

Abstract
While stem cell niches in vivo are complex three-dimensional (3D) microenvironments, the relationship between the dimensionality of the niche to its function is unknown. We have created a 3D microenvironment through electrospinning to study the impact of geometry and different extracellular proteins on the development of cardiac progenitor cells (Flk-1+) from resident stem cells and their differentiation into functional cardiovascular cells. We have investigated the effect of collagen IV, fibronectin, laminin and vitronectin on the adhesion and proliferation of murine ES cells as well as the effects of these proteins on the number of Flk-1+ cells cultured in 2D conditions compared to 3D system in a feeder free condition. We found that the number of Flk-1+ cells was significantly higher in 3D scaffolds coated with laminin or vitronectin compared to colIV-coated scaffolds. Our results show the importance of defined culture systems in vitro for studying the guided differentiation of pluripotent embryonic stem cells in the field of cardiovascular tissue engineering and regenerative medicine.
Keywords: Extracellular matrix, Niche, Cardiovascular tissue engineering, Stem cell, Scaffold
1. Introduction
Stem cells are defined by their ability to self-renew and differentiate into different cell types. They are the most promising cell source for transplantation therapy, tissue regeneration and drug development. However, despite the remarkable potential clinical applications of different stem-cell populations, their use is currently hindered by different hurdles that must be addressed [1]. Thus, a major goal is to develop new culture-based approaches, using advanced biomaterials that more closely mimic what the body already does so well and promotes differentiation of pluripotent cells or propagation of specialized adult stem cells without loss of ‘stemness.’
The relative importance of specific substrate components for stem cell adhesion, survival, and undifferentiated growth is still insufficiently characterized. However, an increasing emphasis is on designing biomaterials, based on basic mechanisms of cell-matrix interactions and cell signaling for applications in stem cell biology. This application has the potential to revolutionize our understanding of extrinsic regulators of cell fate, as matrices can be made using technologies that recapitulate the features of stem-cell microenvironments, or niches, down to the molecular level [2].
During embryonic development, the extracellular matrix (ECM) plays a critical role in regulating stem cell differentiation into different lineages, as well as in cell migration and proliferation [3–7]. In vivo, stem cells reside within instructive, tissue-specific niches that physically localize them and maintain their stem-cell fate [8–10]. Within the niche, stem cells are exposed to complex, spatially and temporally controlled biochemical mixtures of soluble chemokines, cytokines and growth factors, as well as insoluble transmembrane receptor ligands and ECM molecules. While an important function of the ECM is to provide the structural framework to support cellular functions, this scaffold of proteins, proteoglycans, and glycosaminoglycans also provides cell adhesion sites and important signaling cues [10–12]. The ECM interacts with cells via cell surface receptors such as integrins; serves as a reservoir for growth factors; and provides a substrate for cell attachment and spreading, contact guidance for cell migration, and a scaffold for building tissues. The morphology of cells determined by their contact with ECM or with nonbiological surfaces may be associated with particular patterns of cell differentiation and proliferation [13–15].
The geometry of the matrix (i.e., 2D versus 3D) also plays an important role in determining how a cell will respond to biochemical and mechanical cues, since in many native tissues cells are completely surrounded by ECM [16,17]. Conventional 2D cell culture has provided important insight into how cells interact with their environment. The use of 3D culture systems is gaining popularity due to their promise as improved models of tissue physiology and because such systems can potentially be developed into engineered tissues for the treatment of the disease. The field of tissue engineering therefore is in need of a better understanding of how cells interact with 3D matrices and how cell function can be controlled via cell-matrix interactions.
In an effort to elucidate the mechanism through which the complex 3D ECM microenvironment enhances cardiovascular differentiation of ES cells, we have investigated the effect of collagen IV, fibronectin, laminin and vitronectin on the adhesion and proliferation of mES cells in 2D and 3D feeder free condition. Further, we have isolated Flk-1+ cells from partially differentiating mES cultured on vitronectin-coated substrates and investigated their ability to differentiate into cardiovascular lineage i.e. cardiac myocytes (CMs), smooth muscle cells (SMCs) and endothelial cells (ECs).
2. Materials and methods
2.1. Human procurement and processing
First-trimester (7–12 week) human hearts were purchased from Novogenix laboratories (Los Angeles, CA). All heart tissues were fixed in 10% buffered formalin for 12 h and transferred to 70% ethanol prior to receiving. The fixed specimens were embedded in paraffin and cut into 5 μm sections by the UCLA Translational Pathology Core Laboratory (TPCL).
2.2. Mouse ES cell cultures, In vitro differentiation assays and magnetic cell sorting in 2D condition
Unless otherwise noted all reagents were purchased from Sigma Aldrich (St. Louis, MO). Murine Flk-1 GFP-labeled embryonic stem cells (mES) were a kind gift from Dr. MacLellan’s laboratory at the Department of Medicine/Cardiology at the University of California Los Angeles. mES cells were maintained in an undifferentiated state on mitomycin-C-treated primary mouse embryonic stem fibroblasts (MEF) in leukemia inhibitory factor (LIF) supplemented medium (Knockout Dulbecco’s modified Eagle’s medium (Invitrogen, Carlsbad, CA) supplemented with 10% ES-FCS (Invitrogen), 0.1 mM β-mercaptoethanol, 2 mM glutamine (Invitrogen), 0.1 mM nonessential amino acids (Invitrogen) and 1000 U/ml recombinant LIF (Chemicon, Temecula, CA) and HEPES (2 mM, Invitrogen). For adaptation of the cells to a feeder free condition, the cells were detached from the culture dish using accutase (Chemicon) and cultured in 90% LIF-medium as described before and 10% ESGRO Complete medium (Chemicon) on gelatin coated (0.1% gelatin in PBS, coated for 2 h at 37 °C) T-75 flasks at 37 °C, 5% CO2, in a humidified incubator. All cells were passaged every other day and cultured in a reducing LIF-medium and increasing ESGRO combination. After several passagings all mES cells were cultured in 100% ESGRO medium (Chemicon). The feeder free mES cells were then expanded for two additional passages before being used in experiments.
For differentiation assays, the mES cells were either introduced into a dynamic suspension culture system for generating embryoid bodies (EBs) or cultured on coated plates with collagen type IV (ColIV, 5 μg/cm2, BD Biosciences, San Jose, CA), vitronectin (50 ng/cm2, Chemicon), fibronectin (5 μg/cm2), laminin (5 μg/cm2, BD Biosciences) or Matrigel (BD, 4/μg/cm2, used as a positive control for cell attachement). Briefly, for EB formation, the cells were dissociated, resuspended in α-minimum essential medium (Invitrogen, Carlsbad, CA) supplemented with 10% ES-FCS (Invitrogen), 0.1 mM β-mercaptoethanol, 2 mM glutamine (Invitrogen), and 0.1 mM nonessential amino acids (Invitrogen) HEPES (2 mM, Invitrogen) and transferred into 60-mm ultralow-attachment dishes (4 × 105 cells per dish; Corning Life Sciences, Acton, MA), placed onto an orbital rotary shaker (Stovall Belly Button; ATR, Laurel, MD), and cultured under continuous shaking at approximately 50 rpm for up to two weeks. For morphometric analysis, phase-contrast images of mES-derived EBs were acquired every day during the course of culture.
