

Abstract
Polyploid giant cancer cells (PGCC) constitute a dangerous subpopulation of cancer cells and are a driving force in cancer recurrence. These unique cells arise from diploid tumor cells in response to stress encountered in the tumor microenvironment or during cancer therapy. PGCC are greatly dedifferentiated, acquire pluripotency, and are able to replicate through a form of asymmetric division called neosis, which results in new populations that are themselves able to differentiate into new cell types or to re-establish tumors. Progeny tend to be more genetically unstable than the founding population due to the dysregulation required to transition through a PGCC state. Therefore, cancers that escape stressors through this mechanism tend to re-emerge with a more aggressive phenotype that is therapy resistant. This review focuses on the clinical significance of PGCC, the need for standardized nomenclature and molecular markers, as well as possible avenues to develop therapies aimed at PGCC and the process of neosis. The biology underlying the development of PGCC including cell cycle checkpoint dysregulation, stress responses, dedifferentiation, stemness and epithelial-mesenchymal transition is discussed.
Keywords: cancer, polyploid giant cancer cells, depolyploidization, dedifferentiation, neosis, cancer therapy, tumor microenvironment, cancer recurrence
Introduction
Traditional cancer therapies, such as radiation or chemotherapy, aim to disrupt the dysregulated and overactive mitotic processes of malignant cells with the intention to preferentially lead to their demise. While therapy response varies, most patients respond at least initially, and in some cases bulk tumor is eliminated to levels below clinical detection. However, even the declaration of a “cure” is frequently temporary and an eventual relapse suggests that residual malignant cells must have escaped therapy and formed a seed population that enabled tumor recurrence. In this review we discuss the ability of polyploid cancer cells to fuel cancer recurrence. The sprawling, strangely multinucleated appearance of this small yet dangerous subpopulation has spawned a colorful and wide-ranging nomenclature including “giant cells” and “monster cells”, which inspired the title of this review. First, we will discuss the clinical significance of polyploid cancer cells. Their morphology is consistently described as the flattened, large shape typical of senescent cells with abnormal nuclei. However, polyploid cancer cells are neither irreversibly senescent nor do their abnormal nuclei indicate that mitotic catastrophe is imminent. The danger associated with polyploid cancer cells is their ability to reproduce asymmetrically in an amitotic fashion involving yeast-like budding or bursting. Offspring exhibit increased drug resistance, stemness, metastasis or a combination thereof. This danger has historically been overlooked but is now increasingly being recognized. Second, we will address the terminology used to describe these unusual cancer cells and their progeny. The lack of standardized nomenclature has resulted in a body of literature that is currently fragmented and difficult to search. This presents a major challenge for communication among researchers and clinicians who encounter and wish to study these cells. Throughout this review we will use the abbreviation PGCC for polyploid giant cancer cells and the term “neosis” for generation of their progeny. The third major challenge facing the field is the lack of specific markers to identify PGCC. Lack of molecular markers can, especially in clinical samples, lead to misinterpretation of cell lineage. In addition, PGCC are by definition a transient phenomenon, since they arise from a subset of mitotic cancer cells in response to stress, with offspring resuming mitosis when conditions are more favorable. Through temporary senescence, PGCC evade attacks on the mitotic machinery to ensure survival of the tumor as a whole until conditions are favorable for neosis, which results in respawning of a more aggressive tumor. This brings us to the final challenge of developing novel therapeutic approaches to target PGCC and their progeny. The success of a sustained, multi-disciplinary focus on treatment approaches to facilitate a solution to this final challenge will require the adoption of a common nomenclature, identification of reliable markers, and innovative clinical trials.
Clinical significance of PGCC and neosis
About 20 years ago the laboratory of Jekaterina Erenpreisa first reported that polyploidy provides a survival mechanism following DNA damage of cells with mutant p53 (Illidge, Cragg, Fringes, Olive, & Erenpreisa, 2000). A few years later Rajaraman introduced the concept of neosis, yet it took nearly another 10 years before PGCCs were clearly declared as critical drug targets in the war on cancer (Coward & Harding, 2014; Rajaraman, Rajaraman, Rajaraman, & Guernsey, 2005). A 2015 mini-review warned “that docetaxel induces these giant cells….should give pause to clinicians” (Ogden, Rida, Knudsen, Kucuk, & Aneja, 2015). PGCCs have subsequently been described as a “keystone species” without which the remainder of the tumor environment would collapse and as the “evil roots of cancer” because their offspring have overlapping characteristics of drug resistance, stemness, and metastatic potential (Amend et al., 2019; Chen et al., 2019). Here we summarize the clinical significance of PGCC in some common cancers, although evidence for PGCC exists in a wide range of malignancies (Table 1).
Table 1. A sampling of literature with references to functional PGCC using varied terminology.
The presence of polynuclear cells with pluripotent and malignant potential have been reported for many years, yet a cohesive field of shared terminology is only beginning to appear. These references represent some of the papers that form a large but sporadic body of work that underlies the emerging PGCC field.
term of growing consensus
Colorectal cancer
Colorectal cancer (CRC) is the third most common cancer in both women and men. In laboratory studies PGCC can be generated in colon cancer cell cultures following a variety of stressors including hypoxia, radiation or chemotherapy (Lopez-Sanchez et al., 2014; Mirzayans, Andrais, Scott, et al., 2017; Puig et al., 2008). Cisplatin- and radiation-induced colon cancer PGCC both show dysregulation of p53 and p21 (Mirzayans, Andrais, Scott, et al., 2017; Puig et al., 2008). Analysis of 159 clinical cases of CRC showed that the presence of stromal PGCC with evidence of budding correlated inversely with the degree of differentiation (D. Zhang et al., 2017). PGCC undergoing neosis were observed in 27.5% of well differentiated, 50% of moderately differentiated and 90.2% of poorly differentiated CRC (D. Zhang et al., 2017). A recent study investigated the role of PGCC formation in locally advanced rectal cancer which is treated with neoadjuvant chemoradiation therapy (nCRT) prior to surgery (Fei, Zhang, et al., 2019). Comparison of samples from patients receiving nCRT (n=304) and those who did not (n=301) showed that the presence of PGCC was significantly higher in the nCRT group compared to the non-nCRT group. In the nCRT group, PGCC appeared after therapy and exhibited morphologies that were consistent with tumor invasion and metastasis. Although additional long-term follow up studies are needed, the study found significant differences in survival depending on the rest interval between nCRT and surgery. Since nCRT induces PGCC and generation of aggressive progeny, a rest interval of less than 50 days was suggested as it may improve survival.
Ovarian cancer
Similar to colorectal cancer, treatment of patients with ovarian cancer increases PGCC. Analysis of paired specimens isolated before and after chemotherapy showed relative homogeneous cells prior to chemotherapy whereas specimens isolated post-chemotherapy contained PGCC with varying and bizarre nuclei as well as evidence of neosis (Niu, Mercado-Uribe, & Liu, 2017). An increase in the number of PGCC was observed with tumor grade and stage (S. Zhang, I. Mercado-Uribe, Z. Xing, et al., 2014). To study this in more detail, a total of 80 serous ovarian tissue samples (21 primary high-grade carcinomas with patient-matched metastatic tumors, 26 cases of low grade tumors, and 12 cases of borderline cystadenoma) were analyzed. Results showed that high-grade carcinomas contained a significantly higher number of single PGCC with evidence of neosis in the stroma compared to samples from patients with low-grade tumors (18/21 vs. 6/26) (Lv et al., 2014). In addition, the number of PGCC positively correlated with vascular mimicry in these samples (L. Zhang et al., 2014). Dissection of cell cycle regulation in ovarian cancer-derived PGCC has implicated cyclin D1 as a key driver with the additional upregulation of Cyclins B1 and E reported in a xenograft model (Niu et al., 2016; S. Zhang, I. Mercado-Uribe, Z. Xing, et al., 2014). In addition, whether cyclin B1 remains in the cytoplasm or accumulates in the nucleus, which is determined by its phosphorylation status and rates of nuclear import and export, is critical in committing the cell to mitosis (Walsh, Margolis, & Kornbluth, 2003). Fei, et al. demonstrated that Cyclin B1 is excluded from PGCC nuclei but is present in the nuclei of mitotic daughter cells (Fei, Qu, et al., 2019). Ovarian cancer has also been studied in a model of hypoxia and, as in colon cancer, hypoxic conditions induced the formation of PGCC and were accompanied by the overexpression of HIF1α (S. Zhang, Mercado-Uribe, Hanash, & Liu, 2013). Such models suggest that the hypoxic tumor microenvironment may be sufficient to induce PGCC.
Breast cancer
In breast cancer, treatment with doxorubicin or taxol induces PGCC formation independently of estrogen receptor status (Parekh et al., 2018). Breast PGCC have been shown to transcribe multiple stemness genes, such as ALDH1A and CD24, and show increased migratory potential (Pirsko V, 2019; Xuan, Ghosh, Cheney, Clifton, & Dawson, 2018). Interestingly, breast cancer PGCCs stabilize the hypoxia response gene HIF1α even when the stress applied is not hypoxia as chemotherapeutic stress has also been observed to upregulate the gene (Parekh et al., 2018). Analysis of 167 breast cancer tissues including 52 patients with paired primary tumor and lymph node metastasis, 52 patients without metastases and 11 patients with benign tumors showed that the number of PGCC increased with malignant grade and was highest in the lymph node metastases (Fei et al., 2015).
Prostate cancer
Prostate cancer is usually a slow growing malignancy such that mortality even in patients with the highest possible Gleason score is only 5% at 2 years (Alharbi, De Marzo, Hicks, Lotan, & Epstein, 2018). However, if PGCC are detected in the biopsy, mortality increases to 37% (7/19) at 8 months (Alharbi et al., 2018). In the laboratory, the castration resistant prostate cancer cell lines, PC3 and its derivative PPC1, have been used extensively to trace the kinetics of PGCC arising in response to clinically relevant dosages of docetaxel or radiation (Lin et al., 2019; Mittal et al., 2017; White-Gilbertson, Lu, Norris, & Voelkel-Johnson, 2019). Increases in the PGCC population are highly dose and time dependent but occur in response to clinically relevant concentrations of docetaxel, a drug that is commonly used to treat castration resistant prostate cancer despite development of resistance within 7–8 months (Lin et al., 2019; Tannock et al., 2004; White-Gilbertson et al., 2019). Interestingly, PGCC are now suspected as the culprit driving docetaxel resistance in prostate cancer patients (Mittal et al., 2017).
The lysosomal enzyme acid ceramidase, which is frequently overexpressed in prostate cancer, increases drug resistance and oncogenic signaling (Camacho et al., 2013; Norris et al., 2006; Saad et al., 2007). We recently demonstrated that PGCC resulting from radiation or docetaxel therapy induced stress express high levels of ASAH1 and that this enzyme was required for neosis (White-Gilbertson et al., 2019). Increased expression of ASAH1 has also been observed in patient specimens following radiation treatment failure, but unfortunately the presence of PGCCs was not specifically examined or correlated to ASAH1 expression (Cheng et al., 2013). However, if PGCCs are detected in a biopsy after a prostate cancer recurrence the mortality rate increases to 57% at 7 months (Alharbi et al., 2018). In summary, the presence of PGCC is indicative of a poor prognosis across multiple cancers and associated with a more aggressive tumor phenotype.
