

Abstract
Background:
2′-C-methyl and 4′-azido nucleosides have previously demonstrated inhibition of hepatitis C virus (HCV) replication by targeting the RNA-dependent RNA polymerase NS5B. In an effort to discover new and more potent anti-HCV agents, we envisioned synthesizing nucleoside analogues by combining the 2′-C-methylmoiety with the 4′-azido-moiety into one molecule.
Methods:
2′-C-methyl-4′-azido pyrimidine nucleosides were synthesized by first converting 2′-C-methyl ribonucleosides to the corresponding 4′-exocyclic methylene nucleosides. Treatment with iodine azide, benzoylation of the 2′- and 3′-hydroxy groups, oxidative displacement of the 5′-iodo group with meta-chloroperoxybenzoic acid, and debenzoylation gave the desired 2′-C-methyl-4′-azido uridine and thymidine analogues in good yield. Standard conversion of uridine to cytidine via the 4-triazole yielded 2′-C-methyl-4′-azido cytidine. In addition, 5′-phosphoramidate derivatives of 2′-C-methyl-4′-azido uridine and cytidine were synthesized to bypass the initial phosphorylation step.
Results:
The prepared nucleosides and their 5′- monophosphate prodrugs were evaluated for their ability to inhibit replication of the hepatitis C virus in a subgenomic replicon cell based assay. Cytotoxicity in Huh7 cells was determined simultaneously with anti-HCV activity by extraction and amplification of both HCV RNA and ribosomal RNA. Among the newly synthesized compounds, only the 5′-monophosphate nucleoside prodrugs had modest and selective anti-HCV activity. All prepared pyrimidine nucleosides and 5′-monophosphate nucleoside prodrugs displayed no evidence of cytotoxicity at high concentrations.
Conclusions:
This work is the first example of both inactive uridine and cytidine analogues of a nucleoside being converted to active anti-HCV nucleosides via 5′-monophosphate prodrugs.
Introduction
Twenty years ago, hepatitis C virus (HCV) was discovered by scientists at Chiron (now Novartis) and the Centers for Disease Control and Prevention to be the causative agent of non-A and non-B hepatitis in humans. Today, this virus has infected an estimated 3% of the global population and 3–4 million individuals become newly infected each year [1,2]. HCV infections often lead to reduced liver function, cirrhosis, and hepatocellular carcinoma, and eventually liver transplantation. The current approved therapy based on pegylated interferon alone or in combination with ribavirin is effective in only approximately half of the genotype 1 population [3,4]. Moreover, this limited efficacy is often associated with significant side effects leading to discontinuation of treatment [5–7]. Therefore, there is a need for the development of more effective therapeutic agents for the treatment of HCV infection.
The HCV RNA-dependent RNA polymerase (NS5B) and NS3/4A protease are currently the most promising targets for the development of novel treatments [8–11]. The activity of these virally encoded enzymes is essential for HCV replication, and antiviral agents targeting these enzymes are in both preclinical and clinical development. To date, most of the reported nucleoside analogues that inhibit HCV polymerase have modifications at the 2′ or 4′ positions of the sugar [1–12]. Several 2′-C-methyl nucleoside analogues [13–16] and 4′-azido cytidine analogues [17,18] have been identified as potent inhibitors of HCV NS5B polymerase (Figure 1).
