

Calmodulin controls KCNQ1 potassium channel pore opening by competing with PIP2 in the KCNQ1 voltage-sensing domain.
Abstract
Calmodulin (CaM) and phosphatidylinositol 4,5-bisphosphate (PIP2) are potent regulators of the voltage-gated potassium channel KCNQ1 (KV7.1), which conducts the cardiac IKs current. Although cryo–electron microscopy structures revealed intricate interactions between the KCNQ1 voltage-sensing domain (VSD), CaM, and PIP2, the functional consequences of these interactions remain unknown. Here, we show that CaM-VSD interactions act as a state-dependent switch to control KCNQ1 pore opening. Combined electrophysiology and molecular dynamics network analysis suggest that VSD transition into the fully activated state allows PIP2 to compete with CaM for binding to VSD. This leads to conformational changes that alter VSD-pore coupling to stabilize open states. We identify a motif in the KCNQ1 cytosolic domain, which works downstream of CaM-VSD interactions to facilitate the conformational change. Our findings suggest a gating mechanism that integrates PIP2 and CaM in KCNQ1 voltage-dependent activation, yielding insights into how KCNQ1 gains the phenotypes critical for its physiological function.
INTRODUCTION
The KCNQ (KV7) voltage-gated potassium channel family consists of five members (KCNQ1 to KCNQ5), which provide K+ currents critical for physiological function of both excitable and nonexcitable tissues (1–4). All KCNQ channels require the association of calmodulin (CaM) (5, 6) and membrane lipid phosphatidylinositol 4,5-bisphosphate (PIP2) (7, 8) for voltage-dependent opening. In the heart, the predominant cardiac isoform KCNQ1 (KV7.1) also associates with the auxiliary subunit KCNE1 (1, 2) to carry the slow delayed rectifier K+ current (IKs). IKs facilitates repolarization of the cardiac action potential, especially during β-adrenergic stimulation (9). Hundreds of mutations in KCNQ1 are linked to arrhythmias such as atrial fibrillation and long QT syndrome (LQTS), predisposing patients to sudden cardiac death (10, 11).
Structurally, KCNQ1 channels comprise four identical subunits, each with six transmembrane (S1 to S6) and four cytoplasmic [helix A (HA) to HD] α-helices, with S1 to S4 forming the voltage-sensing domain (VSD) and S5 and S6 the pore domain (Fig. 1, A and B) (12, 13). Upon membrane depolarization, the KCNQ1 VSDs activate with two experimentally resolvable steps: first from the resting state to an intermediate state and then to the fully activated state (14–19). The VSD conformational changes are communicated to the pore domain via a series of residue-residue interactions, leading to pore opening (14, 16, 19). KCNQ1 features a unique two-open-state gating mechanism (Fig. 1C). Previous work showed that KCNQ1 VSD can trigger pore opening when activated to both the intermediate and the fully activated states, yielding two open states: the intermediate-open (IO) and activated-open (AO) states (16, 18–22). The IO and AO states have been shown to give rise to biexponential current activation kinetics (16, 20, 21), with the ratio of the fast and slow components approximating the relative stability of IO and AO states, respectively (Fig. 1C and fig. S1, A to C). The IO and AO states exhibit different functional properties important for KCNQ1 physiological function, with regulation of IO and AO by different auxiliary subunits leading to tissue-specific current phenotypes (18). For example, although KCNQ1 conducts predominantly at the IO state as seen by the large fraction of the fast-current component (Fig. 1C and fig. S1A), the auxiliary subunit KCNE1 eliminates the kinetically fast IO state and enhances the slow AO state, contributing to the kinetically slow IKs suitable for cardiac tissue needs (16, 18, 19).
Fig. 1. The S2-S3 linker within the KCNQ1 VSD modulates KCNQ1 currents.
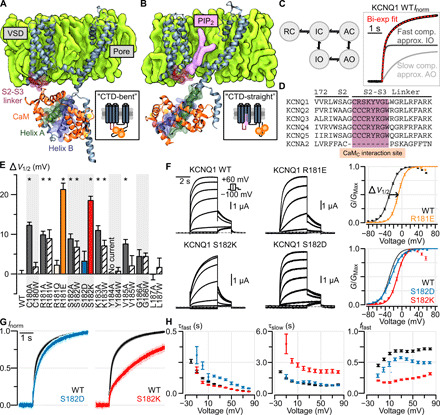
(A and B) Structural depiction of one monomer of the tetrameric KCNQ1/CaM complex when determined in the absence (A) and presence (B) of PIP2 [i.e., CTD-bent (A) and CTD-straight (B) conformations]. (C) Left: A simplified KCNQ1 gating model (16, 18–21) with two open states: IO and AO. Horizontal transitions correspond to VSD activation, vertical transitions correspond to pore opening. RC, resting closed; IC, intermediate closed; AC, activated closed. Right: Exemplar biexponential activation kinetics fitting of a Q1-WT normalized current. Voltage was stepped from −80 to 0 mV. (D) Sequence alignment of the KCNQ family and KCNA2 at the S2-S3 linker region predicted to interact with CaM. (E) Mutagenesis screen of the KCNQ1 S2-S3 linker region shown in (D). Y axis: ΔV1/2 from WT channel [see (F) for example]. X axis: residue position. Stars indicate significant ΔV1/2 from WT (table S1). Error bars are SEM. (F) Left: Exemplar recordings from KCNQ1 WT and S2-S3 linker mutant channels. Voltage protocol is shown in inset. Currents were collected with 10-mV increments at the test pulse but shown in 20-mV increments to avoid crowding. Right: Average G-V relationship for the respective channels. An example of ΔV1/2 measurement is shown for mutant R181E. (G) Average normalized KCNQ1 channel activation kinetics, stepping from −80 to 0 mV, for Q1-WT (black, n = 10), Q1-S182D (blue, n = 7), and Q1-S182K (red, n = 10) channels. The light shadings show SEM. (F) Average biexponential kinetic fit parameters for Q1-WT (black), Q1-S182D (blue), and Q1-S182K (red) channels at different test voltages. Error bars are SEM. τfast and τslow are the time constants for the fast and slow components, respectively. ffast is fraction of total current contributed by the fast component.
KCNQ regulation by CaM (5, 6, 12, 13) is still not clearly understood. CaM features a two-lobe structural architecture with the N-terminal domain (N-lobe) and the C-terminal domain (C-lobe), and each lobe harbors two Ca2+ binding sites (23). CaM regulation of KCNQ channels is still debated, some studies suggested that CaM confers Ca2+-dependent inhibition of KCNQ2/3 and KCNQ4 currents but Ca2+-dependent enhancement of KCNQ1 currents (24–29). Other studies found that CaM C-lobe bound to KCNQ1 does not coordinate Ca2+ ions under 1 mM EGTA or 5 mM Ca2+ conditions (30). CaM binding by itself may therefore be sufficient to affect KCNQ1 function, similar to other voltage-gated channels (31). Structurally, CaM binds the KCNQ1 C-terminal domain (CTD) such that CaM N-lobe associates with KCNQ1 HB and CaM C-lobe interacts with KCNQ1 HA (Fig. 1A) (5, 6, 12, 13, 30). Recent cryo–electron microscopy (EM) structural studies of the full-length KCNQ1/CaM complex (12, 13) revealed intricate interactions between CaM, the KCNQ1 VSD, and the membrane lipid PIP2. In the absence of PIP2, the KCNQ1 CTD “bends” at the S6-HA linker to enable CaM interaction with the VSD S2-S3 linker (Fig. 1A) (12, 13). In the presence of PIP2, the KCNQ1 CTD “straightens” to disengage CaM from the VSD, and the S2-S3 linker instead interacts with PIP2 (Fig. 1B) (13). We will refer to these two cryo-EM structural states as the “CTD-bent” and the “CTD-straight” conformations (Fig. 1, A and B). The VSDs in both conformations adopt the fully activated state (12, 13, 18). The CTD-bent conformation features a closed pore, while the CTD-straight conformation exhibits a dilated pore (12, 13).
