

Abstract
Aryl hydrocarbon receptor (AhR) is a transcription factor, which can be activated by a plethora of structure-diverse ligands. Historically, AhR is known for its involvements in regulation of metabolism of xenobiotics. However, normal physiological roles of AhR have been defined in other essential biological processes, including vascular growth and function, reproduction, and immunoresponses. In contrast, aberrant expression and activation of the AhR signaling pathway occur in a variety of human diseases, many of which (e.g., preeclampsia, atherosclerosis, and hypertension) could be associated with endothelial dysfunction. Indeed, emerging evidence has shown that either exogenous or endogenous AhR ligands can induce endothelial dysfunction in either an AhR-dependent or AhR-independent manner, possibly reliant on the blood vessel origin (artery and vein) of endothelial cells. Given that the AhR signaling pathway has broad impacts on endothelial and cardiovascular function, AhR ligands, AhR, and their downstream genes could be considered novel therapeutic targets for those endothelial-related diseases. This review will discuss the current knowledge of AhR’s mediation on endothelial function and potential mechanisms underlying these actions with a focus on placental endothelial cells.
Keywords: AhR, AhR ligands, angiogenesis, endothelial cells
Aryl hydrocarbon receptor critically mediates endothelial function.
Introduction
The endothelium is an essential component for vascular growth, which is required for supporting tissue growth occurring either under physiological (e.g., placental and fetal growth) or under pathological (e.g., solid tumor growth) states [1]. Even after completion of vascular growth, maintaining normal endothelial function is still vital for vascular homeostasis [1–3]. In contrast, endothelial dysfunction is associated with severe vascular disorders such as preeclampsia (PE), intrauterine growth restriction (IUGR), placental chorioangioma (PC), and hypertension (HTN) [1–5]. Thus, any factor that disrupts normal endothelial growth and function could have significant impacts on the vascular function, which in turn can critically affect individual health and well-being.
Aryl hydrocarbon receptor (AhR) is a ligand-activated transcription factor, which was first identified in mouse hepatic cytosol [6]. Over the last four decades, AhR has been extensively investigated for its participations in metabolizing environmental toxicants such as 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) [7]. Additional normal physiological roles of AhR have also been recognized in other essential biological processes including vascular growth and function, reproduction, and immunoresponses, largely based on animal knockout studies or the discovery of a variety of structure-diverse endogenous AhR ligands [7–14].Given many of these AhR-regulated biological processes are associated with vascular growth and function, this review will elaborate the comprehensive regulation of AhR ligands in endothelial responses with a focus on placental endothelial responses. In addition, the potential cellular and molecular mechanisms underlying this regulation in endothelial cells will also be discussed.
Regulation of fetoplacental angiogenesis
The placenta contains extremely rich blood vessels, which are resulted from an extensive angiogenesis (neovascular formation from preexisting blood vessels) after the completion of placental vasculogenesis (the differentiation of precursor cells into endothelial cells and the de novo formation of a primitive vascular network) [4]. Fetoplacental vasculogenesis and angiogenesis are tightly controlled by peptide growth factors [2–4]. Among these growth factors, fibroblast growth factor 2 (FGF2) and vascular endothelial growth factor A (VEGFA) are potent endothelial regulators, which exert their actions primarily via binding and activating their tyrosine kinases receptors [2, 15, 16]. These kinase receptors of FGF2 include FGF receptor 1 (FGFR1), 2 (FGFR2), 3 (FGFR3), and 4 (FGFR4) [16], among which FGFR1 is the predominant form in the human umbilical cord endothelial cells [17, 18]. The major VEGFA receptors consist of VEGF receptor 1 (VEGFR1) and 2 (VEGFR2) [15, 16].
Accumulating evidence has clearly shown that FGF2 and VEGFA promote placental angiogenesis [2, 17–19]. FGF2 primarily synthesized and secreted from placental trophoblasts regulates many endothelial functions (e.g., proliferation, migration, and production of nitric oxide and prostacyclin) [2, 16]. The deficiency of FGF2 in mice impairs capillarogenesis [20]. However, knockout of FGFR1 and FGFR2 in mice is embryonic lethal [21]. Conversely, endothelial cell-specific FGFR1 and FGFR2 knockout has no significant effects on vascular formation in embryos, although it leads to impaired angiogenesis after injury in adult mice [21]. Similar to FGF2, VEGFs are primarily expressed in trophoblast cells and stromal cells [19]. The deficiency of VEGFA and its receptors causes many deleterious effects. For example, VEGFA knockout mice showed impaired myocardial angiogenesis and ischemic cardiomyopathy [22]. Knockout of either Vegfr1 or Vegfr2 gene is lethal to embryo in mice due to impaired vascular formation and growth [15], indicating the essential roles of VEGFs signaling in vasculogenesis and angiogenesis. Moreover, neuropilin-1 (NP-1) and 2 (NP-2), two additional VEGF receptors, are also critical to VEGFs signaling, as knockout of either Np1, Np2 or both severely impairs developmental yolk sac and embryonic angiogenesis [23]. In human fetoplacental endothelial cells, FGF2 and VEGFA regulate cell proliferation, migration, and permeability [17, 18, 24–27]. Collectively, these data indicate the importance of FGF2 and VEGFA signaling pathways in placental endothelial responses.
Impaired placental endothelial function and angiogenesis are tightly associated with aberrant expression and activation of angiogenesis-related factors including VEGFA, placental growth factor, and their receptors in the placenta and maternal circulation [3, 24–27]. Thus, similar to many angiogenesis-related diseases (e.g., solid tumor), dysregulating expression and activation of angiogenic factors in placentas could be a major contributing factor to the development of pregnancy complications such as preeclampsia (PE) and placental chorioangioma (PC) [1, 3–5].
Beside FGF2 and VEGFA, numerous other peptide factors also regulate vascular and endothelial function during pregnancy. One of these potent factors is endothelin (ET), which can be synthesized and released by trophoblast cells and endothelial cells as recently summarized by several investigators [28–30]. ET-1, one member of the ET family, is considered one of most potent vasoconstrictor and mitogens to vascular smooth muscle. ET-1 also acts on endothelial cells and enhances production of vasodilators (e.g., nitric oxide and prostacyclin) [28–30]. As nitric oxide is a key mediator of fetal endothelial function [2], ET-1 may play an important role in regulating maternal and fetal endothelial function during normal pregnancy. This notion is supported by the observations that maternal ET-1 levels are elevated in normal pregnancy, along with much high levels of ET-1 present in fetal circulation as compared with maternal serum levels [29–31]. Conversely, higher maternal ET-1 levels are associated with PE [29, 30]. Thus, the dysfunction of ET system in fetal endothelial cells may contribute to fetal endothelial dysfunction in PE, particularly since VEGFA can stimulate ET-1 production in human umbilical veins (HUVECs) [32]. To date, the limited information on the role of the AhR singling pathway in ET system is primarily derived from the AhR knockout mice model. Specifically, the AhR null mice exhibited increased circulating ET-1 levels in association with hypertension (HTN) [33–35], suggesting developmental inhibition of AhR on ET system. However, activation of the AhR signaling pathway by dioxin (TCDD) promotes fetoplacental ET-1 expression along with suppressed placental vascular remodeling in wild-type rats [36]. Thus, it is highly likely that the AhR signaling pathway could mediate fetal endothelial function, including angiogenesis partially via ET system.
AhR and its ligands
Distributions of AhR
The AhR gene is evolutionarily highly conserved as its expression is found in numerous species spanning from amphibians, reptiles, and birds to mammals [12, 13]. On the other hand, AhR is highly polymorphic, varying in molecular weight by about 30 kDa across different species (e.g., 95 kDa in C57 mouse; 113 kDa in monkey; 124 kDa in hamster; 106 kDa in human) [13].
Expression of AhR has been identified in a wide range of organs with different abundances [37]. For example, AhR mRNA is highly expressed in the human placenta, lung, heart, liver, and pancreas, but is much lower in the brain, kidney, and skeletal muscle [38]. For a given organ, AhR mRNA levels can also vary significantly between species. Specifically, AhR mRNA expression is higher in human placentas compared with rat placentas [38–40]. In human placentas, AhR protein is present primarily in endothelial cells of large blood vessels, cytotrophoblast and syncytiotrophoblast in villi as well as HUVECs and human umbilical artery endothelial cells (HUAECs) [41, 42]. The endothelial expression of AhR protein has also been detected in human fetuses [41] and lungs [43]. AhR is expressed in other cell types including uterine epithelial cells, ovary theca, granulosa, luteal cells immune T cells, and dendritic cells [44–46]. This wide distribution of AhR in various types of cells and tissues suggests the systemic importance of AhR in cellular function. This concept is supported by previous literature that demonstrates the aberrant expression of AhR is associated with many disorders [46–48]. For instance, AhR expression is increased in placentas of mild PE [48] and in the villi of unexplained miscarriage [49], as well as in lung adenocarcinoma and breast cancer tissues [47, 50].