Figure 1.
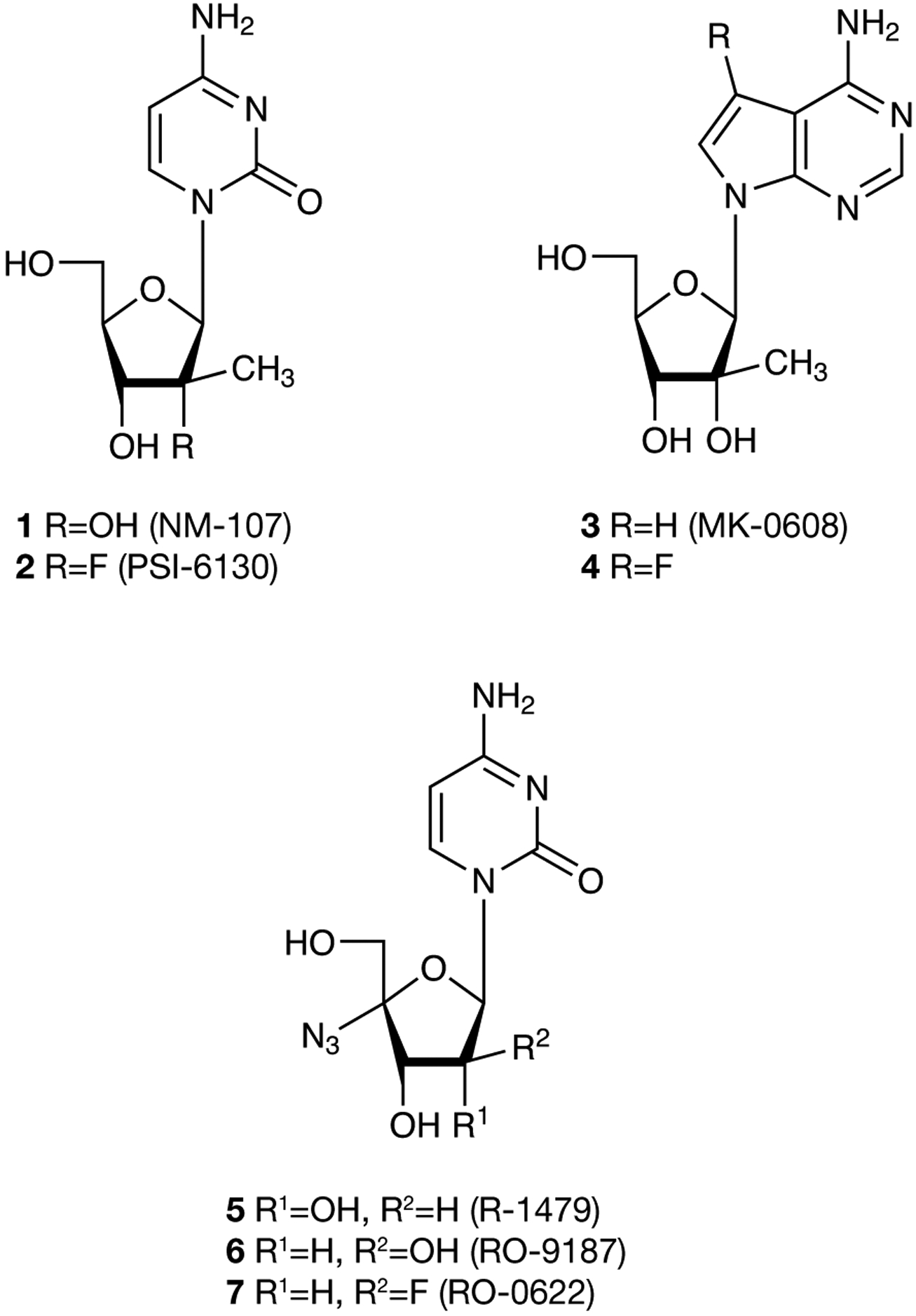
Structures of 2′-C-methyl and 4′-azido nucleosides
In an effort to discover new and more potent anti-HCV agents, we envisioned synthesizing a nucleoside analogue by combining the 2′-C-methyl moiety of NM-107 (1) [15] with the 4′-azido moiety of R-1479 (5) [17,18] into one molecule. Herein, we report the synthesis, anti-HCV activity and toxicity data for several 2′-C-methyl-4′-azido pyrimidines, which were synthesized as intermediates for the synthesis of the corresponding phosphoramidate derivatives. The main potential advantage of these phosphoramidate prodrugs is their ability to bypass the first phosphorylation step that is usually the rate-limiting step towards the 5′-triphosphate necessary for antiviral activity [19].
Methods
Chemistry
Thin layer chromatography was carried out on precoated silica gel thin layer sheets 60 F254 from EMD Merck (Darmstadt, Germany). Plate layer chromatography (PLC) from Analtech, Inc. (Newark, DE, USA) was employed for purification of products. 1H NMR (400.14 MHz) was recorded on Varian VNMR 400 spectrometer (Varian, Inc., Palo Alto, CA, USA). Mass spectral analyses were performed on a Micromass TOF instrument (Waters Corp., Milford, MA, USA) and HPLC (Hewlett–Packard, Palo Alto, CA, USA) driven electrospray MS instrument. Analytical HPLC analyses were performed on a Hewlett–Packard HPLC with a Phenomenex Gemini-NX column (2×50 mm, 3 μm, C18, 110 Å). Mobile phase flow was 0.7 ml/min with a 3.5 min gradient from 96% aqueous media (0.05% formic acid) to 96% CH3CN (0.05% formic acid) with a 5.5 min total acquisition time and 190–360 nm photodiode array detection).
1-(2-Methyl-5-iodo-β-d-ribofuranosyl)uracil (9a) and 1-(2-methyl-5-iodo-β-d-ribofuranosyl)thymine (9b)
2′-C-Methyluridine (5 g, 200 mmol), triphenylphosphine (8 g, 307 mmol) and imidazole (2.09 g, 307 mmol) were slurried in anhydrous tetrahydrofuran (THF). A solution of I2 (5.7 g, 220 mmol) in THF was added slowly to the slurry while the reaction temperature was maintained below 28°C. The reaction mixture was stirred at room temperature for 18 h. The reaction was quenched with water and extracted with ethyl acetate. After evaporation of the solvent, the residue was purified by column chromatography using 5% methanol in dichloromethane as eluent to obtain 9a in 85% yield. 1H NMR (CD3OD), δ: 1.12 (s, 3H, −CH3), 3.48–3.76 (m, 4H, H-3′, H-4′, H-5′), 5.73 (d, 1H, J=8.0 Hz, H-5), 5.93 (s, 1H, H-1′), 7.74 (d, 1H, J=8.0 Hz, H-6); 13C NMR δ: 4.8, 19.5, 78.0, 78.6, 80.1, 92.4, 101.5, 141.4, 150.9, 164.6.
In a similar manner from 8b we obtained 9b in 81% yield. 1H NMR (CD3OD), δ: 1.12 (s, 3H, −CH3), 1.88 (s, 3H, −CH3), 3.53–3.71 (m, 4H, H-3′, H-4′, H-5′), 5.92 (s, 1H, H-1′), 7.62 (d, 1H, H-6); 13C NMR δ: 5.5, 11.2, 19.4, 78.0, 78.4, 79.4, 92.3, 110.3, 137.0, 151.1, 164.9.