Although the KCNQ1/CaM structures have provided fresh insight into the conformational ensemble of KCNQ1, it is still unknown how these structures correlate to the channel’s voltage-dependent activation process. First, the CTD-straight structure likely corresponds to the AO state (Fig. 1C) based on its activated VSD and dilated pore (12, 13, 18), but functional data are lacking to support this. Moreover, the CTD-straight structure additionally features binding by the auxiliary subunit KCNE3 (13), which may lead to conformational differences compared to KCNQ1/CaM alone. Second, it is unknown whether the CTD-bent conformation corresponds to a functional state in the KCNQ1 activation pathway, since PIP2 is present under physiological conditions and the structure was solved without PIP2. By extension, it is unclear whether the CaM-VSD interactions seen in the CTD-bent conformation occur during gating. No studies have shown simultaneous CaM and PIP2 binding to the KCNQ1 VSD. The available studies thus suggest mutually exclusive CaM or PIP2 binding to the S2-S3 linker (Fig. 1, A and B) (12, 13), with PIP2 outcompeting CaM for the activated VSD. Third, if the CTD-bent structure represents a functional state in the KCNQ1 activation pathway, what is the role of CaM-VSD interactions in KCNQ1 gating? In summary, the relationship between KCNQ1/CaM structures and voltage-dependent gating, specifically the role of CaM-VSD interactions, remains unresolved.
Here, we combine electrophysiology experiments and network analysis of molecular dynamics (MD) simulations to show that CaM interacts with the KCNQ1 VSD during voltage-dependent activation to modulate ionic current, despite PIP2 presence in the membrane. We demonstrate that both the CTD-straight and CTD-bent conformations are relevant in the KCNQ1 voltage-dependent gating process. We further provide evidence that the transition between the CTD-bent and CTD-straight conformations during gating is driven by VSD state-dependent interactions between the S2-S3 linker, CaM, and PIP2. Functionally, we find that the loss of CaM-VSD interactions during gating controls channel occupancy of the AO state while having no effect on the properties of the IO state. Last, we map a CTD motif, formed by the S6-HA and HB-HC linkers, that works together with the CaM-VSD interface to affect pore opening to the AO state. On the basis of our results, together with the cryo-EM structural data, we suggest a gating model in which CaM acts as a VSD state-dependent switch to alter pore opening via the KCNQ1 CTD, thereby tuning the KCNQ1 current related to the cardiac repolarizing currents.
RESULTS
The cryo-EM studies showed two KCNQ1 structures with large conformational differences, with the CTD-straight structure solved in the more physiological conditions featuring lipid nanodiscs in the presence of PIP2 and the CTD-bent structure solved in the absence of PIP2 (12, 13). The CTD-straight structure is likely to reflect the AO state based on its activated VSD and dilated pore (13), but whether the CTD-bent structure corresponds to any gating state in voltage-dependent activation is not clear. We designed experiments and molecular dynamic simulations to address the following questions: (i) Is the CTD-bent structure part of the voltage-dependent activation process? (ii) Is there any functional evidence to support that the CTD-straight structure corresponds to the AO state? (iii) What is the mechanism and role of the conformational switch between the CTD-bent and CTD-straight in the gating process of KCNQ1?
The S2-S3 linker within the KCNQ1 VSD modulates KCNQ1 currents
The S2-S3 linker region is conserved among the KCNQ family (Fig. 1D) but not in the wider KV superfamily. We began by examining the functional role of the KCNQ1 S2-S3 linker region close to either CaM or PIP2 in the cryo-EM structures (12, 13). To this end, we undertook a dual alanine/tryptophan mutational scan of the S2-S3 linker (Fig. 1, A and B, maroon), designed to remove side chain properties and induce bulky steric effects, respectively. We assayed the mutant human KCNQ1 channels by two-electrode voltage clamp (TEVC) in Xenopus oocytes, in which the oocytes endogenously supplied PIP2 and CaM, since endogenous Xenopus CaM shares 100% amino acid identity with human CaM. Each mutant channel was characterized by the half-activation voltage (V1/2) of the measured conductance-voltage (G-V) relationship, which indicates the voltage dependence of channel open probability. The mutational scan revealed significant depolarizing shifts in the V1/2 when mutating residues C180-K183 and V185 but not residues Y184 and G186-L187 (Fig. 1E and table S1). These results show the functional impact of S2-S3 linker residues C180-K183 on KCNQ1 activation. As the S2-S3 linker may participate in electrostatic interactions with CaM or PIP2, we further made charged mutations at two positions (R181 and S182) featuring clear effects. We found charge-dependent effects on KCNQ1 currents at positions R181 and S182: the mutants R181E and S182K induced ~20 mV depolarizing shifts in the V1/2 (Fig. 1F, yellow and red). Conversely, the mutants R181Q and S182D caused little change from the wild-type (WT) G-V relationship (Fig. 1, E and F). As previous functional studies and the CTD-straight structure suggested that residue R181 interacts with PIP2 (8, 13), the functional effects of the R181 mutants is likely due to altered PIP2 binding. On the other hand, the CTD-bent structure showed that S182 interacts with CaM (12, 13); therefore, the S182 mutation effects may be due to disrupted CaM-VSD interactions.
We also noted that the S182K mutant exhibited a major slowdown in current activation kinetics compared to the WT channels, while the S182D mutant exhibited similar kinetics as the WT channel (Fig. 1, G and H). As the fast and slow components of the KCNQ1 current kinetics approximate the IO and AO states (Fig. 1C), we quantified current activation by fitting a biexponential function with fast and slow time constants (τfast and τslow). We also calculated the fraction of total current carried by the fast versus the slow component (ffast), which approximates the relative contribution of the IO versus the AO states to the total current (fig. S1, A to C). The S182D mutant exhibited small changes in τfast and ffast compared to WT (Fig. 1, G and H), which correlated with the minimal shift in V1/2 induced by the S182D mutation. By contrast, the S182K mutant revealed a substantial slowing in current activation kinetics compared to WT channels (Fig. 1, G and H), which correlated with the large shift in V1/2 caused by S182K. The slow time course of S182K currents resulted from specific increases in both the τslow and fraction of the slow component (Fig. 1H: τslow and reduction in ffast) but not the fast kinetic component (Fig. 1H: τfast). The specific effect on the slow component of the current kinetics suggests that S182K may shift KCNQ1 to favor the AO state over the IO state (fig. S1, A to C), which may arise from destabilization of the IO state or stabilization of the AO state. The correlation between kinetics changes and significant V1/2 shifts is preserved in other S2-S3 linker mutants (fig. S2A). In contrast to S182K, the R181E mutant did not affect the slow time constant or the fraction of the slow component but instead prolonged the fast time constant (fig. S3, A and B). These distinct kinetic effects are further suggestive that S182 and R181 might differentially affect KCNQ1 by interacting with CaM or PIP2, respectively. Overall, the mutational scan provided functional evidence that the CaM- and PIP2-interacting regions of the KCNQ1 S2-S3 linker, as seen in the cryo-EM structures, functionally affects KCNQ1 activation.