The terminology problem
Polyploid cells within tumors have been described since the invention of the microscope but the significance of this rare subpopulation has been underestimated (Amend et al., 2019). Interpreting a morphology that mimics other cell types and represents a rather bizarre biology, has led to descriptions that are sporadic and idiosyncratic. A collection of terms found in the last several decades of literature, all of which may refer to a single phenomenon, has been summarized in Table 1. Here we briefly address how the significant contribution of PGCCs to therapy resistance and recurrence can unintentionally be missed in many standard assays and list the use of different terminologies by research scientists and pathologists in describing PGCC to emphasize how the disconnect in terminologies can hamper translational efforts.
The Research Scientist View
Laboratory studies involving in vitro assays of cancer cells are usually focused on inducing cell death with typical observation time frames of 72 hours or less. When a majority of cells become non-viable within this time frame after exposure to a clinically relevant therapeutic approach, cultures are declared sensitive. If observed microscopically at the conclusion of the experiment the residual cells are often polyploid but can leave the impression that they are senescent or that mitotic catastrophe is imminent. Indeed, there is no discernible difference between PGCC that are doomed and those that have the potential to undergo neosis. Continued analysis of cultures for a significantly longer amount of observation time is required before the reproductive capability of PGCC becomes evident. One study found that PGCC remain senescent for as long as 2 months (Puig et al., 2008).
PGCCs can also be studied separately through enrichment by size selection using flow cytometry or filtration, and isolated PGCC can spawn a halo of progeny via neosis sufficient to regrow a fully mitotic culture (Kaur et al., 2015; Puig et al., 2008; White-Gilbertson et al., 2019). Throughout the years, researchers have used a variety of terms to try and capture the biology and dangers associated with PGCC. The group who originally described neosis used the term multinuclear/polyploid giants (MN/PGs), while multinucleate cell (MNC), multinucleated and giant cells (MNGCs) or mononucleated giant cells (MOGC), endopolyploidy tumor cells (ETCs), giant cells, and depolyploidizing cells are terms used by different groups at different times to describe similar cell behavior (Table 1). Since single PGCC are capable of initiating tumors, they may also be described as tumor initiating cells. There is currently no commonly encountered term to describe the PGCC progeny, although one group appropriately called the PGCC progeny “escape cells” and another referred to them as “Raju cells” (Puig et al., 2008; Rajaraman et al., 2005). Since PGCC progeny resume mitosis, they cannot be easily distinguished from the parental mitotic cells, increasing the challenge of identifying, studying, and naming them.
The Pathologist View
Pathologists have used a variety of terms such as monstrocellular, osteoclast-like, anaplastic, pleomorphic, syncytial-type, and giant to describe the general phenotype of polyploid cells encountered within a clinical specimen. Representative references for how these terms are used are included in Table 1. It is in the clinical setting that definitive markers of PGCC would be the most immediately impactful, because as discussed above, the presence of PGCC implies a more concerning prognosis. However, studying these cells presents as many challenges to pathologists as to research scientists, again due to the unexpected, dynamic, and plastic nature of PGCC.
One initial challenge encountered by pathologists is to determine the origin of large polyploid cells within a sample. Since polyploidy is uncommon in mammalian cells, the appearance of multinucleated cells within clinical specimens could be interpreted as having derived from the few lineages that typically produce polyploid cells, rather than being an adaptation of the cancer cells themselves (Williamson et al., 2014). For example, multinuclear cells of the trophoblast lineage share many features of cancer cells, including high expression of PDL1 to inhibit host immunity, high levels of angiogenesis, and the ability to invade host tissue (Costanzo, Bardelli, Siena, & Abrignani, 2018). During development, trophoblasts use these characteristics to implant placental tissue into the uterine wall, attract a blood supply, and protect a growing fetus from maternal immune surveillance, all of which could conceivably drive cancer.
Polyploid cells could also be derived from stem cells that differentiate into cells of the monocyte/macrophage or dendritic cell lineages, such as histiocytes and osteoclasts, which are formed through fusion. The abnormal tumor environment may attract immune cells, which could be a favorable development and portend improved outcomes for the patient. Whether polyploid cells seen in cancer are functional immune cells or are cancer PGCC could greatly affect a disease course. However, even identification of polyploid cells as a differentiated cell type can be misleading. Osteoclasts of the macrophage lineage may appear in tumors not because of recruitment, but because the PGCC differentiate into osteoclast-like cells (Wasserman, Sekhon, & Ayroud, 2015). Since osteoclasts specialize in lysing surrounding tissue, the tumor may co-opt this ability to facilitate migration. Hence, determining that polyploid cells are osteoclasts, or “osteoclast-like” may still leave significant prognostic confusion, depending on the derivation of osteoclast-like cells.
The difficulty to identify clear markers for polyploid cells in situ is exemplified by CD68 (LAMP4). CD68 is often used to identify cells of the monocyte lineage such as histiocytes and osteoclasts, but expression has also been detected in polyploid cells of clearly neoplastic origin (Jo, 2014; Sekulic, Gilles, Amin, & Stewart, 2016). This may be because PGCC can give rise to osteoclast-like cells, or because the massive size of PGCC requires metabolic changes that upregulate lysosomal markers such as CD68/LAMP4 (Williamson et al., 2014). The use of p53 expression as a marker can also be misleading. When diploid tumor cells stain positive for p53, lack of p53 staining in polyploid cells is sometimes cited as evidence that the polyploid cell is derived from a tissue other than the malignant cells (Baydar, Amin, & Epstein, 2006; Jo, 2014). As discussed in an upcoming section, loss of p53 expression maybe required for polyploid to occur. Thus interpreting p53 status of a polyploid cell is quite ambiguous as lack of p53 expression may identify a cell that originated outside an otherwise p53 positive tumor or a PGCC that derived from a diploid tumor cell.
Ideally, as the field of PGCC studies begins to cohere, nomenclature will become more specific and insights will be synthesized into a panel of useful markers. Toward that end, the following sections of this review will summarize the current understanding of PGCC development and fate (summarized in Figure 1).
Figure 1. The generation and fate of PGCCs.
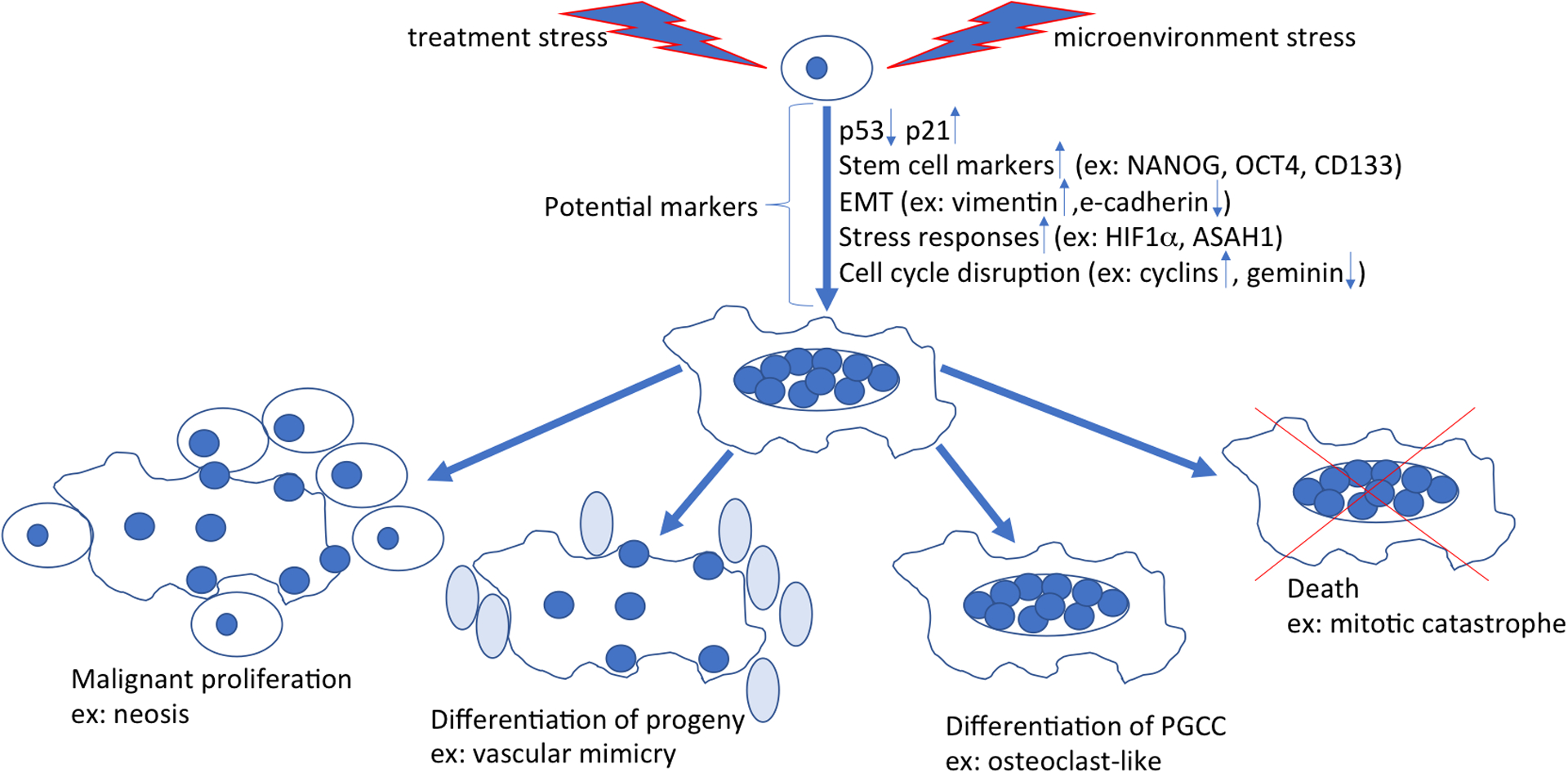
Cancer cells (top, center) stressed by factor such as hypoxia, chemotherapy, or radiation can undergo a complex series of adaptations resulting in a large, multinucleated morphology. The process of adaptation may offer clues about possible markers for PGCC. The factors that determine the ultimate fate of PGCC are currently unknown. PGCC may produce a new generation of malignant mitotic cells through neosis. Progeny can differentiate into different types of cells, including erythroid-like cells that facilitate the binding of oxygen and contribute to vascular mimicry or osteoclast-like cells that facilitate migration of tumor cells by lysis of the dense stroma. Some PGCC may also become permanently senescent or undergo death through mitotic catastrophe. The eventual fate of PGCC in a given cancer patient may closely relate to the clinical outcome.
