

Abstract
The majority of clinical deaths in TNBC patients are due to chemoresistance and aggressive metastases, with high prevalence in younger women of African ethnicity. While tumorigenic drivers are numerous and varied, the drivers of metastatic transition remain largely unknown. Here, we uncovered a molecular dependence of TNBC tumors on the TRIM37 network which enables tumor cells to resist chemotherapeutic as well as metastatic stress. TRIM37-directed histone H2A monoubiquitination enforce changes in DNA repair that rendered TP53-mutant TNBC cells resistant to chemotherapy. Chemotherapeutic drugs triggered a positive feedback loop via ATM/E2F1/STAT signaling, amplifying the TRIM37 network in chemoresistant cancer cells. High expression of TRIM37 induced transcriptomic changes characteristic of a metastatic phenotype, and inhibition of TRIM37 substantially reduced the in vivo propensity of TNBC cells. Selective delivery of TRIM37-specific antisense oligonucleotides using anti-folate receptor 1-conjugated nanoparticles in combination with chemotherapy suppressed lung metastasis in spontaneous metastatic murine models. Collectively, these findings establish TRIM37 as a clinically relevant target with opportunities for therapeutic intervention.
Introduction
Triple negative breast cancer (TNBC) is an aggressive breast cancer subtype that accounts for ~20% of all breast cancer cases with the annual incidence rate in the United States estimated to be 40,000 (1). TNBC patients are disproportionately associated with the highest frequency of chemoresistance, relapse, and metastasis. Consequentially, the 5-year survival rate of TNBC is 77% relative to 93% for other breast cancer subtypes (1). Despite the high mortality rate in women worldwide, chemotherapy remains the standard of care for TNBC patients. Although chemotherapy is effective initially in TNBC patients, it is often accompanied by resistance, relapse, and severe side effects. Therefore, new and effective targeted therapies to prevent and ultimately cure TNBC are a clinical priority. While lifestyle, epidemiologic, and cultural factors shape TNBC clinical outcome, the disease etiology is also dependent on biogeographical ancestry (2). Thus, lack of targeted therapies for TNBC is fraught with multiple challenges attributed to limited understanding of genetic complexities, metastatic biology, and drivers of metastatic traits.
An unresolved question in cancer biology is what drives a primary tumor to become metastatic? This is a clinically relevant question because metastatic, not primary tumors, are fatal. In general, numerous oncogenes and tumor suppressors are genetically or epigenetically altered in cancer and accumulate during tumorigenesis. But whether drivers of tumorigenesis are also the causal factor of the metastatic transition remains to be addressed. To this end, extensive transcriptomic and genetic scans of evolving carcinomas revealed mutations that were represented in premalignant biopsies but not in tumor biopsies, suggesting divergence of genetic alterations during the transition from primary to regional metastases (3,4). Additionally, dynamic epigenetic mechanisms are also intimately linked to metastatic transitions (5,6). For example, TNBC tumors harbor a high frequency of hypermethylated promoters in commonly targetable drivers, such as TP53, BRAF, KRAS, and EGFR (7). Alterations in epigenetic factors causing neomorphic mutations (e.g., EZH2, DNMT3A) or translocations (e.g., NSD2, MMSET) are also frequent in cancer patients (6). As such, several small molecules targeting epigenetic regulators have entered clinical trials, for example, Estinosat, Belinostat, and Panobinostat (8). Understandably, these drug treatments are not mutation-specific and thus, pose a significant toxicity risk to untransformed cells, underscoring the critical need for targeted therapies.
We have originally described tripartite motif-containing protein 37 (TRIM37) as a breast cancer oncoprotein that can epigenetically silence tumor suppressors (9,10). Clinically, high-TRIM37 associate with poor overall survival (9). Mechanistically, TRIM37 mono-ubiquitinates histone H2A at Lys119 (H2Aub) to down-regulate target genes (9). Functionally, TRIM37 over-expression renders non-transformed breast cells tumorigenic, and inhibition of TRIM37 function reduces tumor growth (9). While TRIM37 promotes tumorigenesis, its function in breast cancer metastasis and the therapeutic implications of TRIM37 targeting remain to be demonstrated.
Metastasis is a multistep process that includes pathways regulating the epithelial-mesenchymal transition, infiltration of distant sites, and metastatic growth (11). Given the majority of TNBC patients receive chemotherapy, the ability to resist therapy-induced cell death is perhaps the first step towards a metastatic phenotype. Indeed, recent evidence revealed synchronized expression of genes involved in surviving the stress of chemotherapy as well as overcoming the natural barriers of metastatic growth. For example, a CXCL1/2 paracrine pathway (12) and MTDH (13) were recently identified with dual functionality in metastasis and chemoresistance. Here, we uncover that TRIM37 alters DNA damage response to prevent therapy-induced cell death, and enforces a transcriptional program favoring metastasis. In particular, we report that selective TRIM37 inhibition in TNBC tumors suppress lung metastases in vivo. Together, these data reveal that TRIM37 is a new epigenetic driver of aggressive TNBC biology, which can be targeted to simultaneously increase chemotherapy efficacy and reduce metastasis risk in TNBC patients.
Materials and Methods
Cell lines and cell culture
MDA-MB-231-luc-D3H2LN-BMD2b (provided by Takahiro Ochiya), MDA MB 231, MDA MB 468 and HCC1806 (provided by Michael J. Lee) cells were maintained in RPMI medium supplemented with 10% fetal bovine serum (FBS, Invitrogen) at 37°C and 5% CO2. MCF10A, MCF7, MCF10AT, HCC1806RR (provided by Sophia Ran) and TP53−/− MCF10A (provided by David Weber and Michele Vitolo) were cultured as described previously (9,14,15). Cells cultured at the same time were pooled together and then seeded after counting in a 6-well or 10-cm dish. Cells were then subjected, in a random order, to treatment with a control or test different biologics, which included shRNA, sgRNA, vectors, and small molecule inhibitors. Cells were routinely tested for mycoplasma using PlasmoTest kit from (Invivogen).
Animal Care
NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ and Balb/cJ (Jackson Laboratory) were housed in a specific-pathogen-free facility accredited by the American Association of Laboratory Animal Care. All animal studies were approved (#4112 and #4222) by the Institutional Animal Care and Use Committee.
CRISPR/Cas9 targeting
The Gapdh sgRNAs were cloned in pLentiCrispr v2 plasmid (Addgene) and packaged into virus as recommended by the manufacturer. Cells were infected with packaged virus as described previously (9).
Recombinant Antibody Cloning
Farletuzumab (anti-human FOLR1) and LK26 (anti-mouse FOLR1) antibodies were cloned, engineered, expressed, and purified as described previously (16). To generate an antibody conjugate platform for nanoparticles linkage, a novel linker sequence [(X)3Cys(X)3 amino acid sequence] was engineered in continuation of the carboxy terminal of heavy chain (called Fc-Linkered). The knob chain is exactly similar to hole chain except for the presence of this unique cysteine residue.
Recombinant Antibody Expression
Free style CHO-S cells (Invitrogen) were cultured and maintained according to supplier’s recommendations (Life technologies). A ratio of 2:1 (light chain, VL: heavy chain, VH) DNA was transfected using 1 mg/ml polyethylenimine and cultured at 37°C. After 24 hrs., transfected cells were cultured at 32°C for additional 9 days. Cells were fed every 2nd day with 1:1 ratio of Tryptone feed and CHO Feed B and antibodies were purified as described previously (16). An Autodesk Inventor Professional 2020 was used to draw the design of antibody.
Nanoparticle synthesis and packaging
Cholesterol, 1,2-dioleoyl-3-trimethylammonium-propane chloride salt (DOTAP), 1,2-dioleoyl-sn-glycero-3-phosphate (DOPA), and 1,2-distearoryl-sn-glycero-3-phosphoethanolamine-N-[maleimide-(polyethyleneglycol-2000)] ammonium salt (DSPE-PEG2000-Maleimide) were purchased from Avanti Polar Lipids, Inc. (Alabaster).
TRIM37-ASO (IDT) and control-ASO (IDT) loaded bilayer nanoparticles were prepared as previously described with modification (17). Briefly, 150mM CaCl2 with 0.5mM ASO were dispersed in Cyclohexane/Igepal CO-520 (70:30 v/v) solution to form water-in-oil reverse micro-emulsion (Solution A). The phosphate phase was prepared by mixing 1.5 μM NaHPO4 (pH=9.0) and 6.9 μM DOPA in Cyclohexane/Igepal CO-520 (70:30 v:v) solution (Solution B). Calcium phosphate-alginate (CaP) core was prepared by mixing Solutions A and B. For assembly of outer leaflet, CaP core was mixed with 0.5 μM DOTAP/Cholesterol (1:1), and 0.15 μM DSPE-PEG-2000-Maleimide. CaP-bilipids nanoparticles were mixed with 0.56 μM engineered Farletuzumab solutions and incubated overnight at 4°C. Finally, CaP-bilipids nanoparticles were sterile filtered for subsequent experiments. An Autodesk Inventor Professional 2020 was used to draw the design of smart nanoparticle.
NSG tumor xenograft studies
For indicated cell lines, weight and aged matched female NSG mice were injected subcutaneously with 2x106 TNBC cells in their right flank as described previously (9). For in vivo Dox-induced TRIM37 upregulation studies, mice bearing ~200 mm3 HCC1806 tumors weight matched animals were randomly assigned into groups and injected with 2 mg/kg Dox. Tumors were harvested 24 hrs. following Dox treatment. For smart nanoparticles efficiency in vivo, mice bearing ~200 mm3 231-2b tumors weight matched animals were randomly assigned into groups and injected with control or smart nanoparticles. At the endpoint, RNA and protein lysates were prepared from isolated tumors.
Bioluminescent imaging (BLI)
150 mg/kg luciferin (Perkin Elmer Inc.) was administered to mice intraperitoneally and mice were imaged as described previously (16).
Spontaneous metastatic tumor studies
2×106 4T1 or 231-2b cells were injected into the inguinal mammary fat pad or right flank of 6 to 8-week-old female Balb/cJ or NSG mice and tumor growth was monitored as previously described (9). Where indicated, primary tumors were resected and animals were allowed to develop lung metastases. Mice bearing TNBC tumors were weight matched and randomly assigned into groups that received 1.2 mg/kg smart or control nanoparticles at the indicated times. The lung metastases in the animals were monitored by BLI. At the termination of the experiment, animals were euthanized and indicated tissues were harvested and processed for histological examination and immunohistochemical staining or for qRT-PCR analysis. Metastatic burden was calculated either as the number of visible metastatic lesion in each organ or as the relative luminescence signal from gross organ tissue.
Experimental metastasis in vivo
3x105 231-2b cells were inoculated directly into the left cardiac ventricle. Metastatic growth was monitored using BLI. Lungs, liver, femurs and brains were harvested post-mortem and processed for histological examination and gross analysis.
Bioinformatic analysis
cBioPortal was used to obtain TCGA expression z-scores for genes in Molecular Taxonomy of Breast Cancer International Consortium (METABRIC) cohort (18). FireBrowse (Broad Institute) was used to obtain normal breast tissue expression z-scores. Cooccurrence and mutual exclusivity analysis were performed with cBioPortal. Hazard ratios were assessed with Cox proportional hazard model for which TNBC and non-TNBC patients were stratified into high- and low-TRIM37 as third and first quartile. For Kaplan-Meier analysis, the p-value was calculated with log-rank test. For boxplot analysis, TRIM37-regulated genes were stratified according to TRIM37 expression using z score thresholds (z>0.5, z<−0.5).
