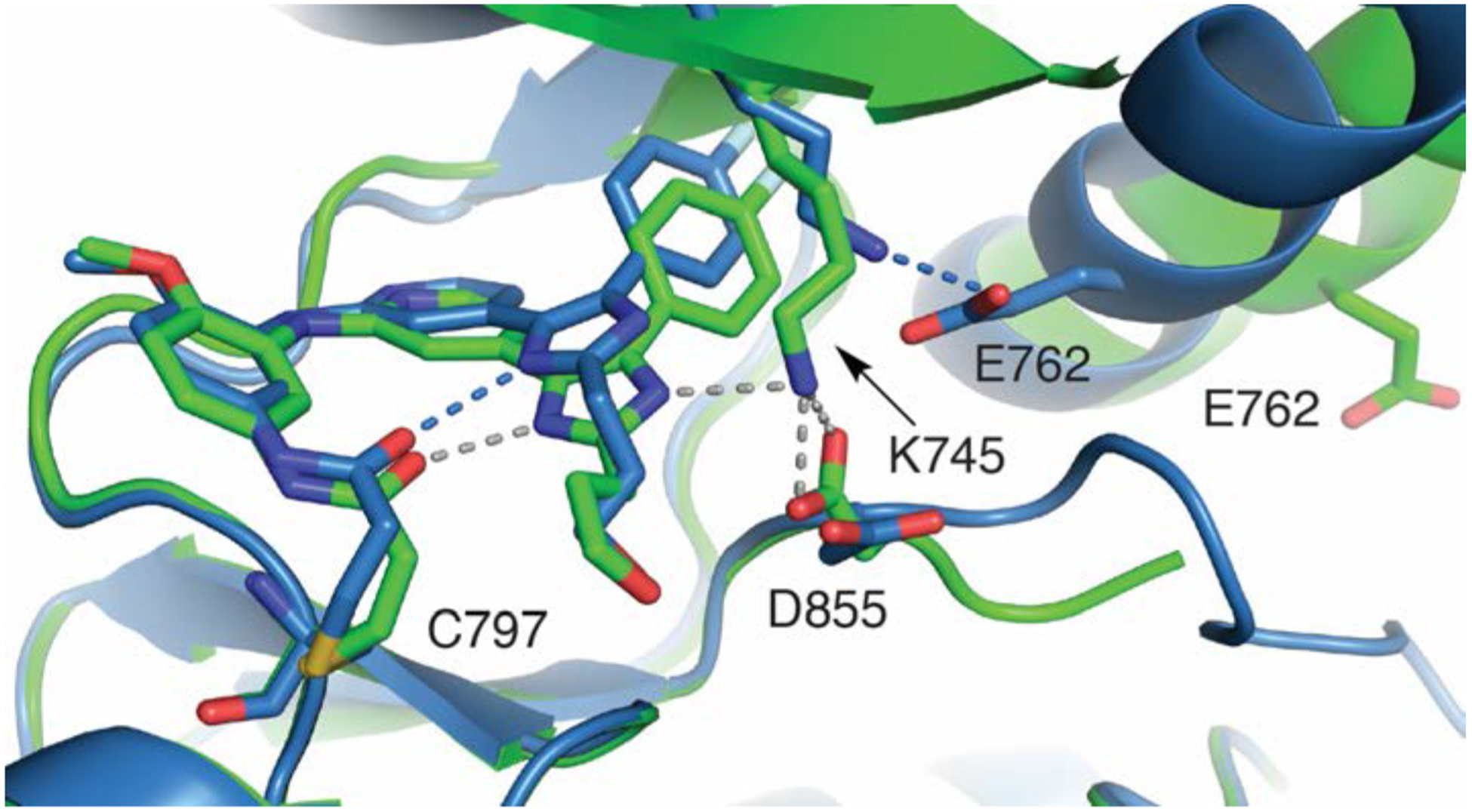
Structural overlay of 3 crystalized in complex with EGFR(T790M/V948R) (green, PDB 6V6O) in the inactive state and WT EGFR (blue, PDB 6VH4) in the active state.
To assess the functional relevance of the imidazole-K745 H-bond for inhibitor potency, we prepared both regioisomeric N-methylimidazole derivatives based on compound 4. To this end, we have synthesized compounds 6 and 7 according to our previously reported procedures.13 For the N3-regioselective introduction of the methyl group, an N1-SEM protected precursor was methylated with methyl trifluoromethanesulfonate according to Scheme 2. Subsequent acidic deprotection of the intermediate imidazolium trifluoromethanesulfonate yielded the desired methylated precursor in one-pot. The identity of the desired N-methyl regioisomer was determined by X-ray crystallography (Table S3 and Figure S4). Subsequent Suzuki cross-coupling reaction, followed by a deprotection step and a Buchwald-Hartwig aryl amination yielded the final compound analogous to our previously described route.13
Scheme 2.
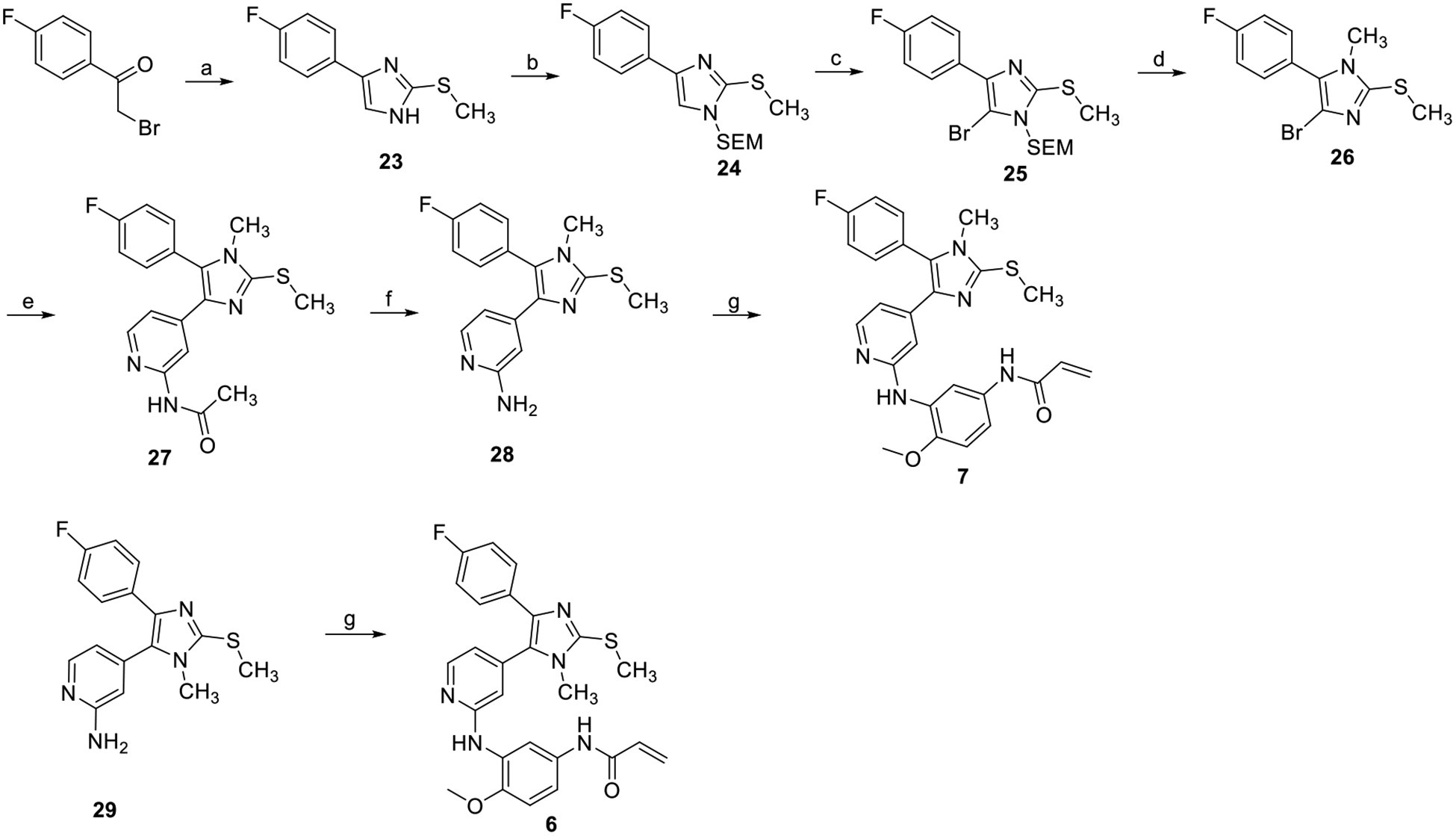
Reagents and conditions: (a) S-methylisothiourea, NaHCO3, THF/water, reflux (50%); (b) NaH, SEM-Cl, THF, 0°C to rt (91%); (c) NBS, THF, −30°C (85%); (d) MeOtf, DCM, TFA, rt (70%); (e) N-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridin-2-yl)acetamide, P(t-bu)3 Pd G3, K3PO4, 1,4-dioxane/water, 50°C (39%); (f) NaOH, MeOH/water, 50°C (88%); (g) N-(3-bromo-4-methoxyphenyl)acrylamide, Brettphos Pd G3, Cs2CO3, t-BuOH/1,4-dioxane, rf (67%).
Biochemical EGFR inhibitory capacities at 50% activity (IC50) measurements were performed with 4 and selectively N-methylated derivatives 6 and 7 (Table 1 and S4). Methylation of N1 is found to significantly impair the potency of the inhibitor against EGFR(L858R/T790M) indicating that the intramolecular H-bond formed between the imidazole and the acrylamide is structurally required for inhibitor binding. Correspondingly, methylation of N3, the nitrogen involved in H-bonding to K745 in the inactive “αC-helix out” kinase, does not significantly alter inhibition of EGFR(L858R/T790M), showing that the potency of 4 does not depend on the K745 H-bond and more likely driven by the formation of the C797-mediated covalent bond. It should be noted that methylation at N3 produces an imidazole derivative that would not form an identical intramolecular H-bond with the acrylamide as seen in the crystal structures of 4. The impact of this alternative isomer is likely a minor effect, which is likely since 7 exhibits low nanomolar potency against EGFR(L858R/T790M) (Table 1). In the case of EGFR(L858R/T790M/C797S), where 4 inhibits entirely through reversible binding, it is found that N-methylation at either N1 or N3 completely abrogates compound potency demonstrating the critical role for both imidazole nitrogens. Our inactive “αC-helix out” crystal structures (Figure 2) demonstrate that the N3 imidazole is engaged in H-bonding with K745 whereas this interaction is not present in the active kinase state (Figure 3–4). Additionally, there is a possibility for N-methylation of these inhibitors to have diminished binding affinity due to steric hinderance with the EGFR protein. The binding mode of 4 (Figures 1D and 2B) indicates that N-methylation at N1 will not interfere with EGFR, as this nitrogen is pointed away from EGFR and out into solvent, while N-methylation at N3 could sterically clash with K745 in the inactive “αC-helix out” state. Since the H-bond to K745 is a conserved feature in all of our inactive “αC-helix out” structures, we expect any resulting steric hinderance from the N3 methylation to be minimal and the domain component of inhibitor reversible binding is the H-bond. Therefore, these collective findings demonstrate that reversible binding trisubstituted imidazole inhibitors acquire stronger affinity to the EGFR kinase through an H-bond to K745 of the inactive kinase state, which enables inhibitor potency against EGFR(L858R/T790M/C797S).
Discussion.