The S2-S3 linker interacts with CaM in KCNQ1 voltage-dependent gating
Is the current effect induced by the mutation S182K (Fig. 1, E and F) due to disruption of CaM-VSD interactions? We next examined whether these CaM-VSD interactions occur during physiological gating, when PIP2 is available in the membrane. We undertook a second mutational scan of CaM residues close to the S2-S3 linker within the CTD-bent conformation (Fig. 2, A and B). We coexpressed each CaM mutant with human Q1-WT channels and compared their V1/2 with that of Q1-WT coexpressed with CaM-WT. We note that KCNQ1 supplied with either endogenous or exogeneous CaM WT yielded near identical phenotype (fig. S4). We found coexpression of CaM mutants at several positions (Y100, N138, E140, E141, and F142) induced significant shifts in the V1/2 (Fig. 2B and table S2). Similar to Q1-S182 mutations (Fig. 1E), mutations at CaM-Y100 demonstrated charge-dependent effects. CaM-Y100D induced a significant depolarizing shift in the V1/2, while CaM-Y100K yielded no change in the V1/2 (Fig. 2, B to D). In addition, CaM-Y100D altered current activation kinetics by increasing the fraction of the slow component current compared to WT (Fig. 2, E and F: black to purple, reduction in ffast), but had no effects on τslow (Fig. 2F). Kinetics analysis of KCNQ1 coexpressed with other CaM mutants demonstrated similar activation kinetics compared to WT (fig. S2B). This specific CaM-Y100D effect on the fraction of slow current component is similar to that seen with Q1-S182K mutation (Fig. 1H) and further suggests that CaM-Y100D may selectively shift KCNQ1 to favor AO state occupancy relative to IO state. Together, the mutational scan revealed that CaM residues close to the S2-S3 linker within the CTD-bent conformation also affect KCNQ1 function.
Fig. 2. CaM interacts with the KCNQ1 S2-S3 linker during voltage-dependent gating.
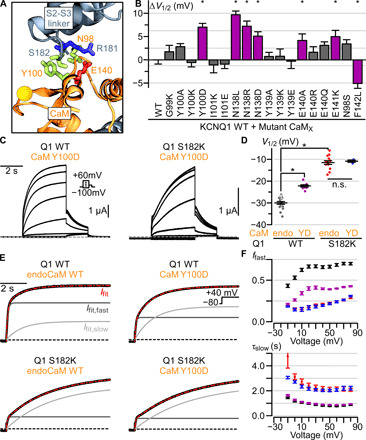
(A) Structural depiction of CaM residues predicted to interact with Q1-S182 in the CTD-bent structure. Residues depicted with sticks are colored according to residue type: positively (negatively) charged, blue (red); polar, green. Ca2+-ions are shown as yellow spheres. (B) Summary ΔV1/2 of human Q1-WT coexpressed with mutant CaM from Q1-WT coexpressed with CaM-WT (table S2). CaM-N98S and CaM-F142L are LQTS-associated CaM mutants. Purple bars and stars indicate significant ΔV1/2. Error bars are SEM. (C) Exemplar currents from coexpression of CaM-Y100D with Q1-WT (left) or Q1-S182K (right) channels. Voltage protocol in inset. (D) Average V1/2 for Q1-WT and -S182K channels associated with either CaM-Y100D (YD) or endogenously supplied CaM-WT (endo). Long bar, mean; short bars, SEM. Star, significantly different. P values, calculated by t test, are 2.18 × 10−8 (Q1-WT/CaM-WT to Q1-WT/CaM-Y100D), 5.25 × 10−17 (Q1-WT/CaM-WT to Q1-S182K/CaM-WT), and 0.73 (Q1-S182K/CaM-WT to Q1-S182K/CaM-Y100D). n.s., not significant. (E) Exemplar biexponential fits of activation kinetics for Q1-WT and -S182K channels associated with either CaM-Y100D or endogenously supplied CaM-WT. Black curve, ionic current; red curve, overall biexponential current fit (weighted sum of fast and slow components); light gray, slow component of the current fit; dark gray, fast component of the current fit; black dashed line, 0 current. (F) Average population data for ffast and 𝛕slow for Q1-WT + endogenous CaM-WT (black, n = 10), Q1-WT + CaM-Y100D (purple, n = 9), Q1-S182K + endogenous CaM-WT (red, n = 10), and Q1-S182K + CaM-Y100D (blue, n = 3). Error bars are SEM.
We next tested whether the S2-S3 linker and CaM interfaces interact during channel gating by combining KCNQ1 and CaM mutants. Because CaM-Y100 and Q1-S182 residues featured similar alteration to current phenotype, we coexpressed CaM-Y100D and Q1-S182K to test for interaction between the two residues. Although CaM-Y100D induced a depolarizing shift in the Q1-WT channel V1/2, CaM-Y100D failed to cause a depolarizing shift in the V1/2 of Q1-S182K channel (Fig. 2, C and D). Likewise, while CaM-Y100D increased the slow component of the Q1-WT current (a reduction in ffast), CaM-Y100D did not induce any further enhancement in the slow component of Q1-S182K channels (Fig. 2, E and F). These findings indicate that the single mutation Q1-S182K confers the full functional impact of the double mutations Q1-S182K and CaM-Y100D. Given that Q1-S182 and CaM-Y100 are in proximity within the CTD-bent structure, the nonadditive shifts of mutant V1/2 are consistent with a functional interaction between Q1-S182 and CaM-Y100 during KCNQ1 activation. By contrast, CaM-Y100D induced a significant depolarizing shift in the Q1-R181E channel V1/2 (fig. S3C), indicating that Q1-R181 and CaM-Y100 may affect channel gating by different mechanisms. Together, these data suggest a specific Q1-S182 and CaM-Y100 interaction during KCNQ1 gating, providing evidence that the mutation effects at Q1-S182 and CaM-Y100 are due to disrupted CaM-VSD interactions. This finding also shows that CaM-VSD interactions observed in the CTD-bent conformation indeed occur during the physiological KCNQ1 activation process despite the presence of PIP2 in the membrane.
To further investigate the interactions at the CaM-VSD interface, we performed MD simulations of KCNQ1/CaM movements at physiological temperature. Statistics calculations from the multiple frames in the simulations allowed modeling of dynamic interactions among residues that are not revealed by the structure itself. We analyzed MD simulations of the human and Xenopus KCNQ1/CaM CTD-bent structures (12, 13), in the absence of PIP2. All MD simulations ran for aggregated time of at least 1 μs (fig. S5, A to C). Potential interactions between CaM and the S2-S3 linker were examined by computing the minimum distance between nonhydrogen atoms of pairwise CaM-VSD residues in each simulation frame. The MD simulations of both the human and Xenopus KCNQ1/CaM structures revealed interaction distances consistent with close interactions (~3.5 Å) between the pairs Q1-S182/CaM-Y100 and Q1-S182/CaM-N138 (fig. S6). Our results therefore showed that the CaM-VSD interactions seen in the CTD-bent structure remain intact after relaxation by MD simulations. Notably, these key residues at the CaM-VSD interface found in the simulations are consistent with the mutagenesis scan (Figs. 1 and 2). We also noticed possible interactions between residues Q1-R181/CaM-E140 and Q1-S182/CaM-N98 in the simulations (fig. S7). CaM-N98S has been previously shown in CHO cell expression system to induce a depolarizing shift in the KCNQ1 V1/2 (12). In our Xenopus oocyte expression system, however, CaM-E140 or CaM-N98 coexpressed with the WT KCNQ1 channel showed more modest effects (Fig. 2B), rendering functional detection and validation of these interactions difficult. Overall, the MD simulations generally agreed with our mutagenesis results and showed that CaM-VSD interactions seen in the CTD-bent conformation are stable in the absence of PIP2 binding at the S2-S3 linker. Together, the combined functional and simulation results suggest CaM interacts with the KCNQ1 S2-S3 linker during gating.