Statistical analysis
All experiments were performed at least in triplicate and the results presented are the mean of at least three different biological replicates. The comparisons between the two groups were done by unpaired t-test; comparisons between multiple treatment groups were done by one-way or two-way ANOVA with indicated multiple comparisons post hoc tests. The enrichment of genes positively correlating with TRIM37 were calculated with two-tailed Fisher’s exact test. The distributions in correlation between GO terms was calculated by Kolmogorov–Smirnov test. All statistical analyses were performed using R/Bioconductor (version 2.15.2).
Results
TRIM37 associates with double strand break (DSB) repair machinery in TNBC
Several observations provided a rationale to explore the potential role of TRIM37 in promoting resistance to chemotherapeutic drugs that induce high level of DNA damage. TRIM37 catalyzes mono-ubiquitination of H2A (9), a chromatin modification enriched at transcriptionally repressed gene promoters (19) as well as at DNA damage sites (20). Furthermore, our analysis of the METABRIC cohorts revealed TRIM37 association with DNA repair genes (Fig.1A, Supplemental Table I), which was significantly stronger than genes involved in cellular proliferation (Supplementary Fig.1A, Supplemental Table I). Double strand break (DSB) repair is one of the major pathways to repair damaged DNA in cancer cells and its kinetics predicts resistance to therapy (21,22). We therefore limited the analysis to repair proteins that participate in homologous recombination (HR) and non-homologous end-joining repair pathways (NHEJ), the two subtypes of DSB repair. RAD51C, XRCC5 (Ku80), RNF8, XRCC6BP1 (Ku70 binding protein), RNF168 and MRE11 were among the DSB genes whose expression most strongly associated with TRIM37 (Fig.1A). RAD51-associated proteins generally promote HR repair (23). XRCC5 and XRCC6BP1 are involved in NHEJ repair pathway (23). MRE11 forms complex with NBS1 and RAD50 to regulate response at DSB (24). RNF8 and RNF168 are E3 ligases that promote binding of DSB genes (23). Additionally, TRIM37 also correlated with the overexpression of a family of other DSB factors, including RAD51AP1, SFR1, DDX1, RAD51, ERCC1 and CHEK2. Together, these findings identified 28 DSB genes that significantly correlated with TRIM37 in breast cancer patients (Pearson’s coefficient>0.2; Supplemental Table I).
Figure 1: TRIM37 associates with DSB repair proteins in TNBC.
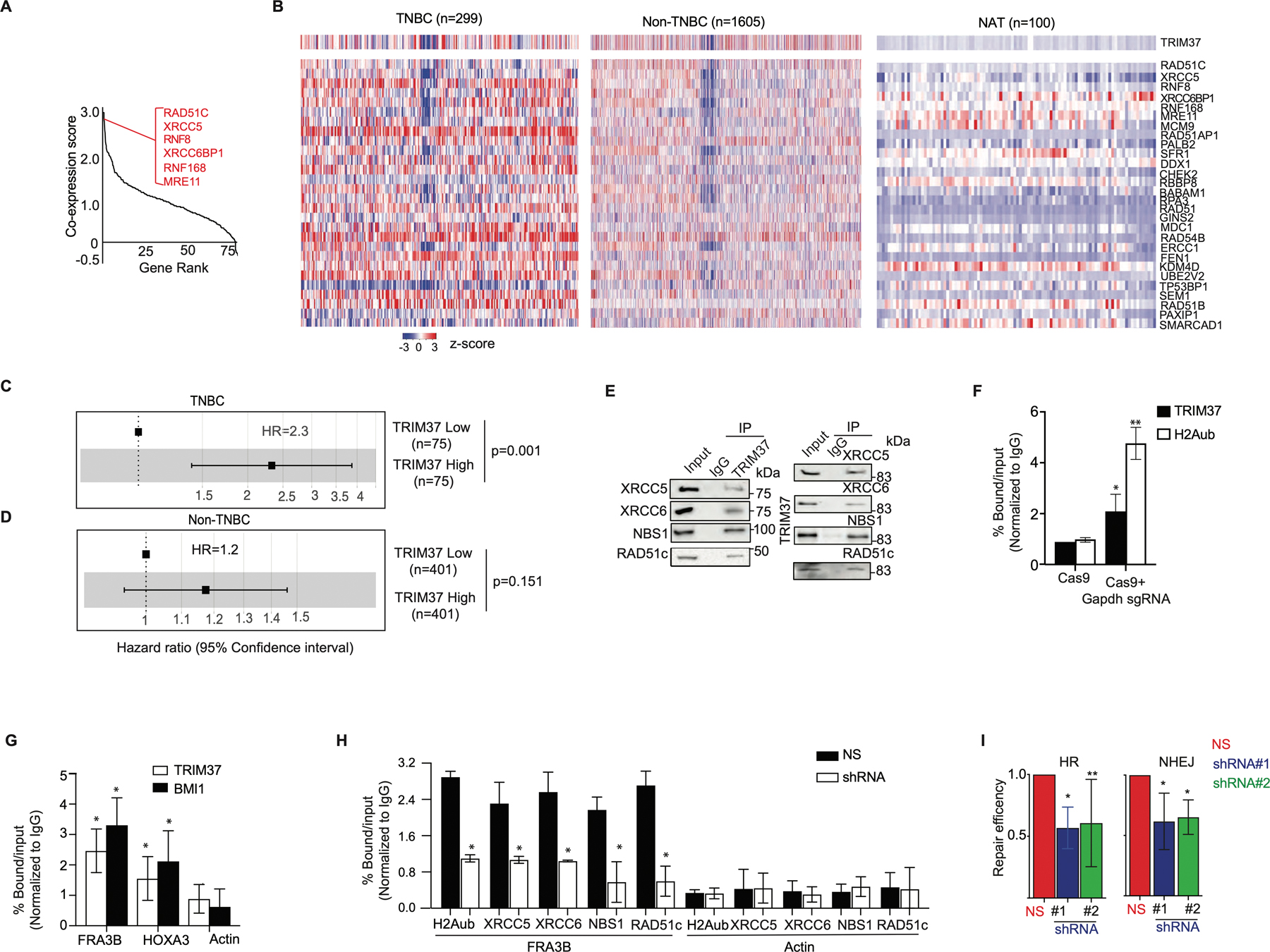
(A) Gene ranking of DNA repair genes according to cooccurrence with high-TRIM37 in breast cancer patients. The inset shows genes that have the highest correlation with TRIM37. A complete list of DNA repair genes that correlate with TRIM37 is presented in Supplemental Table I. (B) Heat map for the expression of TRIM37 and 28 DSB repair genes in breast cancer patients stratified by TNBC (n=299), non-TNBC (n=1605), and NAT (n=100). n, number of samples. (C-D) Forest plot of HR in TNBC (C) and non-TNBC (D) patients stratified for high- and low-TRIM37 expression using METABRIC cohorts. (E) Immunoblot monitoring XRCC5, XRCC6, NBS1, RAD51C, and TRIM37 in protein complexes pulled down by either anti-TRIM37 (Left), the indicated DSB proteins (Right), or an IgG control. Input, ~1–5% of whole cell lysates. (F) ChIP monitoring TRIM37 and H2Aub binding at Gapdh in MDA MB 468 cells expressing either Cas9 alone or with Gapdh site-specific sgRNA. (G) ChIP monitoring TRIM37 and BMI1 binding at FRA3B, HOXA3, and Actin in MDA MB 468 cells treated with doxorubicin (Dox). (H) ChIP monitoring H2Aub, XRCC5, XRCC6, NBS1, and RAD51c binding at FRA3B and Actin in MDA MB 468 cells expressing a non-silencer (NS) or TRIM37 shRNA. (I) HR (Left) and NHEJ (Right)-mediated DSB-repair activity in MDA MB 468 cells expressing control or TRIM37 shRNA (#1, #2). Error bars indicate standard deviation and range of at least three biological replicates. * p<0.05; ** p<0.01; *** p<0.001.
The analysis of METABRIC cohorts stratified by breast cancer subtypes revealed statistically significant correlation between TRIM37 and DSB genes in TNBC patients but not in non-TNBC patients or histologically normal breast tissue adjacent to tumor (NAT, Fig.1B and Supplementary Fig.1B). As shown in Fig.1C, the hazard ratio (HR) was ~2.3-fold higher for TNBC patients with high- compared to low-TRIM37 expression, linking TRIM37 to poor prognosis in TNBC patients. In contrast, no significant association between TRIM37 levels and survival was observed in non-TNBC patients (Fig.1D). Likewise, higher expression of a subset of DSB genes analyzed predicted poor overall-survival in TNBC patients as determined by HR>~1 (Supplementary Fig.1C).
We next performed a series of functional experiments to determine the molecular mechanisms underlying the biological activity of TRIM37 in DSB repair using several human TNBC cell lines (HCC1806, MDA MB 468 and MDA MB 231; Supplementary Fig.1D). We first asked whether TRIM37 was physically associated with DSB genes. To test this idea, HCC1806 whole cell extract was fractionated by sucrose gradient sedimentation, and individual fractions were analyzed for TRIM37 and a representative subset of DSB proteins identified in Fig.1A. Results shown in Supplementary Fig.1E demonstrated that TRIM37 co-sediments with XRCC5, XRCC6, RAD51c, MRE11, and NBS1. Physical interactions between TRIM37 and DSB proteins could be confirmed by co-immunoprecipitation (Fig.1E and Supplementary Fig.1F) and immunofluorescence analysis (Supplementary Fig.1G) in MDA MB 468 cells treated with doxorubicin (Dox), a first-line chemotherapeutic agent. No significant changes in the expression of DSB genes were observed in HCC1806 cells expressing TRIM37-specific short hairpin RNA (shRNA), excluding TRIM37-mediated transcriptional regulation of these DSB genes (Supplementary Fig.1H–I, Supplemental Table II).
Prompted by these findings, we interrogated TRIM37 recruitment to DSB using two independent experimental systems. We first enzymatically-induced DSB at Gapdh using sequence-specific guide RNA (sgRNA) to promote Cas9-nuclease binding. Chromatin immunoprecipitation (ChIP) assay confirmed TRIM37 as well as H2Aub enrichment at the DSB in Gapdh (Fig.1F). Next, we analyzed recruitment of TRIM37 and BMI1, a component of the polycomb complex that participates in DSB repair (25), to the endogenous fragile site, FRA3B. Consistently, ChIP analysis confirmed TRIM37 and BMI1 binding to FRA3B following Dox treatment (Fig.1G). In marked contrast to the control cells, knockdown of TRIM37 substantially decreased H2Aub enrichment as well as DSB proteins binding to FRA3B in Dox-treated MDA MB 468 cells (Fig.1H). As expected, repair efficiencies of NHEJ and HR pathways were significantly reduced in TRIM37-knockdown cells compared to control cells (Fig.1I). Together, these results demonstrate that TRIM37 interacts with DSB repair factors and functionally contributes to the repair of therapy-induced DNA damage.
TRIM37 catalyzed H2Aub is required for its function in chemoresistance
We next directly examined the impact of TRIM37-knockdown on chemotherapy-induced DNA damage and clonogenic growth. Knockdown of TRIM37 resulted in ~6-fold increase in median tail length in comet assay (Fig.2A and Supplementary Fig.1H,2A) and ~5-fold increase in the median nuclear coverage of phosphorylated histone H2AX (γ-H2AX, Fig.2B) in Dox-treated cells. TRIM37-knockdown in TNBC cell lines also markedly increased Caspase 3 activity and PARP cleavage (Fig.2C), hallmarks of cell death. By contrast, Dox-treated MCF7, a hormone receptor positive breast cancer cell line, did not augment PARP cleavage or Caspase 3 activity (Fig.2C), suggesting TRIM37 function in chemoresistance was limited to TNBC cells. As expected, knockdown of TRIM37 sensitized MDA MB 468 cells to chemotherapeutic stress without affecting proliferation as indicated by substantially decreased clonogenic growth relative to the control cells (Fig.2D and Supplementary Fig.2B).