Further for differentiation assay, mES cells were detached from the culture dishes and transferred to colIV- vitronectin-, fibronectin-, and laminin-coated plates for 2D culture. After 4 days, the cells were either harvested for FACS analysis, or they were detached and the Flk-1-positive cells were isolated by indirect magnetic cell sorting using a purified rat anti-mouse Flk-1 antibody (BD Pharmingen, San Diego) and magnetic microbeads (Miltenyi Biotec, Auburn, CA). The Flk-1-positive progenitor (Flk-1+) cells were then plated on fibronectin-coated culture slides (BD Biosciences) in either α-MEM for cardiac differentiation, smooth muscle growth medium (SMGM-2; Lonza, Walkersville, MD) supplemented with 10 ng/ml platelet-derived growth factor-BB (PDGF-BB, R&D Systems Inc., Minneapolis) for SMC differentiation, or endothelial growth medium (EGM-2; Lonza) supplemented with 50 ng/ml vascular endothelial growth factor (VEGF, R&D Systems, Minneapolis, MN) for EC differentiation for up to 12 days at 37 °C and 5% CO2. To expand the mES cell-derived SMCs or ECs, cells were grown to >80% confluence in either SMGM-2 or EGM-2 and passaged into gelatin-coated plates with a 1:3 ratio every 3–4 days. Bright-field images and movies of undifferentiated and differentiated mES cells, as well as EBs, were acquired using the Olympus microscope (Center Valley, PA).
2.3. Alkaline phosphatase activity
The alkaline phosphatase (AP) activity of mES cells cultured in the ESGRO medium for 4 days on gelatin coated plates was detected with a Fast Red substrate kit (Chemicon) according to manufacturer’s protocol. Briefly, the cells were fixed in 4% glutaraldehyde for 1 min before incubating with the staining mix (Fast Red Violet:Naphthol:water (2:1:1)) for 15 min in the dark. The cells were then rinsed with PBS and the red stem cell colonies were detected using Olympus microscope.
2.4. Scaffold fabrication, cell culture and in vitro differentiation assays in 3D condition
Electrospinning has been used to produce a scaffold with nano- to microdiameter fibers with similar structural properties to the ECM as described before [18]. Briefly, gelatin type B (bovine skin, 10% w/v) and PCL (10% w/v) were mixed together and dissolved in 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP). The solution was then loaded into a 10 ml-syringe, to which to an 18-gauge blunt ended needle (spinning nuzzle) was attached. A core solution of 5% w/v PU dissolved in HFIP was loaded into a 3 ml-syringe, to which a 25-gauge needle was attached. This syringe and needle was then loaded into the 10 ml-syringe containing the sheath solution. The entire syringe system was then loaded into a modified syringe pump. The positive output lead of a high voltage supply (25 kV; Glassman High Voltage Inc., NJ, USA) was attached to the needle on the 10 ml-syringe, spinning nuzzle. In the created electric field, a thin jet was ejected from the solution in the syringe at a speed of 70 μL/min. The grounded copper target (5 cm × 5 cm) was placed ∼15 cm under the needle tip and upon introduction of the electric field Taylor cone formation at the base of the spinning nuzzle was observed. A dry fibrous scaffold was collected in the form of a flat 3D mat (100–200 μm thick). The electrospun scaffolds were (1 cm × 1 cm) then sterilized by soaking scaffolds in 70% EtOH for 30 min, and then washed with sterile PBS three times and coated with colIV, fibronectin, laminin, vitronectin, Matrigel and gelatin for 3D in vitro studies.
2.5. Scanning electron microscopy
For ultrastructural analysis, unseeded, seeded scaffolds as well as EBs were processed for characterization by scanning electron microscopy (SEM) as described previously [19]. Briefly, cell-seeded samples and EBs were rinsed with SEM buffer (0.1 M sodium cacodylate buffer, pH 7.2, supplemented with 5% sucrose) for 10 min. The samples were then fixed for 30 min in 2% paraformaldehyde/2% glutaraldehyde in SEM buffer, followed by dehydration through grades of ethanol, 30, 50, 70, 80 and 95% for 10 min each, followed by 3 incubations in 100% ethanol for 10 min and a final incubation in 100% ethanol for 40 min. The samples were dried by incubating in one-half volume 100% ethanol and one-half volume hexamethyldisilazane for 20 min followed by 100% hexamethyldisilazane for 20 min. Finally, the 100% hexamethyldisilazane solution was evaporated during 20 min air-drying. Once dry, the samples were mounted onto stubs and sputter coated by gold/palladium (Au/Pd, thickness of ∼10 nm) using JEOL JSM-6490 (JEOL USA, Inc. Peabody, MA) scanning electron microscope.
2.6. Immunofluorescent staining
The Flk-1+ cells, plated on fibronectin-coated culture slides, as well as undifferentiated, mES cells were washed and fixed with 4% paraformaldehyde in PBS, for 20 min and rinsed with PBS. The EBs were also fixed and mixed with 50 μL Histogel (Fisherscientific, Pittsburgh, PA) prior paraffin embedding and sectioning at TPCL. All the sections were deparaffinized using xylene for 10 min, rehydrated in an ethanol gradient by incubating for 5 min in 100%, 90%, 85%, and 70% ethanol solutions and washed in deionized water for 3 min. All the samples were then blocked with 1% bovine serum albumin (BSA) and 2% goat serum in PBS for 30 min at room temperatue, followed by incubation with primary antibodies (ES markers: Nanog, Oct-4, Sox-2, SSEA-1 (Abcam, Cambrige, MA)); smooth muscle specific markers: SM-α-actin (Dako, Carpinteria, CA), h-caldesmon (Dako), basic calponin (Dako), SM-myosin (Dako); endothelial specific markers: CD31 (Dako), VE-cadherin (CD144, Santa Cruz Biotechnology, Santa Cruz, CA) and von Willebrand Factor (vWF, Dako); cardiomyocyte specific markers: MF20 (Developmental Studies Hybridoma Bank (DSHB), Iowa City, IA), connexin-43 (Santa Cruz Biotechnology, Inc), Troponin-C (Santa Cruz), Nkx2.5 (Santa Cruz) overnight at 4 °C followed by several washes with PBS. The feeder free mES cells were also stained for early markers for ectodermal (nestin (Abcam)), endodermal (β-catenin (Abcam), α-fetoprotein (Santa Cruz Biotechnology)), and mesodermal (brachyury (Santa Cruz Biotechnology), SM-α-actin (Dako)) markers. Alexa Fluor 488- or 546-conjugated secondary antibodies (Molecular Probes, Eugene, OR) were applied to the samples and incubated for 30 min at room temperature. After several washes, the cells were counterstained with 4′−6-diamidino-2-phenylindole (DAPI) followed by adding ProLong Gold antifade mounting medium (Molecular Probes, Carlsbad, CA). Staining without primary antibodies served as controls. Digital images were acquired using a Leica DM IRB inverted microscope system equipped with 20× (0.40 numerical aperture (NA)) and 40× (0.75 NA) objectives (Leica Microsystems Inc., Bannockburn, IL).