Characteristics of PGCC that may lead to identification of molecular markers
Morphology
Until molecular markers of PGCC are established, morphology may offer some opportunities for differentiation. Polyploid cells can arise through various mechanisms including fusion or endoreplication. Evidence for fusion has been reported in glioblastoma cells, in which a lethal dose of radiation resulted in selection for an innately resistant subpopulation (7–10% of cells) that transiently arrested in G2/M and survived therapy. These cells appeared to have formed by fusion, since exposure of mixed cultures containing cells labeled with green or red fluorescent reporters to radiation, demonstrated that the majority of surviving cells emitted a yellow signal (Kaur et al., 2015). However, in breast and ovarian cancer cells, only 10–20% of PGCC in cultures formed by fusion and in myxofibrosarcoma cells only one among many PGCCs formed through this mechanism (Ariizumi et al., 2009; S. Zhang, I. Mercado-Uribe, Z. Xing, et al., 2014). Thus while there is evidence for fusion, endoreplication seems to be more prevalent (Shu, Row, & Deng, 2018; Weihua, Lin, Ramoth, Fan, & Fidler, 2011). During endoreplication, mitosis may be skipped altogether resulting in a mononuclear cell that has multiple copies of the genome. Alternatively, if mitosis is initiated and karyokinesis is completed but cytokinesis fails, a multinuclear cell develops. In our experience, nuclei of PGCC arising due to cytokinesis failure are housed within the same nuclear membrane and may even comprise one massive nucleus rather than multiple discrete nuclei. Thus multinuclear cells arising by endoreplication could potentially be differentiated from fused cells based on analysis of the nuclear membrane. However, the nuclei fated to become daughter cells via neosis move to the periphery of the cell during budding and could closely resemble the appearance of histiocytes or osteoclasts formed by fusion. Diaz-Carbello et al. who used etoposide to induce PGCC from neuro- and glioblastoma cells performed a detailed morphological study (Diaz-Carballo et al., 2014). This study led to the discovery of an astonishing variety of atypical PGCC morphologies, including “spiral cells” in which nuclei were arranged in a spiral fashion in different planes, “pregnant” cells that contained progeny of different sizes embedded in the cytoplasm of the mother cell, “monastery” cells that exhibited a wall structure inside which cell replication took place with mitochondria arranged around daughter cells, and the rarer “shepherd cells” that were always associated with clusters of smaller cells.
Biology
The aberrant biology of PGCC includes dysregulation of the cell cycle, induction of stress responses, and dedifferentiation, all of which are likely accompanied by adaptations in biophysical properties and metabolic activity. With this array of abnormalities, it should be possible to identify a set of molecular markers that is unique to PGCCs, although the transient and plastic nature of the cells complicates the situation considerably.
Cell cycle checkpoint dysregulation
DNA replication is carefully controlled at cell-cycle checkpoints to ensure integrity during cell division. Therefore, one or more cell-cycle checkpoint controls must have been compromised for PGCC generation to occur by endoreplication. Before a cell can replicate its DNA, integrity is checked at the G1/S transition. In the G1 phase the cell prepares for DNA replication and certain intracellular components such as centrosomes undergo replication. CDT1 (Chromatin Licensing And DNA Replication Factor 1) cooperates with CDC6 and the origin recognition complex (ORC) to load the mini-chromosome maintenance (MCM) complex onto DNA to generate pre-replication complexes (Cook, Chasse, & Nevins, 2004). Licensing of DNA replication in S-phase is negatively regulated by geminin, which binds CDT1 and prevents loading of the MCM complex to avoid inappropriate initiation of replication. Lack of geminin during embryonic development results in premature endoreplication, commitment to the trophoblast lineage, and formation of trophoblast giant cells without formation of inner cell mass (Gonzalez et al., 2006). Premature endoreplication may occur because accumulation of CDT1 is permissive for multiple rounds of DNA replication during the G1 phase of the cell cycle. Geminin also binds to CDT1 during mitosis, when CDT1 is stabilized and required to promote kinetochore-microtubule attachments (independent of chromatin binding) (Varma et al., 2012). While lack of geminin contributes to polyploidy, it is important to point out that regulation of this protein is complex, that it has multiple binding partners other than CDT1, and that overexpression of geminin itself appears to be oncogenic as it serves as an independent indicator of poor outcome (Kushwaha, Rapalli, & Kumar, 2016).
Animal studies have shown that polyploid cells from p53-deficient mice are tumorigenic while the corresponding diploid cell is not (Fujiwara et al., 2005). p53 is well known as the “guardian of the genome” for its role in regulation of cell cycle progression. The N-terminal transcriptional activation domain of p53 is targeted by kinases of the MAPK family (JNK1/2/3, ERK1/2 and p38 MAPK) that are activated in response to various stresses including oxidative stress, heat stress, or membrane damage, and by kinases activated in response to DNA damage following genotoxic stress (ATM/ATR, CHK1/2, DNA-PK, CAK, TP53RK). In response to DNA damage, p53 becomes phosphorylated, more stable, and regulates gene expression to interfere with expression of cyclin B and activity of cyclin B complexes, which are necessary for the progression of cells into and out of M phase. An increase in p53 arrests cell cycle progression to allow DNA damage to be repaired or if damage is too severe apoptosis can be initiated. Recent evidence suggests that the primary response triggered by p53 in response to clinically relevant concentrations of therapeutic agents is prolonged growth arrest rather than apoptosis (Mirzayans, Andrais, Kumar, & Murray, 2017). Of additional concern is the observation that p53-deficient cells, which develop into PGCC following radiation exposure, remain adherent and viable for an extended period of time with a majority maintaining DNA synthesis, presumably in the absence of p53-directed repair mechanisms (Mirzayans, Andrais, Scott, et al., 2017). Recently, a gain-of-function genome-wide screen identified 23 miRNAs whose overexpression was permissive for proliferation of tetraploid cells. Two top hits, including miR-523–3p and miR191–3p, enhanced S-phase entry with efficiency comparable to loss of p53 (Vittoria et al., 2018). The E6 protein of human papilloma virus type 16, a type of HPV type responsible for a majority of cervical cancers, also binds and inactivates p53. Together with the HPV E7 protein that binds to Rb, E6 causes polyploidy in normal keratinocytes due to spindle checkpoint failure (Patel, Incassati, Wang, & McCance, 2004).
The miRNA screen also identified the oncogenic miR-191–3p as a significant stimulator of the MAPK pathway, suggesting that strong mitogenic signals may promote cell cycle progression in tetraploid cells. This was corroborated by Liu et al., who used cobalt chloride as a model for hypoxia-induced PGCCs to demonstrate that p38MAPK and ERK phosphorylate CDC25C on Ser216 (K. Liu et al., 2019). CDC25C is a highly conserved gene that controls mitosis. When CDC25C is phosphorylated, it dephosphorylates tyrosine residues on cyclin B-bound CDK1 driving cells in G2 towards the prophase of mitosis.
A key target gene of p53 is the cyclin-dependent kinase inhibitor p21. By binding to cyclin-CDK2, cyclin-CDK1 and cyclin-CDK4/6 complexes, p21 regulates cell cycle progression at the G1/S transition. In MCF-7 or T47D breast cancer cells, overexpression of p21 increased cell size 50–100-fold over several days with karyokinesis progressing in the absence of cytokinesis and proliferation (Sheikh, Rochefort, & Garcia, 1995). How the increase in p21 impacts on cell fate is p53-dependent (Georgakilas, Martin, & Bonner, 2017). In the presence of wild-type p53, the increase in p21 is transient, resulting in cell cycle arrest with subsequent senescence or apoptosis. In contrast, chronic overexpression of p21 in a p53-deficient environment, allows cells to escape control mechanisms resulting in re-replication, error prone repair, and high levels of genomic instability that are associated with an aggressive cancer phenotype.
Stress responses
The cancer therapies aimed at killing rapidly proliferating cells also induce stress responses that promote the formation of PGCC. To more closely mimic the spatially varying drug concentrations that are likely encountered in vivo, Lin and co-workers used a microfabricated “evolution accelerator” environment with increasing concentrations of docetaxel (0–10nM). Cell density was not significantly decreased at lower drug concentrations but in the 2–4nM range of docetaxel, the density of PGCC increased nearly 10-fold with cell size changing by a order of magnitude from 10–15 μm to 100–200 μm (Lin et al., 2019).
The sheer increase in size and nuclear load can be predicted to require adaptations. A study on the biophysical properties of PGCC showed an increased appearance of stress fibers and abnormal microtubule organization, which was associated with dysregulation of the RhoA-Rock1 pathway (Xuan et al., 2018). The size of PGCC also results in metabolic reprogramming such that additional oxidative phosphorylation is required at baseline (Sirois et al., 2019). This finding dovetails with a prior report that PGCC are preferentially dependent on mTOR signaling, particularly the activity of the mTORC2 complex which influences autophagy (Zeng et al., 2014). Taken together, results across cancer cell types suggest that the morphology of PGCC necessitates custom physiology and constitutes its own form of stress.
Another major player in the stress response is the sphingolipid ceramide. Ceramide increases in response to various stresses including chemotherapy and radiation used in cancer therapy. The accumulation of ceramide can lead to cell death, such as apoptosis, necroptosis and/or induction of autophagy and ER stress response pathways (Ogretmen, 2018). Ceramide can also directly activate the promoter of the acid ceramidase (ASAH1) gene, whose protein product hydrolyzes ceramide to sphingosine. Sphingosine can be recycled into ceramide but can also serve as substrate for sphingosine kinases that through phosphorylation generate sphingosine-1-phoshate that functionally opposes ceramide. We have shown that ASAH1 is transcriptionally induced in PGCC and elevated at the protein level (White-Gilbertson et al., 2019). However, the function of the bioactive lipids ceramide and sphingosine-1-phosphate in PGCC remains to be studied in detail.
Stressors encountered in the tumor microenvironment can promote PGCC formation even without the added stress of therapy. The tumor microenvironment contains many types of cells including fibroblasts and immune cells that closely interact with cancer cells via extracellular signals. Compared to normal tissue the tumor microenvironment is quite abnormal with low and uneven blood flow, elevated interstitial fluid pressure, and an often dense and stiff extracellular matrix. The abnormal vasculature within tumors leads to hypoxia, which promotes PGCC formation. Treatment of cells with cobalt chloride is used to model the effects of hypoxia in vitro. This model has been used extensively to demonstrate the formation of PGCC (Lopez-Sanchez et al., 2014; S. Zhang, Mercado-Uribe, Hanash, et al., 2013; S. Zhang, Mercado-Uribe, & Liu, 2013; S. Zhang, I. Mercado-Uribe, Z. Xing, et al., 2014). To mimic the pressure that cells encounter in the tumor interstitial fluid, Arun and co-workers recently used simulated microgravity and demonstrated this also increased formation of PGCC (Arun, Sivanesan, Patra, Varadaraj, & Verma, 2019). We found that subcutaneous implantation of PPC1 prostate cancer cells resulted in tumors that had a significantly higher PGCC content than the parental line when propagated in vitro (White-Gilbertson, unpublished observation). Thus the stress encountered in the tumor microenvironment appears sufficient to promote the PGCC subpopulation. This idea is supported by a study in which repeated cycles of in vivo selection for metastatic potential coincided with enrichment for PGCC in castration resistant PC3 prostate cancer cells (L. Zhang, Wu, & Hoffman, 2015).