Figure 2. TRIM37-catalyzed H2Aub is required for chemoresistance in TNBC.
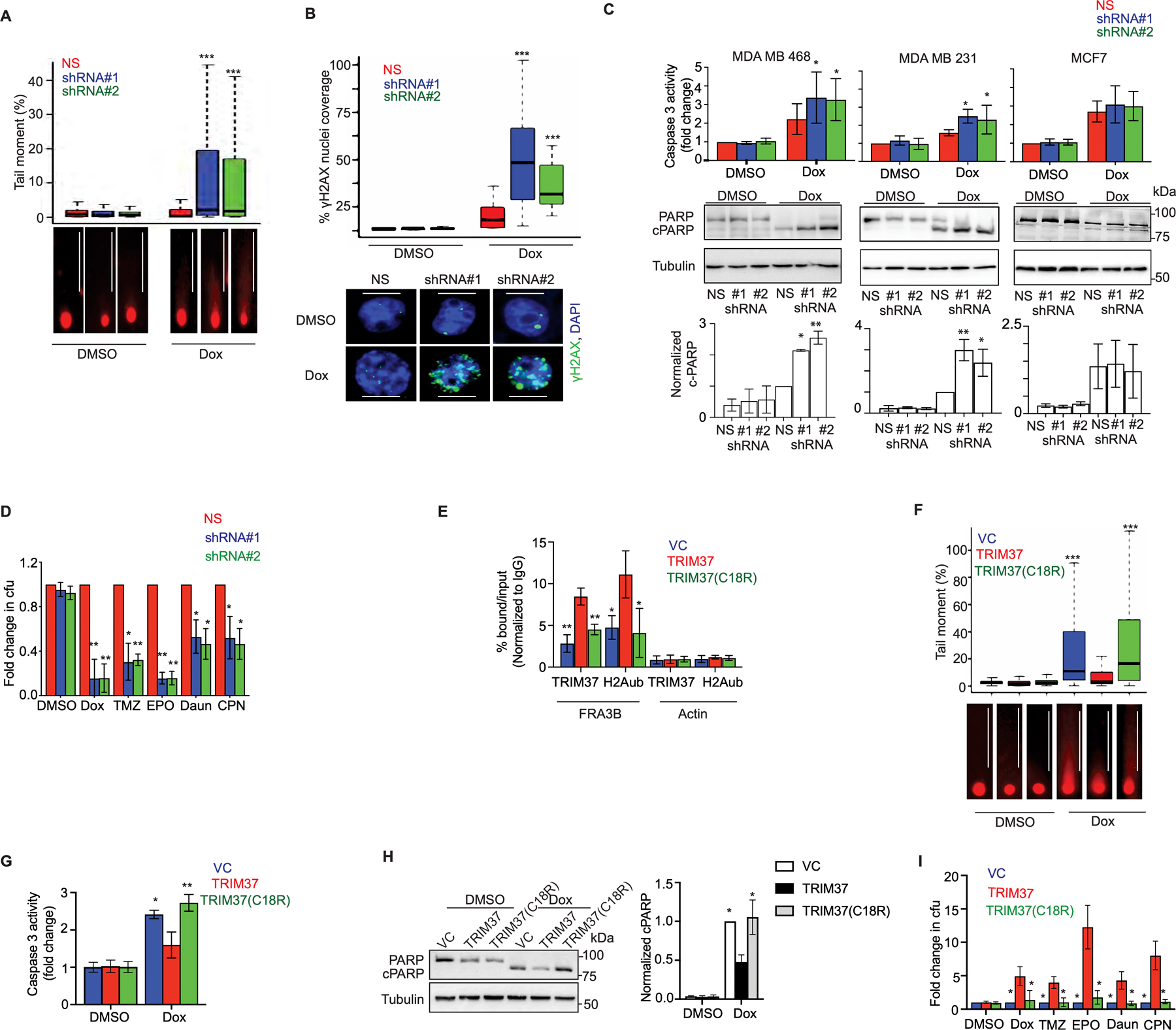
(A) Top, Tail moment in DMSO or Dox-treated MDA MB 468 cells expressing non-silencer (NS) or TRIM37 shRNA (#1, #2). Bottom, Representative images of the tails for each group are shown. Scale bars, 100 μm. (B) Top, Quantification of γ-H2AX foci in MDA MB 468 cells expressing NS or TRIM37 shRNA (#1, #2) following treatment with Dox. Bottom, Representative immunofluorescence images of γ-H2AX foci (Green) in Dox-treated MDA MB 468 cells expressing NS or TRIM37 shRNA (#1, #2). DAPI (Blue) stains the nucleus. Scale bars, 50 μm. (C) Caspase 3 activity assay (Top) and immunoblot for PARP and cleaved PARP (c-PARP) (Middle) in DMSO or Dox-treated MDA MB 468, MDA MB 231, and MCF7 cells expressing NS or TRIM37 shRNA (#1, #2). Quantification of PARP cleavage relative to total PARP is shown (Bottom). (D) Quantification of the fold change in chemotherapeutic drug-resistant colonies obtained for MDA MB 468 cells expressing either NS or TRIM37 shRNA (#1, #2) by a clonogenic assay. Cells were treated with DMSO, Dox, temozolomide (TMZ), etoposide (EPO), daunorubicin (Daun), and cisplatin (CPN). Results were normalized to the colony forming unit (cfu) for DMSO. (E) ChIP monitoring TRIM37 and H2Aub binding at FAR3B and Actin in MCF10AT cells expressing vector control (VC), TRIM37, or mutant TRIM37 (TRIM37(C18R)). (F) Top, Tail moment in DMSO or Dox-treated VC-, TRIM37-, or TRIM37(C18R)-expressing MCF10AT cells. Bottom, Representative images of the tail moment for each group are shown. Scale bars, 100 μm. (G-H) Caspase 3 activity assay (G) and immunoblot for PARP and c-PARP (H, Left) in DMSO or Dox-treated VC, TRIM37 or TRIM37(C18R) expressing MCF10AT cells. Quantification of PARP cleavage relative to total PARP is shown (Right). (I) Quantification of the fold change in chemotherapeutic drug resistant colonies obtained for VC, TRIM37, or TRIM37(C18R) by clonogenic assay. Cells were treated with DMSO, Dox, temozolomide (TMZ), etoposide (EPO), daunorubicin (Daun), and cisplatin (CPN). Results were normalized to the cfu for DMSO. Error bars indicate standard deviation and range of at least three biological replicates. * p<0.05; ** p<0.01; *** p<0.001.
Given ubiquitin is critical for DSB factor recruitment to damaged DNA (26), we next asked whether TRIM37-catalyzed H2Aub is required for its function in chemoresistance. To test this idea, we ectopically expressed either wild type TRIM37 (TRIM37) or catalytically-dead TRIM37 (TRIM37(C18R)) in MCF10AT, a premalignant K-RAS transformed triple negative breast cell line (Supplementary Fig.2C). While Dox-treatment induced significant enrichment of H2Aub at FRA3B in TRIM37 overexpressing cells, TRIM37(C18R) failed to promote H2Aub enrichment at FRA3B (Fig.2E). Consequentially, Dox-treatment of TRIM37(C18R)-expressing cells caused substantially longer comet tails (Fig.2F) as well as increased Caspase 3 activity (Fig.2G) and PARP cleavage (Fig.2H) relative to TRIM37-expressing cells. Finally, substantially fewer colonies were observed for cells expressing TRIM37(C18R) compared to TRIM37 following chemotherapeutic stress (Fig.2I and Supplementary Fig.2D).
TRIM37 promotes chemoresistance in the absence of functional p53
Wild type TP53 represents a barrier to chemoresistance by altering DSB repair responses, activating checkpoints and the stress responses (27). Strikingly, TNBC tumors frequently harbor disrupting TP53 mutations (Supplementary Fig.3A), which primarily cause the loss of its wild type function (28). Surprisingly, analysis of representative TRIM37-associated DSB genes showed a striking correlation between TRIM37 and DSB genes expression in TNBC patients carrying mutant TP53 but not in wild type TP53 (Fig.3A and Supplementary Fig.3B–D). Consistently, TP53 mutant, but not wild type, TNBC patients with high-TRIM37 were at ~2.3-fold higher risk of death relative to low-TRIM37 (Fig.3B–C).
Figure 3. TRIM37 reduces cytotoxicity of chemotherapy in the absence of functional p53.
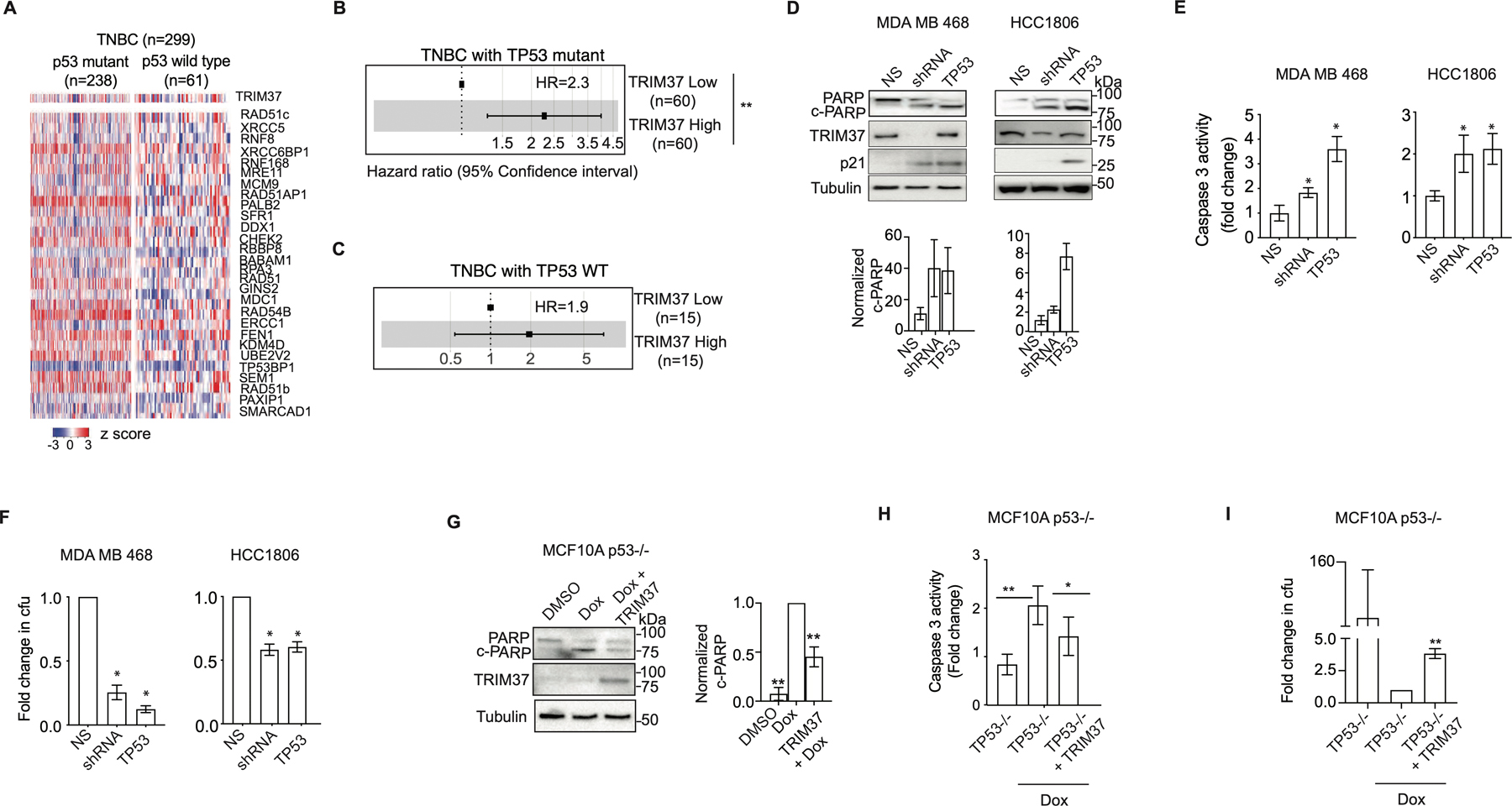
(A) Heat map for expression of TRIM37 and DSB genes in TNBC patients stratified by TP53 status (wild type (n=238) and mutant (n=61)). n, number of patients. (B-C) Forest plot of HR for TNBC patients with mutant TP53 (B), or wild type TP53 (C) stratified for high- and low-TRIM37 expression. (D) Immunoblots in Dox-treated MDA MB 468 and HCC1806 cells expressing NS, TRIM37 shRNA, or TP53. Tubulin is the loading control. Bottom, Quantification of c-PARP relative to total PARP. (E) Caspase 3 activity assay in Dox-treated MDA MB 468 and HCC1806 expressing NS, TRIM37 shRNA, or TP53. (F) Quantification of the fold change in cfu for Dox-treated MDA MB 468 and HCC1806 cells expressing NS, TRIM37 shRNA, or TP53 by clonogenic assay. (G) Immunoblots in Dox-treated p53−/− MCF10A cells expressing TRIM37. Tubulin is the loading control. Right, Quantification of c-PARP relative to total PARP. (H) Caspase 3 activity assay in Dox-treated p53−/− MCF10A cells expressing either vector control or TRIM37. (I) Quantification of the fold change in cfu for p53−/− MCF10A cells expressing TRIM37 plated after Dox treatment by clonogenic assay. Error bars indicate standard deviation and range of at least three biological replicates. * p<0.05; ** p<0.01; *** p<0.001.
