

Abstract
Purpose of the Review
Clonal heterogeneity is a significant obstacle to successful treatment of patients with acute myeloid leukemia (AML). Here we review new advances in the understanding of genetic heterogeneity in AML using single cell DNA sequencing technology.
Recent Findings
New genomics and immunologic discovery tools have provided single cell resolution maps of the clonal architecture of AML. The use of these technologies reveals the mutational landscape of AML at diagnosis, during treatment, and at relapse has an enormous degree of clonal complexity and diversity that is poised to adapt and evolve under environmental pressures.
Summary
AML is a complex ecosystem of competing and cooperating clones undergoing constant evolution and selection.
Keywords: single cell sequencing, AML, tumor heterogeneity, clonal dynamics, clonal evolution
Introduction
Acute myeloid leukemia (AML) is a highly complex and heterogeneous group of diseases that arise as a consequence of accumulation of aberrant genetic alterations involving whole or parts of chromosomes giving rise to copy number variants (CNV) or fusion gene products, various sized insertions and deletions (in/del), and single nucleotide variants (SNV). Over the last decade numerous next generation sequencing (NGS) studies on bulk collections of AML patient peripheral blood and bone marrow biopsies have detailed recurrent SNVs and small in/dels in over 270 different genes in AML, of which 76 are putative cancer driver gene mutations [1–3]. Newer single cell DNA sequencing (scDNA-seq) technologies have revealed each AML case represents a combination of genetically distinct clonal populations resulting in profound inter- and intra-tumoral heterogeneity [4**, 5**]. This genetic and clonal heterogeneity presents a significant challenge in development of new effective therapies for treatment of AML and underscores the improbability of a “one size fits all” treatment approach.
This review focuses on our current understanding of the genetic and immunophenotypic clonal architecture of AML at single cell resolution. We define “clone” as a group of cells containing identical gene mutations. Clones can be further classified into three groups: founding clone (defined as a clone containing the initiating gene mutation that is present in all clones within the same tumor), subclone (any clone within a tumor containing more than one clone with shared clonal origin), and dominant clone (clone that comprises the largest fraction of the total tumor). These definitions classify clones based on genetic identity; however, a given subclone or even the dominant clone may not contain all the mutations found within the tumor as a whole, demonstrating AML has a complex clonal structure. Thus, constructing more precise clonal landscapes at single cell resolution informs novel, clinically meaningful, biologic consequences, and will be highlighted in this review. We will discuss recent advances in our understanding of mechanisms of genetic heterogeneity and gene combinations on the clonal milieu and dynamics in AML patients. Finally, we will emphasize studies evaluating the clonal dynamics during therapeutic treatment illustrating that individual AML clonal uniqueness, diversity, and complexity contribute to therapeutic sensitivity or resistance.
Clonal Evolution
Single cell DNA mutation profiling has revealed two main types of clonal evolution in AML, linear (Fig. 1a) and branched (Fig. 1b), which occur during disease initiation and progression as well as in response to treatment and at relapse. Following a linear evolution trajectory, new clones arise with the sequential acquisition of new mutations. In branching evolution, two or more daughter clones arise from one parental clone upon acquisition of a new distinct mutation in each of the daughter clones, all of which further evolve in parallel. In general, less complex AML structures follow a linear evolution, resulting in less subclones and fewer overall numbers of unique mutations, while AML with branching evolution tends to consist of more subclones and greater overall numbers of mutations [4–6**]. One common factor contributing to the complexity of clonal evolution in AML, is the acquisition of mutations in the same gene along different branches resulting in more subclones, without adding to the overall mutational burden of the tumor (Fig. 1c). Examples of this include NPM1 W288Cfs*12 (NPM1 L287inc(TCTG)* or NPM1c) and NRAS G12D and G12V mutations, which are recurrently detected in distinct subclones evolving in parallel branches within the same AML patient [4**, 5**]. Another observed example contributing to further clonal complexity is AML patients who have independent initiating clones that subsequently evolve by either linear or branching trajectories (Fig. 1d) [4**]. The majority of mutations detected by scDNA-seq are heterozygous; the most frequently mutated genes include ASXL1, FLT3-non-ITD, DNMT3A, EZH2, IDH1/2, KIT, KRAS, NRAS, PTPN11, SF3B1, NPM1, TP53, U2AF1, and WT1 [4**, 5**]. Conversely, JAK2 and GATA2 are often homozygous; while NPM1c, FLT3-ITD, RUNX1, and SRFS2 are heterozygous in some cases but homozygous in a minority of clones [4**, 5**]. In sum, linear evolution results in fewer subclones and lower mutational burden as compared to branching evolution trajectories.