The CTD-straight conformation features properties specific for the AO state
The experimental results so far suggest that the CaM-VSD interface and the CTD-bent conformation exist during KCNQ1 channel gating (Figs. 1 and 2). To further correlate the CTD-bent and CTD-straight structures to functional states, and to better understand the role of CaM-VSD interactions in transitioning between the two conformations, we turned to MD simulations and network analysis. In KCNQ1 gating, the movements of the VSD S4 gating charges are sensed by nearby interacting residues and transmitted to the pore domain via a chain of residue interactions. This chain of interactions effectively constitutes VSD-pore allosteric, or coupling, pathways for the channel to open. The CaM-VSD interactions are present in the CTD-bent structure but absent in the CTD-straight structure (Fig. 1, A and B) (12, 13). To investigate how this change in the CaM-VSD interactions affects VSD-pore allostery, we used MD simulations to track all residue interactions within both structures, followed by network analysis to identify VSD-pore allosteric pathways in the two conformations (Fig. 3A). Consequently, this enabled us to contrast the specifics of how the CaM-VSD interactions influence KCNQ1 VSD-pore allostery.
Fig. 3. Network analysis of MD simulations demonstrates that the CaM-VSD interactions modulate KCNQ1 VSD-pore allosteric pathways.
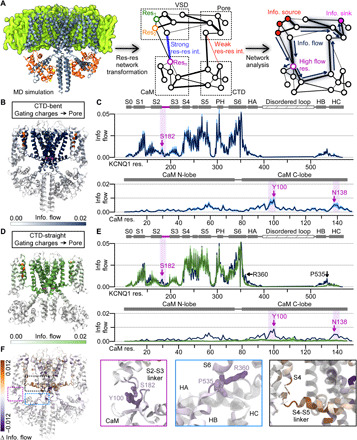
(A) Flowchart of MD simulation analysis pipeline. The KCNQ1/CaM complex cryo-EM structure was inserted into a membrane and simulated with MD in explicit solvent. The protein was transformed into a network; each node is a residue while each edge weight corresponds to interaction strength between residues. Network analysis was applied to track information flow from defined source residues (nodes) to sink residues. High–information flow nodes carry key allosteric pathways from the source to sink residues. (B) CTD-bent information flow (VSD gating charges to the pore) through each residue projected with color intensity onto the KCNQ1/CaM CTD-bent structure. Dark color, high information flow (capped at 0.02). Source residues (gating charges), orange spheres; sink residues (pore), magenta spheres. Two full subunits out of four are shown for clarity; the VSDs, HA/HB, and CaMs in the front and back subunits are omitted. (C) Information flow profiles of human KCNQ1 and CaM along residue sequence. Light purple shadings, rough position of the S2-S3 linker/CaM interface. Purple arrows, peaks in the S2-S3 linker/CaM interface. Protein-specific domains and helices are labeled above the plots. PH, pore helix. (D) Structural depiction of the information flow in the CTD-straight structure, similar to (B). (E) Information flow profile of CTD-straight conformation along the KCNQ1 sequence (green). Dark blue, same as in (C). (F) Left: Δinformation flow projected on the structure (CTD-straight versus CTD-bent information flow). Right: Detailed views of the main interfaces exhibiting large increase (brown) or decrease (purple) in information flow.
Network analysis is an approach that previously has proven effective in protein allostery identification (32–35). First, the MD trajectories of the KCNQ1/CaM complex were converted into a residue interaction network representation (Fig. 3A). In this network, each node corresponded to an individual amino acid residue within the KCNQ1/CaM complex. The weights on the connections (edges) between nodes capture interaction strength between the residues, which were defined by spatial proximity and correlated residue movements within the MD trajectories. The final network thus encodes all residues (nodes) and interactions (edges) within the KCNQ1/CaM complex. Next, we extracted allosteric pathways between the KCNQ1 VSD and the pore by measuring how information flows through the network. We defined the VSD gating charges as the “source,” from which information originates, and tracked how information flows to arrive at the “sink,” the pore-gate residue S349 (Fig. 3, A and B: orange and magenta spheres, respectively). The underlying idea is that a perturbation of residue interactions, such as those induced by the movements of S4 gating charges (the source), spreads to other residues (nodes) via diffusion along the KCNQ1/CaM complex (network) before they arrive at the pore (the sink). We use the method most commonly referred to as “current flow betweenness” (32, 36, 37) to account for all pathways from the source to the sink. Here, we refer to this method as “information flow analysis” to avoid confusion with ion channel terminologies. Critically, the ability of one residue to transmit information to another depends on the edge weight, which encodes the strength of their interaction. The flow across an edge in opposing directions are cancelled out, leaving the net positive information flow. Thus, key residues and pathways that participate in productive information transmission from the gating charges to the pore gate are systematically highlighted by strong information flow. This information flow analysis is carried by the dynamic interactions among all residues such that it is not readily shown by the structural data that only capture one or a few frames of the dynamic motion of the protein. However, we emphasize that this analysis, based on structural data, identifies allosteric pathways specific to the simulated channel state.
We applied network analysis on MD simulations of the human KCNQ1/CaM CTD-bent and CTD-straight structures (Fig. 3, B to E). To visualize the predicted spatial pattern of major VSD-pore allosteric pathways within each state, we projected the calculated information flow at each residue onto the respective structures (Fig. 3, B and D), such that darker color signifies higher flow. Figure 3 (C and E) displays the information flow along the KCNQ1 and CaM residue sequences. As expected, the highest information flow residues in both conformations were observed at the S4-S5 linker/S6 and the S4/S5 interfaces within the transmembrane domains. These two interfaces are well-studied in domain-swapped KV channels as critical for VSD-pore coupling (34, 38), including KCNQ1 (16). The network analysis thus recapitulated the importance of these transmembrane motifs in KCNQ1 VSD-pore coupling. We also performed the same analysis on simulations of the Xenopus KCNQ1/CaM CTD-bent structure. The results were close to identical to the analysis of the human KCNQ1/CaM complex (fig. S8, A to D), reinforcing the findings from the human KCNQ1/CaM simulations.
To examine how the switch from the S2-S3 linker binding CaM to binding PIP2 influences VSD-pore allosteric pathways, we calculated the difference between the CTD-bent and CTD-straight information profiles. The resulting change in information flow (Δinformation flow) is projected onto the cryo-EM structure as shown in Fig. 3F, with brown and purple indicating enhanced and reduced flow strength in the CTD-straight over the CTD-bent conformation. This revealed flow strength differences at distinct locations due to the transition from the CTD-bent to the CTD-straight conformation.
First, the CTD-straight conformation exhibited a clear reduction in information flow at the CaM/VSD interface compared to the CTD-bent conformation (Fig. 3F, purple box). The CTD-bent flow profile featured clear peaks at the S2-S3 linker and CaM (Fig. 3, B and C, purple shading), while the CTD-straight flow profile lacked the same peaks (Fig. 3, D and E, and fig. S8E). This loss of the CaM/VSD peaks is associated with the loss of CaM-VSD interactions, consistent with the notion that the S2-S3 linker/CaM peaks in the CTD-bent flow profile are attributed to CaM-VSD interactions. Notably, the residues corresponding to the S2-S3 linker/CaM flow peaks were Q1-S182, Q1-R181, CaM-N98, CaM-Y100, and CaM-E140 (Fig. 3C), generally consistent with our mutational scans (Figs. 1 and 2). The S2-S3 linker peaks in the CTD-bent conformation are weaker compared to those at the transmembrane interfaces, suggesting that the coupling pathways through the CaM-VSD interface are less critical than those via the transmembrane interfaces. Together, these results show that the CaM-VSD interactions play a role, albeit small, in the VSD-pore allostery pathways in the CTD-bent conformation. This pathway is lost upon the transition to the CTD-straight conformation.
