

Abstract
Mutations in the mitochondrial fusion protein mitofusin (MFN) 2 cause the chronic neurodegenerative condition Charcot-Marie-Tooth Disease type 2A (CMT2A), for which there is currently no treatment. Small molecule activators of MFN1 and MFN2 enhance mitochondrial fusion and offer promise as therapy for this condition, but prototype compounds have poor pharmacokinetic properties. Herein, we describe rational design of a series of 6-phenylhexanamide derivatives whose pharmacokinetic optimization yielded a 4-hydroxy cyclohexyl analog, 13, with the potency, selectivity, and oral bioavailability of a preclinical candidate. Studies of 13 cis- and trans- 4-hydroxy cyclohexyl isostereomers unexpectedly revealed functionality and protein engagement exclusively for the trans- form, 13B. Preclinical ADME and in vivo target engagement studies of 13B support further development of 6-phenylhexanamide derivatives as therapeutic agents for human CMT2A.
Graphical Abstract
INTRODUCTION
Mitofusins (MFN) 1 and 2, named for their central roles in mitochondrial fusion1, are attractive drug targets because their regulatory functioning in mitochondrial dynamics and quality/quantity control is perturbed in several neurodegenerative disorders2, 3. In particular, genetic mutations that abrogate or impair MFN2 functioning, and therefore suppress mitochondrial fusion, cause the rare autosomal dominant neurodegenerative condition Charcot-Marie-Tooth disease type 2A (CMT2A) for which there is currently no clinically available disease-altering therapy.4, 5 Moreover, accumulating experimental evidence supports an important role for MFN1 and MFN2 in heart disease.6–11 A pharmacological means of enhancing mitofusin function has the potential to correct the underlying cause of CMT2A and other cardiac or neurodegenerative diseases caused by mitochondrial dys-dynamism.2
MFN1 and MFN2 form homo- (MFN1-MFN1 or MFN2-MFN2) or hetero- (MFN1-MFN2) trans-dimers between mitochondria. This process, referred to as mitochondrial tethering, is the requisite first step in mitochondrial fusion and is essential for metabolic cellular health.12–14 Mitochondrial tethering and fusion depend upon a shift in MFN protein conformation, from a closed resting state to an open active state.15, 16 MFN conformation is regulated by intra-molecular peptide-peptide interactions (PPI) between alpha helices in the stalk region of the protein.17 Phosphorylation of human (h)MFN2 serine 378 by mitochondrial PINK1 kinase promotes a tight alpha helix within the interacting peptide, directing critical Val372, Met 376, and His380 amino acid side chains to their interacting partners, Leu727, Leu723, and Lys720, respectively.18 A strong PPI promotes a folded protein conformation that decreases the probability of trans MFN-MFN dimer formation between mitochondria, i.e. is unfavorable for mitochondrial tethering/fusion. When serine 378 is not phosphorylated the interacting peptide alpha helix partially unwinds, weakening the PPI and increasing the probability that the protein will unfold to permit MFN trans-dimerization and subsequent mitochondrial fusion.18
Franco et al17 described an 18 amino acid peptide, modified from hMFN2 amino acids 367–384, that competitively inhibited intra-molecular PPI in MFN1 and MFN2, thus promoting the open protein conformation favoring mitochondrial fusion. By understanding the critical interacting amino acids within this activator peptide (Figure 1A), Gavathiotiis developed a pharmacophore model leading to identification of a prototype peptidomimetic small molecule mitofusin activator, Chimera B-A/l (Figure 1B; 1)18. 1 mimicked mitofusin activator peptide effects by promoting mitochondrial fusion after topical application to mitofusin-deficient cultured mouse cells and by increasing mitochondrial motility in ex vivo mouse CMT2A nerves18. Clinical application of small molecule mitofusin activators having drug-like properties promises the first disease-altering therapy for CMT2A and may open the door to a novel therapeutic approach of enhancing mitochondrial fusion in multiple diseases with impaired mitochondrial dynamics.
Figure 1.

Mechanism of action of mitofusin activators. A. Computational model of PPI-regulated conformational changes in human MFN2. (inset) Published pharmacophore model described by E. Gavathiotis17, 18 positing specific amino acid interactions of the green MFN2-derived activator minipeptide. B. A prototype small molecule mitofusin activator, 1, was developed that mimics characteristics of critical amino acid side chains of the mitofusin activating minipeptide.18
Here, pharmacokinetic characterization of 1 and structurally-diverse chemical derivatives revealed a reciprocal relationship between in vitro hepatic microsomal stability and in vitro passive permeability that are central to the target product profile of a putative clinically applicable mitofusin activator. Accordingly, pharmacophore-based rational design was employed to develop a series of 6-phenylhexanamide derivative mitofusin activators having simplified chemical structures, thereby identifying candidate 13 as possibly suitable for preclinical in vivo evaluation in CMT2A rodent models. In vitro and in vivo pharmacokinetic studies show that 13 crosses the blood-brain barrier, engages mitochondrial targets in neuronal axons, and is otherwise pharmaceutically acceptable19, suggesting that 6-phenylhexanamide based mitofusin activators may be optimized as disease-altering therapeutics for CMT2A.
RESULTS AND DISCUSSION
Pharmacokinetic Properties of Chimera B-A/l (1) derivatives.
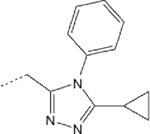
The published description of Chimera B-A/l as the prototype small molecule mitofusin activator indicated low nM potency to promote mitochondrial fusion in vitro measured as an increase in mitochondrial aspect ratio (length/width; EC50 = 3±1 nM), and the ability to rapidly promote mitochondrial trafficking after topical application to mouse CMT2A neurons ex vivo.18 A preliminary analysis of 1 pharmacokinetics revealed it to be stable in mouse and human plasma in vitro with good passive artificial blood brain barrier permeability (PAMPA-BBB Pe),20 but it was rapidly degraded when incubated in vitro with mouse or human liver microsomes (Table 1). Replacing the potentially reactive sulfur in 1 with either carbon (2) or oxygen (3) did not impair fusogenic activity, but neither did it meaningfully improve microsomal instability (Table 1).
Table 1.
Fusogenic potency and in vitro pharmacokinetic characteristics of prototype mitofusin activator, 1, and S-substituted analogsα
| Cpd | Structure | EC50 (nM) | Solubility | t1/2 HLM (min) | t1/2 MLM (min) | Hplasma stability (% @ 120 min) | Mplasma stability (% @ 120 min) | PAMPA-BBB Pe (nm/sec) |
|---|---|---|---|---|---|---|---|---|
| 1 | 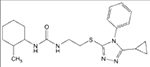 |
6.61 (3.67–11.07) | 67 | 3 | 1 | 95 | 94 | 96.3 |
| 2 | 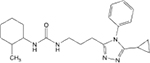 |
4.86 (4.05–5.82) | >200 | 6 | 2 | 95 | 100 | 21.7 |
| 3 | 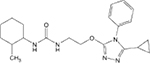 |
17.12 (9.78–27.57) | 24 | 8 | 3 | 98 | 84 | 43.2 |
EC50 values are for mitochondrial elongation in cultured MFN2-deficient mouse fibroblasts; mean with 95% confidence limits of 3–5 determinations. Solubility is in aqueous solution, pH 7.4, HLM and MLM indicate stability in human and mouse liver microsomes, respectively, PAMPA-BBB measures passive membrane permeability. All PK results are means of assays performed in duplicate or triplicate.
We posited that the terminal cyclohexyl or phenyl groups of 1–3 might be the targets of microsomal degradation. Fluorination of the phenyl group at the para-position (4) did not improve microsomal stability, whereas adding fluorine to the fourth carbon of the cyclohexyl group (5) markedly increased compound half-life in hepatic microsomes (Table 2). This suggested that the cyclohexyl group was the source of microsomal instability, a notion supported by emergence of tetrahydropyran derivatives having excellent potency (EC50 for mitochondrial elongation measured as increased aspect ratio) and high stability in both human and mouse liver microsome tests (Table 2; 6). By contrast, altering the position of the phenyl group on the triazol ring (7, 8) had little positive effect on microsomal stability, although mitofusin activator functionality could be preserved (7, 8) (Table 2). Indeed, the most potent and stable derivative of 1 consisted of a simple tetrahydropyran-4-yl group at R1 linked by propyl-urea to a 3-cyclopropyl-4-ethyl 1,2,4 triazol group at R2 (9). However, tetrahydropyran analogs exhibited very poor PAMPA-BBB passive permeability (6 and 9), which we envisioned would impair CNS penetration (Table 2)21,22. Moreover, comparative in vivo PK studies of 2, 6, and 9 demonstrated that increased microsomal stability conferred by the tetrahydropyran moiety did not translate to increased plasma half-life (Table 2). An inverse relationship between stability in the in vitro liver microsome assay and membrane permeability in the PAMPA-BBB assay was observed for 53 analogs of 1 as shown in Figure S1.
Table 2.
Fusogenic potency and pharmacokinetic properties of 1 analogs bearing cyclohexane and triazol group substitutionsα
 |
|||||||
|---|---|---|---|---|---|---|---|
| Cpd | R1 | R2 | EC50 (nM) | t1/2 HLM (min) | t1/2 MLM (min) | PAMPA-BBB Pe (nm/sec) | Plasma t1/2 (hours) |
| 2 | 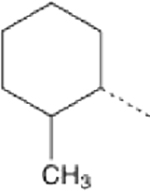 |
 |
4.86 (4.05–5.82) | 6 | 2 | 21.7 | 0.21 |
| 4 | 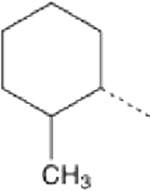 |
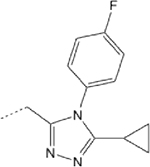 |
76.02 (33.15-ND) | 5 | 1 | 36.6 | ND |
| 5 | 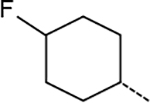 |
 |
5.13 (2.64–9.11) | 62 | 17 | 1.4 | ND |
| 6 | 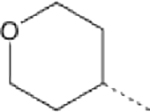 |
 |
10.11 (3.49–28.47) | >145 | 91 | 1 | 0.19 |
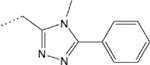
| 7 | 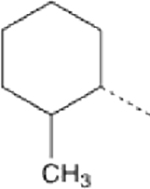 |
 |
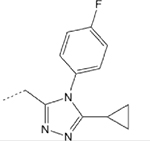
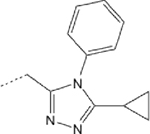
>1000 | 43 | 6 | 10.4 | ND |
| 8 | 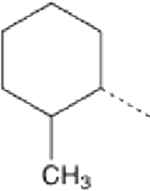 |
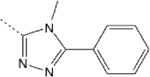 |
5.26 (3.12–8.37) | 8 | 28 | 10.3 | ND |
| 9 | 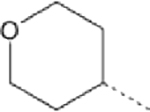 |
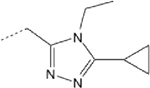 |
2.26 (1.01–4.65) | >145 | >145 | 0.03 | 0.38 |
EC50 values are mean with 95% confidence limits. Plasma t1/2 is in mice after IV bolus administration. ND means not determined.
Pharmacophore-Guided Optimization of Compound 8.
The published pharmacophore model developed by Gavathihotis,18 together with the above SAR studies, suggested leeway in the identity of the R2 group of small molecule mitofusin activators. This series of compounds were fusogenic as long as R2 was intensely hydrophobic (mimicking side chains of hMFN2 Val372 and Met 376; Figure 1B) and optimally spaced from the cyclohexyl/urea moiety (~10.8 Å, or the total distance between three consecutive alpha helix vertices). For example, in 8 the 4-phenyl group is shifted to the 3 position of the triazol ring, replacing the cyclopropyl group (Table 2). After adjusting the spacing between R1 and R2 by deleting one carbon from the urea linker (8 vs 7; Table 2), 8 exhibited comparable fusogenicity to 1 (EC50 ~5 nM) with improved mouse liver microsomal stability (t1/2 28 min vs 2 min) and adequate PAMPA-BBB permeability (10.3 vs 21.7 nm/sec). For these reasons 8 was selected as a lead for further optimization.
Because the pharmacophore model assigned no clear role for the triazol group we designed analogs of 8 lacking this moiety, but added 2 carbons to the linker to optimize functional group spacing (10; Table 3). Compound 10 was a weak mitofusin activator (EC50 ~400 nM), but exhibited 15 times greater PAMPA-BBB permeability than 8. As the cyclohexyl/urea group of small moleculer mitofusin activators mimics the hydrogen bond donor/acceptor ring structure of the hMFN2 His380 side chain in the parental mitofusin activator peptide (Figure 1B), we introduced a hydroxyl on carbon 4 of the 2-methyl-cyclohexyl group to improve hydrogen bond donor activity (11), markedly enhancing fusogenecity and increasing microsomal stability at the cost of reducing PAMPA-BBB by an order of magnitude (Table 3).
Table 3.
Fusogenic potency and pharmacokinetic properties of 8 analogs lacking the triazol ringα
| Cpd | Structure | EC50 (nM) | t1/2 HLM (min) | t1/2 MLM (min) | PAMPA-BBB Pe (nm/sec) |
|---|---|---|---|---|---|
| 8 |  |
5.26 (3.12–8.37) | 8 | 28 | 10.29 |
| 10 | 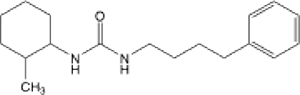 |
383.16 (105.66-ND) | 13.3 | 1.9 | 160 |
| 11 | 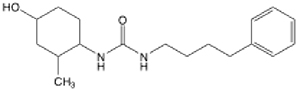 |
9.95 (6.99–14.05) | 40.1 | 24.2 | 15.7 |
| 12 | 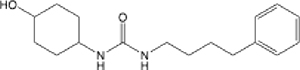 |
40.84 (21.44–71.75) | >145 | >145 | 7.96 |
| 13 | 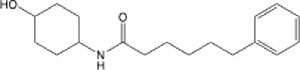 |
8.18 (6.55–10.15) | >145 | 92.4 | 26.28 |
EC50 values are mean with 95% confidence limits.
The 1 SAR studies reported in Table 2 revealed the 2-methyl residue on the cyclohexyl group to be dispensable for mitofusin activation (6 and 9; Table 2). Accordingly, we deleted this methyl group to make 12, which stabilized the compound in the liver microsome assay and maintained marginal PAMPA-BBB permeability. Compound 12 had the most favorable combination of microsomal stability and PAMPA-BBB permeability of any mitofusin activator, but suffered from a modest reduction in mitofusin agonist potency compared to 1 and 8, and its immediate precursor, 11 (Table 3).
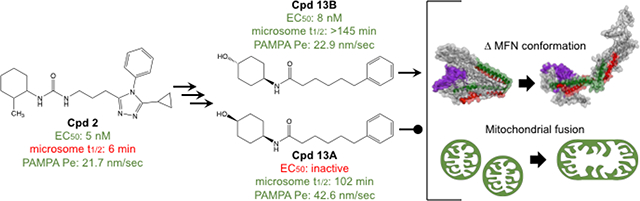
According to the pharmacophore model, a urea group linked to the R1 cyclohexanol mimics hydrogen bond donor/acceptor activity of the hMFN2 His380 side chain.18 However, it was not clear that both NH groups were necessary for mitofusin activator function. Moreover, the greater topological polar surface area of the urea (12, tPSA = 61.36) compared to the amide (13; tPSA = 49.33) could impair passive membrane permeability and CNS penetration.23.24 In support of this notion, the amide 13 exhibited 4-fold greater potency for mitofusin activation and PAMPA-BBB permeability than its urea parent (12), while retaining a high degree of liver microsome stability (Table 3).