Activation of AhR
Most unliganded AhR reside in the cytoplasm [13] (Figure 1). Upon binding to its ligands, the AhR–ligand complex translocates into the nucleus where the complex binds to aryl hydrocarbon receptor nuclear translocator (ARNT or termed as hypoxia-inducible factor-1-beta) to form a heterodimer. This heterodimer binds and activates the dioxin response element (DRE), inducing expression of many downstream genes including cytochrome P450, family 1, subfamily A, polypeptide 1 (CYP1A1); cytochrome P450, family 1, subfamily B, polypeptide 1 (CYP1B1); and other cell cycle regulators such as p21 and p53 [7, 8, 51]. After activation, AhR is degraded in the nucleus or is transported back to the cytoplasm and degraded by the 26S proteasome system [7]. Thus, a decrease in AhR protein expression in cells upon AhR ligand stimulation could be considered indicative of AhR activation [7, 37]. In addition, AhR activity can be suppressed by AhR repressor, which competitively binds to ARNT, reducing availability of ARNT for AhR [7].
Figure 1.
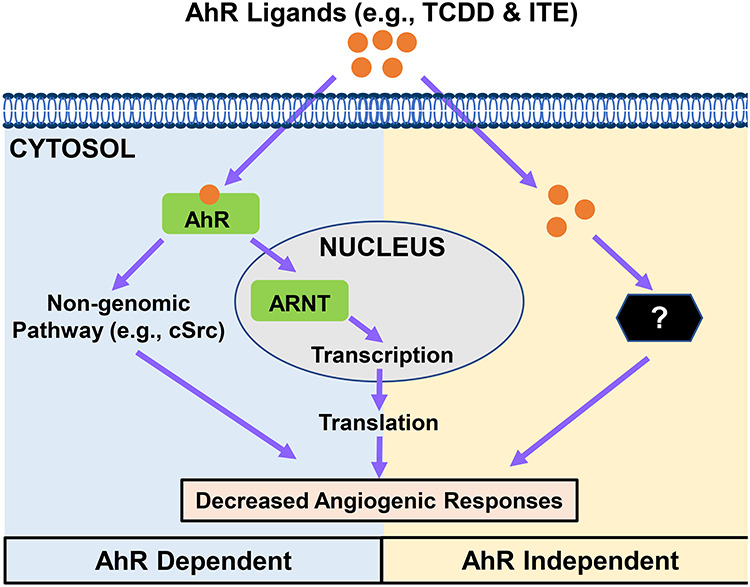
Schematic of proposed mechanisms underlying AhR ligands-induced endothelial angiogenic responses. We propose that AhR ligands at aberrantly high levels induce dysregulation of gene expression and defective activation of diverse signaling pathways either in an AhR-dependent or AhR-independent manner. Such dysregulation leads to inhibition of angiogenic responses and other cellular function in endothelial cells.
The binding affinity of AhR for the same AhR ligand varies between different species, possibly due to species-specific biochemical and physiological characteristics of AhR (i.e., differences in amino acid sequences of the ligand-binding domain) [52]. In addition, even in the same species, fetuses are more sensitive to TCDD than adults, suggesting that different developmental stages have distinct sensitivities in response to the same AhR ligands [53].
AhR ligands
Artificial/exogenous AhR ligands
AhR can be activated by a range of structurally divergent exogenous, natural, and endogenous chemicals with various binding affinities [12]. The majority of high-affinity AhR ligands are synthesized artificially or generated in the environment during the combustion of chemicals during waste incineration, metal production, and fossil fuel or wood burning [12]. These exogenous AhR ligands include planar, hydrophobic halogenated aromatic hydrocarbons (HAHs) (i.e., polyhalogenated dibenzo-p-dioxins and biphenyls) and polycyclic aromatic hydrocarbons (PAHs) (e.g., 3-methylcholanthrene [3MC] and benzoflavones) [8]. These HAHs and PAHs are metabolically stable and resistant to degradation [54]. Among these, TCDD is one of the most potent AhR ligands [12].
Natural/endogenous ligands
Diverse types of natural and endogenous AhR ligands with structures and physiochemical characteristics dramatically different from HAH and PAH have been identified [12, 14]. These ligands include bilirubin, the yellow breakdown product of normal heme catabolism detected in human bile, urine, and blood, as well as in many plants (e.g., corn, wheat, rice, potato, apple, and pears) [55, 56]. Other notable natural ligands [12] include flavonoids, detected in fruits and vegetables (e.g., grape, tomato, cherry, strawberry blackberry, blueberry, onion peel, and potato peel); indole-3-carbinol (I3C), found in many cruciferous plants such as broccoli and the brussels sprouts; indigo, detected in human urine and bovine serum; 7-ketocholesterol, a major sterol constituent of plasma in humans; kynurenine (Kyn), a tryptophan catabolite, found in human tissues and circulation; 6-formylindolo[3,2-b]carbazole (FICZ) and 6,12-diformylindolo[3,2-b]carbazole (dFICZ), the two most active tryptophan photoproducts produced by exposing skin cells to UV light and visible lights [12]; and 2-(1’H-indole-3′-carbonyl)-thiazole-4-carboxylic acid methyl ester (ITE), another tryptophan catabolite, which was first isolated from porcine lung tissues and likely derived from tryptophan and cysteine via a condensation reaction [57]. These AhR ligands after synthesized or ingested might act collectively to alter cellular function in vivo.
These natural and endogenous ligands generally have relatively low affinity for AhR as compared with exogenous ligands and are degraded quickly by the AhR ligand-induced detoxification/metabolism enzymes (e.g., CYP1A1) [12]. Interestingly, though physiological concentrations of the majority of these endogenous AhR ligands are unknown in human, the circulating Kyn/tryptophan ratio is higher in pregnant vs. nonpregnant women and is correlated positively with gestational age [58], implicating the importance of the AhR signaling pathway in pregnancy.
Roles of AhR
Metabolism, bioaccumulation, and actions of TCDD
TCDD, the most potent member of the HAHs family, is metabolized by the cytochrome P-450-monooxygenases and its metabolites are excreted in the feces of animals and humans [59]. However, most environmental toxicants including TCDD resist metabolic breakdown [54]. For example, the half-life of TCDD in human is ~7–10 years [59]. Thus, after ingestion by animals and humans, these exogenous AhR ligands are fairly stably stored in the body [54].
TCDD and its related compounds are highly harmful to human and animal health [7, 54, 59]. Acute or chronic TCDD exposure induces many disorders (e.g., HTN ocular vascular changes diabetes, neurotoxicity, atherosclerosis, and tumors) [7, 59–61].
Perinatal exposure to TCDD and its related chemicals causes embryonic and fetal loss in parallel with increased neonatal mortality in mice [53, 62]. These toxic effects are mediated via AhR since AhR null mice are not susceptible to these adverse effects [63, 64]. Importantly, TCDD-induced fetotoxicity can be partially attributed to abnormal vascular formation, growth, and function. Specifically, prenatal exposure to TCDD impairs coronary vasculogenesis in chicken embryos by attenuating responsiveness to angiogenic factors (e.g., VEGFA) [65, 66]. In rats, a single dose of TCDD does not significantly affect placental angiogenesis, but is sufficient to suppress the placental vascular remodeling along with increased expression of several angiogenic factors and their receptors [e.g., VEGFA, angiopoietin-2, endoglin, placental growth factor (PIGF), VEGFR1, VEGFR2, soluble Flt1, and tunica interna endothelial cell kinase 2 (Tie2)] [36]. In vitro studies have also demonstrated that TCDD and other exogenous AhR ligands inhibit endothelial angiogenic activities (Table 1) [43, 67–77]. Further studies are needed to determine if repeated exposures to AhR ligands will cause different vascular responses in placentas. This is because that humans and animals are more likely to be recurrently exposed to exogenous AhR ligands during occupational exposure or to endogenous AhR ligands naturally synthesized or ingested in AhR ligand-rich diets by the body. Overall, these data indicate that AhR ligands regulate vascular functions, possibly via disturbing normal expression of angiogenic factors and their receptors, which in turn may control the blood flow to these tissues.
Table 1.