Beyond the CaM-VSD interface, the CTD also exhibited reduced flow strengths from the CTD-bent conformation to the CTD-straight conformation. The CTD region with the most reduced flow strengths involved the S6-HA and HB-HC linkers (Fig. 3F, blue box). The concurrent flow reductions at the CaM/VSD interface and the CTD hint that these two regions might be linked allosterically and work together to affect KCNQ1 pore opening. This phenomenon will be further explored later in this study.
In contrast to the reduced flow strengths at the CaM/VSD interface, the CTD-straight conformation interestingly featured enhanced information flow in C terminus of S4 and N terminus of the S4-S5 linker (Fig. 3F, black box). This result correlates with prior functional and simulation studies, which further supports the suggestion that the CTD-straight structure corresponds to the AO state. We previously found that KCNQ1 VSD activation triggers a two-stage VSD-pore coupling mechanism, such that two sets of VSD-pore interactions induce channel opening with the VSD movements to the intermediate state and the activated state (16). In particular, the VSD-pore coupling interactions specific for the AO state involve the C terminus of S4, the N terminus of the S4-S5 linker, and part of S5 and S6 (16). These critical coupling motifs for the AO state are closely correlated with the locations featuring enhanced information flow in the CTD-straight conformation (Fig. 3F, black box). This suggests that the CTD-straight conformation more efficiently transmits allosteric signal through structural regions known to be important for VSD-pore coupling of the AO state, providing another line of evidence that the CTD-straight conformation corresponds to the functional AO state. The flow strength enhancement at these AO coupling motifs is accompanied by the loss of CaM-VSD interface, suggesting that the loss of CaM-VSD interactions reinforces AO-state coupling interactions.
Together, the comparative network analysis provided two insights on the role of CaM-VSD interactions in KCNQ1 VSD-pore coupling. First, the presence of CaM-VSD interface in the CTD-bent state provides a minor VSD-pore coupling pathway (Fig. 3, B to E). Second, the loss of CaM-VSD interactions in the CTD-straight state enhances AO-state specific coupling interactions in the channel transmembrane domains (Fig. 3F). This result suggests that the disruption of CaM-VSD interactions play a role in reinforcing key AO state coupling interactions during the transition from the CTD-bent to CTD-straight conformation, thereby promoting channel occupancy of the AO state.
CaM-VSD interactions control steady state occupancy of the AO state
The results so far partially answered the questions that we raised at the beginning of Results. First, the CTD-bent structure corresponds to a functional state in the gating pathway, as we detected CaM-VSD interactions during activation (Figs. 1 and 2). Second, combined functional and simulation data provide evidence that the CTD-straight structure reflects the AO state. Disruption of the CaM-VSD interface facilitates occupancy and VSD-pore coupling of the AO state (Figs.1 to 3). This indicates that the AO state conformation lacks CaM-VSD interactions, consistent with the CTD-straight structure. Yet, the CTD-bent and CTD-straight structures both feature VSDs at the activated state with seemingly mutually exclusive CaM-VSD and PIP2-VSD interactions. How can both conformations occur during gating? A likely explanation is that CaM and PIP2 compete for binding to the S2-S3 linker in a state-dependent manner, with the exchange in binding partner facilitating transition between the CTD-bent and CTD-straight conformations.
Then, when does the S2-S3 linker switch binding partner during gating? The exclusive CaM-VSD and PIP2-VSD interactions seen in the two structures (12, 13) indicate that the activated VSD binds one partner at steady-state. We therefore hypothesize that the CTD-bent structure reflects a transition state before PIP2 binding the S2-S3 linker, but after the VSD has just moved to the fully activated state. Although this transition state is likely transient during the gating process, the CTD-bent structure captured this state because it was determined at a depolarized voltage in the absence of PIP2. In this hypothesis, CaM-VSD interactions are favored when VSD adopts the resting or intermediate state, and VSD movement to the activated state triggers the S2-S3 linker to switch from binding CaM to binding PIP2 (fig. S1D).
To test this hypothesis, we probed how disrupting CaM-VSD and PIP2-VSD interactions affect distinct aspects of KCNQ1 gating mechanism using voltage-clamp fluorometry (VCF). The KCNQ1 VSDs are fluorescently labeled in VCF to enable simultaneous monitoring of VSD movements and pore opening during activation. We applied VCF on the Q1-R181E and Q1-S182K mutants, which disrupt the S2-S3 linker interaction with PIP2 and CaM, respectively. Figure 4A illustrates VCF recordings from KCNQ1 pseudoWT (psWT) channels, which carries three mutations (C214A/G219C/C331A, hereby denoted Q1*) to enable fluorescent labeling specifically to G219C. This Q1* psWT channel construct has been extensively validated in prior studies across many laboratories as the WT control for KCNQ1 VCF studies (8, 15–17, 19). The Q1*-psWT channels featured robust changes in fluorescence signal upon membrane depolarization corresponding to VSD activation, which tracks closely with ionic currents correlating with pore opening (Fig. 4A, blue and black exemplars). Next, we applied VCF to the Q1*-R181E mutant to assay how disrupted PIP2 binding to the S2-S3 linker affects VSD activation. We found a significant depolarizing shift in the voltage dependence of VSD activation of Q1*-R181E [fluorescence-voltage (F-V); fig. S3, D and E, F1 curve]. This result draws a connection between PIP2 binding to the S2-S3 linker and VSD activation movement. If PIP2 binding is state dependent for when the VSD is activated, as suggested by the CTD-straight structure (13), then PIP2 binding also facilitates VSD activation. The disruption of PIP2-VSD interactions by the Q1-R181E mutation can thus induce depolarizing shifts in VSD activation. This state-dependent PIP2-VSD interaction is consistent with the idea that PIP2 competes with CaM at the S2-S3 linker upon VSD movement into the activated state during gating, leading to a state-dependent loss of CaM-VSD interactions.
Fig. 4. CaM-VSD interactions control AO state occupancy.
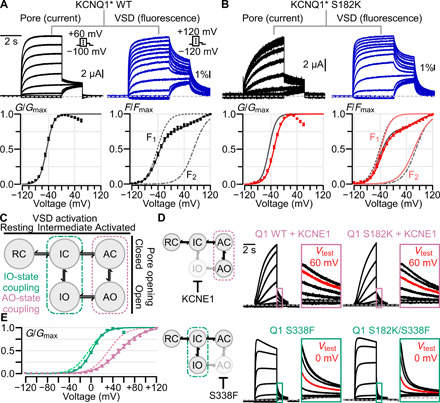
(A) VCF experiments using KCNQ1* psWT. Left and right columns: Exemplar ionic current/VSD fluorescence signal measurements (voltage protocols in inset) and average G-V/F-V relationships, respectively. The F-V relationship (solid black curve) is fitted with a double Boltzmann equation (F1 and F2, dotted black curves), see Materials and Methods. (B) Same as (A), but for KCNQ1* S182K mutant channel. In G-V and F-V plots: black, psWT; red, S182K. (C) KCNQ1 gating model with two open states, IO and AO. The KCNQ1 VSD transitions into the intermediate and activated conformations engage distinct sets of VSD-pore coupling interactions (green and violet boxes) to trigger pore opening. (D) Left: Cartoon schematic illustrating the effect of KCNE1 coexpression (top) and Q1-S338F mutation (bottom) to ablate the IO and AO states respectively to leave KCNQ1 to conduct only at one open state. Right: Current exemplars for Q1-WT and mutant channels designed to probe whether Q1-S182K mutation specifically affects IO or AO state. Each box shows enlarged tail currents, with the red trace indicating the tail current associated with the labeled test voltage, which shows the shift of voltage dependence when comparing the WT and mutant. Voltage protocol same as in (A), except for exemplar currents with KCNE1 coexpression test pulses range from −120 to +100 mV. (E) Average G-V curve and Boltzmann fits for Q1-WT + KCNE1 (dotted pink, n = 5), Q1-S182K + KCNE1 (solid pink, n = 7), Q1-S338F (dotted green, n = 6), and Q1-S182K/S338F (solid green, n = 11).