To investigate the relationship between p53 and TRIM37, we transiently expressed wild type p53 in MDA MB 468 (carries transcriptionally inactive p53 R273H) and HCC1806 (carries p53 T256Kfs*90 deletion) cells. For each cell line, p53-reconstituted TNBC cells showed significantly higher PARP cleavage (Fig.3D) and Caspase 3 activity (Fig.3E) compared to the control cells following Dox-treatment. A clonogenic assay confirmed that p53 over-expression sensitized MDA MB 468 (~4-fold) and HCC1806 (~2-fold) cells to Dox despite high levels of TRIM37 (Fig.3F and Supplementary Fig.3E). Reciprocally, ectopic expression of TRIM37 in genetically ablated p53 null (p53−/−) MCF10A cells (14) substantially reduced PARP cleavage, and Caspase 3 activity relative to empty vector (Fig.3G–H). As expected, TRIM37-expressing p53−/− MCF10A showed an ~4-fold increase in colony formation in comparison to control cells (Fig.3I and Supplementary Fig.3F). In summary, consistent with previous results for MDM2, KRAS, and ARID1 A (29), we find that TRIM37 requires loss of p53 to drive the chemoresistant phenotype in TNBC cells.
Chemotherapy amplifies a TRIM37 survival axis in TNBC
Most TNBC patients receive chemotherapy, which is effective in early stages of the disease but ~30–50% patients develop resistance (1). While the exact mechanisms of chemoresistance remain to be understood, chemotherapeutic drugs often induce genomic and transcriptomic reprogramming of resistant signatures (30), including alterations in DNA repair capacity (31). Previous studies have suggested that accumulation of such changes accompany selection and expansion of resistant TNBC cells (32). We therefore analyzed the expression of TRIM37 in MDA MB 468, HCC1806 and MDA MB 231 cells following chemotherapy. Surprisingly, we found that TRIM37 is upregulated in all the three TNBC cell lines tested in a time-dependent manner (Fig.4A). In contrast, no significant increase in TRIM37 was observed in p53 wild type MCF7 or MCF10A, an immortalized breast epithelial cell (Fig.4A). The analysis of TRIM37 upregulation kinetics in MCF7 and MCF10a revealed quick and robust p53 activation following Dox treatment (Supplementary Fig.4A), supporting our previous findings that p53 overrides TRIM37 function in chemoresistance (Fig.3). Indeed, ectopic expression of p53 in MDA MB 468 cells obliterated Dox-induced TRIM37 upregulation (Supplementary Fig.4B). As expected, TNBC cells treated with additional chemotherapeutic drugs also increased TRIM37 expression post-treatment (Fig.4B). Finally, increased TRIM37 level in xenograft tumors following dox-treatment revealed therapy-induced transcriptional upregulation of TRIM37 in vivo (Fig.4C–D).
Figure. 4. Chemotherapy amplifies oncogenic TRIM37 network in TNBC.
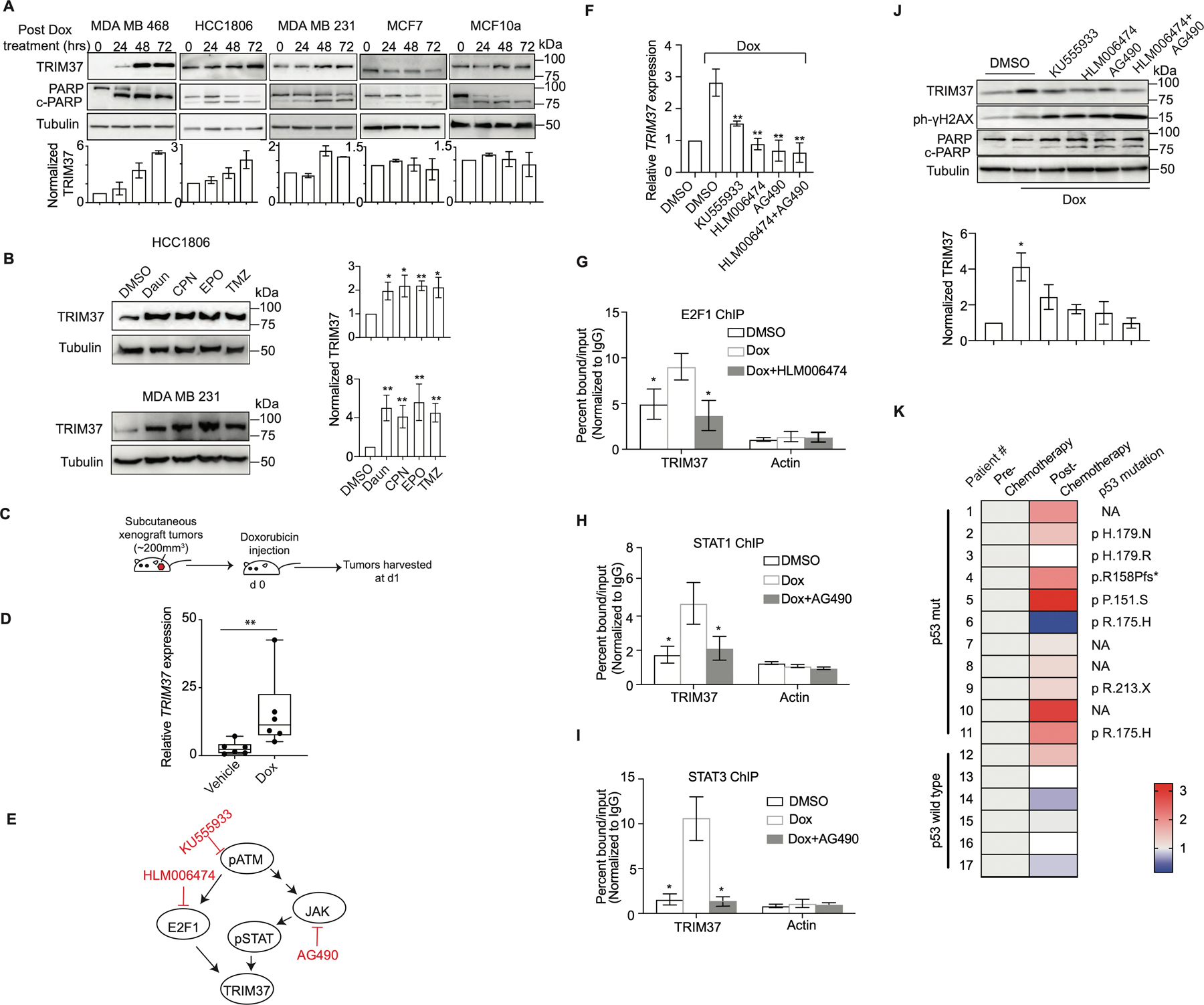
(A) Top, Immunoblots in MDA MB 468, HCC1806, MDA MB 231, MCF7, and MCF10A cells treated with Dox for 0, 24, 48 and 72 hrs. Tubulin is the loading control. Bottom, Quantification of TRIM37 relative to Tubulin. (B) Immunoblot monitoring TRIM37 in HCC1806 (Top) or MDA MB 231 (Bottom) cells treated with daunorubicin (Daun), cisplatin (CPN), etoposide (EPO) or temozolomide (TMZ). Tubulin is the loading control. Right, Quantification of TRIM37 relative to Tubulin. (C) Schematic showing that NSG mice were treated with intraperitoneal injection of 2 mg/kg Dox once tumor reached the size of ~200 mm3. Tumors were harvested postmortem. (D) qRT-PCR monitoring TRIM37 expression in HCC1806 subcutaneous tumors-derived from NSG mice following treatment with Dox. n=6 animals per group. (E) Schematic of ATM signaling with the downstream effectors of ATM, E2F1, and STAT. Specific small molecule inhibitors of the ATM signaling are also indicated (Red). (F) qRT-PCR monitoring TRIM37 expression in MDA MB 468 cells following treatment with KU555933, HLM006474 or AG490 in combination with Dox. (G-I) ChIP analysis monitoring the binding of E2F1 (G), STAT1 (H), and STAT3 (I) to TRIM37 and Actin in MDA MB 468 cells treated with HLM006474 or AG490 in combination with Dox. (J) Top, Immunoblots in MDA MB 468 cells treated with Dox and the indicated inhibitors of ATM signaling. Tubulin is the loading control. Bottom, Quantification of TRIM37 relative to Tubulin. (K) Heat map for TRIM37 expression in mutant and wild type TP53 TNBC tumor tissue samples pre- and post-chemotherapy. The type of TP53 mutation is indicated on the right. NA, not available. (n = 17). Error bars indicate standard deviation and range of at least three biological replicates. * p<0.05; ** p<0.01; ***p<0.001.
We next sought to determine the mechanistic basis for chemotherapy-induced burst in TRIM37 levels in TNBC tumors. A previous study identified TRIM37 association with ataxia-telangiectasia-mutated (ATM) kinase, a DNA damage sensor (33). Moreover, TRIM37 promoter harbors regulatory elements for STAT and E2F1, downstream effectors of ATM kinase (Supplementary Fig.4C–E). We therefore investigated the potential role of ATM signaling in the transcriptional regulation of TRIM37 by utilizing small molecule inhibitors of either ATM or its downstream effectors, E2F1 and JAK (Fig.4E and Supplementary Fig.4F–J). JAK is a tyrosine kinase that phosphorylates STAT. qRT-PCR analysis revealed that pharmacological inhibition of ATM or its downstream effectors blocks TRIM37 upregulation following Dox-treatment (Fig.4F). Similarly, E2F1- and STAT1/3-knockdown abolished Dox-induced TRIM37 upregulation (Supplementary Fig.4K–L), whereas their overexpression significantly increased TRIM37 levels (Supplementary Fig.4M–N). ChIP analysis confirmed Dox-induced STAT1, STAT3, and E2F1 recruitment to TRIM37, which substantially decreased following pharmacological inhibition of JAK or E2F1 activation (Fig.4G–I). Consequently, inhibition of ATM signaling in MDA MB 468 cells induced significantly higher DNA damage and cell death relative to control cells as determined by γ-H2AX and PARP cleavage (Fig.4J).