Effects of AhR ligands on angiogenic activities of human endothelial cells
| Ligands1 | Treatment2 | Cell type3 | Cellular responses | AhR dependency4 | Ref. # | |
|---|---|---|---|---|---|---|
| Exogenous AhR ligands | ||||||
| 1. | TCDD | 10 nM, 5 days | HUVECs | Cell proliferaton and tube formation ↓ | ND | [67] |
| 2. | TCDD | 10–100 nM | HUVECs | Cell proliferaton, migration, and viability ↓ | Yes | [68] |
| daily, 2–6 days | Cell cycle progression and apoptosis ↔ | (AhR siRNA) | ||||
| HUAECs | Cell proliferaton and viability ↓ | |||||
| Cell migration, cycle progression and apoptosis ↔ | No | |||||
| 3. | 3MC | 5–500 nM | HUVECs | Cell proliferation, adhesion, migration, and tube formation ↓ | Yes | [69–71] |
| 4–24 h | (AhR antagonist) | |||||
| 4. | HCB | 0.005–5 μM up to 24 day | HMEC-1 | Cell migration and tube formation ↑ | Yes | [72] |
| (AhR antagonist) | ||||||
| 5. | BaP | 1 μM, 18–24 h | HPVECs | Cell tube formation and migration ↓ | Yes | [73] |
| (AhR antagonist) | ||||||
| 6. | BaP | 3–10 μM | HUVECs | Cell migration, tube formation, and integrin expression ↓ | Yes | [74] |
| 24 h | Cell proliferation ↔ | (AhR shRNA) | ||||
| Endogenous AhR ligands | ||||||
| 7. | ITE | 1 nM–1 μM | HUVECs | Cell proliferaton, viability, and migration ↓ | Yes | [75] |
| Daily, 2–6 days | Cell cycle progression and apoptosis ↔ | (AhR siRNA) | ||||
| HUAECs | Cell proliferaton and viability ↓ | |||||
| Cell migration, cycle progression, and apoptosis ↔ | No | |||||
| 8. | ITE | 10 μM, every 2 days, 2–6 days | HPAECs | Cell proliferaton and viability ↓ | No | [43] |
| (AhR siRNA) | ||||||
| 9. | ITE | 5 μM, 1–7 days 1 and 5% O2 | HPAECs | Cell proliferaton and viability ↑ | ND | [76] |
| 10. | I3C | 5–50 μM, 24 hr | HUVECs | Cell tube formation ↓ | ND | [77] |
1HCB: hexachlorobenzene; BaP: benzo[α]pyrene.
2A single dose, unless specifically stated.
3HPVECs: human placental villous endothelial cells.
4ND: not determined. The text in brackets indicates the method used to determine the AhR dependence.
↑: increase; ↓: decrease; ↔: no change.
Normal physiological roles of AhR
Several lines of evidence have shown that AhR has important normal physiological roles in other biological processes [7–9, 51, 78]. First, this is mostly evidenced by the fact that AhR is highly conserved in phylogeny from invertebrates to vertebrates [12, 13]. Second, the Ahr-mutant mice exhibit a variety of anomalous phenotypes including patent ductus venosus, hyaloid artery, and altered limbal vasculature within the developing eye, hypotension, smaller liver size, and defective T cell differentiation [7–9, 51, 78]. Third, AhR and DRE-responsive gene Cyp1a1 are widely expressed in many tissues including animal and human fetal tissues [12, 41, 79, 80]. In terms of AhR’s roles in pregnancy, AhR knockout mice have difficulties in maintaining pregnancy, leading to decreases in litter size and increases in fetal morbidity and mortality as well as neonatal lethality [7, 81]. All of these defects are similar to pathological outcomes induced by the perinatal exposure to TCDD [53, 62]. Finally, the discovery of many natural and endogenous AhR ligands in humans, animals, and plants that are routinely encountered [12–14] also firmly support the physiological importance of AhR in many essential biological processes.
The AhR ligands/AhR signaling in fetoplacental endothelial cells
AhR signaling in placentas
It has been reported that AhR is constitutively activated in human placentas as indicated by the persistent presence of nuclear AhR in trophoblast cells and expression of AhR downstream genes (e.g., Cyp1a1/b1) in villi throughout pregnancy, suggesting an intrinsic activation of AhR by its endogenous ligands [42]. No definitive endogenous AhR ligand has yet been identified in any placental and fetal tissues. However, the endogenous AhR ligand Kyn is likely to be synthesized in placentas because indoleamine 2,3 dioxygenase (IDO), one of the rate-limiting enzymes for catalyzing Kyn formation [82] is expressed in human placentas and at the maternal–fetal interface (e.g., in vascular endothelium of villous chorion, endothelium of spiral arteries of the decidua, and decidual glandular epithelium in mice) [83]. In humans, the elevated Kyn/tryptophan ratio in maternal circulation during pregnancy [58] has been implicated the hyperactivities of tryptophan-derived AhR ligands in pregnant women [83]. In contrast, these increases in Kyn/tryptophan ratio in the maternal circulation are markedly reduced in pregnant women with PE, probably due to the attenuated IDO activity in placentas [84]. Thus, as many tryptophan catabolites including ITE and Kyn are AhR ligands [12, 14, 37], the impaired metabolism of tryptophan in PE might alter the levels of many other tryptophan-derived AhR ligands in placentas either after local synthesis or transport from the maternal circulation.
Roles of endogenous AhR ligands
Of many tryptophan-derived endogenous AhR ligand, ITE initially detected in porcine lung is recently found to be present in human tumor cells in vitro [85]. Based on its activity on DRE, the biological potency of ITE is ~ 100-fold lower than TCDD [57]. Previous studies have demonstrated that ITE regulates function of multiple cell types including endothelial cells [75], immune cells [86, 87] [88], human ovarian cancer cells [45], and stem-like cancer cells [85]. Specifically, similar to the inhibitory effects of most other AhR ligands on angiogenic responses of human endothelial cells (Table 1), ITE inhibits cell proliferation and viability of HUVECs and HUAECs, as well as migration of HUAECs, without affecting the cell cycle progression and cell apoptosis [75]. More importantly, ITE-suppressed angiogenic responses are AhR-independent and AhR-dependent in HUVECs and HUAECs, respectively, and are not associated with dysregulation of major angiogenic factors and their receptors (e.g., VEGFA, VEGFC, VEGFR1, and VEGFR2, NP-2, and FGFR1) [75]. The antiangiogenic activity of endogenous AhR ligands is supported by another report that shows I3C inhibits the tube formation of HUVECs [77] (Table 1). These data are in line with previous notions that endogenous AhR ligands may prevent excessive angiogenesis in tissues, including placentas under physiological (e.g., pregnancy and estrous cycle) or pathological states (e.g., solid cancer growth and pulmonary arterial HTN) as suggested [43, 45, 89].
Although antiangiogenic activities of AhR ligands are identified in the majority of cells studied (Table 1), their proangiogenic activities have also been described in human microvascular endothelial (HMEC-1) [72] and human pulmonary artery endothelial (HPAECs) cells [76] (Table 1). It is unknown what cause these contradictory effects of AhR ligands on endothelial angiogenic activities. However, the vascular origin of endothelial cells might be a contributing factor since HMEC-1 [72] is the only endothelial cell preparation derived from microvasculature (Table 1). In addition, it is noted that antiangiogenic [43] and proangiogenic [72] activities of ITE have been reported in HPAECs, which were cultured and treated under an ambient (21% O2) and hypoxic (1 and 5% O2) condition, respectively. Thus, different culture conditions could differentially affect cellular responses to the same AhR ligand.
AhR signaling pathways in endothelial cells
Upon binding to AhR, AhR ligands are able to promote expression of many genes via the genomic pathway [90, 91] (Figure 1). These AhR signaling targets include phase I and II drug metabolism enzymes (e.g., CYP1A1) [90, 91]. In addition, AhR signaling increases expression of key protein regulators such as p27 and p21 (cyclin-dependent kinase inhibitor in HUVECs via AhR for cell growth and differentiation [70, 92–94] (Figure 2).
Figure 2.