In summary, PGCC can develop through the influence of stress inherent in the tumor microenvironment, which may explain why these cells are sometimes detected in patients who have not yet undergone any form of cancer therapy. Cancer therapies can exacerbate the stress and further promote PGCC formation. Additionally, the PGCC state itself is a source of stress, and modifications to cellular structure and metabolism are required to accommodate the extreme size of the cells. Consequently, cancer cells adapt to stressors through increased genomic instability, dedifferentiation, stemness, and epithelial-mesenchymal transition, which are associated with treatment resistance and metastasis.
Dedifferentiation and stemness
As mentioned earlier, polyploidy is not commonly encountered in mammalian cells as they typically have a diploid genome and proliferate via mitosis. Polyploidy in humans is limited in differentiated cells of muscle, liver, or cells of the macrophage or trophoblast lineages. However, at less differentiated stages, such as the blastomeres present during early embryonic development, polyploidy is more routine (Chen et al., 2019). The pioneering cancer researcher Robert Weinberg once stated that “deadly secondary tumors happen when cancer cells don’t act their age”, referring to their ability to resurrect programs that occur during embryogenesis (Bourzac, 2007). Although Weinberg was talking about epithelial-mesenchymal transition, the statement also applies to polyploid cancer cells. In 2018, elegant work by Niu, et al. demonstrated that PGCC recapitulate a blastomere-like phenotype (Niu et al., 2017). The formation of PGCCs requires a remarkable de-differentiation and reversion to an embryonic stage, which is discussed in detail in several publications by Jinsong Liu, whose recently proposed “life code” theory highlights the parallels between the origins of human life and tumor development (Chen et al., 2019; J. Liu, 2018, 2019). Briefly, the life code theory posits that cellular stress reactivates embryonic developmental stages and that PGCC, which are observed in nearly 40% of solid tumors, are the somatic equivalent of blastomeres.
To demonstrate stemness properties of PGCC, Niu et al. examined the phenotype of cells generated following paclitaxel treatment (Niu et al., 2017). In addition to PGCC, recovering cultures contained mixtures of budded daughter cells that resembled epithelial cells, fibroblasts, mixtures of epithelial and mesenchymal cells, neurons, and very small spore/yeast-like cells. This plasticity has now been demonstrated in PGCC derived from various tumor cell origins. For example, PGCC derived from ovarian cancer cells can be differentiated into adipose tissue, cartilage, or bone (S. Zhang, I. Mercado-Uribe, Z. Xing, et al., 2014). Breast cancer PGCCs derived following paclitaxel stress were capable of differentiation into cells found in the tumor microenvironment, including myoepithelial, endothelial, and erythroid cells (S. Zhang, Mercado-Uribe, & Liu, 2014). Colon cancer PGCC can also generate erythroid cells (D. Zhang et al., 2017). The formation of erythroid cells from PGCC is of particular interest, since erythroid cells in adults develop from hematopoietic stem cells in the bone marrow. However, during early embryonic development when mature erythrocytes are absent, oxygen can be obtained through the formation of blood islands that contain embryonic hemoglobin and this program may be re-activated in PGCC. PGCC have been shown to generate erythrocytes that express fetal and embryonic hemoglobins, which bind oxygen with high affinity, and likely provide PGCC with significant survival advantages in the tumor microenvironment (S. Zhang, Mercado-Uribe, & Liu, 2013). It has been suggested that this mechanism of vascular mimicry could explain the relative ineffectiveness of anti-angiogenic therapies in cancer patients (Lupo et al., 2016; Yang et al., 2018).
PGCC can also express various embryonic markers in a temporal manner. When benign mullerian epithelial cells are immortalized with H-Ras, the resulting PGCC subpopulation could be grown as spheroids that expressed the self-renewal markers OCT4, Nanog, and SOX-2 (S. Zhang, Mercado-Uribe, Sood, Bast, & Liu, 2016). In PGCC derived from ovarian cancer cells following paclitaxel treatment expression of these markers as well as SSEA1 was also detected (Niu et al., 2017). Marker expression occurred in a temporal fashion with SSEA1 expression limited to subnuclei and cytoplasm beginning on day 1 and decreasing by day 14. OCT4 expression appeared on day 3 and remained expressed until day 14 in a subset of nuclei, whereas Nanog and SOX2 expression were detected as early as day 1 but also remained activated until day 14 in a subset of PGCC. Having observed earlier that PGCC appear post-chemotherapy, the group next determined if these markers are detected in clinical specimens. None of the pre-therapy specimens were positive for OCT4 or Nanog and only 2/38 stained positive for SOX2, whereas post-chemotherapy samples had increased staining of all markers (3/38 OCT4; 12/38 Nanog; 17/38 SOX2)(Niu et al., 2017). In addition, nuclear expression of YAP, a critical Hippo pathway protein involved in the regulation of stem cells and cancer development, was detected in 3/38 cases post-therapy (Niu et al., 2017). Nuclear localization of YAP was also observed in PGCC forming in response to simulated microgravity (Arun et al., 2019). The expression of embryonic stem cell markers in a subset of post-chemotherapy tissues supports the theory that PGCC have undergone dedifferentiation to a primitive stage. Rohnalter and co-workers studied the emergence of PGCC in ovarian cancer cells and also found that PGCC exhibited features of stemness as well as a pro-inflammatory secretory phenotype (Rohnalter et al., 2015).
Stem cells are defined by their potential for self-renewal and pluripotency. According to the cancer stem cell hypothesis, tumors contain a rare and distinct subpopulation of cancer stem cells (CSC) with self-renewal potential and the ability to regenerate the tumor. The broadly expressed CSC markers CD133 and CD44 have been detected in tumors established from PGCC (S. Zhang, I. Mercado-Uribe, Z. Xing, et al., 2014). PGCC that developed in response to simulation of interstitial fluid pressure included cells that were dual positive for CD133/CD44 (Arun et al., 2019). In nasopharyngeal cancer cells, the majority of CD133+ cells are small but co-existed with a subpopulation of large polynuclear cells (Jiang et al., 2015). Scanning electron microscopy showed that the large CD133+ cells had “indefinite, regular small bodies on the surface of or surrounding the giant cancer cells some of which appeared to be creeping out of the parental cells”. This phenomenon was not seen in the CD133− population and, after ruling out the possibility that protrusions are apoptotic bodies and performing acridine orange staining, the authors concluded that what they observed was neosis from CD133+ PGCC (Jiang et al., 2015) . Even such standard markers are not definitive, unfortunately. Our own examination of PGCC derived from the prostate cancer line PPC1, which already constitutively express CD44, showed that expression of CD133 was not induced, underlining the difficulty of developing a generalized panel of markers for PGCCs (White-Gilbertson et al., 2019)
By definition transplantation of a single CSC should be able to form a new tumor. A study investigating the fate of single PGCCs in a syngeneic fibrosarcoma mouse model showed that the efficacy by which a single PGCC was able to establish a tumor was about 10 times higher than that of mononuclear cells (13/60 injections vs. 1/40 injections) (Weihua et al., 2011). Similar results were obtained by Zhang and co-workers who demonstrated that a single PGCC forming spheroids in vitro was capable of generating tumors in immunodeficient mice (S. Zhang, I. Mercado-Uribe, Z. Xing, et al., 2014). Investigation of cancer relapse in a syngeneic rat model of colon cancer using cisplatin treatment showed that following initial tumor regression, residual tissue contained large multinuclear cells with a senescent phenotype (Puig et al., 2008). Similar to human patients, the rats relapsed and were unresponsive to subsequent therapy. Recapitulation of this scenario in vitro revealed that PGCC remain viable and senescent for as long as two months and that a small fraction of these (estimated at 10−4 to 10−5) produced “escape cells”. Isolation of PGCC by flow cytometry clearly demonstrated that “escape cells” were derived from PGCC (Puig et al., 2008). These studies indicate that a single PGCC has the capacity to form a tumor but that this capacity is not present in every PGCC. Thus there is overlap but not congruency between CSC and PGCC, indicating that the relationship between PGCC and CSC remains to be defined.
Epithelial Mesenchymal Transition and metastasis
Epithelial-mesenchymal transition (EMT) is a process by which stationary epithelial cells lose their cell polarity and adhesion and gain the motile behavior of mesenchymal cells, which in cancer contributes to metastasis. EMT is characterized by loss of epithelial markers such as E-cadherin and the gain of mesenchymal markers like vimentin, N-cadherin, or expression of transcription factors such as SNAI1 and SNAI2. Other EMT players include β-catenin, ZEB1 and the transcription factor TWIST. Multiple studies indicate a role for EMT in PGCC and neosis. For example, PGCC derived tumors exhibit a mesenchymal phenotype (S. Zhang, I. Mercado-Uribe, Z. Xing, et al., 2014). PGCC progeny have reduced expression of epithelial markers and acquire the mesenchymal phenotype associated with tumor progression and metastasis (S. Zhang, Mercado-Uribe, Hanash, et al., 2013; S. Zhang, I. Mercado-Uribe, & J. Liu, 2014; S. Zhang, I. Mercado-Uribe, Z. Xing, et al., 2014). For example, vimentin and/or ZEB were upregulated in PGCC derived from ovarian cancer following hypoxia simulation (S. Zhang, Mercado-Uribe, Hanash, et al., 2013). ZEB1 is also upregulated in prostate cancer cells following docetaxel exposure (Lin et al., 2019). The multi-stage process of PGCC formation and neosis has been studied over an extended time frame in ovarian cancer cells treated with carboplatin (Rohnalter et al., 2015). After an initial phase of cell death, cultures enter a stationary phase during which more than 80% of cells are PGCC that strongly express p21, which is followed by neosis accompanied by upregulation of stemness markers and EMT over several weeks with the eventual emergence of a mitotic population that is migratory and chemoresistant. Similarly, in triptolide-induced PGCC, progeny differentially expressed EMT proteins. While post-treatment cells initially had lower proliferation, migration, and invasion rates than parental cells, these properties increased with time and significantly exceeded parental cell characteristics by passage 10 (Wang et al., 2019). Similar mechanisms could be underlying the observation that B16 melanoma PGCCs, which were artificially generated in vitro through a modified PHA-electronic fusion method, are initially less proliferative but exhibit an increased tendency to form lung metastases (Mi et al., 2016). In the B16 melanoma model, bioinformatic analysis of PGCC revealed an upregulation in the β-tubulin gene group, especially TUBB2B (Mi et al., 2016). TUBB2 mRNA also increases in response to vinblastine with preferential accumulation of the protein around the nucleus (Arai, Matsumoto, Nagashima, & Yagasaki, 2006). Although PGCC have increased cytoplasmic and nuclear stiffness as well as slower velocity than diploid counterparts, their higher directional persistence resembles the behavior of migratory fibroblasts rather than diploid cancer cells (Xuan et al., 2018).