Figure 1. Clonal evolution trajectories and clonal diversity.
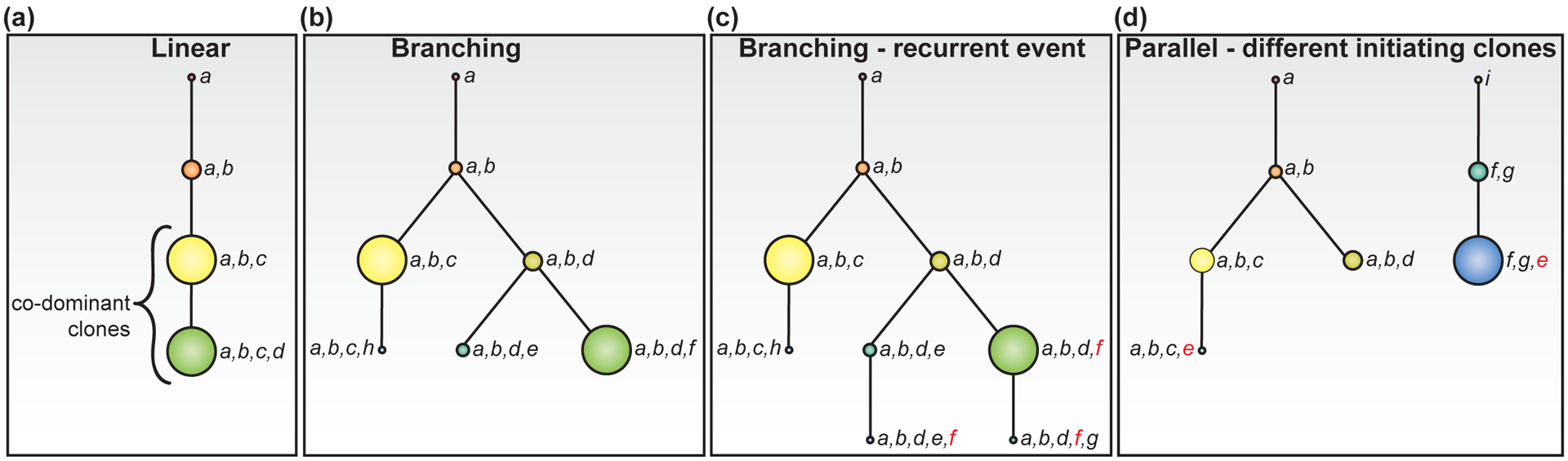
Tree diagrams depict examples of linear (a) and branching (b-d) AML evolution trajectories. The size of each clonal node corresponds to the overall fraction of cells in the clone of the total cells sequenced. (a-c) Each tree starts with the acquisition of mutation in gene “a”. a) In the linear model, sequential acquisition of mutations “b”, “c”, and then “d” in the linear evolution trajectory results in co-mutational cooperation as the clone size expands with new mutations. b) In the branching model, cells from clone “a,b” acquire two different mutations which expand to make clones “a,b,c” and “a,b,d” which evolve through separate clonal trajectories, and one additional branch evolves from clone “a,b,d”. Although there are 4 clones in the linear model (a) and 7 clones in the branching model (c) illustrating greater clonal complexity in the branching model, there is similar clonal diversity in both models as each has two co-dominant clones (yellow and green) that comprise the majority of each tumor. c) Clone “a,b,d,f” and clone “a,b,d,e,f” are in two separate branched clonal trajectories and have acquired the same gene “f” mutation (red) independently late in disease evolution. Clone “a,b,d,f,g” descendent from “a,b,d,f” also carries the gene “f” mutation. d) Two independent clonal trajectories are evolving in parallel from two distinct initiating mutations in “a” and “i” then subsequently evolving through in branched and linear trajectories, respectfully. Mutation “e” (red) is acquired late in both trajectories.