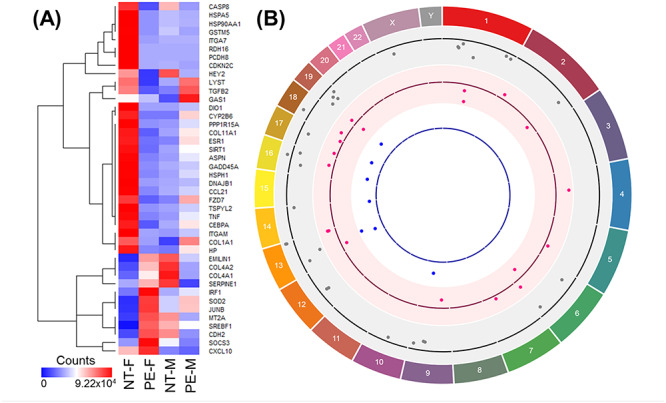
Differentially expressed AhR-regulated genes in female and male unpassaged HUVECs from NT and PE. (A) Heat map showing AhR-regulated genes that are differentially expressed in female (F) and male (M) HUVECs from NT and PE; (B) Circos plot illustrating the chromosomal position of AhR-regulated genes that are differentially expressed in NT-M vs. NT-F (gray dots, 30 genes), PE-F vs. NT-F (pink dots, 19 genes), and PE-M vs. NT-M (blue dots, 7 genes). Each dot represents one gene. The numbers and letters in the outer ring indicate the chromosomal location. For each scatterplot track, dots outside and inside of the centerline are upregulated and downregulated genes, respectively.
AhR ligands can activate diverse signaling molecules (e.g., protein kinases or phospholipases) via a nongenomic pathway, which is important in AhR-mediated activities such as toxicity [90, 91]. In fact, TCDD has been shown to rapidly induce (<15 min) activation of Cellular proto-oncogene tyrosine-protein kinase Src (cSrc) and calcium-triggered Cytosolic phospholipases A2 (cPLA2) activities in MCF10A cells, a normal mammary epithelial cell line [94, 95]. Such activation of cSrc is paralleled with enlarged activation of Focal adhesion kinase (FAK) and Mitogen-activated protein kinases (MAPKs) [90, 91]. Interestingly, TCDD can also activate Extracellular signal-regulated kinase1/2(ERK1/2), p38MAPK, or Jun N-terminal kinase (JUN) via an AhR-independent manner in embryonic fibroblasts and in mouse macrophage cell line (RAW 264.7) [92, 96]. Thus, regardless of AhR involvement, AhR ligands regulate activation of many signaling pathways (e.g., cSrc, ERK1/2, p38 MAPK, and JNK), which are essential for cell mitosis, development, stress, and inflammatory responses [97]. In HUVECs, 3MC activates p38MAPK and Ras homolog family member A (RhoA), whereas it deactivates FAK without affecting ERK1/2 and JNK activation; these actions are all associated with suppression of fetal bovine serum-induced cell angiogenic activities [69–71]. Previous reports from our laboratories have also shown that in HUVECs and HUAECs, neither TCDD nor ITE affects activation of ERK1/2 and v-akt murine thymoma viral oncogene homolog 1 (AKT1) triggered by complete growth media containing fetal bovine serum, heparin, and endothelial cell growth supplement [68, 75]. To date, it remains elusive whether AhR is involved in mediating activation of these kinases (e.g., p38MAPK, RhoA, and FAK) in HUVECs. However, as TCDD- and ITE-inhibited angiogenic responses are either independent or dependent of AhR in HUVECS and HUAECs, respectively [68, 75], such differential regulation might exist in placental endothelial cells. Thus, collectively, these data suggest that p38 MAPK and RhoA, but not ERK1/2 and AKT1 are important in AhR ligand-suppressed placental angiogenesis.
Recently, we have reanalyzed our previously published RNAseq data set (NCBI GEO: GSE116428) [27] derived from unpassaged HUVECs using pathway enrichment analysis. We identified 30 (4 upregulated and 26 downregulated) AhR-regulated genes that are differentially expressed between female and male HUVECs from normotensive (NT) pregnancies (Table 2; Figure 2). In addition, 19 (11 upregulated and 8 downregulated) and 7 (all upregulated) genes were differentially expressed between female and male HUVECs from PE, respectively, of which only 2 of these dysregulated genes were altered in both female and male cells from PE [DnaJ heat shock protein family (DNAJB1) and heat shock protein 105 kDa (HSPH1)], and the dysregulation was in opposite ways (decreased in female and increased in male cells) (Table 2). These sexual differences in AhR-regulated gene expression are probably not linked to the sex chromosome, as only one of these differentially expressed genes [testis-specific Y-encoded-like protein 2 (TSPYL2)] is located on the X-chromosome (Figure 2). All of these data indicate that AhR-regulated gene networks in human fetal endothelial cells are regulated in a fetal sex-specific fashion. Furthermore, such fetal sex-specific dysregulation in PE suggests sexual dimorphisms of PE-dysregulated cell function in response to AhR ligands in fetal endothelial cells.
Table 2.
Differentially expressed AhR-regulated genes in male and female HUVECs from NT and PE1
| Genes in the AhR network | Gene ID | NT-M/NT-F | F_PE/NT | M_PE/NT | |||
|---|---|---|---|---|---|---|---|
| Log2(Fold change) | FDR-adjusted P-value | Log2(Fold change) | FDR-adjusted P-value | Log2(Fold change) | FDR-adjusted P-value | ||
| DNAJB1 | ENSG00000132002 | −4.35 | 0.000 | −4.18 | 0.007 | 3.61 | 0.001 |
| HSPH1 | ENSG00000120694 | −2.61 | 0.001 | −2.43 | 0.039 | 2.38 | 0.007 |
| COL1A1 | ENSG00000108821 | −2.78 | 0.003 | 0.01 | 1.000 | 3.13 | 0.000 |
| GAS1 | ENSG00000180447 | −2.79 | 0.015 | −0.74 | 1.000 | 3.18 | 0.005 |
| ITGAM | ENSG00000169896 | −3.50 | 0.001 | −2.26 | 0.197 | 2.86 | 0.019 |
| PCDH8 | ENSG00000136099 | −7.11 | 0.002 | −4.85 | 0.150 | 6.03 | 0.047 |
| GSTM5 | ENSG00000134201 | −5.11 | 0.004 | −7.42 | 0.007 | 2.27 | 0.463 |
| COL4A1 | ENSG00000187498 | 2.37 | 0.001 | 2.88 | 0.014 | −0.33 | 1.000 |
| MT2A | ENSG00000125148 | 1.44 | 0.024 | 2.14 | 0.006 | 0.10 | 1.000 |
| COL4A2 | ENSG00000134871 | 2.70 | 0.001 | 3.43 | 0.004 | −0.15 | 1.000 |
| COL11A1 | ENSG00000060718 | −1.92 | 0.062 | −2.77 | 0.049 | 2.22 | 0.237 |
| JUNB | ENSG00000171223 | 0.67 | 0.553 | 2.18 | 0.001 | 1.06 | 0.309 |
| CASP8 | ENSG00000064012 | −0.44 | 0.826 | −2.19 | 0.003 | −0.95 | 0.530 |
| SOD2 | ENSG00000112096 | 1.49 | 0.239 | 3.09 | 0.003 | 1.10 | 0.875 |
| IRF1 | ENSG00000125347 | 0.09 | 1.000 | 1.51 | 0.040 | 0.41 | 1.000 |
| CCL21 | ENSG00000137077 | −1.69 | 0.318 | −3.56 | 0.044 | 0.98 | 1.000 |
| CDH2 | ENSG00000170558 | 2.33 | 0.050 | 2.76 | 0.044 | −0.74 | 1.000 |
| SERPINE1 | ENSG00000106366 | 0.94 | 0.073 | 1.44 | 0.026 | −0.37 | 1.000 |
| HEY2 | ENSG00000135547 | −0.17 | 1.000 | −4.22 | 0.041 | −0.41 | 1.000 |
| SOCS3 | ENSG00000184557 | 0.63 | 0.481 | 1.34 | 0.017 | 0.24 | 1.000 |
| CXCL10 | ENSG00000169245 | −0.55 | 1.000 | 5.59 | 0.001 | 0.43 | 1.000 |
