

Abstract
The aggregation of amyloid-β (Aβ) peptides into senile plaques is a hallmark of Alzheimer’s disease (AD) and is hypothesized to be the primary cause of AD related neurodegeneration. Previous studies have shown the ability of curcumin to both inhibit the aggregation of Aβ peptides into oligomers or fibrils and reduce amyloids in vivo. Despite the promise of curcumin and its derivatives to serve as diagnostic, preventative, and potentially therapeutic AD molecules, the mechanism by which curcumin and its derivatives bind to and inhibit Aβ fibrils’ formation remains elusive. Here, we investigated curcumin and a set of curcumin derivatives in complex with a hexamer peptide model of the Aβ1–42 fibril using nearly exhaustive docking, followed by multi-ns molecular dynamics simulations, to provide atomistic-detail insights into the molecules’ binding and inhibitory properties. In the vast majority of the simulations, curcumin and its derivatives remain firmly bound in complex with the fibril through primarily three different principle binding modes, in which the molecules interact with residue domain 17LVFFA21, in line with previous experiments. In a small subset of these simulations, the molecules partly dissociate the outermost peptide of the Aβ1–42 fibril by disrupting β-sheets within the residue domain 12VHHQKLVFF20. A comparison between binding modes leading or not leading to partial dissociation of the outermost peptide suggests that the latter is attributed to a few subtle key structural and energetic interaction-based differences. Interestingly, partial dissociation appears to be either an outcome of high affinity interactions or a cause leading to high affinity interactions between the molecules and the fibril, which could partly serve as a compensation for the energy loss in the fibril due to partial dissociation. In conjunction with this, we suggest a potential inhibition mechanism of Αβ1–42 aggregation by the molecules, where the partially dissociated 16KLVFF20 domain of the outermost peptide could either remain unstructured or wrap around to form intramolecular interactions with the same peptide’s 29GAIIG33 domain, while the molecules could additionally act as a patch against the external edge of the second outermost peptide’s 16KLVFF20 domain. Thereby, individually or concurrently, these could prohibit fibril elongation.
Introduction
Alzheimer’s disease (AD) is characterized by the pathological hallmarks of extracellular amyloid-β (Aβ) plaques and intraneuronal tau-containing neurofibrillary tangles in the brain.1 According to the amyloid cascade hypothesis, Αβ-related toxicity is the primary cause of synaptic dysfunction and neurodegeneration underlying the progression characteristic of AD.2−4 Targeting the production, aggregation, and toxicity of Aβ with small molecule drugs, peptides, sequestering proteins, or antibodies is an active area of AD research due to the general acceptance of this hypothesis, but thus far, several drugs targeting Aβ have faced challenges.5 Despite the fact that this raised skepticism in recent years, the genetic evidence for the causative role of Aβ in AD is strong, including both familial AD-causing mutations6 and the recently discovered rare, protective mutation in an Icelandic kindred.7−9 Thus, targeting Aβ aggregation is a potentially attractive therapeutic approach.8
While monomeric Aβ is nontoxic, upon self-assembly, its toxicity increases substantially as oligomers form and then decreases with formation of fibrils.8 Self-assembly is associated with toxicity, and oligomers ranging from dimers to protofibrils are toxic.10−12 This adds to the complexity of the problem and the difficulty of developing effective inhibitors of Aβ oligomer toxicity. Since the realization of Αβ as a potential therapeutic target, several inhibitors have been suggested, ranging from natural products, peptides, peptidomimetics, and various synthetic compounds, including curcumin derivatives, as potential modulators of Aβ aggregation and inhibitors of its toxicity.8,13−28 In addition, larger molecules have been suggested as amyloid inhibitors, including human monoclonal antibodies (e.g., aducanumab that selectively reacts with Aβ aggregates, including soluble oligomers and insoluble fibrils crossing the blood–brain barrier, engaging its target, and clearing Aβ from plaque-bearing transgenic mouse brains4), or β-wrapin proteins, engineered to bind and sequester amyloid monomeric proteins, including Αβ, and thereby reduce their toxicity.29−37
The challenge of studies tackling Aβ amyloid formation and translating to successful drugs for AD, despite years of research, could be associated with several factors.5 Among others, two critical factors can be the source of these challenges: (1) clinical trials targeting amyloid are taking place too late—modulating Aβ action after clinical diagnosis may be too late in the disease process to have a beneficial effect. This underlines the importance in diagnosis and prevention and the potential ability to reverse the effects induced by fibrils. As for reversing the effects, recent studies showed that Alzheimer’s disease-related learning and memory deficits in asymptomatic transgenic mouse model of the disease are ameliorated by EPPS, an agent capable of disaggregating Aβ fibrils,38,39 which could suggest the importance of amyloid disassembly as a promising therapeutic avenue. (2) Many other proteins are involved in AD apart from Aβ, and perhaps drugs should also be targeting tau pathology rather than solely Aβ.5
In vitro studies have suggested the capability of curcumin or curcumin derivatives to significantly reduce the β-sheet content of the peptide in a time dependent manner,40 destabilize41 and disaggregate42 preformed Aβ fibrils, block the toxicity of Aβ oligomers,42 and disintegrate preformed tau filaments.43 Furthermore, in vivo studies have suggested the ability of curcumin to reduce amyloid plaque burden,42 reduce insoluble Aβ deposits,44 and disassemble tau oligomeric structures.45 Combined with its additional promising properties in the diagnosis, prevention, and treatment of AD (reviewed in ref (46)), curcumin can be considered a highly promising molecule for future investigation and improvement. Since 2005, when curcumin was shown to inhibit formation of Aβ oligomers and fibrils, block toxicity of Aβ oligomers, bind plaques, and suggested to reduce amyloid in vivo,42 a series of experimental and computational studies aimed to highlight its preventive/potential therapeutic properties,42,45,47−50 uncover its mechanism of action in atomistic accuracy,51,52 and propose new curcumin derivatives.44,53−58 The first clinical trials investigating the effect of curcumin on AD patients showed no such promising results, where no significant difference was observed between the curcumin and placebo group after 6 and 12 months of oral administration.59−61 According to recent studies, cells incubated with Aβ that were pre- and post-treated with curcumin lessen mitochondrial dysfunction and retain cell viability and mitochondrial dynamics, mitochondrial biogenesis, and synaptic activity.61 Recently, good acute and chronic activities were demonstrated in test subjects given low doses (80–180 mg/day) of novel curcumin formulations (Longvida and Theracurmin), which were optimized to ensure higher bioavailability.62,63 For instance, Longvida improved sustained attention and working memory tasks immediately after a single dose, and after four-week administration, enhanced memory, mood, alertness, and contentedness.62 Furthermore, a recent double-blind, placebo-controlled trial found that twice-daily oral consumption of 90 mg of a bioavailable form of curcumin led to memory and attention benefits in nondemented adults over 60 years of age.63 Based on positron emission tomography (PET) imaging, the observed benefits were associated with a combination of decreased tau accumulation and amyloid burden.63,64 Additionally, beyond its ability to protect against Aβ toxicity, curcumin has also been shown to protect against lead neurotoxicity65 and oxidative damage.66 All in all, curcumin appears to be a promising diagnostic, preventive, and potentially therapeutic AD molecule.
Nevertheless, despite the important insights gained by computational studies on curcumin’s interactions with primarily Aβ monomers,51,67 dimers,52 trimers,51 and tetramers,52 understanding the mechanism of action of curcumin and its derivatives to bind to and inhibit the elongation of Αβ fibrils is still not clear. The lack of such knowledge significantly impedes the design/discovery of novel molecules with potentially improved properties compared to those of curcumin. Previous experimental studies have investigated the Aβ aggregation inhibition and fibril disassembly by curcumin and its derivatives.44,53,54,63,64 Motivated by such a study investigating a series of curcumin-based molecules for their inhibitory effects on Aβ1–42 aggregation and their ability to induce lower molecular size Aβ1–42 species that have weaker cell toxicity,54 here, we computationally examined a subset of curcumin-based molecules54 in complex with a hexamer Aβ1–42 fibril. The studied set of molecules which were selected here for investigation comprised SY12, SY31, SY5, and curcumin (Figure 1). These were selected based on their capacity to inhibit Aβ1–42 aggregation and induced the formation of lower molecular size Aβ1–42 species.54 Specifically, SY12 and SY31 were proposed to be potential therapeutic candidates for preventing AD,54 and in addition, mice fed a chow diet containing SY5 for six months showed a reduction in insoluble Aβ deposits and a reduction in cognitive deficits when compared to mice fed a control diet.44 Our computational studies comprised of docking studies followed by multi-ns all-atom molecular dynamics (MD) simulations and in-depth structural and energetic analysis of curcumin molecules in complex with a hexamer peptide model of the Aβ1–42 fibril. Our investigation aimed to provide fundamental atomistic-detail insights into the interactions formed by curcumin and curcumin derivatives in complex with Aβ1–42 fibrils and ultimately shed light into their binding and inhibitory properties.
Figure 1.
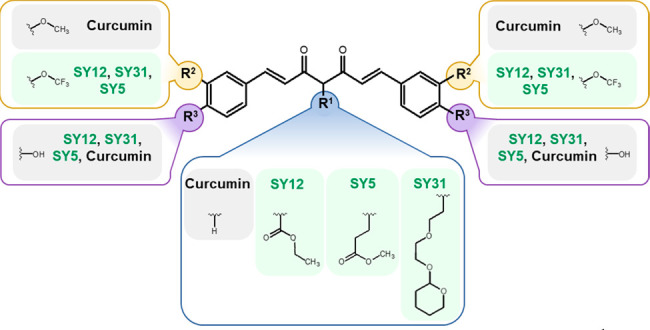
Description of each molecules’ respective R1 (bottom, encapsulated in blue lines), R2 (top-left and top-right, encapsulated in yellow lines), and R3 (bottom-left and bottom-right, encapsulated in purple lines) functional groups with reference to curcumin.
Methods
Docking of the Molecules to a Hexamer Peptide Model of the Aβ1–42 Fibril
We initially performed docking studies to generate initial complex structures of the selected molecules in complex with an Aβ1–42 fibril. We aimed to produce a nearly exhaustive search of docked poses of the molecules that span the space of probable binding within the Aβ1–42 fibril region of interest. For this purpose, we used SwissDock,68 which gave us the capacity to produce numerous clustered docked poses ranked by CHARMM69 energies. Each molecule, SY12, SY31, SY5, and curcumin, was initially docked to an experimentally resolved structure of an Aβ1–42 fibril. The initial structure of each molecule was built using MarvinSketch and UCSF Chimera.70 An Aβ1–42 fibril, in contrast to an Aβ1–40 fibril, was used, in accordance with experiments investigating the same molecules by Yanagisawa et al.54 Additional experimental evidence from Masuda et al. indicated that curcumin interacts with residues 12, 17–21 of Aβ1–42 fibrils.71 Based on this, the experimentally resolved structure of Aβ1–42 fibril reported by Xiao et al. (PDB ID: 2MXU(72)), with residues 11–42 resolved, was preferred over other resolved structures in the PDB (e.g., 5OQV,73 2NAO74). This is due to the fact that in the latter, the flexible N-terminal 1–10 domain is tightly packed against residues Leu17, Ile31, and Phe19 which, according to additional docking studies (not shown), hinders the docking of curcumin to the aforementioned region. In contrast, in 2MXU,72 the terminal 1–10 domain is not resolved; thus, the structure allowed for the docking of molecules to the experimentally determined expected binding site. The experimentally resolved Aβ1–42 fragment used in our study is sufficiently large; it includes residues 11–16, 16–22, and 22–28, which have been identified as amyloid seeds75 and the key interacting residues with curcumin according to previous experiments.71 Here, the Aβ1–42 fragment was modeled as a hexamer using the first six chains of peptides from the first conformation from the ensemble of NMR structures (PDB ID: 2MXU,72Figure S1). The hexamer peptide model of the fibril was considered beneficial and an optimum compromise, as it is sufficiently large to represent a fibril and sufficiently small to reduce computational burden. We acetylated the truncated N-terminal of each of the six modeled Aβ1–42 peptides in the hexamer peptide model of the Aβ1–42 fibril to eliminate artificial charges at the N-termini due to the absence of experimentally unresolved residues. Henceforth, this Aβ1–42 fragment will be denoted as the Aβ1–42 fibril. This Aβ1–42 fibril was used as the starting structure for the docking of all molecules.
To eliminate the possibility of highly similar docked poses generated for each of the investigated molecules, we performed an additional clustering analysis on the docked poses generated by SwissDock68 and sorted by energetic favorability. The clustering analysis was performed in WORDOM76 using leader clustering based on the heavy atoms of each molecule using a 3 Å root-mean-square deviation (RMSD) cutoff. The specific clustering method allowed us to extract the most energetically favorable docked pose per cluster per molecule, as in our previous study,77 and eliminate poses with an RMSD less than 3 Å to the leaders.
Simulations of Molecules in Complex with the Modeled Aβ1–42 Fibrils
We performed 100 ns MD simulations for each of the 94 distinct docked poses of the molecules in complex with the Aβ1–42 fibril. The starting structures for the 94 simulations comprised each of the 94 distinct docked poses extracted in the previous section (Table 1, column 2), independently solvated in a cubic water box. The setup and simulations for all modeled systems were performed in CHARMM,69 version c39b2. We used CHARMM3678 topology and parameters for all simulations, with topologies and parameters for the molecules generated through CGENFF.79 After a 1.0 ns equilibration stage in which the fibril and docked molecule were lightly constrained, all constraints were released, and each of the 94 systems was simulated for 100 ns. Simulation snapshots were extracted every 200 ps for subsequent analysis, focusing on peptide:peptide and molecule:peptide interactions. Additionally, we performed 10 independent, 100 ns MD simulations of the Αβ1–42 fibril in the absence of any molecule (referred to as uncomplexed fibril). The simulations of the uncomplexed fibril were performed to investigate the structure of the modeled Αβ1–42 fibril in the absence of molecules and were used as a comparison reference point. Additional details regarding the preparation and implementation of the MD simulations are described in the Supporting Information.
Table 1. Summary of Investigated Molecules, Their Corresponding Simulations, and Their Binding Properties.a.
| no. of simulations in which the molecule is stable |
no. of simulations in which the molecule
adopted each binding mode |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| molecule | no. of docked poses used as initial structures for MD simulations | Aβ1–42fibril dissociation is absent | Aβ1–42fibril dissociation is initiated | binding mode 1 | binding mode 2 | binding mode 3 | uncategorized binding mode | ||||
| nd | d | nd | d | nd | d | nd | d | ||||
| SY12 | 25 | 13 | 5 | 6 | 2 | 3 | 1 | 1 | 1 | 3 | 1 |
| SY31 | 25 | 14 | 5 | 5 | 2 | 5 | 2 | 1 | 1 | 3 | 0 |
| SY5 | 23 | 14 | 4 | 4 | 2 | 3 | 2 | 3 | 0 | 4 | 0 |
| curcumin | 21 | 10 | 1 | 10 | 1 | 0 | 0 | 0 | 0 | 0 | 0 |
| total | 94 | 50 | 15 | 25 | 7 | 11 | 5 | 5 | 2 | 10 | 1 |
Column 1 corresponds to the investigated molecules. Column 2 corresponds to the total number of distinct docked poses and simulations per molecule. Column 3 corresponds to the number of MD simulations in which the molecule remains stable in its binding to the modeled fibril. Column 4 the number of simulations in which the molecule adopts binding modes 1, 2, 3, or an uncategorized binding mode. nd denotes no dissociation. pd denotes partial dissociation.