1-(4-Azido-2,3-O-dibenzoyl-2-methyl-5-iodo-β-d-ribofuranosyl)uracil (12a) and 1-(4-azido-2,3-O-dibenzoyl-2-methyl-5-iodo-β-d-ribofuranosyl)thymine (12b)
A solution of 9a (210 mg, 0.57 mmol) and 0.5 M sodium methoxide solution (2.84 ml, 1.42 mmol) was stirred at 60°C for 2 h. The reaction mixture was then added to a solution of N-methylmorpholinium mesylate in MeOH [prepared in situ by adding N-methylmorpholine (0.15 ml, 1.42 mmol) to a solution of methanesulfonic acid (0.09 ml, 1.42 mmol)]. The reaction mixture was concentrated under reduced pressure and the residue was partially purified by silica gel column chromatography (10% MeOH in CH2Cl2) to give a white solid product 10a (76%). A mixture of benzyl triethylammonium chloride (194 mg, 0.855 mmol) and NaN3 (55 mg, 0.855 mmol) was slurried in acetonitrile (5 ml). The insoluble NaCl was removed by filtration and the filtrate washed with acetonitrile. The acetonitrile solution of benzyltriethylammonium azide was added to a solution of 10a and N-methylmorpholine (0.02 ml) in THF (5 ml), and a clear solution was formed. A solution of I2 (217 mg, 0.855 mmol) in THF was added slowly at 0°C. The reaction mixture was stirred at 5–10°C for 2 h. After removal of the solvents, the residue was purified by silica gel column chromatography with 10% MeOH in CH2Cl2 to afford the product 11a (65% yield). The benzoylation of 11a was carried out by reaction with benzoyl chloride (0.23 ml, 1.995 mmol), triethylamine (0.277 ml, 1.995 mmol) and DMAP (14 mg, 0.114 mmol) in THF (10 ml) at room temperature overnight. The solvent was removed under reduced pressure and the residue was purified by silica gel column chromatography (1–5% MeOH in CH2Cl2) to give 12a as a foam (82%). 1H NMR (CD3OD), δ 1.30 (s, 3H, −CH3), 3.32 (s, 1H, H-3′), 3.70 (d, 1H, J=11.2 Hz, H-5′), 3.79 (d, 1H, J=11.2 Hz, H-5″), 5.74 (d, 1H, J=8.0 Hz, H-5), 5.98 (s, 1H, H-1′), 7.41–7.62 (m, 6H, Ar-H), 7.65 (d, 1H, J=8.0 Hz, H-6), 7.93–8.16 (m, 4H, Ar-H). 12b was obtained using similar procedures in good yield, as shown in Figure 2, from 9b. 1H NMR (CD3OD), δ 1.82 (s, 3H, −CH3), 2.02 (s, 3H, −CH3), 3.34 (s, 1H, H-3′), 3.76 (d, 1H, J=11.2 Hz, H-5′), 3.83 (d, 1H, J=11.2 Hz, H-5″), 6.22 (s, 1H, H-1′), 7.38–8.16 (m, 11H, Ar-H, H-6).
Figure 2.
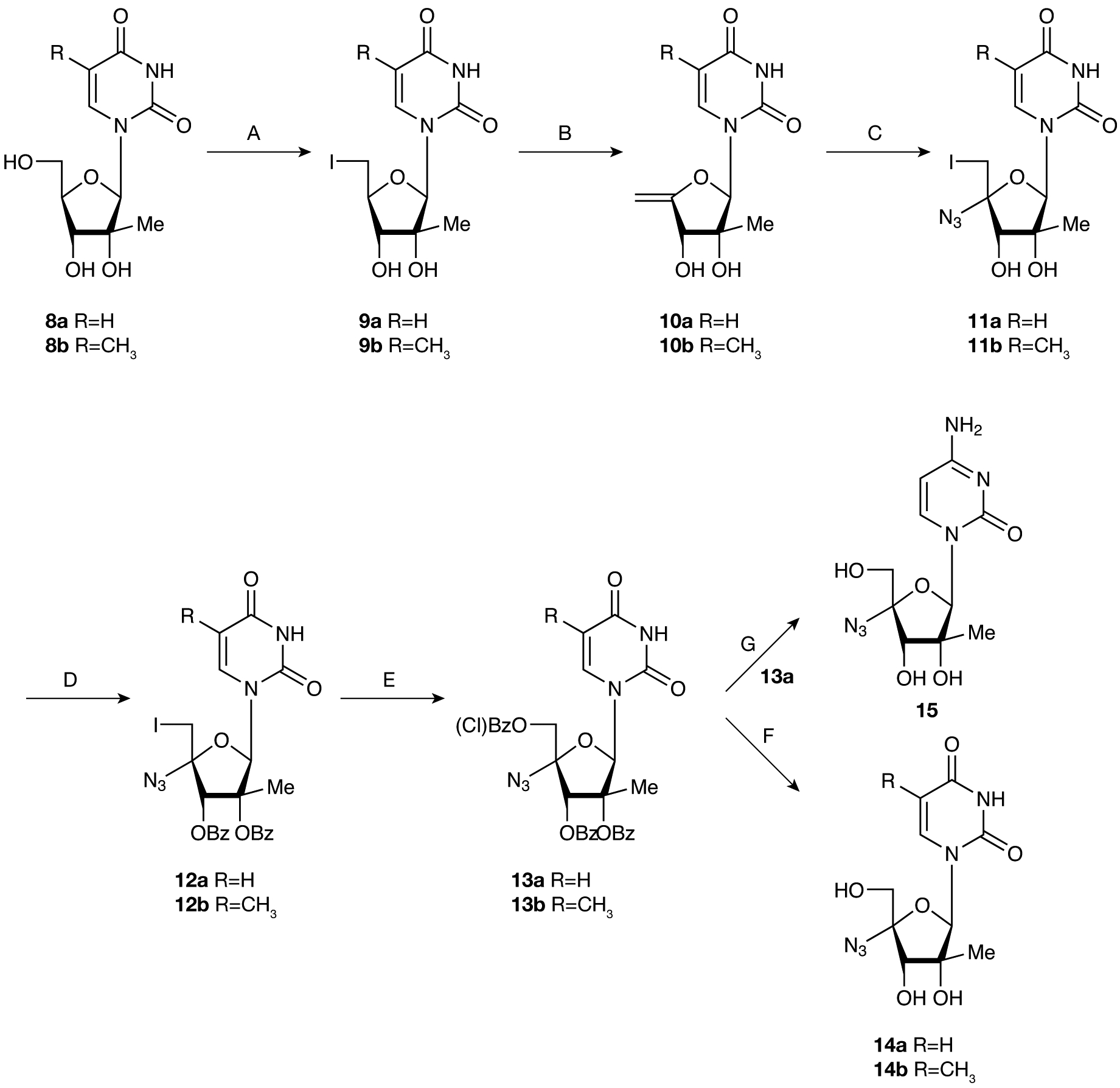
Synthetic route to 2′ C-methyl-4′azido pyrimidine nucleosides
A. I2, Ph3P, imidazole, tetrahydrofuran (THF), rt, (85%; 9a), (81%; 9b). B. NaOCH3, CH3OH, 60°C, (76%; 10a), (78%; 10b). C. ICI, NaN3, THF, (65%; 11a), (61%; 11b). D. BzCl, DMAP, Et3N, CH3CN, rt, (82%; 12a), (85%; 12b). E. meta-Chloroperoxybenzoic acid, CH2Cl2, rt, (62%; 13a), (66%; 13b). F. NH3/MeOH, rt, (86%; 14a), (83%; 14b). G. Step 1: 1,2,4-triazole, Et N, POCl3, CH2Cl2, 0°C to rt. Step 2: NH4OH/CH3CN/MeOH, rt, 42%.