Finally, to clinically validate chemotherapy-induced burst in TRIM37, we analyzed TRIM37 expression in a panel of matched pre- and post-neoadjuvant chemotherapy-treated tumor biopsies from TNBC patients (see also Methods). qRT-PCR analysis showed that chemotherapy treatment increased TRIM37 expression in ~82% of TNBC tumors carrying mutation in TP53 (n=11, Fig.4K). Consistent with TNBC cellular models, no significant change in TRIM37 was observed in TP53-wild type TNBC tumors (n=6; Fig.4K). Collectively, our results show that chemotherapeutic stress increases TRIM37 expression in TNBC tumors in an ATM-dependent manner.
TRIM37 remodels transcriptional program favoring TNBC metastasis
While the emergence of chemoresistance is closely related to metastasis, the ability of a cancer cell to survive and proliferate under continuous standard chemotherapy does not warrant metastasis. Our analysis of previously published expression profiling of PDX mammary fat pad primary and corresponding lung metastatic tumors (34) revealed higher TRIM37 in metastatic lesions (Supplementary Fig.5A). These results indicated that higher TRIM37 levels are maintained throughout the metastatic transition.
TRIM37 in association with polycomb complex alters gene expression to promote tumorigenesis (9). To test whether TRIM37 causes transcriptional misregulation of genes involved in metastasis, we knocked down TRIM37 in MDA-MB-231-D3H2LN-2b (35), hereafter referred to as 231-2b, using TRIM37-specific ASO (TRIM37-ASO, Supplementary Fig.5B) and performed transcriptomic analysis. Of the ~2,600 genes whose expression differed significantly between TRIM37-knockdown and control cells (GSE136617; Fig.5A–B), ~71 tumor and metastases suppressors, such as KISS1 and BRMS1, were significantly down-regulated by TRIM37 (Supplemental Table III). As robust metastatic suppressors, KISS1 and BRMS1 reciprocally correlate with increased tumor recurrence, metastatic foci and reduced disease-free survival (36,37). The anti-metastatic function is mediated by altered gene expression through cell signaling pathways (38,39) as well as transcriptional regulation (40,41). Additionally, gene set enrichment analysis (GSEA) revealed that TRIM37-knockdown downregulates hypoxia, EMT transition, glycolysis, angiogenesis, inflammatory, and immune response-related genes in TNBC cells, indicating TRIM37-dependent activation of a pro-metastatic transcriptional program (Fig.5C and Supplementary Fig.5C). Likewise, KEGG pathway analysis identified TRIM37 target genes that associated with focal adhesion, pathways in cancer, actin cytoskeleton, ECM interaction, and signaling pathways (Supplementary Fig.5D).
Figure 5. TRIM37 alters the transcription program to favor metastatic growth of TNBC tumors.
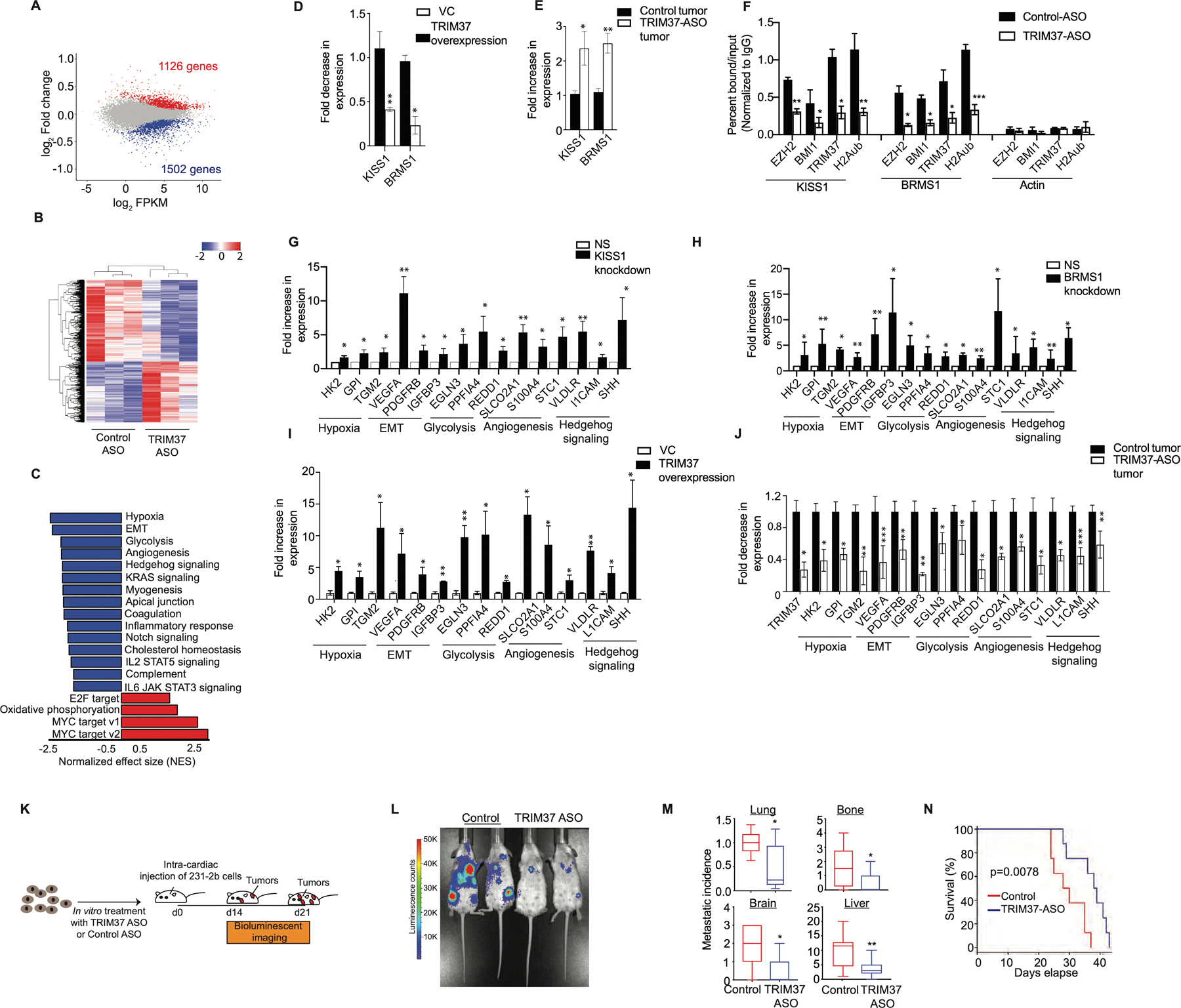
(A) MA plot illustrates differential gene expression in TRIM37-ASO-treated compared to control 231-2b cells. Red are significantly upregulated genes (n=1126), blue are significantly downregulated genes (n=1502) and grey are genes not significantly changed (n=12,440). FDR < 0.05. (B) Hierarchical clustering of median-centered gene expression in control or TRIM37-ASO treated 231-2b cells. Each colored line in the dendrogram identifies a different gene. n=3 (C) Pathways significantly downregulated (Blue) or upregulated (Red) in TRIM37-ASO treated cells relative to control 231-2b cells identified by GSEA. (D-E) qRT-PCR monitoring TRIM37-regulated metastasis suppressor genes in TRIM37 overexpressing p53−/− MCF10A cells relative to vector control (D) and TRIM37-ASO treated 231-2b tumors relative to control tumors (E). (F) ChIP monitoring BMI1, EZH2, TRIM37, and H2Aub binding at KISS1, BRMS1, and Actin in control and TRIM37-ASO treated 231-2b cells. (G-H) qRT-PCR monitoring TRIM37 target genes in 231-2b cells expressing KISS1 shRNA (G) and BRMS1 shRNA (H) relative to NS shRNA. (I-J) qRT-PCR monitoring TRIM37 target genes in TRIM37 overexpressing p53−/− MCF10A cells relative to vector control (I) and TRIM37-ASO treated 231-2b tumors relative to control tumors (J). (K) Schematic showing that mice were injected with control or TRIM37-ASO treated 231-2b cells intracardially and monitored for metastatic tumor burden. n=8 animals per group. (L) Representative ventral BLI of 231-2b expressing control or TRIM37-knockdown (TRIM37-ASO) at day 21. (M) Analysis of metastatic tumor growth in mice tissues measured by relative luciferase signal for lungs and number of metastatic lesions in lungs, bones, brain, and liver. n=8 animals per group. (N) Kaplan-Meier survival curve for mice injected with control or TRIM37-ASO-treated 231-2b cells. n=8 animals per group. Error bars indicate standard deviation and range of at least three biological replicates. * p<0.05; ** p<0.01; ***p<0.001.
To validate the RNA-seq results, we analyzed expression of representative genes in p53−/− MCF10A cells ectopically expressing TRIM37. Expression of KISS1 and BRMS1 was significantly lower in cells ectopically expressing TRIM37 compared with empty vector (Fig.5D). Conversely, TRIM37-knockdown tumors expressed KISS1 and BRMS1 at significantly higher levels relative to control xenograft tumors (Fig.5E). To investigate the mechanism by which TRIM37 regulate KISS1 and BRMS1, we analyzed binding of polycomb complex components, BMI1 and EZH2, to BRMS1 and KISS1 by directed-ChIP assays. Both the gene promoters were enriched for BMI1 and EZH2 which was diminished after TRIM37 knockdown (Fig.5F). These gene promoters were also enriched for H2Aub, which was reduced after TRIM37 knockdown (Fig.5F). As expected, knockdown of BMI1 and EZH2 resulted in increased expression of these genes (Supplementary Fig.5E–F).
Our results raised the possibility that TRIM37-mediated repression of metastases suppressors induces transcriptional program favoring metastasis. To test this idea, we analyzed a representative set of 15 genes in the top GSEA categories based on statistical analysis and their known biological functions in multiple steps of metastasis (Supplemental Table IV). For all 15 genes analyzed, knockdown of BRMS1 or KISS1 resulted in their increased expression (Fig.5G–H and Supplementary Fig.5G). Consistently, ectopic expression of TRIM37 in p53−/− MCF10A cells significantly increased expression of all the TRIM37 target genes compared with empty vector (Fig.5I), indicating reciprocal relationship between TRIM37 and metastases suppressors. To validate RNA-seq results in vivo, a subset of TRIM37 target genes were analyzed in TRIM37-knockdown tumors. As expected, all the 15 genes analyzed were significantly reduced in TRIM37-knockdown tumors relative to control tumors (Fig.5J). Moreover, knockdown of TRIM37 also decreased expression of TRIM37 target genes in MCF7 cells (Supplementary Fig.5H).