SAR of 13 and its analogs.
We reasoned that the optimal screening profile for a clinically translatable small molecule mitofusin activator included high potency for inducing mitochondrial fusion (EC50 < 30 nM), stability (t1/2 > 100 min) in human and mouse liver microsome assays, and high passive artificial blood brain barrier membrane permeability (PAMPA-BBB Pe> 10 nm/sec). The results described above pointed to the R2 phenyl group of this class of mitofusin activators as a central mediator of PAMPA-BBB permeability, while the R1 cyclohexanol group appeared to be a major determinant of liver microsomal stability. Accordingly, our early SAR studies of 13 analogs maintained the phenylhexanamide portion constant and generated variants of the cyclohexyl moiety.
We first examined how surface charge impacted functional potency and microsomal stability by replacing the 4-hydroxyl cyclohexyl of 13 with groups conferring a range of tPSA (Table 4). Among compounds having higher tPSA values (38 to 55), the tetrahydropyran (14) and 4-amino (15) analogs were as potent as, but less stable than, parent 13. The 4-amide (16) was similarly potent and stable to 13, but both the 4-amine and 4-amide analogs exhibited greatly reduced PAMPA-BBB permeability (Table 4). By comparison, the two compounds having lower tPSA values (< 30), the 4-methyl (17) and 4,4-difluoro (18) analogs, were poor mitofusin activators and were unstable in the liver microsome assays (Table 4). Together, these results suggest that phenylhexanamides with 4-hydroxyl cyclohexyl groups at the R1 position are functionally and pharmacokinetically optimized for mitofusin activation.
Table 4.
Structure-activity relationships of cyclohexane group substitutions in phenylhexanamidesα
| Cpd | R3 | tPSA | EC50 (nM) | t1/2 HLM (min) | t1/2 MLM (min) | PAMPA-BBB Pe(nm/sec) |
|---|---|---|---|---|---|---|
| 13 | 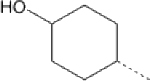 |
49.33 | 8.18 (6.55–10.15) | >145 | 92.4 | 26.28 |
| 14 | 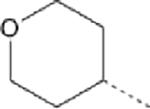 |
38.33 | 19.41 (13.28–27.53) | 53.6 | 9.9 | 240.14 |
| 15 | 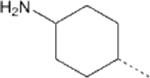 |
55.12 | 6.34 (4.48–8.71) | 23.7 | 111.9 | 9.64 |
| 16 | 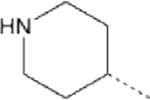 |
41.13 | 2.71 (1.79–3.93) | >145 | 137.5 | 7.1 |
| 17 | 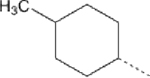 |
29.1 | >5 ∝M | 7 | 1.4 | 86.7 |
| 18 | 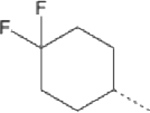 |
29.1 | 46.93 (36.29–59.75) | 10.2 | 2.8 | 214.6 |
EC50 values are mean with 95% confidence limits.
To assess functional plasticity of the 13 R2 phenyl group we created a series of pyridine and pyrimidine substitutions (Table 5). Consistent with prior observations that the phenyl group improves PAMPA-BBB permeability in mitofusin activators (6 and 9, Table 2), pyridine and pyrimidine variants of 13 exhibited very low Pe values (i.e. <1.0 nm/sec; Table 5). Moreover, plasma protein binding was markedly reduced for the pyridines (19–22, Table 5). Thus, although N2 and N3 pyridines and the pyrimidine analog of 13 exhibit excellent activity as mitofusin activators, they suffer from the same reciprocal relationship between microsomal stability and PAMPA-BBB permeability as did the parent, triazol group-containing compounds.
Table 5.
Functional and pharmacokinetic properties of pyridine and pyrimidine analogs of 13α
| Cpd | Structure | EC50 (nM) | hPPB (%) | mPPB (%) | t1/2 HLM (min) | t1/2 MLM (min) | PAMPA-BBB Pe (nm/sec) |
|---|---|---|---|---|---|---|---|
| 13 | 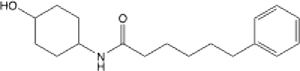 |
8.18 (6.55, 10.15) | 90.4 | 96.7 | >145 | 127 | 22.9 |
| 19 | 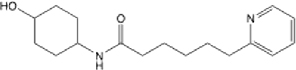 |
23.15 (6.68, ND) | 42.9 | 29.1 | >145 | >145 | 0.441 |
| 20 | 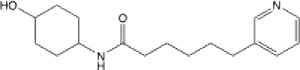 |
4.14 (3.32–5.11) | 27.5 | 40.9 | >145 | 125.3 | 0.523 |
| 21 | 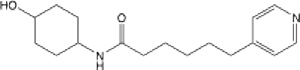 |
1.27 (0.97–1.64) | 24.8 | 18 | >145 | >145 | 0.744 |
| 22 | 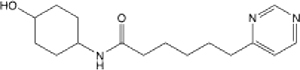 |
4.58 (3.40–6.02) | 92.5 | 96.7 | >145 | >145 | 0.053 |
EC50 values are mean with 95% confidence limits.
Mitofusin activating activity of 13 is isostereomer-specific.
Of the mitofusin activators created, the combined functional and pharmacokinetic characteristics of 13 seemed best suited for potential clinical application; it was therefore selected as an advanced lead for further evaluation. The chemical syntheses of candidate mitofusin activators for initial screening sourced starting materials as a mixture of stereoisomers. Since target recognition and biological activity can differ between stereoisomers25,26, the cis- and trans- diastereomers of 13 (13A and 13B) were individually synthesized, validated, and functionally characterized. Remarkably, 13A (the cis form) exhibited no measurable mitofusin activating activity measured as mitochondrial elongation (increase in aspect ratio) in either Mfn1 or Mfn2 null murine embryonic fibroblasts (Figure 2A, 2B). By contrast, 13B (the trans form), which was the dominant form obtained from the original synthesis, was equipotent with the prototype mitofusin agonist, 2, exhibiting comparable fusogenic activity in cells expressing only Mfn2 (Figure 2A) or only Mfn1 (Figure 2B). Fusion-impaired mitochondria typically exhibit dissipation of the inner mitochondrial membrane electrochemical gradient that drives ATP production, referred to as “depolarization”14. Compounds 2 and 13B, but not 13A, also reversed this respiratory defect (Figure 2C, 2D). The relative abilities of 13A and 13B to provoke the characteristic open/active MFN2 conformation17, 18 likewise paralleled their fusogenic activities (Figure 3), mechanistically linking biological effects to target protein engagement. The cis- and trans- stereoisomers had similar in vitro pharmacokinetic profiles (Table 6).
Figure 2.

Diastereomer-selective correction of mitochondrial fragmentation and depolarization in Mfn knockout cells for 13. A and B. Dose-response curves for 13 stereoisomers increasing mitochondrial aspect ratio in cells expressing only Mfn2 (A) or only Mfn1 (B). Prototype mitofusin activator 2 data are shown for comparison. C. Effects of compounds on mitochondrial inner membrane polarization in Mfn1 knockout (KO; top) and Mfn2 KO (bottom) cells. D. Representative confocal imaging of mitochondrial morphology and polarization status in Mfn2 null MEFs treated with different compounds (1 μM, 24 h). Green mitochondria are depolarized and have damaged respiratory function. Scale bars are 10 μm. MT Green (mitotracker green), TMRE (tetramethylrhodamine ethyl ester, red) and nuclear Hoescht (blue) were used for staining. Data are means ± SEM of three independent experiments.
Figure 3.

Diastereomer-selective MFN2 conformational switching by 13. A. FRET emission spectra of MFN2 in open active (white) and closed inactive (black) conformation. Schematic to the right depicts FRET-labelled MFN2 protein in closed (top) and open (bottom) conformations. C is N-terminus fluorphore cerulean; V is C-terminus fluorophore mVenus. B. FRET analysis of conformational switching provoked by mitofusin activators. “Peptide” is MP1 mitofusin agonist peptide described in reference17 and depicted in Fig 1A inset, used as a positive control. Data are mean ± SEM of six experiments.
Table 6.
In vitro functional and pharmacokinetic properties of 13 diastereomersα
| Cpd | Structure | EC50 (nM) Mfn1 KO | EC50 (nM) Mfn2 KO | hPPB (%) | mPPB (%) | t1/2 HLM (min) | t1/2 MLM (min) | PAMPA-BBB Pe (nm/sec) |
|---|---|---|---|---|---|---|---|---|
| 13A | 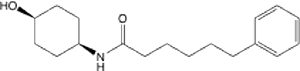 |
inactive | inactive | 93.9 | 93.8 | 102 | 70 | 42.6 |
| 13B | 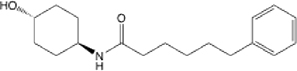 |
9.53 (6.35–14.02) | 7.69 (5.40–10.74) | 90.4 | 96.7 | >145 | 127 | 22.9 |
EC50 values are mean with 95% confidence limits.
Effects of Oxy-Substituted Linkers on Compound 13B SAR.
Our early SAR studies of 1 revealed that an ether linker (3) had similar potency for mitofusin activation with greater PAMPA-BBB permeability, compared to the straight carbon linker (2; Table 1). Accordingly, we synthesized the complete series of oxy-substituted linker analogs for 13B as trans- diastereomers (Table 7). All of these analogs retained excellent fusogenic activity, but the position of the oxygen within the linker had substantial effects on passive membrane permeability, with 27, the carbamate, having 10-fold greater PAMPA-BBB permeability compared to parent 13B
Table 7.
Functional and pharmacokinetic properties of oxy-substituted linker 13B analogsα
| Cpd | Structure | EC50 (nM) | hPPB (%) | mPPB (%) | t1/2 HLM (min) | t1/2 MLM (min) | PAMPA-BBB Pe (nm/sec) |
|---|---|---|---|---|---|---|---|
| 13B | 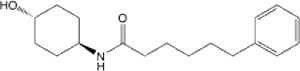 |
7.7 (5.40–10.74) | 90.4 | 96.7 | >145 | 127 | 22.9 |
| 23 | 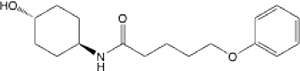 |
9.63 (6.59–13.84) | 80.2 | 81.8 | >145 | >145 | 3.66 |
| 24 | 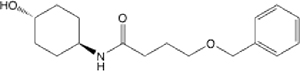 |
12.92 (8.56–18.81) | 57.1 | 60.4 | >145 | >145 | 1.176 |
| 25 | 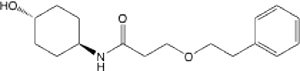 |
3.31 (1.87–5.52) | 38.2 | 48.5 | >145 | >145 | 2.228 |
| 26 | 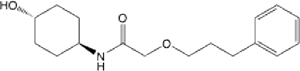 |
7.31 (3.33–23.51) | 55.2 | 66.4 | >145 | >145 | 23.799 |
| 27 | 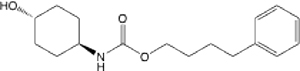 |
6.28 (4.24–9.07) | 93.3 | 94.8 | 131.3 | 48.9 | 210.149 |
EC50 values are mean with 95% confidence limits.
In Vitro and In Vivo Functional and Pharmacological Profiling of 13B.
The above results demonstrate that 13B engages its mitofusin protein target, exhibits low nM fusogenic potency in cultured cells, and has sufficiently high microsomal stability and passive membrane permeability to enter the central and peripheral nervous systems. 13B also exhibited a low efflux ratio in NIH MDR1 cells that did not change in the presence of the P-gp inhibitor GF120918 (Table S9), indicating that it is not a P-gp substrate.
Closely related mitofusin agonists 11, 12, and 13 exhibited a range of in vitro mouse liver microsomal stabilities (from 24 to > 145 seconds) and passive membrane permeability (PAMPA-BBB Pe from 7.96 to 26.28 nm/sec) (see Table 3). We had envisioned that microsomal stability would correlate with in vivo plasma t1/2, and passive permeability with CNS levels. This notion was examined by in vivo PK studies of the trans- diastereomers of these three compounds. In vivo PK was performed in mice, the only species in which pre-clinical models of human CMT2A have been published27–29. We assigned target therapeutic brain levels of 10 times the in vitro EC50 for each compound, or 30 ng/g (~100 nM) for 11 and 13B and 120 ng/g (~400 nM) for 12.
Comparative mouse plasma and brain levels were determined at increasing times after a single 10 mg/kg intravenous (IV) dose of the three compounds; calculated PK results are in Table 8. Consistent with its more rapid microsomal degradation, 11 plasma t1/2 was shorter than that of the other two compounds, evoking lower peak plasma and brain levels; peak 11 levels in the brain were only 366 ng/g and fell below the target level of 30 ng/g after 1 hour. 12, which was stable in the liver microsome assay but had the lowest PAMPA-BBB Pe of the three compounds, achieved a ~4-fold higher peak brain level of 1268 ng/g. However, because it was less potent than the other two compounds as a mitofusin activator (EC50 of ~40 nM vs ~10 nM), it also fell below its target therapeutic level after 1 hour. 11 and 12 exhibited similar volumes of distribution (Vdss), ~2 L/kg, while the Vdss of 13B was only 0.35 L/kg. Moreover, 13B peak brain levels after single dose IV bolus were 2,793 ng/g and therapeutic levels (of 30 ng/g) were maintained for greater than 2 hours after the single IV dose (Figure 4A).
Table 8.
In vivo pharmacokinetic properties of mitofusin activators in mice.
| Cpd 11 single dose; 10 mg/kg IV | |||||
| Sample site | t1/2 (h) | Cmax (ng/ml or ng/g) | AUC (ng.h/ml or ng.h/g) | CL (ml/min/kg) | Brain/plasma (total) |
| Plasma | 0.64 | 3647 | 2564 | 63.9 | |
| Brain | 1.62 | 366 | 207 | 0.081 | |
| Cpd 12 single dose; 10 mg/kg IV | |||||
| Sample site | t1/2 (h) | Cmax (ng/ml or ng/g) | AUC (ng.h/ml or ng.h/g) | CL (ml/min/kg) | Brain/plasma (total) |
| Plasma | 1.15 | 6100 | 4860 | 29.5 | |
| Brain | 1.74 | 1268 | 829 | 0.171 | |
| Cpd 13 single dose; 10 mg/kg IV | |||||
| Sample site | t1/2 (h) | Cmax (ng/ml or ng/g) | AUC (ng.h/ml or ng.h/g) | CL (ml/min/kg) | Brain/plasma (total) |
| Plasma | 1.1 | 15400 | 11657 | 14.3 | |
| Brain | 1.06 | 2793 | 1199 | 0.1 | |
| Cpd 13 steady state; 60 mg/kg/day × 3 days S.C. | |||||
| Sample site | t1/2 (h) | Cmax (ng/ml or ng/g) | AUC (ng.h/ml or ng.h/g) | Brain/plasma (total) | |
| Plasma | 1.33 | 710 | 926 | ||
| Brain | 3.37 | 340 | 342 | 0.37 | |
Figure 4.

13B PK studies in mice. A. Total plasma and brain concentrations after single dose IV injections. B. Steady state elimination kinetics after 3 days continuous subcutaneous infusion. Data are means ± differences of results from two mice of each time point.