1-(4-Azido-5-O-(4-chloro)benzoyl-2,3-O-dibenzoyl-2-methyl-β-d-ribofuranosyl)uracil (13a) and 1-(4-azido-5-O-(4-chloro)benzoyl-2,3-O-dibenzoyl-2-methyl-β-d-ribofuranosyl)thymine (13b)
To a solution of compound 12a (170 mg, 0.275 mmol) in CH2Cl2-H2O (5:1, 24 ml), tetrabutyl ammonium sulfate (94 mg, 0.275 mmol), potassium hydrogen phosphate (94 mg, 0.412 mmol) and m-chloroperbenzoic acid (308 mg, 1.37 mmol) were added. The reaction mixture was stirred at room temperature overnight and then quenched with a solution of sodium thiosulfate. After stirring 5 min, a solution of sodium carbonate was added. The organic layer was separated and evaporated. The residue was purified by silica gel column chromatography using 2% MeOH in CH2Cl2 to give 13a (62% yield). 1H NMR (CD3OD), δ 1.31 (s, 3H, −CH3), 3.32 (s, 1H, H-3′), 4.72 (s, 2H, H-5′), 5.65 (d, 1H, J=8.0 Hz, H-5), 5.81 (s, 1H, H-1′), 7.22–7.61 (m, 10H, Ar-H), 7.66 (d, 1H, J=8.0 Hz, H-6), 7.74–8.10 (m, 4H, Ar-H). 13b was obtained by using same procedure with 66% yield. 1H NMR (CD3OD), δ: 1.80 (s, 3H, −CH3), 2.01 (s, 3H, −CH3), 3.33 (s, 1H, H-3′), 3.78 (d, 1H, J=11.2 Hz, H-5′), 3.84 (d, 1H, J=11.2 Hz, H-5″), 6.23 (s, 1H, H-1′), 7.39–8.14 (m, 15H, Ar-H, H-6).
1-(4-Azido-2-methyl-β-d-ribofuranosyl)uracil (14a) and 1-(4-azido-2-methyl-β-d-ribofuranosyl)thymine (14b)
Compound 13a (100 mg, 0.15 mmol) was dissolved in saturated NH3 in MeOH (20 ml) and the mixture was stirred at room temperature overnight. The solvent was evaporated under reduced pressure and the residue was purified by silica gel column chromatography using 10% MeOH in CH2Cl2 to give white foam solid (40 mg, 86%). 1H NMR (CD3OD), δ 1.19 (s, 3H, −CH3), 3.67 (d, 1H, J=12.0 Hz, H-5′), 3.78 (d, 1H, J=12.0 Hz, H-5″), 4.03 (s, 1H, H-4′), 5.70 (d, 1H, J=8.0 Hz, H-5), 6.22 (s, 1H, H-1′), 8.04 (d, 1H, J=8.0 Hz, H-6); 13C NMR δ 19.9, 61.8, 73.2, 77.7, 93.2, 98.2, 101.7, 140.9, 151.0, 164.6. MS (ESI): 298 (M-H)−. 14b was obtained with same method in 83% yield. 1H NMR (CD3OD), δ 1.15 (s, 3H, −CH3), 1.84 (s, 3H, −CH3), 3.64 (d, 1H, J=12.4 Hz, H-5′), 3.78 (d, 1H, J=11.6 Hz, H-5″), 4.06 (s, 1H, H-4′), 6.21 (s, 1H, H-1′), 7.93(s, 1H, H-6); 13C NMR δ 11.3, 19.8, 61.7, 73.1, 77.7, 93.1, 98.2, 110.5, 136.5, 151.2, 164.9. MS (ESI): 312 (M-H)−.
1-(4-Azido-2-methyl-β-d-ribofuranosyl)cytosine (15)
To a solution of 13a (200 mg, 0.31 mmol), 1,2,4-triazole (339 mg, 4.91 mmol) and triethylamine (0.68 ml, 4.91 mmol) in anhydrous CH2Cl2 (30 ml) was added POCl3 (0.09 ml, 0.98 mmol). The reaction mixture was stirred at room temperature for 48 h and then quenched with water. The organic layer was separated. After evaporation of the solvent under reduced pressure, the residue was dissolved in acetonitrile. A quantity of 10 ml of concentrated NH4OH was added. The reaction mixture was stirred for 2 h. The solvent was evaporated and the residue was dissolved in MeOH. A quantity of 10 ml of concentrated NH4OH was added and the reaction mixture was stirred overnight. After removed the solvent, the residue was purified by silica gel column chromatography by using MeOH/CH2Cl2 (5–30%) as eluent to give 15 in 42% yield. 1H NMR (CD3OD), δ 1.20 (s, 3H, −CH3), 3.70 (d, 1H, J=12.0 Hz, H-5′), 3.80 (d, 1H, J=12.0 Hz, H-5′), 4.05 (s, 1H, H-4′), 6.15 (d, 1H, J=7.6 Hz, H-5), 6.20 (s, 1H, H-1′), 8.50 (d, 1H, J=7.6 Hz, H-6); 13C NMR δ 19.7, 61.6, 72.8, 78.0, 93.7, 94.0, 98.6, 144.6, 147.4, 159.8. MS (ESI): 299 (M+H)+.