2.7. Fluorescence-activated cell sorter analysis
Cells were harvested from different 2D and 3D conditions using accutase (Chemicon), pelleted by centrifugation, washed in PBS, and stained with the purified rat anti-mouse Flk-1 antibody (BD Pharmingen, San Diego) followed by fluorescein isothiocyanate (FITC) or phycoerythrin (PE). Nonspecific fluorochrome- and isotype matched IgGs (BD Pharmingen) served as controls. The cells were gated by forward scatter (FSC) versus side scatter (SSC) to eliminate debris. A minimum of 10,000 events was counted for each analysis. All analyses were performed using a Becton Dickinson FACScan analytic flow cytometer (BD Bioscience, San Jose, CA) with FCS Express software (DeNovo Software, Thornhill, Ontario, Canada) at the UCLA Flow Cytometry Laboratory.
2.8. Smooth muscle and endothelial cell functionality assays
To assess the functionality of the cells, the contraction of SMC, EC uptake of acetylated low-density lipoprotein (acLDL) and ability to form tubes in vitro on Matrigel (BD Biosciences) were determined. ES cell-derived SMCs were subjected to the effect of 10−5 M carbamoylcholine chloride (carbachol) in SMGM-2 medium (Lonza) for up to 45 min. The contraction was observed by bright-field imaging.
ES cell-derived ECs were incubated with 10 μg/ml Alexa Fluor 594-labeled acLDL (Molecular Probes) for 4 h at 37 °C, washed in PBS, fixed with 4% paraformaldehyde, counterstained with 4′−6-diamidino-2-phenylindole (DAPI) and visualized. Also, Matrigel (BD Biosciences) was added to a few wells of a 24-well plate in 200-μl volumes and allowed to solidify for 30 min at 37 °C. After the Matrigel solidified, 50,000 ES cell-derived ECs were suspended in EC medium and added to each Matrigel-coated wells. The cells were then incubated at 37 °C and 5% CO2 for 24 h and observed for tube-like formations with a phase-contrast microscope. Human umbilical vein endothelial cells (HUVEC) and smooth muscle cells (SMC) (American Type Culture Collection (ATCC), Manassas, VA) cultured in either SMGM-2 or EGM-2 (Lonza) served as controls.
2.9. Proliferation assays
Undifferentiated mES cells were detached from the culture dish and seeded in flat-bottom 96-well plates for 2D experiments and on the scaffold (the scaffold covered the bottom of the wells in flat-bottom 96-well plates) for 3D experiments. The culture plates and the scaffolds were previously coated with colIV, fibronectin, laminin, and vitronectin as described before. Approximately 35,000 cells/well in triplicate per condition, in a mixture of α-minimum essential medium (Invitrogen) supplemented with 10% ES-FCS (Invitrogen), 0.1 mM β-mercaptoethanol, 2 mM glutamine (Invitrogen), and 0.1 mM nonessential amino acids (Invitrogen) and alamar blue (Serotec, Raleigh, NC; in an amount equal to 10% of the total culture volume) were seeded on each well. Samples were incubated with medium-alamar blue mix for 72 h. The metabolism levels were evaluated on a Benchmark Plus Microplate Spectrofluorometer (Bio Rad, Hercules, CA) at wavelengths of 570 and 600 nm (the amount of reduced alamar blue is Absorbance 570nm - Absorbance 600nm) after 2, 6, 24, 48 and 72 h.
2.10. Statistical analyses
All results are presented as mean values ± standard error of mean (SEM). Statistical significance was assessed by Student’s t-test or ANOVA with Tukey’s multiple comparison tests. P-values less than 0.05 were defined as statistically significant.
3. Results
3.1. Endogenous Flk-1+ cardiac progenitor cells and ECM proteins
Immunofluorescence staining of human first trimester (7–12 weeks) hearts showed the present of the endogenous Flk-1+ cells in the developing hearts (Fig. 1). To determine the present of different endogenous ECM proteins in the microenvironment surrounding these Flk-1+ cells we performed immunofluorescence staining. ColIV, laminin, fibronectin and vitronectin were found in the microenvironment. The basement membrane proteins ColIV and laminin were found within and around the Flk1+ (Fig. 1A, C, E, G). In contrast, fibronectin and vitronectin were predominantly found within the myocardium (Fig. 1B, D, F, H).
Fig. 1.
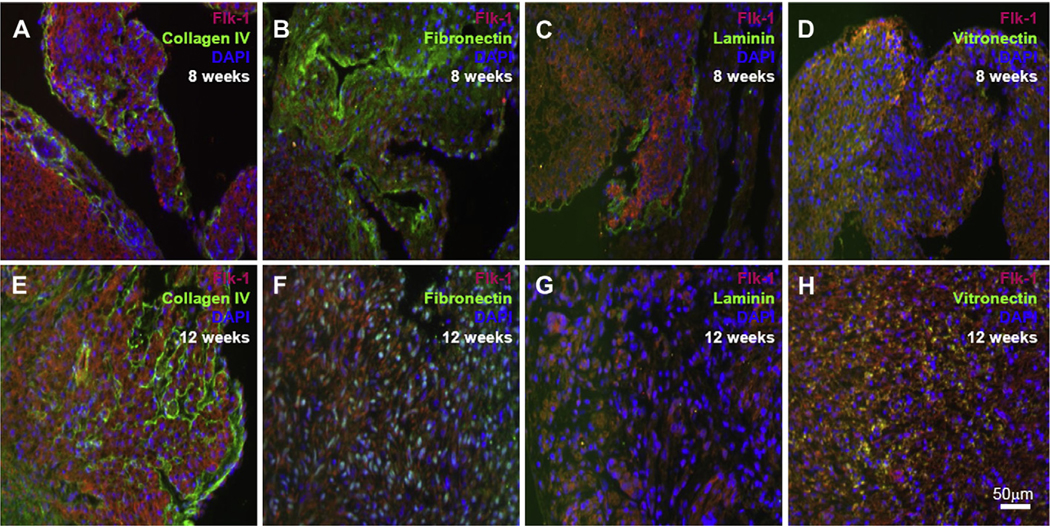
Immunofluorescence staining of first-trimester human heart tissues shows the endogenous Flk-1 expressing CPC (red) as well as the expression of ColIV (A, E), fibronectin (B, F), laminin (C, G) and vitronectin (D, H) all in green. Cell nuclei are identified with DAPI staining (blue). Scale bars 50 μm.