Because of its low Vdss, high Pe, and low efflux in NIH MDR1 cells we surmised that 13B might accumulate in brains over time. If this were so, then brain levels after single dose administration might not reflect therapeutic efficacy during repeated dosing. To test this idea we used osmotic mini-pumps to deliver 13B subcutaneously (s.c.) at a daily dose of 60 mg/kg/day for three days to achieve steady state, removed the mini-pumps, and defined its elimination kinetics thereafter (Figure 4B). 13B plasma half-lives were similar after bolus IV and chronic s.c. administration (1.1h and 1.33h, respectively), but brain half-life was substantially longer after chronic infusion (3.37h vs 1.06h after IV). Accordingly, the brain/(total) plasma ratio was 3-fold higher for 13B than for 11 and 12 (Table 8). Because it is 96.7% plasma protein bound in mice (see Table 5), the unbound fraction (fu) of 13B is 0.033. Using this factor to convert plasma [13B]total to plasma [13B]free/unbound, the calculated ratio of brain to free plasma level is 10.8 after chronic infusion.
Together, the above results suggested that 13B might have properties enabling it to activate mitofusins of neuronal mitochondria in CMT2A mice in vivo. In the only published study of a mitofusin activator, the prototype compound chimera B-A/l (1) increased neuronal mitochondrial motility after topical ex vivo application to mouse sciatic nerves in short term organ culture18. In vivo effects of a mitofusin activator on CMT2A neuronal mitochondria have not been reported, likely because this class of mitofusin agonists has a very short in vivo plasma half-life of ~0.2 hour (see Table 2). Thus, we determined that 13B was >75% orally bioavailable (Figure 5A) and tested its ability to engage mitochondrial targets in vivo after a single oral dose. Transgenic mice expressing the human MFN2 T105M mutation in motor neurons18 received a single 50 mg/kg dose of 13B (5 mg/ml in 10% DMSO/90% [30%HP-b-CP]) administered by oral gavage. Mitochondrial motility in sciatic nerve neuronal axons, assessed by a blinded investigator 6 hours thereafter, was markedly increased as measured both the number and velocity of motile mitochondria (Figure 5B).
Figure 5.

Oral bioavailability and in vivo target engagement of 13B in mice. A. Plasma levels after single dose IV (closed circles) or oral (open circles) administration. Data are means ± SD of results from three mice for each condition. B. (left) Kymographs demonstrating mitochondrial motility in sciatic nerves of CMT2A mice 6 hours after oral administration as in A. Motile mitochondria exhibit horizontal displacement. To the right are group quantitative data. Results are means ± SD, each dot representing one axon from two mice per condition.
Off-target, specificity, and safety studies performed to determine the suitability of 13B for possible clinical translation (Table 9) demonstrated that it is a potent and selective mitofusin activator with a favorable drug profile. It has sub-10 nM potency for both Mfn1 and Mfn2, with very low inhibitory activity for cytochrome P450 enzymes, indicating a small likelihood for drug-drug interactions. Activity screening against hERG, hNAV1.5, hKCNQ, and a panel of 44 receptors/kinases revealed only mild inhibition of dopamine amino transferase (DAT) and monoamine oxidase (MAO-A; ~30% inhibition at 10 μM) (Table S1), indicating a safety window of 1,000-fold compared to on-target efficacy, and a correspondingly limited potential for off-target side-effects.
Table 9.
Summary of key properties of 13Bα.
| On-target potency | |
| EC50 Mfn1 KO cells (nM) | 9.5 (7.0–12.7) |
| EC50 Mfn2 KO cells (nM) | 7.7 (5.4–10.7) |
| Physical properties | |
| MW/cLogP/TPSA | 289/3.22/49.33Å |
| kinetic solubility (uM) | 175 |
| fsp3 (%) | 61 |
| Selectivity and safety profiles | |
| hNav1.5 (% inhib @10 uM) | 8.3 |
| hKCNQ (% inhib @10 uM) | 0 |
| hERG patch clamp IC50 (uM) | >30 |
| 44 receptor/kinase panel (10uM) | DAT and MAO-A > 30% inhib |
| CYP1A2/2C9/2C19/2D6/3A4-M IC50 (uM) | >50/>50/>50/>50/>50 |
| AMES test | negative |
EC50 are reported as mean value with 95% confidence limits of fitting curve from three independent experiments, AMES was done by triplicate.
CHEMISTRY
The syntheses of the compounds were accomplished as outlined in Schemes 1–6. Scheme 1 includes compounds 2 and 3 isosters of 1, the prototype small molecule mitofusin activator18. Compounds 2 and 3 were synthesized by a similar route as reported for 1 but with replacement of the sulfur linked intermediate 1b with either carbon or oxygen linked analogs. For 3, synthesis of intermediate 3b was required and it was prepared from commercially available 3c, 3H-1,2,4-triazol-3-one, 3-cyclopropyl-4-phenyl-4,5-dihydro-1H-1,2,4-triazol-5-one by coupling with a protected 2-hydroxyethanamine, as outlined. Commercially available 2b and intermediate 3b were coupled with 1-isocyanato-2-methylcyclohexane to afford the targets.
Scheme 1. Synthesis of 1 and Its Backbone Variantsα.

αReagents and conditions: (a) tert-butyl (2-hydroxyethyl)carbamate, DEAD, PPh3, THF, r.t., 1h. (b) HCl/Dioxane, r.t., 18 h. (c) 1 and 2: DCM, r.t., 18 h, 3: DCM, r.t., 30 min.
Scheme 6. Synthesis of 23–27α.

αReagents and conditions: (a) 23: DMF, DIPEA, HOBt, EDCI, 25 °C, 2 h. 24,25: HOBt, EDCI, DIPEA, DMF, 25 °C, 1 h; 26: T3P, TEA, DMF, 25 °C, 1 h; 27: DIPEA, DCM, 25 °C, 1 h;
The syntheses of the urea derivatives 4–9 outlined in Scheme 2 were all accomplished in one step by coupling of amino-triazoles with amines as indicated. The amino-triazoles are either commercially available 6a, 7a, 8a, 9a, or were prepared according to a reported procedure 4a, 5a.31 The two cyclohexanamines 4b/7b/8b or 5b and the 4-amino-tetrahydropyran 6b/9b are all commercially available. The coupling conditions used either Troc-Cl and DIPEA or CDI and TEA and proceeded via formation of an intermediate carbamoyl chloride that was not isolated.
Scheme 2. Synthesis of 4–9α.

αReagents and conditions: (a) 4: Troc-Cl, DIPEA, DMAP, DCM, 40 °C, 12 h, N2, 95 °C, 10h. 5: TEA, DCM, CDI, 0–20 °C, 14 h. 6,7: TEA, DCM, CDI, 5–10 °C, 3 h, 10–15 °C, 12 h. 8: Troc-Cl, DIEA, DCM, 0–20 °C, 1 h, DMF, 90 °C, 12h; 9: TEA, DCM, CDI, 5–10 °C, 3 h, 10–15 °C, 2 h;
The syntheses of urea derivatives 10, 11 and 12 were achieved as shown in Scheme 3. In all cases coupling of 4-phenylbutane-1-amine with the appropriate commercially available cyclohexanamine to directly afford the target compounds was achieved by sequential treatment of one amine with CDI and then the second amine, all in the presence of DIPEA.
Scheme 3. Synthesis of 10–12α.

αReagents and conditions: (a) 10–11: THF, DIPEA, CDI, 25 °C, 16 h, 12: THF, DIPEA, CDI, 25 °C, 23.5 h.
The syntheses of 6-phenylhexanamide 13, its pure stereoisomers and its cyclohexanamine analogs are shown in Scheme 4. In all cases the amides were formed by coupling of commercially available 6-phenylhexanoic acid and the specified cyclohexanamine with HOBt and EDCI in the presence of DIEA. For compounds 13, 13A, 13B, 14, 15, 16, 17 and 18 the required cyclohexanamines, 13a, 13Aa, 13Ba, 14a, 15a, 16a, 17a and 18a respectively, were commercially available. For compounds 13, 13A, 13B, 14, 17, 18 this reaction directly afforded the target compounds. As specified in Scheme 4, preparation of 15 and 16 additionally required treatment of the amide coupling product with methanolic HCl.
Scheme 4. Synthesis of 13–18α.

αReagents and conditions: (a) 13, 13A, 13B, 14, 17, 18: DIEA, DMF, HOBt, EDCI, 25 °C, 3–16 h; 15, 16: DIEA, DMF, HOBt, EDCI, 25 °C 12–16h, followed by HCl/MeOH 15 – 25 °C, 2–16 h.
The syntheses of compounds 19–22 are shown in Scheme 5. These compounds incorporate pyridine and pyrimidine replacements for the phenyl group of 13. In all cases the desired compounds are formed by coupling of the appropriate 6-heteroaryl substituted hexanoic acid, 19d-22d, with 4-aminocyclohexan-1-ol via use of HOBt, EDCI and DIEA in DMF. The three isomeric 6-[pyridin-X-yl]hexanoic acids 19d-21d were commercially available. However acid 22d, 6-(pyrimidin-4-yl)hexanoic acid, needed to be synthesized as shown in this Scheme. Coupling of 4-chloropyrimidine (22a) with methyl hex-5-ynoate in the presence of catalytic amounts of CuI and Pd(PPh3)2Cl2 afforded methyl hex-5-ynoate (22b), catalytic hydrogenation of which over Pd/C, followed by hydrolysis yielded 22d.
Scheme 5. Synthesis of 19–22α.

αReagents and conditions: (a) Pd(PPh3)2Cl2, Cul, TEA, DMF, 20 °C,16 h; (b) H2, Pd/C, MeOH, 20 °C, 4 h; (c) LiOH.H2O, THF, MeOH, 20 °C, 16 h; (d) 4-amino-cyclohexan-1-ol, HOBt, EDCI, DIEA, DMF, 25 °C, 16 h.
The syntheses of Compounds 23–27, analogs of 13 in which a single methylene in the pentanyl chain is replaced with oxygen are shown in Scheme 6. Compounds 23–26 were prepared by coupling the commercially available acids 23a-26a with (trans)-4-aminocyclohexan-1-ol under the conditions specified in the Scheme. The carbamate 27 was formed by the coupling of commercially available 4-phenylbutyl chloroformate 27a with (trans)-4-aminocyclohexan-1-ol in the presence of DIPEA.
CONCLUSIONS
In summary, a novel series of 6-phenylhexanamide derivative mitofusin activators was identified as having mitofusin activating properties. Pharmacophore-based rational design guided functional and pharmacokinetic optimization, leading to compound 13 that exhibited potent and stereoselective fusogenic effects. The active trans- diastereomer, compound 13B, had excellent cell permeability, microsomal stability, and in vivo PK, providing greatly improved pharmaceutical properties compared to its precursors. When systemically administered to a pre-clinical mouse model of CMT2A, compound 13B reversed characteristic mitochondrial dysmotility in sciatic nerve neurons, demonstrating in vivo target engagement in the pathophysiological relevant model and tissues. Preclinical profiling of compound 13B supports its potential to be the first disease-modifying treatment for CMT2A caused by MFN2 mutations. The discovery and development of pharmaceutically-acceptable mitofusin activators unlocks the possibility of a previously unexplored therapeutic approach for other mitochondrial diseases, improving overall mitochondrial function by enhancing mitochondrial dynamics.
EXPERIMENTAL SECTION
General Procedures and Instrumentation.
1–3 were synthesized and analyzed by Paraza Pharm, Inc18. 4–27 were synthesized and analyzed by WuXi Apptec Co. Ltd. NMR spectrometry method for 1–3 was described previously18. NMR spectrometry for 4–27 was carried out on a Bruker AVANCE NEO 400MHz with a 5 mm PABBO BB/19F-1H/D Z-GRD probe. Analytical HPLC for 1–3 used a XBridge C18 column (3.5μm, 4.6 × 30mm); Gradient: 0.2min at 95%A: 5%B, 5% B to 100% B in 1.8min, 100%B for 1min; the flow rate was 3 ml/min; Mobile phase A: 10mM ammonium formate in water (pH = 3.8); Mobile phase B: acetonitrile (no additive)18,31. Analytical HPLC for 4–27 used a Kinetex C18 LC Column (4.6X50 mm,5 μm) that ran at 50°C: Mobile phase A: 0.0375% TFA in water (v/v), B: 0.01875% TFA in Acetonitrile (v/v). Preparative HPLC methods are described in synthesis procedures for each compound. LC-MS of 1–3 was performed by Paraza Pharm, Inc as described previously.18 LC-MS/MS (ESI) of 4–27 was performed by using two different systems: 1) SHIMADZU LC-MS-2020 instrument and data analyzed with LabSolution Version 5.72 software. HPLC used a Chromolith@Flash RP-18E 25*2.0 MM column that ran at 50°C and a PDA (220&254nm) detector. Data was acquired in scan MS Mode (positive mode) and selected with a scan range of m/z = 100–1000. The drying Gas (N2) flow was 15 L/min, the DL Voltage was 120(V) and the Qarray DC Voltage 20(V). 2) Agilent 1200\G6110A instrument and data analyzed with Agilent ChemStation Rev. B. 04.03[54] software. The HPLC contained a Xbridge C18 2.1*50 mm, 5 μm column that ran at 40 °C and a DAD (220 nm) / ELSD detector. Data was acquired in scan MS Mode (positive mode) and selected with a scan range of m/z = 100–1000. The drying gas (N2) flow was 10 L/min, the capillary voltage was 2500(V), and the drying gas temperature was 350 °C and the nebulizer pressure 35 psi. All compounds are least 95% pure. 1–5 and 8–27 were validated from HPLC 220 nm absorbance peaks, 6 and 7 were validated from LC-MS 220 nm absorbance peaks as shown in Supporting Information. tPSA values were calculated using ChemDraw 19.0.
Synthesis and Characterization of Compounds 1–27
Starting materials whose preparation is not reported in this experimental section are known compounds purchased by Paraza Pharm, Inc and WuXi Apptec Co. Ltd from third party suppliers or were prepared by them as reported.31,32
1-(2-((5-cyclopropyl-4-phenyl-4H-1,2,4-triazol-3-yl)thio)ethyl)-3-(2-methylcyclohexyl)urea (1).
The synthetic procedure of 1 has been described previously.18 HPLC: Rt: 1.52 min; Purity: 99.4%. LCMS: MS cal.: 399.55; Mass found: [M+H] +: 400.2. 1H NMR (500 MHz, CDCl3) δ 7.59 – 7.51 (m, 3H), 7.34 – 7.30 (m, 2H), 5.63 (s) and 5.56 (s, 1H), 5.04 (s, 1H), 4.75 (s) and 3.79 (s, 1H), 3.57 (dq, J = 12.7, 6.5 Hz, 2H), 3.29 – 3.09 (m, 2H), 1.98 – 1.02 (m, 12H), 0.98 – 0.90 (m, 4H), 0.85 (d, J = 7.0 Hz, 1H). 13C NMR (126 MHz, CDCl3) δ 158.4, 158.2, 157.9, 151.2, 151.0, 133.3, 133.3, 130.0, 127.2, 127.2, 55.0, 50.3, 40.7, 40.6, 38.9, 34.7, 34.5, 34.0, 32.8, 32.5, 30.2, 26.0, 25.8, 19.4, 8.10, 6.14.
1-(3-(5-cyclopropyl-4-phenyl-4H-1,2,4-triazol-3-yl)propyl)-3-(2-methylcyclohexyl)urea (2).