While osimertinib is a highly effective therapeutic for NSCLC driven by sensitive EGFR mutants, including the treatment-acquired T790M resistance mutation, the C797S mutation abolishes its efficacy as well as that of other third-generation EGFR TKIs. Inhibitors based on a trisubstituted imidazole scaffold, initially optimized to target EGFR(L858R/T790M), were found to unexpectedly exhibit low nanomolar activity against EGFR(L858R/T790M/C797S).13–15 To determine the structural basis for their unique ability to target this drug resistant EGFR mutation through a non-covalent mechanism, we determined the binding modes of several trisubstituted imidazole inhibitors in complex with the EGFR kinase domain using X-ray crystallography. While all inhibitors bind to the EGFR kinase through conserved H-bonding to the kinase hinge, as expected, marked differences with respect to interactions with the imidazole are observed depending on the state of the kinase. In the inactive “αC-helix out” state, the imidazole inhibitors all are directly involved in H-bonding to the catalytic lysine (K745) through the imidazole N3 nitrogen whereas the inward rotation of the αC-helix in the active state results in a H-bond with E862, which apparently outcompetes K745 from the inhibitor. Overall, these structures are consistent with previous crystal structures of related pyridinylimidazole inhibitors in complex with p3824–25 and JNK3.26 To assess the dependence of inhibitor potency on the ability of K745 to form an H-bond with the imidazole, we synthesized a variant of 4 with selective N-methylation at either of the two imidazole nitrogens. Biochemical potencies with selective N-methylation at the imidazole N3 nitrogen of 4 was found to minimally impact inhibitor binding to EGFR(L858R/T790M), while entirely ablating inhibitor efficacy for EGFR(L858R/T790M/C797S). Collectively, our findings suggest that the imidazole N3-K745 H-bond, present only in the inactive “αC-helix out” kinase, is responsible for driving reversible binding and inhibition of EGFR(L858R/T790M/C797S).
Our structures demonstrate the key molecular features of the imidazole compounds in directing reversible inhibition of the EGFR(L858R/T790M/C797S), which resists inhibition by third-generation EGFR TKIs. Third-generation EGFR TKIs such as WZ4002 and osimertinib lack the ability to make this H-bond and reversibly bind EGFR more weakly compared to imidazole-based inhibitors rendering them ineffective against the C797S mutation (Figure 5A). This is consistent as the imidazole inhibitors are found to inhibit EGFR with enhanced Ki ~ 15-fold compared to osimertinib.14 While reversible inhibitors that bind EGFR more tightly may be able to overcome the drug-resistant C797S mutant, it is more so a liability with respect to developing a targeted drug since strong interactions with conserved amino acids will diminish the ability for the inhibitor to preferentially bind to the mutant compared to the wild-type kinase. Since this is the case for the trisubstitued imidazole inhibitors in this study, further optimization is required to achieve mutant selectivity.
Figure 5.
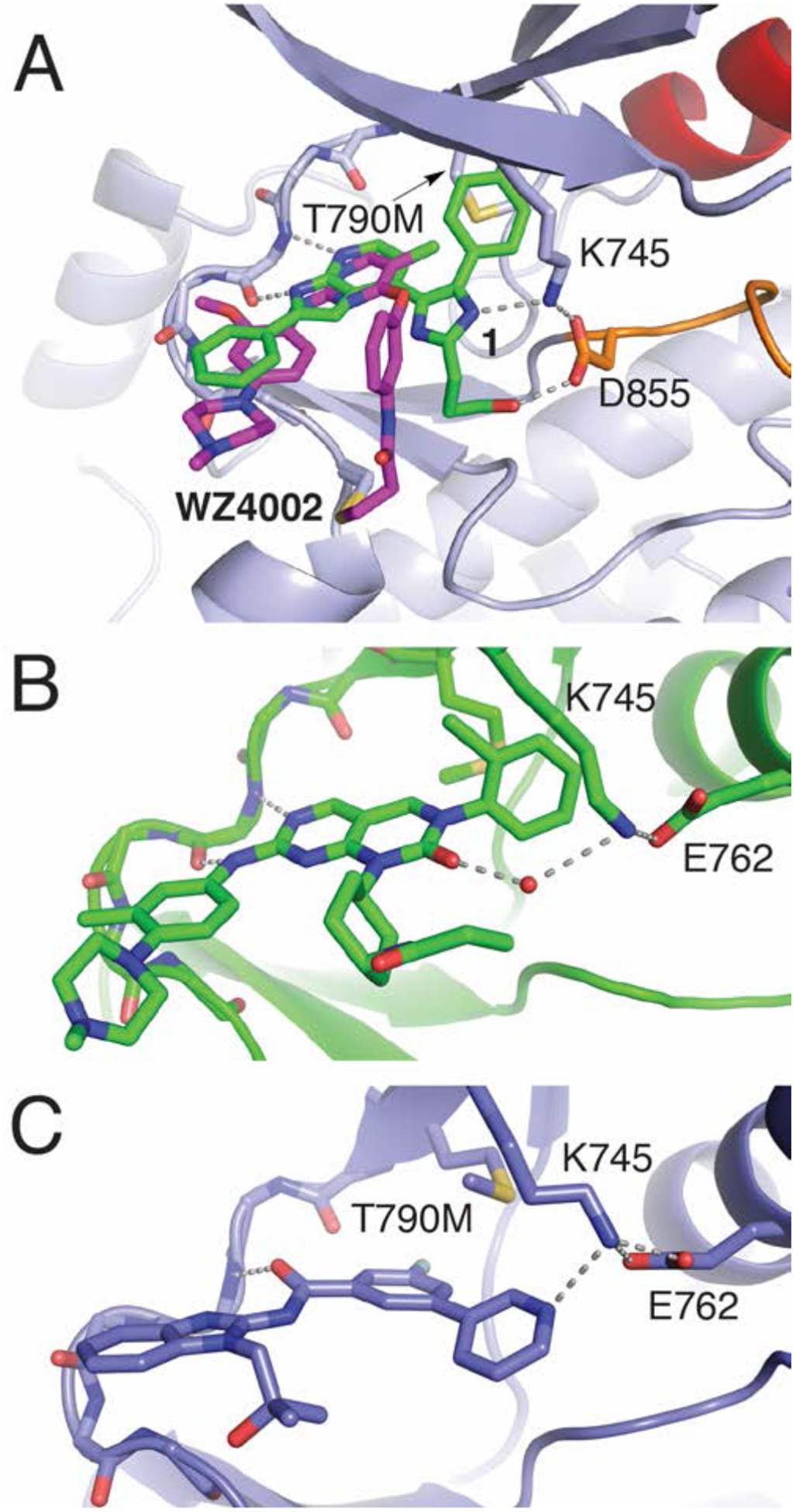
A) Structural overlay of EGFR crystals structures in complex with 1 (green, 6V5N) and WZ4002 (magenta, 3IKA). B) EGFR crystal structures of JN3229 (5ZT0) and C) aminobenzimidazole compound 1 (6S9B) from ref22.
In addition to the trisubstituted imidazoles, several small molecules have been subsequently reported to inhibit C797S-containing EGFR mutants operating through non-covalent mechanisms.11 Analysis of all structurally-characterized inhibitor binding modes reveals that many of these compounds function as a H-bond acceptor for K745. The inhibitor Brigatinib, originally developed as an ALK inhibitor, was shown to exhibit off-target inhibition of other kinases, including EGFR(del/T790M/C797S), which is maximally effective if dosed in combination with an anti-EGFR antibody cetuximab.16 While no structure of Brigatinib bound to EGFR is reported to our knowledge, the crystal structure of Brigatinib in complex with ALK in the active state shows that the inhibitor phosphine oxide moiety is pointing toward the catalytic lysine (K1150), potentially confirming enhanced potency in a similar mechanism as the trisubstituted imidazoles.27 Related compounds containing a sulfonamide in the position of the phosphine oxide effectively inhibit ALK and EGFR and, based on docking models, are expected to form H-bonds with the respective EGFR/ALK catalytic lysine in the active state.28 Additionally, pyrimidopyrimidinone inhibitors were developed to target EGFR(C797S) and co-crystal structures indicate that the inhibitors ketone is positioned as a H-bond acceptor with K745 through a bridging water molecule in the active kinase (Figure 5B).18–19 It is likely that in the inactive “αC-helix out” kinase state such molecules form a stronger, more direct H-bond with K745 operating through a similar mechanism to the trisubstituted imidazole inhibitors (1–2). A more distinct example is the recent report of a macrocyclic aminobenzimidazole inhibitor (BI-4020) that selectively inhibits T790M/C797S-containing EGFR due to the compounds macrocyclic structure with marked efficacy for regressing EGFR(del/T790M/C797S) tumor xenografts in vivo.22 While the best performing macrocycle inhibitor has not been structurally characterized, crystal structures of several precursor molecules indicate that the mutant-selective aminobenzimidazole obtains their strong potency through direct H-bonds with K745 and T854 in the active kinase state and is potentially made mutant selective due to limited steric interference with T790M (Figure 5C).22 Compared to our structures here, the lack of mutant-selectivity in the imidazole inhibitors maybe due to steric clashing between T790M and the fluorophenyl group on C4 of the imidazole inhibitor requiring the methionine to adopt a forced conformation (Figure 5A and C).22 In addition, a distinct strategy has also emerged, which involves designing EGFR drug molecules that purposefully form strong H-bonds with the mutant C797S serine alcohol. Inhibitors consisting of 2-aryl-4-aminoquinazoline17 and 9-heterocyclyl substituted 9H-purine21 scaffolds have been reported to exhibit selectivity for EGFR(T790M/C797S) mutations. It is clear that an EGFR inhibitor operating through a non-covalent mechanism can overcome the C797S mutation through engineering of additional intermolecular interactions with the kinase, although with the caveat that targeting conserved residues, such as K745, will negatively impact the inhibitor mutant selectivity as well as overall kinase selectivity.
As an alternative to ATP-competitive TKIs, a new class of highly mutant-selective EGFR allosteric inhibitors (EAIs) has been developed to bind to an allosteric pocket of the inactive EGFR kinase made accessible by the oncogenic EGFR(L858R/T790M) mutations.29–31 Importantly, the most effective single agent of the EAIs (JBJ-04-125-02) exhibits synergy in vivo with the third-generation TKI osimertinib, potentially in a co-operative mechanism involving simultaneous inhibitor binding the EGFR kinase domain.30 Efforts to completely span these two sites have resulted in a compound consisting of a chimeric fusion of an EAI scaffold (EAI045) with a TKI (vandetinib) that exhibited low nanomolar potency against EGFR(L858R/T790M/C797S), although at similar potencies for the WT kinase.32 Although this is a single example, it is probable that compounds consisting of elements of effective TKIs and mutant-selective allosteric inhibitors may represent a productive avenue to produce a C797S-targeting reversible inhibitor with enhanced mutant selectivity.
Conclusions
We have employed a combination of structural and biochemical studies to define the molecular basis for non-covalent inhibition of EGFR(L858R/T790M/C797S) by trisubstituted imidazole inhibitors. The reversible binding imidazole inhibitors overcome the C797S mutation through stabilization of the inactive kinase state anchored by an imidazole-dependent H-bond with K745. Insights from this work demonstrate the diversity of avenues available for the development of next generation therapies in EGFR mutant NSCLC. Future directions of these inhibitors are dependent on further optimization for compounds with selectivity for the oncogenic mutants and sparingly limited potency against the WT EGFR kinase.
Experimental Section
General Information.