| SREBF1 | ENSG00000072310 | 0.90 | 0.122 | 1.59 | 0.006 | 0.22 | 1.000 |
| EMILIN1 | ENSG00000138080 | 1.40 | 0.057 | 2.10 | 0.007 | 0.08 | 1.000 |
| HSP90AA1 | ENSG00000080824 | −2.24 | 0.003 | −2.00 | 0.085 | 1.91 | 0.050 |
| TSPYL2 | ENSG00000184205 | −2.29 | 0.003 | −1.89 | 0.117 | 2.17 | 0.052 |
| HSPA5 | ENSG00000044574 | −1.90 | 0.010 | −1.61 | 0.173 | 1.68 | 0.080 |
| SIRT1 | ENSG00000096717 | −1.38 | 0.018 | −0.99 | 0.373 | 1.45 | 0.114 |
| TGFB2 | ENSG00000092969 | −1.24 | 0.033 | 0.58 | 1.000 | 1.43 | 0.120 |
| CEBPA | ENSG00000245848 | −3.00 | 0.028 | −3.19 | 0.153 | 2.30 | 0.132 |
| PPP1R15A | ENSG00000087074 | −1.97 | 0.007 | −1.60 | 0.132 | 1.84 | 0.137 |
| FZD7 | ENSG00000155760 | −2.74 | 0.015 | −2.79 | 0.099 | 2.48 | 0.141 |
| ASPN | ENSG00000106819 | −2.96 | 0.031 | −1.98 | 0.400 | 3.19 | 0.170 |
| ITGA7 | ENSG00000135424 | −3.04 | 0.005 | −3.20 | 0.071 | 2.25 | 0.196 |
| HP | ENSG00000257017 | −2.46 | 0.033 | −2.13 | 0.320 | 1.95 | 0.203 |
| TNF | ENSG00000232810 | −3.44 | 0.009 | −2.25 | 0.442 | 2.20 | 0.243 |
| ESR1 | ENSG00000091831 | −2.75 | 0.002 | −2.36 | 0.081 | 1.82 | 0.280 |
| CYP2B6 | ENSG00000197408 | −4.44 | 0.022 | −6.27 | 0.054 | 3.13 | 0.297 |
| DIO1 | ENSG00000211452 | −2.71 | 0.046 | −2.37 | 0.253 | 2.79 | 0.344 |
| GADD45A | ENSG00000116717 | −1.25 | 0.049 | −0.72 | 0.635 | 1.21 | 0.431 |
| LYST | ENSG00000143669 | −1.50 | 0.022 | −1.30 | 0.317 | 1.03 | 0.533 |
| CDKN2C | ENSG00000123080 | −2.38 | 0.006 | −2.35 | 0.065 | 0.80 | 1.000 |
| RDH16 | ENSG00000139547 | −2.32 | 0.040 | −1.66 | 0.529 | 0.79 | 1.000 |
The AhR signaling pathway can crosstalk with many other cell signaling pathways including the estrogen (E2)/estrogen receptor (ER) pathway [98]. For instance, it has been reported that TCDD inhibits a broad spectrum of E2-induced cellular responses in rodents and human breast cancer cell lines [99–101]. Reciprocally, E2 also impedes the TCDD-induced cellular responses in human endometrial cancer cells-1 [101]. These data suggest that TCDD and E2 may antagonize each other, leading to alternations of cellular responses. To date, the crosstalk between TCDD and E2 in placental endothelial cells is still not fully understood. However, given that the E2/ER pathway mediates placental angiogenesis [102, 103], it is possible that the AhR signaling pathway might interact with the E2/ER signaling pathway in placental endothelial cells, controlling fetoplacental angiogenesis.
Conclusions
In conclusion, current evidence is clear that AhR ligands regulate function of placental endothelial (Table 1) and trophoblast cells [66], and perhaps immune cells too [7–12, 51]. Given that trophoblast and immune cells are two major sources of angiogenic factors in placentas [19, 104] and the AhR signaling pathway mediates expression of these angiogenic factors [36, 65, 66], AhR may regulate placental endothelial function through directly acting on endothelial cells or indirectly on nonendothelial cells. Such pivotal roles of the AhR signaling pathway in placental endothelial cells suggest its potential as a therapeutic target for various endothelial dysfunction-associated diseases (e.g., PE, IUGR, PC, and miscarriage) during pregnancy.
To date, though much progress has been made on the impacts of the AhR signaling pathway on placental endothelial growth and function, many challenges lie ahead of us. In particular, it will be critical to determine what are the major types of endogenous AhR ligands synthesized and present in human placental tissues, and whether and how the different levels of these AhR ligands affect cellular function under either physiological or pathological states. In addition, we know a little about how AhR ligands regulate cellular function independently of AhR. Omic approaches could be very useful to profile the extremely complex gene and signaling networks that participate AhR ligands-induced endothelial responses before dissecting each individual gene and signaling pathway. Moreover, the identity of other factors (e.g., sex and oxygen levels) that may significantly impact cellular and systemic responses to AhR ligands remains unknown. These unanswered questions warrant further investigations.
Acknowledgments
Authors thank their collaborators and colleagues (Drs. Ian M. Bird, Dong-bao Chen, Cai-feng Dai, Shi-An Huang, Yi-zhou Jiang, and Ronald R. Magness) for their valuable contributions to this work. Authors thank Laura H. Hogan, Ph.D., a Science Writer/Editor with the UW ICTR, for critically reading and editing this manuscript.
Footnotes
† Grant Support: This study was supported by the US National Institutes of Health grants (PO1 HD38843, PO1 HD038843-14S1 to JZ), and (RO3 HD100778 to CZ), as well as American Heart Association awards (17POST33670283, 19CDA34660348 to CZ). The project was also supported by Translational Basic and Clinical Pilot Award (JZ and CZ) from the UW Institute for Clinical and Translational Research (ICTR) and the Clinical and Translational Science Award program, through the US National Institutes of Health National Center for Advancing Translational Sciences, grant (UL1TR002373).
Contributor Information
Yan Li, Department of Obstetrics and Gynecology, University of Wisconsin-Madison, Madison, Wisconsin, USA.
Chi Zhou, Department of Obstetrics and Gynecology, University of Wisconsin-Madison, Madison, Wisconsin, USA.
Wei Lei, Department of Cardiovascular Medicine, Affiliated Hospital of Guangdong Medical University, Zhanjiang, Guangdong, China.
Kai Wang, Shanghai First Maternity and Infant Hospital, Tongji University School of Medicine, Shanghai, China.
Jing Zheng, Department of Obstetrics and Gynecology, University of Wisconsin-Madison, Madison, Wisconsin, USA; Department of Cardiovascular Medicine, Affiliated Hospital of Guangdong Medical University, Zhanjiang, Guangdong, China.
Author contributions
YL and JZ conceived the idea, wrote, and revised the manuscript. CZ and JZ reanalyzed the RNAseq data set. KW, CZ, and WL wrote and revised the manuscript.
Conflict of interest
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The authors have declared that no conflict of interest exists.