A DCM (5.84 mL) solution of 1-isocyanato-2-methylcyclohexane (406 mg, 2.92 mmol) was added to a DCM (5.84 mL) solution of 3-(5-cyclopropyl-4-phenyl-4H-1,2,4-triazol-3-yl)propan-1-amine (2b, 708 mg, 2.92 mmol) and the reaction was stirred for 18 h at rt, then water (20 mL) was added, the organic layer was separated, dried over anhydrous Na2SO4 and concentrated under reduced pressure. The crude was purified by flash column chromatography (silica 50 g, 5 to 50% MeOH/DCM) affording 2 (750 mg, 67 % yield) as a white powder. HPLC: Rt: 1.43 min; Purity: 98.8%; LCMS: MS cal.: 381.51; Mass found: [M+H] +: 382.2. 1H NMR: (500 MHz, CDCl3) δ 7.59 – 7.51 (m, 3H), 7.28 – 7.26 (m, 1H), 7.26 – 7.25 (m, 1H), 5.67 (s) and 5.61 (s, 1H), 5.07 (d, J = 8.0 Hz) and 4.87 (d, J = 8.9 Hz, 1H), 3.80 (m) and 3.31 – 3.16 (m, 3H), 2.60 (td, J = 7.2, 5.1 Hz, 2H), 1.98 – 1.01 (m, 14H), 0.94 – 0.86 (m, 4H), 0.84 (d, J = 7.0 Hz, 1H). 13C NMR: (126 MHz, CDCl3) δ 158.8, 158.7, 156.5, 154.8, 154.7, 133.7, 133.7, 130.1, 129.9, 127.2, 54.69, 39.5, 39.4, 39.1, 34.7, 34.6, 28.0, 26.0, 25.8, 22.8, 19.4, 7.92, 5.97.
1-(3-(5-cyclopropyl-4-phenyl-4H-1,2,4-triazol-3-yl)propyl)-3-(2-methylcyclohexyl)urea (3).
Tert-butyl (2-hydroxyethyl)carbamate (65.1 μL, 0.412 mmol) was added to a THF (2.06 mL) solution of 5-cyclopropyl-4-phenyl-4H-1,2,4-triazol-3-ol (3c, 83.0mg, 0.412mmol). Then PPh3 (162 mg, 0.619 mmol) and DEAD (100 μL, 0.619 mmol) were added and the reaction was stirred for 1 h at r.t. The resulting mixture was concentrated under reduced pressure and the crude was purified by flash column chromatography affording tert-butyl (2-((5-cyclopropyl-4-phenyl-4H-1,2,4-triazol-3-yl)oxy)ethyl)carbamate (3a, 110 mg, 77 %) as a yellow oil. A solution of hydrochloric acid (4 M in dioxane, 798 μL, 3.19 mmol) was added to a dioxane (1.60 mL) solution of tert-butyl (2-((5-cyclopropyl-4-phenyl-4H-1,2,4-triazol-3-yl)oxy)ethyl)carbamate (3a) and the reaction was stirred at r.t. for 18 h, the resulting mixture was concentrated under reduced pressure, before using the material in the next step, an excess of Et3N was added in the crude mixture dissolved in DCM, a DCM (1.0 mL) solution of 1-isocyanato-2-methylcyclohexane (44.4 mg, 0.319 mmol) was added to a DCM (1.0 mL) solution of deprotected amine 2-((5-cyclopropyl-4-phenyl-4H-1,2,4-triazol-3-yl)oxy)ethan-1-amine (3b, 77.9 mg, 0.319 mmol) and the reaction was stirred for 30 min at r.t., then water (10 mL) was added and the organic layer was separated, dried over anhydrous Na2SO4 and concentrated under reduced pressure, the residue was purified by flash column chromatography (silica 25 g, 2 to 50% MeOH/DCM) affording 3 (90 mg, 74 % yield) as a white powder. HPLC: Rt: 1.47 min; Purity: 98.6%; LCMS: MS cal.: 383.49; Mass found: [M+H] +: 384.1. 1H NMR: (500 MHz, CDCl3) δ 7.55 – 7.39 (m, 5H), 5.27 (s) and 5.22 (s, 1H), 4.33 (d, J = 9.0 Hz) and 4.07 (d, J = 9.2 Hz, 1H), 3.99 – 3.92 (m, 2H), 3.73 (s, 0.18H), 3.63 – 3.54 (m, 2H), 3.20 – 3.07 (m, 0.69H), 2.01 – 0.81 (m, 17H). 13C NMR: (101MHz, CDCl3) δ 158.01, 157.96, 153.80, 147.71, 132.84, 129.47, 128.74, 126.84, 54.97, 50.28, 46.04, 45.99, 39.77, 38.84, 34.40, 34.37, 33.90, 30.13, 30.01, 25.71, 25.51, 23.43, 22.16, 19.17, 16.21, 7.21, 6.69.
1-(3-(5-cyclopropyl-4-(4-fluorophenyl)-4H-1,2,4-triazol-3-yl)propyl)-3-(2-methylcyclohexyl)urea (4):
A mixture of 3-(5-cyclopropyl-4-(4-fluorophenyl)-4H-1,2,4-triazol-3-yl)propan-1-amine (4a, 0.600 g, 1.92 mmol, HCl), Troc-Cl (488 mg, 2.30 mmol, 309 uL), DIPEA (745 mg, 5.76 mmol, 1.00 mL), DMAP (11.7 mg, 96.0 umol, 0.05 eq) and DCM (20 mL) was stirred at 40 °C for 12 h, then filtered and concentrated in vacuo to afford intermediate product as a gray solid (0.600 g, 1.28 mmol, 66.5% yield, 92.8% purity), to this was then added 2-Methylcyclohexanamine (4b, 181 mg, 1.60 mmol, 211 μL, 1.5 eq) in DMSO (3 mL), the mixture was degassed and purged with N2 for 3 times, and then stirred at 95 °C for 10 h under N2, the mixture was concentrated and purified by Reverse Phase HPLC (column: Phenomenex Synergi Max-RP 250*50 mm*10 μm; mobile phase: [water (0.225%FA)-ACN]; B%: 10ACN%−50ACN%, 35 min), the eluent was concentrated to remove most of MeCN, the aqueous phase was adjusted to pH = 8 by addition NaHCO3 solution and extracted with DCM (50 mL*3), the combined organic phases were dried over Na2SO4, filtered and concentrated in vacuo to afford 4 (0.180 g, 448 μmol, 42.1% yield) as a yellow solid. HPLC: Rt: 2.42 min; Purity: 99.4%; LCMS: MS cal.: 399.55; Mass found: [M+H] +: 400.2. Structure was confirmed by 1H NMR and 13C NMR. 1HNMR: (400 MHz DMSO-d6) δ 7.61 – 7.53 (m, 2H), 7.48 – 7.39 (m, 2H), 5.80 – 5.53 (m, 2H), 2.99 – 2.91 (m, 2H), 2.48 – 2.40 (m, 2H), 1.73 – 1.44 (m, 6H), 1.41 – 1.07 (m, 5H), 1.02 – 0.77 (m, 8H), 0.74 (d, J = 6.8 Hz, 1H). (d, J = 6.8 Hz, 1H).
1-(3-(5-cyclopropyl-4-(4-fluorophenyl)-4H-1,2,4-triazol-3-yl)propyl)-3-(4-fluorocyclohexyl)urea (5):
To a solution of 4-fluorocyclohexan-1-amine (5b, 101 mg, 866 μmol, 1.00 eq, HCl) and TEA (438 mg, 4.33 mmol, 602 μL) in DCM (10.0 ml) was added CDI (182 mg, 1.13 mmol). The reaction mixture was stirred at 0 °C for 2 h, then to a solution of 3-(5-cyclopropyl-4-(4-fluorophenyl)-4H-1,2,4-triazol-3-yl)propan-1-amine (5a, 257 mg, 866 μmol, HCl) and TEA (438 mg, 4.33 mmol, 602 μL) in DCM (10.0 mL) was added to the reaction mixture and stirred at 20 °C for 12 h, then the reaction mixture was filtered and concentrated under reduced pressure to give a residue, that was purified Reverse Phase HPLC (column: Phenomenex Synergi C18 150*25 mm*10 μm; mobile phase: [water (0.225%FA)-ACN]; B%: 30%−60%, 9min) to obtain 5 (190 mg, 54.1% yield) as a yellow solid. HPLC: Rt: 1.596 min, purity: 99.37%. LCMS: Rt = 0.805min, MS (M+H+): 404.2. HNMR: 400 MHz DMSO.δ7.54–7.58 (m, 2H), 7.41–7.46 (m, 2H), 5.80–5.82 (d, J = 7.6Hz, 1H), 6.75 (s, 1H), 5.71 (s, 1H), 4.64–4.77 (d, J = 48.8Hz, 1H), 2.92–2.97 (q, J1 = 6.4 Hz, J2 = 12.8Hz, 2H), 2.42–2.46 (t, J = 8.0Hz, 2H), 1.80 (m, 2H), 1.60–1.63 (m, 8H), 1.48–1.54 (m, 2H), 0.88–0.90 (m, 2H), 0.81–0.83 (m, 2H). 13CNMR: DMSO, 13C δ163.87, 161.41, 157.82, 155.98, 154.19, 130.61, 130.59, 130.32, 130.23, 117.34, 117.15, 89.74, 88.08, 46.51, 38.93, 29.42, 29.22, 28.02, 27.83, 22.65, 22.47, 7.40, 5.97.
1-(3-(5-cyclopropyl-4-phenyl-4H-1,2,4-triazol-3-yl)propyl)-3-(tetrahydro-2H-pyran-4-yl)urea (6):
To a solution of tetrahydro-2H-pyran-4-amine (6b, 130 mg, 1.29 mmol, 320 uL) and TEA (370 mg, 3.66 mmol, 509 μL) in DCM (5.00 mL) was added CDI (217 mg, 1.34 mmol) and the reaction mixture was stirred at 5 ~ 10 °C for 3 h, then 3-(5-cyclopropyl-4-phenyl-4H-1,2,4-triazol-3-yl)propan-1-amine (6a, 380 mg, 1.36 mmol, HCl) was added to the reaction mixture and stirred at 10 ~ 15°C for 12 h, the reaction mixture was concentrated under reduce pressure to give a residue that was purified by Reverse Phase HPLC (column: Xtimate C18 150*25 mm*10 μm; mobile phase: [water (0.05% ammonia hydroxide v/v)-ACN]; B%: 15%−45%, 8min) to obtain 6 (85.96 mg, 233 μmol, 17.1% yield) as a white gum. LCMS: MS cal.: 369.5; Mass found: [M+H] +: 370.2, Rt: 1.38 min; Purity: 99.4% at 220 nm absorbance. 1H NMR: (400 MHz DMSO) δ 7.64–7.57 (m, 3H), 7.49–7.47 (t, J = 4.0 Hz, 3H), 5.81–5.72 (m, 3H), 3.80–3.75 (m, 2H), 3.55–3.45 (m, 1H), 3.32–3.28 (m, 2H), 2.97–2.92 (m, 2H), 2.47–2.43 (t, J = 7.6 Hz, 2H), 1.68–1.61 (m, 2H), 1.30–1.20 (m, 2H), 0.91–0.82 (m, 2H). 13C NMR: (101 MHz, DMSO-d6) δ 6.05 (s, 1 C) 7.55 (s, 1 C) 22.52 (s, 1 C) 27.76 (s, 1 C) 33.90 (s, 1 C) 38.67–41.26 (m, 1 C) 45.81 (s, 1 C) 66.41 (s, 1 C) 127.85 (s, 1 C) 129.63–130.77 (m, 1 C) 134.28 (s, 1 C) 153.71–158.20 (m, 1 C).
1-(3-(4-methyl-5-phenyl-4H-1,2,4-triazol-3-yl)propyl)-3-(2-methylcyclohexyl)urea (7):
To a solution of 2-methylcyclohexan-1-amine (7b, 246 mg, 2.17 mmol, 286 μL) and TEA (627 mg, 6.20 mmol, 862 μL) in DCM (10.0 mL) was added CDI (368 mg, 2.27 mmol) and the reaction mixture was stirred at 5 ~ 10°C for 3 h, then 3-(4-methyl-5-phenyl-4H-1,2,4-triazol-3-yl)propan-1-amine (7a, 500 mg, 2.31 mmol, HCl) was added to the reaction mixture and stirred at 10 ~ 15°C for 12 h, the reaction mixture was concentrated under reduce pressure to give a residue, that was purified by Reverse Phase HPLC (column: Phenomenex Synergi C18 150*25 mm*10 μm; mobile phase: [water (0.05% HCl)-ACN]; B%: 15%−35%, 10min) to obtain 7 (171.84 mg, 20.9% yield) as an off-white solid. HPLC: Rt: 2.76 min, purity: 99.7%. LCMS: Rt: 2.761 min, purity: 99.71% at 220 nm absorbance; Rt: 2.761 min, purity: 99.41% at 254 nm absorbance.1H NMR: 400 MHz DMSO δ 7.82–7.80 (t, J = 5.6 Hz, 2H), 7.80–7.68 (m, 3H), 3.76–3.75 (d, J = 4.0 Hz, 3H), 3.17–3.13 (m, 2H), 3.06–3.01 (m, 2H), 1.96–1.90 (m, 2H), 1.66–1.15 (m, 10H), 0.85–0.77 (m, 3H).
1-(2-(4-methyl-5-phenyl-4H-1,2,4-triazol-3-yl)ethyl)-3-(2 methylcyclohexyl)urea (8):
To a solution of Troc-Cl (976 mg, 4.61 mmol, 617 μL) and DIEA (1.35 g, 10.4 mmol, 1.82 mL) in DCM (20 mL) was added 2-(4-methyl-5-phenyl-4H-1,2,4-triazol-3-yl)ethan-1-amine (8a,1 g, 4.19 mmol, HCl) at 0°C, then stirred at 20°C for 1 h to give an intermediate product, to which was added 2-methylcyclohexan-1-amine (8b, 44.9 mg, 397 μmol, 52.3 μL), in DMF (2 mL), and then stirred at 90°C for 12 h, the reaction mixture was filtered, concentrated to afford a residue that was purified by Reverse Phase HPLC (column: Phenomenex Synergi C18 150*25 mm*10 μm; mobile phase: [water (0.225% FA)-ACN]; B%: 35%−49%, 10 min) to give 8 (0.152 g, 445 μmol, 77.0% yield) as a white solid. HPLC: Rt: 1.62 min, purity: 99.3%. 1H NMR: 400 MHz DMSO-d6 δ = 7.61 – 7.73 (m, 2 H), 7.49 – 7.59 (m, 3 H), 5.80 – 6.10 (m, 2 H), 3.59 (s, 3 H), 3.41 – 3.45 (m, 1 H), 2.95 – 3.07 (m, 1 H), 2.87 (t, J = 6.75 Hz, 2 H), 0.95 – 1.77 (m, 10 H), 0.83 (d, J = 6.50 Hz, 2 H), 0.74 – 0.79 (m, 1 H).