4-Azido-2-C-methyluridine-5′-O-[Phenyl(ethyloxyl-alaninyl)]phosphate (16) and 4-azido-2-C-methylcytidine-5′-O-[phenyl(ethyloxy-l-alaninyl)]phosphate (17)
To a solution of 14a (70 mg, 0.23 mmol) in THF at 0°C, t-BuMgCl (0.47 ml, 0.47 mmol) was added and the mixture was stirred at room temperature for 20 min. A solution of phenyl-(ethoxy-l-alaninyl)-phosphorochloridate in THF (0.47 ml, 0.47 mmol) was added at 0°C and stirring was continued for 72 h. The reaction was quenched with methanol followed by aqueous NH4Cl. The solvents were evaporated and the residue was purified by PLC to give prodrug 16 (3 mg, 2.3%) and 30 mg of recovered 14a. 1H NMR (CD3OD), δ 1.14–1.22 (m, 6H, CH3-ethyl, CH3-alanine), 1.33 (s, 3H, CH3-2′), 3.92–4.29 (m, 6H, CH2-ethyl, CH-alanine, H-3′, H-5′), 5.61 (d, 1H, J=8.0 Hz, H-5), 6.17 (s, 1H, H-1′), 7.17–7.37 (m, 5H, phenyl-H), 7.58 (d, 1H, J=8.0 Hz, H-6). MS (ESI): 553 (M-H)−. The same procedure was employed for 15 (35 mg, 0.12 mmol) to prepared prodrug 17 (3 mg, 4.6%). 1H NMR (CD3OD), δ 1.12–1.33 (m, 9H, CH3-ethyl, CH3-alanine, CH3-2′), 3.89–4.48 (m, 6H, CH2-ethyl, CH-alanine, H-3′, H-5′), 5.83 (d, 1H, J=7.6 Hz, H-5), 6.28 (s, 1H, H-1′), 7.17–7.38 (m, 5H, phenyl-H), 7.60 (d, 1H, J=7.6 Hz, H-6). MS (ESI): 554 (M+H)+.
Virology
HCV replicon assays
Huh 7 clone B cells containing HCV replicon RNA were seeded in a 96-well plate at 5,000 cells/well, and the compounds tested initially at 10 μM in triplicate immediately after seeding. Following 5 days incubation (37°C, 5% CO2), total cellular RNA was isolated by using the VersaGene RNA purification kit (Gentra, Minneapolis, MN, USA). Replicon RNA and an internal control (TaqMan rRNA control reagents, Applied Biosystems, Foster City, CA, USA) were amplified in a single step multiplex real time reverse transcriptase (RT)-PCR assay. A dose–response curve was determined for nucleosides demonstrating antiviral activity below 10 μM. The antiviral effectiveness of the nucleosides was calculated by subtracting the threshold RT-PCR cycle of the test compound from the threshold RT-PCR cycle of the no-drug control (ΔCt HCV). A ΔCt of 3.3 equals a 1 log10 reduction (equal to 90% less starting material) in replicon RNA levels. The cytotoxicity of the compounds was also calculated by using the ΔCt ribosomal RNA (rRNA) values. (2′-Me-C) was used as the control. To determine 90% effective concentration, 50% effective concetration and 50% cytotoxicity concentrations, ΔCt values were first converted into fraction of starting material and were then used to calculate the percentage inhibition.
Results
During the preparation of this paper, an alternate synthesis appeared for 2′-C-methyl-4′-azido cytidine (15) [20]. The general procedure used for the synthesis of 2′-C-methyl-4′-azido nucleosides was an extension of that previously reported in the literature (Figure 2) [17,18,21,22].
2′-C-Methyl ribonucleosides 8a–b [23] were converted to the corresponding 5′-iodo compounds 9a–b and then treated with sodium methoxide to yield the 4′-exocyclic methylene nucleosides 10a–b. 4′-Azido-nucleosides 11a–b were obtained in good yield by treatment of olefins 10a–b with in situ generated iodine azide. Benzoylation of the 2′ and 3′-hydroxy groups followed by oxidative displacement of 5′-iodo group with meta-chloroperoxybenzoic acid afforded the protected 4′-azido nucleosides 13a–b. Deprotection with NH3 in MeOH gave the desired 2′-C-methyl-4′-azido uridine 14a and thymidine analogue 14b in good yield. 2′-C-Methyl-4′-azido cytidine (15) was prepared, for comparison, in two steps by treatment of compound 13a with 1,2,4-triazole and phosphorous oxychloride in the presence of triethyl amine. Subsequent exposure of the crude 4-(1H-1,2,4-triazol-1-yl)-2′-C-methyl-4′-azido pyrimidinone to NH4OH furnished the desired 2′-C-methyl-4′-azido cytidine, (15; Figure 2). This synthesis represents an alternate and higher yielding approach to this class of compounds when compared with previous work [20].
The 2′-C-methyl-4′-azido nucleosides 14a and 14b were evaluated for their ability to inhibit HCV RNA replication using a Huh7-cell-based subgenomic replicon assay [24] and compared with the known cytidine analogue 15. Cytotoxicity in Huh7 cells was determined simultaneously with anti-HCV activity by extraction and amplification of both HCV RNA and rRNA. The anti-HCV replicon activity and cytotoxicity of these compounds are summarized in Table 1. It is interesting to note that NM-107 displayed low toxicity in peripheral blood mononuclear (PBM) and CEM cells, whereas R-1479 showed some toxicity only in the more intensively proliferative CEM cell line. Although the lack of anti-HCV activity might have been predicted for the uridine analogue 14a and the thymidine analogue 14b, we were surprised to find that the cytidine analogue 15 was also inactive at concentrations up to 10 μM [20]. The inactivity of the cytidine analogue 15 is in clear contrast to the trend that is seen in the R-1479 [25], NM-107 [26] and PSI-6130 [27,28] compound families where the cytidine nucleoside analogues display anti-HCV activity, whereas the uridine nucleosides are devoid of anti-HCV activity. Indeed, these three new analogues displayed no inhibition of HCV replicon RNA replication or toxicity to Huh7. Further evaluation in PBM, CEM, and Vero cells up to 100 μM indicated no cytotoxicity. The lack of cytotoxicity or activity of these nucleosides as anti-HCV agents might be the result of inefficient uptake and/or their inability to be anabolized to the corresponding nucleoside triphosphates.