Despite the complex evolutionary trajectories observed in AML, patterns with respect to mutation order are observed. Bulk NGS and scDNA-seq studies concur that mutations in epigenetic modifiers, including DNMT3A, IDH1/2, ASXL1, and SRSF2 tend to be acquired early in disease progression often in the founding clone, while mutations in NPM1, FLT3, and RAS tend to be acquired later [4–8]. These patterns, however, are not exclusive as NPM1c and TET2 can be early or later events in clonal evolution [4**, 6**]. Even independently acquired mutations in the same gene that arise in separate AML subclones through a branching trajectory maintain this general order. These mutation order trends suggest gene-specific biologic mechanisms and genotype-specific bottlenecks which can be overcome by specific additional mutations. Only very recently have studies begun to understand the consequences of mutation order using mouse models on leukemia pathogenesis. Loberg et. al, generated transgenic mice with differentially inducible Dnmt3a R878H under the control of interferon-inducible cre recombinase and Npm1c controlled by tamoxifen-inducible flipase recombinase [9*]. Starting with the induction of Dnmt3a R878H, immediate subsequent induction of Npm1c mice eventually developed myeloid malignancies. However, if mice were allowed to live with Dnmt3a R878H for a longer period of time, as is observed in human patients that progress from clonal hematopoiesis or myeloproliferative neoplasms to AML, the delayed induction of Npm1c resulted in significantly faster disease onset [9*]. This work demonstrates some of the nuances of mutation order and timing during clonal evolution that are not yet well understood. Altogether, these studies reveal the order of mutation acquisition and well as the evolutionary trajectory of subclones are biologically and clinically meaningful aspects of AML pathogenesis.
Genotype Phenotype Clonal Complexity
Combination of scDNA-seq with sequence tagged antibody labeling of cell surface proteins can define immunophenotypically similar populations and compare genotype with immunophenotype at the single cell level [5**]. This approach can help distinguish normal hematopoietic cells by the absence of AML gene mutations [4–6**] and/or non-myeloid (e.g. CD3+) immunophenotype [5**]. Single cell genotype/phenotype studies, although few in number, have revealed that clonal immunophenotypes also evolve with mutational acquisition. For example, IDH1 mutant clones express high levels of CD34 [5**], and the addition of DNMT3A mutations further enhances CD34 levels in IDH1/DNMT3A co-mutant clones. Conversely, the addition of either an NPM1c or NRAS mutation results in a CD34 low-negative IDH1/NPM1c and IDH1/NRAS clonal immunophenotype [5**, 10]. Different RAS family mutations differentially alter immunophenotype of clones with coincident DNMT3A mutations. DNMT3A/NRAS clones contain dramatically more CD11b+ cells, while the DNMT3A/KRAS clones contain substantially more CD19+ cells [5**], indicative of different biologic mechanisms for different RAS family members in AML. FLT3 mutations, which are highly common in AML, strongly specify clonal immunophenotype. For example, DNMT3A-mutant clones are largely CD90+CD45RA+, while FLT3-mutant and DNMT3A/FLT3 co-mutant clones contain more CD38+ cells [5**]. Overall, this work demonstrates substantial immunophenotypic heterogeneity within genetically defined AML clones and AML immunophenotypes have some plasticity specified to genotype combinations. In contrast, these genetic and immunophenotypic features revealed by single cell approaches are not apparent in bulk sequencing analyses in the absence of detailed genomic analysis of flow cytometric sorted immunophenotype-defined populations. Future studies aimed at characterizing clonal immunophenotypes, when also accounting for genotype, may shed light on how genotype and differentiation status collectively impact therapeutic response.