References
- 1. Goel S, Duda DG, Xu L, Munn LL, Boucher Y, Fukumura D, Jain RK. Normalization of the vasculature for treatment of cancer and other diseases. Physiol Rev 2011; 91:1071–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wang K, Zheng J. Signaling regulation of fetoplacental angiogenesis. J Endocrinol 2012; 212:243–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Karumanchi SA, Granger JP. Preeclampsia and pregnancy-related hypertensive disorders. Hypertension 2016; 67:238–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Burton GJ, Charnock-Jones DS, Jauniaux E. Regulation of vascular growth and function in the human placenta. Reproduction 2009; 138. [DOI] [PubMed] [Google Scholar]
- 5. Fan M, Skupski D. Placental chorioangioma: literature review. J Perinat Med 2013; 42:273. [DOI] [PubMed] [Google Scholar]
- 6. Poland A, Glover E, Kende AS. Stereospecific, high affinity binding of 2,3,7,8-tetrachlorodibenzo-p-dioxin by hepatic cytosol. Evidence that the binding species is receptor for induction of aryl hydrocarbon hydroxylase. J Biol Chem 1976; 251:4936–4946. [PubMed] [Google Scholar]
- 7. Denison MS, Soshilov AA, He G, DeGroot DE, Zhao B. Exactly the same but different: promiscuity and diversity in the molecular mechanisms of action of the aryl hydrocarbon (dioxin) receptor. Toxicol Sci 2011; 124:1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Safe S, McDougal A. Mechanism of action and development of selective aryl hydrocarbon receptor modulators for treatment of hormone-dependent cancers (review). Int J Oncol 2002; 20:1123–1128. [PubMed] [Google Scholar]
- 9. Walisser JA, Bunger MK, Glover E, Bradfield CA. Gestational exposure of Ahr and Arnt hypomorphs to dioxin rescues vascular development. Proc Natl Acad Sci U S A 2004; 101:16677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Stockinger B, Meglio PD, Gialitakis M, Duarte JH. The aryl hydrocarbon receptor: Multitasking in the immune system. Annu Rev Immunol 2014; 32:403–432. [DOI] [PubMed] [Google Scholar]
- 11. Rothhammer V, Quintana FJ. The aryl hydrocarbon receptor: an environmental sensor integrating immune responses in health and disease. Nat Rev Immunol 2019; 19:184–197. [DOI] [PubMed] [Google Scholar]
- 12. Nguyen LP, Bradfield CA. The search for endogenous activators of the aryl hydrocarbon receptor. Chem Res Toxicol 2008; 21:102–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Busbee PB, Rouse M, Nagarkatti M, Nagarkatti PS. Use of natural AhR ligands as potential therapeutic modalities against inflammatory disorders. Nutr Rev 2013; 71:353–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Roager HM, Licht TR. Microbial tryptophan catabolites in health and disease. Nat Commun 2018; 9:3294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ferrara N, Gerber H-P, LeCouter J. The biology of VEGF and its receptors. Nat Med 2003; 9:669–676. [DOI] [PubMed] [Google Scholar]
- 16. Turner N, Grose R. Fibroblast growth factor signalling: from development to cancer. Nat Rev Cancer 2010; 10:116–129. [DOI] [PubMed] [Google Scholar]
- 17. Jiang Y-Z, Wang K, Li Y, Dai C-F, Wang P, Kendziorski C, Chen D-B, Zheng J. Transcriptional and functional adaptations of human endothelial cells to physiological chronic low oxygen. Biol Reprod 2013; 88:114–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jiang Y-Z, Wang K, Li Y, Dai C-F, Wang P, Kendziorski C, Chen D-B, Zheng J. Enhanced cellular responses and distinct gene profiles in human fetoplacental artery endothelial cells under chronic low oxygen1. Biol Reprod 2013; 89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zygmunt M, Herr F, Münstedt K, Lang U, Liang OD. Angiogenesis and vasculogenesis in pregnancy. Eur J Obstet Gynecol Reprod Biol 2003; 110:S10–S18. [DOI] [PubMed] [Google Scholar]
- 20. Amann K, Faulhaber J, Campean V, Balajew V, Dono R, Mall G, Ehmke H. Impaired myocardial capillarogenesis and increased adaptive capillary growth in FGF2-deficient mice. Lab Invest 2006; 86:45–53. [DOI] [PubMed] [Google Scholar]
- 21. Yang X, Liaw L, Prudovsky I, Brooks PC, Vary C, Oxburgh L, Friesel R. Fibroblast growth factor signaling in the vasculature. Curr Atheroscler Rep 2015; 17:509–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Carmeliet P, Ng Y-S, Nuyens D, Theilmeier G, Brusselmans K, Cornelissen I, Ehler E, Kakkar VV, Stalmans I, Mattot V, Perriard J-C, Dewerchin M et al. Impaired myocardial angiogenesis and ischemic cardiomyopathy in mice lacking the vascular endothelial growth factor isoforms VEGF164 and VEGF188. Nat Med 1999; 5:495–502. [DOI] [PubMed] [Google Scholar]
- 23. Takashima S, Kitakaze M, Asakura M, Asanuma H, Sanada S, Tashiro F, Niwa H, Miyazaki J, Hirota S, Kitamura Y, Kitsukawa T, Fujisawa H et al. Targeting of both mouse neuropilin-1 and neuropilin-2 genes severely impairs developmental yolk sac and embryonic angiogenesis. Proc Natl Acad Sci U S A 2002; 99:3657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhou C, Zou Q-Y, Li H, Wang R-F, Liu A-X, Magness RR, Zheng J. Preeclampsia downregulates microRNAs in fetal endothelial cells: roles of miR-29a/c-3p in endothelial function. J Clin Endocrinol Metab 2017; 102:3470–3479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zou Q, Zhao Y, Li H, Wang X, Liu A, Zhong X, Yan Q, Li Y, Zhou C, Zheng J. GNA11 differentially mediates fibroblast growth factor 2- and vascular endothelial growth factor A-induced cellular responses in human fetoplacental endothelial cells. J Physiol 2018; 596:2333–2344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zou Q, Zhao Y, Zhou C, Liu A, Zhong X, Yan Q, Li Y, Yi F, Bird IM, Zheng J. G protein α subunit 14 mediates fibroblast growth factor 2-induced cellular responses in human endothelial cells. J Cell Physiol 2019; 234:10184–10195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhou C, Yan Q, Zou Q, Zhong XQ, Tyler CT, Magness RR, Bird IM, Zheng J. Sexual dimorphisms of preeclampsia-dysregulated transcriptomic profiles and cell function in fetal endothelial cells. Hypertension 2019; 74:154–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Davenport AP, Hyndman KA, Dhaun N, Southan C, Kohan DE, Pollock JS, Pollock DM, Webb DJ, Maguire JJ. Endothelin. Pharmacol Rev 2016; 68:357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Granger JP, Spradley FT, Bakrania BA. The endothelin system: a critical player in the pathophysiology of preeclampsia. Curr Hypertens Rep 2018; 20:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hitzerd E, Neuman RI, Mirabito Colafella KM, Reiss IKM, van den Meiracker AH, Danser AHJ, Visser W, Versmissen J, Saleh L. Endothelin receptor antagonism during preeclampsia: a matter of timing? Clin Sci (Lond) 2019; 133:1341–1352. [DOI] [PubMed] [Google Scholar]
- 31. Sand AE, Ostlund E, Andersson E, Fried G. Endothelin-induced contractions in placental arteries is mediated by both ETA- and ETB-receptors. Acta Physiol Scand 1998; 163:227–234. [DOI] [PubMed] [Google Scholar]
- 32. Lee K-J, Kim M-K, Park Y-H, Seol H-J, Lim J-E, Lee JN, Oh M-J. Vascular endothelial growth factor induces endothelin-1 production via matrix metalloproteinase-2 rather than endothelin-converting enzyme-1. Hypertens Pregnancy 2007; 26:189–199. [DOI] [PubMed] [Google Scholar]
- 33. Lund AK, Goens MB, Kanagy NL, Walker MK. Cardiac hypertrophy in aryl hydrocarbon receptor null mice is correlated with elevated angiotensin II, endothelin-1, and mean arterial blood pressure. Toxicol Appl Pharmacol 2003; 193:177–187. [DOI] [PubMed] [Google Scholar]
- 34. Lund AK, Goens MB, Nuñez BA, Walker MK. Characterizing the role of endothelin-1 in the progression of cardiac hypertrophy in aryl hydrocarbon receptor (AhR) null mice. Toxicol Appl Pharmacol 2006; 212:127–135. [DOI] [PubMed] [Google Scholar]
- 35. Lund AK, Peterson SL, Timmins GS, Walker MK. Endothelin-1-mediated increase in reactive oxygen species and NADPH oxidase activity in hearts of aryl hydrocarbon receptor (AhR) null mice. Toxicol Sci 2005; 88:265–273. [DOI] [PubMed] [Google Scholar]