Reagents and solvents were purchased from Sigma Aldrich, Acros, CombiBlocks or Fluorochem. Commercially available reagents and solvents were used as received without any further purification or drying procedures unless otherwise noted. All NMR spectra were measured with a Bruker Avance 200, with a Bruker Avance 400 or with a Bruker Avance 600 MHz (research group Prof. Dr. Pichler, Dep. of Preclinical Imaging and Radio pharmacy, Tübingen). Solvents for NMR are noted in the experimental procedures for each compound. Residual solvent peaks were used to calibrate the chemical shifts. Chemical shifts (δ) are reported in parts per million (ppm). LRMS were determined either by TLC-MS (Advion expressions) (ESI) or FAB-MS at the Mass Spectrometry Department, Institute of Organic Chemistry, Eberhard Karls Universität Tübingen. ESI-HRMS was obtained from the research group of Prof. Dr. Lämmerhofer (Pharmaceutical (Bio)-Analyses), University of Tübingen. TLC analyses were performed on fluorescent silica gel 60 F254 plates (Merck). Compound spots were identified either via UV illumination at 254 nm and 366 nm or developed with a suitable stain. For Column chromatography either Davisil LC60A 20–45 micron silica from Grace Davison or Geduran Si60 63–200 micron silica from Merck was used as stationary phase. VWR LaFlash automated flash chromatography system or standard glass columns were used for flash chromatography. The purity of tested compounds was determined via reverse phase high performance liquid chromatography (HPLC) on Agilent 1100 LC equipped with a UV diode array detector (DAD, detection at 230 nm and 254 nm) and in all cases was determined to be > 95%. The chromatographic separation was performed on a Phenomenex Luna 5u C8 column (150 mm × 4.6 mm, 5 μm) at 35 °C oven temperature. The injection volume was 5 μL and the gradient of the used method was (Flow: 1.5 mL/min): 0.01 M KH2PO4, pH 2.3 (Solvent A), methanol (Solvent B): 40 % B to 85 % B in 8 min, 85 % B for 5 min, 85% to 40 % B in 1 min, 40 % B for 2 min, stop time 16 min.
Synthesis
2-(4-fluorophenyl)-2-oxoacetaldehyde (8):
Selenium dioxide (14.89 g, 134.2 mmol) in 200 ml 1,4-dioxane and 6 ml water was heated to 60°C until a clear solution formed. Then 4’-Fluoroacetophenone (16.85 g, 14.8ml, 122.0 mmol) was added and the solution was refluxed for 16 hours. After cooling to room temperature, the solid was filtered. The filtrate was evaporated in vacuum to yield a yellow oil that was distilled at 85°C at 1 mbar to give the pure product in 73% yield (13.52 g, 88.9 mmol). 1H-NMR (200 MHz, CDCl3) δ 9.62 (s, 1H), 8.47 – 7.84 (m, 2H), 7.19 (t, J = 8.7 Hz, 2H). 13C-NMR (50 MHz, CDCl3) δ 189.6, 186.0, 167.1 (d, J = 258.8 Hz), 133.6 (d, J = 9.8 Hz), 128.3 (d, J = 3.0 Hz), 116.5 (d, J = 22.0 Hz). FAB: Calculated: 152.03 for C13H10N2. Found: 153.0 [M+H]+.
4-(4-fluorophenyl)-2-(3-methoxypropyl)-1H-imidazole (9):
A solution of 8 (3.09 g, 20.3 mmol) in methanol was added dropwise over 30 minutes to a stirred solution of 4-methoxybutanal (2.07 g, 20.3 mmol) and NH4OAc (7.51 g, 97.4 mmol) in methanol. The mixture was stirred overnight. The reaction was quenched by the addition of a saturated aqueous NaHCO3 solution and stirred for 30 minutes. Methanol was removed by rotary evaporation and the residual aqueous phase extracted three times with ethyl acetate. The combined organic layers were dried over Na2SO4, filtered and the solvents removed in vacuum. The crude product was purified by flash chromatography (ethyl acetate + 2% NH3 in MeOH (7M)) to give an orange gum in 16% yield. (761 mg, 3.25 mmol). 1H-NMR (200 MHz, CDCl3) δ 9.92 (s, 1H), 7.71–7.55 (m, 2H), 7.13 (s, 1H), 7.10–6.96 (m, 2H), 3.47 (t, J = 5.8 Hz, 2H), 3.37 (s, 3H), 2.88 (t, J = 6.9 Hz, 2H), 2.04–1.90 (m, 2H). 13C-NMR (50 MHz, CDCl3) δ 161.8 (d, J = 245.0 Hz), 149.2, 129.9 (d, J = 2.7 Hz), 126.3 (d, J = 7.9 Hz), 115.6 (d, J = 21.5 Hz), 72.6, 58.8, 28.0, 26.3. TLC-MS (ESI+) (m/z): Calculated: 234,12 for C13H15FN2O. Found: 234,9 [M+H]+.
4-(4-fluorophenyl)-1-(methoxymethyl)-2-(3-methoxypropyl)-1H-imidazole (10):
A cooled solution of 9 (719 mg, 3.06 mmol) in 20 ml dry DMF was treated portion wise with sodium hydride (60%, 3.52 mmol) and stirred for 30 minutes at 0°C. MOM-Cl (2.5 M in toluene, 3.06 mmol, 1.22 ml) was added dropwise to the stirred solution. The mixture was stirred for 10 minutes at 0°C and for 30 minutes at room temperature. The reaction was quenched by the addition of a saturated aqueous NH4Cl solution and stirred for 30 minutes. The aqueous mixture was extracted three times with ethyl acetate. The combined organic layers were dried over Na2SO4, filtered and the solvents removed in vacuum. The crude product was purified by gradient flash chromatography with 100% DCM to DCM/MeOH 93:7 to give the pure product as a yellow oil in 77% yield (657 mg, 2.36 mmol). 1H-NMR (200 MHz, CDCl3) δ 7.77–7.64 (m, 2H), 7.14 (s, 1H), 7.10–6.95 (m, 2H), 5.20 (s, 2H), 3.46 (t, J = 6.1 Hz, 2H), 3.34 (s, 3H), 3.31 (s, 3H), 2.91–2.79 (m, 2H), 2.17–1.99 (m, 2H). 13C-NMR (50 MHz, CDCl3) δ 162.0 (d, J = 244.9 Hz), 149.2, 139.5, 130.5 (d, J = 3.2 Hz), 126.5 (d, J = 7.9 Hz), 115.4 (d, J = 21.5 Hz), 114.9, 76.7, 71.7, 58.6, 56.1, 28.3, 23.5. TLC-MS (ESI+): calculated: 278,14 for C15H19FN2O2. Found: 278,9 [M+H]+.
4-(4-fluorophenyl)-5-iodo-1-(methoxymethyl)-2-(3-methoxypropyl)-1H-imidazole (11):
To a cooled solution of 10 (284 mg, 1.09 mmol) in 10 ml MeCN was added dropwise at 0°C N-Iodosuccinimide (281 mg, 1.25 mmol) dissolved in 5 ml MeCN. The solution was stirred for 10 minutes at 0°C, the ice bath was removed and stirring continued overnight at room temperature. The solvent was removed in vacuum and the oily residue purified by gradient flash chromatography (from 100 % DCM to DCM/MeOH 97:3) to give the pure product as an orange oil in 94% yield (412mg, 1.02 mmol). 1H-NMR (200 MHz, CDCl3) δ 7.90–7.79 (m, 2H), 7.15–7.02 (m, 2H), 5.30 (s, 2H), 3.47 (t, J = 6.0 Hz, 2H), 3.38 (s, 3H), 3.34 (s, 3H), 3.00–2.89 (m, 2H), 2.16–2.00 (m, 2H). 13C-NMR (50 MHz, CDCl3) δ 162.4 (d, J = 246.5 Hz), 152.6, 142.9, 130.1 (d, J = 3.2 Hz), 129.6 (d, J = 8.1 Hz), 115.2 (d, J = 21.5 Hz), 77.4, 71.6, 67.7, 58.7, 56.2, 28.1, 24.5. TLC-MS (ESI+) (m/z): Calculated: 404,04 for C15H18FIN2O2. Found: 404,7 [M+H]+.
4-(4-fluorophenyl)-1-(methoxymethyl)-2-(3-methoxypropyl)-5-(tributylstannyl)-1H-imidazole (12):
A cooled solution of 11 (513mg, 1.27 mmol) in 15 ml dry THF was treated with a solution of i-PrMgCl*LiCl (1.3M in THF, 1.65 mmol, 1.27 ml) dropwise at 0°C. The mixture was stirred for 30 minutes at 0°C. Next, tributyltin chloride (1.78mmol, 482μl) was added and the reaction stirred for 10 minutes at 0°C. The ice bath was removed and the solution stirred at room temperature overnight. The reaction was quenched by the addition of an aqueous NaHCO3 solution. The aqueous mixture was extracted three times with ethyl acetate. The combined organic layers were dried over Na2SO4, filtered and the solvents removed in vacuum. The crude product was used in the next step without further purification or characterization of the mixture.
4-(4-(4-fluorophenyl)-1-(methoxymethyl)-2-(3-methoxypropyl)-1H-imidazol-5-yl)-1-(methoxymethyl)-2-phenyl-1H-pyrrolo[2,3-b]pyridine (13):
A mixture of Pd(OAc)2 (2 mol%) and XPhos (6 mol%) was dissolved in dry and degassed 1,4-dioxane (5 ml) under an atmosphere of argon and stirred for 30 minutes at room temperature. To this solution, crude 12 and 4-chloro-1-(methoxymethyl)-2-phenyl-1H-pyrrolo[2,3-b]pyridine (690 mg, 2.54 mmol) dissolved in 10 ml 1,4-dioxane was added. The mixture was refluxed for until complete consumption of the azaindol. After cooling to room temperature, a 40% aqueous KF solution was added and the mixture stirred for 1 hour at room temperature. The aqueous mixture was extracted three times with ethyl acetate. The combined organic layers were dried over Na2SO4, filtered and the solvents removed in vacuum. The crude product was purified with flash chromatography (DCM/MeOH 97:3 + 2 % AcOH) to give the pure product as a yellow gum in 52% yield (170 mg, 0.33 mmol). 1H-NMR (200 MHz, CDCl3) δ 8.66 (s, 2H), 8.43 (d, J = 5.0 Hz, 1H), 7.62–7.51 (m, 2H), 7.48–7.32 (m, 5H), 7.16 (d, J = 5.0 Hz, 1H), 6.92–6.78 (m, 2H), 6.18 (s, 1H), 5.63 (dd, J = 26.9, 10.3 Hz, 2H), 5.08 (dd, J = 37.3, 10.6 Hz, 2H), 3.58–3.46 (m, 5H), 3.37 (s, 3H), 3.10 (s, 3H), 3.00 (t, J = 7.5 Hz, 2H), 2.25–2.11 (m, 2H). 13C-NMR (50 MHz, CDCl3) δ 162.0 (d, J = 245.7 Hz), 150.6, 150.2, 143.3, 137.3, 131.4, 130.4, 130.0 (d, J = 2.8 Hz), 129.3, 129.1, 129.0, 128.9, 124.8, 120.7, 119.1, 115.2 (d, J = 21.4 Hz), 100.5, 74.4, 73.0, 71.9, 58.6, 56.7, 55.9, 28.3, 23.8. TLC-MS (ESI+) (m/z): Calculated: 514,24 for C30H31FN4O3. Found: 514,9 [M+H]+.
4-(4-(4-fluorophenyl)-2-(3-methoxypropyl)-1H-imidazol-5-yl)-2-phenyl-1H-pyrrolo[2,3-b]pyridine (2):
A solution of 13 (170 mg, 0,33 mmol) in 5ml methanol and 5 ml concentrated hydrochloric acid was stirred under reflux for 24 hours. After complete conversion, the mixture was cooled to room temperature and neutralized with a saturated aqueous solution of K2CO3. The aqueous mixture was extracted three times with ethyl acetate. The combined organic layers were dried over Na2SO4, filtered and the solvents removed in vacuum. The crude product was purified with gradient flash chromatography (from 100% DCM to DCM/MeOH 90:10) to give the pure product as a yellow solid in 58% yield (82 mg, 0.19 mmol). 1H-NMR (400 MHz, DMSO-d6) δ 12.36 (s, 1H), 12.17 (s, 1H), 8.16 (s, 1H), 7.77 (d, J = 7.4 Hz, 2H), 7.49–7.37 (m, 4H), 7.35–7.28 (m, 1H), 7.22–7.08 (m, 2H), 7.04 (d, J = 4.7 Hz, 1H), 6.55 (s, 1H), 3.45 (t, J = 5.9 Hz, 2H), 3.28 (s, 3H), 2.79 (t, J = 7.4 Hz, 2H), 2.09–1.95 (m, 2H). 13C-NMR (100 MHz, DMSO-d6) δ 161.1 (d, J = 243.8 Hz), 150.5, 148.8, 142.7, 137.7, 131.5, 129.3, 129.3, 128.8, 127.9, 125.1, 118.3, 115.1 (d, J = 21.4 Hz), 114.9, 97.9, 71.3, 57.8, 27.9, 24.6.. TLC-MS (ESI−) (m/z): Calculated: 426,19 for C26H23FN4O. Found: 425,0 [M−H]−.