We next studied the Q1*-S182K mutant to measure how disruption of CaM-VSD interactions affects VSD activation. VCF recordings of the Q1*-S182K channels showed a depolarizing shift in G-V and slowdown in current activation kinetics (Fig. 4B). However, little change in the voltage dependence or kinetics of VSD activation was observed by the mutation (Fig. 4B and table S3). This finding indicates that Q1-S182K did not directly modulate VSD activation. Therefore, the mutation changed pore openings by altering either the VSD-pore coupling or the pore directly. Since the mutation is in the S2-S3 linker, spatially far from the pore domain, it is likely that the mutation altered VSD-pore coupling. This result is consistent with the suggestion that the disruption of CaM-VSD interactions promotes the AO state occupancy by enhancing VSD-pore coupling pathways specifically important for the AO state (Fig. 3F) (16).
We next examined further if CaM-VSD interactions indeed control AO state occupancy. We thus disrupted the CaM-VSD interactions with Q1-S182K mutation in modified KCNQ1 channels that selectively open only to the AO or IO state. Our previous work showed that coexpression of the auxiliary subunit KCNE1 with KCNQ1 suppresses the IO state (16, 18, 19), forcing KCNQ1 to conduct only in the AO state (Fig. 4D, top). Conversely, the KCNQ1 mutation S338F selectively abolishes the AO state by disabling the VSD-pore coupling when the VSD is at the activated state, thus leaving the channel to only conduct at the IO state (Fig. 4D, bottom) (16, 20, 21). We first tested how disrupting CaM-VSD interactions affects the AO state by KCNE1 coexpression with Q1-S182K channels. The G-V relationships (Fig. 4, D and E, and table S3) showed that Q1-S182K + KCNE1 maintained a 20-mV right shift in V1/2 compared to Q1-WT + KCNE1, consistent with the shift observed between Q1-S182K and Q1-WT channels without KCNE1 (Fig. 1E). These data provide another line of evidence that the CaM-VSD interactions modulate AO state occupancy. This result also indicates that the S182K mutation may affect intrinsic properties, such as voltage dependence of the closed/AO state equilibrium. We next probed whether CaM-VSD interactions modulate the IO state with the Q1-S182K/S338F double mutant. Distinct from the KCNE1 coexpression experiments, Q1-S338F strongly reduced the depolarizing shift in the V1/2 induced by Q1-S182K (Fig. 4, D and E, and table S3). Introduction of the Q1-S182K mutation also failed to cause a slowdown of Q1-S338F current activation kinetics. These results indicate that CaM-VSD interactions exert little modulation on the properties of the IO state. This lack of IO state modulation is also consistent with the lack of S182K effect on the KCNQ1 fast-kinetic current component (Fig. 1H), which approximates the IO state. Together, these results further confirm that the CaM-VSD interactions modulates the AO state occupancy. Along with the finding of state-dependent PIP2-VSD interaction, all these results suggest that the loss of CaM-VSD interactions during KCNQ1 gating, as driven by PIP2 competition, destabilizes the CTD-bent conformation to stabilize the AO state. Furthermore, these findings are consistent with the hypothesis that the CTD-straight structure corresponds to the AO state, while the CTD-bent structure with the activated VSD reflects a short-lived transition state.
A hub in the KCNQ1 CTD cooperates with the CaM-VSD interactions to control AO state occupancy
Does the CaM-VSD interactions work alone or with other structural motifs to control AO state occupancy? Our MD network analysis identified a reduction in information flow strength in the CTD that accompanied the loss of CaM-VSD interactions (Fig. 3F), hinting that the CTD might be allosterically downstream of CaM-VSD interactions. Moreover, the cryo-EM structures showed that the loss of CaM-VSD interactions was accompanied by major conformational rearrangements in the CTD (Fig. 1, A and B). Specifically, the “RQKH” motif within the KCNQ1 S6-HA linker was shown to undergo a loop-to-helix transformation from the CTD-bent to the CTD-straight conformation (13). These data together hint that the CaM-VSD interactions may work together with the CTD to affect channel transition into the AO state. We thus further pursued this hypothesis and turned back to MD network analysis to systematically identify key residues within the CTD which work together with the CaM-VSD interface.
To identify CTD structural motifs that might be allosterically downstream of the CaM-VSD interaction, we performed an alternate network analysis on the simulations of the CTD-bent conformation. The information flow in the CTD-bent conformation was computed using all residues within the CaM C-lobe, as opposed to the S4 gating charges, as source residues (Fig. 5A). As described previously, the information profile calculated with the S4 gating charges as the source corresponds to VSD-pore allosteric pathways in the CTD-bent structure. By contrast, the profile calculated with the CaM C-lobe as the source revealed how perturbation in CaM is propagated to the pore domain residue Q1-S349. Thus, residues that are specifically sensitive to changes in CaM, such as the loss of CaM-VSD interactions, should feature higher information flow when CaM is defined as the source versus the gating charges. The Δinformation flow between the CaM C-lobe and gating charge information profiles is projected onto the CTD-bent structure in Fig. 5B. When using CaM C-lobe as the source, there was a general reduction in flow strengths through the transmembrane domains but a general enhancement of flow strengths through the CTD (Fig. 5, A and B, and fig. S9A). This indicates that perturbation in CaM is preferentially propagated to the pore through the CTD, consistent with the idea that loss of CaM-VSD interactions might work with the CTD to affect the pore. The CTD residues featuring the greatest flow enhancement included Q1-R360 (S6-HA linker) and Q1-P535 (HB-HC linker) (fig. S9A, green shading). Similar results were obtained with the same analysis on the Xenopus KCNQ1/CaM complex (fig. S9, B and C), further validating the conserved importance of Q1-R360 and Q1-P535. Moreover, in our previous Δinformation flow comparison between the CTD-bent to CTD-straight conformation, these same two CTD residues (Q1-R360 and Q1-P535) exhibited simultaneous reduction in information flow with the CaM/VSD interface (Fig. 3F, black box). Together, these results suggest that residues Q1-R360 and Q1-P535 are allosterically coupled to the CaM-VSD interactions. Consequently, these CTD residues may be important in conjunction with the CaM-VSD interactions to affect occupancy of the AO state.
Fig. 5. A hub in the KCNQ1 CTD cooperates with the CaM-VSD interactions to control AO state occupancy.
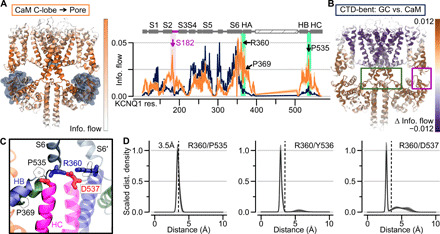
(A) Left: Information flow projection onto the CTD-bent structure, capped at 0.02. For clarity, VSDs, HA/HB, and CaMs in the front and back subunits are omitted. Right: Orange, information flow profile along KCNQ1 sequence (CaM C-lobe used as the source) in the CTD-bent structure; dark blue, information flow profile with gating charges as source. Purple shading, information flow peak in the S2-S3 linker/CaM interface. Green shading, peak between the HB/HC and HA/S6 loops. (B) Δinformation flow projected on the CTD-bent structure (CaM C-lobe versus the gating charges as the source). Dark violet residues, decreased information flow as a result of using CaM C-lobe as source; brown, increased information flow. (C) Structural depiction of residues in the CTD hub. Green ribbons, HA; blue ribbons, HB; pink ribbons, HC; gray ribbons, S6. S6′ is from a neighboring subunit. Residues depicted with sticks are colored according to residue type: positively (negatively) charged, blue (red); hydrophobic, white. (D) Scaled distance density of interacting residues in the CTD hub.