To investigate directly the potential function of TRIM37 in metastasis, we compared the in vivo propensity of control and TRIM37-knockdown cells in NSG mice (Fig.5K). Knockdown of TRIM37 showed dramatic reduction in the metastatic burden in comparison to control tumors that developed in sites comparable to human breast cancer metastases, such as the brain, lung, liver, lymph nodes, and bone (Fig.5L and Supplementary Fig.5I–J). TRIM37-knockdown reduced the metastatic tumor burden in lungs by ~2-fold, bone by ~2-fold, brain by ~3-fold, and liver by ~3-fold compared to the control animals 21-days after TNBC cell injection (Fig.5M). Further, histological analysis of tumors confirmed the distant tumor growth of the control and TRIM37-knockdown 231-2b cells in lung and liver (Supplementary Fig.5K). Finally, TRIM37-knockdown in 231-2b resulted in a modest but significant improvement of post-injection survival (Fig.5N). Collectively, these results demonstrate that TRIM37 overexpression enforces transcriptional program in TNBC tumors that promotes metastatic progression.
Design, construction and characterization of molecularly targeted nanoparticles to deliver TRIM37-ASO in vivo
We show that TRIM37 alters chromatin modification to resist chemotherapy and enforces changes in gene expression to favor metastatic transition. These results formed the underlying rationale for targeting TRIM37 as a therapeutic strategy for treating TNBC. To systematically assess the therapeutic effectiveness of inhibiting TRIM37, we engineered liposome-based nanoparticles consisting of DOPA, DOTAP, and DSPE-PEG2000-Maleimide with TRIM37-ASO encapsulated in the core (see also Methods). We functionalized the nanoparticles by incorporating an investigational FOLR1 antibody (Farletuzumab). To this end, we site-specifically, and covalently conjugated DSPE-PEG2000-Maleimide to a cysteine-containing Fc-linkered sequence at the C-terminus of a knob heavy chain in Farletuzumab (Fig.6A and Supplementary Fig.6A–C). For convenience, these mono-disperse complexes are referred to as “smart nanoparticles” (Fig.6B and Supplementary Fig.6D). Fig.6C confirmed that ~67% of the maleimide groups present on the outer surface of the smart nanoparticles were functionalized with Farletuzumab. No significant differences in the binding affinity of Fc-linkered and monomeric Farletuzumab were observed (Fig.6D). As a control, we generated and characterized Farletuzumab-conjugated nanoparticles with control-ASO as a payload; hereafter, referred to as “control nanoparticles”. As expected, the diameter of 106 ± 21 nm (Fig.6E), and the overall charge of −4.72 ± 0.30 mV (Fig.6F) were not significantly different between smart and control nanoparticles.
Figure 6. Design, structural and functional characterization of smart nanoparticles in TNBC cellular and xenograft mouse models.
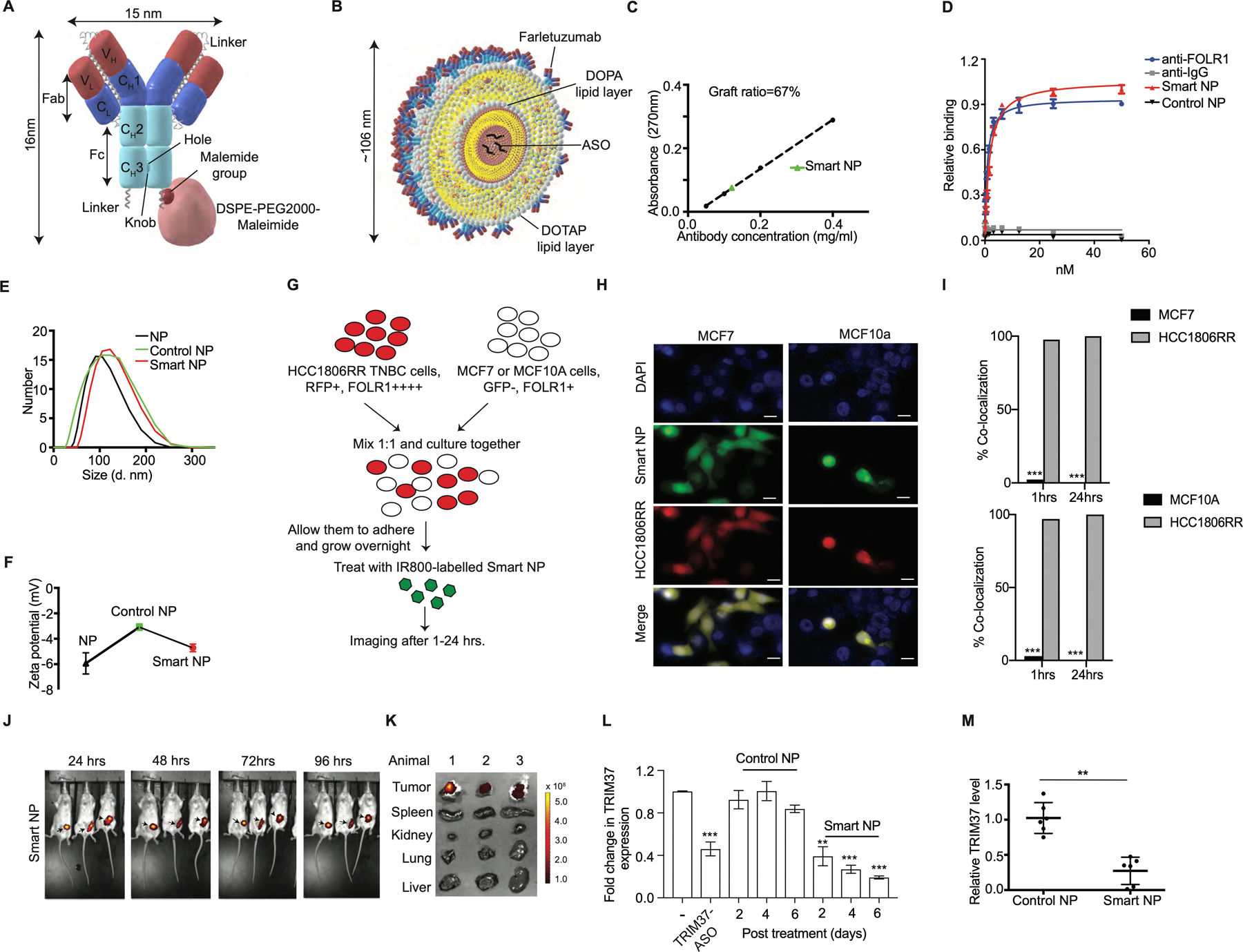
(A) A scheme of site-specific covalent conjugation of Farletuzumab to DSPE-PEG2000-Maleimide. An Fc-linkered sequence harboring a Cys at the extended C-terminal hole chain in Farletuzumab and conjugated to DSPE-PEG2000-Maleimide. (B) Structural model of smart nanoparticles with TRIM37-ASO encapsulated in the core. (C) Density of Farletuzumab on smart nanoparticles (Smart NP). (D) Binding assay for relative avidity index of Farletuzumab or nanoparticles. (E-F) Dynamic light scattering (E) and zeta potential (F) for nanoparticles. (G) Schematic of co-culturing experiments described in (H-I). HCC1806RR (Red) and MCF7 or HCC1806RR (Red) and MCF10A cells were co-cultured and treated with the IR800-labeled smart nanoparticles. (H-I) Representative images for the uptake of smart nanoparticles by HCC1806RR co-cultured with either MCF7 or MCF10A as described in (G). Scale bars represent 50 μm. Results are quantified (I). (J) Representative fluorescent images of tumor bearing mice after treatment with IR800-labeled smart nanoparticles at indicated times. The color scale depicts the fluorescence counts emitted from the tumor cells. (K) Necropsies from animals in (J) were analyzed by fluorescent imaging for detailed organ-specific distribution of IR800-labeled smart nanoparticles. (L) qRT-PCR monitoring expression of TRIM37 in 231-2b cells treated with either TRIM37-ASO or control-ASO or nanoparticles for indicated times. (M) qRT-PCR monitoring expression of TRIM37 in 231-2b tumors treated with control or smart nanoparticles. Error bars indicate standard deviation and range of at least three biological replicates. n=6 animals per group. * p<0.05; ** p<0.01; ***p<0.001.
Because higher levels of FOLR1 associate with advanced metastatic stage and recurrence in TNBC (42), we rationalized that smart nanoparticles will preferentially deliver TRIM37-ASO to TNBC cells. To test this idea, HCC1806RR (15) was co-cultured with either MCF7 or MCF10A cells (Fig.6G). Immunoblot analysis confirmed that FOLR1 is expressed at significantly higher levels in HCC1806RR compared to MCF7 or MCF10A cells (Supplementary Fig.6E). Interestingly, we observed ~100% uptake of IR800-labeled nanoparticles by TNBC cells compared to MCF7 or MCF10A cells post-mixing (Fig.6H–I). Finally, to test the TNBC cells selectivity of smart nanoparticles in vivo, we injected smart nanoparticles into the 231-2b xenograft-bearing mice. As expected, the smart nanoparticles selectively accumulated in the xenograft tumors within 24 hrs. and remained localized to the tumor up to 96 hrs. (Fig. 6J). The accumulation of smart nanoparticles in xenograft tumors was confirmed by the detailed tissue distribution using mice necropsies (Fig.6K).
Next, we evaluated the biological activity of the smart nanoparticles, which demonstrated sustained TRIM37-ASO release over a period of 48 hours with a biphasic release pattern (Supplementary Fig.6F). Treatment of MDA MB 231 cells with smart nanoparticles decreased TRIM37 expression by ~80% in a time-dependent manner relative to control nanoparticles (Fig.6L). To investigate smart nanoparticles-mediated TRIM37 targeting in vivo, we challenged 231-2b-derived xenografts with smart nanoparticles. qRT-PCR and immunoblot analysis confirmed TRIM37 inhibition in smart nanoparticle-treated tumors compared to control nanoparticle-treated tumors (Fig.6M and Supplementary Fig.6G). Together, these results confirmed that smart nanoparticles-mediated delivery of TRIM37-ASO effectively decrease TRIM37 expression in TNBC tumors.
Targeting TRIM37 to prevent metastasis in TNBC
Next, we asked whether TRIM37 targeting will reduce metastatic lesions in syngeneic spontaneous metastasis murine model. To this end, Balb/c mice bearing mammary fat pad tumors derived from murine 4T1 cells were administered 1.2 mg/kg of control or smart nanoparticles (Fig.7A). To inhibit TRIM37, we utilized smart nanoparticles conjugated with murine cross-reactive anti-FOLR1 (Supplementary Fig.7A–C). While ~90% of the control nanoparticle treated mice developed overt lung metastasis, smart nanoparticles-treated animals showed a dramatic decrease in lung metastasis, with ~60% of smart nanoparticles-treated animals showing no detectable lung metastases (Fig.7B and Supplementary Fig.7D). Animals were sacrificed at day 30 post-treatment due to the moribund condition of control nanoparticles-treated mice, which correlated with more rapid tumor growth at the primary and metastatic sites. The lung metastases were confirmed by H&E staining (Fig.7C and Supplementary Fig.7E), the gross lung tissue isolated post-mortem (Fig.7D–E), and a high proliferative index as determined by Ki67 staining (Fig.7F). Furthermore, TRIM37 was significantly reduced in the metastatic lesions isolated from the smart nanoparticle-treated animals compared to control animals (Supplementary Fig.7F). No significant changes in liver histology (Supplementary Fig.7G) or serum AST and ALT levels (Supplementary Fig.7H) between the control and smart nanoparticles-treated animals indicated a lack of hepatotoxicity.
Figure 7. Targeting of TRIM37 suppresses metastatic lung tumors in vivo.