1-(3-(5-cyclopropyl-4-ethyl-4H-1,2,4-triazol-3-yl)propyl)-3-(tetrahydro-2H-pyran-4-yl)urea (9):
To a solution of 5-methyl-3,6-dihydro-2H-pyran-4-amine (9b, 260 mg, 2.26 mmol, 320 μL) and TEA (645 mg, 6.37 mmol, 887 μL) in DCM (10.0 mL) was added CDI (379 mg, 2.34 mmol) was added and the reaction mixture was stirred at 5 ~ 10°C for 3 h, then 3-(5-cyclopropyl-4-methyl-4H-1,2,4-triazol-3-yl)propan-1-amine (9a, 400 mg, 1.73 mmol, HCl) was added to the reaction mixture and stirred at 10 ~ 15°C for 12 h, the reaction mixture was concentrated under reduce pressure to give a residue, that was purified by Reverse Phase HPLC to obtain 9 (162.59 mg, 19.7% yield) as an off-white gum. HPLC: Rt: 1.29 min, purity: 99.0%. 1H NMR: (400 MHz DMSO) δ 3.77 (s, 3H), 3.66–3.64 (m, 1H), 3.43–3.42 (d, J = 4.4 Hz, 2H), 3.11–3.08 (t, J = 6.0 Hz, 2H), 2.28–2.26 (m 1H), 1.85–1.80 (m, 4H), 1.52–1.46(m, 2H), 1.21–1.14 (m, 4H), 0.80–0.74(m, 4H). 13C NMR: (101MHz, CDCl3) δ 159.52, 157.03, 154.67, 73.08, 70.74, 67.19, 65.22, 53.40, 48.61, 45.89, 38.58, 37.31, 33.74, 31.69, 29.78, 25.87, 21.47, 13.89, 12.12, 8.63, 8.21, 4.96.
1-(2-methylcyclohexyl)-3-(4-phenylbutyl)urea (10):
To a solution of 2-methylcyclohexan-1-amine (10a, 0.759 g, 6.70 mmol) and DIPEA (0.866 g, 6.70 mmol) in THF (10 mL) was added CDI (1.09 g, 6.70 mmol), the mixture was stirred at 25°C for 10 min, and to it was added 4-phenylbutan-1-amine (1.00 g, 6.70 mmol), stirred mixture at 25°C for 16 h. The crude product was purified by Reverse Phase HPLC, then the eluent was concentrated under reduced pressure, then the mixture was filtered, the filter cake was washed with saturated solution of NaHCO3 (20 mL x 2), then the filter cake was washed with H2O (20 mL x 2) to give 10 (110.12 mg, 379.50 μmol, 5.72% yield, 99.4% purity) as a white solid. HPLC: Rt: 2.273 min, purity: 99.4%. LC-MS: RT = 0.823 min, m/z = 289.0 (M + 1) +. 1H NMR: 400 MHz MeOD δ: 7.26–7.22 (m, 2H), 7.18–7.13 (m, 3H), 3.14 (t, J = 3.60 Hz 3H), 2.63 (t, J = 7.6 Hz, 2H), 1.89–1.86 (m, 1H), 1.78–1.53 (m, 5H), 1.53–1.46 (m, 2H), 1.35–1.01 (m, 5H), 1.07–0.91 (m, 3H). 13C NMR: 13C MeOD 161.222, 143.825, 129.509, 126.884, 56.169–48.164, 40.875, 40.093, 36.717, 35.935, 35.625, 31.149, 30.106, 27.122, 26.975, 19.809.
1-(4-hydroxy-2-methylcyclohexyl)-3-(4-phenylbutyl)urea (11):
To a solution of 4-amino-3-methylcyclohexan-1-ol (11a, 1.50 g, 11.6 mmol) and DIPEA (1.00 g, 7.74 mmol, 1.35 mL) in THF (10 mL), was added CDI (1.26 g, 7.74 mmol) the reaction mixture was stirred at 25°C for 10 min, then 4-phenylbutan-1-amine (1.16 g, 7.74 mmol, 1.22 mL) was added to the reaction mixture, stirred at 25°C for 16 h, the reaction mixture was poured into H2O (10 mL) and extracted with EtOAc (5 mL*3), the organic layers were dried over Na2SO4, filtered and concentrated under reduced pressure to give a residue, the residue was purified by Reverse Phase HPLC to get 11 (1.63 g, 5.16 mmol, 66.6% yield) as a light yellow solid. HPLC: Rt: 2.22 min, purity: 96.3%. LCMS: Rt : 1.988 min, m/z = 305.2(M+H)+. 1HNMR: 400 MHz CDCl3 δ: 7.20 – 7.06 (m, 5H), 5.34 (s, 1H), 3.94 – 3.73 (m, 1H), 3.65 – 3.50 (m, 1H), 3.10 (t, J = 7.2 Hz, 2H)), 2.55 (t, J = 7.2 Hz, 2H), 1.74 – 1.09 (m,10H), 0.83 (d, J = 6.8 Hz, 3H). 13CNMR: 400 MHz CDCl3 δ:142.13, 142.06, 128.37, 125.80, 77.45 – 76.61, 69.85, 48.46, 40.46, 38.44, 35.53, 33.79, 29.75, 28.67, 18.41, 18.37.
1-(4-hydroxycyclohexyl)-3-(4-phenylbutyl)urea (12):
To a solution of 4-phenylbutan-1-amine (1.0 g, 6.70 mmol 1.06 mL) and DIPEA (866 mg, 6.70 mmol, 1.17 mL) in THF (10 mL) was added CDI (1.09 g, 6.70 mmol, 1.0 eq) and the mixture stirred at 25°C for 15 min. To the reaction mixture was added 4-aminocyclohexan-1-ol (12a, 772 mg, 6.70 mmol) stirred at 25 °C for 23.5 h, the reaction mixture was concentrated and it was purified by Reverse Phase HPLC (column: Phenomenex luna C18 250*50 mm*10 μm; mobile phase: [water (0.05%HCl)-ACN]; B%: 25%−55%, 25 min) twice, the mixture was concentrated under reduced pressure and filtered, the filter cake was washed with NaHCO3 to give 12 (620 mg, 1.62 mmol, 69.0% yield) as white solid. HPLC: Rt: 1.73 min, purity: 99.8%. LCMS: Rt = 0.842 min, m/z = 291.1 (M+1)+. 1H NMR: 400 MHz MeOD δ: 7.26–7.22 (m, 2H), 7.18–7.13 (m, 2H), 3.53–3.42 (m, 2H), 3.12 (t, J = 7.0Hz,2H), 2.61 (d, J = 7.6Hz, 2H), 1.91 (d, J = 10,0Hz, 3H), 1.65–1.61 (m, 2H), 1.50–1.46 (m, 2H), 1.35–1.33(m, 2H), 1.25–1.20(m, 2H). 13C NMR: 400 MHz MeOD δ: 160.800, 143.788, 129.554, 129.430, 126.875, 70.654, 40.842, 36.688, 34.998, 32.558, 31.083, 30.069.
N-(4-hydroxycyclohexyl)-6-phenylhexanamide (13):
To a solution of 4-aminocyclohexan-1-ol (13a, 6.00 g, 39.6 mmol, HCl salt) and DIEA (14.0 g, 108 mmol, 18.8 mL) in DMF (60.0 mL) was added 6-phenylhexanoic acid (6.92 g, 36.0 mmol, 6.78 mL, 1.00 eq), HOBt (5.83 g, 43.2 mmol, 1.20 eq) and EDCI (10.3 g, 54.0 mmol, 1.50 eq), the mixture was stirred at 25 °C for 16 h, the mixture was filtered and concentrated the residue was purified by Reverse Phase HPLC (column: Phenomenex luna C18 250*80 mm*10 μm; mobile phase: [water (10 mM NH4HCO3)– CAN ]; B%: 38ACN%−68ACN%, 8.5 min), the appropriate fractions were concentrated and the residue was triturated with petroleum ether / ethyl acetate = 5 / 1 (240 mL) at 25 °C for 5 mins to obtain 13 as an off-white solid (3.53 g, 12.0 mmol, 33.9% yield). HPLC: Rt = 1.863 min; purity = 96.6% [220 nm, top], 97.3% [254 nm, bottom] as shown in Figure S2. HRMS: Rt = 1.884 min, m/z = 290.2128 (M+H)+ as shown in Figure S2. 1H NMR: 400 MHz MeOD δ 7.25 – 7.22 (m, 2H), 7.16 – 7.11 (m, 3H), 3.59 – 3.48 (m, 2H), 2.60 (t, J = 7.6 Hz, 2H), 2.13 (t, J = 7.4 Hz, 2H), 1.92 – 1.65 (m, 4H), 1.65 – 1.59 (m, 4H), 1.35 – 1.29 (m, 6H) as shown in Figure S2. 13C NMR: 400 MHz MeOD δ 175.669, 143.887, 129.595, 126.817, 70.605, 37.240, 36.869, 34.973, 32.517, 31.668, 29.814, 27.102 as shown in Figure S2.
N-(cis-4-hydroxycyclohexyl)-6-phenylhexanamide (13A):
To a solution of 6-phenylhexanoic acid (200 mg, 1.04 mmol, 196 μL, 1.00 eq) and DIEA (403 mg, 3.12 mmol, 544 μL, 3.00 eq) in DMF (2.00 mL) of (cis)-4-aminocyclohexan-1-ol (13A-1, 132 mg, 1.14 mmol), HOBt (169 mg, 1.25 mmol) and EDCI (299 mg, 1.56 mmol) was added, the mixture was stirred at 25 °C for 16 h, LCMS showed that 13A-1 was consumed and the desired product MS (Rt = 1.884 min) was detected, the mixture was filtered and purified over a Reverse Phase HPLC (column: Waters Xbridge 150*25 mm*5 μm; mobile phase: [water (10 mM NH4HCO3)-ACN]; B%: 26%−56%,10 min), 13A was obtained as a light yellow gum (209.9 mg, 701 μmol, 67.4% yield). HPLC: Rt = 1.860 min; purity = 96.8% [220 nm, top], 98.4% [254 nm, bottom] as shown in Figure S3. LCMS: Rt = 1.884 min, m/z = 290.1 (M+H)+ as shown in Figure S3. 1H NMR: 400 MHz MeOD δ 7.25 – 7.22 (m, 2H), 7.17 – 7.11 (m, 3H), 3.84 (s, 1H), 3.70 – 3.65 (m, 1H), 2.60 (t, J = 7.6 Hz, 2H), 2.16 (t, J = 7.2 Hz, 2H), 1.73 – 1.65 (m, 3H), 1.65 – 1.59 (m, 9H), 1.36 – 1.34 (m, 2H) as shown in Figure S3. 13C NMR: 400 MHz MeOD δ 175.628, 143.895, 129.579, 129.414, 126.809, 66.945, 37.182, 36.885, 32.558, 32.179, 29.863, 28.074, 27.168 as shown in Figure S3.
N-(trans-4-hydroxycyclohexyl)-6-phenylhexanamide (13B):
To a solution of (trans)-4-aminocyclohexan-1-ol (13B-1, 200 mg, 1.04 mmol, 196 μL,) and DIEA (403 mg, 3.12 mmol, 544 μL, 3.00 eq) in DMF (2.00 mL) was added 6-phenylhexanoic acid (132 mg, 1.14 mmol, 1.10 eq), HOBt (169 mg, 1.25 mmol, 1.20 eq) and EDCI (299 mg, 1.56 mmol, 1.50 eq). The mixture was stirred at 25 °C for 16 h. The mixture was filtered and purified by Reverse Phase HPLC (column: Waters Xbridge 150*25 mm*5 μm; mobile phase: [water (10 mM NH4HCO3)-ACN]; B%: 26%−56%, 10 min). 13B (243.82 mg, 810 μmol, 77.8% yield) was obtained as a white solid. HPLC: Rt = 1.857 min; purity = 96.1% [220 nm, top], 99.0% [254 nm, bottom] as shown in Figure S3. LCMS: Rt = 1.886 min, m/z = 290.2 (M+H)+ as shown in Figure S3. 1H NMR: EW17569–19-P1B3 400 MHz MeOD δ 7.25 – 7.22 (m, 2H), 7.16 – 7.13 (m, 3H), 3.63 – 3.48 (m, 2H), 2.60 (t, J = 7.2 Hz, 2H), 2.13 (t, J = 7.2 Hz, 2H), 1.95 – 1.83 (m, 4H), 1.63 – 1.59 (m, 4H), 1.35 – 1.28 (m, 6H). 13C NMR: EW17569–19-P1C1 400 MHz MeOD δ 175.661, 143.887, 129.587, 129.405, 126.809, 70.597, 37.240, 36.851, 34.973, 32.493, 31.660, 29.814, 27.085.
6-phenyl-N-(tetrahydro-2H-pyran-4-yl)hexanamide (14):
To a solution of 6-phenylhexanoic acid (0.30 g, 1.56 mmol, 294 μL) in DMF (3.0 mL) of HOBt (253 mg, 1.87 mmol) and EDCI (448 mg, 2.34 mmol), DIEA (403 mg, 3.12 mmol, 543 μL) was added, then tetrahydro-2H-pyran-4-amine (14a, 173 mg, 1.72 mmol) was added to the mixture, the mixture was stirred at 25°C for 3 h, the mixture was concentrated under reduced pressure to give the residue, the residue was purified by Reverse Phase HPLC (column: Phenomenex luna C18 150*40 mm*15 μm; mobile phase: [water (0.225%FA)-ACN]; B%: 28%−58%, 8.5 min) to obtain 14 (169.36 mg, 713 umol, 38.7% yield) was obtained as a white solid. HPLC: Rt: 2.56 min, purity: 98.3%. LCMS: Rt = 0.861 min, m/z = 276.0 (M+H) +. 1H NMR: 400 MHz DMSO-d6 δ: 7.72 (d, J =7.58 Hz, 1 H) 7.08 – 7.33 (m, 5 H) 3.65 – 3.86 (m, 3 H) 3.31 – 3.36 (m, 2 H) 2.55 (t, J =7.64 Hz, 2 H) 2.03 (t, J =7.40 Hz, 2 H) 1.64 (m, 2 H) 1.53 (m, 4 H) 1.20 – 1.40 (m, 4 H). 13C NMR: 400 MHz DMSO δ: 171.713, 142.682, 128.725 – 128.651, 126.043, 66.383, 45.129, 35.811 – 35.526, 33.039, 31.213, 28.661, 25.629.
N-(4-aminocyclohexyl)-6-phenylhexanamide (15):
To a solution of 6-phenylhexanoic acid (500 mg, 2.60 mmol, 490 uL) and tert-butyl (4-aminocyclohexyl)carbamate (15a, 613 mg, 2.86 mmol) in DMF (5.00 mL) of HOBt (422 mg, 3.12 mmol), DIEA (672 mg, 5.20 mmol, 906 μL) and EDCI (748 mg, 3.90 mmol) was added at 25°C, the mixture was stirred at 25°C for 12 h, tert-butyl (4-(6-phenylhexanamido)cyclohexyl)carbamate (782 mg, 2.01 mmol, 77.4% yield) was obtained as a yellow solid. To solution of tert-butyl (4-(6-phenylhexanamido)cyclohexyl)carbamate (300 mg, 772 μmol, 1.00 eq) in MeOH (3.0 mL) of HCl/MeOH (4 M, 6.0 mL, 31.1 eq) was added, the mixture was stirred at 15°C for 2 h, LCMS showed that tert-butyl (4-(6-phenylhexanamido)cyclohexyl)carbamate was consumed and desired MS (Rt = 0.921 min) was detected, the mixture was concentrated under reduced pressure to give the residue, the residue was purified by Reverse Phase HPLC (column: Waters Xbridge 150*25 mm*5 μm; mobile phase: [water (10 mM NH4HCO3)-ACN]; B%: 14%−44%, 10 min) to obtain 15 (89.4 mg, 309 μmol, 40.1% yield) was obtained as a white gum. HPLC: Rt: 1.35 min, purity: 99.8%. LCMS: Rt = 0.712 min, m/z = 289.1 (M+H) +. SFC: Rt = 1.761 min, Rt = 1.945 min. 1H NMR: 400 MHz CDCl3 δ: 7.19 – 7.16 (m, 5H), 5.57 – 5.21 (m, 1H), 3.96 – 3.70 (m, 1H), 2.95 – 2.91 (m, 1H), 2.63 – 2.59 (m, 2H), 2.16 – 2.12 (m, 2H), 1.68 – 1.61 (m, 12H), 1.40 – 1.34 (m, 4H). 13C NMR: 400 MHz CDCl3 δ: 172.155, 142.488, 128.354 −128.224, 125.167, 45.217, 36.962, 35.706, 31.271 – 31.112, 28.823, 27.935, 25.674.