Clonal Dominance and Diversity
Clonal dominance is defined by the largest clone present at any given time in AML progression. Single cell genomic studies suggest certain AML mutations are more frequently found in the dominant clone [4–6**]. IDH1/2 (~75%) and NPM1 (~90%) mutations, for example, are preferentially found in the dominant AML clones [4**, 5**]. Interestingly, mutations in signaling factors such as FLT3, NRAS, and KIT are less likely to be present in the dominant clone, observed in only ~25% of cases [5**, 6**]. Thus, specific mutations confer a greater fitness advantage than others, but genetic contribution to dominant clones is likely also influenced by leukemic contexts such as clonal diversity and therapeutic selection.
Mutational cooperativity is inherent to the transformation and progression of AML, as >85% of AML harbor two or more driver gene mutations [2]. Predictably then certain co-occurring mutations should also contribute to clonal dominance. Single cell genomics revealed more than half of AML cases exhibit co-incident mutations in epigenetic modifiers DNMT3A, TET2, ASXL1, and/or IDH1/2 [5**]. Moreover, this clonal subtype comprised the dominant clone in greater than 80% of cases [5**, 7*]. These data suggest mutant epigenetic modifiers may have functional cooperativity in a cell-autonomous manner that increases the fitness advantage of this clonal subtype. In agreement with this, a recent study using mouse Dnmt3a knockout hematopoietic cells as a model of DNMT3A loss-of-function mutations in human AML, found that expression of Idh2 R140Q synergized with Dnmt3a loss to promote the accumulation of immature myeloid progenitors and significantly shorten survival as compared to mice with either Idh2 R140Q or Dnmt3a loss alone [11*]. Another study using mice with functional deletions of both Dnmt3a and Tet2 observed that the loss of these two epigenetic modifiers led to the expansion of hematopoietic stem and progenitor compartments, and significantly shortened survival compared to single mutant mice [12]. These mouse model studies demonstrate the cooperative nature of some epigenetic modifiers and support the hypothesis that co-occurrence of these mutations in the dominant clone confer a fitness advantage. On the other hand, mutations in signaling factors KRAS, NRAS, KIT, and FLT3 were found almost exclusively in different clones in single cell studies [4**, 5**, 7*, 13], and in rare cases where signaling factor mutations did co-occur, it was never the dominant clone [5**]. This mutual exclusivity of signaling mutations suggests a functional redundancy that does not provide a fitness advantage. In sum, single cell genomics most definitively resolves the co-mutational events at the single cell and clonal levels, which will ultimately guide future mechanistic studies to explain the biologic requirements for certain complex mutation combinations that are observed in AML.
How is clonal dominance balanced by clonal diversity? Clonal diversity considers both the number of unique clones and the relative abundance of each clone per sample. By bulk NGS the average AML patient sample contains between 1–4 unique AML clones [3]. However, in striking contrast, scDNA-seq studies have exposed staggering clonal diversity with >30 clones in some extreme cases and up to 7 different gene mutations within a single patient tumor [5**]. While this high degree of clonal diversity is uncommon, the majority of AML patients harbor between 3–13 unique clones and 3–7 different gene mutations demonstrating overall higher clonal diversity within individual AML samples than previously appreciated [4**, 5**, 7*, 10, 14]. Despite numerous subclones per AML, generally 1–2 clones comprise the majority of the tumor [4**, 5**]. The relative abundance of each clone tends to grow with the acquisition of a new mutation; however, this is not true for all clones suggesting some mutation combinations are more synergistic conferring a greater fitness advantage than others, as described above. The size of the dominant clone varies from patient to patient, but tends to be smaller as the total number of AML subclones increases [5*]. In many cases, the dominant clone is easy to discern by scDNA-seq; significantly outnumbering the cellular sum of other subclones; however, there are many AML cases that are less clear containing two or more subclones nearly equal in size [4**, 5**]. This discovery has led to emerging paradigms of clonal cooperativity whereby multiple subclones, in addition to the dominant clone, contribute to the overall progression of leukemia, and the existence of co-dominant clones provides additional stability to the tumor under stress of chemotherapy. A potential mechanism by which clones may cooperate and/or interact is through secreted factors, such as cytokines, chemokines, or growth factors, that mutually benefit one another or provide defense against anti-tumor immune response or therapeutic pressures. The converse wherein clones are in direct competition with one another is likely also a contributing factor to leukemic progression. Overall, these new discoveries better define the mutational impact on AML clonal dominance and diversity and present new challenges to understand how the interplay between subclones contributes to leukemogenesis either competitively in equilibrium or cooperatively.