Conformational Analysis of the Modeled Aβ1–42 Fibril within the Simulations
We inspected the structural preservation of the modeled Aβ1–42 fibril in each of the modeled systems through secondary structure, RMSD, root-mean-square fluctuation (RMSF), and β-sheet content calculations. The preservation of β-sheet conformations between the individual Aβ1–42 monomers within the Αβ1–42 fibril was inspected using the STRIDE algorithm80 implemented in VMD.81 Specifically, the degree of structural preservation of the Αβ1–42 fibril within the simulations was evaluated by calculating the backbone RMSD of the entire fibril with respect to their initial structure, by calculating the RMSF of all Aβ1–42 fibril Cα atoms, and by calculating the β-sheet content as a function of time of the outermost peptide within the simulations. Based on the analyses evaluating structural preservation of the simulated fibrils, within the majority of the simulations of the molecules in complex with the Aβ1–42 fibril and all the simulations of the uncomplexed Aβ1–42 fibril, the structure of the Aβ1–42 fibril is preserved. Interestingly, in a small subset of simulations of the molecules in complex with the Aβ1–42 fibril, a partial dissociation of the (first) outermost peptide in the Aβ1–42 fibril is observed (see Results). The calculated RMSD values as a function of time are plotted in Figures S2 and S3. The calculated RMSF values per Aβ1–42 fibril Cα atom are plotted in Figures S4–S6. The β-sheet content for the outermost peptide as a function of time is plotted in Figure S7.
Determination of the Stability of the Bound Molecules in Complex with the Modeled Αβ1–42 Fibril within the Simulations
For each simulation, we assessed the stability of the molecules in complex with the modeled Αβ1–42 fibril through RMSD calculations. Prior to all RMSD calculations, the trajectories were aligned based on the backbone atoms of all peptides within the Aβ1–42 fibril. The RMSD calculations were performed based on the molecules’ heavy atoms with respect to their average structure. Within simulations in which no partial dissociation of the (first) outermost peptide of the Aβ1–42 fibril is observed, the binding of a molecule was considered stable in the simulations if the RMSD of a molecule’s heavy atoms with respect to the average position of the molecule was less than 4.5 Å. Within simulations in which partial dissociation of the outermost peptide of the Aβ1–42 fibril is observed, the binding of a molecule was considered stable in the simulations if the RMSD of a molecule’s heavy atoms with respect to the average position of the molecule was less than a relaxed cutoff of 5.5 Å. The RMSD criterion for the latter was intentionally more relaxed as the partial dissociation is expected to inherently lead to some conformational instability in the bound molecule, and in some cases, the partial readjustment of the molecule after the partial dissociation may occur. The simulations in which the bound molecules were considered stable were structurally and energetically analyzed as follows to identify and delineate key binding modes leading to (pd) or not leading to (nd) partial dissociation of the outermost peptide of the Aβ1–42 fibril.
Structural and Energetic Characterization of the Molecules’ Interactions with Aβ1–42 Residues and Binding Properties in Complex with the Modeled Αβ1–42 Fibril
We calculated the average contact propensity and the average interaction energies between the functional groups of each molecule and the residues of each peptide of the Aβ1–42 fibril within the entire simulation trajectories (nd) or the binding stage leading to partial dissociation (pd). For the analyses, each molecule was divided into nine groups of atoms or functional groups (Figure S8). In both the simulations in which partial dissociation is observed and no dissociation is observed, the molecules adopt conformations in which a portion of the molecule’s functional groups comprising the “head” or “heads” (as defined in Results) are in contact with residues 32IGL34, and the remaining portion of the molecule’s functional groups comprising the “tail” or “tails” (as defined in Results) are in contact with residue domain 12VHHQKLVFF20. Due to the symmetry of the Aβ1–42 fibril, we defined the edge outermost Aβ1–42 peptide nearest to the tail of the molecule as the first outermost peptide (henceforth also referred to as the outermost peptide), the adjacent peptide as the second outermost peptide, the third removed peptide as the third outermost peptide, the fourth removed peptide as the fourth outermost peptide, the fifth removed peptide as the fifth outermost peptide, and the peptide on the opposite end of the fibril as the sixth outermost peptide. Due to the symmetry of the molecules, we defined the R3 group nearest to the first outermost peptides as functional group 1. Through these definitions, in the simulations in which partial dissociation is observed, the first outermost peptide always corresponds to the partially dissociated peptide.
In the structural analysis, we determined the average propensity of a contact between a molecule’s functional group and an Aβ1–42 residue side chain, analogously to ref (82). A contact was considered if any atom of a molecule’s functional group and any atom of an Aβ1−42 residue side chain was within 5 Å of each other, similarly to ref (83). In the energetic analysis, we calculated the average interaction energy of polar and nonpolar interactions between a molecule’s functional group and an Aβ1–42 residue belonging to different peptides, following the numbering described above. The functional group:Aβ1–42 residue pairwise interaction energy values were decomposed into polar and nonpolar contributions, analogously to refs (36, 37, and 84−87), to identify important polar interactions (e.g., hydrogen bonds) and nonpolar interactions (π–π or van der Waals interactions). The pairwise interaction energies for each individual production run were calculated using CHARMM,69 WORDOM,76 and in-house FORTRAN programs, and are detailed in the Supporting Information. For the simulations in which no partial dissociation occurs (nd), both the structural and energetic analyses were performed in 20 ps intervals for the entire 100 ns duration for simulations; within these simulations, the molecules are firmly bound to the fibril throughout the duration of their respective simulations, and the binding modes’ stability was relatively high. For the simulations in which partial dissociation occurs (pd) (Table 1, column 3), the structural and energetic analyses of the binding of the molecules to the Aβ1–42 fibril were performed in 20 ps intervals, focusing on the simulation stages (one per simulation) prior to the initiation of partial dissociation of the outermost peptide of the Aβ1–42 fibril. The determination of how simulation stages were defined per simulation is described in the Supporting Information. This enabled us to clearly focus and analyze the specific interactions between the molecules and the fibril that could lead to partial dissociation given the fact that the molecules’ configuration and orientation in the entire trajectory can fluctuate due to partial dissociation of the (first) outermost peptide of the Aβ1–42 fibril.
We created maps entailing the probability of a contact between a molecule’s functional group and an Aβ1–42 residue side chain belonging to different peptides as well as the average polar and nonpolar energy between a molecule’s functional group and an Aβ1–42 residue for all simulations. The maps enabled the structural and energetic characterization of binding modes and subsequently the categorization of binding modes of all molecules into three principle binding modes based on common and recurring interactions between each of the molecules’ functional groups and Aβ1–42 fibril residues as well as the molecule’s orientation with respect to the Aβ1–42 fibril. In this perspective, it is worth noting that the binding modes were similar across the entire simulation trajectories in which no partial dissociation is observed and the simulation stages defined above in which partial dissociation is observed. Accordingly, the structural and energetic analyses were performed for all molecules, for the entire simulations (nd) or the simulation stages (pd). We present a portion of such maps, which are representative for simulations encompassing binding modes 1, 2, and 3 (Figures S9–S14).
Binding Energy Calculations
We calculated the binding energy of SY5, SY12, SY31, and curcumin in complex with the Aβ1–42 fibril for the entire simulation trajectories in which the molecule was stable according to the RMSD criteria described above (RMSD of a molecule’s heavy atoms with respect to the average position of the molecule was less than 4.5 Å in simulations in which no dissociation occurs or 5.5 Å in which partial dissociation occurs). The binding energies between the molecules and the modeled Αβ1–42 fibrils were calculated through two independent approaches, the Molecular Mechanics Generalized Born Surface Area (MM-GBSA) approximation88 and AutoDock Vina’s scoring function.89 All calculations were performed in 20 ps intervals to improve accuracy. The binding energy calculations were performed to identify the most energetically favorable binding modes of each molecule in complex with the Aβ1–42 fibril as well as to observe the energies as a function of time.
In the first approach, we used the one-trajectory MM-GBSA approximation,90,91 according to which we assumed that the structures of the Aβ1–42 fibril and the molecule were identical in both their bound and free states, neglecting intramolecular energy contributions due to structural relaxation, which may introduce large uncertainties if the relative affinities are neglected.90−92 Due to the use of the MM-GBSA one-trajectory approximation, the association energy values are systemically large in magnitude due to the combination of the omission of the entropic effect due to structural relaxation and the approximations of the continuum solvation model.37,90 The MM-GBSA one-trajectory approximation was preferred over more computationally demanding methods, as they are computationally efficient,93 which is important when investigating a large number of simulated complexes and has proven successful in assessing the relative affinities of different binding modes of a given molecule in a number of studies.77,86,94 Due to the use of the one-trajectory approximation, the calculated energies are referred to as association energies and were used to compare and rank the relative energetic favorability of the binding conformations per molecule in complex with the Aβ1–42 fibril rather than to compare the energetic favorability across different molecules; the above energy calculations were performed in CHARMM.69 In the second approach, we calculated the absolute binding energies of the molecules in complex with the Aβ1–42 fibril using AutoDock Vina’s knowledge-based and empirically derived scoring function.89 AutoDock Vina’s scoring function was shown to have the best scoring power among the programs evaluated in a comprehensive study95 and can be considered advantageous for fast implementation as well as the ability to calculate absolute binding energy values. We showed that there is a high correlation between the two methods (see Supporting Information), and the energy values calculated through AutoDock Vina’s scoring function are used henceforth as a metric of binding energy (Figure S15).
In addition, we used the MM-GBSA approximation, as detailed above, to calculate the association energy between the (first) outermost peptide and the rest of the peptides in the modeled fibril in simulation when partial dissociation occurs (Figure S16).
Results
Docking of the Molecules to a Hexamer Peptide Model of the Aβ1–42 Fibril
The docking procedure resulted in 25 docked poses of SY12, 25 docked poses of SY31, 23 docked poses of SY5, and 21 docked poses of curcumin (Table 1, column 2). Each of the 94 distinct docked poses of the molecules was subsequently used as a starting structure in independent MD simulations investigating each molecule in complex with the Aβ1–42 fibril.77 The number of distinct docked poses, and thus the number of simulations performed, was different for each molecule (Table 1, column 1), but overall emphasis was given to the docked poses to sufficiently span the space of probable binding, per molecule, within the region of interest (in contact with Aβ1–42 residues 12 and 17–21). All docked poses of curcumin were in contact with Aβ1–42 residues that were experimentally characterized to interact with curcumin, as defined in the Methods.71 Structures of the 94 distinct docked poses used as starting structures for subsequent MD simulations are provided as Supporting Information in PDB format.
Conformational Analysis of the Modeled Αβ1–42 Fibril within the Simulations
Of the 94 simulations of different molecules in complex with the Aβ1–42 fibril, we observed that in 20 simulations, the first outermost peptide (henceforth referred to as the outermost peptide) is partially dissociated through the disruption of all its β-sheet interactions with the second outermost within residue domain 12VHHQKLVFF20. The complete disruption of β-sheet specific interactions between the outermost peptide and the second outermost peptide within the residue domain 12VHHQKLVFF20 was not observed in the uncomplexed fibril runs, which served as an indicator that this could be an outcome of the presence of the molecules interacting with the fibril. The complete disruption of the specific interactions was defined by us as partial dissociation, and interestingly, all simulated molecules showed such capacity (Table 1, column 3). The portion of simulations in which the molecule led to partial dissociation of the outermost peptide of the Aβ1–42 fibril could also be considered as an indicator that this event is not driven by highly nonequilibrium conditions due to the presence of curcumin within the Αβ1–42 fibril.
Within the specific 20 simulations in which the outermost peptide is partially dissociated as defined above, we observed that the Aβ1–42 fibril’s conformation is affected by the bound molecule. The average Aβ1–42 fibrils’ backbone RMSD for the last 20 ns of each simulation calculated with respect to the initial structure is slightly larger (5.1 ± 0.9 Å) in the presence of partial dissociation compared to the simulations of the uncomplexed Aβ1–42 fibril or the complexed Αβ1–42 fibril in the absence of partial dissociation (3.2 ± 0.7 or 4.2 ± 1.2 Å, respectively). The larger average value (5.1 Å) can primarily be attributed to the disruption of β-sheet interactions between the outermost peptide and its adjacent peptide within residue domain 12VHHQKLVFF20, which can be indicated by analogous RMSD calculations focusing only on the specific residue domain (5.9 ± 1.7 Å compared to 2.6 ± 0.9 Å or 3.3 ± 1.2 Å, Figure S2, see Supporting Information). The relatively large RMSD values of the Aβ1–42 fibril with respect to its initial structure, even in the simulations of the uncomplexed Aβ1–42 fibril or the complexed Αβ1–42 fibril in the absence of any dissociation, are primarily attributed to the formation of an amyloid twist96 as well as some deformation of β-sheet interactions within Aβ1–42 residue domains 21AEDVGSNK28 and 39VVIA42 in the two outer peptides within the simulations, as indicated by the relatively larger RMSF values in these domains (Figures S5 and S6). Their deformation, which also occurs in the simulations where partial dissociation within 12VHHQKLVFF20 is observed (Figure S4), could be most presumably attributed to the absence of additional peptides, which is the case in an actual nearly “infinite”-peptide fibril. The larger RMSF values within the residue domain 11EVHH14 compared to other domains excluding 21AEDVGSNK28 and 39VVIA42 (Figures S5 and S6) are attributed to both the twist and the fact that, in some cases, β-sheet interactions between residues in these domains can be deformed and reformed.
We also calculated the β-sheet content of the first outermost peptide of the entire simulation and averaged across simulations in which partial dissociation is observed (Figure S7A) or not (Figure S7B), or simulations of the uncomplexed Aβ1–42 fibril (Figure S7C). The overall β-sheet content of the outermost peptide is reduced in all simulations, the uncomplexed Aβ1–42 fibril (Figure S7C) or the complexed Αβ1–42 fibril simulations in the absence (Figure S7B) or presence of any partial dissociation (Figure S7A) which is primarily an outcome of the loss of β-sheet interactions within residue domains 11EVHH14, 21AEDVGSNK28 and 39VVIA42, in all cases. Importantly, the β-sheet content of the outermost peptide is less in the simulations of a complexed Αβ1–42 fibril in the presence of partial dissociation compared to the other two cases because, in the former, β-sheet interactions are deformed at least in the entire 12VHHQKLVFF20 domain. The β-sheet content of the outermost peptide is less in the simulations of a complexed Αβ1–42 fibril in the absence of any dissociation compared to the uncomplexed simulations due to the fact that a portion of β-sheet interactions in the 12VHHQKLVFF20 could also be deformed due to any perturbations introduced by the molecules binding in the former compared to the latter. Representative structures of the Αβ1–42 fibril extracted at 100 ns from the uncomplexed Αβ1–42 fibril, complexed Αβ1–42 fibril in the absence of dissociation, and complexed Αβ1–42 fibril in which partial dissociation is initiated are shown in Figures 2A–C, respectively.
Figure 2.