2-(4-fluorophenyl)-2-oxoethan-1-aminium chloride (14):
2-Bromo-1-(4-fluorophenyl)ethan-1-one (7.9 g, 36.6 mmol) and Urotropine (5.6 g, 39.9 mmol) were dissolved in 150 ml dry chloroform and stirred at 50°C for 2 hours. The precipitate was removed by filtration and washed with chloroform and ethanol. The solid was dissolved in 150 ml abs. ethanol and 20 ml concentrated hydrochloric acid and refluxed for 2 hours. The mixture was filtered and the filtrate was evaporated to dryness to give the pure product as a white solid in 97% yield (6.7 g, 35.2 mmol). 1H NMR (200 MHz, DMSO-d6) δ 8.60 (s, 3H), 8.22 – 8.05 (m, 2H), 7.47 – 7.38 (m, 2H), 4.58 (s, 2H). 13C NMR (50 MHz, DMSO-d6) δ 191.78, 165.97 (d, J = 253 Hz), 131.75 (d, J = 9.8 Hz), 130.82 (d, J = 3.6 Hz), 116.46 (d, J = 22.2 Hz), 45.08.
4-(4-fluorophenyl)-1,3-dihydro-2H-imidazole-2-thione (15):
A suspension of 14 (2.0 g, 10.7 mmol) and potassium thiocyanate (2.61 g, 26.8 mmol) in 15 ml glacial acid was refluxed for 1 hour. After cooling to 0°C and the addition of water, the precipitated solid was collected by filtration. The solid was washed with water and dried in vacuum overnight to give the product as a pale yellow solid in 71% yield (1.5 g, 7.6 mmol). The obtained product was used without further purification. LRMS (ESI-MS): 193.1 [M-H]−. 1H NMR (200 MHz, DMSO-d6) δ 12.55 (s, 1H), 12.16 (s, 1H), 7.79 – 7.65 (m, 2H), 7.37 (s, 1H), 7.23 (t, J = 8.7 Hz, 2H). 13C NMR (50 MHz, DMSO-d6) δ 162.14, 161.63 (d, J = 245 Hz) 128.52, 126.34 (d, J = 8.3 Hz), 125.34 (d, J = 3.0 Hz), 116.12 (d, J = 21.9 Hz), 112.44.
2-((4-(4-fluorophenyl)-1H-imidazol-2-yl)thio)ethan-1-ol (16):
Intermediate 15 (1.0 g, 5.15 mmol) was dissolved in 20 ml dry methanol. Potassium carbonate (890 mg, 6.44 mmol) was added to the solution at room temperature under vigorous stirring. The resulting suspension was stirred for 30 minutes at room temperature. 2-Bromoethanol (402 μl, 5.67 mmol) was added and the suspension was stirred at room temperature overnight. After complete reaction, water and a saturated NH4Cl solution was added. The aqueous mixture was extracted three times with ethyl acetate. The combined organic layers were dried over Na2SO4, filtered and the solvents removed in vacuum. The obtained pure product was used without further purification. After washing with n-pentane and drying overnight, the product was obtained in quantitative yield. LRMS (ESI-MS): 237.0 [M-H]−. 1H NMR (200 MHz, DMSO-d6) δ 12.39 (s, 1H), 7.86 – 7.70 (m, 2H), 7.65 (s, 1H), 7.26 – 7.13 (m, 2H), 5.14 (s, 1H), 3.76 – 3.60 (m, 2H), 3.16 (t, J = 6.5 Hz, 2H). 13C NMR (50 MHz, DMSO-d6) δ 161.24 (d, J = 243 Hz), 140.67, 140.41, 131.24, 126.28 (d, J = 8.0 Hz), 115.61 (d, J = 21.1 Hz), 114.85, 61.06, 36.11.
2-((4-(4-fluorophenyl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-imidazol-2-yl)thio)ethan-1-ol (17):
To a solution of 16 (965 mg, 4.05 mmol) in dry THF (20 ml) was added sodium hydride (60%, 4.45 mmol) at −15°C under vigorous stirring. The cooling bath was removed and the mixture warmed to room temperature over 30 minutes. The solution was again cooled to −15°C and SEM-Cl (716 μl, 4.05 mmol) was added dropwise. After complete consumption of the starting material, brine was added. The aqueous mixture was extracted three times with ethyl acetate. The combined organic layers were dried over Na2SO4, filtered and the solvents removed in vacuum. The crude mixture of regioisomers was dissolved in MeCN (2 ml) and treated with a catalytic amount of SEM-Cl (5 mol%). The solution was stirred at 70°C overnight. After cooling to room temperature, the solvent was removed in vacuum. The crude product was purified with gradient flash chromatography (from 100% n-hexane to n-hexane/ethyl acetate 3:2) to give the pure product as a pale yellow oil in 67% yield (1.03 g, 2.71 mmol). LRMS (ESI-MS): 369.1 [M+H]+. 1H NMR (200 MHz, Chloroform-d) δ 7.88 – 7.59 (m, 2H), 7.30 (s, 1H), 7.18 – 6.98 (m, 2H), 5.49 – 5.27 (m, 3H), 4.14 – 4.04 (m, 2H), 3.68 – 3.53 (m, 2H), 3.33 – 3.23 (m, 2H), 1.04 – 0.93 (m, 2H), 0.04 (s, 9H). 13C NMR (50 MHz, Chloroform-d) δ 162.10 (d, J = 246 Hz), 143.50, 140.74, 128.88 (d, J = 3.1 Hz), 126.31 (d, J = 8.3 Hz), 116.21, 115.51 (d, J = 21.5 Hz), 66.64, 63.72, 37.02, 17.64, −1.48.
2-((5-bromo-4-(4-fluorophenyl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-imidazol-2-yl)thio)ethan-1-ol (18):
To a cooled solution of 17 (1.0 g, 2.71 mmol) in 30 ml MeCN was added dropwise at −30°C N-Bromosuccinimide (507 mg, 2.85 mmol) dissolved in 15 ml MeCN. The solution was stirred for 1 hour at −30°C, the ice bath was removed and stirring continued at room temperature until complete consumption of the starting material. Saturated NaHCO3 solution and solid sodium thiosulfate was added and the mixture stirred for 10 minutes. The aqueous mixture was extracted three times with ethyl acetate. The combined organic layers were washed with saturated NaHCO3 solution, dried over Na2SO4, filtered and the solvents removed in vacuum. The obtained pure product was used without further purification. The product was isolated as a yellow oil in 64% yield (771 mg, 0.64 mmol). LRMS (ESI-MS): 447.1/449.2 [M+H]+.1H NMR (200 MHz, DMSO-d6) δ 7.81 – 7.54 (m, 2H), 7.54 – 7.26 (m, 2H), 5.55 (s, 2H), 3.96 – 3.40 (m, 7H), 0.85 (t, J = 6.1 Hz, 2H), 0.03 (s, 9H). 13C NMR (50 MHz, DMSO-d6) δ 163.95 (d, J = 252 Hz), 143.32, 138.48, 128.28 (d, J = 8 Hz), 127.88 (d, J = 3 Hz), 116.31 (d, J = 22 Hz), 98.09, 75.29, 66.42, 60.44, 35.33, 18.06, −1.43.
5-bromo-4-(4-fluorophenyl)-2-((2-methoxyethyl)thio)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-imidazole (19):
Intermediate 18 (193 mg, 0.43 mmol) was dissolved in 10 ml dry THF and cooled to −15°C. To the vigorously stirred solution was added sodium hydride (60%, 21 mg, 0.52 mmol) portion wise. After complete addition the reaction was slowly warmed to room temperature and methyl iodide (33 μl, 0.52 mmol) was added. The mixture was stirred for 1 hour at room temperature. After complete reaction, saturated NH4Cl solution was added. The aqueous mixture was extracted three times with DCM. The combined organic layers were dried over Na2SO4, filtered and the solvents removed in vacuum. The crude product was purified with gradient flash chromatography (from 100% n-hexane to n-hexane/ethyl acetate 3:1) to give the pure product as a colorless oil in 75% yield (148 mg, 0.32 mmol). 1H NMR (200 MHz, Chloroform-d) δ 8.14 – 7.87 (m, 2H), 7.23 – 7.01 (m, 2H), 5.41 (s, 2H), 3.85 – 3.55 (m, 4H), 3.46 – 3.36 (m, 5H), 1.14 – 0.86 (m, 2H), 0.04 (s, 9H). 13C NMR (50 MHz, Chloroform-d) δ 162.06 (d, J = 247 Hz), 143.67, 138.43, 128.91 (d, J = 3.3 Hz), 128.35 (d, J = 8.2 Hz), 115.06 (d, J = 21.5 Hz), 100.06, 73.80, 71.00, 66.53, 58.55, 33.30, 17.76, −1.46.
N-(4-(4-(4-fluorophenyl)-2-((2-methoxyethyl)thio)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-imidazol-5-yl)pyridin-2-yl)acetamide (20):
To a solution of 19 (148 mg, 0.32 mmol) and N-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridin-2-yl)acetamide (126 mg, 0.48 mmol) in 4 ml degassed 1,4-dioxane was added a n aqueous 1M K3PO4 solution (1 ml). The biphasic mixture was degassed 3 times, by evacuating and backfilling with argon. Under argon, P(t-Bu)3Pd G3 (5 mol%, 9mg) was added, the reaction flask was sealed and the suspension stirred for 5 hours at 50°C. After complete reaction, the mixture was cooled to room temperature and brine was added. The aqueous mixture was extracted three times with DCM. The combined organic layers were dried over Na2SO4, filtered and the solvents removed in vacuum. The crude product was purified with gradient flash chromatography (from 100% n-hexane to n-hexane/ethyl acetate 1:1) to give the pure product as a colorless oil in 56% yield (92 mg, 0.17 mmol). LRMS (ESI-MS): 539.6 [M+Na]+. 1H NMR (200 MHz, Chloroform-d) δ 9.25 (s, 1H), 8.42 – 8.09 (m, 2H), 7.55 – 7.37 (m, 2H), 7.13 – 6.85 (m, 3H), 5.25 (s, 2H), 3.76 (t, J = 6.2 Hz, 2H), 3.64 – 3.36 (m, 7H), 2.22 (s, 3H), 0.94 (t, J = 8.3 Hz, 2H), −0.00 (s, 9H). 13C NMR (50 MHz, Chloroform-d) δ 168.76, 162.02 (d, J = 246 Hz), 152.25, 147.90, 144.49, 140.87, 139.49, 129.61 (d, J = 3.0 Hz), 129.12 (d, J = 7.9 Hz), 127.42, 121.19, 115.30, 115.05 (d, J = 21.4 Hz), 71.12, 66.32, 58.59, 33.35, 24.77, 24.49, 17.70, −1.53.