Therapeutic approaches to target PGCC and neosis
Multiple studies and reviews have alerted the scientific community about therapy-induced PGCCs and called for eliminating this small but dangerous subpopulation but the means to identify PGCC and strategies to clinically target these cells have been elusive (Amend et al., 2019; Chen et al., 2019; Mirzayans, Andrais, Scott, Wang, & Murray, 2013; Niculescu, 2019; Ogden et al., 2015; Rajaraman, Guernsey, Rajaraman, & Rajaraman, 2006; Rajaraman et al., 2005; Vitale et al., 2011; Yang et al., 2018; D. Zhang, Wang, & Zhang, 2014). While dedifferentiation increases resistance to traditional cancer treatments, the stress-induced adaptations may also create new vulnerabilities that open the possibility for innovative clinical trials. Recent research has suggested that targeting of PGCC or neosis may be possible through immunological or pharmacological approaches.
Immunological approaches
Studies in mice have shown that polyploid cells from p53-deficient animals are tumorigenic while the corresponding diploid cells are not (Fujiwara et al., 2005). Using a colon carcinogenesis model, Guido Kroemer’s laboratory demonstrated that tetraploid p53-negative colonocytes formed neoplastic lesions only in immunocompromised mice, suggesting that the abnormal cells may be recognized through an immunosurveillance mechanism (Boileve et al., 2013). In a separate study the group demonstrated that polyploidy increases ER stress and results in surface expression of the ER-resident protein calreticulin, which is one of the damage-associated molecular patterns (DAMP) that alert the immune system to cells that appear “foreign” and to eliminate them via an immunogenic cell death mechanism (Senovilla et al., 2012). Peptides that are specifically associated with immunogenic cell death were identified through mass spectrometry but injection of these peptides did not elicit a growth-inhibitory tumor immune response. Therefore the enhanced immunogeniticity of polyploid cells is likely due to increased adjuvanticity rather than antigenicity (Bloy et al., 2017).
Pharmacological approaches
The growing understanding of PGCC biology has led to consideration of how to design targeted treatments. One group suggests targeting centrosomes, which are an integral part of PGCC formation (Erenpreisa, Cragg, Salmina, Hausmann, & Scherthan, 2009). Several studies show recent progress in this area but to the best of our knowledge these drugs are not in clinical use (Choe et al., 2018; Johannes et al., 2015; Koo et al., 2017; Pannu et al., 2014; Thomopoulou et al., 2016). A recent paper has suggested that the commonly used diabetic drug metformin could preferentially disrupt the metabolic needs of PGCC (Sirois et al., 2019). Here we will focus on two targets that have translational potential in the near future.
As discussed in the “Cell cycle checkpoint dysregulation” section above, CDK2 activation is an important player in the duplication of the genome. CDK2 is inhibited by mifepristone, a drug typically used as part of an emergency contraception cocktail (Goyeneche, Seidel, & Telleria, 2012). Mifepristone, which is preferentially detrimental to undifferentiated cells, also appears to block a critical aspect of PGCC biology (Kapperman, Goyeneche, & Telleria, 2018). Because the drug is relatively safe, a clinical trial is currently underway to test the use of mifepristone as a salvage therapy for lung cancer patients who have failed at least two previous chemotherapy regimens (NCT02642939). The primary outcome measures of this Phase II clinical trial are overall survival and quality of life. However, the underlying biology for improved overall survival could be due to inhibition of the PGCC cycle that would otherwise drive the progression of their disease (Kapperman et al., 2018). Results for this trial will not be available until 2021, but the trial design could be emulated quickly. Where possible, analysis of PGCCs could be added as an experimental objective.
It may also be possible to target sphingolipid metabolism. Drugs specifically aimed at enzymes in the sphingolipid pathway have entered the clinic (NCT03377179, NCT02834611) but have not yet been studied from the perspective of their effect on PGCCs or neosis (Britten et al., 2017). As mentioned in the “Stress responses” section, upregulation of the sphingolipid enzyme ASAH1 occurs as a consequence to the rise in ceramide levels following chemotherapy and radiation. We have shown that while inhibition of ASAH1 has no effect on the formation of PGCC, it significantly inhibits neosis. Interestingly, like mifepristone treatment, inhibition of ASAH1 is also preferentially detrimental to undifferentiated cells yet fairly safe for developed organisms (Eliyahu, Park, Shtraizent, He, & Schuchman, 2007; Eliyahu, Shtraizent, Shalgi, & Schuchman, 2012). The parallel vulnerabilities of early embryonic development and PGCC physiology are a promising avenue of exploration, and multiple lines of investigation suggest that ASAH1 is potentially a key target. Interfering with ASAH1 expression or function eliminated the tumor-initiating subpopulation within melanoma and glioblastoma cultures (Doan et al., 2017; Lai et al., 2017). Inhibition of ASAH1 also completely abrogated relapse after radiation therapy in a prostate xenograft model, suggesting that the subpopulation responsible for regrowth was eliminated by ASAH1 inhibition (Cheng et al., 2013).
Since specific inhibitors of ASAH1 are not yet clinically available, we recently became interested in the possibility of repurposing FDA approved drugs reported to have off-target effect on the enzyme (Voelkel-Johnson, Norris, & White-Gilbertson, 2018). One such drug is tamoxifen, which is typically used as maintenance therapy in pre-menopausal women to reduce relapse in breast cancer (Morad et al., 2013). Although the intended use of tamoxifen is inhibition of estrogen and progesterone signaling, the off-target effect on ASAH1 may be an underappreciated aspect of the drug. We studied breast cancer survivors who developed second (non-breast) malignancies, and compared the outcomes of these second cancers between women who were taking tamoxifen for the original breast cancer and those who were not. The results showed that tamoxifen was an independent predictor for longer survival for the second cancers, suggesting an important tamoxifen effect that operates independently of estrogen signaling (White-Gilbertson et al., 2020). A brief report that describes long-term remissions in metastatic melanoma when tamoxifen was provided as maintenance therapy following chemotherapy supports this idea (Metzner et al., 2011). However, the success of tamoxifen in other clinical trials has been limited, which may be due to clinical trials design that likely assumed drug activity against bulk tumor that primarily consists of diploid cancer cells (Beguerie, Xingzhong, & Valdez, 2010; Broniscer, Leite, Lanchote, Machado, & Cristofani, 2000; Morad & Cabot, 2015). We suggest that clinical trials be designed to combine anti-mitotic therapies aimed at diploid cancer cells with tamoxifen to target neosis of PGCC.
CONCLUSIONS AND FINAL REMARKS
This review outlined some of the challenges and needs of the emerging field of PGCC research. Recurrent cancer is generally more aggressive, metastatic, and treatment resistant than the original disease, and PGCC may be driving these adverse characteristics. If embryonic-like PGCCs exemplify the functional unit underlying cancer relapse, then designing therapies that target this subpopulation requires innovation both in the lab and in clinical trials. Researchers must trace the biological underpinnings of the PGCC plasticity over time and create nomenclature to facilitate precise discussions. Clinical trials to study PGCC will necessarily focus on relapse rather than initial treatment response, requiring long term studies and engaged pathologists armed with truly specific markers to help correlate the appearance of PGCC to outcome. If the vulnerabilities of PGCC can be targeted, many patients may be spared the difficult news that their cancer has returned. Importantly the approach of targeting embryonic vulnerabilities of PGCC may spare somatic, diploid cells allowing clinicians to do great good without simultaneously doing great harm. The field is at an exciting moment, when bench-to-bedside research has the potential to make a difference in the lives of many cancer patients. For those who are interested in further reading, we suggest three seminal papers that describe neosis (Erenpreisa et al., 2011; Sundaram, Guernsey, Rajaraman, & Rajaraman, 2004) and the self-renewing nature of giant cells and proposed a life cycle for these cells (Niu et al., 2016)
Acknowledgements
We continually discover studies that were published with different terminologies, which was part of the impetus for writing this review, and we apologize to any authors who we have left out due to our ignorance or the constraints of space. This work was supported a grant from the National Cancer Institute [grant number: P01 CA203628].