1-(4-phenylbutyl)-3-(piperidin-4-yl)urea (16):
To a solution of 6-phenylhexanoic acid (500 mg, 2.60 mmol, 490 μL, 1.00 eq) and tert-butyl 4-aminopiperidine-1-carboxylate (16a, 573 mg, 2.86 mmol, 1.10 eq) in DMF (3.00 mL) was adde HOBT (422 mg, 3.12 mmol, 1.20 eq), EDCI (748 mg, 3.90 mmol, 1.50 eq) and DIEA (672 mg, 5.20 mmol, 906 μL, 2.00 eq) at 25°C, the mixture was stirred at 25°C for 12 h, intermediate was (740 mg, 1.98 mmol, 76.0% yield) was obtained as a yellow solid, then added HCl/MeOH (4 M, 12 mL, 24.3 eq). The mixture was stirred at 25°C for 16 h, then was concentrated under reduced pressure to give the residue. The residue was purified Reverse Phase HPLC (column: Waters Xbridge 150*25 mm*5 μm; mobile phase: [water (10 mM NH4HCO3)-ACN]; B%: 11%−41%, 10 min) to obtain 16 (418.27 mg, 1.45 mmol, 73.4% yield) as a white solid. HPLC: Rt: 1.42 min, purity: 95.2%. LCMS: Rt = 0.708 min, m/z = 276.3 (M+H) +. 1H NMR: 400 MHz MeOD δ: 7.26 – 7.22 (m, 2H), 7.18 – 7.12 (m, 3H), 3.59 – 3.55 (m, 1 H), 3.12 (t, J = 6.8 Hz, 2H), 3.03 – 2.99 (m, 2H), 2.67 – 2.60 (m, 4H), 1.88 – 1.84 (m, 2H), 1.66 – 1.62 (m, 2H), 1.50 – 1.47 (m, 2H), 1.47 – 1.32 (m, 2H). 13C NMR: 400 MHz MeOD δ: 160.610, 143.800, 129.713, 129.582, 128.460, 126.900, 45.946, 40.810, 36.709, 34.337, 31.100, 30.114.
N-(4-methylcyclohexyl)-6-phenylhexanamide (17):
To a solution of 4-methylcyclohexan-1-amine (17a, 194 mg, 1.72 mmol, 227 μL) in DMF (5.0 mL) of HOBt (253 mg, 1.87 mmol), EDCI (448 mg, 2.34 mmol) and DIEA (403 mg, 3.12 mmol, 543 μL) was added, then 6-phenylhexanoic acid (0.30 g, 1.56 mmol, 294 μL, 1.00 eq) was added to the mixture, the mixture was stirred at 25°C for 3 h, LCMS showed 6-phenylhexanoic acid was consumed, and desired MS (Rt = 1.036 min) was detected, the reaction mixture was concentrated under reduced pressure to give a residue, the residue was purified by Reverse Phase HPLC (column: Phenomenex luna C18 150*40 mm*15 μm; mobile phase: [water (0.225%FA)-ACN]; B%: 52%−82%, 8.5 min) to obtain 17 (67.98 mg, 237 μmol, 15.2% yield) was obtained as a white solid. HPLC: Rt: 3.44 min, purity: 98.6%. LCMS: Rt = 0.999 min, m/z = 288.0 (M+H) +. SFC: Rt = 1.177 min, Rt = 1.308 min. 1H NMR: 400 MHz DMSO δ: 7.58 (d, J = 7.6 Hz, 5H), 7.27 – 7.13 (m, 5H), 3.47 – 3.40 (m, 1H), 2.56 – 2.50 (m, 2H), 2.10 – 2.01 (m, 2H), 1.56 – 1.49 (m, 9H), 1.30 – 1.25 (m, 5H), 0.89 – 0.84 (m, 4H). 13C NMR: 400 MHz DMSO δ: 171.335 – 171.050, 142.215, 128.233 −128.152, 125.551, 47.434, 35.108, 33.665, 32.450, 31.553, 30.819, 30.795, 29.401, 28.830, 28.227, 25.251, 22.186.
N-(4-fluoro-4-methylcyclohexyl)-6-phenylhexanamide (18) :
To a solution of 6-phenylhexanoic acid (300 mg, 1.56 mmol, 294 μL) in DMF (3.0 mL) of HOBt (253 mg, 1.87 mmol) and EDCI (448 mg, 2.34 mmol) DIEA (403 mg, 3.12 mmol, 543 μL, 2.00 eq) was added, then 4,4-difluorocyclohexan-1-amine (18a, 232 mg, 1.72 mmol) was added to the mixture, stirred at 25°C for 3 h, LCMS showed 18a was consumed and desired MS (Rt = 0.986 min) was detected, mixture was concentrated under reduced pressure to give a residue, the residue was purified by Reverse Phase HPLC (column: Phenomenex Synergi C18 150*25mm*10um; mobile phase: [water (0.225%FA)-ACN]; B%: 48%−78%, 11min) to obtain 18 (98.23 mg, 317 μmol, 20.0% yield) was obtained as a white solid. HPLC: Rt: 3.09 min, purity: 98.3%. LCMS: Rt = 0.933 min, m/z = 310.0 (M+H) +. 1H NMR: 400 MHz DMSO-d6 δ: 7.72 (d, J = 7.58 Hz, 1H), 7.13 – 7.28 (m, 5H), 3.72 (d, J = 7.58 Hz, 1H), 2.55 (t, J = 7.58 Hz, 2H), 2.01 – 2.06 (m, 2H), 1.68 – 2.00 (m, 6H), 1.48 – 1.61 (m, 4H), 1.39 – 1.48 (m, 2H), 1.24 (m, 2H). 13C NMR: 400 MHz DMSO-d6 δ: 171.949, 142.682, 128.733 – 128.659, 126.059, 45.072, 35.778, 35.518, 31.727, 31.197, 28.621, 28.482, 28.384, 25.620.
N-(4-hydroxycyclohexyl)-6-(pyridin-2-yl)hexanamide (19):
To a solution of 6-(pyridin-2-yl)hexanoic acid (19d, 1.09 g, 5.64 mmol) in DMF (10 mL) was added HOBt (914 mg, 6.77 mmol), EDCI (1.62 g, 8.46 mmol) and DIEA (2.19 g, 16.9 mmol, 2.95 mL), 4-aminocyclohexan-1-ol (940 mg, 6.20 mmol, 1.10 eq, HCl) was added to the mixture, stirred at 25 °C for 16 h, the residue was purified by Reverse Phase HPLC (column: Waters Xbridge C18 150*50 mm*10 μm; mobile phase: [water (10 mM NH4HCO3)-ACN]; B%: 7%−37%, 10 min) to obtain 19 (106.1 mg, 355 μmol, 6.30% yield) as an off-white solid. HPLC: Rt: 1.78 min, purity: 97.2%. LCMS: Rt: 0.752 min, m/z = 291.0 (M+H)+. 1H NMR: 400 MHz MeOD, δ 8.42 (d, J = 4.00 Hz, 1H), 7.77 – 7.73 (m, 1H), 7.31 – 7.30 (m, 1H), 7.25 – 7.23 (m, 1H), 3.61 – 3.48 (m, 2H), 2.80 – 2.76 (m, 2H), 2.16 – 2.12 (m, 2H), 1.91 – 1.91 (m, 2H), 1.72 – 1.70 (m, 2H), 1.65 – 1.63 (m, 2H), 1.61 – 1.61 (m, 2H), 1.37 – 1.33 (m, 4H), 1.32 – 1.26 (m, 2H). 13C NMR: 400 MHz MeOD. δ 175.538, 163.333, 149.597, 138.868, 124.829, 122.881, 70.591, 38.666, 37.133, 34.973, 31.671, 30.954, 29.861, 27.016.
N-(4-hydroxycyclohexyl)-6-(pyridin-3-yl)hexanamide (20):
To a solution of 6-(pyridin-3-yl)hexanoic acid (20d, 594 mg, 3.07 mmol) in DMF (10 mL) was added HOBt (498 mg, 3.69 mmol), EDCI (883 mg, 4.61 mmol) and DIEA (1.19 g, 9.22 mmol, 1.61 mL), then 4-aminocyclohexan-1-ol (512 mg, 3.38 mmol, HCl) was added to the mixture, the mixture was stirred at 25 °C for 16 h, LCMS showed that 20d was consumed and desired MS (Rt = 0.790 min) was detected, the reaction was diluted with H2O (20 mL) and extracted with ethyl acetate (10 mL * 3), the combined organic layers were washed with brine (10 mL * 3), dried over Na2SO4, filtered and concentrated under reduced pressure to give a residue, the mixture was purified by Reverse Phase HPLC (column: Waters Xbridge C18 150*50 mm*10 μm; mobile phase: [water (10 mM NH4HCO3)-ACN]; B%: 7%−37%, 10 min) to afford 20 (100.5 mg, 346 μmol, 11.2% yield) as an off-white solid. HPLC: Rt: 1.53 min, purity: 99.9%. LCMS: product: Rt = 0.763 min, m/z = 291.0 (M+H)+. 1H NMR: 400 MHz MeOD, δ 8.38 – 8.37 (m, 1H), 8.35 – 8.34 (m, 1H), 7.70 (d, J = 8.0 Hz, 1H), 7.36 – 7.33 (m, 1H), 3.59 – 3.49 (m, 2H), 2.68 – 2.65 (m, 2H), 2.16 – 2.12 (m, 2H), 1.92 – 1.92 (m, 2H), 1.68 – 1.65 (m, 2H), 1.65 – 1.61 (m, 4H), 1.36 – 1.35 (m, 4H), 1.32 – 1.26 (m, 2H). 13C NMR: 400 MHz MeOD δ 175.513, 150.200, 147.599, 140.262, 138.436, 125.286, 70.575, 37.109, 34.973, 33.734, 32.038, 31.671, 29.690, 26.926.
N-(4-hydroxycyclohexyl)-6-(pyridin-4-yl)hexanamide (21):
To a solution of 6-(pyridin-4-yl)hexanoic acid (21d, 100 mg, 517 μmol) in DMF (1.00 mL) of EDCI (149 mg, 776 umol), HOBt (83.9 mg, 621 μmol) and DIEA (201 mg, 1.55 mmol, 270 μL) was added, then 4-aminocyclohexan-1-ol (86.3 mg, 569 umol, HCl) was added to the mixture, stirred at 25 °C for 16 h, LCMS showed that 21d was consumed and desired MS (Rt = 0.758 min) was detected, mixture was purified by Reverse Phase HPLC (column: Waters Xbridge C18 150*25 mm*5 μm; mobile phase: [water (10 mM NH4HCO3)-ACN]; B%: 8%−38%, 10 min), 21 (43.73 mg, 149 μmol, 28.9% yield) was obtained as white solid. HPLC: Rt: 1.50 min, purity: 99.7%. LCMS: Rt = 0.748 min, m/z = 291.1 (M+H)+. 1H NMR: 400 MHz MeOD, δ 8.40 – 8.38 (m, 2H), 7.29 – 7.27 (m, 2H), 3.61 – 3.48 (m, 2H), 2.67 (t, J = 7.6 Hz, 2H), 2.14 (t, J = 7.2 Hz, 2H), 1.95 – 1.91 (m, 2H), 1.87 – 1.84 (m, 2H), 1.68 – 1.61 (m, 4H), 1.36 – 1.35 (m, 2H), 1.35 – 1.26 (m, 4H). 13C NMR: 400 MHz MeOD, δ 175.505, 154.685, 149.937, 125.853, 125. 853, 70.580, 37.100, 36.045, 34.965, 31.668, 31.182, 29.723, 26.896.
N-(4-hydroxycyclohexyl)-6-(pyrimidin-4-yl)hexanamide (22):
To a solution of 4-chloropyrimidine (22a, 1.00 g, 6.62 mmol, HCl), CuI (63.1 mg, 331 μmol), Pd(PPh3)2Cl2 (232 mg, 331 μmol) and TEA (2.01 g, 19.9 mmol, 2.77 mL) in DMF (10.0 mL) of methyl hex-5-ynoate (835 mg, 6.62 mmol, 1.00 eq) was added, mixture was stirred at 20 °C for 16 h, methyl 6-(pyrimidin-4-yl)hex-5-ynoate (22b, 740 mg, 3.33 mmol, 50.3% yield, 91.9% purity) was obtained as a yellow oil. To a solution of 22b (740 mg, 3.33 mmol, 1.00 eq) in MeOH (10.0 mL) was added Pd/C (74.0 mg, 10% purity) under N2, the mixture was stirred at 25 °C for 16 h under H2 (15 psi), methyl 6-(pyrimidin-4-yl)hexanoate (22c, 734 mg, crude) was obtained as yellow oil. To a solution of 22c (734 mg, 3.52 mmol, 1.00 eq) in THF (3.00 mL), MeOH (3.00 mL) and H2O (3.00 mL) was added LiOH.H2O (296 mg, 7.05 mmol, 2.00 eq), the mixture was stirred at 25 °C for 16 h, 6-(pyrimidin-4-yl)hexanoic acid (22d, 1.20 g, crude) was obtained as a yellow solid. To a solution of 22d (1.20 g, 6.18 mmol) in DMF (12.0 mL) was added EDCI (1.78 g, 9.27 mmol), HOBt (1.00 g, 7.41 mmol) and DIPEA (2.40 g, 18.5 mmol, 3.23 mL), then 4-aminocyclohexan-1-ol (1.03 g, 6.80 mmol, HCl) was added to the mixture, the mixture was stirred at 25 °C for 16 h, LCMS showed that 22d was consumed and desired MS (Rt = 0.689 min) was detected. The mixture was diluted with H2O (50 mL) and extracted with EtOAc (50 mL * 3)., the combined organic layers was dried over Na2SO4, filtered and concentrated under reduced pressure to give the residue, the residue was purified by Reverse Phase HPLC (column: Phenomenex Gemini C18 250*50 mm*10 μm; mobile phase: [water (10 mM NH4HCO3)-ACN]; B%: 10%−30%, 15 min), 22 (90.98 mg, 312 μmol, 5.05% yield) was obtained as off-white solid. HPLC: Rt: 1.14 min, purity: 99.9%. LCMS: Rt = 0.657 min, m/z = 292.1 (M+H)+. 1H NMR: 400 MHz MeOD, δ 9.02 (s, 1H), 8.65 (d, J = 5.2 Hz, 1H), 7.43 (d, J = 5.2 Hz, 1H), 3.59 – 3.48 (m, 2H), 2.79 (t, J = 7.6 Hz, 2H), 2.15 (t, J = 7.6 Hz, 2H), 1.95 – 1.92 (m, 2H), 1.88 – 1.84 (m, 2H), 1.80 – 1.72 (m, 2H), 1.68 – 1.60 (m, 2H), 1.40 – 1.35 (m, 2H), 1.33 – 1.19 (m, 4H). 13C NMR: 400 MHz MeOD, δ 175.475, 172.851, 159.152, 158.089, 122.490, 70.588, 38.344, 37.050, 34.965, 31.676, 29.781, 29.682, 26.888.