3.2. Characterization of feeder free mES cells
The feeder free mES cells in this study have shown a tighter and more closely packed morphology than the cells in a feeder dependent condition (Fig. 2A and B). The pluripotency of these cells after several passages was confirmed by staining for alkaline phosphatase, Oct-4, SOX-2 and Nanog (Fig. 2C–F).
Fig. 2.

Phase-contrast images of the feeder dependent (A) and the feeder free (B) mES cells show a rounder and tighter packed morphology for the cells in the feeder free condition. Alkaline phosphatase (C, pink) and immunofluorescence staining for Oct-4, SOX-2 and Nanog (D–F, green) confirm the pluripotency of these cells after several passages. Cell nuclei are identified with DAPI staining (blue). Scale bars 50 μm.
Murine ES cells are able to differentiate into cell lineages of all three embryonic germ layers (mesoderm, endoderm, and ectoderm) when allowed to aggregate in suspension to form 3D embryoid bodies [20]. To characterize the differentiation potential of the feeder free mES cells, EBs were formed (Fig. 3 A–E). Immunofluorescent staining shows no signals for pluripotency markers such as Nanog and Oct 4 in the differentiating EBs. However, the presence of the early ectodermal (nestin), endodermal (α-fetoprotein), as well as mesodermal marker (brachyury) were detected by immunofluorescent staining (Fig. 3F–I).
Fig. 3.

Phase-contrast images of EB formation and growth characteristics of murine ES day 1 to day 6 (A–E). Immunofluorescence staining confirm the differentiation capacity of mES-derived EBs to endoderm (F, β-catenin (green); I, α-fetoprotein (AFP, red)), mesoderm (G, brachyury (green)) and ectoderm (H, nestin (green)). Cell nuclei are identified with DAPI staining (blue). Scanning electron micrographs of mES-derived EB show the ECM deposition (J–N).
3.3. ECM in mES-derived EBs
Mouse embryonic fibroblasts (MEFs) are most commonly used as feeder layers and produce a complex matrix of many structural ECM proteins, including laminins, different types of collagens, fibronectin, etc [21]. Understanding the composition of ECM components, which sustains undifferentiated proliferation of these cells are a very important issue. Detail is lacking on the exact composition of these ECM components, making it difficult to predict which components are essential and which are redundant in ES differentiation.
In vitro aggregation of murine ES cells initiates the formation of EBs, which structurally resembles the pregastrulation-stage embryo and can facilitate spontaneous, unguided differentiation analogous to that seen in developing mouse embryos. Scanning electron microscopy images of mES cell-derived EB showed the ECM deposition and the embedded cells in ECM which make a smooth outer surface (Fig. 3J–N). Immunofluorescence staining for ECM proteins in the mES-derived EBs showed the presence of ColI, ColIV, fibronectin, vitronectin and laminin within the developing and differentiating EBs (Fig. 4).
Fig. 4.

Immunofluorescence staining of differentiating EBs shows the expression of major ECM proteins such as collagen I (A), collagen IV (B), fibronectin (C), laminin (D) and vitronectin (E) in green. Cell nuclei are stained with DAPI staining (blue). Scale bars 50 μm.
3.4. ECM and mES proliferation in 2D and 3D
To determine if the proliferation of the cells was affected by different ECM proteins and three-dimensionality of the environment, we measured the metabolic activity of the feeder free undifferentiated mES cells cultured on different coated conventional 2D culture dishes as well as on coated 3D electrospun scaffolds, using an Alamar Blue assay.
The cells had similar proliferation rate after 2 h on colIV-, laminin-, fibronectin-and vitronectin-coated surfaces in both 2D and 3D culture condition. The metabolic activity of the cells was significantly increased after 6 h in 2D culture compared to 3D condition on all surfaces. By 24 h post-seeding, the proliferation rate increased in 3D conditions with significantly higher rates on colIV-coated scaffolds compared to vitronectin-coated scaffolds. This trend in metabolic activity changed by 48 h post-seeding and was followed by a higher proliferation rate on vitronectin-coated scaffold in 3D cultures to the end of experiment. The proliferation rate of the cells on 2D colIV-coated wells continued to increase and dropped slightly by 72 h in culture. On vitronectin-coated wells, in 2D condition, the proliferation rate decreased significantly between 24 and 48 h in culture. However, this metabolic activity increased and it was significantly higher after 72 h in culture compared to colIV-, laminin- and fibronectin-coated wells in 2D condition. No significant differences were observed between proliferation rates of the cells cultured on laminin- or fibronectin-coated surfaces in 2D or 3D cultures (Fig. 5).
Fig. 5.

Proliferation of mES cells on colIV, fibronectin, laminin and vitronectin in 2D and 3D culture system for 72 h. The metabolic activity of the cells was significantly increased after 6 h in 2D culture condition on all surfaces. By 24 h post-seeding, the proliferation rate increased in 3D conditions with significantly higher rates on colIV-coated scaffolds. This trend in metabolic activity changed by 48 h post-seeding and was followed by a higher proliferation rate on vitronectin-coated scaffold in 3D cultures to the end of experiment. The proliferation rate of the cells on 2D colIV-coated wells continued to increase and dropped slightly by 72 h in culture. On vitronectin-coated wells, in 2D condition, the proliferation rate decreased significantly between 24 and 48 h in culture. However, this metabolic activity increased and it was significantly higher after 72 h in culture compared to colIV-, laminin- and fibronectin-coated wells in 2D condition.
3.5. Vitronectin and cardiovascular differentiation of mES cells in 3D
It has been demonstrated recently that culturing ES cells on ColIV coated culture dishes is a simple in vitro system to differentiate these cells toward the cardiovascular lineages [22–25] To determine whether our feeder free mES cells have a similar differentiation potential, we cultured undifferentiated mES cells for 4 days on ColIV-, laminin-, fibronectin- and vitronectin-coated culture plates (2D) as well as colIV-, laminin-, fibronectin- and vitronectin-coated electrospun 3D scaffolds. FACS analysis showed the presence of Flk-1+ cells in partially differentiated ES cells cultured on colIV (2D: 7.17 ± 1.49; 3D: 1.43 ± 0.21), laminin (2D: 1.41 ± 0.27; 3D: 3.46 ± 0.80), fibronectin (2D: 1.41 ± 0.27; 3D: 1.67 ± 0.36) and vitronectin (2D: 2.20 ± 0.20; 3D: 5.45 ± 0.91). The number of Flk-1+ cells were significantly higher (7.17 ± 1.49) in 2D colIV-coated dishes compared to other 2D cultures. However, the number of Flk-1+ cells in 3D vitronectin-coated scaffold was significantly higher than in 3D colIV- (1.43 ± 0.21), laminin- (3.46 ± 0.80) and fibronectin-coated scaffolds (1.67 ± 0.36). The number of Flk-1+ cells in 3D vitronectin- and laminin-coated scaffold was also significantly higher compared to 2D condition. ColIV-coated culture plates enhanced the number of Flk-1+ cells significantly in 2D compared to colIV-coated scaffolds in the 3D condition(Fig. 6).
Fig. 6.