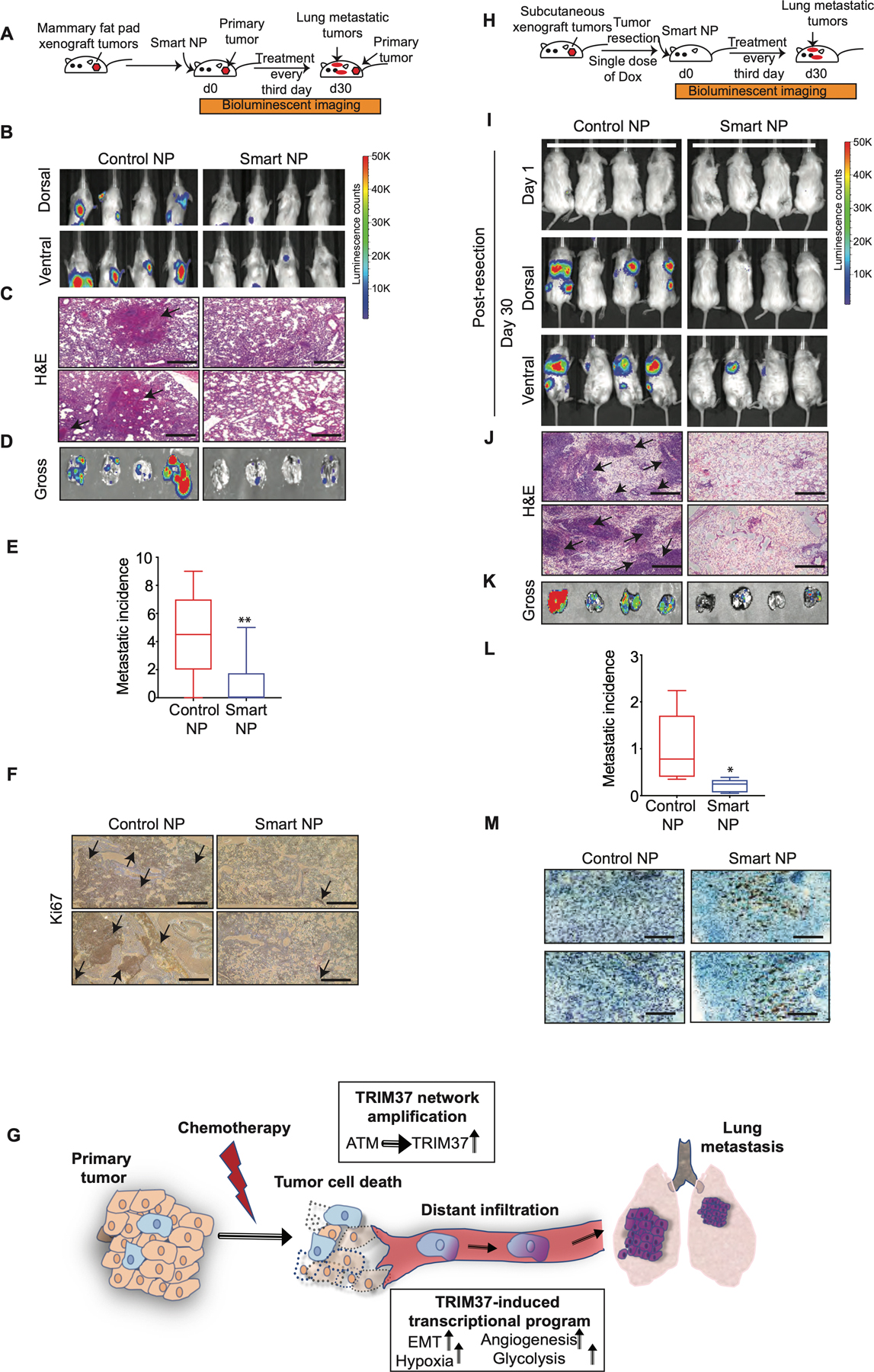
(A) Schematic showing that female Balb/c mice bearing mammary fat pad 4T1 tumors were treated with intranasal and intratumor injections of smart or control nanoparticles. n=8 animals per group. (B) Representative dorsal and ventral BLI images of tumor bearing mice at day 30 after treatment with either control or smart nanoparticles. The color scale depicts the luminescence counts emitted from the metastasis cells. n=4 animals per group. (C) Representative 10X H&E staining images of the lung metastases for control and smart nanoparticle treated animals. Scale bar, 0.5 mm. Arrows indicate lung metastatic nodules. (D) Lung necropsies from animals in (B) were analyzed by fluorescent imaging for tumor burden. (E) Quantification of metastasis incidence in the lung tissue after control or smart nanoparticles treatment. n=8 animals per group. (F) Ki67 staining of lung tumors derived from mice treated with control or smart nanoparticles. Scale bar, 0.5 mm. Arrows indicate highly proliferative lung metastatic nodules. (G) Model depicting TRIM37 function in multiple steps of TNBC metastasis. (H) Schematic showing that NSG mice bearing subcutaneous 231-2b tumors were treated with intranasal injections of smart or control nanoparticles in combination with Dox post-tumor resection. n=8 animals per group. (I) Representative BLI images for tumor-bearing mice at day 1 and day 30 post-primary tumor resection. The color scale depicts the luminescence counts emitted from the metastasis cells. n=4 animals per group. (J) Representative 10X H&E staining images of the lung sections of control and smart nanoparticle treated animals. Scale bar, 0.5 mm. Arrows indicate lung metastatic nodules. (K) Lung necropsies from animals in (I) were analyzed for tumor burden. (L) Quantification of accumulated luciferase signal from the lung tissue after control and smart nanoparticles treatment. n=8 animals per group. (M) Caspase 3 staining of lung tumors derived from mice treated with control or smart nanoparticles in combination with Dox. Scale bar, 0.5 mm. Error bars indicate standard deviation and range of at least three biological replicates. * p<0.05; ** p<0.01; *** p<0.001.
Primary tumors are routinely treated with a combination of surgery and chemotherapy. Our findings revealed that TRIM37 augments the metastatic potential of TNBC tumors by promoting survival under chemotherapeutic stress, and by inducing metastatic effectors (summarized in Fig.7G). These results raised the possibility that combining TRIM37 inhibition with chemotherapy will simultaneously increase chemotherapy efficacy and prevent metastatic progression of TNBC to increase the overall survival in TNBC patients.
To test this idea in a clinically relevant setting, we generated primary tumors by subcutaneously implanting 231-2b lung-tropic cells in female NSG mice (Fig.7H). Animals were treated with either control or smart nanoparticles intranasally in combination with a single dose of 2 mg/kg of Dox intraperitoneally post-tumor resection. Significantly, animals treated with control nanoparticles developed lung metastases, whereas smart nanoparticles treatment dramatically reduced metastatic burden in the lungs (Fig.7I and Supplementary Fig.7I). H&E-stained lung sections (Fig.7J) and luciferase signals from gross lung tissues revealed ~5-fold decrease in metastatic growth in animals treated with smart nanoparticles compared to control nanoparticle treated animals (Fig.7K–L and Supplementary Fig.7J). Tumors from smart nanoparticles and Dox-treated animals showed decreased tumor growth as indicated by significantly higher staining for Caspase 3 (Fig.7M) and lower Ki67 staining (Supplementary Fig.7K) in lung metastatic tumors in comparison to the control tumors.
Discussion
This study identifies a new TRIM37 network, which is amplified by chemotherapeutic drugs, as a unifying mechanism that drives chemoresistant and metastatic phenotype in TNBC tumors. The results are relevant to ~80% of TNBC patients that lack functional p53 and rely on systemic chemotherapeutic treatments due to the unavailability of any targeted TNBC therapy.
Chemoresistance, in general, is accompanied with extensive genetic and epigenetic alterations. However, whether selection of the clonal cancer cells or new mutations drive chemoresistant phenotype remain to be resolved (32). A recent genomic and phenotypic evolution profiling of TNBC tumors identified both pre-existing resistant genotypes as well as transcriptional reprogramming of resistant signatures in TNBC tumors (30). Our findings are in line with Kim et. al showing that pre-existing higher levels of TRIM37 in TNBC tumors promote resistance to chemotherapeutic stress and thus, increase survival of TNBC cells. On the other hand, chemotherapy triggers ATM signaling to transcriptionally upregulate TRIM37, which could further select for aggressive TNBC cells. In summary, TRIM37-positive TNBC tumors are protected and thrive under continued chemotherapy to cause aggressive metastatic disease.
A major hurdle in finding cure for aggressive TNBC is the lack of known drivers of the metastatic transition, in part due to a lack of mechanistic insights in the development of metastatic tumors. A genomics-driven discovery of recurring genetic mutations and epigenetic aberrations in the breast cancer genome has revealed tumorigenic drivers (6,43). However, whether these drivers that were primarily identified in the primary tumors are maintained throughout the chemotherapy regimen and the multistep process of metastasis remain to be evaluated. We used genomic and genetic approaches in relevant TNBC cellular, preclinical murine models and tumor biopsies to establish TRIM37 function in reducing therapy-induced DNA damage, increasing cancer cell survival, and causing transcriptional aberrations (Fig.7G). Our proof-of-concept results thus provide a rationale to target such common molecular effectors in combination with chemotherapy to prevent or significantly delay the metastatic progression in TNBC patients.
While targeted therapies are desperately needed to limit damage to healthy tissues, new delivery mechanism for cancer cell-specific targeting are also required to reduce detrimental side effects in healthy tissues. Molecularly targeted nanoparticles represent one such mechanism and are being aggressively explored for developing new treatment designs. As such, there are four nanoparticle-based therapies in clinical trials - BIND-014 for NSCLC and prostate cancer (44), CALAA-01 for solid tumors (45), SEL-068 for nicotine addiction (46), and Yale BNP for skin cancer (47).
Similar to mechanisms used in antibody-drug conjugates, we utilized a clinically investigative monoclonal antibody, which selectively delivers TRIM37-ASO into TNBC cells. The molecularly targeted nanoparticles offer a significant advantage over antibody-drug conjugates in terms of higher payload concentrations in tumor cells by enhancing retention times and permeability. An important consideration for nanoparticle delivery designs is clearance from the mononuclear phagocytic system. This is particularly critical for therapies designed to treat metastatic TNBC because premature elimination from circulation will prevent uptake by circulating tumor cells, decrease their accumulation in cancer cells and minimize their therapeutic impact. To overcome these issues, we incorporated PEG into our design, which enables steric stabilization of nanoparticles, and prevents interaction of the nanoparticles with immune cells (48). Functionalizing nanoparticles with “self” markers or homing molecules can further improve the systemic delivery of nanoparticles (49).
Notably, the majority of single-agent therapies tested to date for metastatic TNBC achieved an unimpressive response rate of less than 20%, with minimal impact on patient survival (50). The effective and selective delivery of TRIM37-ASO by Farletuzumab-conjugated nanoparticles provides an excellent opportunity to test additional TNBC-enriched surface proteins using monoclonal or bi-specific antibody formats. Some of the targets that can be exploited include TNBC-enriched MUC1, Trop-2 and VEGFR2. Our results also raise the possibility that molecularly targeted nanoparticles can deliver diverse payloads selectively to cancer cells.
In conclusion, our results identify a new driver of metastatic progression in TNBC patients and provide a mechanistic link between the two clinically linked phenotypes: chemoresistance and metastasis. Our findings also raise the possibility of clinically targeting TRIM37 to diminish the resistance to therapy, reduce the dissemination of cancer cells, and infiltration of distant sites. We demonstrate that our therapeutic design selectively inhibits TRIM37 and attenuates metastatic progression of TNBC tumors in vivo.
Supplementary Material
Significance:
TRIM37 drives aggressive TNBC biology by promoting resistance to chemotherapy and inducing a pro-metastatic transcriptional program; inhibition of TRIM37 increases chemotherapy efficacy and reduces metastasis risk in TNBC patients.