4-(4-(4-fluorophenyl)-2-((2-methoxyethyl)thio)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-imidazol-5-yl)pyridin-2-amine (21):
The intermediate 20 (92 mg, 0.17 mmol) was dissolved in 5 ml methanol and 1 ml of an aqueous solution of NaOH (5M) was added under stirring. The suspension was stirred for 3 hours at 50°C. After complete reaction, the mixture was cooled to room temperature and a saturated aqueous NH4Cl solution was added. The aqueous mixture was extracted three times with ethyl acetate. The combined organic layers were dried over Na2SO4, filtered and the solvents removed in vacuum. The crude product was purified with gradient flash chromatography (from 100% n-hexane to n-hexane/ethyl acetate 1:1) to give the pure product as a white solid in 59% yield (50 mg, 0.11 mmol). LRMS (ESI-MS): 475.5 [M+H]+. 1H NMR (200 MHz, Chloroform-d) δ 8.11 (d, J = 5.4 Hz, 1H), 7.60 – 7.39 (m, 2H), 6.97 (t, J = 8.6 Hz, 2H), 6.69 (d, J = 5.2 Hz, 1H), 6.59 (s, 1H), 5.19 (s, 2H), 4.71 (s, 2H), 3.77 (t, J = 6.2 Hz, 2H), 3.60 (t, J = 8.2 Hz, 2H), 3.52 – 3.38 (m, 5H), 0.95 (t, J = 8.3 Hz, 2H), 0.03 (s, 9H). 13C NMR (50 MHz, Chloroform-d) δ 161.93 (d, J = 246 Hz), 159.48, 148.32, 144.04, 140.06, 138.76, 129.75, (d, J = 3.1 Hz), 128.86 (d, J = 8.0 Hz), 127.95, 115.35, 115.07 (d, J = 21.5 Hz), 109.65, 72.87, 71.11, 66.46, 58.59, 33.30, 17.89, −1.48.
N-(3-((4-(4-(4-fluorophenyl)-2-((2-methoxyethyl)thio)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-imidazol-5-yl)pyridin-2-yl)amino)-4-methoxyphenyl)acrylamide (22):
A solution of 21 (50 mg, 0,11 mmol) and N-(3-bromo-4-methoxyphenyl)acrylamide (32 mg, 0.126 mmol) and cesium carbonate (171 mg, 0.525 mmol) in a mixture of 1,4-dioxane/t-BuOH (4ml + 1ml) was degassed three times by evacuating and backfilling with argon under stirring. Under an atmosphere of argon, Brettphos Pd G3 (5 mol%, 5 mg, 0.005 mmol) was added. The solution was stirred under reflux for 3 hours. The mixture was cooled to room temperature and a saturated aqueous NH4Cl solution was added. The aqueous mixture was extracted three times with ethyl acetate. The combined organic layers were dried over Na2SO4, filtered and the solvents removed in vacuum. The crude product was purified with gradient flash chromatography (from 100% n-hexane to 100 % ethyl acetate) to give the pure product as a white solid in 44% yield (30 mg, 0.046 mmol). LRMS (ESI-MS): 651.0 [M+H]+ . 1H NMR (200 MHz, Chloroform-d) δ 8.28 (d, J = 5.3 Hz, 1H), 8.11 (s, 1H), 7.60 – 7.45 (m, 4H), 7.23 (s, 1H), 7.05 – 6.81 (m, 5H), 6.53 – 6.20 (m, 2H), 5.80 – 5.70 (m, 1H), 5.23 (s, 2H), 3.88 (s, 3H), 3.78 (t, J = 6.1 Hz, 3H), 3.64 – 3.44 (m, 6H), 0.98 – 0.89 (m, 2H), 0.02 (s, 9H).
N-(3-((4-(4-(4-fluorophenyl)-2-((2-methoxyethyl)thio)-1H-imidazol-5-yl)pyridin-2-yl)amino)-4-methoxyphenyl)acrylamide (23):
The intermediate 22 (30 mg, 0.046 mmol) was dissolved in an ethanolic HCl solution (1.25 M) and stirred overnight at room temperature. After complete consumption of the starting material, the solution was evaporated to dryness and the oily residue taken up in ethyl acetate. The organic solution was washed with a saturated aqueous NaHCO3 solution, dried over Na2SO4, filtered and the solvents removed in vacuum. The crude product was dissolved in a minimum amount of ethyl acetate and triturated with n-pentane to give the pure product as a pale yellow solid in 70% yield (16 mg, 0.031 mmol). 1H NMR (200 MHz, DMSO-d6) δ 12.73 (s, 1H), 10.00 (s, 1H), 8.31 (d, J = 9.7 Hz, 1H), 8.19 – 7.95 (m, 2H), 7.62 – 7.40 (m, 3H), 7.37 – 7.12 (m, 3H), 6.95 (dd, J = 8.9, 3.5 Hz, 1H), 6.73 – 6.18 (m, 3H), 5.80 – 5.61 (m, 1H), 3.81 (s, 3H), 3.64 (t, J = 6.3 Hz, 2H), 3.32 – 3.24 (m, 5H). 13C NMR (50 MHz, DMSO-d6) δ 163.07, 162,20 (d, J = 249 Hz), 156.74, 147.32, 146.03, 143.33, 141.78, 140.92, 135.31, 132.54, 132.27, 130.95 (d, J = 8.3 Hz), 130.51, 129.97, 129.76, 126.45, 116.11 (d, J = 21.6 Hz), 113.22, 112.41, 111.22, 108.32, 71.15, 58.26, 56.23, 32.35. LRMS(ESI-MS): 517.9 [M-H]−.
Synthesis of 4-(4-fluorophenyl)-2-(methylthio)-1H-imidazole (23):
3.00 g (13.82 mmol) 2-Bromo-1-(4- fluorophenyl)ethan-1-one in 15 ml THF was added dropwise to a refluxing mixture of 1.92 g (13.82 mmol) 2-methyl-2-thiopseudourea hemi sulfate salt and 3.82 g (27.64 mmol) K2CO3 in THF/H2O (8 ml/2 ml). The mixture was refluxed for an additional h until TLC indicated complete consumption of the starting material. The mixture was evaporated to dryness and the residue dissolved in EtOAc and water. The layers were separated. The organic layer was washed twice with brine, dried over Na2SO4, filtered and evaporated to dryness. The crude mixture was purified via flash chromatography (n-hexane/EtOAc 1:1) to obtain the title product as a beige solid in 50 % yield (1.44 g, 6.91 mmol). 1H-NMR (200 MHz, d6-DMSO) δ 12.61 – 12.10 17 (m, 1H), 7.83 – 7.66 (m, 2H), 7.62 (s, 1H), 7.28 – 7.05 (m, 2H), 2.55 (s, 3H). 13C-NMR (50 MHz, d6-DMSO) δ 161.2(d, J = 242 Hz) 141.5, 140.5, 131.3 (d, J = 3 Hz), 126.3, 126.2 (d, J = 8 Hz), 115.5 (d, J = 21 Hz), 114.6, 15.8. ESI-MS: 209.1 [M+H]+.
4-(4-fluorophenyl)-2-(methylthio)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-imidazole (24):
710 mg (3.41 mmol) 23 was dissolved in 10 ml dry THF and cooled to −15°C with an ice/acetone mixture under an argon atmosphere. 150 mg NaH (3.75 mmol, 60 % dispersion in mineral oil) was added in small portions. The resulting suspension was stirred for 20 min at −15°C after which, 663 μl (3.75 mmol) SEM-Cl was added dropwise to the mixture. The mixture was allowed to warm to room temperature slowly. The reaction was quenched by the addition of saturated aqueous NH4Cl solution and the crude product was extracted three times with DCM. The organic layer was dried over Na2SO4, filtered and evaporated to dryness. The resulting mixture of regioisomeres (60:40) was dissolved in 10 ml dry acetonitrile under an argon atmosphere. 28 μl (0.171 mmol) SEM-Cl was added and the biphasic mixture was stirred for 4 h at 80°C to perform the “SEM-switch”. The reaction was quenched by the addition of a few drops of 10 % aqueous ammonia solution. The mixture was evaporated to dryness and the crude product was purified by flash chromatography (from n-hexane 100 % to n-hexane/EtOAc 3:1) to obtain the title compound as a colorless oil in 91% yield (1050 mg, 3.11 mmol). 1H-NMR (200 MHz, d6 -DMSO) δ 7.88 – 7.67 (m, 3H), 7.31 – 7.02 (m, 2H), 5.26 (s, 2H), 3.52 (t, J = 7.9 Hz, 2H), 2.58 (s, 3H), 0.81 – 0.90 (t, J = 7.9 Hz, 2H), 0.01 (s, 9H). 13C-NMR (50 MHz, d6 -DMSO) δ 170.6, 161.0 (d, J = 243 Hz), 143.5, 139.9, 130.4 (d, J = 3 Hz), 126.2 (d, J = 8 Hz), 118.5, 18 115.6 (d, J = 22 Hz), 74.9, 65.9, 17.5, 16.2, −1.0. ESI-MS: 339.1 [M+H]+.
5-Bromo-4-(4-fluorophenyl)-2-(methylthio)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-imidazole (25):
895 mg (2.64 mmol) 24 was dissolved in 20 ml dry acetonitrile under argon atmosphere. The solution was cooled to −30°C. 470 mg (2.64 mmol) N-Bromosuccinimide dissolved in 5 ml dry acetonitrile was added dropwise under vigorous stirring maintaining −30°C. The reaction mixture was stirred for 1h at −30°C and let allowed to slowly warm to room temperature. TLC indicated complete consumption of the starting material. The reaction was quenched by the addition of an aqueous sodium thiosulfate solution (10 %). The product was partitioned between water and DCM. The organic layer was washed with brine, dried over Na2SO4, filtered and evaporated to dryness. The crude product was purified via flash chromatography (hexane/EtOAc 5:1) to yield the title product as a colorless oil in 85 % yield (940 mg, 2.25 mmol). 1H-NMR (200 MHz, d6-DMSO) δ 7.97 – 7.85 (m, 2H), 7.34 – 7.19 (m, 2H), 5.30 (s, 2H), 3.58 (t, J = 7.9 Hz, 2H), 2.60 (s, 3H), 0.95 – 0.77 (t, J = 7.9 Hz, 2H), - 0.05 (s, 9H). 13C-NMR (50 MHz, d6 -DMSO) δ 161.7 (d, J = 244 Hz), 145.3, 137.5, 129.5 (d, J = 3 Hz), 128.4 (d, J = 8 Hz), 115.7 (d, J = 22 Hz), 100.8, 100.8, 74.0, 66.3, 17.5, 15.8, −1.03. ESI-MS: Found: 417/418 [M+H]+.
4-bromo-5-(4-fluorophenyl)-1-methyl-2-(methylthio)-1H-imidazole (26):
25 (675 mg, 1.62 mmol) was dissolved in dry DCM and stirred at room temperature. To this solution, methyl trifluoromethanesulfonate (230 μl, 2.10 mmol) was added and stirring continued for 2 hours. 1 ml trifluoroacetic acid was added and the solution stirred for 30 minutes at room temperature. After complete consumption of the imidazolium intermediate, the reaction was quenched by the addition of concentrated ammonia. The aqueous mixture was extracted three times with DCM. The combined organic layers were dried over Na2SO4, filtered and the solvents removed in vacuum. The crude product was purified with flash chromatography (100% DCM) to give the pure product as colorless crystals in 70% yield (252 mg, 1.13 mmol). 1H NMR (200 MHz, Chloroform-d) δ 7.37 – 7.26 (m, 2H), 7.17 – 7.07 (m, 2H), 3.53 (s, 3H), 2.65 (s, 3H). LRMS (ESI-MS): not detected.