References
- Albawardi AS, Awwad AA, & Almarzooqi SS (2014). Mammary carcinoma with osteoclast-like giant cells: a case report. Int J Clin Exp Pathol, 7(12), 9038–9043. [PMC free article] [PubMed] [Google Scholar]
- Alharbi AM, De Marzo AM, Hicks JL, Lotan TL, & Epstein JI (2018). Prostatic Adenocarcinoma With Focal Pleomorphic Giant Cell Features: A Series of 30 Cases. Am J Surg Pathol, 42(10), 1286–1296. doi: 10.1097/PAS.0000000000001112 [DOI] [PubMed] [Google Scholar]
- Amend SR, et al. (2019). Polyploid giant cancer cells: Unrecognized actuators of tumorigenesis, metastasis, and resistance. Prostate, 79(13), 1489–1497. doi: 10.1002/pros.23877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arai K, Matsumoto Y, Nagashima Y, & Yagasaki K (2006). Regulation of class II beta-tubulin expression by tumor suppressor p53 protein in mouse melanoma cells in response to Vinca alkaloid. Mol Cancer Res, 4(4), 247–255. doi: 10.1158/1541-7786.MCR-05-0183 [DOI] [PubMed] [Google Scholar]
- Ariizumi T, et al. (2009). Multinucleation followed by an acytokinetic cell division in myxofibrosarcoma with giant cell proliferation. J Exp Clin Cancer Res, 28, 44. doi: 10.1186/1756-9966-28-44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arun RP, Sivanesan D, Patra B, Varadaraj S, & Verma RS (2019). Simulated microgravity increases polyploid giant cancer cells and nuclear localization of YAP. Sci Rep, 9(1), 10684. doi: 10.1038/s41598-019-47116-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baydar D, Amin MB, & Epstein JI (2006). Osteoclast-rich undifferentiated carcinomas of the urinary tract. Mod Pathol, 19(2), 161–171. doi: 10.1038/modpathol.3800521 [DOI] [PubMed] [Google Scholar]
- Beguerie JR, Xingzhong J, & Valdez RP (2010). Tamoxifen vs. non-tamoxifen treatment for advanced melanoma: a meta-analysis. Int J Dermatol, 49(10), 1194–1202. doi: 10.1111/j.1365-4632.2010.04529.x [DOI] [PubMed] [Google Scholar]
- Bergmann F, et al. (2010). Expression of L1CAM, COX-2, EGFR, c-KIT and Her2/neu in anaplastic pancreatic cancer: putative therapeutic targets? Histopathology, 56(4), 440–448. doi: 10.1111/j.1365-2559.2010.03499.x [DOI] [PubMed] [Google Scholar]
- Bloy N, et al. (2017). Immunogenic stress and death of cancer cells: Contribution of antigenicity vs adjuvanticity to immunosurveillance. Immunol Rev, 280(1), 165–174. doi: 10.1111/imr.12582 [DOI] [PubMed] [Google Scholar]
- Boileve A, et al. (2013). Immunosurveillance against tetraploidization-induced colon tumorigenesis. Cell Cycle, 12(3), 473–479. doi: 10.4161/cc.23369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourzac K (2007). Rewriting Life: How a Tumor Is Like an Embryo. from https://http://www.technologyreview.com/s/409004/how-a-tumor-is-like-an-embryo/
- Boyd AS, Wu H, & Shyr Y (2005). Monster cells in malignant melanoma. Am J Dermatopathol, 27(3), 208–210. doi: 10.1097/01.dad.0000158294.23630.ef [DOI] [PubMed] [Google Scholar]
- Britten CD, et al. (2017). A Phase I Study of ABC294640, a First-in-Class Sphingosine Kinase-2 Inhibitor, in Patients with Advanced Solid Tumors. Clin Cancer Res, 23(16), 4642–4650. doi: 10.1158/1078-0432.CCR-16-2363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broniscer A, Leite CC, Lanchote VL, Machado TM, & Cristofani LM (2000). Radiation therapy and high-dose tamoxifen in the treatment of patients with diffuse brainstem gliomas: results of a Brazilian cooperative study. Brainstem Glioma Cooperative Group. J Clin Oncol, 18(6), 1246–1253. doi: 10.1200/JCO.2000.18.6.1246 [DOI] [PubMed] [Google Scholar]
- Camacho L, et al. (2013). Acid ceramidase as a therapeutic target in metastatic prostate cancer. J Lipid Res, 54(5), 1207–1220. doi: 10.1194/jlr.M032375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caruso R, et al. (2011). Mitotic catastrophe in malignant epithelial tumors: the pathologist’s viewpoint. Ultrastruct Pathol, 35(2), 66–71. doi: 10.3109/01913123.2010.543753 [DOI] [PubMed] [Google Scholar]
- Chen J, et al. (2019). Polyploid Giant Cancer Cells (PGCCs): The Evil Roots of Cancer. Curr Cancer Drug Targets, 19(5), 360–367. doi: 10.2174/1568009618666180703154233 [DOI] [PubMed] [Google Scholar]
- Cheng JC, et al. (2013). Radiation-induced acid ceramidase confers prostate cancer resistance and tumor relapse. J Clin Invest, 123(10), 4344–4358. doi: 10.1172/JCI64791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choe MH, et al. (2018). Centrosome Clustering Is a Tumor-selective Target for the Improvement of Radiotherapy in Breast Cancer Cells. Anticancer Res, 38(6), 3393–3400. doi: 10.21873/anticanres.12606 [DOI] [PubMed] [Google Scholar]
- Chung HJ, Wolpowitz D, Scott G, Gilmore E, & Bhawan J (2016). Squamous cell carcinoma with osteoclast-like giant cells: a morphologically heterologous group including carcinosarcoma and squamous cell carcinoma with stromal changes. J Cutan Pathol, 43(2), 148–157. doi: 10.1111/cup.12607 [DOI] [PubMed] [Google Scholar]
- Cook JG, Chasse DA, & Nevins JR (2004). The regulated association of Cdt1 with minichromosome maintenance proteins and Cdc6 in mammalian cells. J Biol Chem, 279(10), 9625–9633. doi: 10.1074/jbc.M311933200 [DOI] [PubMed] [Google Scholar]
- Costanzo V, Bardelli A, Siena S, & Abrignani S (2018). Exploring the links between cancer and placenta development. Open Biol, 8(6). doi: 10.1098/rsob.180081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coward J, & Harding A (2014). Size Does Matter: Why Polyploid Tumor Cells are Critical Drug Targets in the War on Cancer. Front Oncol, 4, 123. doi: 10.3389/fonc.2014.00123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daneshbod Y, Khademi B, Kadivar M, & Ganjei-Azar P (2008). Fine needle aspiration of salivary gland lesions with multinucleated giant cells. Acta Cytol, 52(6), 671–680. doi: 10.1159/000325620 [DOI] [PubMed] [Google Scholar]
- Deeken-Draisey A, Yang GY, Gao J, & Alexiev BA (2018). Anaplastic thyroid carcinoma: an epidemiologic, histologic, immunohistochemical, and molecular single-institution study. Hum Pathol, 82, 140–148. doi: 10.1016/j.humpath.2018.07.027 [DOI] [PubMed] [Google Scholar]
- Defty CL, Segen J, Carter JJ, Ahmed I, & Carr RA (2011). Basaloid squamous cell carcinoma with ‘monster’ cells: a mimic of pleomorphic basal cell carcinoma. J Cutan Pathol, 38(4), 354–356. doi: 10.1111/j.16000560.2010.01627.x [DOI] [PubMed] [Google Scholar]
- Diaz-Carballo D, et al. (2014). Atypical cell populations associated with acquired resistance to cytostatics and cancer stem cell features: the role of mitochondria in nuclear encapsulation. DNA Cell Biol, 33(11), 749–774. doi: 10.1089/dna.2014.2375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doan NB, et al. (2017). Acid ceramidase and its inhibitors: a de novo drug target and a new class of drugs for killing glioblastoma cancer stem cells with high efficiency. Oncotarget, 8(68), 112662–112674. doi: 10.18632/oncotarget.22637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eliyahu E, Park JH, Shtraizent N, He X, & Schuchman EH (2007). Acid ceramidase is a novel factor required for early embryo survival. FASEB J, 21(7), 1403–1409. doi: 10.1096/fj.06-7016com [DOI] [PubMed] [Google Scholar]
- Eliyahu E, Shtraizent N, Shalgi R, & Schuchman EH (2012). Construction of conditional acid ceramidase knockout mice and in vivo effects on oocyte development and fertility. Cell Physiol Biochem, 30(3), 735–748. doi: 10.1159/000341453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erenpreisa J, Cragg MS, Salmina K, Hausmann M, & Scherthan H (2009). The role of meiotic cohesin REC8 in chromosome segregation in gamma irradiation-induced endopolyploid tumour cells. Exp Cell Res, 315(15), 2593–2603. doi: 10.1016/j.yexcr.2009.05.011 [DOI] [PubMed] [Google Scholar]
- Erenpreisa J, et al. (2008). Endopolyploidy in irradiated p53-deficient tumour cell lines: persistence of cell division activity in giant cells expressing Aurora-B kinase. Cell Biol Int, 32(9), 1044–1056. doi: 10.1016/j.cellbi.2008.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erenpreisa J, et al. (2005). Segregation of genomes in polyploid tumour cells following mitotic catastrophe. Cell Biol Int, 29(12), 1005–1011. doi: 10.1016/j.cellbi.2005.10.008 [DOI] [PubMed] [Google Scholar]
- Erenpreisa J, et al. (2011). Polyploid tumour cells elicit paradiploid progeny through depolyploidizing divisions and regulated autophagic degradation. Cell Biol Int, 35(7), 687–695. doi: 10.1042/CBI20100762 [DOI] [PubMed] [Google Scholar]
- Faragalla H, Al-Haddad S, Stewart R, & Yousef GM (2010). The significance of florid giant cell component in renal cell carcinoma: a case report and review of the literature. Can J Urol, 17(3), 5219–5222. [PubMed] [Google Scholar]
- Fei F, et al. (2019). Syncytin 1, CD9, and CD47 regulating cell fusion to form PGCCs associated with cAMP/PKA and JNK signaling pathway. Cancer Med, 8(6), 3047–3058. doi: 10.1002/cam4.2173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fei F, et al. (2019). The subcellular location of cyclin B1 and CDC25 associated with the formation of polyploid giant cancer cells and their clinicopathological significance. Lab Invest, 99(4), 483–498. doi: 10.1038/s41374-018-0157-x [DOI] [PubMed] [Google Scholar]
- Fei F, et al. (2015). The number of polyploid giant cancer cells and epithelial-mesenchymal transition-related proteins are associated with invasion and metastasis in human breast cancer. J Exp Clin Cancer Res, 34, 158. doi: 10.1186/s13046-015-0277-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fei F, et al. (2019). Formation of Polyploid Giant Cancer Cells Involves in the Prognostic Value of Neoadjuvant Chemoradiation in Locally Advanced Rectal Cancer. J Oncol, 2019, 2316436. doi: 10.1155/2019/2316436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedrichs N, et al. (2005). Clear cell sarcoma-like tumor with osteoclast-like giant cells in the small bowel: further evidence for a new tumor entity. Int J Surg Pathol, 13(4), 313–318. doi: 10.1177/106689690501300402 [DOI] [PubMed] [Google Scholar]
- Fujiwara T, et al. (2005). Cytokinesis failure generating tetraploids promotes tumorigenesis in p53-null cells. Nature, 437(7061), 1043–1047. doi: 10.1038/nature04217 [DOI] [PubMed] [Google Scholar]
- Gaumann A, et al. (2001). The expression of cathepsins in osteoclast-like giant cells of an anaplastic thyroid carcinoma with tracheal perforation. Pathol Res Pract, 197(4), 257–262. doi: 10.1078/0344-0338-00044 [DOI] [PubMed] [Google Scholar]
- Georgakilas AG, Martin OA, & Bonner WM (2017). p21: A Two-Faced Genome Guardian. Trends Mol Med, 23(4), 310–319. doi: 10.1016/j.molmed.2017.02.001 [DOI] [PubMed] [Google Scholar]
- Gherardi R, et al. (1986). Monstrocellular heavily lipidized malignant glioma. Acta Neuropathol, 69(1–2), 28–32. doi: 10.1007/bf00687035 [DOI] [PubMed] [Google Scholar]