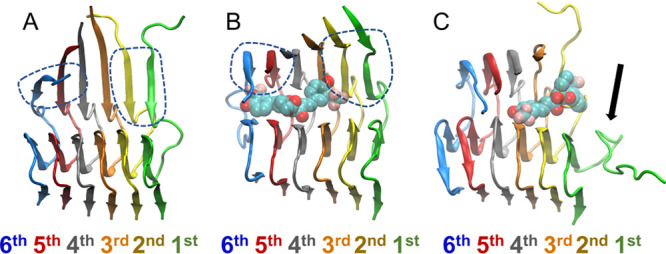
Molecular graphics images of representative modeled Aβ1–42 fibrils after 100 ns (A) in the absence of a molecule, (B) in the presence of a molecule in the absence of dissociation, and (C) in the presence of molecule in which partial dissociation of the outermost peptide of the Aβ1–42 fibril is initiated. SY12 is shown in transparent, licorice representation, and the Aβ1–42 fibril is shown in cartoon representation. (A and B) The β-sheet interactions between residues 12VHHQKLVFF20 in the first outermost peptide and the second outermost peptide of the Aβ1–42 fibril are preserved (circled with blue dotted lines) at 100 ns. (C) The β-sheet interactions between residues 12VHHQKLVFF20 of the first outermost peptide and the second outermost peptide of the Aβ1–42 fibril are completely deformed at 100 ns in the presence of a molecule. The deformation of these β-sheet interactions defines partial dissociation.
Binding Modes Leading to or Not Leading to Partial Dissociation of the Outermost Peptide
Overall, the molecules’ binding to the fibril is relatively stable in the vast majority of the simulations of the molecules in complex with the Αβ1–42 fibril (Table 1, column 3). Within these simulations, the molecules adopt conformations that can be categorized into three principle binding modes. Any additional uncategorizable modes are not analyzed in detail below as they are infrequent and mutually dissimilar. The three principle binding modes are categorized by which of three sets of the molecule’s nine functional groups (as originally determined, Figure S8) act as the head, buried within the Aβ1–42 fibril (in contact with residues 32IGL34 of the fourth, fifth, and sixth outermost peptides) and which of the three sets of functional groups act as the tail, interacting with the edge of the Aβ1–42 fibril (in contact with residue domain 12VHHQKLVFF20 in the first and second outermost peptides) (Figure 3). The three sets of functional groups are (1) one of the molecules’ aromatic groups and its two substituents, (2) the molecule’s central R1 group, and (3) the molecule’s remaining aromatic groups and its two substituents.
Figure 3.
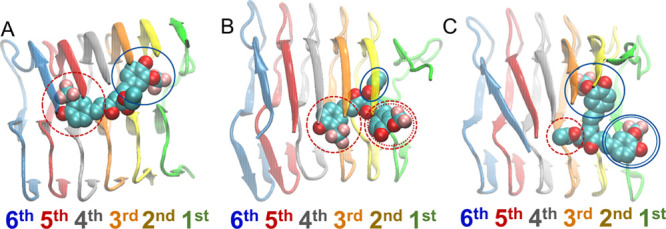
Molecular graphics image of SY12 adopting (A) binding mode 1, (B) binding mode 2, or (C) binding mode 3 in complex with the Aβ1–42 fibril. The molecule is shown in VdW representation, and the Aβ1–42 fibril is shown in cartoon representation.
The three principle binding modes are common across the different compounds and are also common across simulations in which the molecule’s binding does not lead to (nd) or leads to (pd) partial dissociation of the outermost peptide of the Aβ1–42 fibril. In the first binding mode, one aromatic functional group and its substituents act as the head while the remaining aromatic group and its substituents act as the tail (Figure 3A); in the second binding mode, both aromatic functional groups and their substituents act as heads, while the central R1 group acts as the tail (Figure 3B). In the third binding mode, the central R1 group acts as the head, and both aromatic functional groups and their substituents act as tails (Figure 3C).
Interestingly, in the case of binding modes leading to partial dissociation, the heads can also be viewed as “anchors” as they stabilize the molecule to the Aβ1–42 fibril, allowing the tails to act as “breakers” as they disrupt β-sheet interactions of the edge Aβ1–42 peptides of the fibril. The terms anchor and breaker are used according to visual inspection; the former is less mobile compared to the latter. The detailed structural and energetic analysis of interactions formed between the molecules and the different residues of different peptides of the Aβ1–42 fibril enabled us to categorize the three principle nd binding modes and three principle pd binding modes (Figure 3). Interactions between the functional groups of the molecules and the residues of the Aβ1–42 fibril were considered key interactions if their pairwise interaction energies were less than or equal to −2.0 kcal/mol. Representative pairwise interaction energy plots associated with the binding modes are provided as Supporting Information.
Binding Mode 1
In binding mode 1, irrespective of if the binding mode leads to (pd1) or does not lead to (nd1) partial dissociation of the outermost peptide of the fibril, one of a molecule’s aromatic groups and its substituents act as the head and its other aromatic group and its substituents act as the tail (Figure 3A); the central R1 group of a molecule is not involved in particular interactions with the Aβ1–42 fibril, “acting” as neither a head nor a tail. All of the investigated molecules are capable of adopting binding mode 1 (Table 1, column 4). In binding mode 1, the head interacts with the fourth through sixth outermost peptides, and the tail interacts with the first through third outermost peptides (Figures 4A and D). The molecule’s head aromatic group and its substituents stabilize the molecule through favorable nonpolar interactions with Aβ1–42 residues Ile32, Gly33, and Leu34 of the fourth or fifth outermost peptide as well as Val12 and His14 of the fifth and sixth outermost peptides, circled with red dotted lines in Figures 4A and B and indicated by dotted red lines in Figures S9B and D. The tail aromatic group and its substituents form nonpolar interactions with Ile32, Gly33, and Leu34 of the second and third outermost peptides, circled with blue lines in Figures 4A and C and indicated by blue lines in Figures S9B and D.
Figure 4.

Molecular graphics images of the lowest energy binding mode 1 for SY12 not leading to partial dissociation (A–C) and leading to partial dissociation (D–F) of the outermost peptide. SY12 is shown in thick licorice representation, and the Aβ1–42 fibril and residues are shown in cartoon and thin licorice representation, respectively. (A) Bird’s eye view of SY12 in binding mode nd1. The head comprising one of the aromatic functional group and its substituents is circled with a red dotted line. The tail comprising the remaining aromatic functional group and its substituents is circled with a blue line. (B) Key interactions between the head group and Aβ1–42 residues common for all molecules adopting binding mode nd1. (C) Key interactions between the tail group and Aβ1–42 residues common for all molecules adopting binding mode nd1. (D) Bird’s eye view of SY12 adopting binding mode pd1. The anchor is circled with a red dotted line. The breaker is circled with a blue line. (E) Interactions between the anchor group and Aβ1–42 residues stabilizing SY12 common for all molecules adopting binding mode pd1. (F) Interactions between the breaker group and Aβ1–42 residues common for all molecules adopting binding mode pd1.
In the binding modes leading to partial dissociation (pd1), the aforementioned interactions are shifted towards the partially dissociated first outermost peptide, e.g., the interactions of the sixth peptide are shifted to the fifth peptide (Figure 4D and E). In addition, for the binding modes leading to partial dissociation (pd1), the anchor hydroxyl group further stabilizes the molecule through the formation of hydrogen bonds with the backbone atoms of Gly33 or Leu34 of the fourth through sixth outermost peptides, circled with red dotted lines in Figure 4D and E, and indicated by dotted red lines in Figures S10C and D. The additional stability endowed by the anchor allows for the breaker aromatic group and its substituents to form additional nonpolar interactions with His14, Leu17, Ile32, Gly33, and Leu34 of the first and second outermost peptides as well as π–π interactions with His14 and Phe19 of the first and second outermost peptides circled with blue lines in Figure 4D and F, and indicated by blue lines in Figure S10B and D.
Binding mode 2
In binding mode 2, irrespective of if the binding mode leads (pd2) to or does not lead (nd2) to partial dissociation of the outermost peptide of the fibril, both of a molecule’s aromatic groups and their substituents act as heads while its central R1 group acts as the tail (Figure 3B). Only molecules possessing the trifluoromethoxy substituent in place of the methoxy substituent on the aromatic groups (SY5, SY12, SY31, Table 1, column 5) are capable of adopting binding mode 2. In binding mode 2, the molecule adopts a conformation such that the trifluoromethoxy substituents of the two opposing head aromatic functional groups form nonpolar interactions with each other, stabilizing the molecule’s conformation within the Αβ1–42 fibril. The heads interact with the fourth through fifth outermost peptides, and the tail interacts with the first and second outermost peptides (Figures 5A and D). One head aromatic functional group and its substituents stabilize the molecule through nonpolar interactions with Val12 and His14 of the fifth outermost peptide, while the opposing head aromatic functional group and its substituents stabilize the molecule through nonpolar interactions with the Ile32, Gly33, and Leu34 of the fourth outermost peptide, circled with red dotted lines in Figure 5B and indicated with red dotted lines in Figures S11B and D. The central R1 group of the molecule acts as the tail stabilizing the molecule through nonpolar interactions with Leu17 of the second outermost peptide, and Ile32, Gly33, and Leu34 of the first outermost peptide, circled with blue lines in Figure 5A and C and indicated in blue lines in Figures S11C and D.
Figure 5.

Molecular graphics images of the lowest energy binding mode 2 for SY12 not leading to partial dissociation (A–C) and leading to partial dissociation (D–F) of the outermost peptide. SY12 is shown in thick licorice representation, the Aβ1–42 fibril and residues are shown in cartoon and thin licorice representation, respectively. (A) Bird’s eye view of SY12 in binding mode nd2. The heads comprising both of the aromatic functional groups and their substituents are circled with a red dotted line. The tail comprising the central R1 group is circled with a blue line. (B) Key interactions between the head groups and Aβ1–42 residues common for all molecules adopting binding mode nd2. (C) Key interactions between the tail group and Aβ1–42 residues common for all molecules adopting binding mode nd2. (D) Bird’s eye view of SY12 adopting binding mode pd2. The anchors are circled with a red dotted line. The breaker is circled with a blue line. (E) Interactions between the anchor groups and Aβ1–42 residues stabilizing SY12 common for all molecules adopting binding mode pd2. (F) Interactions between the breaker group and Aβ1–42 residues common for all molecules adopting binding mode pd2.
In the binding modes leading to partial dissociation (pd2), the aforementioned interactions are shifted towards the partially dissociated first outermost peptide (Figure 5D and E). In addition, for the binding mode leading to partial dissociation (pd2), the anchor aromatic group and its substituents are further stabilized the molecule through the formation of a hydrogen bond by the hydroxyl group of one anchor aromatic group to the backbone atoms of Gly33 in the third or fourth outermost peptide, circled with red dotted lines in Figure 5D and E, and indicated in Figures S12B and D, or side chain atoms of His14 in the third outermost peptide. The central R1 group of the molecule acts as the breaker partly dissociating the first outermost peptide through additional nonpolar interactions with His14, Leu17, Phe19 of the first and second outermost peptides, circled with blue lines in Figure 5D and F and indicated by blue lines in Figures S12B and D.
Binding Mode 3
In binding mode 3 irrespective of if the binding mode leads to (pd3) or does not lead to (nd3) partial dissociation of the outermost peptide of the fibril, a molecule’s central R1 group acts as the head, and its two aromatic groups and their substituents act as tails (Figure 3C). SY12, SY31, and SY5 are the only molecules capable of adopting binding mode 3 (Table 1, column 6). In binding mode 3, the head interacts with the third through fifth outermost peptides, and the tails interact with the first through third outermost peptides. The head central R1 group stabilizes the molecule through nonpolar interactions with Val12, His14, Ile32, Gly33, and Leu34 of the third through fifth outermost peptides, circled with red dotted lines in Figures 6A and B, and indicated by red dotted lines in Figures S13B and D. One tail aromatic group and its substituents form nonpolar interactions with Phe19, Ile32, and Gly33 of the first outermost peptide, circled with blue lines in Figures 6A and C, and indicated in blue lines in Figures S13B and D. Additionally, the hydroxyl group of the aforementioned tail aromatic functional group and its substituents can also form a hydrogen bond with His14 of the third outermost peptide. The opposing tail aromatic group and its substituents form nonpolar interactions with Leu17, Ile32, and Gly33 of the first outermost peptide, circled with blue lines in Figure 6A and C and indicated in blue lines in Figures S13B and D.
Figure 6.

Molecular graphics images of the lowest energy binding mode 3 for SY12 not leading to partial dissociation (A–C) and leading to partial dissociation (D–F) of the outermost peptide. SY12 is shown in thick licorice representation, and the Aβ1–42 fibril and residues are shown in cartoon and thin licorice representation, respectively. (A) Bird’s eye view of SY12 in binding mode nd3. The head comprising the central R1 group is circled with a red dotted line. The tails comprising both of the aromatic functional groups and their substituents are circled with a blue line. (B) Key interactions between the head group and Aβ1–42 residues common for all molecules adopting binding mode nd3. (C) Key interactions between the tail groups and Aβ1–42 residues common for all molecules adopting binding mode nd3. (D) Bird’s eye view of SY12 adopting binding mode pd3. The anchor is circled with a red dotted line. The breakers are circled with a blue line. (E) Interactions between the anchor group and Aβ1–42 residues stabilizing SY12 common for all molecules adopting binding mode pd3. (F) Interactions between the breaker groups and Aβ1–42 residues common for all molecules adopting binding mode pd3.
In the binding modes leading to partial dissociation (pd3), the aforementioned interactions are shifted towards the partially dissociated first outermost peptide (Figure 6D and E). In addition, for the binding mode leading to partial dissociation (pd3), the anchor central R1 group additionally forms hydrogen bonds with the Gly33 backbone amide atom of the fifth or sixth outermost peptide, further stabilizing the molecule within the Αβ1–42 fibril, indicated by red dotted lines in Figures S14C and D. One breaker aromatic group and its substituents form additional nonpolar interactions with His14 and Leu17 of the first and second outermost peptides, circled with blue lines in Figures 6D and F, and indicated in blue lines in Figures S14B and D. Additionally, the hydroxyl group of the aforementioned breaker aromatic group and its substituents can also form a hydrogen bond with His14 of the second outermost peptide. The opposing breaker aromatic group and its substituents additionally form nonpolar interactions with Leu34 of the first through third outermost peptides, circled with blue lines in Figures 6D and F, and indicated in blue lines in Figures S14B and D. Together, the two breaker aromatic groups and their substituents disrupt β-sheet interactions within Aβ1–42 residue domains 12VHHQKLVFF20 and 32IGLMVGG38, containing the key Αβ amyloidogenic domains 16KLVFF2097 and 29GAIIG3382,98,99 simultaneously. The more disruptive nature of binding mode pd3 is indicated by the backbone RMSD over time of fibrils in complex with molecules adopting binding mode pd3 compared to the backbone RMSD over time of fibrils in complex with molecules adopting binding modes pd1 or pd2 (Figure S4).