N-(4-(5-(4-fluorophenyl)-1-methyl-2-(methylthio)-1H-imidazol-4-yl)pyridin-2-yl)acetamide (27):
To a solution of 26 (158 mg, 0.52 mmol) and N-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridin-2-yl)acetamide (343 mg, 1.31 mmol) in 6 ml degassed 1,4-dioxane was added a n aqueous 1M K3PO4 solution (1.5 ml). The biphasic mixture was degassed 3 times, by evacuating and backfilling with argon. Under argon, P(t-Bu)3Pd G3 (5 mol%, 9 mg) was added, the reaction flask was sealed and the suspension stirred for 5 hours at 50°C. After complete reaction, the mixture was cooled to room temperature and brine was added. The aqueous mixture was extracted three times with DCM. The combined organic layers were dried over Na2SO4, filtered and the solvents removed in vacuum. The crude product was purified with gradient flash chromatography (from 100% n-hexane to n-hexane/ethyl acetate 1:2) to give the pure product as a colorless solid in 39% yield (72 mg, 0.20 mmol). 1H NMR (200 MHz, Chloroform-d) δ 9.46 (s, 1H), 8.27 (s, 1H), 8.03 (d, J = 5.4 Hz, 1H), 7.36 – 7.07 (m, 5H), 3.33 (s, 3H), 2.71 (s, 3H), 2.09 (s, 3H). 13C NMR (50 MHz, Chloroform-d) δ 163.12 (d, J = 250 Hz), 151.90, 146.89, 144.31, 144.27, 135.56, 132.34 (d, J = 8.4 Hz), 132.07, 125.78 (d, J = 3.8 Hz), 116.42 (d, J = 22 Hz), 110.99, 31.15, 24.38, 15.53. LRMS (ESI-MS): not detected.
4-(5-(4-fluorophenyl)-1-methyl-2-(methylthio)-1H-imidazol-4-yl)pyridin-2-amine (7):
The intermediate 27 (70 mg, 0.223 mmol) was dissolved in 5 ml methanol and 1 ml of an aqueous solution of NaOH (5M) was added under stirring. The suspension was stirred for 3 hours at 60°C. After complete reaction, the mixture was cooled to room temperature and a saturated aqueous NH4Cl solution was added. The aqueous mixture was extracted three times with ethyl acetate. The combined organic layers were dried over Na2SO4, filtered and the solvents removed in vacuum. The crude product was purified with gradient flash chromatography (from 100% n-hexane to n-hexane/ethyl acetate 1:1) to give the pure product as a pale yellow solid in 88% yield (62 mg, 0.196 mmol). LRMS (ESI-MS): 315.5 [M+H]+. 1H NMR (200 MHz, Chloroform-d) δ 7.78 (d, J = 5.5 Hz, 1H), 7.34 – 7.23 (m, 2H), 7.22 – 7.09 (m, 2H), 6.76 (d, J = 1.3 Hz, 1H), 6.51 (dd, J = 5.5, 1.5 Hz, 1H), 4.50 (s, 2H), 3.33 (s, 3H), 2.68 (s, 3H). 13C NMR (50 MHz, Chloroform-d) δ 163.01 (d, J = 249.7 Hz), 158.58, 147.25, 143.85, 143.34, 135.83, 132.39 (d, J = 8.3 Hz), 131.44, 126.15 (d, J = 3.5 Hz), 116.28 (d, J = 21.7 Hz), 111.42, 105.22, 31.22, 15.85.
N-(3-((4-(5-(4-fluorophenyl)-1-methyl-2-(methylthio)-1H-imidazol-4-yl)pyridin-2-yl)amino)-4-methoxyphenyl)acrylamide (7):
A solution of 7 (63 mg, 0,20 mmol), N-(3-bromo-4-methoxyphenyl)acrylamide (23 mg, 0.24 mmol) and cesium carbonate (326 mg, 1.0 mmol) in a mixture of 1,4-dioxane/t-BuOH (8ml + 2ml) was degassed three times by evacuating and backfilling with argon under stirring. Under an atmosphere of argon, Brettphos Pd G3 (5 mol%, 9 mg, 0.01 mmol) was added. The solution was stirred under reflux for 3 hours. The mixture was cooled to room temperature and a saturated aqueous NH4Cl solution was added. The aqueous mixture was extracted three times with ethyl acetate. The combined organic layers were dried over Na2SO4, filtered and the solvents removed in vacuum. The crude product was purified with gradient flash chromatography (from 100% n-hexane to 100% ethyl acetate) to give the pure product as a white solid in 67% yield (74 mg, 0.15 mmol). LRMS (ESI-MS): 490[M+H]+.1H NMR (200 MHz, DMSO-d6) δ 9.98 (s, 1H), 8.22 (s, 1H), 8.01 (s, 1H), 7.91 (d, J = 5.4 Hz, 1H), 7.59 – 7.26 (m, 5H), 7.22 (s, 1H), 6.94 (d, J = 8.8 Hz, 1H), 6.62 – 6.12 (m, 3H), 5.71 (d, J = 9.7 Hz, 1H), 3.81 (s, 3H), 3.34 (s, 3H), 2.68 (s, 3H). 13C NMR (50 MHz, DMSO) δ 163.07, 162.85(d, J = 246.7 Hz), 156.72, 147.25, 146.17, 143.38, 142.94, 135.38, 133.22 (d, J = 8.4 Hz), 132.55, 132.30, 131.94, 130.43, 126.54 (d, J = 3.1 Hz), 126.39, 116.55 (d, J = 21.6 Hz), 113.36, 112.53, 111.82, 111.24, 107.27, 56.20, 31.59, 15.72.
N-(3-((4-(4-(4-fluorophenyl)-1-methyl-2-(methylthio)-1H-imidazol-5-yl)pyridin-2-yl)amino)-4-methoxyphenyl)acrylamide (6):
A solution of 4-(4-(4-fluorophenyl)-1-methyl-2-(methylthio)-1H-imidazol-5-yl)pyridin-2-amine (60 mg, 0,190 mmol,synthesized according to WO 2006089798 A1 Aug 31, 2006) and N-(3-bromo-4-methoxyphenyl)acrylamide (63 mg, 0.247 mmol) and cesium carbonate (311 mg, 0.954 mmol) in a mixture of 1,4-dioxane/t-BuOH (8ml + 2ml) was degassed three times by evacuating and backfilling with argon under stirring. Under an atmosphere of argon, Brettphos Pd G3 (5 mol%, 9 mg, 0.01 mmol) was added. The solution was stirred under reflux for 3 hours. The mixture was cooled to room temperature and a saturated aqueous NH4Cl solution was added. The aqueous mixture was extracted three times with ethyl acetate. The combined organic layers were dried over Na2SO4, filtered and the solvents removed in vacuum. The crude product was purified with gradient flash chromatography (from 100% n-hexane to n-hexane/ethyl acetate 1:1) to give the pure product as a white solid in 81% yield (75 mg, 0.154 mmol). LRMS (ESI-MS): 490.5 [M+H]+. 1H NMR (400 MHz, DMSO-d6) δ 10.01 (s, 1H), 8.46 (d, J = 2.6 Hz, 1H), 8.32 (s, 1H), 8.23 (d, J = 5.2 Hz, 1H), 7.46 (dd, J = 8.7, 5.7 Hz, 2H), 7.42 – 7.37 (m, 1H), 7.16 – 7.08 (m, 2H), 7.04 (s, 1H), 6.96 (d, J = 8.8 Hz, 1H), 6.74 – 6.67 (m, 1H), 6.51 – 6.41 (m, 1H), 6.23 (dd, J = 17.0, 2.2 Hz, 1H), 5.71 (dd, J = 10.1, 2.2 Hz, 1H), 3.80 (s, 3H), 3.44 (s, 3H), 2.64 (s, 3H). 13C NMR (101 MHz, DMSO) δ 163.29, 161.51 (d, J = 243.7 Hz), 156.96, 148.52, 146.01, 143.92, 139.53, 137.23, 132.59, 132.35, 131.05, 130.11, 128.77, 128.68 (d, J = 8.0 Hz), 126.55, 115.98, 115.57 (d, J = 21.4 Hz), 113.76, 112.56, 112.41, 111.34, 56.35, 31.98, 15.84.
EGFR protein purification.
Constructs spanning residues 696–1022 of the human EGFR (including WT and T790M/V948R mutant sequences) were prepared in a GST-fusion format using the pTriEX system (Novagen) for expression in Sf9 insect cells as described previously.3, 33 Briefly, EGFR kinase proteins were purified by Ni-NTA and glutathione-affinity chromatography in 50 mM Tris-HCl (pH = 8.0), 500 mM NaCl, 5% glycerol, 5 mM TCEP followed by size-exclusion chromatography after cleavage with Tomato etch virus (TEV) to remove the GST fusion partner following established procedures.
Structure determination.
EGFR(T790M/V948R) pre-incubated with 1 mM AMP-PNP and 10 mM MgCl2 on ice was prepared by hanging-drop vapor diffusion over a reservoir solution containing 0.1 M Bis-Tris (pH = 5.5), 25% PEG-3350, and 5 mM TCEP (buffer A). WT EGFR crystals were prepared by prepared by hanging-drop vapor diffusion over a reservoir solution containing 1.3 M sodium citrate and 0.1 M MES pH = 6.5 (buffer B). Drops containing crystals in buffer A were exchanged with solutions of buffer A containing ~1.0 mM 1–5 three times for an hour and then left overnight. WT crystals were soaked with solutions of buffer B containing 10% DMSO and were soaked for 24 – 48 hours prior to harvesting. In both case, crystals were flash frozen after rapid immersion in a cryoprotectant solution with buffer A or B, for T790M/V948R or WT, respectively, containing 25% Ethylene glycol. X-ray diffraction data was collected at 100K the Advanced Light Source a part of the Northeastern Collaborative Access Team (NE-CAT) on Beamline 24-ID-C or 24-ID-E or at the National Synchrotron Light Source II 17-ID-1. Diffraction data was processed and merged in Xia2 using aimless and XDS. The structure was determined by molecular replacement with the program PHASER using the inactive EGFR kinase from our previous allosteric inhibitor work excluding the EAI001 ligand (PDB 5D41)29 or WT EGFR (PDB 6D8E)34 Repeated rounds of manual refitting and crystallographic refinement were performed using COOT and Phenix. The inhibitor was modeled into the closely fitting positive Fo − Fc electron density and then included in following refinement cycles. Statistics for diffraction data processing and structure refinement are shown in Table S1.
Biochemical assays.
Inhibition data (IC50) against EGFR-wildtype, EGFR-L858R/T790M and EGFR-L858R/T790M/C797S were conducted at Reaction Biology corp (Malvern, PA) via commercial radiolabeled 33P(ATP) kinase assays. IC50 values were measured in duplicates at 5 different concentration with 10-fold dilution steps starting from 5 μM. Staurosporine was tested as control compound in singlicate at 10 different concentrations with a 4-fold dilution starting at 20 μM. A detailed description of the assay format is given in the literature.35–36
Supplementary Material
Scheme 1.