- Glassmann A, et al. (2018). Staurosporine Induces the Generation of Polyploid Giant Cancer Cells in Non-Small-Cell Lung Carcinoma A549 Cells. Anal Cell Pathol (Amst), 2018, 1754085. doi: 10.1155/2018/1754085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez MA, et al. (2006). Geminin is essential to prevent endoreduplication and to form pluripotent cells during mammalian development. Genes Dev, 20(14), 1880–1884. doi: 10.1101/gad.379706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goyeneche AA, Seidel EE, & Telleria CM (2012). Growth inhibition induced by antiprogestins RU-38486, ORG-31710, and CDB-2914 in ovarian cancer cells involves inhibition of cyclin dependent kinase 2. Invest New Drugs, 30(3), 967–980. doi: 10.1007/s10637-011-9655-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa K, et al. (2017). Imaging the Role of Multinucleate Pancreatic Cancer Cells and Cancer-Associated Fibroblasts in Peritoneal Metastasis in Mouse Models. Anticancer Res, 37(7), 3435–3440. doi: 10.21873/anticanres.11711 [DOI] [PubMed] [Google Scholar]
- Hawryluk EB, Baran JL, Gerami P, & Sepehr A (2013). ‘Monster cell’ melanoma with pulmonary metastasis and cyclin D1 amplification. J Cutan Pathol, 40(1), 61–65. doi: 10.1111/cup.12024 [DOI] [PubMed] [Google Scholar]
- Hoorens A, Prenzel K, Lemoine NR, & Kloppel G (1998). Undifferentiated carcinoma of the pancreas: analysis of intermediate filament profile and Ki-ras mutations provides evidence of a ductal origin. J Pathol, 185(1), 53–60. doi: [DOI] [PubMed] [Google Scholar]
- Ianzini F, et al. (2009). Activation of meiosis-specific genes is associated with depolyploidization of human tumor cells following radiation-induced mitotic catastrophe. Cancer Res, 69(6), 2296–2304. doi: 10.1158/0008-5472.CAN-08-3364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Illidge TM, Cragg MS, Fringes B, Olive P, & Erenpreisa JA (2000). Polyploid giant cells provide a survival mechanism for p53 mutant cells after DNA damage. Cell Biol Int, 24(9), 621–633. doi: 10.1006/cbir.2000.0557 [DOI] [PubMed] [Google Scholar]
- Jiang Q, et al. (2015). A Fraction of CD133+ CNE2 Cells Is Made of Giant Cancer Cells with Morphological Evidence of Asymmetric Mitosis. J Cancer, 6(12), 1236–1244. doi: 10.7150/jca.12626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo S (2014). Huge undifferentiated carcinoma of the pancreas with osteoclast-like giant cells. World J Gastroenterol, 20(10), 2725–2730. doi: 10.3748/wjg.v20.i10.2725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johannes JW, et al. (2015). Discovery of AZ0108, an orally bioavailable phthalazinone PARP inhibitor that blocks centrosome clustering. Bioorg Med Chem Lett, 25(24), 5743–5747. doi: 10.1016/j.bmcl.2015.10.079 [DOI] [PubMed] [Google Scholar]
- Kalejs M, et al. (2006). Upregulation of meiosis-specific genes in lymphoma cell lines following genotoxic insult and induction of mitotic catastrophe. BMC Cancer, 6, 6. doi: 10.1186/1471-2407-6-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapperman HE, Goyeneche AA, & Telleria CM (2018). Mifepristone inhibits non-small cell lung carcinoma cellular escape from DNA damaging cisplatin. Cancer Cell Int, 18, 185. doi: 10.1186/s12935-018-0683-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur E, et al. (2019). Molecular features unique to glioblastoma radiation resistant residual cells may affect patient outcome - a short report. Cell Oncol (Dordr), 42(1), 107–116. doi: 10.1007/s13402-018-0411-7 [DOI] [PubMed] [Google Scholar]
- Kaur E, et al. (2015). Radiation-induced homotypic cell fusions of innately resistant glioblastoma cells mediate their sustained survival and recurrence. Carcinogenesis, 36(6), 685–695. doi: 10.1093/carcin/bgv050 [DOI] [PubMed] [Google Scholar]
- Koo CY, et al. (2017). Targeting TAO Kinases Using a New Inhibitor Compound Delays Mitosis and Induces Mitotic Cell Death in Centrosome Amplified Breast Cancer Cells. Mol Cancer Ther, 16(11), 2410–2421. doi: 10.1158/1535-7163.MCT-17-0077 [DOI] [PubMed] [Google Scholar]
- Kushwaha PP, Rapalli KC, & Kumar S (2016). Geminin a multi task protein involved in cancer pathophysiology and developmental process: A review. Biochimie, 131, 115–127. doi: 10.1016/j.biochi.2016.09.022 [DOI] [PubMed] [Google Scholar]
- Lai M, et al. (2017). Complete Acid Ceramidase ablation prevents cancer-initiating cell formation in melanoma cells. Sci Rep, 7(1), 7411. doi: 10.1038/s41598-017-07606-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin KC, et al. (2019). The role of heterogeneous environment and docetaxel gradient in the emergence of polyploid, mesenchymal and resistant prostate cancer cells. Clin Exp Metastasis, 36(2), 97–108. doi: 10.1007/s10585-019-09958-1 [DOI] [PubMed] [Google Scholar]
- Liu J (2018). The dualistic origin of human tumors. Semin Cancer Biol, 53, 1–16. doi: 10.1016/j.semcancer.2018.07.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J (2019). The “life code”: a theory that unifies the human life cycle and the origin of human tumors. Semin Cancer Biol, in press. [DOI] [PubMed] [Google Scholar]
- Liu K, et al. (2019). Association and clinicopathologic significance of p38MAPK-ERK-JNK-CDC25C with polyploid giant cancer cell formation. Med Oncol, 37(1), 6. doi: 10.1007/s12032-019-1330-9 [DOI] [PubMed] [Google Scholar]
- Lopez-Sanchez LM, et al. (2014). CoCl2, a mimic of hypoxia, induces formation of polyploid giant cells with stem characteristics in colon cancer. PLoS One, 9(6), e99143. doi: 10.1371/journal.pone.0099143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luchini C, et al. (2018). PD-1, PD-L1, and CD163 in pancreatic undifferentiated carcinoma with osteoclast-like giant cells: expression patterns and clinical implications. Hum Pathol, 81, 157–165. doi: 10.1016/j.humpath.2018.07.006 [DOI] [PubMed] [Google Scholar]
- Lupo G, et al. (2016). Anti-angiogenic Therapy in Cancer: Downsides and New Pivots for Precision Medicine. Front Pharmacol, 7, 519. doi: 10.3389/fphar.2016.00519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lv H, et al. (2014). Polyploid giant cancer cells with budding and the expression of cyclin E, S-phase kinase-associated protein 2, stathmin associated with the grading and metastasis in serous ovarian tumor. BMC Cancer, 14, 576. doi: 10.1186/1471-2407-14-576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsubayashi H, et al. (2019). Osteoclast-like Giant Cell-type Pancreatic Anaplastic Carcinoma Presenting with a Duodenal Polypoid Lesion. Intern Med, 58(24), 3545–3550. doi: 10.2169/internalmedicine.3271-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzner B, et al. (2011). Long-term remissions in metastatic malignant melanoma following chemotherapy and tamoxifen maintenance. Onkologie, 34(4), 208–209. doi: 10.1159/000327003 [DOI] [PubMed] [Google Scholar]
- Mi R, et al. (2016). Identification of the metastasis potential and its associated genes in melanoma multinucleated giant cells using the PHA-ECM830 fusion method. Oncol Rep, 35(1), 211–218. doi: 10.3892/or.2015.4376 [DOI] [PubMed] [Google Scholar]
- Mirzayans R, Andrais B, Kumar P, & Murray D (2017). Significance of Wild-Type p53 Signaling in Suppressing Apoptosis in Response to Chemical Genotoxic Agents: Impact on Chemotherapy Outcome. Int J Mol Sci, 18(5). doi: 10.3390/ijms18050928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirzayans R, et al. (2017). Multinucleated Giant Cancer Cells Produced in Response to Ionizing Radiation Retain Viability and Replicate Their Genome. Int J Mol Sci, 18(2). doi: 10.3390/ijms18020360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirzayans R, Andrais B, Scott A, Wang YW, & Murray D (2013). Ionizing radiation-induced responses in human cells with differing TP53 status. Int J Mol Sci, 14(11), 22409–22435. doi: 10.3390/ijms141122409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittal K, et al. (2017). Multinucleated polyploidy drives resistance to Docetaxel chemotherapy in prostate cancer. Br J Cancer, 116(9), 1186–1194. doi: 10.1038/bjc.2017.78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morad SA, & Cabot MC (2015). Tamoxifen regulation of sphingolipid metabolism--Therapeutic implications. Biochim Biophys Acta, 1851(9), 1134–1145. doi: 10.1016/j.bbalip.2015.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morad SA, et al. (2013). Novel off-target effect of tamoxifen--inhibition of acid ceramidase activity in cancer cells. Biochim Biophys Acta, 1831(12), 1657–1664. doi: 10.1016/j.bbalip.2013.07.016 [DOI] [PubMed] [Google Scholar]
- Niculescu VF (2019). The reproductive life cycle of cancer: Hypotheses of cell of origin, TP53 drivers and stem cell conversions in the light of the atavistic cancer cell theory. Med Hypotheses, 123, 19–23. doi: 10.1016/j.mehy.2018.12.006 [DOI] [PubMed] [Google Scholar]
- Niu N, Mercado-Uribe I, & Liu J (2017). Dedifferentiation into blastomere-like cancer stem cells via formation of polyploid giant cancer cells. Oncogene, 36(34), 4887–4900. doi: 10.1038/onc.2017.72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu N, et al. (2016). Linking genomic reorganization to tumor initiation via the giant cell cycle. Oncogenesis, 5(12), e281. doi: 10.1038/oncsis.2016.75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norris JS, et al. (2006). Combined therapeutic use of AdGFPFasL and small molecule inhibitors of ceramide metabolism in prostate and head and neck cancers: a status report. Cancer Gene Ther, 13(12), 1045–1051. doi: 10.1038/sj.cgt.7700965 [DOI] [PubMed] [Google Scholar]
- Ogawa K, et al. (2019). Giant cell glioblastoma is a distinctive subtype of glioma characterized by vulnerability to DNA damage. Brain Tumor Pathol. doi: 10.1007/s10014-019-00355-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogden A, Rida PC, Knudsen BS, Kucuk O, & Aneja R (2015). Docetaxel-induced polyploidization may underlie chemoresistance and disease relapse. Cancer Lett, 367(2), 89–92. doi: 10.1016/j.canlet.2015.06.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogretmen B (2018). Sphingolipid metabolism in cancer signalling and therapy. Nat Rev Cancer, 18(1), 33–50. doi: 10.1038/nrc.2017.96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh JE, et al. (2016). Genetic Alterations in Gliosarcoma and Giant Cell Glioblastoma. Brain Pathol, 26(4), 517–522. doi: 10.1111/bpa.12328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palma L, Celli P, Maleci A, Di Lorenzo N, & Cantore G (1989). Malignant monstrocellular brain tumours. A study of 42 surgically treated cases. Acta Neurochir (Wien), 97(1–2), 17–25. doi: 10.1007/bf01577735 [DOI] [PubMed] [Google Scholar]
- Pannu V, et al. (2014). Centrosome-declustering drugs mediate a two-pronged attack on interphase and mitosis in supercentrosomal cancer cells. Cell Death Dis, 5, e1538. doi: 10.1038/cddis.2014.505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parekh A, et al. (2018). Multi-nucleated cells use ROS to induce breast cancer chemo-resistance in vitro and in vivo. Oncogene, 37(33), 4546–4561. doi: 10.1038/s41388-018-0272-6 [DOI] [PubMed] [Google Scholar]