We considered it worthwhile to investigate whether the simulations in which the molecule adopts binding mode 3, disrupting β-sheet interactions within Aβ1–42 residue domains 12VHHQKLVFF20 and 32IGLMVGG38 simultaneously, could potentially lead to complete dissociation. Thus, we additionally extended the simulations in which SY12 and SY31 partly dissociated the Αβ1–42 fibril through adopting binding mode 3 to 200 ns (Figure S4). In both the extended simulations of SY12 and SY31 adopting binding mode pd3, the β-sheet interactions between the first outermost peptide of the Αβ1–42 fibril and the second outermost peptide are nearly completely lost at the end of the 200 ns simulation such that only β-sheet interactions between 3 and 6 residues of the first and second outermost peptides remained. The 200 ns structure of SY12 in complex with the Αβ1–42 fibril is shown in Figure S17. Additional details are provided in Supporting Information.
Key Interactions Differentiating between Binding Modes Leading to or Not Leading to Partial Dissociation of the Outermost Peptide
Irrespective of the molecule or the binding mode it adopts, the molecules initiate partial dissociation of the outermost peptide within the Aβ1–42 fibrils by forming key interactions to specific Aβ1–42 fibril residues. In all three pd binding modes, the molecules lead to partial dissociation by processively disrupting the Αβ1–42 fibril starting from either the residues within the 11EVHH14 motif or the hydrophobic cluster consisting of Leu17, Phe19, and Ile32 of the first outermost peptide and the second outermost peptide (Figures 4–6, bottom panels). Additionally, in all three binding modes leading to partial dissociation, the molecule is anchored to the Αβ1–42 fibril through interactions with residues Val12, His14, Ile32, Gly33, and Leu34 belonging to peptides in the interior of the Αβ1–42 fibril (Figures 4–6, bottom panels).
A comparison across all binding modes not leading to partial dissociation and the simulation stages leading to partial dissociation showed that in all cases the molecule is positioned further away from the first outermost peptide and further into the interior of the Αβ1–42 fibril (toward the second through sixth outermost peptides) in the former compared to the latter. Irrespective of the binding mode and irrespective of the molecule, the absence or weakening of specific interactions by either of the head and tail groups appear to serve as switches preventing the molecules from initiating partial dissociation. Across all molecules and binding modes, in general, binding modes not leading to partial dissociation, the head groups of the molecules do not form key stabilizing hydrogen bond interactions with Gly33 or Leu34 of the third or fourth outermost peptides, as indicated in panel C of Figures S9-S14. Additionally, the tail groups of the molecules do not form key disrupting interactions with either (1) Ile32, Gly33, and Leu34, (2) Leu17 and Phe19, or (3) a combination of (1) and (2) of the first and second outermost peptides, as shown in Figures 4–6, panels C and F.
Binding Energy Calculations
Irrespective of the molecule bound to the Aβ1–42 fibril and irrespective of if the molecules adopt binding modes leading to partial dissociation or not within the simulations, binding mode 1 is overall the most energetically favorable binding mode (Table S1). Thus, binding mode 1 could be considered to be the most probable to naturally occur for all investigated molecules in complex with the Aβ1–42 fibril, at least according to our simulations and calculations. Interestingly, the binding energy of a molecule in binding mode 1 is lower in the case in which the molecule partly dissociates the Aβ1–42 fibril than if the molecule does not dissociate the fibril (SY5 and SY12) or becomes lower after the molecule partly dissociates the Aβ1–42 fibril (SY31), (Table S2, Figure S15). The specific binding energy-based difference between curcumin and the rest of the molecules could presumably be associated with experimental findings suggesting a possible higher propensity for SY5, SY12, and SY31 to increase the number of smaller Aβ1–42 species when incubated with Aβ1–42 aggregates compared to curcumin according to SDS-PAGE profile (Table 2 of ref (54)). Interestingly, for all molecules adopting binding mode 1, partial dissociation appears to be either an outcome of either high affinity interactions (curcumin, SY5, and SY12, Figures S15A, C, and D) or a cause leading to high affinity interactions between the molecules and the fibril (SY31, Figure S15B), which could partly serve as a compensation for the energy loss between the outermost peptide and the rest of the fibril due to partial dissociation (Figure S16). Given the high correlation and the consistency of the two different methods used in evaluating binding energy (see Supporting Information), the results above are reported based on calculations performed through AutoDock Vina, which allows for a direct comparison both across different modes and across different molecules.
Additional Simulations and Analysis of Curcumin’s Enol Form Interacting with the Modeled Aβ1–42 Fibrils
The aforementioned analyses focused on curcumin and a set of curcumin derivatives that were included in a previous experimental study. In addition to the aforementioned simulations, we performed simulations of curcumin’s enol form (Figure S18) in complex with the Aβ1–42 fibril. The enol form of curcumin was additionally investigated both as a means to validate the key interactions acting as switches leading to partial dissociation or not (as identified above) as well as to study the effect of the enol form of curcumin binding to the Aβ1–42 fibril. The enol form of curcumin differs from the investigated form of curcumin in that one of its carbonyl groups is protonated to a hydroxyl group (Figure S18). While curcumin primarily exists in its keto form in water, it can also exist in its enol form100,101, which also binds Aβ1–42 fibrils.101 The procedure used for the docking, MD simulations, and analysis of curcumin’s enol form was identical to that of the other molecules investigated in this study (described above). From the docking procedure, 24 distinct docked poses were generated; thus, 24 MD simulations of curcumin’s enol form in complex with the Aβ1–42 fibril were performed and analyzed. Within all of these simulations, the curcumin’s enol form only adopts binding mode 1, with the exception of one uncategorizable nd binding mode. Of these simulations, partial dissociation of the outermost peptide of the modeled Aβ1–42 fibril occurs in six simulation runs, serving as validation that the presence/absence of specific interactions between the molecule and the fibril (referred to above as switches) are reproducible and are necessary for partial dissociation of the outermost peptide to occur/not occur. There is only one exception in which the molecule forms all of the key interactions expected to lead to partial dissociation and no partial dissociation occurs. However, this can be due to the strict criteria used to define partial dissociation of the fibril, as defined in the Methods, as in the specific exception simulation run, all β-sheets within residue domain 12VHHQKLVFF20 of the outermost peptide are disrupted except for one single β-bridge. Interestingly, our energy calculations suggest that the enol form of curcumin is more energetically favored to bind to the Aβ1–42 fibril than the keto form of curcumin, regardless of if the molecule is adopting a binding mode leading to partial dissociation or not (Table S1). This could be related to previous experimental studies suggesting that the enol form of curcumin has a higher propensity to bind Aβ1–42 aggregates than its keto form.101
Insights into a Potential Inhibitory Mechanism by the Molecules through Partial Dissociation of the Outermost Peptide
We focused on the lowest binding energy simulation runs in which molecules adopt binding mode 1 per molecule, both for cases in which no dissociation or partial dissociation is observed, and extended these simulations to 200 ns. In addition, we extended all uncomplexed simulations, which could serve as control. Within the extended simulations of the uncomplexed Αβ1–42 fibril, the outermost peptides remain firmly bound to the rest of the fibril due to the preservation of most of the β-sheet interactions within the N- and C-terminal domains 12VHHQKLVFF20 and 32IGLMVGG38, and no dissociation (as defined above) occurs.
Within the extended simulations of the molecules adopting binding mode 1 not leading to partial dissociation of the outermost peptide (nd1), the molecules primarily remain in binding mode 1, and the β-sheet interactions of the Αβ1–42 fibril are conserved. The only exception is observed in the extended simulation of SY5 in complex with the Αβ1–42 fibril, in which the molecule actually starts to partly dissociate the outermost peptide by gradually forming interactions with Val12, His14, Ile32, Gly33, and Leu34 of the third through sixth outermost peptides with one aromatic ring and its substituents (anchor), as well as interactions with Leu17, Phe19, Ile32, Gly33, and Leu34 of the first and second outermost peptides with the opposing aromatic ring and its substituents (breaker), which have also been identified above, to be key interactions leading to partial dissociation of the outermost peptide of the Aβ1–42 fibril (Figure S19). This could serve as an additional proof of concept that specific interactions should be considered necessary for partial dissociation to be initiated. Additionally, the binding energy of the molecule in the specific simulation decreases as it shifts to form specific interactions leading to partial dissociation, providing additional evidence that the act of partial dissociation of the outermost peptide of the Aβ1–42 fibril could be driven by a molecule’s tendency toward a more energetically favorable state (Figure S20).
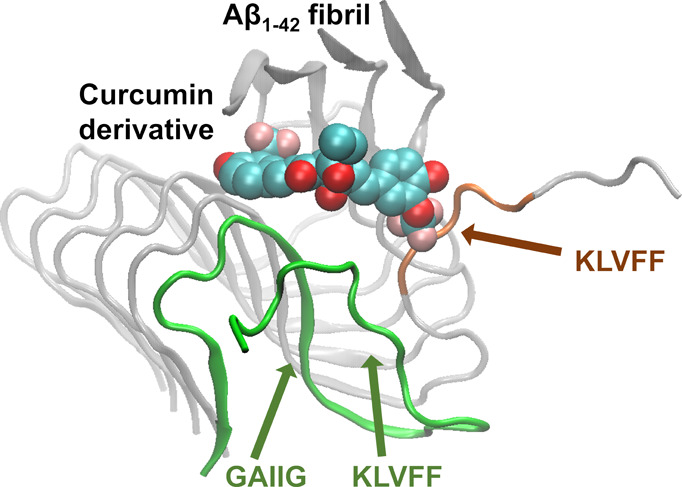
Interestingly, within all the extended simulations of the molecules adopting binding mode 1 leading to partial dissociation of the outermost peptide, the molecules act as a patch against the newly exposed β-sheet forming surface of the second outermost peptide’s 16KLVFF20 domain (Figure 7, circled in blue). In addition, within two of the extended simulations (SY12 and SY31), the partially dissociated outermost peptide’s 16KLVFF20 domain wraps around to form intramolecular interactions with the same peptide’s 29GAIIG33 domain (Figure 7, circled with red dotted lines, Movie S1). Both events, independently or collectively, could contribute to a potential inhibition mechanism, disallowing the further elongation of the fibril through the addition of an extra peptide which could be present in the actual case.
Figure 7.
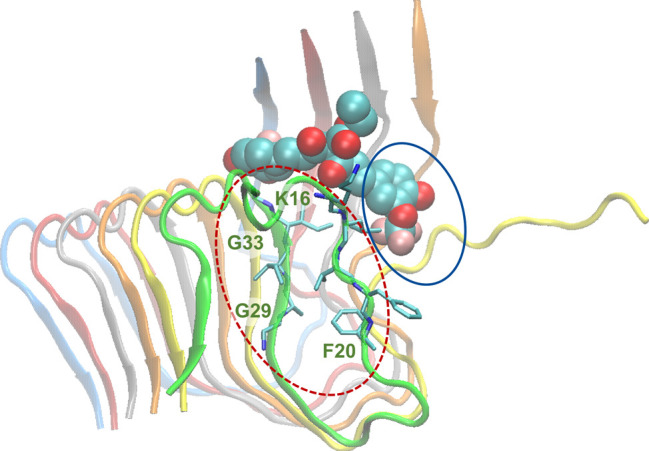
Molecular graphics image of SY12 adopting binding mode pd1 at ∼200 ns. SY12 is shown in vdW representation, and the Aβ1–42 fibril is shown in cartoon representation. The partly dissociated outermost peptide is shown in green cartoon representation. The patching of the KLVFF domain by SY12 is encircled in blue. The wrapping of the partly dissociated outermost peptide’s 16KLVFF20 domain to form intramolecular interactions with the same peptide’s 29GAIIG33 domain is encircled with red dotted lines.
Concluding Remarks
Computational methods have been used to provide insights into molecular docking,22,23,52,102,103 drug discovery,16−20,94 and amyloid formation104−130 and inhibition.27,36,37,86,82,131−149 The thermodynamics of Aβ fibril elongation and dissociation was also investigated in the absence of any molecules, providing outstanding insights into the atomistic origins of the Arrhenius barriers.150,151 Such computational methods have enhanced the understanding of Aβ fibril formation as well as curcumin’s interactions with Aβ peptides, primarily monomers.51,52,152 For example, invaluable insights were gained by Wang et al. through coarse-grained simulations, revealing the binding of curcumin to hydrophobic residues near the N- and C-terminals of Aβ17–36 aggregates, thereby inhibiting Aβ aggregation.25
Several studies have suggested that curcumin and its derivatives can serve as promising diagnostic, preventative, and potentially therapeutic AD molecules, having the capacity to inhibit the elongation of Aβ fibrils through binding Aβ monomers and fibrils as well as reducing amyloids in vivo.42,46−50 Motivated by previous experiments, we computationally investigated curcumin and a subset of experimentally studied curcumin-based molecules54 which were shown to inhibit elongation of Aβ1–42 fibrils. The nearly exhaustive docking performed in our study allowed for the molecules to be initially placed at various positions and orientations within the Aβ1–42 fibril binding site, in contact with Aβ1–42 residues experimentally shown to be key in curcumin binding.71 The use of all clustered multiple binding modes, rather than solely the highest predicted affinity docked mode, as initial structure in multi-ns MD simulations removed, to some extent, the dependence on longer duration MD simulations that would be needed for a molecule to adjust their conformations into appropriate binding modes. This allowed us to investigate the interactions between the molecules and the fibril, which could lead to or not lead to partial dissociation of the outermost peptide of the Aβ1–42 fibril, using relatively short simulations starting from different docked poses.153−156
Within the simulations, we observed that the binding properties of the molecules with Aβ1–42 fibril can vary depending on the initial docked position, and the molecules remain primarily firmly bound to the fibril throughout the trajectories. Interestingly, in specific trajectories in which the molecule is firmly bound to the fibril, the molecules could also partly dissociate the outermost peptide of the Aβ1–42 fibril, which is initiated by disruption of all β-sheets within the residue domain 12VHHQKLVFF20. The specific dissociation was not observed in trajectories of an uncomplexed Aβ1–42 fibril with the same simulation duration. Irrespective of the presence or absence of partial dissociation of the outermost peptide, the binding modes by which the investigated molecules bind the Aβ1–42 fibril can primarily be categorized into three principle binding modes based on the positions and interactions of the key sets of functional groups of the molecules. A given molecule may be able to adopt more than one binding mode, determined by its chemical composition (e.g., SY12 is capable of adopting all three principle binding modes). For all investigated molecules, binding mode 1 is overall the most energetically favorable binding mode, regardless of if the binding mode leads to partial dissociation or not. Although less energetically favorable, and thus less probable to occur than binding mode 1, molecules adopting binding mode 3 leading to partial dissociation disrupt the fibril more significantly than any other binding mode by disrupting residue domains 12VHHQKLVFF20 and 32IGLMVGG38 of the outermost peptide simultaneously. Residues within the aforementioned domains have been identified as key interacting residues for curcumin binding to Aβ1–42 fibrils71 and are key components within the Aβ1–42 amyloidogenic domains 16KLVFF2097 and 29GAIIG33.98,82 The nearly complete dissociation of the outermost peptide in binding mode 3 by SY12 could possibly contribute to the gradual reduction40 or destabilization41 of amyloids, as suggested in previous studies for curcumin; however, this should be investigated further and in more depth, and can be of a focus in future computational and experimental studies.