Acknowledgments
We thank Michael J. Lee, Michael R. Green, Takahiro Ochiya, David Weber, Michele Vitolo, and Sophia Ran for providing reagents; University of Virginia Tissue Histology Core, Biorepository and Tissue Research Facility (P30CA044579), Flow Cytometry Core (P30CA044579), Molecular Electron Microscopy Core, and The Office of Animal Welfare at University of Virginia. Edward Egelman, Ani W. Manichaikul, Marya Dunlap-Brown, and Stefan Bekiranov for technical and scientific discussion of statistical analysis. Christine Siu and Agnieszka Kokot for technical assistance with culturing cells. David Auble and Kristine Zengeler for editorial assistance. M.M. is supported by NCI RO1CA192399. J.T.S. is an Ovarian cancer Early Career Investigator. S.B. is the Hartwell investigator. S.B. is supported by Department of Defense Breast Cancer Research Breakthrough Award (BC170197P1, BC190343P1) and Metavivor Translational Research Award.
Footnotes
Competing interests: Authors include no conflict of interest.
References:
- 1.Pal SK, Childs BH, Pegram M. Triple negative breast cancer: unmet medical needs. Breast Cancer Res Treat 2011;125:627–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bauer KR, Brown M, Cress RD, Parise CA, Caggiano V. Descriptive analysis of estrogen receptor (ER)-negative, progesterone receptor (PR)-negative, and HER2-negative invasive breast cancer, the so-called triple-negative phenotype: a population-based study from the California cancer Registry. Cancer 2007;109:1721–8 [DOI] [PubMed] [Google Scholar]
- 3.Bertucci F, Ng CKY, Patsouris A, Droin N, Piscuoglio S, Carbuccia N, et al. Genomic characterization of metastatic breast cancers. Nature 2019;569:560–4 [DOI] [PubMed] [Google Scholar]
- 4.Yates LR, Knappskog S, Wedge D, Farmery JHR, Gonzalez S, Martincorena I, et al. Genomic Evolution of Breast Cancer Metastasis and Relapse. Cancer Cell 2017;32:169–84 e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ryan RJ, Bernstein BE. Molecular biology. Genetic events that shape the cancer epigenome. Science 2012;336:1513–4 [DOI] [PubMed] [Google Scholar]
- 6.Shen H, Laird PW. Interplay between the cancer genome and epigenome. Cell 2013;153:38–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stephens PJ, Tarpey PS, Davies H, Van Loo P, Greenman C, Wedge DC, et al. The landscape of cancer genes and mutational processes in breast cancer. Nature 2012;486:400–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cheng Y, He C, Wang M, Ma X, Mo F, Yang S, et al. Targeting epigenetic regulators for cancer therapy: mechanisms and advances in clinical trials. Signal Transduct Target Ther 2019;4:62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bhatnagar S, Gazin C, Chamberlain L, Ou J, Zhu X, Tushir JS, et al. TRIM37 is a new histone H2A ubiquitin ligase and breast cancer oncoprotein. Nature 2014;516:116–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bhatnagar S, Green MR. TRIMming down tumor suppressors in breast cancer. Cell Cycle 2015;14:1345–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lambert AW, Pattabiraman DR, Weinberg RA. Emerging Biological Principles of Metastasis. Cell 2017;168:670–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Acharyya S, Oskarsson T, Vanharanta S, Malladi S, Kim J, Morris PG, et al. A CXCL1 paracrine network links cancer chemoresistance and metastasis. Cell 2012;150:165–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hu G, Chong RA, Yang Q, Wei Y, Blanco MA, Li F, et al. MTDH activation by 8q22 genomic gain promotes chemoresistance and metastasis of poor-prognosis breast cancer. Cancer Cell 2009;15:9–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Weiss MB, Vitolo MI, Mohseni M, Rosen DM, Denmeade SR, Park BH, et al. Deletion of p53 in human mammary epithelial cells causes chromosomal instability and altered therapeutic response. Oncogene 2010;29:4715–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Volk-Draper LD, Rajput S, Hall KL, Wilber A, Ran S. Novel model for basaloid triple-negative breast cancer: behavior in vivo and response to therapy. Neoplasia 2012;14:926–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shivange G, Urbanek K, Przanowski P, Perry JSA, Jones J, Haggart R, et al. A Single-Agent Dual-Specificity Targeting of FOLR1 and DR5 as an Effective Strategy for Ovarian Cancer. Cancer Cell 2018;34:331–45 e11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lou S, Zhao Z, Dezort M, Lohneis T, Zhang C. Multifunctional Nanosystem for Targeted and Controlled Delivery of Multiple Chemotherapeutic Agents for the Treatment of Drug-Resistant Breast Cancer. ACS Omega 2018;3:9210–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pereira B, Chin SF, Rueda OM, Vollan HK, Provenzano E, Bardwell HA, et al. The somatic mutation profiles of 2,433 breast cancers refines their genomic and transcriptomic landscapes. Nat Commun 2016;7:11479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhou W, Zhu P, Wang J, Pascual G, Ohgi KA, Lozach J, et al. Histone H2A monoubiquitination represses transcription by inhibiting RNA polymerase II transcriptional elongation. Mol Cell 2008;29:69–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shanbhag NM, Rafalska-Metcalf IU, Balane-Bolivar C, Janicki SM, Greenberg RA. ATM-dependent chromatin changes silence transcription in cis to DNA double-strand breaks. Cell 2010;141:970–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lord CJ, Ashworth A. The DNA damage response and cancer therapy. Nature 2012;481:287–94 [DOI] [PubMed] [Google Scholar]
- 22.Goldstein M, Kastan MB. The DNA damage response: implications for tumor responses to radiation and chemotherapy. Annu Rev Med 2015;66:129–43 [DOI] [PubMed] [Google Scholar]
- 23.Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol Cell 2010;40:179–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Williams RS, Williams JS, Tainer JA. Mre11-Rad50-Nbs1 is a keystone complex connecting DNA repair machinery, double-strand break signaling, and the chromatin template. Biochem Cell Biol 2007;85:509–20 [DOI] [PubMed] [Google Scholar]
- 25.Ismail IH, Andrin C, McDonald D, Hendzel MJ. BMI1-mediated histone ubiquitylation promotes DNA double-strand break repair. J Cell Biol 2010;191:45–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cohn MA, D’Andrea AD. Chromatin recruitment of DNA repair proteins: lessons from the fanconi anemia and double-strand break repair pathways. Mol Cell 2008;32:306–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kastenhuber ER, Lowe SW. Putting p53 in Context. Cell 2017;170:1062–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gasco M, Yulug IG, Crook T. TP53 mutations in familial breast cancer: functional aspects. Hum Mutat 2003;21:301–6 [DOI] [PubMed] [Google Scholar]
- 29.Mina M, Raynaud F, Tavernari D, Battistello E, Sungalee S, Saghafinia S, et al. Conditional Selection of Genomic Alterations Dictates Cancer Evolution and Oncogenic Dependencies. Cancer Cell 2017;32:155–68 e6 [DOI] [PubMed] [Google Scholar]
- 30.Kim C, Gao R, Sei E, Brandt R, Hartman J, Hatschek T, et al. Chemoresistance Evolution in Triple-Negative Breast Cancer Delineated by Single-Cell Sequencing. Cell 2018;173:879–93 e13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.O’Connor MJ. Targeting the DNA Damage Response in Cancer. Mol Cell 2015;60:547–60 [DOI] [PubMed] [Google Scholar]
- 32.Navin NE. Tumor evolution in response to chemotherapy: phenotype versus genotype. Cell Rep 2014;6:417–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wu G, Song L, Zhu J, Hu Y, Cao L, Tan Z, et al. An ATM/TRIM37/NEMO Axis Counteracts Genotoxicity by Activating Nuclear-to-Cytoplasmic NF-kappaB Signaling. Cancer Res 2018;78:6399–412 [DOI] [PubMed] [Google Scholar]
- 34.Echeverria GV, Powell E, Seth S, Ge Z, Carugo A, Bristow C, et al. High-resolution clonal mapping of multi-organ metastasis in triple negative breast cancer. Nat Commun 2018;9:5079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tominaga N, Kosaka N, Ono M, Katsuda T, Yoshioka Y, Tamura K, et al. Brain metastatic cancer cells release microRNA-181c-containing extracellular vesicles capable of destructing blood-brain barrier. Nat Commun 2015;6:6716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ulasov IV, Borovjagin AV, Timashev P, Cristofanili M, Welch DR. KISS1 in breast cancer progression and autophagy. Cancer Metastasis Rev 2019;38:493–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kodura MA, Souchelnytskyi S. Breast carcinoma metastasis suppressor gene 1 (BRMS1): update on its role as the suppressor of cancer metastases. Cancer Metastasis Rev 2015;34:611–8 [DOI] [PubMed] [Google Scholar]
- 38.Cicek M, Fukuyama R, Welch DR, Sizemore N, Casey G. Breast cancer metastasis suppressor 1 inhibits gene expression by targeting nuclear factor-kappaB activity. Cancer Res 2005;65:3586–95 [DOI] [PubMed] [Google Scholar]
- 39.Wu P-Y, Yu I-S, Lin Y-C, Chang Y-T, Chen C-C, Lin K-H, et al. Activation of Aryl Hydrocarbon Receptor by Kynurenine Impairs Progression and Metastasis of Neuroblastoma. Cancer Research 2019:canres.3272.2018 [DOI] [PubMed]
- 40.Cicek M, Fukuyama R, Cicek MS, Sizemore S, Welch DR, Sizemore N, et al. BRMS1 contributes to the negative regulation of uPA gene expression through recruitment of HDAC1 to the NF-kappaB binding site of the uPA promoter. Clin Exp Metastasis 2009;26:229–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hurst DR, Xie Y, Vaidya KS, Mehta A, Moore BP, Accavitti-Loper MA, et al. Alterations of BRMS1-ARID4A interaction modify gene expression but still suppress metastasis in human breast cancer cells. J Biol Chem 2008;283:7438–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ginter PS, McIntire PJ, Cui X, Irshaid L, Liu Y, Chen Z, et al. Folate Receptor Alpha Expression Is Associated With Increased Risk of Recurrence in Triple-negative Breast Cancer. Clin Breast Cancer 2017;17:544–9 [DOI] [PubMed] [Google Scholar]
- 43.Cancer Genome Atlas N. Comprehensive molecular portraits of human breast tumours. Nature 2012;490:61–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sanna V, Pala N, Sechi M. Targeted therapy using nanotechnology: focus on cancer. Int J Nanomedicine 2014;9:467–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zuckerman JE, Gritli I, Tolcher A, Heidel JD, Lim D, Morgan R, et al. Correlating animal and human phase Ia/Ib clinical data with CALAA-01, a targeted, polymer-based nanoparticle containing siRNA. Proc Natl Acad Sci U S A 2014;111:11449–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Desai RI, Bergman J. Effects of the Nanoparticle-Based Vaccine, SEL-068, on Nicotine Discrimination in Squirrel Monkeys. Neuropsychopharmacology 2015;40:2207–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Deng Y, Ediriwickrema A, Yang F, Lewis J, Girardi M, Saltzman WM. A sunblock based on bioadhesive nanoparticles. Nat Mater 2015;14:1278–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Suk JS, Xu Q, Kim N, Hanes J, Ensign LM. PEGylation as a strategy for improving nanoparticle-based drug and gene delivery. Adv Drug Deliv Rev 2016;99:28–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Alqaraghuli HGJ, Kashanian S, Rafipour R. A Review on Targeting Nanoparticles for Breast Cancer. Curr Pharm Biotechnol 2019;20:1087–107 [DOI] [PubMed] [Google Scholar]
- 50.Vidula N, Bardia A. Targeted therapy for metastatic triple negative breast cancer: The next frontier in precision oncology. Oncotarget 2017;8:106167–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.