- Patel D, Incassati A, Wang N, & McCance DJ (2004). Human papillomavirus type 16 E6 and E7 cause polyploidy in human keratinocytes and up-regulation of G2-M-phase proteins. Cancer Res, 64(4), 1299–1306. doi: 10.1158/0008-5472.can-03-2917 [DOI] [PubMed] [Google Scholar]
- Pena-Jaimes L, et al. (2018). Pleomorphic lobular carcinoma of the breast with osteoclast-like giant cells: a case report and review of the literature. Diagn Pathol, 13(1), 62. doi: 10.1186/s13000-018-0744-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirsko V, Čakstiņa I , Nitiša D , Samoviča M , Daneberga Z, Miklaševičs E. (2019). Alterations of The Stem-Like Properties in The Breast Cancer Cell Line MDA-MB-231 Induced by Single Pulsed Doxorubicin Treatment. Proceedings of the Latvian Academy of Sciences, 73(2), 89–99. [Google Scholar]
- Pouryazdanparast P, Newman M, Mafee M, Guitart J, & Gerami P (2009). Malignant melanoma with monster cells showing massive cyclin D1 amplification. Am J Dermatopathol, 31(4), 402–403. doi: 10.1097/DAD.0b013e31819f8316 [DOI] [PubMed] [Google Scholar]
- Puig PE, et al. (2008). Tumor cells can escape DNA-damaging cisplatin through DNA endoreduplication and reversible polyploidy. Cell Biol Int, 32(9), 1031–1043. doi: 10.1016/j.cellbi.2008.04.021 [DOI] [PubMed] [Google Scholar]
- Qu Y, Zhang L, Rong Z, He T, & Zhang S (2013). Number of glioma polyploid giant cancer cells (PGCCs) associated with vasculogenic mimicry formation and tumor grade in human glioma. J Exp Clin Cancer Res, 32, 75. doi: 10.1186/1756-9966-32-75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajaraman R, Guernsey DL, Rajaraman MM, & Rajaraman SR (2006). Stem cells, senescence, neosis and self-renewal in cancer. Cancer Cell Int, 6, 25. doi: 10.1186/1475-2867-6-25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajaraman R, Rajaraman MM, Rajaraman SR, & Guernsey DL (2005). Neosis--a paradigm of self-renewal in cancer. Cell Biol Int, 29(12), 1084–1097. doi: 10.1016/j.cellbi.2005.10.003 [DOI] [PubMed] [Google Scholar]
- Rohnalter V, et al. (2015). A multi-stage process including transient polyploidization and EMT precedes the emergence of chemoresistent ovarian carcinoma cells with a dedifferentiated and pro-inflammatory secretory phenotype. Oncotarget, 6(37), 40005–40025. doi: 10.18632/oncotarget.5552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saad AF, et al. (2007). The functional effects of acid ceramidase overexpression in prostate cancer progression and resistance to chemotherapy. Cancer Biol Ther, 6(9), 1455–1460. [DOI] [PubMed] [Google Scholar]
- Schwarz-Finsterle J, et al. (2013). Volume increase and spatial shifts of chromosome territories in nuclei of radiation-induced polyploidizing tumour cells. Mutat Res, 756(1–2), 56–65. doi: 10.1016/j.mrgentox.2013.05.004 [DOI] [PubMed] [Google Scholar]
- Sekulic M, Gilles S, Amin K, & Stewart J 3rd. (2016). Undifferentiated (anaplastic) carcinoma with osteoclast-like giant cells of the pancreas: a series of 5 cases with clinicopathologic correlation and cytomorphologic characterization. J Am Soc Cytopathol, 5(6), 321–330. doi: 10.1016/j.jasc.2016.04.003 [DOI] [PubMed] [Google Scholar]
- Senovilla L, et al. (2012). An immunosurveillance mechanism controls cancer cell ploidy. Science, 337(6102), 1678–1684. doi: 10.1126/science.1224922 [DOI] [PubMed] [Google Scholar]
- Sheikh MS, Rochefort H, & Garcia M (1995). Overexpression of p21WAF1/CIP1 induces growth arrest, giant cell formation and apoptosis in human breast carcinoma cell lines. Oncogene, 11(9), 1899–1905. [PubMed] [Google Scholar]
- Shu Z, Row S, & Deng WM (2018). Endoreplication: The Good, the Bad, and the Ugly. Trends Cell Biol, 28(6), 465–474. doi: 10.1016/j.tcb.2018.02.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirois I, et al. (2019). A Unique Morphological Phenotype in Chemoresistant Triple-Negative Breast Cancer Reveals Metabolic Reprogramming and PLIN4 Expression as a Molecular Vulnerability. Mol Cancer Res. doi: 10.1158/1541-7786.MCR-19-0264 [DOI] [PubMed] [Google Scholar]
- Sundaram M, Guernsey DL, Rajaraman MM, & Rajaraman R (2004). Neosis: a novel type of cell division in cancer. Cancer Biol Ther, 3(2), 207–218. doi: 10.4161/cbt.3.2.663 [DOI] [PubMed] [Google Scholar]
- Tannock IF, et al. (2004). Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer. N Engl J Med, 351(15), 1502–1512. doi: 10.1056/NEJMoa040720 [DOI] [PubMed] [Google Scholar]
- Thomopoulou P, et al. (2016). New Colchicine-Derived Triazoles and Their Influence on Cytotoxicity and Microtubule Morphology. ACS Med Chem Lett, 7(2), 188–191. doi: 10.1021/acsmedchemlett.5b00418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varma D, et al. (2012). Recruitment of the human Cdt1 replication licensing protein by the loop domain of Hec1 is required for stable kinetochore-microtubule attachment. Nat Cell Biol, 14(6), 593–603. doi: 10.1038/ncb2489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitale I, et al. (2011). Illicit survival of cancer cells during polyploidization and depolyploidization. Cell Death Differ, 18(9), 1403–1413. doi: 10.1038/cdd.2010.145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vittoria MA, et al. (2018). A genome-wide microRNA screen identifies regulators of tetraploid cell proliferation. Mol Biol Cell, 29(13), 1682–1692. doi: 10.1091/mbc.E18-02-0141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voelkel-Johnson C, Norris JS, & White-Gilbertson S (2018). Interdiction of Sphingolipid Metabolism Revisited: Focus on Prostate Cancer. Adv Cancer Res, 140, 265–293. doi: 10.1016/bs.acr.2018.04.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh S, Margolis SS, & Kornbluth S (2003). Phosphorylation of the cyclin b1 cytoplasmic retention sequence by mitogen-activated protein kinase and Plx. Mol Cancer Res, 1(4), 280–289. [PubMed] [Google Scholar]
- Wang X, et al. (2019). EMT-related protein expression in polyploid giant cancer cells and their daughter cells with different passages after triptolide treatment. Med Oncol, 36(9), 82. doi: 10.1007/s12032-019-1303-z [DOI] [PubMed] [Google Scholar]
- Wasserman JK, Sekhon HS, & Ayroud Y (2015). Malignant Melanoma With Osteoclast-Like Differentiation. Int J Surg Pathol, 23(6), 478–482. doi: 10.1177/1066896915592016 [DOI] [PubMed] [Google Scholar]
- Weihua Z, Lin Q, Ramoth AJ, Fan D, & Fidler IJ (2011). Formation of solid tumors by a single multinucleated cancer cell. Cancer, 117(17), 4092–4099. doi: 10.1002/cncr.26021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- White-Gilbertson S, et al. (2020). Tamoxifen improves outcomes for second cancers in breast cancer patients and is a potential candidate anti-neosis drug due to an off target effect on acid ceramidase. Cancer Med, submitted. [Google Scholar]
- White-Gilbertson S, Lu P, Norris JS., & Voelkel-Johnson C (2019). Genetic and pharmacological inhibition of acid ceramidase prevents asymmetric cell division by neosis. J Lipid Res, 60(7), 1225–1235. doi: 10.1194/jlr.M092247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson SR, et al. (2014). Clear cell renal cell carcinoma with a syncytial-type multinucleated giant tumor cell component: implications for differential diagnosis. Hum Pathol, 45(4), 735–744. doi: 10.1016/j.humpath.2013.10.033 [DOI] [PubMed] [Google Scholar]
- Xuan B, Ghosh D, Cheney EM, Clifton EM, & Dawson MR (2018). Dysregulation in Actin Cytoskeletal Organization Drives Increased Stiffness and Migratory Persistence in Polyploidal Giant Cancer Cells. Sci Rep, 8(1), 11935. doi: 10.1038/s41598-018-29817-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, et al. (2018). Generation of erythroid cells from polyploid giant cancer cells: re-thinking about tumor blood supply. J Cancer Res Clin Oncol, 144(4), 617–627. doi: 10.1007/s00432-018-2598-4 [DOI] [PubMed] [Google Scholar]
- Zeng JY, et al. (2014). Synergistic activities of MET/RON inhibitor BMS-777607 and mTOR inhibitor AZD8055 to polyploid cells derived from pancreatic cancer and cancer stem cells. Mol Cancer Ther, 13(1), 37–48. doi: 10.1158/1535-7163.MCT-13-0242 [DOI] [PubMed] [Google Scholar]
- Zhang D, Wang Y, & Zhang S (2014). Asymmetric cell division in polyploid giant cancer cells and low eukaryotic cells. Biomed Res Int, 2014, 432652. doi: 10.1155/2014/432652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D, et al. (2017). Daughter Cells and Erythroid Cells Budding from PGCCs and Their Clinicopathological Significances in Colorectal Cancer. J Cancer, 8(3), 469–478. doi: 10.7150/jca.17012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, et al. (2014). Number of polyploid giant cancer cells and expression of EZH2 are associated with VM formation and tumor grade in human ovarian tumor. Biomed Res Int, 2014, 903542. doi: 10.1155/2014/903542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Wu C, & Hoffman RM (2015). Prostate Cancer Heterogeneous High-Metastatic Multi-Organ-Colonizing Chemo-Resistant Variants Selected by Serial Metastatic Passage in Nude Mice Are Highly Enriched for Multinucleate Giant Cells. PLoS One, 10(11), e0140721. doi: 10.1371/journal.pone.0140721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Mercado-Uribe I, Hanash S, & Liu J (2013). iTRAQ-based proteomic analysis of polyploid giant cancer cells and budding progeny cells reveals several distinct pathways for ovarian cancer development. PLoS One, 8(11), e80120. doi: 10.1371/journal.pone.0080120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Mercado-Uribe I, & Liu J (2013). Generation of erythroid cells from fibroblasts and cancer cells in vitro and in vivo. Cancer Lett, 333(2), 205–212. doi: 10.1016/j.canlet.2013.01.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Mercado-Uribe I, & Liu J (2014). Tumor stroma and differentiated cancer cells can be originated directly from polyploid giant cancer cells induced by paclitaxel. Int J Cancer, 134(3), 508–518. doi: 10.1002/ijc.28319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Mercado-Uribe I, Sood A, Bast RC, & Liu J (2016). Coevolution of neoplastic epithelial cells and multilineage stroma via polyploid giant cells during immortalization and transformation of mullerian epithelial cells. Genes Cancer, 7(3–4), 60–72. doi: 10.18632/genesandcancer.102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, et al. (2014). Generation of cancer stem-like cells through the formation of polyploid giant cancer cells. Oncogene, 33(1), 116–128. doi: 10.1038/onc.2013.96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Zhang D, Yang Z, & Zhang X (2016). Tumor Budding, Micropapillary Pattern, and Polyploidy Giant Cancer Cells in Colorectal Cancer: Current Status and Future Prospects. Stem Cells Int, 2016, 4810734. doi: 10.1155/2016/4810734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, et al. (2018). Fine needle aspiration of giant cell tumor involving thyroid gland: A case report of an unprecedented entity. Diagn Cytopathol, 46(10), 879–882. doi: 10.1002/dc.24053 [DOI] [PubMed] [Google Scholar]