A comparison of the binding of the investigated molecules within high affinity binding modes not leading to partial dissociation to those leading to partial dissociation of the outermost peptide reveal key identified interactions differentiating the two, irrespective of which binding mode the molecules adopt. Also, the binding energies are comparable among the binding modes leading to or not leading to partial dissociation, although binding modes leading to partial dissociation could be considered either as more energetically favorable or the precursor stages leading to a more energetically favorable state. It is also possible that the energy loss between the outermost peptide and the rest of the fibril could be partly compensated by the low binding energies between a molecule and the fibril during or after partial dissociation. Nevertheless, given the fact that the energetic differences in binding modes leading or not to partial dissociation are usually within an error of a standard deviation, and that these modes share overall significant structural similarities, it is quite possible that binding modes not leading to partial dissociation could potentially, given sufficient additional time, eventually adopt slightly different binding modes leading to it, as observed in the extended simulation of SY5 binding to the fibril, or vice versa.
Our study suggests a potential inhibition mechanism of Αβ1–42 aggregation by the molecules, where the partially dissociated 16KLVFF20 domain of the outermost peptide could either remain unstructured or wrap around to form intramolecular interactions with the same peptide’s 29GAIIG33 domain, while the molecules could additionally act as a patch against the external edge of the second outermost peptide’s 16KLVFF20 domain; thereby, individually or concurrently, these could prohibit fibril elongation. This is in accordance with experiments showing that curcumin inhibits the elongation of Aβ1–40 fibrils consistent with a first order kinetic model, which was hypothesized to be due to curcumin destabilizing the conformation of Aβ1–40 peptides at the fibril ends.41 The proposed possible mechanism of Aβ1–42 fibril elongation inhibition through patching is reminiscent of the mechanism by which GAIPIG peptide inhibitors, investigated by us in a previous study,82 were suggested to block Aβ fibril elongation through binding and blocking the 16KLVFF2097 and 29GAIIG33 domains.82 Additionally, the disruption of β-sheets within the residue domain 12VHHQKLVFF20 by the investigated molecules, which is common across all binding modes leading to partial dissociation, could be analogous to how the green tea compound epigallocatechin-3-gallate (EGCG) reduces the toxicity of Aβ1–40 oligomers.157 EGCG’s binding to Aβ1–40 oligomers causes the N-terminus of Aβ1–40 (residues 1–20) to be unstructured, whereas the C-terminal portion (residues 22–39) adopts β-sheet conformations, which is proposed to reduce the oligomer’s toxicity.157 Thus, although our studies investigate the interactions of curcumin and its derivatives to a hexamer peptide model of an Aβ1–42 fibril, curcumin and its derivatives could also potentially reduce the toxicity of Aβ1–42 oligomers through a mechanism similar to that of EGCG, which was believed to immobilize residues 1–20 in the peptide primary structure of the EGCG-induced oligomer.157
The current study is focused on interactions and possible events related to curcumin and a set of curcumin derivatives binding to a model Aβ1–42 fibril. Our study provides insights into the interactions of curcumin and a set of curcumin derivatives with a hexamer model of the Aβ1–42 fibril leading to partial dissociation of the fibril. Additional future in-depth studies are needed to clarify the link between inhibition and potential dissociation effects induced by the studied molecules adopting the principle binding modes using larger scale simulations with longer durations and a larger and wider set of polymorphic fibrils. In addition, regarding the role of metal ions, according to experimental studies, curcumin inhibits the seeding of fibrillation by preventing the peptide–metal complex formation with Cu(II) and Zn(II).40 Additional and longer duration simulations could potentially uncover how certain molecules could have an effect on presumably altering the path of dissociation of an Aβ1–42 peptide from the fibril, which is known to have high Arrhenius barriers.150,151 Our findings can also provide an impetus for the investigation of molecules binding to and inhibiting the formation of other preformed amyloid fibrils (e.g., tau43) as well as the discovery and design of novel highly potent molecules binding to and dissociating Aβ1–42 fibrils as potential diagnostic, preventive, or potentially therapeutic AD molecules.
Acknowledgments
The authors dedicate this work to the memory of Dr. Dimitrios Morikis who, through his devoted mentoring, passed down his love and passion for biophysics and bioengineering to the next generation of scientists and engineers. The authors thank Dr. Wolfgang Hoyer and Dr. Georgios Archontis for useful discussions. This work was supported in part by the Texas A&M University Graduate Diversity Fellowship from the TAMU Office of Graduate and Professional Studies (A.A.O.) and startup funding by the Artie McFerrin Department of Chemical Engineering at Texas A&M University (P.T.). All MD simulations and energy calculations were conducted using the Ada and Curie supercomputing cluster and additional computational resources available to P.T. at the Texas A&M High Performance Research Computing Facility and the Artie McFerrin Department of Chemical Engineering.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jcim.9b00561.
Author Contributions
† J.M.J. and A.A.O. are equally contributing first authors.
The authors declare no competing financial interest.
Supplementary Material
References
- Wang S.; Mims P. N.; Roman R. J.; Fan F. Is Beta-Amyloid Accumulation a Cause or Consequence of Alzheimer’s Disease?. Journal of Alzheimer’s parkinsonism & dementia 2016, 1 (2), 007. [PMC free article] [PubMed] [Google Scholar]
- Hardy J.; Selkoe D. J. The Amyloid Hypothesis of Alzheimer’s Disease: Progress and Problems on the Road to Therapeutics. Science 2002, 297 (5580), 353. 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- Hardy J. A.; Higgins G. A. Alzheimer’s disease: the amyloid cascade hypothesis. Science 1992, 256 (5054), 184. 10.1126/science.1566067. [DOI] [PubMed] [Google Scholar]
- Sevigny J.; Chiao P.; Bussière T.; Weinreb P. H.; Williams L.; Maier M.; Dunstan R.; Salloway S.; Chen T.; Ling Y.; O’Gorman J.; Qian F.; Arastu M.; Li M.; Chollate S.; Brennan M. S.; Quintero-Monzon O.; Scannevin R. H.; Arnold H. M.; Engber T.; Rhodes K.; Ferrero J.; Hang Y.; Mikulskis A.; Grimm J.; Hock C.; Nitsch R. M.; Sandrock A. The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature 2016, 537, 50. 10.1038/nature19323. [DOI] [PubMed] [Google Scholar]
- Doig A. J.; del Castillo-Frias M. P.; Berthoumieu O.; Tarus B.; Nasica-Labouze J.; Sterpone F.; Nguyen P. H.; Hooper N. M.; Faller P.; Derreumaux P. Why Is Research on Amyloid-β Failing to Give New Drugs for Alzheimer’s Disease?. ACS Chem. Neurosci. 2017, 8 (7), 1435–1437. 10.1021/acschemneuro.7b00188. [DOI] [PubMed] [Google Scholar]
- Attar A.; Meral D.; Urbanc B.; Bitan G.. Chapter 38 - Assembly of Amyloid β-Protein Variants Containing Familial Alzheimer’s Disease-Linked Amino Acid Substitutions. In Bio-nanoimaging; Uversky V. N., Lyubchenko Y. L., Eds.; Academic Press: Boston, 2014; pp 429–442. [Google Scholar]
- Jonsson T.; Atwal J. K.; Steinberg S.; Snaedal J.; Jonsson P. V.; Bjornsson S.; Stefansson H.; Sulem P.; Gudbjartsson D.; Maloney J.; Hoyte K.; Gustafson A.; Liu Y.; Lu Y.; Bhangale T.; Graham R. R.; Huttenlocher J.; Bjornsdottir G.; Andreassen O. A.; Jönsson E. G.; Palotie A.; Behrens T. W.; Magnusson O. T.; Kong A.; Thorsteinsdottir U.; Watts R. J.; Stefansson K. A mutation in APP protects against Alzheimer’s disease and age-related cognitive decline. Nature 2012, 488, 96. 10.1038/nature11283. [DOI] [PubMed] [Google Scholar]
- Rahimi F.; Li H.; Sinha S.; Bitan G.. Chapter 6 - Modulators of Amyloid β-Protein (Aβ) Self-Assembly. In Developing Therapeutics for Alzheimer’s Disease; Wolfe M. S., Ed.; Academic Press: Boston, 2016; pp 97–191. [Google Scholar]
- Li H.; Rahimi F.; Bitan G. Modulation of Amyloid β-Protein (Aβ) Assembly by Homologous C-Terminal Fragments as a Strategy for Inhibiting Aβ Toxicity. ACS Chem. Neurosci. 2016, 7 (7), 845–856. 10.1021/acschemneuro.6b00154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T.; Bitan G. Modulating Self-Assembly of Amyloidogenic Proteins as a Therapeutic Approach for Neurodegenerative Diseases: Strategies and Mechanisms. ChemMedChem 2012, 7 (3), 359–374. 10.1002/cmdc.201100585. [DOI] [PubMed] [Google Scholar]
- Sharma A. K.; Pavlova S. T.; Kim J.; Finkelstein D.; Hawco N. J.; Rath N. P.; Kim J.; Mirica L. M. Bifunctional compounds for controlling metal-mediated aggregation of the aβ42 peptide. J. Am. Chem. Soc. 2012, 134 (15), 6625–6636. 10.1021/ja210588m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martins I. C.; Kuperstein I.; Wilkinson H.; Maes E.; Vanbrabant M.; Jonckheere W.; Van Gelder P.; Hartmann D.; D’Hooge R.; De Strooper B.; Schymkowitz J.; Rousseau F. Lipids revert inert Abeta amyloid fibrils to neurotoxic protofibrils that affect learning in mice. EMBO J. 2008, 27 (1), 224–233. 10.1038/sj.emboj.7601953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Härd T.; Lendel C. Inhibition of Amyloid Formation. J. Mol. Biol. 2012, 421 (4), 441–465. 10.1016/j.jmb.2011.12.062. [DOI] [PubMed] [Google Scholar]
- Malishev R.; Shaham-Niv S.; Nandi S.; Kolusheva S.; Gazit E.; Jelinek R. Bacoside-A, an Indian Traditional-Medicine Substance, Inhibits β-Amyloid Cytotoxicity, Fibrillation, and Membrane Interactions. ACS Chem. Neurosci. 2017, 8 (4), 884–891. 10.1021/acschemneuro.6b00438. [DOI] [PubMed] [Google Scholar]
- Frydman-Marom A.; Levin A.; Farfara D.; Benromano T.; Scherzer-Attali R.; Peled S.; Vassar R.; Segal D.; Gazit E.; Frenkel D.; Ovadia M. Orally administrated cinnamon extract reduces β-amyloid oligomerization and corrects cognitive impairment in Alzheimer’s disease animal models. PLoS One 2011, 6 (1), e16564–e16564. 10.1371/journal.pone.0016564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H.; Zhang C.; Dong X. Y.; Zheng J.; Sun Y. Design of nonapeptide LVFFARKHH: A bifunctional agent against Cu2+ -mediated amyloid β-protein aggregation and cytotoxicity. J. Mol. Recognit. 2018, 31 (6), e2697 10.1002/jmr.2697. [DOI] [PubMed] [Google Scholar]
- Xiong N.; Zhao Y.; Dong X.; Zheng J.; Sun Y. Design of a Molecular Hybrid of Dual Peptide Inhibitors Coupled on AuNPs for Enhanced Inhibition of Amyloid β-Protein Aggregation and Cytotoxicity. Small 2017, 13 (13), 1601666. 10.1002/smll.201601666. [DOI] [PubMed] [Google Scholar]
- Ren B.; Jiang B.; Hu R.; Zhang M.; Chen H.; Ma J.; Sun Y.; Jia L.; Zheng J. HP-β-cyclodextrin as an inhibitor of amyloid-β aggregation and toxicity. Phys. Chem. Chem. Phys. 2016, 18 (30), 20476–20485. 10.1039/C6CP03582E. [DOI] [PubMed] [Google Scholar]
- Xiong N.; Dong X. Y.; Zheng J.; Liu F. F.; Sun Y. Design of LVFFARK and LVFFARK-functionalized nanoparticles for inhibiting amyloid β-protein fibrillation and cytotoxicity. ACS Appl. Mater. Interfaces 2015, 7 (10), 5650–5662. 10.1021/acsami.5b00915. [DOI] [PubMed] [Google Scholar]
- Wang Q.; Liang G.; Zhang M.; Zhao J.; Patel K.; Yu X.; Zhao C.; Ding B.; Zhang G.; Zhou F.; Zheng J. De novo design of self-assembled hexapeptides as β-amyloid (Aβ) peptide inhibitors. ACS Chem. Neurosci. 2014, 5 (10), 972–981. 10.1021/cn500165s. [DOI] [PubMed] [Google Scholar]
- Ren B.; Zhang M.; Hu R.; Chen H.; Wang M.; Lin Y.; Sun Y.; Jia L.; Liang G.; Zheng J. Identification of a New Function of Cardiovascular Disease Drug 3-Morpholinosydnonimine Hydrochloride as an Amyloid-β Aggregation Inhibitor. ACS Omega 2017, 2 (1), 243–250. 10.1021/acsomega.6b00397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q.; Yu X.; Patal K.; Hu R.; Chuang S.; Zhang G.; Zheng J. Tanshinones inhibit amyloid aggregation by amyloid-β peptide, disaggregate amyloid fibrils, and protect cultured cells. ACS Chem. Neurosci. 2013, 4 (6), 1004–1015. 10.1021/cn400051e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kai T.; Zhang L.; Wang X.; Jing A.; Zhao B.; Yu X.; Zheng J.; Zhou F. Tabersonine inhibits amyloid fibril formation and cytotoxicity of Aβ(1–42). ACS Chem. Neurosci. 2015, 6 (6), 879–888. 10.1021/acschemneuro.5b00015. [DOI] [PubMed] [Google Scholar]