FACS analysis of differentiating mES cells cultured on colIV, fibronectin, laminin and vitronectin for showing the number of Flk1+ cells in 2D and 3D culture system after 4 days. The number of Flk-1+ cells were significantly higher in 2D colIV-coated dishes. However, the number of Flk-1+ cells in 3D vitronectin-coated scaffold was significantly higher than in 3D colIV-, laminin- and fibronectin-coated scaffolds.
3.6. Differentiation of mES-derived Flk-1+ progenitors cells into functional SMC, EC and cardiac cells
To determine whether vitronectin-differentiated mES cell-derived Flk-1+ cells had the capacity to differentiate into cardiovascular cells, we isolated the Flk-1+ cells and cultured them in differentiation-promoting conditions for smooth muscle, endothelial and cardiac cells (Fig. 7). When treated with VEGF, mES cell-derived Flk-1+ cells differentiated into EC cells and expressed specific markers, including CD31, CD144, vWF with typical cobblestone morphology of EC cells (Fig. 7A–D). These cells showed the ability to take up acLDL(Fig. 7E and F).
Fig. 7.
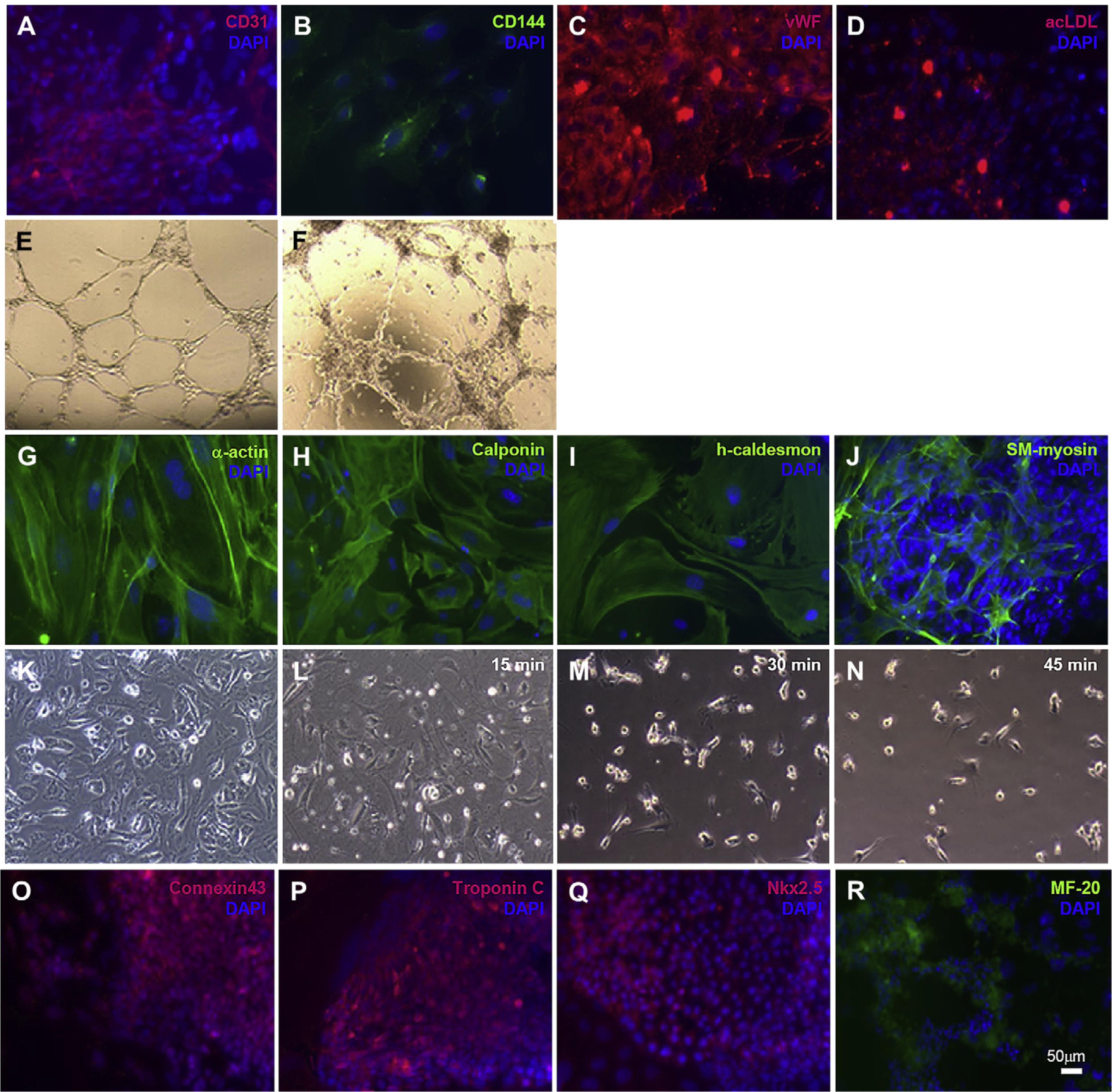
In vitro differentiation of mES-derived Flk1+ progenitor cells into functional cardiovascular cells i.e. endothelial cells CD31 (A, red), CD144 (B, green), vWF (C, red) and acLDL uptake (D, red), tube formation in matrigel (F) (HUVEC as control, E); smooth muscle cells α-actin (G, green), calponin (H, green), caldesmon (I, green), SM-myosin (J, green), cell contraction when incubated with carbachol for 30 and 45 min (K–N); cardiomyocytes connxtin 43 (O, red), troponin C (P, red), Nkx2.5 (Q, red) and MF-20 (R, green). Cell nuclei were stained with DAPI (blue). Scale bars 50 μm.
The mES cell-derived Flk-1+ cells, cultured in PDGF-BB supplemented medium, as described before, differentiated into SMC cells and expressed αSMA, calponin, caldesmon and SM-myosin (Fig. 7G–J). To assess their functional capacity, these cells were exposed to carbachol for a total of 45 min and showed cell contraction after 15 min exposure (Fig. 7K–N).
Flk-1+ cells which cultured in conditions to promote cardiac differentiation, developed spontaneously beating cell clusters after 10–12 days of culture. Immunofluorescent staining of the spontaneously beating areas revealed the presence of cardiac specific markers, including sarcomeric myosin (MF-20), gap junction protein connexin-43, Nkx2.5 and troponin C (Fig. 7O–R, Movie 1).
Supplementary video related to this article can be found at doi: 10.1016/j.biomaterials.2011.11.065.
4. Discussion
The goal of regenerative medicine is to repair or replace damaged or diseased tissues or organs. Different strategies are being investigated for regenerative therapies such as cell-based, tissue-engineered bioscaffolds seeded with selected cells prior to engraftment, to stimulate endogenous repair mechanisms. The source and availability of the cells for tissue engineering (TE) is very critical. The potential of ES cells, which have the capacity to differentiate into all somatic cell types, has attracted interest in the field of TE, regenerative medicine and drug screening [26].