Table 1.
Anti-HCV replicon activity and cellular toxicity of synthesized nucleoside and nucleotide analoguesa
| CC50, μM | ||||||
|---|---|---|---|---|---|---|
| Compound | HCV EC50, μM | HCV EC90, μM | Huh7 | PBM | CEM | Vero |
| NM-107 | 2.8 | 9.6 | >10 | 29.4 | 24.5 | >100 |
| R-1479 | 10.6 | 31.8 | >10 | >100 | 10.5 | >100 |
| 14a | >10 | >10 | >10 | >100 | >100 | >100 |
| 14b | >10 | >10 | >10 | >100 | >100 | >100 |
| 15 | >10 | >10 | >10 | >100 | >100 | >100 |
| 16 | 3.0 ±1.1b | 7.6 ±1.7b | >100 | >100 | >100 | >100 |
| 17 | 4.9 ±2.3b | >10b | >100 | >100 | >100 | >100 |
Values are means of triplicate measurements.
Means of two experiments ±sd. CC50, 50% cytotoxicity concentration; EC50, 50% effective concentration; EC90, 90% effective concentration; HCV, hepatitis C virus; PBM, peripheral blood mononuclear cell.
Because the cytidine analogue 15 is a combination of the 2′-C-methyl moiety of NM107 (1) [15] and the 4′-azido moiety of R1479 (5) [17,18] and it has been reported that the initial phosphorylation of each is the result of different cellular enzymes [18], we hypothesized that disruption in the phosphorylation pathway was a likely cause for loss of anti-HCV activity. To address this possibility, the 5′-monophosphate aryloxy phosphoramidate derivatives of uridine analogue 14a and cytidine analogue 15 were synthesized. This phosphoramidate prodrug approach was successfully applied to various nucleosides [19,29–31] and allowed the nucleoside analogue to bypass the first phosphorylation step by intracellular delivery of monophosphorylated nucleoside. HCV phosphoramidate prodrugs have been successfully utilized to convert the inactive uridine analogue of R-1479 [25]; adenosine analogue of R-1479 [32]; uridine analogue of PSI-6130 [27,28]; and uridine analogue of NM-107 [26] into active compounds in the HCV replicon system.
The target monophosphate prodrugs 16 and 17 were prepared as diastereoisomeric mixtures at the phosphorous centre by the addition of phenyl-(ethoxy-l-alaninyl)-phosphorochloridate to the preformed 5′-alkoxide of nucleosides 14a and 15 in THF (Figure 3).
Figure 3.