- Saini R. K.; Shuaib S.; Goyal B. Molecular insights into Aβ42 protofibril destabilization with a fluorinated compound D744: A molecular dynamics simulation study. J. Mol. Recognit. 2017, 30 (12), e2656. 10.1002/jmr.2656. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Latshaw D. C.; Hall C. K. Aggregation of Aβ(17–36) in the Presence of Naturally Occurring Phenolic Inhibitors Using Coarse-Grained Simulations. J. Mol. Biol. 2017, 429 (24), 3893–3908. 10.1016/j.jmb.2017.10.006. [DOI] [PubMed] [Google Scholar]
- Doig A. J.; Derreumaux P. Inhibition of protein aggregation and amyloid formation by small molecules. Curr. Opin. Struct. Biol. 2015, 30, 50–56. 10.1016/j.sbi.2014.12.004. [DOI] [PubMed] [Google Scholar]
- de Almeida N. E. C.; Do T. D.; LaPointe N. E.; Tro M.; Feinstein S. C.; Shea J.-E.; Bowers M. T. 1,2,3,4,6-penta-O-galloyl-β-d-glucopyranose binds to the N-terminal metal binding region to inhibit amyloid β-protein oligomer and fibril formation. Int. J. Mass Spectrom. 2017, 420, 24–34. 10.1016/j.ijms.2016.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F.; Du W.; Sun Y.; Zheng J.; Dong X. Atomistic characterization of binding modes and affinity of peptide inhibitors to amyloid-β protein. Front. Chem. Sci. Eng. 2014, 8 (4), 433–444. 10.1007/s11705-014-1454-6. [DOI] [Google Scholar]
- Hoyer W.; Härd T. Interaction of Alzheimer’s A beta peptide with an engineered binding protein--thermodynamics and kinetics of coupled folding-binding. J. Mol. Biol. 2008, 378, 398–411. 10.1016/j.jmb.2008.02.040. [DOI] [PubMed] [Google Scholar]
- Hoyer W.; Grönwall C.; Jonsson A.; Ståhl S.; Härd T. Stabilization of a beta-hairpin in monomeric Alzheimer’s amyloid-beta peptide inhibits amyloid formation. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 5099–5104. 10.1073/pnas.0711731105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirecka E. A.; Shaykhalishahi H.; Gauhar A.; Akgül Ş.; Lecher J.; Willbold D.; Stoldt M.; Hoyer W. Sequestration of a β-hairpin for control of α-synuclein aggregation. Angew. Chem., Int. Ed. 2014, 53, 4227–4230. 10.1002/anie.201309001. [DOI] [PubMed] [Google Scholar]
- Shaykhalishahi H.; Mirecka E. A.; Gauhar A.; Grüning C. S.; Willbold D.; Härd T.; Stoldt M.; Hoyer W. A β-hairpin-binding protein for three different disease-related amyloidogenic proteins. ChemBioChem 2015, 16, 411–414. 10.1002/cbic.201402552. [DOI] [PubMed] [Google Scholar]
- Mirecka E. A.; Gremer L.; Schiefer S.; Oesterhelt F.; Stoldt M.; Willbold D.; Hoyer W. Engineered aggregation inhibitor fusion for production of highly amyloidogenic human islet amyloid polypeptide. J. Biotechnol. 2014, 191, 221–7. 10.1016/j.jbiotec.2014.06.006. [DOI] [PubMed] [Google Scholar]
- Lindberg H.; Härd T.; Löfblom J.; Ståhl S. A truncated and dimeric format of an Affibody library on bacteria enables FACS-mediated isolation of amyloid-beta aggregation inhibitors with subnanomolar affinity. Biotechnol. J. 2015, 10, 1707–1718. 10.1002/biot.201500131. [DOI] [PubMed] [Google Scholar]
- Grüning C. S. R.; Mirecka E. A.; Klein A. N.; Mandelkow E.; Willbold D.; Marino S. F.; Stoldt M.; Hoyer W. Alternative Conformations of the Tau Repeat Domain in Complex with an Engineered Binding Protein. J. Biol. Chem. 2014, 289 (33), 23209–23218. 10.1074/jbc.M114.560920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr A. A.; Wördehoff M. M.; Hoyer W.; Tamamis P. Uncovering the Binding and Specificity of β-Wrapins for Amyloid-β and α-Synuclein. J. Phys. Chem. B 2016, 120 (50), 12781–12794. 10.1021/acs.jpcb.6b08485. [DOI] [PubMed] [Google Scholar]
- Orr A. A.; Shaykhalishahi H.; Mirecka E. A.; Jonnalagadda S. V. R.; Hoyer W.; Tamamis P. Elucidating the multi-targeted anti-amyloid activity and enhanced islet amyloid polypeptide binding of beta-wrapins. Comput. Chem. Eng. 2018, 116, 322–332. 10.1016/j.compchemeng.2018.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H. Y.; Kim H. V.; Jo S.; Lee C. J.; Choi S. Y.; Kim D. J.; Kim Y. EPPS rescues hippocampus-dependent cognitive deficits in APP/PS1 mice by disaggregation of amyloid-β oligomers and plaques. Nat. Commun. 2015, 6, 8997. 10.1038/ncomms9997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee W.; Lee S. W.; Lee G.; Yoon D. S. Atomic Force Microscopy Analysis of EPPS-Driven Degradation and Reformation of Amyloid-β Aggregates. ADR 2018, 2 (1), 41–49. 10.3233/ADR-170024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee R. Effect of Curcumin on the metal ion induced fibrillization of amyloid-β peptide. Spectrochim. Acta, Part A 2014, 117, 798–800. 10.1016/j.saa.2013.09.064. [DOI] [PubMed] [Google Scholar]
- Ono K.; Hasegawa K.; Naiki H.; Yamada M. Curcumin Has Potent Anti-Amyloidogenic Effects for Alzheimer’s β-Amyloid Fibrils In Vitro. J. Neurosci. Res. 2004, 75, 742–750. 10.1002/jnr.20025. [DOI] [PubMed] [Google Scholar]
- Yang F. S.; Lim G. P.; Begum A. N.; Ubeda O. J.; Simmons M. R.; Ambegaokar S. S.; Chen P. P.; Kayed R.; Glabe C. G.; Frautschy S. A.; Cole G. M. Curcumin Inhibits Formation of Amyloid beta Oligomers and Fibrils, Binds Plaques, and Reduces Amyloid in vivo. J. Biol. Chem. 2005, 280, 5892–5901. 10.1074/jbc.M404751200. [DOI] [PubMed] [Google Scholar]
- Rane J. S.; Bhaumik P.; Panda D. Curcumin Inhibits Tau Aggregation and Disintegrates Preformed Tau Filaments in vitro. J. Alzheimer's Dis. 2017, 60 (3), 999–1014. 10.3233/JAD-170351. [DOI] [PubMed] [Google Scholar]
- Yanagisawa D.; Ibrahim N. F.; Taguchi H.; Morikawa S.; Hirao K.; Shirai N.; Sogabe T.; Tooyama I. Curcumin derivative with the substitution at C-4 position, but not curcumin, is effective against amyloid pathology in APP/PS1 mice. Neurobiol. Aging 2015, 36 (1), 201–210. 10.1016/j.neurobiolaging.2014.07.041. [DOI] [PubMed] [Google Scholar]
- Morales I.; Cerda-Troncoso C.; Andrade V.; Maccioni R. B. The Natural Product Curcumin as a Potential Coadjuvant in Alzheimer’s Treatment. J. Alzheimer's Dis. 2017, 60 (2), 451–460. 10.3233/JAD-170354. [DOI] [PubMed] [Google Scholar]
- Chen M.; Du Z. Y.; Zheng X.; Li D. L.; Zhou R. P.; Zhang K. Use of curcumin in diagnosis, prevention, and treatment of Alzheimer’s disease. Neural Regener. Res. 2018, 13 (4), 742–752. 10.4103/1673-5374.230303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy P. H.; Manczak M.; Yin X.; Grady M. C.; Mitchell A.; Kandimalla R.; Kuruva C. S. Protective effects of a natural product, curcumin, against amyloid β induced mitochondrial and synaptic toxicities in Alzheimer’s disease. J. Invest. Med. 2016, 64 (8), 1220–1234. 10.1136/jim-2016-000240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandra B.; Mithu V. S.; Bhowmik D.; Das A. K.; Sahoo B.; Maiti S.; Madhu P. K. Curcumin Dictates Divergent Fates for the Central Salt Bridges in Amyloid-β(40) and Amyloid-β(42). Biophys. J. 2017, 112 (8), 1597–1608. 10.1016/j.bpj.2017.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S. Y.; Kim H. S.; Cho E. K.; Kwon B. Y.; Phark S.; Hwang K. W.; Sul D. Curcumin Protected PC12 Cells against Beta-Amyloid-Induced Toxicity through the Inhibition of Oxidative Damage and Tau Hyperphosphorylation. Food Chem. Toxicol. 2008, 46, 2881–2887. 10.1016/j.fct.2008.05.030. [DOI] [PubMed] [Google Scholar]
- Frautschy S. A.; Hu W.; Kim P.; Miller S. A.; Chu T.; Harris-White M. E.; Cole G. M. Phenolic Anti-inflammatory Antioxidant Reversal of A beta-induced Cognitive Deficits and Neuropathology. Neurobiol. Aging 2001, 22, 993–1005. 10.1016/S0197-4580(01)00300-1. [DOI] [PubMed] [Google Scholar]
- Ngo S. T.; Li M. S. Curcumin Binds to Abeta1–40 Peptides and Fibrils Stronger than Ibuprofen and Naproxen. J. Phys. Chem. B 2012, 116, 10165–10175. 10.1021/jp302506a. [DOI] [PubMed] [Google Scholar]
- Rao P. P.; Mohamed T.; Teckwani K.; Tin G. Curcumin Binding to Beta Amyloid: A Computational Study. Chem. Biol. Drug Des. 2015, 86 (4), 813–820. 10.1111/cbdd.12552. [DOI] [PubMed] [Google Scholar]
- Ngo S. T.; Fang S.-T.; Huang S.-H.; Chou C.-L.; Huy P. D. Q.; Li M. S.; Chen Y.-C. Anti-arrhythmic Medication Propafenone a Potential Drug for Alzheimer’s Disease Inhibiting Aggregation of Aβ: In Silico and in Vitro Studies. J. Chem. Inf. Model. 2016, 56 (7), 1344–1356. 10.1021/acs.jcim.6b00029. [DOI] [PubMed] [Google Scholar]
- Yanagisawa D.; Taguchi H.; Morikawa S.; Kato T.; Hirao K.; Shirai N.; Tooyama I. Novel curcumin derivatives as potent inhibitors of amyloid β aggregation. Biochemistry and biophysics reports 2015, 4, 357–368. 10.1016/j.bbrep.2015.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orlando R. A.; Gonzales A. M.; Royer R. E.; Deck L. M.; Vander Jagt D. L. A chemical analog of curcumin as an improved inhibitor of amyloid Abeta oligomerization. PLoS One 2012, 7 (3), e31869 10.1371/journal.pone.0031869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L.; Fiala M.; Cashman J.; Sayre J.; Espinosa A.; Mahanian M.; Zaghi J.; Badmaev V.; Graves M. C.; Bernard G.; Rosenthal M. Curcuminoids Enhance Amyloid-beta Uptake by Macrophages of Alzheimer’s Disease Patients. J. Alzheimer's Dis. 2006, 10, 1–7. 10.3233/JAD-2006-10101. [DOI] [PubMed] [Google Scholar]
- Lakey-Beitia J.; González Y.; Doens D.; Stephens D. E.; Santamaría R.; Murillo E.; Gutiérrez M.; Fernández P. L.; Rao K. S.; Larionov O. V.; Durant-Archibold A. A. Assessment of Novel Curcumin Derivatives as Potent Inhibitors of Inflammation and Amyloid-β Aggregation in Alzheimer’s Disease. J. Alzheimer's Dis. 2017, 60, S59–S68. 10.3233/JAD-170071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orteca G.; Tavanti F.; Bednarikova Z.; Gazova Z.; Rigillo G.; Imbriano C.; Basile V.; Asti M.; Rigamonti L.; Saladini M.; Ferrari E.; Menziani M. C. Curcumin derivatives and Aβ-fibrillar aggregates: An interactions’ study for diagnostic/therapeutic purposes in neurodegenerative diseases. Bioorg. Med. Chem. 2018, 26, 4288–4300. 10.1016/j.bmc.2018.07.027. [DOI] [PubMed] [Google Scholar]
- Ringman J. M.; Frautschy S. A.; Teng E.; Begum A. N.; Bardens J.; Beigi M.; Gylys K. H.; Badmaev V.; Heath D. D.; Apostolova L. G.; Porter V.; Vanek Z.; Marshall G. A.; Hellemann G.; Sugar C.; Masterman D. L.; Montine T. J.; Cummings J. L.; Cole G. M. Oral Curcumin for Alzheimer’s Disease: Tolerability and Efficacy in a 24-week Randomized, Double Blind, Placebo-Controlled Study. Alzheimer's Res. Ther. 2012, 4, 43. 10.1186/alzrt146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baum L.; Lam C. W. K.; Cheung S. K. K.; Kwok T.; Lui V.; Tsoh J.; Lam L.; Leung V.; Hui E.; Ng C.; Woo J.; Chiu H. F. K.; Goggins W. B.; Zee B. C. Y.; Cheng K. F.; Fong C. Y. S.; Wong A.; Mok H.; Chow M. S. S.; Ho P. C.; Ip S. P.; Ho C. S.; Yu X. W.; Lai C. Y. L.; Chan M. H.; Szeto S.; Chan I. H. S.; Mok V. Six-month Randomized, Placebo-controlled, Double-blind, Pilot Clinical Trial of Curcumin in Patients with Alzheimer’s Disease. J. Clin. Psychopharmacol. 2008, 28, 110–113. 10.1097/jcp.0b013e318160862c. [DOI] [PubMed] [Google Scholar]
- Salehi B.; Stojanović-Radić Z.; Matejić J.; Sharifi-Rad M.; Anil Kumar N. V.; Martins N.; Sharifi-Rad J. The therapeutic potential of curcumin: A review of clinical trials. Eur. J. Med. Chem. 2019, 163, 527–545. 10.1016/j.ejmech.2018.12.016. [DOI] [PubMed] [Google Scholar]
- Cox K. H.; Pipingas A.; Scholey A. B. Investigation of the effects of solid lipid curcumin on cognition and mood in a healthy older population. J. Psychopharmacol. 2015, 29 (5), 642–651. 10.1177/0269881114552744. [DOI] [PubMed] [Google Scholar]
- Small G. W.; Siddarth P.; Li Z.; Miller K. J.; Ercoli L.; Emerson N. D.; Martinez J.; Wong K. P.; Liu J.; Merrill D. A.; Chen S. T.; Henning S. M.; Satyamurthy N.; Huang S. C.; Heber D.; Barrio J. R. Memory and Brain Amyloid and Tau Effects of a Bioavailable Form of Curcumin in Non-Demented Adults: A Double-Blind, Placebo-Controlled 18-Month Trial. Am. J. Geriatr Psychiatry 2018, 26 (3), 266–277. 10.1016/j.jagp.2017.10.010. [DOI] [PubMed] [Google Scholar]
- Schelke M. W.; Attia P.; Palenchar D. J.; Kaplan B.; Mureb M.; Ganzer C. A.; Scheyer O.; Rahman A.; Kachko R.; Krikorian R.; Mosconi L.; Isaacson R. S. Mechanisms of Risk Reduction in the Clinical Practice of Alzheimer’s Disease Prevention. Front. Aging Neurosci. 2018, 10, 96–96. 10.3389/fnagi.2018.00096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla P. K.; Khanna V. K.; Khan M. Y.; Srimal R. C. Protective effect of curcumin against lead neurotoxicity in rat. Hum. Exp. Toxicol. 2003, 22, 653–658. 10.1191/0960327103ht411oa. [DOI] [PubMed] [Google Scholar]
- Nery-Flores S. D.; Mendoza-Magaña M. L.; Ramírez-Herrera M. A.; Ramírez-Vázquez J. J.; Romero-Prado M. M. J.; Cortez-Álvarez C. R.; Ramírez-Mendoza A. A. Curcumin Exerted Neuroprotection against Ozone-Induced Oxidative Damage and Decreased NF-κB Activation in Rat Hippocampus and Serum Levels of Inflammatory Cytokines. Oxid. Med. Cell. Longevity 2018, 2018, 9620684. 10.1155/2018/9620684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozmon S.; Tvaroška I. Molecular dynamic studies of amyloid-beta interactions with curcumin and Cu2+ ions. Chemical Papers 2015, 69 (9), 1262–1276. 10.1515/chempap-2015-0134. [DOI] [Google Scholar]