The development and normal function of stem cells are likely to depend on interactions with molecules in their microenvironment, referred to as the stem cell “niche”. However, a major challenge in stem cell biology remains in defining the components of those niches crucial for stem cell regulation and exploiting this knowledge for therapeutic potential. Niches harboring stem or progenitor cells, have been described as anatomically protected 3D sites, consisting of neighboring cells that regulate the stem cell population through direct contact, secretion of soluble factors, and the production of specialized extracellular matrices with physical, structural or mechanical properties of the tissues they inhabit [27–29]. The development of simple tools for efficient ES cell differentiation into a particular lineage is critically important for all these applications. Today, such tools are not yet available. The ECM can be used as one such tool to guide ES cell differentiation into a particular lineage in vitro, since the ECM plays pivotal roles in cell differentiation, as well as in cell migration and proliferation in vivo. Unfortunately, neither the patterns of ES cell differentiation triggered by different ECM components nor the mechanisms mediating this differentiation are well known.
Cells, including stem cells, cultured in plastic dishes are typically exposed to soluble factors in liquid media. These culture conditions are very different from the conditions experienced by cells in the body, where they are associated with anchored molecules presented in close proximity to surrounding cell surfaces and contained within an ECM that creates a relatively soft microenvironment. The constraints imposed on stem cells within the 3D niche have effects that are still being explored and should not be ignored.
Cell-cell (through cadherins and cell adhesion molecules) and cell-matrix (through integrins) interactions have been proven to play a crucial role during embryogenesis. For example, the compaction of the inner cell mass requires E-cadherin [30], laminin appeared as early as the 2-cell stage, entactin/nidogen appeared at the 16-cell stage [31], and fibronectin and type ColIV appeared later in the inner cell mass of 3–4- day-old blastocysts [32]. The existence of these various ECM components makes it clear that at a given time and place, the ECM has the potential to provide specific environmental information to cells. Remarkably, regardless of whether ES cells are cultured on a biological ECM or on a nonbiological substrate, production of an endogenous ECM is required for cell survival.
Cells in their natural environment are anchored by discrete attachements to proteins in the ECM. Similarly, cell attachment to culture surfaces in vitro is usually mediated by adhesion proteins contained in serum-supplemented culture medium [33–35]. Because cells depend on specific proteins for anchorage and extracellular instructions, the composition of the adsorbed layer is a key mediator of cell behavior. In this manner, the required proteins, correctly presented, can stimulate a constructive cell response, favoring wound repair and tissue integration, whereas proteins in an unrecognizable state may indicate a foreign material to be removed or isolated.
To develop a cell culture platform based on defined ECM protein Ludwig et al used human ColIV, laminin, fibronectin and vitronectin to expand hES cells in a defined cell culture medium [36–41]. Vitronectin is a multifunctional ECM protein that promotes cell adhesion and spreading [42,43]. It has been shown that vitronectin adsorbed to tissue culture culture polystyrene is a viable substrate capable of supporting the long-term propagation of multiple hES cell lines [44].
With the emergence of a defined substrate as a realistic possibility, Braam et al and Rowland et al identified that surface-presented vitronectin enables the adhesion of hES cells and activates a key integrin which is believed to play a role in supporting the long-term propagation of hES cells [45,46].
It has been shown that mES cells cultured on ColI and IV or poly-D-lysine remained undifferentiated, whereas laminin or fibronectin induce differentiation [47].
When exposed to colIV, laminin, fibronectin and vitronectin, the mES cells partially differentiated into Flk-1 expressing progenitor cells in both 2D and 3D culture system, however, the proliferation rate in the 3D systems was lower than in 2D systems. The number of these Flk-1+ cells was significantly higher in 3D scaffolds coated with laminin or vitronectin compared to colIV-coated scaffolds. Although this work demonstrates that three-dimensionality is sufficient to induce a similar number of progenitor cells such as Flk-1+ cells compared to conventional 2D in vitro culture systems, understanding the mechanisms underlying the effect that three-dimensionality has on cell fate decisions will require further studies. Enhanced cell-cell and cell-matrix interactions and improved cell signaling in 3D cultures may play an important role [48]. ColIV-coating in the 2D culture system described here enhanced the number of Flk-1+ cells significantly compared to laminin and vitronectin. Vitronectin is a widely distributed high molecular weight glycoprotein found in most extracellular matrices and blood plasma that is known to promote cell adhesion and affect cell morphology, migration, differentiation, and cytoskeletal organization. It is also known to synergize with numerous growth factors to maintain both embryonic and adult stem cells in an undifferentiated state [49,50].
In the presented study, the isolated Flk-1+ cells from vitronectin-coated surfaces most likely represents a population of multipotent mesodermal progenitor cells which give rise to the cardiomyocyte, vascular endothelial, and smooth muscle lineages [51–54]. A cell capable of differentiating into all cardiovascular cell types has a theoretical advantage for more complete tissue regeneration, as has been demonstrated for the ES cells in the presented study [26]. Also, partially differentiated cardiovascular progenitor cells, such as the Flk-1+ cells described here, will likely reduce the tumor formation seen when transplanting undifferentiated ES cells into the heart [55]. To confirm that these Flk-1+ cells were capable of generating all cardiovascular cell types, we isolated Flk-1+ cells from vitronectin-coated dishes and exposed them to cardiac, smooth muscle, and endothelial cell-specific differentiation media. The presence of different cardiovascular markers as well as cell contraction, uptake of acLDL, in vitro tube formation and spontaneously beating cell clusters showed the differentiation capacity of mES-derived Flk-1+ cells into functional SMC, EC and cardiac cells similar to previously reported studies [22,23].
We have showed that the feeder free mES cells build colonies when maintained in undifferentiated conditions similar to the feeder dependent mES cells. We observed that feeder free mES cells reproducibly formed spherical EBs. They are able to differentiate into all three germ layers (mesoderm, endoderm, and ectoderm) when allowed to aggregate in suspension and form embryoid bodies. Further, we determined the effect of different ECM proteins on cardiovascular differentiation potential of feeder free murine ES cells in a 2D versus 3D culture system. The ongoing studies in our laboratory will determine the importance of cell-cell and cell-ECM interactions, via integrins, in pluripotency and self-renewal of embryonic stem cells that can be further used in directing the differentiation of ES cells for different clinical applications.
5. Conclusion
We characterized mouse ES cells in a feeder free culture system and their differentiation capacity into cardiovascular lineages in the presented study. We reported the effect of three-dimensionality on the differentiation of Flk-1+ progenitor cells using in vitro culture systems. The existence of various ECM components makes it clear that at a given defined microenvironment within the ECM has the potential to provide specific environmental information to cells. Remarkably, regardless of whether ES cells are cultured on a biological ECM or on a nonbiological substrate, production and existence of an ECM is required for cell survival and related to cell differentiation. The development of defined culture systems in vitro that exhibit features of natural 3D niche microenvironments, are critical issues for studying the role of the ECM in the guided differentiation of pluripotent embryonic stem cells in the field of tissue engineering and regenerative medicine. The present study provides a way of designing in vitro systems for directing mES cell differentiation into cells of cardiovascular lineages.