Does the spatial arrangement of Q1-R360 (S6-HA linker) and Q1-P535 (HB-HC linker) support the hypothesis that these CTD residues are allosterically downstream of the CaM-VSD interactions? Although these residues are far apart in terms of primary sequence, we found a cluster of residues, including Q1-R360 and Q1-P535, which formed a structural motif that pinches the S6-HA and HB-HC linkers together in the CTD-bent simulations (Fig. 5C). These interactions included a close interaction between Q1-P535 and Q1-R360, a salt-bridge between Q1-D537 and Q1-R360, as well as hydrogen bonding between Q1-Y536 and Q1-R360 (Fig. 5D). Notably, these residues and interactions are conserved between the human and Xenopus KCNQ1 channels (figs. S9, D and E, and S10). Q1-R360 interacts with Q1-Y536 and Q1-D537 of the neighboring subunit (i.e., intersubunit) in the CTD-bent structure, but these interactions switch to intrasubunit in the CTD-straight structure. The residue Q1-R360 further corresponds to the arginine in the RQKH motif that undergoes a loop-to-helix transition between the CTD-bent and CTD-straight structures (13). Overall, the key residues found by network analysis are spatially organized in a critical cluster, or hub (“CTD hub”), which may facilitate CTD conformational rearrangements to control AO state occupancy and VSD-pore coupling together with the CaM-VSD interface.
Functional validation that the CTD hub cooperates with CaM-VSD interactions in KCNQ1 gating
To test the prediction that the CTD hub is allosterically downstream of the CaM-VSD interactions, we performed functional electrophysiology experiments. Specifically, we examined the two key CTD residues identified by network analysis, Q1-R360 and Q1-P535. We additionally tested another HA residue, Q1-P369, which showed a slightly increased peak of information flow when CaM C-lobe was used as the source in the CTD-bent conformation (Fig. 5A). Notably, P369 is well conserved in the KCNQ family and mutation at the analogous position in KCNQ5 (P369R) is linked to intellectual disability (39). If the CaM-VSD interactions and the CTD hub are allosterically coupled to control channel occupancy of the AO state, then double mutations at the S2-S3 linker and key residues in the CTD should feature nonadditive changes to shift in V1/2, and likely nonadditive effects on current kinetics. We thus characterized S2-S3 linker S182K and CTD double mutants to test this hypothesis (Fig. 6A).
Fig. 6. Functional validation that the CTD cooperates with CaM-VSD interactions in KCNQ1 gating.
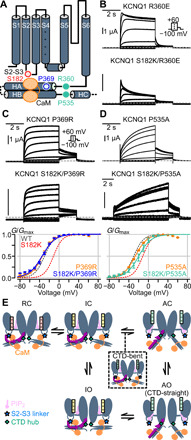
(A) Cartoon depiction of KCNQ1 channel with key CTD residues (blue and green circles) predicted to be allosterically downstream of the CaM-VSD interaction (red circle). (B) Current exemplars for Q1-R360E and Q1-S182K/R360E mutants. Voltage protocol in inset. (C and D) Top and middle: Current exemplars for mutants involving the S2-S3 linker (S182K), the proximal HA (P369R), and the HB-HC loop (P535A). Bottom: Average G-V relationships calculated for the respective mutants. Dashed lines are G-V fits for Q1-WT (gray) and Q1-S182K (red). N = 7 (Q1-P369R), 5 (Q1-S182K/P369R), 8 (Q1-P535A), and 3 (Q1-S182K/P535A). Q1-P369R versus WT ΔV1/2 = −4.77 ± 1.24 mV, P < 0.01. Q1-P535A versus WT ΔV1/2 = 0.75 ± 0.79 mV, P = 0.35. P values are calculated with t test assuming unequal variance. (E) KCNQ1 voltage-dependent gating model integrating CaM and PIP2. Color designations: KCNQ1 channel (dark gray), KCNQ1 S2-S3 linker (light blue star), KCNQ1 CTD hub (green diamond), CaM (orange), PIP2 (violet), and S4 in the VSD is colored red, yellow, and green for resting, intermediate, and activated states, respectively. HC and HD are not drawn for clarity. PIP2 interaction with the KCNQ1 S2-S3 linker is dependent on the VSD state. PIP2 does not interact with the S2-S3 linker when the VSD is in the resting (R) or intermediate (I) states, thus CaM favorably interacts with the S2-S3 linker. Upon VSD transition into the activated (A) state, PIP2 favors interaction with the S2-S3 linker and competes with CaM. This competition disengages CaM from the S2-S3 linker and allows the CTD to undergo large conformational rearrangement pivoted at the CTD hub (green diamond) to induce the AO state. The CTD-straight structure corresponds to the AO state, and the CTD-bent structure represents a short-lived transition state (black dashed box).
We first probed the CTD S6-HA linker single mutation Q1-R360E, which yielded robust ionic currents with slight inactivation (Fig. 6B), consistent with prior studies (8). In contrast to the slowly activating Q1-S182K single mutant current, the double mutant Q1-S182K/R360E exhibited a rapid inactivation current phenotype (Fig. 6B). We did not calculate the V1/2 of Q1-S182K/R360E double mutant due to the strong current inactivation. Nevertheless, the marked switch in current activation kinetics from the single to double mutant channel is consistent with allosteric interaction between S182 and R360. Next, we made the HA Q1-P369R mutant, corresponding to the analogous KCNQ5 pathogenic variant (39), and the Q1-P369A mutant to remove its side-chain properties. As no disease variant is found to link to Q1-P535, we created the Q1-P535A mutation. The Q1-P369R mutant (HA) featured rapid current activation kinetics and a hyperpolarized V1/2 compared to WT (Fig. 6C), with Q1-P369A showing similar phenotype (fig. S11, A and B). These results suggest that the mutational effect at P369 is likely attributed to the loss of proline. On the other hand, the Q1-P535A mutant (HB-HC linker) exhibited a markedly slowed activation kinetics with a similar V1/2 compared to WT (Fig. 6D). Together, these findings indicate that both CTD residues Q1-P369 and Q1-P535 are important for KCNQ1 gating. We then probed whether these two residues are allosterically linked to the CaM-VSD interface with Q1-S182K/P389R and Q1-S182K/P535A. Both double mutants featured hyperpolarized V1/2 compared to Q1-S182K (Fig. 6, C and D), reversing the V1/2 shift induced by the Q1-S182K mutation and demonstrating nonlinear shifts in V1/2. Moreover, the double mutant Q1-S182K/P369R featured a rapidly activating current kinetics, completely reversing the slowly activating phenotype of the Q1-S182K single mutant. Together, we found residues in the KCNQ1 CTD (P369 and P535) that interfered with the ability of the CaM-VSD interface to affect AO state occupancy. To further test whether the position P369 is specifically critical, we probed the Q1-A370W and Q1-S182K/A370W mutants. We found that Q1-A370W mutation was unable to interfere with the S182K mutation, with Q1-A370W resembling WT channel and Q1-A370W/S182K featuring a phenotype similar to the Q1-S182K single mutant (fig. S11, C and D). This finding suggests that specific residues in the CTD, such as Q1-P369 and residues at the CTD hub, are important to propagate changes at the CaM-VSD interface to the pore. Together, these results provide experimental evidence that the CaM-VSD interface works in concert with the CTD hub. As mentioned, these residues are dispersed in terms of primary sequence (S6-HA linker, HA, and HB-HC linker). This indicates that a switch in CaM-VSD interactions extensively uses the tertiary structure of the CTD to control KCNQ1 opening at the AO state.