- Grosdidier A.; Zoete V.; Michielin O. SwissDock, a protein-small molecule docking web service based on EADock DSS. Nucleic Acids Res. 2011, 39, W270–W277. 10.1093/nar/gkr366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks B. R.; Brooks C. L. 3rd; Mackerell A. D. Jr.; Nilsson L.; Petrella R. J.; Roux B.; Won Y.; Archontis G.; Bartels C.; Boresch S.; Caflisch A.; Caves L.; Cui Q.; Dinner A. R.; Feig M.; Fischer S.; Gao J.; Hodoscek M.; Im W.; Kuczera K.; Lazaridis T.; Ma J.; Ovchinnikov V.; Paci E.; Pastor R. W.; Post C. B.; Pu J. Z.; Schaefer M.; Tidor B.; Venable R. M.; Woodcock H. L.; Wu X.; Yang W.; York D. M.; Karplus M. CHARMM: the biomolecular simulation program. J. Comput. Chem. 2009, 30 (10), 1545–614. 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen E. F.; Goddard T. D.; Huang C. C.; Couch G. S.; Greenblatt D. M.; Meng E. C.; Ferrin T. E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25 (13), 1605–1612. 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Masuda Y.; Fukuchi M.; Yatagawa T.; Tada M.; Takeda K.; Irie K.; Akagi K.-i.; Monobe Y.; Imazawa T.; Takegoshi K. Solid-state NMR analysis of interaction sites of curcumin and 42-residue amyloid β-protein fibrils. Bioorg. Med. Chem. 2011, 19 (20), 5967–5974. 10.1016/j.bmc.2011.08.052. [DOI] [PubMed] [Google Scholar]
- Xiao Y.; Ma B.; McElheny D.; Parthasarathy S.; Long F.; Hoshi M.; Nussinov R.; Ishii Y. Aβ(1–42) fibril structure illuminates self-recognition and replication of amyloid in Alzheimer’s disease. Nat. Struct. Mol. Biol. 2015, 22 (6), 499–505. 10.1038/nsmb.2991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gremer L.; Schölzel D.; Schenk C.; Reinartz E.; Labahn J.; Ravelli R. B. G.; Tusche M.; Lopez-Iglesias C.; Hoyer W.; Heise H.; Willbold D.; Schröder G. F. Fibril structure of amyloid-β(1–42) by cryo-electron microscopy. Science (Washington, DC, U. S.) 2017, 358 (6359), 116–119. 10.1126/science.aao2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wälti M. A.; Ravotti F.; Arai H.; Glabe C. G.; Wall J. S.; Böckmann A.; Güntert P.; Meier B. H.; Riek R. Atomic-resolution structure of a disease-relevant Aβ(1–42) amyloid fibril. Proc. Natl. Acad. Sci. U. S. A. 2016, 113 (34), E4976–E4984. 10.1073/pnas.1600749113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dulak D.; Banach M.; Gadzala M.; Konieczny L.; Roterman I. Structural analysis of the Abeta(15–40) amyloid fibril based on hydrophobicity distribution. Acta Biochim Pol 2018, 65 (4), 595–604. 10.18388/abp.2018_2647. [DOI] [PubMed] [Google Scholar]
- Seeber M.; Felline A.; Raimondi F.; Muff S.; Friedman R.; Rao F.; Caflisch A.; Fanelli F. Wordom: a user-friendly program for the analysis of molecular structures, trajectories, and free energy surfaces. J. Comput. Chem. 2011, 32 (6), 1183–1194. 10.1002/jcc.21688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamamis P.; Kieslich C. A.; Nikiforovich G. V.; Woodruff T. M.; Morikis D.; Archontis G. Insights into the mechanism of C5aR inhibition by PMX53 via implicit solvent molecular dynamics simulations and docking. BMC Biophys. 2014, 7 (1), 5. 10.1186/2046-1682-7-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J.; MacKerell A. D. Jr. CHARMM36 all-atom additive protein force field: validation based on comparison to NMR data. J. Comput. Chem. 2013, 34 (25), 2135–2145. 10.1002/jcc.23354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanommeslaeghe K.; Hatcher E.; Acharya C.; Kundu S.; Zhong S.; Shim J.; Darian E.; Guvench O.; Lopes P.; Vorobyov I.; Mackerell A. D. CHARMM general force field: a force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2009, 31, 671–690. 10.1002/jcc.21367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frishman D.; Argos P. Knowledge-based protein secondary structure assignment. Proteins: Struct., Funct., Genet. 1995, 23 (4), 566–579. 10.1002/prot.340230412. [DOI] [PubMed] [Google Scholar]
- Humphrey W.; Dalke A.; Schulten K. VMD: Visual molecular dynamics. J. Mol. Graphics 1996, 14 (1), 33–38. 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- Kokotidou C.; Jonnalagadda S. V. R.; Orr A. A.; Seoane-Blanco M.; Apostolidou C. P.; van Raaij M. J.; Kotzabasaki M.; Chatzoudis A.; Jakubowski J. M.; Mossou E.; Forsyth V. T.; Mitchell E. P.; Bowler M. W.; Llamas-Saiz A. L.; Tamamis P.; Mitraki A. A novel amyloid designable scaffold and potential inhibitor inspired by GAIIG of amyloid beta and the HIV-1 V3 loop. FEBS Lett. 2018, 592 (11), 1777–1788. 10.1002/1873-3468.13096. [DOI] [PubMed] [Google Scholar]
- Saez N. J.; Deplazes E.; Cristofori-Armstrong B.; Chassagnon I. R.; Lin X.; Mobli M.; Mark A. E.; Rash L. D.; King G. F. Molecular dynamics and functional studies define a hot spot of crystal contacts essential for PcTx1 inhibition of acid-sensing ion channel 1a. British journal of pharmacology 2015, 172 (20), 4985–4995. 10.1111/bph.13267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamamis P.; Floudas C. A. Molecular recognition of CXCR4 by a dual tropic HIV-1 gp120 V3 loop. Biophys. J. 2013, 105 (6), 1502–1514. 10.1016/j.bpj.2013.07.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamamis P.; Pierou P.; Mytidou C.; Floudas C. A.; Morikis D.; Archontis G. Design of a modified mouse protein with ligand binding properties of its human analog by molecular dynamics simulations: The case of C3 inhibition by compstatin. Proteins: Struct., Funct., Genet. 2011, 79 (11), 3166–3179. 10.1002/prot.23149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamamis P.; López de Victoria A.; Gorham R. D. Jr.; Bellows-Peterson M. L.; Pierou P.; Floudas C. A.; Morikis D.; Archontis G. Molecular dynamics in drug design: new generations of compstatin analogs. Chem. Biol. Drug Des. 2012, 79 (5), 703–718. 10.1111/j.1747-0285.2012.01324.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- A. Kieslich C.; Tamamis P.; D. Gorham R. Jr.; Lopez de Victoria A.; U. Sausman N.; Archontis G.; Morikis D. Exploring Protein-Protein and Protein-Ligand Interactions in the Immune System using Molecular Dynamics and Continuum Electrostatics. Current Physical Chemistry 2012, 2 (4), 324–343. 10.2174/1877947611202040324. [DOI] [Google Scholar]
- Kollman P. A.; Massova I.; Reyes C.; Kuhn B.; Huo S.; Chong L.; Lee M.; Lee T.; Duan Y.; Wang W.; Donini O.; Cieplak P.; Srinivasan J.; Case D. A.; Cheatham T. E. Calculating Structures and Free Energies of Complex Molecules: Combining Molecular Mechanics and Continuum Models. Acc. Chem. Res. 2000, 33 (12), 889–897. 10.1021/ar000033j. [DOI] [PubMed] [Google Scholar]
- Trott O.; Olson A. J. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2009, 31 (2), 455–461. 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gohlke H.; Case D. A. Converging free energy estimates: MM-PB(GB)SA studies on the protein–protein complex Ras–Raf. J. Comput. Chem. 2004, 25 (2), 238–250. 10.1002/jcc.10379. [DOI] [PubMed] [Google Scholar]
- Page C. S.; Bates P. A. Can MM-PBSA calculations predict the specificities of protein kinase inhibitors?. J. Comput. Chem. 2006, 27 (16), 1990–2007. 10.1002/jcc.20534. [DOI] [PubMed] [Google Scholar]
- Hayes J. M.; Archontis G.. MM-GB (PB) SA Calculations of Protein-Ligand Binding Free Energies. In Molecular Dynamics – Studies of Synthetic and Biological Macromolecules; Wang L., Ed.; Tech, 2012; pp 171–190. [Google Scholar]
- Hou T.; Wang J.; Li Y.; Wang W. Assessing the Performance of the MM/PBSA and MM/GBSA Methods. 1. The Accuracy of Binding Free Energy Calculations Based on Molecular Dynamics Simulations. J. Chem. Inf. Model. 2011, 51 (1), 69–82. 10.1021/ci100275a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohan R. R.; Wilson M.; Gorham R. D. Jr.; Harrison R. E. S.; Morikis V. A.; Kieslich C. A.; Orr A. A.; Coley A. V.; Tamamis P.; Morikis D. Virtual Screening of Chemical Compounds for Discovery of Complement C3 Ligands. ACS Omega 2018, 3 (6), 6427–6438. 10.1021/acsomega.8b00606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z.; Sun H.; Yao X.; Li D.; Xu L.; Li Y.; Tian S.; Hou T. Comprehensive evaluation of ten docking programs on a diverse set of protein-ligand complexes: the prediction accuracy of sampling power and scoring power. Phys. Chem. Chem. Phys. 2016, 18 (18), 12964–12975. 10.1039/C6CP01555G. [DOI] [PubMed] [Google Scholar]
- Periole X.; Huber T.; Bonito-Oliva A.; Aberg K. C.; van der Wel P. C. A.; Sakmar T. P.; Marrink S. J. Energetics Underlying Twist Polymorphisms in Amyloid Fibrils. J. Phys. Chem. B 2018, 122 (3), 1081–1091. 10.1021/acs.jpcb.7b10233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao K.; Wang J.; Zhou P.; Wang C.; Xu H.; Zhao X.; Lu J. R. Self-assembly of short aβ(16–22) peptides: effect of terminal capping and the role of electrostatic interaction. Langmuir 2011, 27, 2723–2730. 10.1021/la1034273. [DOI] [PubMed] [Google Scholar]
- de Groot N. S.; Aviles F. X.; Vendrell J.; Ventura S. Mutagenesis of the central hydrophobic cluster in Aβ42 Alzheimer’s peptide. FEBS J. 2006, 273 (3), 658–668. 10.1111/j.1742-4658.2005.05102.x. [DOI] [PubMed] [Google Scholar]
- Cheng P. N.; Liu C.; Zhao M.; Eisenberg D.; Nowick J. S. Amyloid β-sheet mimics that antagonize protein aggregation and reduce amyloid toxicity. Nat. Chem. 2012, 4, 927–933. 10.1038/nchem.1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koo H. J.; Shin S.; Choi J. Y.; Lee K. H.; Kim B. T.; Choe Y. S. Introduction of Methyl Groups at C2 and C6 Positions Enhances the Antiangiogenesis Activity of Curcumin. Sci. Rep. 2015, 5, 14205. 10.1038/srep14205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanagisawa D.; Shirai N.; Amatsubo T.; Taguchi H.; Hirao K.; Urushitani M.; Morikawa S.; Inubushi T.; Kato M.; Kato F.; Morino K.; Kimura H.; Nakano I.; Yoshida C.; Okada T.; Sano M.; Wada Y.; Wada K. N.; Yamamoto A.; Tooyama I. Relationship between the tautomeric structures of curcumin derivatives and their Abeta-binding activities in the context of therapies for Alzheimer’s disease. Biomaterials 2010, 31 (14), 4179–4185. 10.1016/j.biomaterials.2010.01.142. [DOI] [PubMed] [Google Scholar]
- Kynast P.; Derreumaux P.; Strodel B. Evaluation of the coarse-grained OPEP force field for protein-protein docking. BMC Biophys. 2016, 9 (1), 4. 10.1186/s13628-016-0029-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagarde N.; Carbone A.; Sacquin-Mora S. Hidden partners: Using cross-docking calculations to predict binding sites for proteins with multiple interactions. Proteins: Struct., Funct., Genet. 2018, 86 (7), 723–737. 10.1002/prot.25506. [DOI] [PubMed] [Google Scholar]
- Zhao B.; Cohen Stuart M. A.; Hall C. K. Dock ’n roll: folding of a silk-inspired polypeptide into an amyloid-like beta solenoid. Soft Matter 2016, 12 (16), 3721–3729. 10.1039/C6SM00169F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latshaw D. C. 2nd; Hall C. K. Effects of hydrophobic macromolecular crowders on amyloid β (16–22) aggregation. Biophys. J. 2015, 109 (1), 124–134. 10.1016/j.bpj.2015.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagoner V. A.; Cheon M.; Chang I.; Hall C. K. Impact of sequence on the molecular assembly of short amyloid peptides. Proteins: Struct., Funct., Genet. 2014, 82 (7), 1469–1483. 10.1002/prot.24515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagoner V. A.; Cheon M.; Chang I.; Hall C. K. Computer simulation study of amyloid fibril formation by palindromic sequences in prion peptides. Proteins: Struct., Funct., Genet. 2011, 79 (7), 2132–2145. 10.1002/prot.23034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen H. D.; Hall C. K. Molecular dynamics simulations of spontaneous fibril formation by random-coil peptides. Proc. Natl. Acad. Sci. U. S. A. 2004, 101 (46), 16180–16185. 10.1073/pnas.0407273101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Y.; Dinkel P.; Yu X.; Margittai M.; Zheng J.; Nussinov R.; Wei G.; Ma B. Molecular insights into the reversible formation of tau protein fibrils. Chem. Commun. (Cambridge, U. K.) 2013, 49 (34), 3582–3584. 10.1039/c3cc00241a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren B.; Hu R.; Zhang M.; Liu Y.; Xu L.; Jiang B.; Ma J.; Ma B.; Nussinov R.; Zheng J. Experimental and Computational Protocols for Studies of Cross-Seeding Amyloid Assemblies. Methods Mol. Biol. 2018, 1777, 429–447. 10.1007/978-1-4939-7811-3_27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu R.; Zhang M.; Chen H.; Jiang B.; Zheng J. Cross-Seeding Interaction between β-Amyloid and Human Islet Amyloid Polypeptide. ACS Chem. Neurosci. 2015, 6 (10), 1759–1768. 10.1021/acschemneuro.5b00192. [DOI] [PubMed] [Google Scholar]
- Shea J. E.; Levine Z. A. Studying the Early Stages of Protein Aggregation Using Replica Exchange Molecular Dynamics Simulations. Methods Mol. Biol. 2016, 1345, 225–250. 10.1007/978-1-4939-2978-8_15. [DOI] [PubMed] [Google Scholar]