Supplementary Material
Acknowledgments
This work was supported by the Ruth Kirschstein National Research Service Award (T32HL69766 to J.M.G.) and the Department of Surgery at UCLA.
References
- [1].Daley GQ, Scadden DT. Prospects for stem cell-based therapy. Cell 2008;132: 544–8. [DOI] [PubMed] [Google Scholar]
- [2].Lutolf MP, Hubbell JA. Synthetic biomaterials as instructive extracellular microenvironments for morphogenesis in tissue engineering. Nat Biotechnol 2005;23:47–55. [DOI] [PubMed] [Google Scholar]
- [3].Flaim CJ, Chien S, Bhatia SN. An extracellular matrix microarray for probing cellular differentiation. Nat Methods 2005;2:119–25. [DOI] [PubMed] [Google Scholar]
- [4].Campbell A, Wicha MS, Long M. Extracellular matrix promotes the growth and differentiation of murine hematopoietic cells in vitro. J Clin Invest 1985;75: 2085–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Haylock DN, Nilsson SK. Stem cell regulation by the hematopoietic stem cell niche. Cell Cycle 2005;4:1353–5. [DOI] [PubMed] [Google Scholar]
- [6].Naugle JE, Olson ER, Zhang X, Mase SE, Pilati CF, Maron MB, et al. Type VI collagen induces cardiac myofibroblast differentiation: implications for postinfarction remodeling. Am J Physiol Heart Circ Physiol 2006;290:H323–30. [DOI] [PubMed] [Google Scholar]
- [7].Suzuki A, Iwama A, Miyashita H, Nakauchi H, Taniguchi H. Role for growth factors and extracellular matrix in controlling differentiation of prospectively isolated hepatic stem cells. Development 2003;130:2513–24. [DOI] [PubMed] [Google Scholar]
- [8].Scadden DT. The stem-cell niche as an entity of action. Nature 2006;441: 1075–9. [DOI] [PubMed] [Google Scholar]
- [9].Morrison SJ, Spradling AC. Stem cells and niches: mechanisms that promote stem cell maintenance throughout life. Cell 2008;132:598–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Ekblom P, Timpl R. Cell-to-cell contact and extracellular matrix. A multifaceted approach emerging. Curr Opin Cell Biol 1996;8:599–601. [DOI] [PubMed] [Google Scholar]
- [11].Discher DE, Mooney DJ, Zandstra PW. Growth factors, matrices, and forces combine and control stem cells. Science 2009;324:1673–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Guilak F, Cohen DM, Estes BT, Gimble JM, Liedtke W, Chen SC. Control of stem cell fate by physical interactions with the extracellular matrix. Cell Stem Cell 2009;5:17–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Bissell MJ, Barcellos-Hoff MH. The influence of extracellular matrix on gene expression: is structure the message? J Cell Sci Suppl 1987;8:327–43. [DOI] [PubMed] [Google Scholar]
- [14].Huet C, Pisselet C, Mandon-Pepin B, Monget P, Monniaux D. Extracellular matrix regulates ovine granulosa cell survival, proliferation and steroidogenesis: relationships between cell shape and function. J Endocrinol 2001;169:347–60. [DOI] [PubMed] [Google Scholar]
- [15].Watt FM, Jordan PW, O’Neill CH. Cell shape controls terminal differentiation of human epidermal keratinocytes. Proc Natl Acad Sci U S A 1988;85:5576–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Xu J, Clark RA. A three-dimensional collagen lattice induces protein kinase C-zeta activity: role in alpha2 integrin and collagenase mRNA expression. J Cell Biol 1997;136:473–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Grinnell F. Fibroblast biology in three-dimensional collagen matrices. Trends Cell Biol 2003;13:264–9. [DOI] [PubMed] [Google Scholar]
- [18].Gluck JM, Rahgozar P, Ingle NP, Rofail F, Petrosian A, Cline MG, et al. Hybrid coaxial electrospun nanofibrous scaffolds with limited immunological response created for tissue engineering. J Biomed Mater Res B Appl Biomater 2011;99:180–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Heydarkhan-Hagvall S, Schenke-Layland K, Dhanasopon AP, Rofail F, Smith H, Wu BM, et al. Three-dimensional electrospun ECM-based hybrid scaffolds for cardiovascular tissue engineering. Biomaterials 2008;29:2907–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Keller G. Embryonic stem cell differentiation: emergence of a new era in biology and medicine. Genes Dev 2005;19:1129–55. [DOI] [PubMed] [Google Scholar]
- [21].Horák V, Fléchon JE. Immunocytochemical characterisation of rabbit and mouse embryonic fibroblasts. Reprod Nutr Dev 1998;38:683–95. [DOI] [PubMed] [Google Scholar]
- [22].Nishikawa SI, Nishikawa S, Hirashima M, Matsuyoshi N, Kodama H. Progressive lineage analysis by cell sorting and culture identifies FLK1+VE-cadherin+ cells at a diverging point of endothelial and hemopoietic lineages. Development 1998;125:1747–57. [DOI] [PubMed] [Google Scholar]
- [23].Yamashita J, Itoh H, Hirashima M, Ogawa M, Nishikawa S, Yurugi T, et al. Flk-1positive cells derived from embryonic stem cells serve as vascular progenitors. Nature 2000;408:92–6. [DOI] [PubMed] [Google Scholar]
- [24].McCloskey KE, Stice SL, Nerem RM. In vitro derivation and expansion of endothelial cells from embryonic stem cells. Methods Mol Biol 2006;330: 287–301. [DOI] [PubMed] [Google Scholar]
- [25].Schenke-Layland K, Angelis E, Rhodes KE, Heydarkhan-Hagvall S, Mikkola HK, Maclellan WR. Collagen IV induces trophoectoderm differentiation of mouse embryonic stem cells. Stem Cells 2007;25:1529–38. [DOI] [PubMed] [Google Scholar]
- [26].Dai W, Kloner RA. Myocardial regeneration by embryonic stem cell transplantation: present and future trends. Expert Rev Cardiovasc Ther 2006;4: 375–83. [DOI] [PubMed] [Google Scholar]
- [27].Xu Y, Shi Y, Ding S. A chemical approach to stem-cell biology and regenerative medicine. Nature 2008;453:338–44. [DOI] [PubMed] [Google Scholar]
- [28].Mitsui K, Tokuzawa Y, Itoh H, Segawa K, Murakami M, Takahashi K, et al. The homeoprotein Nanog is required for maintenance of pluripotency in mouse epiblast and ES cells. Cell 2003;113:631–42. [DOI] [PubMed] [Google Scholar]