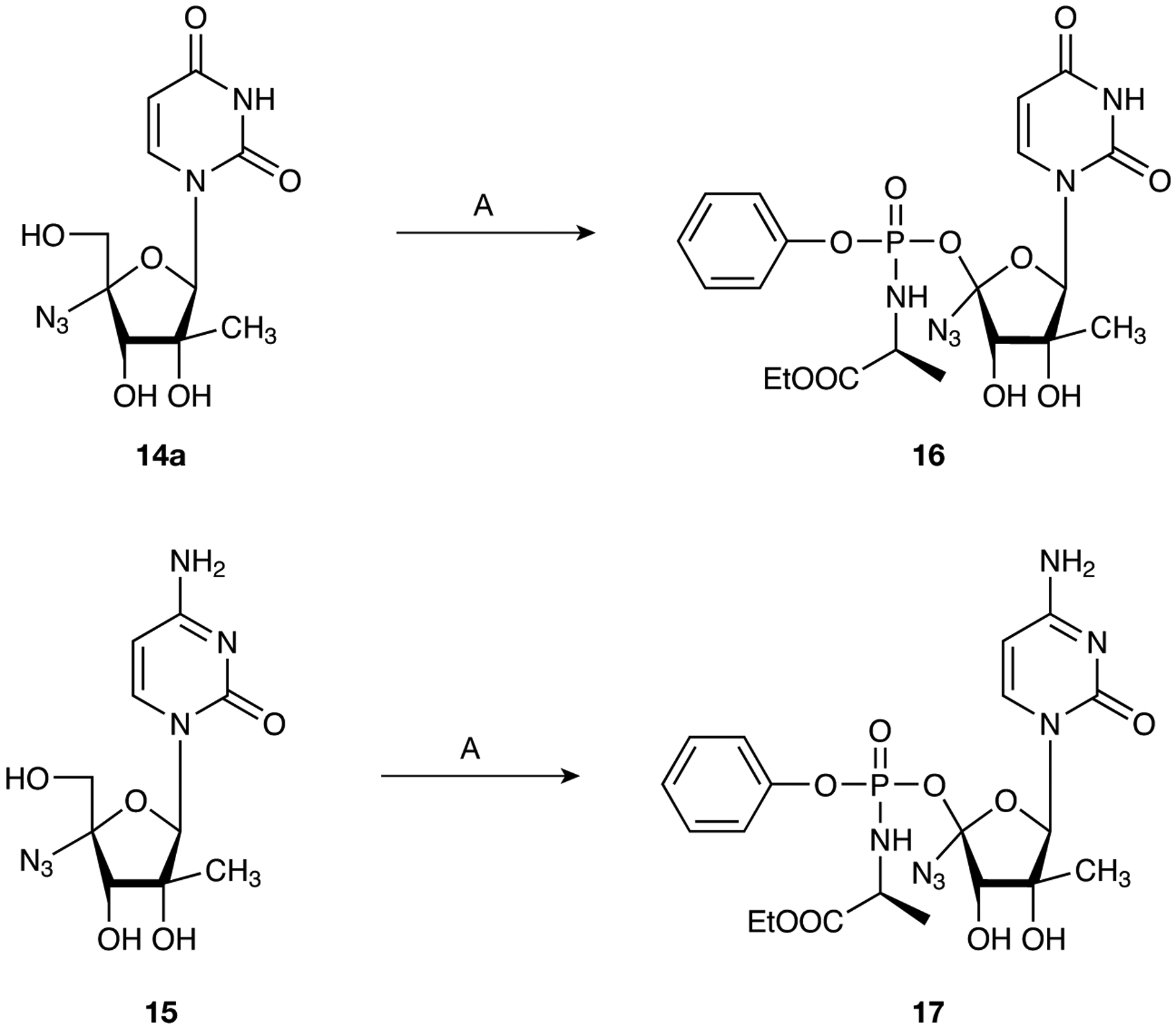
Reagents and conditions
A. t-BuMgCl, phenyl-(ethoxy-l-alaninyl)-phosphorochloridate, tetrahydrofuran, 0°C to rt, 72 h (2.3%; 16), (4.6%; 17).
In contrast to the parent nucleosides 14a and 15 that were inactive, the prodrugs 16–17 exhibited anti-HCV activity with 50% effective concentration values of 3.0 μM and 4.9 μM, respectively. Additionally, these 5′-monophosphate prodrugs display no observable toxicity toward Huh7, PBM, CEM, or Vero cells up to 100 μM. These results suggest that the poor activity of parent nucleosides was probably the result of insufficient conversion to their corresponding 5′-monophosphate nucleoside.
Discussion
Three 2′-C-methyl-4′-azido-pyrimidine nucleosides and two novel 5′-monophosphate prodrugs were synthesized and evaluated for their antiviral activity in an HCV replicon system. As expected, 2′-C-methyl-4′-azido cytidine 15 did not exhibit anti-HCV activity up to 10 μM [20]. Furthermore, we prepared and tested the corresponding uridine and thymidine 2′-C-methyl-4′-azido analogues and found them also to be devoid of anti-HCV activity and all three pyrimidine analogues displayed no evidence of cytotoxicity at high concentrations. We demonstrated that the 5′-monophosphate prodrugs of the uridine analogue 14a and cytidine analogue 15 exhibited good anti-HCV activity without apparent cytotoxicity. This work is the first example of both inactive analogues of a nucleoside being converted to active anti-HCV nucleosides via 5′-monophosphate prodrugs. Ongoing work is focused on optimizing the 5′-monophosphate prodrug portion of these novel nucleoside analogues to improve their potency.
Acknowledgements
This work is supported in part by NIH grants 2P30-AI-50409 (CFAR), 5R37-AI-041980, and by the Department of Veterans Affairs.
Footnotes
Disclosure statement
RFS is the principal founder and a Director of RFS Pharma, LLC. His laboratory received no funding from RFS Pharma for this work. All other authors declare no competing interests.
References
- 1.Wasley A, Alter MJ. Epidemiology of hepatitis C: geographic differences and temporal trends. Semin Liver Dis 2000; 20:1–16. [DOI] [PubMed] [Google Scholar]
- 2.Schinazi RF, Sommadossi JP, Rice C (Editors). Frontiers in viral hepatitis. Amsterdam: Elsevier; 2003. [Google Scholar]
- 3.Wu JZ, Yao N, Walker M, Hong Z. Recent advances in discovery and development of promising therapeutics against hepatitis C virus NS5B RNA-dependent RNA polymerase. Mini Rev Med Chem 2005; 5:1103–1112. [DOI] [PubMed] [Google Scholar]
- 4.McHutchison JG, Patel K. Future therapy of hepatitis C. Hepatology 2002; 36 Suppl 1:S245–S252. [DOI] [PubMed] [Google Scholar]
- 5.Pawlotsky JM. Mechanisms of antiviral treatment efficacy and failure in chronic hepatitis C. Antiviral Res 2003; 59:1–11. [DOI] [PubMed] [Google Scholar]
- 6.McHutchison JG, Manns M, Patel K, et al. Adherence to combination therapy enhances sustained response in genotype-1-infected patients with chronic hepatitis C. Gastroenterology 2002; 123:1061–1069. [DOI] [PubMed] [Google Scholar]
- 7.Fried MW. Side effects of therapy of hepatitis C and their management. Hepatology 2002; 36:S237–S244. [DOI] [PubMed] [Google Scholar]
- 8.Tan SL, Pause A, Shi Y, Sonenberg N. Hepatitis C therapeutics: current status and emerging strategies. Nat Rev Drug Discov 2002; 1:867–881. [DOI] [PubMed] [Google Scholar]
- 9.De Francesco R, Migliaccio G. Challenges and successes in developing new therapies for hepatitis C. Nature 2005; 436:953–960. [DOI] [PubMed] [Google Scholar]
- 10.Pawlotsky JM, Gish RG. Future therapies for hepatitis C. Antivir Ther 2006; 11:397–408. [PubMed] [Google Scholar]
- 11.Schinazi RF, Coats SJ, Bassit LC, et al. Approaches for the development of antiviral compounds: the case of hepatitisC virus In Kräusslich H-G, Bartenschlager R (Editors). Handbook of experimental pharmacology: antiviral strategies. Vol. 189 Berlin Heidelberg: Springer–Verlag; 2009; pp. 25–51. [DOI] [PubMed] [Google Scholar]
- 12.Carroll SS, Tomassini JE, Bosserman M, et al. Inhibition of hepatitis C virus RNA replication by 2′-modified nucleoside analogs. J Biol Chem 2003; 278:11979–11984. [DOI] [PubMed] [Google Scholar]
- 13.Olsen DB, Eldrup AB, Bartholomew L, et al. A 7-deaza-adenosine analog is a potent and selective inhibitor of hepatitis C virus replication with excellent pharmacokinetic properties. Antimicrob Agents Chemother 2004; 48:3944–3953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Eldrup AB, Prhavc M, Brooks J, et al. Structure-activity relationship of heterobase-modified 2′-C-methyl ribonucleosides as inhibitors of hepatitis C virus RNA replication. J Med Chem 2004; 47:5284–5297. [DOI] [PubMed] [Google Scholar]