N-((1r,4r)-4-hydroxycyclohexyl)-5-phenoxypentanamide (23):
To a mixture of 5-phenoxypentanoic acid (23a, 400 mg, 2.06 mmol) and (trans)-4-aminocyclohexan-1-ol (266 mg, 2.27 mmol) in DMF (4.00 mL) was added DIPEA (532 mg, 4.12 mmol, 717 μL), HOBt (333 mg, 2.47 mmol) and EDCI (592 mg, 3.09 mmol), and then the mixture was stirred at 25 °C for 2 h, LCMS showed 23a was consumed and the desired MS (Rt = 0.848 min) was detected, the reaction mixture was quenched by addition water (0.50 mL) and concentrated under vacuum to get a residue, that was purified by Reverse Phase HPLC (column: Waters Xbridge C18 150*50 mm*10 μm; mobile phase: [water (10 mM NH4HCO3)-ACN]; B%: 20%−50%, 11.5 min) to give 23 (40.0 mg, 1.48 mmol, 7.17% yield) as an off - white solid. HPLC: Rt: 2.17 min, purity: 99.9%. LCMS: Rt = 0.830 min, m/z = 292.3 (M+H)+. 1H NMR: 400 MHz, MeOD. δ 7.25 (t, J = 8.0 16.4 Hz, 2H), 6.9 (d, J = 8.4Hz, 3H), 3.9 (m, 2H), 3.51 – 3.63 (m, 2H), 2.24 (s, 2H), 1.92 – 1.97 (m, 4H), 1.77 – 1.70 (m, 4H), 1.30 – 1.37 (m, 4H). 13C NMR: (400 MHz, MeOD) δ (176, 161, 131, 122, 116, 71.2, 69.2, 37.6, 35.6, 32.3, 30.7, 24.5) ppm.
4-(benzyloxy)-N-((1r,4r)-4-hydroxycyclohexyl)butanamide (24):
The mixture of 4-(benzyloxy)butanoic acid (24a, 500 mg, 2.57 mmol, 454 μL), DIPEA (664 mg, 5.14 mmol, 895 μL), HOBt (416 mg, 3.08 mmol), EDCI (739 mg, 3.86 mmol) and (trans)-4-aminocyclohexan-1-ol (325 mg, 2.83 mmol) in DMF (2.00 mL) was stirred at 25 °C for 1 h, LCMS showed that the 24a was consumed and desired MS (Rt = 0.786 min) was detected, the reaction mixture was quenched with water (0.10 mL), adjusted pH to 7 with 2 N HCl, filtered and the filtrate was concentrated, the crude product was purified by Reverse Phase HPLC (column: Waters Xbridge C18 150*50mm*10um; mobile phase: [water (10 mM NH4HCO3)-ACN]; B%: 16%−46%,11.5 min) and freeze-dried to afford 24 (40.0 mg, 137 umol, 5.33% yield) as an off-white solid. HPLC: Rt:1.80 min, purity: 99.8%. LCMS: Rt = 0.802 min, m/z = 292.3 (2M+Na)+. 1H NMR: (400 MHz, MeOD) δ 7.25 – 7.33 (m, 5H), 4.48 (s, 2H), 3.46 – 3.59 (m, 4H), 2.24 (t, J = 7.2, 2H), 1.82 – 1.92 (m, 6H), 1.20 – 1.33 (m, 4H). 13C NMR: (400 MHz, MeOD) δ (175, 139, 130, 129, 128, 74.0, 70.6, 70.5, 34.9, 34.1, 31.6, 27.2) ppm.
N-((1r,4r)-4-hydroxycyclohexyl)-3-phenethoxypropanamide (25):
The mixture of 3-phenethoxypropanoic acid (25a, 700 mg, 3.60 mmol), (trans)-4-aminocyclohexan-1-ol (621 mg, 5.40 mmol), HOBt (583 mg, 4.32 mmol), EDCI (1.04 g, 5.40 mmol), DIPEA (930 mg, 7.20 mmol, 1.25 mL) in DMF (7.00 mL) was stirred at 25 °C for 1 h, LCMS showed that 25a was consumed and the desired MS (Rt = 0.794 min) was detected, the reaction mixture was quenched with water (0.10 mL), and adjusted pH to 7 with 2 N HCl, filtered and the filtrate was concentrated, the crude product was purified by Reverse Phase HPLC (column: Waters Xbridge C18 150*50 mm*10 μm; mobile phase: [water (10 mM NH4HCO3)-ACN]; B%: 15%−45%,11.5 min) and freeze-dried to afford 25 (40.0 mg, 133 μmol, 3.72% yield) as a white solid. HPLC: Rt: 1.86 min, purity: 97.6%. LCMS: Rt = 0.789 min, m/z = 292.4 (M+H)+. 1H NMR: (400 MHz, MeOD) δ 7.17 – 7.27 (m, 5H), 3.66 – 3.70 (m, 4H), 3.49 – 3.64 (m, 2H), 2.84 (t, J = 6.8, 2H), 2.37 (t, J = 6.0, 2H), 1.82 – 1.94 (m, 4H), 1.17 – 1.34 (m, 4H). 13C NMR: (400 MHz, MeOD) δ (173, 140, 130, 129, 127, 73.0, 70.5, 68.1, 37.9, 37.2, 34.9, 31.5) ppm.
N-((1trans,4trans)-4-hydroxycyclohexyl)-2-(3-phenylpropoxy)acetamide (26):
The mixture of 2-(3-phenylpropoxy)acetic acid (26a, 1.00 g, 5.15 mmol), (trans)-4-aminocyclohexan-1-ol (652 mg, 5.67 mmol, 1.10 eq), T3P (2.29 g, 7.21 mmol, 2.14 mL, 1.40 eq) and TEA (1.56 g, 15.4 mmol, 2.15 mL, 3.00 eq) in DMF (10.0 mL) was stirred at 25 °C for 1 h, LCMS showed that 26a was consumed and desired MS (Rt = 0.797 min) was detected, the mixture was quenched with water (1.00 mL), adjusted pH to 7 with 2 N HCl, filtered and the filtrate was concentrated, the crude product was purified by Reverse Phase HPLC (column: Phenomenex Luna C18 75*30 mm*3 μm; mobile phase: [water (0.05%HCl)-ACN]; B%: 29%−49%,7.5 min) and freeze-dried to afford 26 (40.0 mg, 134 μmol, 2.62% yield) as a white solid. HPLC: Rt: 2.24 min, purity: 98.3%. LCMS: Rt = 0.825 min, m/z = 292.1 (M+H)+. 1H NMR: (400 MHz, MeOD) δ 7.24 – 7.28 (m, 2H), 7.15 – 7.20 (m, 3H), 3.88 (s, 2H), 3.68 – 3.70 (m, 1H), 3.53 – 3.56 (m, 1H), 3.50 (t, J = 6.4 Hz, 2H), 2.70 (t, J = 7.6 Hz, 2H), 1.91 – 1.96 (m, 4H), 1.86 – 1.87 (m, 2H), 1.33 – 1.38 (m, 4H). 13C NMR: (400 MHz, MeOD) δ (172, 143, 130, 129, 127, 72.1, 71.1, 70.4, 34.9, 33.3, 32.3, 31.4) ppm.
4-phenylbutyl ((1trans,4trans)-4-hydroxycyclohexyl)carbamate (27):
A solution of 4-phenylbutyl carbonochloridate (27a, 14.2 g, 66.7 mmol), (trans)-4-aminocyclohexan-1-ol (11.5 g, 100 mmol, 1.50 eq) and DIPEA (17.2 g, 133 mmol, 23.2 mL, 2.00 eq) in DCM (142 mL) was stirred at 25 °C for 1 h, LCMS showed that 27a was consumed and the desired MS (Rt = 0.884 min) was detected, the reaction mixture was quenched with water (150 mL) and extracted with DCM (100 mL * 3), the organic layer was washed with brine (200 mL * 4), and adjusted pH to 7 with 1 N HCl, dried over Na2SO4, filtered and the filtrate was concentrated, the crude product was purified by Reverse Phase HPLC (column: Phenomenex luna C18 150*40 mm* 15 μm; mobile phase: [water (0.05%HCl)-ACN]; B%: 31%−61%,11 min) to give 27 (35.0 mg, 119 μmol, 0.20% yield) was obtained as a white solid. HPLC: Rt: 2.54 min, purity: 99.7%. LCMS: Rt = 0.886 min, m/z = 292.2 (M+H)+. 1H NMR: (400 MHz, MeOD) δ 7.12 – 7.26 (m, 5H), 4.02 (t, J = 6.0, 2H), 3.47 – 3.52 (m, 1H), 3.33 – 3.35 (m, 1H), 2.63 (t,. J =7.2, 2H), 1.88 – 1.94 (m, 4H), 1.64 – 1.66 (m, 4H), 1.1.23 – 1.31 (m, 4H). 13C NMR: (400 MHz, MeOD) δ (158, 143, 130, 129, 126, 70.5, 65.6, 50.7, 36.5, 34.9, 31.9, 29.9, 29.1) ppm.
PAINS Analysis.
All reported compounds were screened for PAINS through FAF-Drugs4 website tool (https://mobyle.rpbs.univ-paris-diderot.fr/cgi-bin/portal.py#welcome) and none were identified as pan assay interference compounds.
Animals.
Rosa-STOP-mMFN Thr105Met (T105M) mice (C57BL/6 Gt(ROSA)26 Sortm1 (CAG-MFN2*T105M)Dple/J) from The Jackson Laboratory (Bar Harbor, Maine, USA; Stock No: 025322) were crossed to HB9-Cre mice (B6.129S1-Mnx1tm4(cre)Tmj/J) from The Jackson Laboratory (Stock No: 006600) to generate motor neuron-targeted MFN2 T105M mice. Mouse procedures were approved by the Institutional Animal Care and Use Committee of Washington University in St. Louis, Protocol ID: 19–0910. In vivo and in vitro pharmacokinetic analyses involved mouse and human tissues were approved by Institutional Committee Animal Care and Use Committee, Shanghai Site (IACUC-SH); (WuXi Corporate Committee for Animal Research Ethics (WX-CCARE)) and done by WuXi Apptec Co. Ltd. (Shanghai, China), 2mg/ml compounds were in 10%DMSO/90% (30%HP-b-CD, (2-Hydroxyproryl-ß-cyclodextrin; Sigma, 332607)) solution and administered to 7–9 week male CD-1 mice from SIPPR/BK Laboratory Animal Co. Ltd., Shanghai, China (15 mice per compound) by tail vein (10 mg/kg) or subcutaneously via osmotic mini-pump (60 mg/kg/day x three days).
Functional evaluation of mitofusin agonist fusogenicity was performed in MFN1- or MFN2-deficient MEFs purchased from ATCC (Cat #: CRL-2992, CRL-2993) and cultured at 37°C, 5% CO2-95% air in Dulbecco’s minimal essential medium (DMEM) containing glucose (4.5 g/l) with 10% (v/v) fetal bovine serum (FBS; Gibco, 26140–079), 1x nonessential amino-acids (Gibco, 11130051), 2 mM L-glutamine (Corning, 34717007), and 1% (v/v) penicillin/streptomycin (Gibco, 15140–122). For screening assays cells were seeded on day1 in 6 well plates at a density of 1×104 cells/ml and small molecules added at a final concentration of 1 μM (1:1,000 dilution from 1mM stick in DMSO) overnight; 1 μM 2 was included in all experiments as positive control. Mitochondria were stained with MitoTracker Orange (200 nM; M7510; Invitrogen, Carlsbad, CA, USA), nuclei were stained with Hoescht (10 μg/ml; Invitrogen, Thermo Fisher Scientific Cat: # H3570). Images were acquired at room temperature as previously described18 on a Nikon Ti Confocal microscope using a 60 X 1.3 NA oil- immersion objective in Krebs-Henseleit buffer (138 NaCl, 3.7 nM KCL, 1.2 nM KH2PO4, 15 nM Glucose, 20 nM HEPES pH: 7.2–7.5, and 1 mM CaCl2). Laser excitation was 549 nm with emission at 590 nm for MitoTracker Orange and excitation at 306 nm with emission at 405 nm for Hoescht. Images were analyzed using ImageJ30 and fusogenicity quantified as mitochondrial aspect ratio (length/width).
Dose-response studies were performed using 8 compound concentrations (0.5 nM-5 μM). Mitochondrial aspect ratios were indexed to the maximal response elicited by 2 or 13 and response curves interpolated using the sigmoidal model using Prism 8 software. EC50 values are reported as mean with 95% confidence limits for 3 or 4 experiments assessing ~300 mitochondria per condition.
Functional evaluation of mitofusin agonist effect on mitochondrial depolarization.
Cultured WT, Mfn2 KO, Mfn1 KO and DKO MEFs (Cat #: CRL-2994) treated with different compounds with 1 μM for 24 hours, then were stained with Tetramethylrhodamine ethyl ester (TMRE, 200 nM, Invitrogen Thermo Fisher Scientific Cat:# T-669), MitoTracker Green (200 nM; Invitrogen, Thermo Fisher Scientific Cat:# M7514) and Hoechst (10 ug/ml; Invitrogen, Thermo Fisher Scientific Cat:# H3570) for 30 min at 37°C in 5% CO2-95% air, washed twice in PBS. Images were acquired at room temperature on a Nikon Ti Confocal microscope using either 60 X 1.3 NA oil- immersion objective, in Krebs-Henseleit buffer (138 NaCl, 3.7 nM KCL, 1.2 nM KH2PO4, 15 nM Glucose, 20 nM HEPES pH: 7.2–7.5, and 1mM CaCl2): laser excitation was 488 nm with emission at 510 nm for MitoTracker Green, 549 nm with emission at 590 nm for TMRE, and 306 nm with emission 405 nm for Hoecsht. Mitochondrial depolarization was reported as % number of green mitochondria/ number of yellow+green mitochondria using Image J.
HB9-Cre/Mfn2 T105M mouse nerve motility studies.
With all investigators blinded, 12–14 week old male HB9-Cre/Mfn2 T105M CMT2A mice18 received 13 (50 mg/kg, 10%DMSO/90% (30% HP-b-CD), N = 2) or vehicle (N= 2) by oral gavage using sterile animal feeding needles (20Gx1.2”, Cadence Science, Cat#: 9931). After six hours the mice were sacrificed and their sciatic nerves dissected and placed in organ culture (Neurobasal Medium without phenol red, Thermo Fisher Scientific; room temp). Axonal mitochondria were labelled with tetramethylrhodamine ethyl ester (TMRE, Thermo Fisher Scientific, Cat#: T669; 200 μM, 30 mins, room temp) and time-lapse (1 frame/5 seconds) confocal microscopic images acquired at 37°C on a Nikon A1Rsi Confocal Microscope using a 40x oil immersion objective. Kymographs and quantitative data were generated using an Image-J plug-in18. Data are mean ± SD; p values were calculated using Student’s t-test.