- [29].Nichols J, Zevnik B, Anastassiadis K, Niwa H, Klewe-Nebenius D, Chambers I, et al. Formation of pluripotent stem cells in the mammalian embryo depends on the POU transcription factor Oct4. Cell 1998;95:379–91. [DOI] [PubMed] [Google Scholar]
- [30].Kemler R, Ozawa M, Ringwald M. Calcium-dependent cell adhesion molecules. Curr Opin Cell Biol 1989;1:892–7. [DOI] [PubMed] [Google Scholar]
- [31].Dziadek M, Timpl R. Expression of nidogen and laminin in basement membranes during mouse embryogenesis and in teratocarcinoma cells. Dev Biol 1985;111:372–82. [DOI] [PubMed] [Google Scholar]
- [32].Leivo I, Vaheri A, Timpl R, Wartiovaara J. Appearance and distribution of collagens and laminin in the early mouse embryo. Dev Biol 1980;76:100–14. [DOI] [PubMed] [Google Scholar]
- [33].Grinnell F, Feld MK. Fibronectin adsorption on hydrophilic and hydrophobic surfaces detected by antibody binding and analyzed during cell adhesion in serum-containing medium. J Biol Chem 1982;257:4888–93. [PubMed] [Google Scholar]
- [34].Hayman EG, Pierschbacher MD, Suzuki S, Ruoslahti E. Vitronectinea major cell attachment-promoting protein in fetal bovine serum. Exp Cell Res 1985;160: 245–58. [DOI] [PubMed] [Google Scholar]
- [35].Wilson CJ, Clegg RE, Leavesley DI, Pearcy MJ. Mediation of biomaterial-cell interactions by adsorbed proteins: a review. Tissue Eng 2005;11:1–18. [DOI] [PubMed] [Google Scholar]
- [36].Bale MD, Wohlfahrt LA, Mosher DF, Tomasini B, Sutton RC. Identification of vitronectin as a major plasma protein adsorbed on polymer surfaces of different copolymer composition. Blood 1989;74:2698–706. [PubMed] [Google Scholar]
- [37].Underwood PA, Bennett FA. A comparison of the biological activities of the cell-adhesive proteins vitronectin and fibronectin. J Cell Sci 1989;93:641–9. [DOI] [PubMed] [Google Scholar]
- [38].Fabrizius-Homan DJ, Cooper SL. Competitive adsorption of vitronectin with albumin, fibrinogen, and fibronectin on polymeric biomaterials. J Biomed Mater Res 1991;25:953–71. [DOI] [PubMed] [Google Scholar]
- [39].McFarland CD, Thomas CH, DeFilippis C, Steele JG, Healy KE. Protein adsorption and cell attachment to patterned surfaces. J Biomed Mater Res 2000;49: 200–10. [DOI] [PubMed] [Google Scholar]
- [40].Ludwig TE, Levenstein ME, Jones JM, Berggren WT, Mitchen ER, Frane JL, et al. Derivation of human embryonic stem cells in defined conditions. Nat Biotechnol 2006;24:185–7. [DOI] [PubMed] [Google Scholar]
- [41].Li J, Bardy J, Yap LY, Chen A, Nurcombe V, Cool SM, et al. Impact of vitronectin concentration and surface properties on the stable propagation of human embryonic stem cells. Biointerphases 2010;5:FA132–142. [DOI] [PubMed] [Google Scholar]
- [42].Felding-Habermann B, Cheresh DA. Vitronectin and its receptors. Curr Opin Cell Biol 1993;5:864–8. [DOI] [PubMed] [Google Scholar]
- [43].Schvartz I, Seger D, Shaltiel S. Vitronectin. Int J Biochem Cell Biol 1999;31: 539–44. [DOI] [PubMed] [Google Scholar]
- [44].Yap LY, Li J, Phang IY, Ong LT, Ow JZ, Goh JC, et al. Defining a threshold surface density of vitronectin for the stable expansion of human embryonic stem cells. Tissue Eng Part C Methods 2011;17:193–207. [DOI] [PubMed] [Google Scholar]
- [45].Rowland TJ, Miller LM, Blaschke AJ, Doss EL, Bonham AJ, Hikita ST, et al. Roles of integrins in human induced pluripotent stem cell growth on Matrigel and vitronectin. Stem Cells Dev 2010;19:1231–40. [DOI] [PubMed] [Google Scholar]
- [46].Van Hoof D, Braam SR, Dormeyer W, Ward-van Oostwaard D, Heck AJ, Krijgsveld J, et al. Feeder-free monolayer cultures of human embryonic stem cells express an epithelial plasma membrane protein profile. Stem Cells 2008; 26:2777–81. [DOI] [PubMed] [Google Scholar]
- [47].Hayashi Y, Furue MK, Okamoto T, Ohnuma K, Myoishi Y, Fukuhara Y, et al. Integrins regulate mouse embryonic stem cell self-renewal. Stem Cells 2007; 25:3005–15. [DOI] [PubMed] [Google Scholar]
- [48].Vorotnikova E, McIntosh D, Dewilde A, Zhang J, Reing JE, Zhang L, et al. Extracellular matrix-derived products modulate endothelial and progenitor cell migration and proliferation in vitro and stimulate regenerative healing in vivo. Matrix Biol 2010;29:690–700. [DOI] [PubMed] [Google Scholar]
- [49].Yamada KM, Even-Ram S. Integrin regulation of growth factor receptors. Nat Cell Biol 2002;4:E75–6. [DOI] [PubMed] [Google Scholar]
- [50].Woei Ng K, Speicher T, Dombrowski C, Helledie T, Haupt LM, Nurcombe V, et al. Osteogenic differentiation of murine embryonic stem Cells is mediated by fibroblast growth factor receptors. Stem Cells Dev 2007;16:305–18. [DOI] [PubMed] [Google Scholar]
- [51].Kattman SJ, Huber TL, Keller GM. Multipotent flk-1+ cardiovascular progenitor cells give rise to the cardiomyocyte, endothelial, and vascular smooth muscle lineages. Dev Cell 2006;11:723–32. [DOI] [PubMed] [Google Scholar]
- [52].Wu SM, Fujiwara Y, Cibulsky SM, Clapham DE, Lien CL, Schultheiss TM, et al. Developmental origin of a bipotential myocardial and smooth muscle cell precursor in the mammalian heart. Cell 2006;15:1137–50. [DOI] [PubMed] [Google Scholar]
- [53].Laugwitz KL, Moretti A, Lam J, Gruber P, Chen Y, Woodard S, et al. Postnatal isl1+ cardioblasts enter fully differentiated cardiomyocyte lineages. Nature 2005;433:647–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Moretti A, Caron L, Nakano A, Lam JT, Bernshausen A, Chen Y, et al. Multipotent embryonic Isl1+ progenitor cells lead to cardiac, smooth muscle, and endothelial cell diversification. Cell 2006;127:1151–65. [DOI] [PubMed] [Google Scholar]
- [55].Behfar A, Perez-Terzic C, Faustino RS, Arrell DK, Hodgson DM, Yamada S, et al. Cardiopoietic programming of embryonic stem cells for tumor-free heart repair. J Exp Med 2007;204:405–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.