Clonal Changes in Response to Treatment
The ability to track clonal dynamics from diagnosis through treatment and relapse with single cell resolution provides a unique opportunity to further dissect the clinical impact and biologic intersection of mutational subsets and therapeutic response on clone size, evolution, and diversity during the disease course of AML. Generally, in the upfront setting standard induction chemotherapy is given for patients under 70 years old with de novo AML (7+3 chemotherapy) [15, 16]. Unfortunately, this form of treatment is associated with non-specific cytotoxicity, a high rate of relapse, low overall survival, and is not suitable for most patients >70 years old or with comorbidities. Many studies have attempted to understand why patients are refractory to induction chemotherapy or relapse after a period of remission. Bulk NGS studies suggest patients who eventually relapse tend to have a higher mutational burden at diagnosis than patients who exhibit a longer relapse free survival [8]. However, correlating the number of mutations with relapse is a gross over-simplification of AML responsiveness to chemotherapy, and has no clinical prognostic or predictive power. Using scDNA-seq to evaluate the leukemic clonal architecture at diagnosis, remission, and relapse, Ediriwickrema et al. discerned minimal residual disease (MRD) clones that contributed to relapse, and identified patterns in clonal evolution and heterogeneity that might predispose patients to relapse after standard chemotherapy [6**]. Comparing patients with a similar number of clones at diagnosis, a greater decrease in clonal diversity at remission was associated with a longer relapse-free survival, suggesting that a stable clonal landscape or increase in clonal diversity after treatment portends resistance/relapse [6**]. For example, an AML patient with co-dominant clones NPM1/IDH1/KRAS (36%) and NPM1/IDH2/FLT3-ITD (22%) at diagnosis received treatment with Cytarabine and FLT3 inhibitor Quizartinib. The NPM1/IDH2/FLT3-ITD clone was reduced to less than 1%, but the NPM1/IDH1/KRAS clone was still present at 21% of total cells. Additionally, a subclone NPM1/IDH1/PTPN11 (7% at diagnosis), expanded to become the dominant clone (48%) [4**]. This demonstrates the need to target a greater amount of clonal diversity with effective combinatorial therapeutic regimens and the need for additional studies to better understand the relationship between dominant/subclones and how their stoichiometric balance and/or physical or chemical interaction contributes to relapse. At present, the number of patients assessed using single cell genomics at diagnosis, remission, and relapse are limited, thus it is too early to tell which mutations or clones may provide the greatest treatment resistance. However, the implications of these findings further chip away at the widely subscribed hypothesis that specific surface markers can be used to define a leukemia stem cell population, a subset of which is quiescent and therefore resistant to chemotherapy, ultimately giving rise to disease relapse [17]. By contrast, chemotherapy may impose a genetic selection on AML clones leading to relapse rather than selecting for leukemia stem cells. Thus, a newly favored hypothesis is that relapse is dependent on the clonal architecture of AML as defined by specific gene mutation combinations. It will be critical to delineate if clonal architecture has prognostic value in AML and if different features of the AML clonal repertoire can predict for response to induction chemotherapy or to newer AML therapeutic regimens such as venetoclax plus hypomethylating agent combination therapy [18].