FRET studies of MFN2 conformation were performed as described18 using hMFN2-FRET expressing mitochondria isolated from Mfn1, Mfn2 double null MEFs in a total volume of 100 μl 10 mM Tris-MOPS (pH 7.4), 10 mM EGTA/Tris, 200 mM sucrose in 96 well assay plate (Costar 3916). 5 μM of mitofusin agonist/ antagonist peptides and in H20, or 1 μM of 2, 13A, or 13B in DMSO were added for 1 and 4 hours; untreated mitochondria were controls. FRET (normalized to Cerulean) signals were acquired on a Tecan Safire II multi-mode plate reader: FRET – Excitation 433/8 nm, Emission 528/8 nm; Cerulean – Excitation 433/8 nm, Emission – 475/8 nm.
In vitro pharmacokinetic analyses of mitofusin agonists were performed in duplicate using standard methods by WuXi Apptec Co. Ltd. (Shanghai, China). Kinetic solubility was determined by UV absorbance of 1, 20, 200 μM solutions in 50 mM phosphate buffer, pH 7.4 shaken for 24 h at room temperature. Plasma protein binding was measured by equilibrium dialysis; % bound = (1-[free compound in dialysate]/[total compound in retentate]) X 100. Plasma stability of 2 μM compounds in clarified freeze-thawed plasma was assessed by LC-MS/MS of supernatants after protein precipitation; 120 min data are reported for studies including 0, 10, 30, 60, and 120 min. Microsome stability of 1 μM compounds in liver microsomes (0.5 mg/ml) after 0, 5, 10, 20, 30, 60 min. incubation was assessed by LC/MS/MS of reaction extracts. Passive artificial blood brain barrier membrane permeability assay (PAMPA-BBB) were performed using 150 μL of 10 μM compounds (5% DMSO) added to PVDF membranes pre-coated with 5 μL of 1% brain polar lipid extract (Porcine) /dodecane mixture and incubated for 4h at room temperature with shaking at 300 rpm. Donor and acceptor samples were analyzed by LC-MS/MS.
In vivo pharmacokinetic analyses were performed in duplicate or triplicate using standard methods by WuXi Apptec Co. Ltd. (Shanghai, China). Compounds were dissolved (2 mg/ml) in 10%DMSO/90% (30%HP-b-CD) and administered by tail vein (10mg/kg) or subcutaneously via osmotic mini-pump (60 mg/kg/day x three days) to 7–9 week male CD-1 mice (SLAC Laboratory Animal Co. Ltd., Shanghai, China or SIPPR/BK Laboratory Animal Co. Ltd., Shanghai, China, 15 mice were used for each compound). Blood and brain samples were analyzed in duplicate by LC-MS/MS. Plasma and brain concentration versus time data were analyzed by non-compartmental approaches using the Phoenix WinNonlin 6.3 software program, data are presented as mean from at least 3 mice for each condition.
In vitro bi-directional permeability across MDR1-MDCKI cell monolayer study:
Test compounds were diluted with the transport buffer (HBSS with 10 mM Hepes) from DMSO stock solution to a concentration of 2 μM (DMSO<1%) and applied to the apical or basolateral side of the cell monolayer. Reference compounds were diluted with transport buffer (HBSS with 10 mM Hepes, pH7.4) from stock solution to a concentration of 2 μM (DMSO<1%) and applied to the apical or basolateral side of the cell monolayer. Permeation of the test compounds from A to B direction or B to A direction was determined in duplicate with/without P-gp inhibitor (GF120918, 10 μM). Digoxin was tested at 10 μM in the presence or absence of 10 μM GF120918 bi-directionally as well, while nadolol and metoprolol were tested at 2 μM in the absence of GF120918 in A to B direction in duplicate. The plate was incubated for 1.5 hours in CO2 incubator at 37±1°C, with 5% CO2 at saturated humidity without shaking. In addition, the efflux ratio of each compound was also determined. Test and reference compounds were quantified by LC/MS/MS analysis based on the peak area ratio of analyte/IS. After transport assay, Lucifer yellow rejection assay are applied to determine the cell monolayer integrity. Buffers are removed from both apical and basolateral chambers, followed by the addition of 75 μL of 100 μM lucifer yellow in transport buffer and 250 μL transport buffer in apical and basolateral chambers, respectively. The plate is incubated for 30 minutes at 37°C with 5% CO2 and 95% relative humidity without shaking. After 30 minutes incubation, 20 μL of lucifer yellow samples are taken from the apical sides, followed by the addition of 60 μL of Transport Buffer. And then 80 μL of lucifer yellow samples are taken from the basolateral sides. The relative fluorescence unit (RFU) of lucifer yellow is measured at 425/528 nm (excitation/emission) with an Envision plate reader, data are presented as mean from two independent experiments.
Supplementary Material
ACKNOWLEDGEMENTS
Funding: Supported by the China Scholarship Council to XD and NIH R41NS113642, R41NS115184, R35135736, and Research Grant 628906 from the Muscular Dystrophy Association to GWD. GWD is the Philip and Sima K. Needleman-endowed Professor at Washington University in St. Louis and a Scholar-Innovator awardee of the Harrington Discovery Institute.
ABBREVIATIONS USED
- ADME
absorption, distribution, metabolism and excretion
- MFN
mitofusin
- CMT2A
Charcot-Marie-Tooth Disease type 2A
- PPI
peptide-peptide interactions
- PAMPA-BBB
passive artificial blood brain barrier membrane permeability assay
- CNS
central nervous system
- PK
pharmacokinetic
- tPSA
topological polar surface area
- IV
intravenous
- s.c.
subcutaneous
- DMSO
dimethyl sulfoxide
- DAT
dopamine amino transferase
- MAO-A
monoamine oxidase
- DCM
dichloromethane
- PPH3
triphenylphosphine
- DMF
dimenthylformamide
- r.t.
room temperature
- THF
Tetrahydrofuran
- DEAD
Diethyl azodicarboxylate
- DIPEA
N,N-Diisopropylethylamine
- CDI
1,1’-carbonyldiimidazole
- TEA
triethylamine
- EDCI
N-Ethyl-N′-(3-dimethylaminopropyl)carbodiimide hydrochloride
- MEFs
Mouse Embryonic Fibroblasts
- FRET
förster resonance energy transfer
- PBS
phosphate-buffered saline
- LC-MS
liquid chromatography tandem mass spectrometry
- NMR
nuclear magnetic resonance
- HPLC
high performance liquid chromatography
- HRMS
high resolution mass spectrometry
Footnotes
Competing interests: G.W.D. is an inventor on patent applications PCT/US18/028514 submitted by Washington University and PCT/US19/46356 submitted by Mitochondria Emotion, Inc that cover the use of small molecule mitofusin agonists to treat chronic neurodegenerative diseases, and is the founder of Mitochondria in Motion, Inc., a Saint Louis based biotech R&D company focused on enhancing mitochondrial trafficking and fitness in neurodegenerative diseases. The other authors declare no competing interests. Studies were performed under terms of an MTA between Mitochondria in Motion, Inc. and Washington University in St. Louis.
ASSOCIATED CONTENT
Supporting Information
Figure S1 (Correlation Between In Vitro Human Liver Microsome Stability and PAMPA-BBB Pe of 1 analogs); Figure S2 HPLC HRMS NMR chromatography of 13; Figure S3 HPLC LC-MS NMR chromatography of 13A and 13B; Table S1 (13B Selectivity Mini-panel Assay), Table S2-S7 (CYP Inhibition by 13B in Human Liver Microsomes), Table S8 (13B AMES Test), Table S9 (P-gp studies of 13B).
HPLC (1–5 and 8–27) or LC-MS (6 and 7) chromatography.
Molecular formula string for all the final compounds (CSV).
REFERENCES
- (1).Chen H; Chan DC Emerging functions of mammalian mitochondrial fusion and fission. Hum Mol Genet. 2005, 14, R283–289. [DOI] [PubMed] [Google Scholar]
- (2).Zorzano A; Sebastian D; Segales J; Palacin M The molecular machinery of mitochondrial fusion and fission: An opportunity for drug discovery? Curr Opin Drug Disc Deve.l 2009, 12, 597–606. [PubMed] [Google Scholar]
- (3).Muyderman H; Chen T Mitochondrial dysfunction in amyotrophic lateral sclerosis - a valid pharmacological target? Brit J Pharmacol. 2014, 171, 2191–2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Zuchner S; Mersiyanova IV; Muglia M; Bissar-Tadmouri N; Rochelle J; Dadali EL; Zappia M; Nelis E; Patitucci A; Senderek J; Parman Y; Evgrafov O; Jonghe PD; Takahashi Y; Tsuji S; Pericak-Vance MA; Quattrone A; Battaloglu E; Polyakov AV; Timmerman V; Schroder JM; Vance JM Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A. Nat. Genet. 2004, 36, 449–451. [DOI] [PubMed] [Google Scholar]
- (5).Feely SM; Laura M; Siskind CE; Sottile S; Davis M; Gibbons VS; Reilly MM; Shy ME MFN2 mutations cause severe phenotypes in most patients with CMT2A. Neurology. 2011, 76, 1690–1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Chen Y; Liu Y; Dorn GW 2nd,. Mitochondrial fusion is essential for organelle function and cardiac homeostasis. Circ. Res. 2011, 109, 1327–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Papanicolaou KN; Kikuchi R; Ngoh GA; Coughlan KA; Dominguez I; Stanley WC; Walsh K Mitofusins 1 and 2 are essential for postnatal metabolic remodeling in heart. Circ. Res. 2012, 111, 1012–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Kasahara A; Cipolat S; Chen Y; Dorn GW 2nd,; Scorrano L Mitochondrial fusion directs cardiomyocyte differentiation via calcineurin and Notch signaling. Science. 2013, 342, 734–737. [DOI] [PubMed] [Google Scholar]
- (9).Gong G; Song M; Csordas G; Kelly DP; Matkovich SJ; Dorn GW 2nd. Parkin-mediated mitophagy directs perinatal cardiac metabolic maturation in mice. Science. 2015, 350, aad2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Song M; Mihara K; Chen Y; Scorrano L; Dorn GW 2nd,. Mitochondrial fission and fusion factors reciprocally orchestrate mitophagic culling in mouse hearts and cultured fibroblasts. Cell Metabol. 2015, 21, 273–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Song M; Franco A; Fleischer JA; Zhang L; Dorn GW 2nd. Abrogating mitochondrial dynamics in mouse hearts accelerates mitochondrial senescence. Cell Metabol. 2017, 26, 872–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Koshiba T; Detmer SA; Kaiser JT; Chen H; McCaffery JM; Chan DC Structural basis of mitochondrial tethering by mitofusin complexes. Science. 2004, 305, 858–862. [DOI] [PubMed] [Google Scholar]
- (13).Chan DC Mitochondria: dynamic organelles in disease, aging, and development. Cell. 2006, 125, 1241–1252. [DOI] [PubMed] [Google Scholar]
- (14).Chan DC Fusion and fission: interlinked processes critical for mitochondrial health. Ann. Rev. Genet. 2012, 46, 265–287. [DOI] [PubMed] [Google Scholar]
- (15).Knott AB; Perkins G; Schwarzenbacher R; Bossy-Wetzel E Mitochondrial fragmentation in neurodegeneration. Nat. Rev. Neurosci. 2008, 9, 505–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Dorn GW 2nd,. Evolving Concepts of Mitochondrial Dynamics. Ann. Rev. Physiol. 2019, 81, 1–17. [DOI] [PubMed] [Google Scholar]
- (17).Franco A; Kitsis RN; Fleischer JA; Gavathiotis E; Kornfeld OS; Gong G; Biris N; Benz A; Qvit N; Donnelly SK; Chen Y; Mennerick S; Hodgson L; Mochly-Rosen D; Dorn GW II. Correcting mitochondrial fusion by manipulating mitofusin conformations. Nature. 2016, 540, 74–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Rocha AG; Franco A; Krezel AM; Rumsey JM; Alberti JM; Knight WC; Biris N; Zacharioudakis E; Janetka JW; Baloh RH; Kitsis RN; Mochly-Rosen D; Townsend RR; Gavathiotis E; Dorn GW 2nd., MFN2 agonists reverse mitochondrial defects in preclinical models of Charcot-Marie-Tooth disease type 2A. Science. 2018, 360, 336–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Hefti FF. Requirements for a lead compound to become a clinical candidate. BMC neuroscience. 2008, 9(3), S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Di L, Kerns EH, Fan K, McConnell OJ, Carter GT. High throughput artificial membrane permeability assay for blood–brain barrier. European journal of medicinal chemistry. 2003. 38(3), 223–232. [DOI] [PubMed] [Google Scholar]
- (21).Kansy M; Senner F; Gubernator K Physicochemical high throughput screening: parallel artificial membrane permeation assay in the description of passive absorption processes. J. Med. Chem. 1998, 41, 1007–1010. [DOI] [PubMed] [Google Scholar]
- (22).Avdeef A; Bendels S; Di L; Faller B; Kansy M; Sugano K; Yamauchi Y PAMPA--critical factors for better predictions of absorption. J. Pharmaceut. Sci. 2007, 96, 2893–2909. [DOI] [PubMed] [Google Scholar]
- (23).Ertl P; Rohde B; Selzer P Fast calculation of molecular polar surface area as a sum of fragment-based contributions and its application to the prediction of drug transport properties. J. Med. Chem. 2000, 43, 3714–3717. [DOI] [PubMed] [Google Scholar]
- (24).Wager TT; Hou X; Verhoest PR; Villalobos A Moving beyond rules: the development of a central nervous system multiparameter optimization (CNS MPO) approach to enable alignment of druglike properties. ACS Chem. Neurosci 2010, 1, 435–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Sunden H; Ma JN; Hansen LK; Gustavsson AL; Burstein ES; Olsson R Design of a highly selective and potent class of non-planar estrogen receptor beta agonists. Chem. Med. Chem. 2013, 8, 1283–1294. [DOI] [PubMed] [Google Scholar]
- (26).Jafurulla M; Rao BD; Sreedevi S; Ruysschaert JM; Covey DF; Chattopadhyay A Stereospecific requirement of cholesterol in the function of the serotonin1A receptor. Biochim. Biophys. Acta. (BBA)-Biomembranes. 2014, 1838, 158–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Cartoni R; Arnaud E; Medard JJ; Poirot O; Courvoisier DS; Chrast R; Martinou JC Expression of mitofusin 2(R94Q) in a transgenic mouse leads to Charcot-Marie-Tooth neuropathy type 2A. Brain. 2010, 133, 1460–1469. [DOI] [PubMed] [Google Scholar]
- (28).Bannerman P; Burns T; Xu J; Miers L; Pleasure D Mice hemizygous for a pathogenic mitofusin-2 allele exhibit hind limb/foot gait deficits and phenotypic perturbations in nerve and muscle. PloS One. 2016, 11, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Zhou Y; Carmona S; Muhammad A; Bell S; Landeros J; Vazquez M; Ho R; Franco A; Lu B; Dorn GW 2nd,; Wang S; Lutz CM; Baloh RH Restoring mitofusin balance prevents axonal degeneration in a Charcot-Marie-Tooth type 2A model. J. Clin. Invest. 2019, 130, 1756–1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Schneider CA; Rasband WS; Eliceiri KW NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Dorn GW II; Mitofusin Modulation Agents and Methods of Use Thereof, WO 2019/094830 Al, 2019.
- (32).Dorn GW II; Small Molecule Regulators of Mitochondrial Fusion and Methods of Use Thereof, U.S. Patent 20190169138A1, 2019.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.