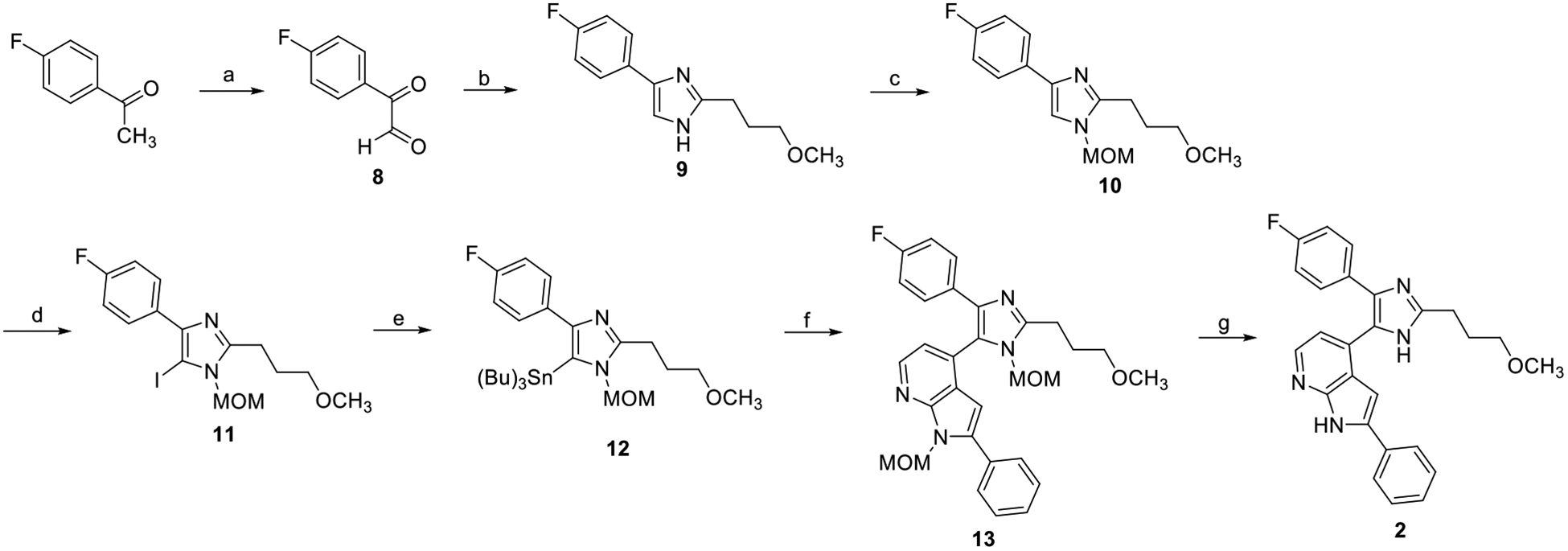
Reagents and conditions: (a) SeO2, 1,4-dioxane, H2O, 60°C to reflux (73%); (b) 4-methoxybutanal, NH4OAc, MeOH, rt (16%); (c) NaH, MOM-Cl, THF, 0°C to rt (77%); (d) N-iodosuccinimide, MeOH, 0°C (94%); (e) iPrMgCl*LiCl, Sn(Bu)3Cl, THF (quant.), (f) 4-chloro-1-(methoxymethyl)-2-phenyl-1H-pyrrolo[2,3-b]pyridine, Pd(OAc)2, XPhos, 1,4-dioxane, reflux (52%); (g) HCl(aq), MeOH, reflux (58%).
Scheme 3.
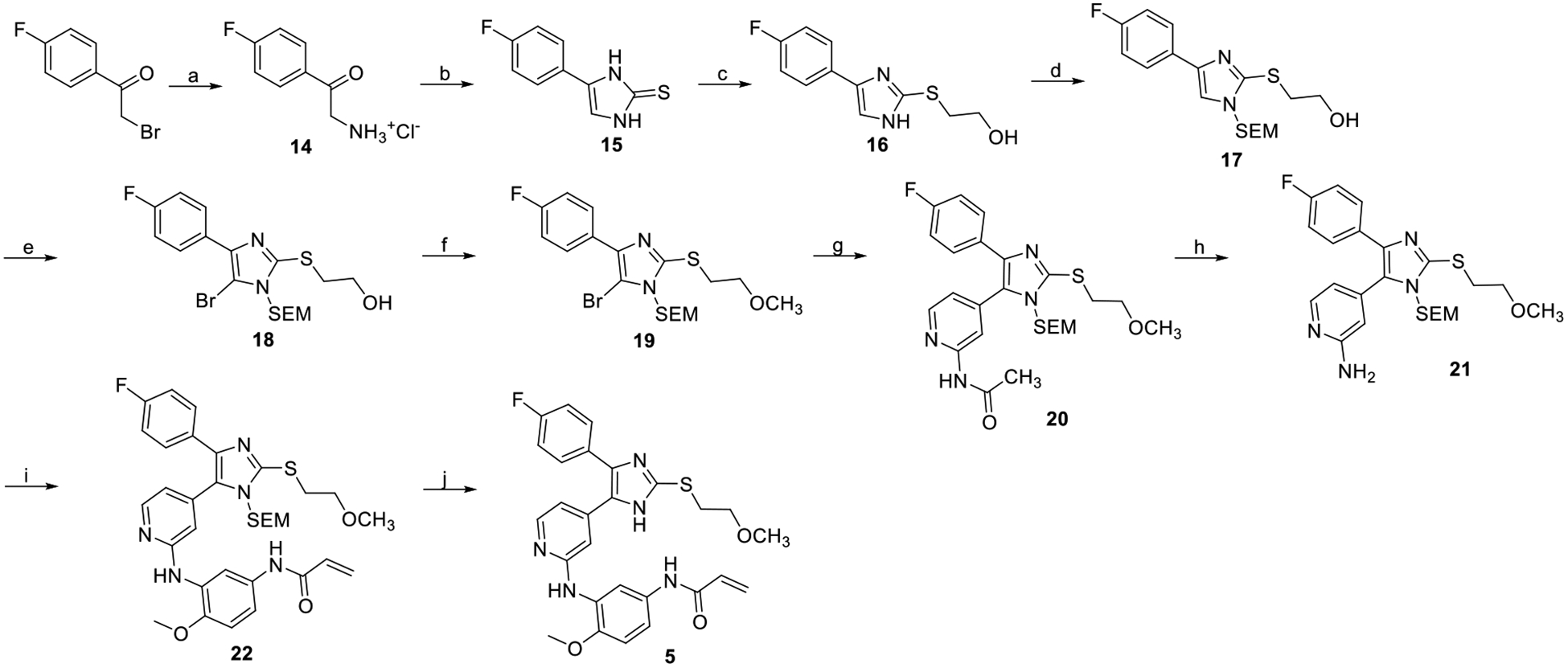
Reagents and conditions: (a) urotropine, chloroform, 50°C then EtOH, HCl(aq), rf (97%); (b) KSCN, HOAc, rf (71%); (c) 2-bromoethan-1-ol, K2CO3, MeOH, rt (quant.); (d) NaH, SEM-Cl, THF, 0°C (67%); (e) NBS, MeCN, −30°C (quant.);(f) NaH, MeI,THF, −15°C (75%); (g) N-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridin-2-yl)acetamide, P(t-bu)3 Pd G3, K3PO4, 1,4-dioxane/water, 50°C (56%%); (h) NaOH, THF/water, 50°C (59%); (i) N-(3-bromo-4-methoxyphenyl)acrylamide, Brettphos Pd G3, Cs2CO3, t-BuOH/1,4-dioxane, rf (44%); (j) HCl in EtOH 2.5 M, rt (70%).
ACKNOWLEDGMENT
We thank Dr. Tyler S. Beyett for technical assistance and helpful discussions.
Funding Sources
This work was supported by the National Institutes of Health grants R01CA201049 and R01CA116020 (to M.J.E). Funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany’s Excellence Strategy - EXC 2180 - 390900677 (to S.A.L). This work is based upon research as a part of the Cluster of Excellence iFIT (EXC 2180) “Image-Guided and Functionally Instructed Tumor Therapies”, University of Tübingen, Germany. This work is based upon research conducted at the Northeastern Collaborative Access Team (NECAT) beamlines, which are funded by the National Institute of General Medical Sciences from the National Institutes of Health (P30 GM124165). The Pilatus 6M detector on 24-ID-C beam line is funded by a NIH-ORIP HEI grant (S10 RR029205). This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC020-6CH11357. This research used resources 17-ID FMX and AMX of the National Synchrotron Light Source II, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Brookhaven National Laboratory under Contract No. DE-SC0012704. The Life Science Biomedical Technology Research resource is primarily supported by the National Institute of Health, National Institute of General Medical Sciences (NIGMS) through a Biomedical Technology Research Resource P41 grant (P41GM111244), and by the DOE Office of Biological and Environmental Research (KP1605010).
ABBREVIATIONS
- TKIs
Tyrosine kinase inhibitors
- EGFR
Epidermal growth factor receptor
- NSCLC
non-small cell lung cancer
- H-bond
hydrogen bond
Footnotes
Conflicts of Interest
M.J.E. reports receiving a commercial research grant from Novartis Institutes for Biomedical Research, reports receiving other commercial research support from Takeda, and has been a consultant for Novartis Institutes for Biomedical Research.
Supporting Information
A listing of the contents of each file supplied as Supporting Information should be included. For instructions on what should be included in the Supporting Information as well as how to prepare this material for publication, refer to the journal’s Instructions for Authors.
The Supporting Information is available free of charge on the ACS Publications website.
Crystallography and biochemical data (file type, i.e., PDF)
The supporting information is available free of charge on the ACS Publications Website at DOI:
X-ray crystallography statistics; supporting structural images; biochemical potencies of 1–3 and 5; additional chemistry for precursor.
SMILES molecular formula strings.
Accession Codes
X-ray coordinates and structures factors have been deposited in the Protein Data Bank. PDB accession codes: EGFR(T790M/V948R): 6V5N (1), 6V5P (2), 6V6O (3), 6V6K (4), 6V66 (5); wildtype EGFR: 6VH4 (3), 6VHN (4), 6VHN (5).
Authors will release the atomic coordinates upon article publication.