In the relapsed refractory AML, there is an emerging role for molecularly targeted therapy, including IDH inhibitors for IDH mutant disease and FLT3 inhibitors for FLT3 mutant AML. Specifically, in the area of FLT3 inhibitors, single cell genomics has helped to inform mechanisms of therapeutic action and resistance [19**, 20*]. Small molecule FLT3 inhibitors gilteritinib, which is approved for treatment of relapsed and/or refractory AML with FLT3 mutations, and crenolanib, which is a new investigational drug in AML, each inhibit both FLT3-internal tandem duplication (ITD) and FLT3-D835 tyrosine kinase domain (TKD) mutations. With these treatment approaches, secondary mutations in FLT3 are relatively uncommon mechanism of resistance, with exception of the gatekeeper FLT3-F691L mutation which enhances the competition between ATP and small molecular inhibitors for the ATP binding pocket and confers resistance to all clinically utilized FLT3 inhibitors [19**, 20*]. Bulk NGS on samples collected from patients after gilteritinib monotherapy identified newly acquired off-target mutations in the RAS pathway, including NRAS, KRAS, PTPN11, BRAF, and CBL in expanded leukemic clones, as well as the persistence of FLT3-mutant clones [19**]. Single cell studies clarified that the newly acquired RAS pathway mutations co-occurred in the FLT3-mutant cells and were not independently expanded subclones [19**]. In the case of crenolanib treatment, bulk NGS analyses suggested upfront resistance of FLT3/TET2-mutant clones, and expansion of FLT3 wild-type NRAS, KRAS, and IDH2-mutant subclones and IDH1/FLT3-mutant subclones in patients with acquired resistance [20*]. Altogether, these studies suggest the newly acquired mutations and subsequent clonal expansion as well as clonal shifts during treatment confer resistance and are viable targets for combinatorial therapy. In proof-of-concept studies, the addition of IDH1 inhibitor AG5198 enhanced the sensitivity of IDH1/FLT3-mutant AML to FLT3 inhibitor crenolanib [20*], and in the case of NRAS/FLT3-mutant AML, the MEK inhibitor tremetinib overcame the resistance to gilteritinib [19**]. The efficacy of these targeted therapeutic combinations to overcome treatment resistance clones underscores the importance of drug treatment selection based on AML clonal architecture.
Single cell RNA sequencing for mutation detection and beyond
scDNA-seq, the main focus of this review, provides single cell level mutation information for a targeted panel of genes, which can include deleterious and pathogenic mutations in both coding and non-coding DNA regions of interest. Single cell RNA sequencing (scRNA-seq) technology can also be used to identify mutations in transcribed coding regions of genes, as well as whole transcriptome information. Caveats to using scRNA-seq to describe the clonal architecture of AML are 3’- or 5’-end biased cDNA libraries that do not have the full-length cDNA sequences, absence of non-transcript encoding DNA information, high levels of allelic dropout due to low transcript abundance, and limited throughput. Recent studies have improved the detection of low-level transcripts, longer read lengths to detect mutations distantly located from transcript ends, as well as increased throughput to sequence thousands of cells such as GoT (genotyping of transcriptomes) and other single cell method adaptations [13, 14, 21]. One promising new approach called TARGET-seq simultaneously captures whole transcriptomes and genomic DNA from single cells overcoming many of the limitations of scRNA-seq mutation profiling approaches [22]. In sum, the future of single cell sequencing to describe the AML ecosystem aims to deliver both DNA and RNA sequence information filling the links between mutations and transcriptional output that ultimately influence and describe the behavior of individual cells within a clone, interactions between clones, and therapeutic responsiveness.