- 36. Wu Y, Chen X, Zhou Q, He Q, Kang J, Zheng J, Wang K, Duan T. ITE and TCDD differentially regulate the vascular remodeling of rat placenta via the activation of AhR. PLoS One 2014; 9:e86549–e86549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Denison MS, Nagy SR. Activation of the aryl hydrocarbon receptor by structurally diverse exogenous and endogenous chemicals. Annu Rev Pharmacol Toxicol 2003; 43:309–334. [DOI] [PubMed] [Google Scholar]
- 38. Dolwick KM, Schmidt JV, Carver LA, Swanson HI, Bradfield CA. Cloning and expression of a human Ah receptor cDNA. Mol Pharmacol 1993; 44:911. [PubMed] [Google Scholar]
- 39. Carver LA, Hogenesch JB, Bradfield CA. Tissue specific expression of the rat Ah-receptor and ARNT mRNAs. Nucleic Acids Res 1994; 22:3038–3044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Li W, Donat S, Dohr O, Unfried K, Abel J. Ah receptor in different tissues of C57BL/6J and DBA/2J mice: use of competitive polymerase chain reaction to measure Ah-receptor mRNA expression. Arch Biochem Biophys 1994; 315:279–284. [DOI] [PubMed] [Google Scholar]
- 41. Jiang Y, Wang K, Fang R, Zheng J. Expression of aryl hydrocarbon receptor in human placentas and fetal tissues. J Histochem Cytochem 2010; 58:679–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wakx A, Nedder M, Tomkiewicz-Raulet C, Dalmasso J, Chissey A, Boland S, Vibert F, Degrelle SA, Fournier T, Coumoul X, Gil S, Ferecatu I. Expression, localization, and activity of the aryl hydrocarbon receptor in the human placenta. Int J Mol Sci 2018; 19:3762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Pang L-P, Li Y, Zou Q-Y, Zhou C, Lei W, Zheng J, Huang S-A. ITE inhibits growth of human pulmonary artery endothelial cells. Exp Lung Res 2017; 43:283–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Frericks M, Meissner M, Esser C. Microarray analysis of the AHR system: tissue-specific flexibility in signal and target genes. Toxicol Appl Pharmacol 2007; 220:320–332. [DOI] [PubMed] [Google Scholar]
- 45. Wang K, Li Y, Jiang Y-Z, Dai C-F, Patankar MS, Song J-S, Zheng J. An endogenous aryl hydrocarbon receptor ligand inhibits proliferation and migration of human ovarian cancer cells. Cancer Lett 2013; 340:63–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kim HO, Kim JH, Chung BY, Choi MG, Park CW. Increased expression of the aryl hydrocarbon receptor in patients with chronic inflammatory skin diseases. Exp Dermatol 2014; 23:278–281. [DOI] [PubMed] [Google Scholar]
- 47. Lin P, Chang H, Tsai W-T, Wu M-H, Liao Y-S, Chen J-T, Su J-M. Overexpression of aryl hydrocarbon receptor in human lung carcinomas. Toxicol Pathol 2003; 31:22–30. [DOI] [PubMed] [Google Scholar]
- 48. Hao K, Zhou Q, He Q, Zheng J, Wang K. Protein expression of aryl hydrocarbon receptors in human placentas from mild preeclamptic and early pregnancies In: Recent Advances in Research on the Human Placenta, vol. 2012 InTech-Open Access Publisher; 119–123. [Google Scholar]
- 49. Fan H, Su X, Yang B, Zhao A. Aryl hydrocarbon receptor and unexplained miscarriage. J Obstet Gynaecol Res 2017; 43:1029–1036. [DOI] [PubMed] [Google Scholar]
- 50. Wong PS, Li W, Vogel CF, Matsumura F. Characterization of MCF mammary epithelial cells overexpressing the arylhydrocarbon receptor (AhR). BMC Cancer 2009; 9:234–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Quintana FJ, Sherr DH. Aryl hydrocarbon receptor control of adaptive immunity. Pharmacol Rev 2013; 65:1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hahn ME. Aryl hydrocarbon receptors: diversity and evolution. Chem Biol Interact 2002; 141:131–160. [DOI] [PubMed] [Google Scholar]
- 53. Peterson RE, Theobald HM, Kimmel GL. Developmental and reproductive toxicity of dioxins and related compounds: cross-species comparisons. Crit Rev Toxicol 1993; 23:283–335. [DOI] [PubMed] [Google Scholar]
- 54. Hites RA. Dioxins: an overview and history. Environ Sci Technol 2011; 45:16–20. [DOI] [PubMed] [Google Scholar]
- 55. Pirone C, Quirke JME, Priestap HA, Lee DW. Animal pigment bilirubin discovered in plants. J Am Chem Soc 2009; 131:2830–2830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Pirone C, Johnson JV, Quirke JME, Priestap HA, Lee D. Bilirubin present in diverse angiosperms. AoB Plants 2010; 2010:plq020–plq020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Song J, Clagett-Dame M, Peterson RE, Hahn ME, Westler WM, Sicinski RR, DeLuca HF. A ligand for the aryl hydrocarbon receptor isolated from lung. Proc Natl Acad Sci U S A 2002; 99:14694–14699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Schröcksnadel H, Baier-Bitterlich G, Dapunt O, Wachter H, Fuchs D. Decreased plasma tryptophan in pregnancy. Obstet Gynecol 1996; 88:47–50. [DOI] [PubMed] [Google Scholar]
- 59. Gasiewicz T, Olson J, Geiger L, Neal R. Absorption, distribution and metabolism of 2,3,7,8-tetrachlorodibenzodioxin (TCDD) in experimental animals In: Human and Environmental Risks of Chlorinated Dioxins and Related Compounds, vol. 26 Boston, MA: Springer; 1983: 495–525. [Google Scholar]
- 60. Xue P, Fu J, Zhou Y. The aryl hydrocarbon receptor and tumor immunity. Front Immunol 2018; 9:286–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. International Agency for Research on Cancer Polychlorinated Dibenzo-para-dioxins and Polychlorinated Dibenzofurans, vol. 69 Lyon, France: IARC Working Group on the Evaluation of Carcinogenic Risk to Humans; 1997: 33–343. [PMC free article] [PubMed] [Google Scholar]
- 62. Hernández-Ochoa I, Karman BN, Flaws JA. The role of the aryl hydrocarbon receptor in the female reproductive system. Biochem Pharmacol 2009; 77:547–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Fernandez-Salguero PM, Hilbert DM, Rudikoff S, Ward JM, Gonzalez FJ. Aryl-hydrocarbon receptor-deficient mice are resistant to 2,3,7,8-tetrachlorodibenzo-p-dioxin-induced toxicity. Toxicol Appl Pharmacol 1996; 140:173–179. [DOI] [PubMed] [Google Scholar]
- 64. Mimura J, Yamashita K, Nakamura K, Morita M, Takagi TN, Nakao K, Ema M, Sogawa K, Yasuda M, Katsuki M, Fujii-Kuriyama Y. Loss of teratogenic response to 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in mice lacking the Ah (dioxin) receptor. Genes Cells 1997; 2:645–654. [DOI] [PubMed] [Google Scholar]
- 65. Ivnitski-Steele ID, Walker MK. Vascular endothelial growth factor rescues 2,3,7,8-tetrachlorodibenzo-p-dioxin inhibition of coronary vasculogenesis. Birth Defects Res A Clin Mol Teratol 2003; 67:496–503. [DOI] [PubMed] [Google Scholar]
- 66. Ivnitski-Steele ID, Friggens M, Chavez M, Walker MK. 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) inhibition of coronary vasculogenesis is mediated, in part, by reduced responsiveness to endogenous angiogenic stimuli, including vascular endothelial growth factor A (VEGF-A). Birth Defects Res A Clin Mol Teratol 2005; 73:440–446. [DOI] [PubMed] [Google Scholar]
- 67. Ivnitski-Steele I, Walker M. Inhibition of neovascularization by environmental agents. Cardiovasc Toxicol 2005; 5:215–226. [DOI] [PubMed] [Google Scholar]
- 68. Li Y, Wang K, Zou Q-Y, Magness RR, Zheng J. 2,3,7,8-Tetrachlorodibenzo-p-dioxin differentially suppresses angiogenic responses in human placental vein and artery endothelial cells. Toxicology 2015; 336:70–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Juan S-H, Lee J-L, Ho P-Y, Lee Y-H, Lee W-S. Antiproliferative and antiangiogenic effects of 3-methylcholanthrene, an aryl-hydrocarbon receptor agonist, in human umbilical vascular endothelial cells. Eur J Pharmacol 2006; 530:1–8. [DOI] [PubMed] [Google Scholar]
- 70. Pang P-H, Lin Y-H, Lee Y-H, Hou H-H, Hsu S-P, Juan S-H. Molecular mechanisms of p21 and p27 induction by 3-methylcholanthrene, an aryl-hydrocarbon receptor agonist, involved in antiproliferation of human umbilical vascular endothelial cells. J Cell Physiol 2008; 215:161–171. [DOI] [PubMed] [Google Scholar]