REFERENCES
- 1.Paez JG; Jänne PA; Lee JC; Tracy S; Greulich H; Gabriel S; Herman P; Kaye FJ; Lindeman N; Boggon TJ; Naoki K; Sasaki H; Fujii Y; Eck MJ; Sellers WR; Johnson BE; Meyerson M, EGFR Mutations in Lung Cancer: Correlation with Clinical Response to Gefitinib Therapy. Science 2004, 304 (5676), 1497–1500. [DOI] [PubMed] [Google Scholar]
- 2.Lynch TJ; Bell DW; Sordella R; Gurubhagavatula S; Okimoto RA; Brannigan BW; Harris PL; Haserlat SM; Supko JG; Haluska FG; Louis DN; Christiani DC; Settleman J; Haber DA, Activating Mutations in the Epidermal Growth Factor Receptor Underlying Responsiveness of Non-Small-Cell Lung Cancer to Gefitinib. New England Journal of Medicine 2004, 350 (21), 2129–2139. [DOI] [PubMed] [Google Scholar]
- 3.Yun C-H; Mengwasser KE; Toms AV; Woo MS; Greulich H; Wong K-K; Meyerson M; Eck MJ, The T790M Mutation in EGFR Kinase Causes Drug Resistance by Increasing the Affinity for ATP. Proceedings of the National Academy of Sciences 2008, 105 (6), 2070–2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kobayashi S; Boggon TJ; Dayaram T; Jänne PA; Kocher O; Meyerson M; Johnson BE; Eck MJ; Tenen DG; Halmos B, EGFR Mutation and Resistance of Non-Small-Cell Lung Cancer to Gefitinib. New England Journal of Medicine 2005, 352 (8), 786–792. [DOI] [PubMed] [Google Scholar]
- 5.Zhou W; Ercan D; Chen L; Yun C-H; Li D; Capelletti M; Cortot AB; Chirieac L; Iacob RE; Padera R, Novel Mutant-Selective EGFR Kinase Inhibitors Against EGFR T790M. Nature 2009, 462 (7276), 1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cross DA; Ashton SE; Ghiorghiu S; Eberlein C; Nebhan CA; Spitzler PJ; Orme JP; Finlay MRV; Ward RA; Mellor MJ, AZD9291, An Irreversible EGFR TKI, Overcomes T790M-Mediated Resistance to EGFR Inhibitors in Lung Cancer. Cancer Discovery 2014, 4 (9), 1046–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jänne PA; Yang JC-H; Kim D-W; Planchard D; Ohe Y; Ramalingam SS; Ahn M-J; Kim S-W; Su W-C; Horn L; Haggstrom D; Felip E; Kim J-H; Frewer P; Cantarini M; Brown KH; Dickinson PA; Ghiorghiu S; Ranson M, AZD9291 in EGFR Inhibitor-Resistant Non-Small-Cell Lung Cancer. New England Journal of Medicine 2015, 372 (18), 1689–1699. [DOI] [PubMed] [Google Scholar]
- 8.Soria J-C; Ohe Y; Vansteenkiste J; Reungwetwattana T; Chewaskulyong B; Lee KH; Dechaphunkul A; Imamura F; Nogami N; Kurata T, Osimertinib in Untreated EGFR-Mutated Advanced Non–Small-Cell Lung Cancer. New England Journal of Medicine 2018, 378 (2), 113–125. [DOI] [PubMed] [Google Scholar]
- 9.Thress KS; Paweletz CP; Felip E; Cho BC; Stetson D; Dougherty B; Lai Z; Markovets A; Vivancos A; Kuang Y, Acquired EGFR C797S Mutation Mediates Resistance to AZD9291 in Non–Small Cell Lung Cancer Harboring EGFR T790M. Nature Medicine 2015, 21 (6), 560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Niederst MJ; Hu H; Mulvey HE; Lockerman EL; Garcia AR; Piotrowska Z; Sequist LV; Engelman JA, The Allelic Context of the C797S Mutation Acquired upon Treatment with Third-Generation EGFR Inhibitors Impacts Sensitivity to Subsequent Treatment Strategies. Clinical Cancer Research 2015, 21 (17), 3924–3933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen L; Fu W; Zheng L; Liu Z; Liang G, Recent Progress of Small-Molecule Epidermal Growth Factor Receptor (EGFR) Inhibitors against C797S Resistance in Non-Small-Cell Lung Cancer. Journal of Medicinal Chemistry 2018, 61 (10), 4290–4300. [DOI] [PubMed] [Google Scholar]
- 12.Selig R; Goettert M; Schattel V; Schollmeyer D; Albrecht W; Laufer S, A Frozen Analogue Approach to Aminopyridinylimidazoles Leading to Novel and Promising p38 MAP Kinase Inhibitors. Journal of Medicinal Chemistry 2012, 55 (19), 8429–8439. [DOI] [PubMed] [Google Scholar]
- 13.Günther M; Juchum M; Kelter G; Fiebig H; Laufer S, Lung Cancer: EGFR Inhibitors with Low Nanomolar Activity Against a Therapy-Resistant L858R/T790M/C797S mutant. Angewandte Chemie International Edition 2016, 55 (36), 10890–10894. [DOI] [PubMed] [Google Scholar]
- 14.Günther M; Lategahn J; Juchum M; Döring E; Keul M; Engel J; Tumbrink HL; Rauh D; Laufer S, Trisubstituted Pyridinylimidazoles as Potent Inhibitors of the Clinically Resistant L858R/T790M/C797S EGFR Mutant: Targeting of Both Hydrophobic Regions and the Phosphate Binding Site. Journal of Medicinal Chemistry 2017, 60 (13), 5613–5637. [DOI] [PubMed] [Google Scholar]
- 15.Juchum M; Günther M; Döring E; Sievers-Engler A; Lämmerhofer M; Laufer S, Trisubstituted Imidazoles with a Rigidized Hinge Binding Motif Act As Single Digit nM Inhibitors of Clinically Relevant EGFR L858R/T790M and L858R/T790M/C797S Mutants: An Example of Target Hopping. Journal of Medicinal Chemistry 2017, 60 (11), 4636–4656. [DOI] [PubMed] [Google Scholar]
- 16.Uchibori K; Inase N; Araki M; Kamada M; Sato S; Okuno Y; Fujita N; Katayama R, Brigatinib Combined with Anti-EGFR Antibody Overcomes Osimertinib Resistance in EGFR-Mutated Non-Small-Cell Lung Cancer. Nature communications 2017, 8, 14768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Park H; Jung H-Y; Mah S; Hong S, Discovery of EGF Receptor Inhibitors That Are Selective for the d746–750/T790M/C797S Mutant through Structure-Based de Novo Design. Angewandte Chemie International Edition 2017, 56 (26), 7634–7638. [DOI] [PubMed] [Google Scholar]
- 18.Lu X; Zhang T; Zhu S-J; Xun Q; Tong L; Hu X; Li Y; Chan S; Su Y; Sun Y; Chen Y; Ding J; Yun C-H; Xie H; Ding K, Discovery of JND3229 as a New EGFRC797S Mutant Inhibitor with In Vivo Monodrug Efficacy. ACS Medicinal Chemistry Letters 2018, 9 (11), 1123–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shen J; Zhang T; Zhu S-J; Sun M; Tong L; Lai M; Zhang R; Xu W; Wu R; Ding J; Yun C-H; Xie H; Lu X; Ding K, Structure-Based Design of 5-Methylpyrimidopyridone Derivatives as New Wild-Type Sparing Inhibitors of the Epidermal Growth Factor Receptor Triple Mutant (EGFRL858R/T790M/C797S). Journal of Medicinal Chemistry 2019, 62 (15), 7302–7308. [DOI] [PubMed] [Google Scholar]
- 20.Lategahn J; Keul M; Klövekorn P; Tumbrink HL; Niggenaber J; Müller MP; Hodson L; Flaßhoff M; Hardick J; Grabe T; Engel J; Schultz-Fademrecht C; Baumann M; Ketzer J; Mühlenberg T; Hiller W; Günther G; Unger A; Müller H; Heimsoeth A; Golz C; Blank-Landeshammer B; Kollipara L; Zahedi RP; Strohmann C; Hengstler JG; van Otterlo WAL; Bauer S; Rauh D, Inhibition of Osimertinib-Resistant Epidermal Growth Factor Receptor EGFR-T790M/C797S. Chemical Science 2019, 10, 10789–10801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lei H; Fan S; Zhang H; Liu Y-J; Hei Y-Y; Zhang J-J; Zheng AQ; Xin M; Zhang S-Q, Discovery of Novel 9-Heterocyclyl Substituted 9H-Purines as L858R/T790M/C797S Mutant EGFR Tyrosine Kinase Inhibitors. European Journal of Medicinal Chemistry 2019, 111888. [DOI] [PubMed] [Google Scholar]
- 22.Engelhardt H; Böse D; Petronczki M; Scharn D; Bader G; Baum A; Bergner A; Chong E; Döbel S; Egger G; Engelhardt C; Ettmayer P; Fuchs JE; Gerstberger T; Gonnella N; Grimm A; Grondal E; Haddad N; Hopfgartner B; Kousek R; Krawiec M; Kriz M; Lamarre L; Leung J; Mayer M; Patel ND; Simov BP; Reeves JT; Schnitzer R; Schrenk A; Sharps B; Solca F; Stadtmüller H; Tan Z; Wunberg T; Zoephel A; McConnell DB, Start Selective and Rigidify: The Discovery Path toward a Next Generation of EGFR Tyrosine Kinase Inhibitors. Journal of Medicinal Chemistry 2019, 62 (22), 10272–10293. [DOI] [PubMed] [Google Scholar]
- 23.Zhang X; Gureasko J; Shen K; Cole PA; Kuriyan J, An Allosteric Mechanism for Activation of the Kinase Domain of Epidermal Growth Factor Receptor. Cell 2006, 125 (6), 1137–1149. [DOI] [PubMed] [Google Scholar]
- 24.Wilson KP; McCaffrey PG; Hsiao K; Pazhanisamy S; Galullo V; Bemis GW; Fitzgibbon MJ; Caron PR; Murcko MA; Su MSS, The Structural Basis for the Specificity of Pyridinylimidazole Inhibitors of p38 MAP Kinase. Chemistry & Biology 1997, 4 (6), 423–431. [DOI] [PubMed] [Google Scholar]
- 25.Fitzgerald CE; Patel SB; Becker JW; Cameron PM; Zaller D; Pikounis VB; O’Keefe SJ; Scapin G, Structural Basis for p38α MAP Kinase Quinazolinone and Pyridol-Pyrimidine Inhibitor Specificity. Nature Structural & Molecular Biology 2003, 10 (9), 764–769. [DOI] [PubMed] [Google Scholar]
- 26.Ansideri F; Macedo JT; Eitel M; El-Gokha A; Zinad DS; Scarpellini C; Kudolo M; Schollmeyer D; Boeckler FM; Blaum BS; Laufer SA; Koch P, Structural Optimization of a Pyridinylimidazole Scaffold: Shifting the Selectivity from p38α Mitogen-Activated Protein Kinase to c-Jun N-Terminal Kinase 3. ACS Omega 2018, 3 (7), 7809–7831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huang W-S; Liu S; Zou D; Thomas M; Wang Y; Zhou T; Romero J; Kohlmann A; Li F; Qi J; Cai L; Dwight TA; Xu Y; Xu R; Dodd R; Toms A; Parillon L; Lu X; Anjum R; Zhang S; Wang F; Keats J; Wardwell SD; Ning Y; Xu Q; Moran LE; Mohemmad QK; Jang HG; Clackson T; Narasimhan NI; Rivera VM; Zhu X; Dalgarno D; Shakespeare WC, Discovery of Brigatinib (AP26113), a Phosphine Oxide-Containing, Potent, Orally Active Inhibitor of Anaplastic Lymphoma Kinase. Journal of Medicinal Chemistry 2016, 59 (10), 4948–4964. [DOI] [PubMed] [Google Scholar]
- 28.Jang J; Son JB; To C; Bahcall M; Kim SY; Kang SY; Mushajiang M; Lee Y; Jänne PA; Choi HG, Discovery of a Potent Dual ALK and EGFR T790M Inhibitor. European Journal of Medicinal Chemistry 2017, 136, 497–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jia Y; Yun C-H; Park E; Ercan D; Manuia M; Juarez J; Xu C; Rhee K; Chen T; Zhang H; Palakurthi S; Jang J; Lelais G; DiDonato M; Bursulaya B; Michellys P-Y; Epple R; Marsilje TH; McNeill M; Lu W; Harris J; Bender S; Wong K-K; Jänne PA; Eck MJ, Overcoming EGFR(T790M) and EGFR(C797S) Resistance with Mutant-Selective Allosteric Inhibitors. Nature 2016, 534, 129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.To C; Jang J; Chen T; Park E; Mushajiang M; De Clercq DJH; Xu M; Wang S; Cameron MD; Heppner DE; Shin BH; Gero TW; Yang A; Dahlberg SE; Wong K-K; Eck MJ; Gray NS; Janne PA, Single and Dual Targeting of Mutant EGFR with an Allosteric Inhibitor. Cancer Discovery 2019, 9 (7), 926–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.De Clercq DJH; Heppner DE; To C; Jang J; Park E; Yun C-H; Mushajiang M; Shin BH; Gero TW; Scott DA; Jänne PA; Eck MJ; Gray NS, Discovery and Optimization of Dibenzodiazepinones as Allosteric Mutant-Selective EGFR Inhibitors. ACS Medicinal Chemistry Letters 2019, 10 (11), 1549–1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li Q; Zhang T; Li S; Tong L; Li J; Su Z; Feng F; Sun D; Tong Y; Wang X; Zhao Z; Zhu L; Ding J; Li H; Xie H; Xu Y, Discovery of Potent and Noncovalent Reversible EGFR Kinase Inhibitors of EGFRL858R/T790M/C797S. ACS Medicinal Chemistry Letters 2019, 10 (6), 869–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yun C-H; Boggon TJ; Li Y; Woo MS; Greulich H; Meyerson M; Eck MJ, Structures of Lung Cancer-Derived EGFR Mutants and Inhibitor Complexes: Mechanism of Activation and Insights into Differential Inhibitor Sensitivity. Cancer Cell 2007, 11 (3), 217–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jang J; Son J; Park E; Kosaka T; Saxon JA; De Clercq DJH; Choi HG; Tanizaki J; Eck MJ; Jänne PA; Gray NS, Discovery of a Highly Potent and Broadly Effective Epidermal Growth Factor Receptor and HER2 Exon 20 Insertion Mutant Inhibitor. Angewandte Chemie International Edition 2018, 57 (36), 11629–11633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Duong-Ly KC; Peterson JR, A High-Throughput Radiometric Kinase Assay. Methods in Molecular Biology 2016, 1360, 87–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Anastassiadis T; Deacon SW; Devarajan K; Ma H; Peterson JR, Comprehensive Assay of Kinase Catalytic Activity Reveals Features of Kinase Inhibitor Selectivity. Nature Biotechnology 2011, 29 (11), 1039–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.