Conclusion
Applications of single cell DNA sequencing technologies are at the forefront of discovery, pushing the limits of our understanding of the genetic complexity and intra- and inter-tumor heterogeneity of AML. These new technologic advances clarify important implications of clonal diversity and the competitive outgrowth of dominant clones as critical biologic mechanisms contributing to disease progression and relapse. Single cell analyses also reveal significant clonal architecture shifts in AML upon the acquisition of mutations during disease evolution and in response to therapeutic pressures suggesting the clonal architecture of AML is highly dynamic lending to treatment escape. Even in cases where targeted therapies against FLT3 and IDH1 where inhibitors suppress mutant clonal expansion, RAS pathway mutations play a major role in relapse both in FLT3 and IDH1 mutant and non-mutant clones [19**, 20*]. Thus, the sensitivity and accuracy of defining minimal residual disease at the clonal level is significantly enhanced with single cell information. One surprising finding from the combined use of single cell DNA and cell surface protein analyses is the immunophenotypic heterogeneity within a genetically defined AML clone. Future studies will help to determine whether this is due to the transformed state of the cells, therapeutic influence, or if the cells are individually selecting for the expression of cell surface proteins required for survival in their immediate environment. Overall, the growing number of single cell technologies and studies applying these methods will lead to a better understanding of AML initiation, progression, and newer strategies for treatment of this heterogeneous disease. Moreover, these studies pave the way to a better mechanistic and translational view of leukemia which has broad implications for clonal evolution in different human cancers.
Key Points.
Most, but not all, AML consist of 2 – 4 similar sized clones that coexist at diagnosis.
Clonal cooperativity and minor subclones contribute to leukemia progression and relapse.
Specific pairs of recurrent coincident mutations provide a clonal fitness advantage.
Genotype influences immunophenotype, but cell surface protein expression is heterogeneous within AML clones.
Single cell DNA sequencing reveals clonal evolutionary trajectories dependent upon cell autonomous (e.g. mutations) and non-autonomous (e.g. therapeutic selection) factors.
Financial support and sponsorship
This work is supported by NCI T32 CA236736 (J.S.R.), NCI R37 CA226433 (S.E.M.), and Sidney Kimmel Cancer Center Support Grant NIH P30 CA056036.
Footnotes
Conflicts of interest
None.
References and recommended reading
Papers of particular interest published within the annual period of review have been highlighted as: *of special interest, **of outstanding interest.
- 1.Cancer Genome Atlas Research N, Ley TJ, Miller C, et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013;368(22):2059–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Papaemmanuil E, Dohner H, Campbell PJ. Genomic Classification in Acute Myeloid Leukemia. N Engl J Med. 2016;375(9):900–1. [DOI] [PubMed] [Google Scholar]
- 3.Ding L, Ley TJ, Larson DE, et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature. 2012;481(7382):506–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. **.Morita KWF, Jahn K, Kuipers J, et al. Clonal Evolution of Acute Myeloid Leukemia Revealed by High-Throughput Single-Cell Genomics. 2020. [DOI] [PMC free article] [PubMed]; This report used scDNA-sequencing to detail clonal evolutionary trajectories, zygosity, and mapping genetic evolution following therapy.
- 5. **.Miles L, Bowman RL, Merlinsky TR, et al. Single cell mutational profiling delineates clonal trajectories in myeloid malignancies. 2020.; Using scDNA-sequencing, this study detailed clonal evolutionary trajectories, mutational co-occurrence/exclusivity in the dominant clone, and genotype/immunophenotype.
- 6. **.Ediriwickrema A, Aleshin A, Reiter JG, et al. Single-cell mutational profiling enhances the clinical evaluation of AML MRD. Blood Adv. 2020;4(5):943–52. [DOI] [PMC free article] [PubMed] [Google Scholar]; Using scDNA-sequencing, this study followed clonal evolution in sequential samples at diagnosis, remission, and relapse.
- 7. *.Potter N, Miraki-Moud F, Ermini L, et al. Single cell analysis of clonal architecture in acute myeloid leukaemia. Leukemia. 2019;33(5):1113–23. [DOI] [PMC free article] [PubMed] [Google Scholar]; Genetic mutations were identified in diagnostic patient samples and then these samples were transplanted into mice where individual cells were sorted and assessed for mutations using multiplex qPCR.