- 71. Chang C, Tsai S, Lin H, Li H, Lee Y, Chou Y, Jen C, Juan S. Aryl-hydrocarbon receptor-dependent alteration of FAK/RhoA in the inhibition of HUVEC motility by 3-methylcholanthrene. Cell Mol Life Sci 2009; 66:3193–3205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Pontillo C, Español A, Chiappini F, Miret N, Cocca C, Alvarez L, Kleiman de Pisarev D, Sales ME, Randi AS. Hexachlorobenzene promotes angiogenesis in vivo, in a breast cancer model and neovasculogenesis in vitro, in the human microvascular endothelial cell line HMEC-1. Toxicol Lett 2015; 239:53–64. [DOI] [PubMed] [Google Scholar]
- 73. Palatnik A, Xin H, Su EJ. Dichotomous effects of aryl hydrocarbon receptor (AHR) activation on human fetoplacental endothelial cell function. Placenta 2016; 44:61–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Li C-H, Cheng Y-W, Hsu Y-T, Hsu Y-J, Liao P-L, Kang J-J. Benzo[a]pyrene inhibits angiogenic factors–induced αvβ3 integrin expression, neovasculogenesis, and angiogenesis in human umbilical vein endothelial cells. Toxicol Sci 2010; 118:544–553. [DOI] [PubMed] [Google Scholar]
- 75. Li Y, Wang K, Zou Q-Y, Jiang Y-Z, Zhou C, Zheng J. ITE suppresses angiogenic responses in human artery and vein endothelial cells: differential roles of AhR. Reprod Toxicol 2017; 74:181–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Wang J, Yan G, Guo H, Zhu Y, Shui X, He Y, Chen C, Lei W. ITE promotes hypoxia-induced transdifferentiation of human pulmonary arterial endothelial cells possibly by activating transforming growth factor-β/Smads and MAPK/ERK pathways. J Cell Biochem 2019; 120:19567–19577. [DOI] [PubMed] [Google Scholar]
- 77. Wang M-L, Lin S-H, Hou Y-Y, Chen Y-H. Suppression of lipid accumulation by indole-3-carbinol is associated with increased expression of the aryl hydrocarbon receptor and CYP1B1 proteins in adipocytes and with decreased adipocyte-stimulated endothelial tube formation. Int J Mol Sci 2016; 17:1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Lahvis GP, Pyzalski RW, Glover E, Pitot HC, McElwee MK, Bradfield CA. The aryl hydrocarbon receptor is required for developmental closure of the ductus venosus in the neonatal mouse. Mol Pharmacol 2005; 67:714. [DOI] [PubMed] [Google Scholar]
- 79. Omiecinski C, Redlich C, Costa-Mallen P. Induction and developmental expression of cytochrome P450IA1 messenger RNA in rat and human tissues: detection by the polymerase chain reaction. Cancer Res 1990; 50:4315–4321. [PubMed] [Google Scholar]
- 80. Choudhary D, Jansson I, Schenkman JB, Sarfarazi M, Stoilov I. Comparative expression profiling of 40 mouse cytochrome P450 genes in embryonic and adult tissues. Arch Biochem Biophys 2003; 414:91–100. [DOI] [PubMed] [Google Scholar]
- 81. Abbott BD, Schmid JE, Pitt JA, Buckalew AR, Wood CR, Held GA, Diliberto JJ. Adverse reproductive outcomes in the transgenic Ah receptor-deficient mouse. Toxicol Appl Pharmacol 1999; 155:62–70. [DOI] [PubMed] [Google Scholar]
- 82. Nishizawa H, Suzuki M, Pryor-Koishi K, Sekiya T, Tada S, Kurahashi H, Udagawa Y. Impact of indoleamine 2,3-dioxygenase on the antioxidant system in the placentas of severely pre-eclamptic patients. Syst Biol Reprod Med 2011; 57:174–178. [DOI] [PubMed] [Google Scholar]
- 83. Sedlmayr P, Blaschitz A, Stocker R. The role of placental tryptophan catabolism. Front Immunol 2014; 5:230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Kudo Y, Boyd CAR, Sargent IL, Redman CWG. Decreased tryptophan catabolism by placental indoleamine 2,3-dioxygenase in preeclampsia. Am J Obstet Gynecol 2003; 188:719–726. [DOI] [PubMed] [Google Scholar]
- 85. Cheng J, Li W, Kang B, Zhou Y, Song J, Dan S, Yang Y, Zhang X, Li J, Yin S, Cao H, Yao H et al. Tryptophan derivatives regulate the transcription of Oct4 in stem-like cancer cells. Nat Commun 2015; 6:7209–7209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Quintana FJ, Murugaiyan G, Farez MF, Mitsdoerffer M, Tukpah A-M, Burns EJ, Weiner HL. An endogenous aryl hydrocarbon receptor ligand acts on dendritic cells and T cells to suppress experimental autoimmune encephalomyelitis. Proc Natl Acad Sci U S A 2010; 107:20768–20773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Yeste A, Nadeau M, Burns EJ, Weiner HL, Quintana FJ. Nanoparticle-mediated codelivery of myelin antigen and a tolerogenic small molecule suppresses experimental autoimmune encephalomyelitis. Proc Natl Acad Sci U S A 2012; 109:11270–11275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Wei P, Hu G, Kang H, Yao H, Kou W, Liu H, Zhang C, Hong S. An aryl hydrocarbon receptor ligand acts on dendritic cells and T cells to suppress the Th17 response in allergic rhinitis patients. Lab Invest 2014; 94:528–535. [DOI] [PubMed] [Google Scholar]
- 89. Murray IA, Patterson AD, Perdew GH. Aryl hydrocarbon receptor ligands in cancer: friend and foe. Nat Rev Cancer 2014; 14:801–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Backlund M, Ingelman-Sundberg M. Regulation of aryl hydrocarbon receptor signal transduction by protein tyrosine kinases. Cell Signal 2005; 17:39–48. [DOI] [PubMed] [Google Scholar]
- 91. Larigot L, Juricek L, Dairou J, Coumoul X. AhR signaling pathways and regulatory functions. Biochim Open 2018; 7:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Tan Z, Chang X, Puga A, Xia Y. Activation of mitogen-activated protein kinases (MAPKs) by aromatic hydrocarbons: role in the regulation of aryl hydrocarbon receptor (AHR) function. Biochem Pharmacol 2002; 64:771–780. [DOI] [PubMed] [Google Scholar]
- 93. Jin D-Q, Jung JW, Lee YS, Kim J-A. 2,3,7,8-Tetrachlorodibenzo-p-dioxin inhibits cell proliferation through arylhydrocarbon receptor-mediated G1 arrest in SK-N-SH human neuronal cells. Neurosci Lett 2004; 363:69–72. [DOI] [PubMed] [Google Scholar]
- 94. Dong B, Matsumura F. Roles of cytosolic phospholipase A2 and Src kinase in the early action of 2,3,7,8-Tetrachlorodibenzo-p-dioxin through a nongenomic pathway in MCF10A cells. Mol Pharmacol 2008; 74:255. [DOI] [PubMed] [Google Scholar]
- 95. Dong B, Cheng W, Li W, Zheng J, Wu D, Matsumura F, Vogel CFA. FRET analysis of protein tyrosine kinase c-Src activation mediated via aryl hydrocarbon receptor. Biochim Biophys Acta 2011; 1810:427–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Park S, Yoon W, Kim H, Son H, Cho S, Jeong K, Kim T, Kim S, Kim S, Ryu S. 2,3,7,8-Tetrachlorodibenzo-p-dioxin activates ERK and p38 mitogen-activated protein kinases in RAW 264.7 cells. Anticancer Res 2005; 25:2831–2836. [PubMed] [Google Scholar]
- 97. Henklová P, Vrzal R, Ulrichová J, Dvořák Z. Role of mitogen-activated protein kinases in aryl hydrocarbon receptor signaling. Chem Biol Interact 2008; 172:93–104. [DOI] [PubMed] [Google Scholar]
- 98. Carlson DB, Perdew GH. A dynamic role for the Ah receptor in cell signaling? Insights from a diverse group of Ah receptor interacting proteins. J Biochem Mol Toxicol 2002; 16:317–325. [DOI] [PubMed] [Google Scholar]
- 99. Safe SH. Modulation of gene expression and endocrine response pathways by 2,3,7,8-tetrachlorodibenzo-p-dioxin and related compounds. Pharmacol Ther 1995; 67:247–281. [DOI] [PubMed] [Google Scholar]
- 100. Safe S, Wormke M. Inhibitory aryl hydrocarbon receptor−estrogen receptor α cross-talk and mechanisms of action. Chem Res Toxicol 2003; 16:807–816. [DOI] [PubMed] [Google Scholar]
- 101. Wormke M, Castro-Rivera E, Chen I, Safe S. Estrogen and aryl hydrocarbon receptor expression and crosstalk in human Ishikawa endometrial cancer cells. J Steroid Biochem Mol Biol 2000; 72:197–207. [DOI] [PubMed] [Google Scholar]
- 102. Morales DE, McGowan KA, Grant DS, Maheshwari S, Bhartiya D, Cid MC, Kleinman HK, Schnaper HW. Estrogen promotes angiogenic activity in human umbilical vein endothelial cells in vitro and in a murine model. Circulation 1995; 91:755–763. [DOI] [PubMed] [Google Scholar]
- 103. Oviedo PJ, Sobrino A, Laguna-Fernandez A, Novella S, Tarín JJ, García-Pérez M-A, Sanchís J, Cano A, Hermenegildo C. Estradiol induces endothelial cell migration and proliferation through estrogen receptor-enhanced RhoA/ROCK pathway. Mol Cell Endocrinol 2011; 335:96–103. [DOI] [PubMed] [Google Scholar]
- 104. Pratt A, Da Silva Costa F, Borg A, Kalionis B, Keogh R, Murthi P. Placenta-derived angiogenic proteins and their contribution to the pathogenesis of preeclampsia. Angiogenesis 2014; 18. [DOI] [PubMed] [Google Scholar]