- 15.Pierra C, Amador A, Benzaria S, et al. Synthesis and pharmacokinetics of valopicitabine (NM283), an efficient prodrug of the potent anti-HCV agent 2′-C-methylcytidine. J Med Chem 2006; 49:6614–6620. [DOI] [PubMed] [Google Scholar]
- 16.Clark JL, Hollecker L, Mason JC, et al. Design, synthesis, and antiviral activity of 2′-deoxy-2′-fluoro-2′-C-methylcytidine, a potent inhibitor of hepatitis C virus replication. J Med Chem 2005; 48:5504–5508. [DOI] [PubMed] [Google Scholar]
- 17.Smith DB, Martin JA, Klumpp K, et al. Design, synthesis, and antiviral properties of 4′-substituted ribonucleosides as inhibitors of hepatitis C virus replication: the discovery of R1479. Bioorg Med Chem 2007; 17:2570–2576. [DOI] [PubMed] [Google Scholar]
- 18.Klumpp K, Kalayanov G, Ma H, et al. 2′-deoxy-4′-azido nucleoside analogs are highly potent inhibitors of hepatitis C virus replication despite the lack of 2′-alpha-hydroxyl groups. J Biol Chem 2008; 283:2167–2175. [DOI] [PubMed] [Google Scholar]
- 19.Hecker SJ, Erion MD. Prodrugs of phosphates and phosphonates. J Med Chem 2008; 51:2328–2345. [DOI] [PubMed] [Google Scholar]
- 20.Smith DB, Kalayanov G, Sund C, et al. The design, synthesis, and antiviral activity of monofluoro and difluoro analogues of 4′-azidocytidine against hepatitis C virus replication: the discovery of 4′-azido-2′-deoxy-2′-fluorocytidine and 4′-azido-2′-dideoxy-2′,2′-difluorocytidine. J Med Chem 2009; 52:2971–2978. [DOI] [PubMed] [Google Scholar]
- 21.Moffatt JG. Chemical transformations of the sugar moiety of nucleosides In Walker RT, De Clercq E, Eckstein F (Editors). Nucleoside analogues. New York: Plenum Publishing Corporation; 1979; pp. 71–164. [Google Scholar]
- 22.Verheyden JPH, Moffat JG. 4′-Substituted nucleosides. I. Synthesis of 4′-methoxyuridine and related compounds. J Am Chem Soc 1975; 97:4386–4395. [DOI] [PubMed] [Google Scholar]
- 23.Benzaria S, Bardiot D, Bouisset T, et al. 2′-C-Methyl branched pyrimidine ribonucleoside analogues: potent inhibitors of RNA virus replication. Antivir Chem Chemother 2007; 18:225–242. [DOI] [PubMed] [Google Scholar]
- 24.Stuyver LJ, Whitaker T, McBrayer T, et al. Ribonucleoside analogue that blocks replication of bovine viral diarrhea and hepatitis C viruses in culture. Antimicrob Agents Chemother 2003; 47:244–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Perrone P, Luoni GM, Kelleher MR, et al. Application of the phosphoramidate ProTide approach to 4′-azidouridine confers sub-micromolar potency versus hepatitis C virus on an inactive nucleoside. J Med Chem 2007; 50:1840–1849. [DOI] [PubMed] [Google Scholar]
- 26.Sommadossi J-P, Gosselin G, Pierra C, Perigaud C, Peyrottes S, inventors; Idenix Pharmaceuticals Inc., assignee. Compounds and pharmaceutical compositions for the treatment of viral infections. WO 2008/082601 28 December 2008.
- 27.Murakami E, Niu C, Bao H, et al. The mechanism of action of beta-d-2′-deoxy-2′-fluoro-2′-C-methylcytidine involves a second metabolic pathway leading to beta-d-2′-deoxy-2′-fluoro-2′-C-methyluridine 5′-triphosphate, a potent inhibitor of the hepatitis C virus RNA-dependent RNA polymerase. Antimicrob Agents Chemother 2008; 52:458–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sofia MJ, Du J, Wang P, Nagarathnam D, inventors; Pharmasset Inc., assignee. Nucleoside phosphoramidate prodrugs. WO 2008/121634 26 March 2008.
- 29.Cahard D, McGuigan C, Balzarini J. Aryloxy phosphoramidate triesters as pro-tides. Mini Rev Med Chem 2004; 4:371–381. [DOI] [PubMed] [Google Scholar]
- 30.McGuigan C, Cahard D, Sheeka HM, De Clercq E, Balzarini J. Aryl phosphoramidate derivatives of d4T have improved anti-HIV efficacy in tissue culture and may act by the generation of a novel intracellular metabolite. J Med Chem 1996; 39:1748–1753. [DOI] [PubMed] [Google Scholar]
- 31.McGuigan C, Hassan-Abdallah A, Srinivasan S, et al. Application of phosphoramidate ProTide technology significantly improves antiviral potency of carbocyclic adenosine derivatives. J Med Chem 2006; 49:7215–7226. [DOI] [PubMed] [Google Scholar]
- 32.Perrone P, Daverio F, Valente R, et al. First example of phosphoramidate approach applied to a 4′-substituted purine nucleoside (4′-azidoadenosine): conversion of an inactive nucleoside to a submicromolar compound versus hepatitis C virus. J Med Chem 2007; 50:5463–5470. [DOI] [PubMed] [Google Scholar]