- 8.Dunlap JB, Leonard J, Rosenberg M, et al. The combination of NPM1, DNMT3A, and IDH1/2 mutations leads to inferior overall survival in AML. Am J Hematol. 2019;94(8):913–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. *.Loberg MA, Bell RK, Goodwin LO, et al. Sequentially inducible mouse models reveal that Npm1 mutation causes malignant transformation of Dnmt3a-mutant clonal hematopoiesis. Leukemia. 2019;33(7):1635–49. [DOI] [PMC free article] [PubMed] [Google Scholar]; Using a mouse model of independently inducible Dnmt3a loss and mutant Npm1, this group identified that the timing of mutation acquisition has an effect on disease progression.
- 10.Mason EF, Hasserjian RP, Aggarwal N, et al. Blast phenotype and comutations in acute myeloid leukemia with mutated NPM1 influence disease biology and outcome. Blood Adv. 2019;3(21):3322–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. *.Zhang X, Wang X, Wang XQD, et al. Dnmt3a loss and Idh2 neomorphic mutations mutually potentiate malignant hematopoiesis. Blood. 2020;135(11):845–56. [DOI] [PMC free article] [PubMed] [Google Scholar]; Using a mouse model of Dnmt3a loss and mutant IDH1, this report demonstrates cooperativity in disease progression between two epigenetic modifiers.
- 12.Zhang X, Su J, Jeong M, et al. DNMT3A and TET2 compete and cooperate to repress lineage-specific transcription factors in hematopoietic stem cells. Nat Genet. 2016;48(9):1014–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.van Galen P, Hovestadt V, Wadsworth Ii MH, et al. Single-Cell RNA-Seq Reveals AML Hierarchies Relevant to Disease Progression and Immunity. Cell. 2019;176(6):1265–81 e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Petti AA, Williams SR, Miller CA, et al. A general approach for detecting expressed mutations in AML cells using single cell RNA-sequencing. Nat Commun. 2019;10(1):3660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Marando L, Huntly BJP. Molecular Landscape of Acute Myeloid Leukemia: Prognostic and Therapeutic Implications. Curr Oncol Rep. 2020;22(6):61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dombret H, Itzykson R. How and when to decide between epigenetic therapy and chemotherapy in patients with AML. Hematology Am Soc Hematol Educ Program. 2017;2017(1):45–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Boyd AL, Aslostovar L, Reid J, et al. Identification of Chemotherapy-Induced Leukemic-Regenerating Cells Reveals a Transient Vulnerability of Human AML Recurrence. Cancer Cell. 2018;34(3):483–98 e5. [DOI] [PubMed] [Google Scholar]
- 18.DiNardo CD, Pratz K, Pullarkat V, et al. Venetoclax combined with decitabine or azacitidine in treatment-naive, elderly patients with acute myeloid leukemia. Blood. 2019;133(1):7–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. **.McMahon CM, Ferng T, Canaani J, et al. Clonal Selection with RAS Pathway Activation Mediates Secondary Clinical Resistance to Selective FLT3 Inhibition in Acute Myeloid Leukemia. Cancer Discov. 2019;9(8):1050–63. [DOI] [PubMed] [Google Scholar]; This study followed the changes in genetic mutations and clones following treatment with giltinirib using scDNA-seq, identified which new mutations occurred in the dominant clone, and demonstrated that targeted drug treatment resensitized cells to giltinirib.
- 20. *.Zhang H, Savage S, Schultz AR, et al. Clinical resistance to crenolanib in acute myeloid leukemia due to diverse molecular mechanisms. Nat Commun. 2019;10(1):244. [DOI] [PMC free article] [PubMed] [Google Scholar]; The genetic response to crenolanib treatment was interrogated using NGS, identifed the new mutations likely to confer therapy resistance, and demonstrated targeted drug treatment resensitized cells to crenolanib.
- 21.Nam AS, Kim KT, Chaligne R, et al. Somatic mutations and cell identity linked by Genotyping of Transcriptomes. Nature. 2019;571(7765):355–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rodriguez-Meira A, Buck G, Clark SA, et al. Unravelling Intratumoral Heterogeneity through High-Sensitivity Single-Cell Mutational Analysis and Parallel RNA Sequencing. Mol Cell. 2019;73(6):1292–305 e8. [DOI] [PMC free article] [PubMed] [Google Scholar]