- Do T. D.; LaPointe N. E.; Nelson R.; Krotee P.; Hayden E. Y.; Ulrich B.; Quan S.; Feinstein S. C.; Teplow D. B.; Eisenberg D.; Shea J. E.; Bowers M. T. Amyloid β-Protein C-Terminal Fragments: Formation of Cylindrins and β-Barrels. J. Am. Chem. Soc. 2016, 138 (2), 549–557. 10.1021/jacs.5b09536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong A. G.; Wu C.; Hannaberry E.; Watson M. D.; Shea J. E.; Raleigh D. P. Analysis of the Amyloidogenic Potential of Pufferfish (Takifugu rubripes) Islet Amyloid Polypeptide Highlights the Limitations of Thioflavin-T Assays and the Difficulties in Defining Amyloidogenicity. Biochemistry 2016, 55 (3), 510–518. 10.1021/acs.biochem.5b01107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Do T. D.; Economou N. J.; Chamas A.; Buratto S. K.; Shea J. E.; Bowers M. T. Interactions between amyloid-β and Tau fragments promote aberrant aggregates: implications for amyloid toxicity. J. Phys. Chem. B 2014, 118 (38), 11220–11230. 10.1021/jp506258g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larini L.; Shea J. E. Role of β-hairpin formation in aggregation: the self-assembly of the amyloid-β(25–35) peptide. Biophys. J. 2012, 103 (3), 576–586. 10.1016/j.bpj.2012.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C.; Shea J. E. The Structure of Intrinsically Disordered Peptides Implicated in Amyloid Diseases: Insights from Fully Atomistic Simulations. Computational Modeling of Biological Systems. 2012, 215–227. 10.1007/978-1-4614-2146-7_9. [DOI] [Google Scholar]
- Wei G.; Jewett A. I.; Shea J. E. Structural diversity of dimers of the Alzheimer amyloid-beta(25–35) peptide and polymorphism of the resulting fibrils. Phys. Chem. Chem. Phys. 2010, 12 (14), 3622–3629. 10.1039/c000755m. [DOI] [PubMed] [Google Scholar]
- Chiricotto M.; Tran T. T.; Nguyen P. H.; Melchionna S.; Sterpone F.; Derreumaux P. Coarse-grained and All-atom Simulations towards the Early and Late Steps of Amyloid Fibril Formation. Isr. J. Chem. 2017, 57 (7), 564–573. 10.1002/ijch.201600048. [DOI] [Google Scholar]
- Nguyen P. H.; Sterpone F.; Pouplana R.; Derreumaux P.; Campanera J. M. Dimerization Mechanism of Alzheimer Aβ40 Peptides: The High Content of Intrapeptide-Stabilized Conformations in A2V and A2T Heterozygous Dimers Retards Amyloid Fibril Formation. J. Phys. Chem. B 2016, 120 (47), 12111–12126. 10.1021/acs.jpcb.6b10722. [DOI] [PubMed] [Google Scholar]
- Sterpone F.; Melchionna S.; Tuffery P.; Pasquali S.; Mousseau N.; Cragnolini T.; Chebaro Y.; St-Pierre J. F.; Kalimeri M.; Barducci A.; Laurin Y.; Tek A.; Baaden M.; Nguyen P. H.; Derreumaux P. The OPEP protein model: from single molecules, amyloid formation, crowding and hydrodynamics to DNA/RNA systems. Chem. Soc. Rev. 2014, 43 (13), 4871–4893. 10.1039/C4CS00048J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Côté S.; Laghaei R.; Derreumaux P.; Mousseau N. Distinct dimerization for various alloforms of the amyloid-beta protein: Aβ(1–40), Aβ(1–42), and Aβ(1–40)(D23N). J. Phys. Chem. B 2012, 116 (13), 4043–4055. 10.1021/jp2126366. [DOI] [PubMed] [Google Scholar]
- Qiao Q.; Qi R.; Wei G.; Huang X. Dynamics of the conformational transitions during the dimerization of an intrinsically disordered peptide: a case study on the human islet amyloid polypeptide fragment. Phys. Chem. Chem. Phys. 2016, 18 (43), 29892–29904. 10.1039/C6CP05590G. [DOI] [PubMed] [Google Scholar]
- Nguyen P. H.; Li M. S.; Derreumaux P. Amyloid oligomer structure characterization from simulations: a general method. J. Chem. Phys. 2014, 140 (9), 094105 10.1063/1.4866902. [DOI] [PubMed] [Google Scholar]
- Magno A.; Pellarin R.; Caflisch A. Mechanisms and Kinetics of Amyloid Aggregation Investigated by a Phenomenological Coarse-Grained Model. Computational Modeling of Biological Systems. 2012, 191–214. 10.1007/978-1-4614-2146-7_8. [DOI] [Google Scholar]
- Ilie I. M.; Caflisch A. Disorder at the Tips of a Disease-Relevant Aβ42 Amyloid Fibril: A Molecular Dynamics Study. J. Phys. Chem. B 2018, 122 (49), 11072–11082. 10.1021/acs.jpcb.8b05236. [DOI] [PubMed] [Google Scholar]
- Feige M. J.; Groscurth S.; Marcinowski M.; Yew Z. T.; Truffault V.; Paci E.; Kessler H.; Buchner J. The structure of a folding intermediate provides insight into differences in immunoglobulin amyloidogenicity. Proc. Natl. Acad. Sci. U. S. A. 2008, 105 (36), 13373–13378. 10.1073/pnas.0802809105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paci E.; Gsponer J.; Salvatella X.; Vendruscolo M. Molecular dynamics studies of the process of amyloid aggregation of peptide fragments of transthyretin. J. Mol. Biol. 2004, 340 (3), 555–569. 10.1016/j.jmb.2004.05.009. [DOI] [PubMed] [Google Scholar]
- Jonnalagadda S. V. R.; Kokotidou C.; Orr A. A.; Fotopoulou E.; Henderson K. J.; Choi C.-H.; Lim W. T.; Choi S. J.; Jeong H.-K.; Mitraki A.; Tamamis P. Computational Design of Functional Amyloid Materials with Cesium Binding, Deposition, and Capture Properties. J. Phys. Chem. B 2018, 122 (30), 7555–7568. 10.1021/acs.jpcb.8b04103. [DOI] [PubMed] [Google Scholar]
- Deidda G.; Jonnalagadda S. V. R.; Spies J. W.; Ranella A.; Mossou E.; Forsyth V. T.; Mitchell E. P.; Bowler M. W.; Tamamis P.; Mitraki A. Self-Assembled Amyloid Peptides with Arg-Gly-Asp (RGD) Motifs As Scaffolds for Tissue Engineering. ACS Biomater. Sci. Eng. 2017, 3 (7), 1404–1416. 10.1021/acsbiomaterials.6b00570. [DOI] [PubMed] [Google Scholar]
- Nath A.; Schlamadinger D. E.; Rhoades E.; Miranker A. D. Structure-Based Small Molecule Modulation of a Pre-Amyloid State: Pharmacological Enhancement of IAPP Membrane-Binding and Toxicity. Biochemistry 2015, 54 (22), 3555–3564. 10.1021/acs.biochem.5b00052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorham R. D. Jr.; Forest D. L.; Khoury G. A.; Smadbeck J.; Beecher C. N.; Healy E. D.; Tamamis P.; Archontis G.; Larive C. K.; Floudas C. A.; Radeke M. J.; Johnson L. V.; Morikis D. New compstatin peptides containing N-terminal extensions and non-natural amino acids exhibit potent complement inhibition and improved solubility characteristics. J. Med. Chem. 2015, 58 (2), 814–826. 10.1021/jm501345y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittayanarakul K.; Hannongbua S.; Feig M. On the correlation between drug-resistant pattern of HIV-1 protease inhibitors and binding free energy and structural nike giannis freak 1 changes. BMC Syst. Biol. 2007, 1 (1), P58. 10.1186/1752-0509-1-S1-P58. [DOI] [Google Scholar]
- Leonard S. R.; Cormier A. R.; Pang X.; Zimmerman M. I.; Zhou H. X.; Paravastu A. K. Solid-state NMR evidence for β-hairpin structure within MAX8 designer peptide nanofibers. Biophys. J. 2013, 105 (1), 222–230. 10.1016/j.bpj.2013.05.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cormier A. R.; Pang X.; Zimmerman M. I.; Zhou H. X.; Paravastu A. K. Molecular structure of RADA16-I designer self-assembling peptide nanofibers. ACS Nano 2013, 7 (9), 7562–7572. 10.1021/nn401562f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurry T.; Fisher C. K.; Schmidt M.; Stultz C. M.. Analyzing Ensembles of Amyloid Proteins Using Bayesian Statistics. In Protein Amyloid Aggregation: Methods and Protocols, Eliezer D., Ed.; Springer New York: New York, NY, 2016; pp 269–280. [DOI] [PubMed] [Google Scholar]
- Gurry T.; Stultz C. M. The Mechanism of Amyloid-Beta Fibril Elongation. Biochemistry 2014, 53, 6981–6991. 10.1021/bi500695g. [DOI] [PubMed] [Google Scholar]
- Fisher C. K.; Ullman O.; Stultz C. M. Comparative studies of disordered proteins with similar sequences: application to Aβ40 and Aβ42. Biophys. J. 2013, 104 (7), 1546–1555. 10.1016/j.bpj.2013.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurry T.; Ullman O.; Fisher C. K.; Perovic I.; Pochapsky T.; Stultz C. M. The Dynamic Structure of α-Synuclein Multimers. J. Am. Chem. Soc. 2013, 135 (10), 3865–3872. 10.1021/ja310518p. [DOI] [PubMed] [Google Scholar]
- Ma S.; Zhang H.; Dong X.; Yu L.; Zheng J.; Sun Y. Head-to-tail cyclization of a heptapeptide eliminates its cytotoxicity and significantly increases its inhibition effect on amyloid β-protein fibrillation and cytotoxicity. Front. Chem. Sci. Eng. 2018, 12 (2), 283–295. 10.1007/s11705-017-1687-2. [DOI] [Google Scholar]
- Zhang H.; Dong X.; Liu F.; Zheng J.; Sun Y. Ac-LVFFARK-NH2 conjugation to β-cyclodextrin exhibits significantly enhanced performance on inhibiting amyloid β-protein fibrillogenesis and cytotoxicity. Biophys. Chem. 2018, 235, 40–47. 10.1016/j.bpc.2018.02.002. [DOI] [PubMed] [Google Scholar]
- Zhang M.; Ren B.; Chen H.; Sun Y.; Ma J.; Jiang B.; Zheng J. Molecular Simulations of Amyloid Structures, Toxicity, and Inhibition. Isr. J. Chem. 2017, 57 (7–8), 586–601. 10.1002/ijch.201600075. [DOI] [Google Scholar]
- Qiao Y.; Zhang M.; Liang Y. n.; Zheng J.; Liang G. A computational study of self-assembled hexapeptide inhibitors against amyloid-β (Aβ) aggregation. Phys. Chem. Chem. Phys. 2017, 19 (1), 155–166. 10.1039/C6CP07341G. [DOI] [PubMed] [Google Scholar]
- Wang Q.; Yu X.; Li L.; Zheng J. Inhibition of amyloid-β aggregation in Alzheimer’s disease. Curr. Pharm. Des. 2014, 20 (8), 1223–1243. 10.2174/13816128113199990068. [DOI] [PubMed] [Google Scholar]
- Zheng X.; Wu C.; Liu D.; Li H.; Bitan G.; Shea J.-E.; Bowers M. T. Mechanism of C-Terminal Fragments of Amyloid β-Protein as Aβ Inhibitors: Do C-Terminal Interactions Play a Key Role in Their Inhibitory Activity?. J. Phys. Chem. B 2016, 120 (8), 1615–1623. 10.1021/acs.jpcb.5b08177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Susa A. C.; Wu C.; Bernstein S. L.; Dupuis N. F.; Wang H.; Raleigh D. P.; Shea J.-E.; Bowers M. T. Defining the Molecular Basis of Amyloid Inhibitors: Human Islet Amyloid Polypeptide–Insulin Interactions. J. Am. Chem. Soc. 2014, 136 (37), 12912–12919. 10.1021/ja504031d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bleiholder C.; Do T. D.; Wu C.; Economou N. J.; Bernstein S. S.; Buratto S. K.; Shea J.-E.; Bowers M. T. Ion Mobility Spectrometry Reveals the Mechanism of Amyloid Formation of Aβ(25–35) and Its Modulation by Inhibitors at the Molecular Level: Epigallocatechin Gallate and Scyllo-inositol. J. Am. Chem. Soc. 2013, 135 (45), 16926–16937. 10.1021/ja406197f. [DOI] [PubMed] [Google Scholar]
- Frydman-Marom A.; Convertino M.; Pellarin R.; Lampel A.; Shaltiel-Karyo R.; Segal D.; Caflisch A.; Shalev D. E.; Gazit E. Structural Basis for Inhibiting β-Amyloid Oligomerization by a Non-coded β-Breaker-Substituted Endomorphin Analogue. ACS Chem. Biol. 2011, 6 (11), 1265–1276. 10.1021/cb200103h. [DOI] [PubMed] [Google Scholar]
- Ren B.; Liu Y.; Zhang Y.; Cai Y.; Gong X.; Chang Y.; Xu L.; Zheng J. Genistein: A Dual Inhibitor of Both Amyloid β and Human Islet Amylin Peptides. ACS Chem. Neurosci. 2018, 9 (5), 1215–1224. 10.1021/acschemneuro.8b00039. [DOI] [PubMed] [Google Scholar]
- Cohen S. I.; Cukalevski R.; Michaels T. C. T.; Šarić A.; Törnquist M.; Vendruscolo M.; Dobson C. M.; Buell A. K.; Knowles T. P. J.; Linse S. Distinct thermodynamic signatures of oligomer generation in the aggregation of the amyloid-β peptide. Nat. Chem. 2018, 10 (5), 523–531. 10.1038/s41557-018-0023-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez R. A.; Chen L. Y.; Plascencia-Villa G.; Perry G. Thermodynamics of Amyloid-β Fibril Elongation: Atomistic Details of the Transition State. ACS Chem. Neurosci. 2018, 9 (4), 783–789. 10.1021/acschemneuro.7b00409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin T. D.; Malagodi A. J.; Chi E. Y.; Evans D. G. Computational Study of the Driving Forces and Dynamics of Curcumin Binding to Amyloid-β Protofibrils. J. Phys. Chem. B 2019, 123 (3), 551–560. 10.1021/acs.jpcb.8b09185. [DOI] [PubMed] [Google Scholar]
- Tamamis P.; Floudas C. A. Elucidating a key component of cancer metastasis: CXCL12 (SDF-1α) binding to CXCR4. J. Chem. Inf. Model. 2014, 54 (4), 1174–1188. 10.1021/ci500069y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamamis P.; Floudas C. A. Elucidating a key anti-HIV-1 and cancer-associated axis: the structure of CCL5 (Rantes) in complex with CCR5. Sci. Rep. 2015, 4, 5447. 10.1038/srep05447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamamis P.; Floudas C. A. Molecular recognition of CXCR4 by a dual tropic HIV-1 gp120 V3 loop. Biophys. J. 2013, 105 (6), 1502–1514. 10.1016/j.bpj.2013.07.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamamis P.; Floudas C. A. Molecular Recognition of CCR5 by an HIV-1 gp120 V3 Loop. PLoS One 2014, 9 (4), e95767 10.1371/journal.pone.0095767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez del Amo J.; Fink U.; Dasari M.; Grelle G.; Wanker E. E.; Bieschke J.; Reif B. Structural Properties of EGCG-Induced, Nontoxic Alzheimer’s Disease Aβ Oligomers. J. Mol. Biol. 2012, 421 (4–5), 517–524. 10.1016/j.jmb.2012.01.013. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.