DISCUSSION
With increasing number of resolved atomic structures of full-length ion channels, correlating these structures to their gating states is becoming a widespread question. In the KCNQ1/CaM structure determined without PIP2, which we called the CTD-bent conformation, the VSD is activated and the pore is closed (12, 13). The S6-HA linker bends to bring CaM near the VSD and enable CaM C-lobe interaction with the S2-S3 linker (Fig. 1A) (12, 13, 18). The KCNQ1/CaM structure solved with PIP2, which we termed CTD-straight conformation, features activated VSD and dilated pore (13, 18). Notably, the S6-HA linker straightened to rotate CaM away from the VSD, and the CaM-VSD interactions are replaced by S2-S3 linker interaction with PIP2 (Fig. 1B) (13). Functionally, KCNQ1 gating involves a two-step VSD activation, from the resting state to intermediate state and lastly to the activated state (15–22). Each VSD activation step engages unique VSD-pore coupling interactions and triggers pore opening when the VSD adopts both the intermediate and activated states, leading to the IO and AO open states (Fig. 1C) (16, 18–21). In this study, we provided functional and simulation analyses to connect the KCNQ1/CaM cryo-EM structures (12, 13) to the KCNQ1 voltage-dependent activation pathway.
First, our results provide functional evidence that the CTD-straight conformation corresponds to the AO state. MD network analysis revealed that VSD-pore coupling pathways specific to the AO state are emphasized in the CTD-straight structure (Fig. 3). Moreover, mutational disruption of CaM-VSD interactions, which is expected to destabilize the CTD-bent structure and promote the CTD-straight conformation, exhibited specific enhancement of AO state occupancy (Figs. 1, 2, and 4). These results substantiate the suggestion that the CTD-straight conformation reflects the AO state. Second, our results provide answers to whether the CTD-bent structure occurs during KCNQ1 gating, despite structural determination under nonphysiological conditions. Our results show that disruption of the S2-S3 linker/CaM interface affects KCNQ1 gating and alters current kinetics (Figs. 1, 2, and 4). Moreover, double mutant experiments revealed an interaction between the S2-S3 linker and CaM during gating (Fig. 2). These findings show that the CaM-VSD interface is functionally important during gating and suggest that the CTD-bent conformation exists during activation. Third, our results delineate how CaM and PIP2 affect transition between the CTD-bent and CTD-straight conformations. We find a direct relationship between VSD movement and PIP2 binding to the S2-S3 linker (fig. S3, D and E). By contrast, disrupting CaM-VSD interactions does not affect VSD movement (Fig. 4, A and B) but enhances AO state occupancy and VSD-pore coupling (Figs. 3F and 4, C to E). Together, these results suggest that PIP2 and CaM competition for the S2-S3 linker during gating modulates AO state occupancy. These findings further resolve the question of how the S2-S3 linker in the two cryo-EM structures interacts exclusively with either CaM or PIP2 (12, 13). Specifically, the results presented here are consistent with the idea that the KCNQ1 CTD-bent conformation represents a short-lived transition state, which is only stabilized in the absence of PIP2.
In this study, we used MD simulations and network analysis to map a critical CTD hub, which is allosterically downstream of the CaM-VSD interactions. Together, the two interfaces affect channel occupancy of the AO state (Figs. 3, 5, and 6). The CTD hub not only contains the S6-HA “RQKH” motif thought to be important for CTD rearrangement in the cryo-EM structural studies (13), but also HB-HC residues. We note that although the cryo-EM structures clearly show a large conformational change at the CaM-VSD interface, the identification of the CTD hub and its role in converting the channel to the CTD-straight conformation with an allosteric link to the CaM-VSD interactions are nonobvious from the structures. The MD network analysis approach thus proved to be a powerful tool to scrutinize structural data and guide functional experiments.
On the basis of the above conclusions, we propose a voltage-dependent KCNQ1 gating scheme integrating CaM and PIP2 (Fig. 6E), in which the VSD conformation controls CaM and PIP2 binding to the S2-S3 linker, providing conceptual improvement and structural basis to the transitions within the gating model. We emphasize that this gating model represents a starting point that accounts for all currently available structural and functional data. Although the model could quantitatively simulate the currents of KCNQ1 channels (19–21), more work is required to validate our proposed gating scheme. When the VSD adopts the resting or the intermediate state, S2-S3 linker binds CaM, while PIP2 is present in the membrane (Fig. 6E: RC, IC, and IO states). The CTD accordingly adopts the conformation seen in the CTD-bent structure. When the VSD transitions into the activated state, the S2-S3 linker starts interacting with PIP2, which disengages CaM. The CTD-bent structure with the activated VSDs reflects a transient state during this step (Fig. 6E). The loss of CaM-VSD interactions then works together with the CTD hub (Fig 6E, green diamonds) to trigger transition into CTD-straight conformation and subsequent pore domain opening into the AO state.
PIP2 plays a vital role in our model by interaction with the S2-S3 linker to facilitate channel transition into the AO state. This is consistent with previous studies suggesting that PIP2 binds the S2-S3 linker (8, 13, 40–42). However, the S2-S3 linker may not be the only PIP2 binding site. As we showed in this study, PIP2 binding to the S2-S3 linker is VSD state dependent (fig. S3, D and E), indicating that PIP2 may bind elsewhere when the VSD is in the resting or intermediate state. Previous studies found that PIP2 is required for KCNQ1 VSD-pore coupling in both the IO and AO states, which we attributed to PIP2 binding to the S4-S5 linker and the bottom of S5/S6 (8, 40). These regions have also been suggested to bind PIP2 in other KCNQ channels (42). The S4-S5 linker, S5, and S6 may form a second PIP2 binding site specifically important for VSD-pore coupling in both the IO and AO states. This idea is further supported by the recent discovery of a PIP2-mimetic compound, which enhances KCNQ1 VSD-pore coupling through occupancy of this second site (43). This second PIP2-binding site may be important for KCNQ1 VSD-pore coupling, while the S2-S3 linker PIP2 binding site is state dependent and modulates channel transition into the AO state. Characterization of other PIP2 binding sites in KCNQ1 will be important in future studies.
With potential alternate PIP2 binding sites in mind, we note the caveat that the pore in the CTD-straight conformation, while dilated, is not fully open for current conduction (13). Thus, although the structure may capture the major features of the AO state, such as the loss of CaM-VSD interactions, the structure may reflect a conformation that is not exactly the AO state. The fact that the pore is not fully open could be because PIP2 in nanodiscs failed to occupy the second binding site crucial for VSD-pore coupling. A second reason could be because the structure was solved without ATP, which is required for KCNQ1 pore opening (44).
In this study, we did not explicitly control for intracellular Ca2+ levels. Several structural studies invariably revealed that the EF-hand motif 3 (EF3) within CaM C-lobe is uncalcified over wide-ranging intracellular Ca2+ levels (12, 13, 30). Given that the CaM-VSD interactions mostly involved residues near EF3, we expect that CaM C-lobe adopted the same calcification state as seen in the structures with the oocytes intracellular Ca2+ levels in our experiments. How changing Ca2+ levels may affect the CaM-VSD interactions and KCNQ1 gating remains a subject for future studies. We further note that we did not suppress the endogenous CaM in experiments involving mutant CaM coexpression, similar to other studies of CaM and KCNQ1 (5, 30). The endogenous CaM WT may thus compete with mutant CaM for KCNQ1, leading to potential differences in the observed V1/2 shift magnitude in our study compared to other expression systems.
CaM has long been known to modulate voltage-gated ion channels by binding to their intracellular domains (5, 6, 24, 26, 28, 30, 45). Yet, understanding how CaM modulates gating in context of the channel transmembrane domains has remained a challenge. Here, we show that the CaM-VSD interactions act as a switch to facilitate a CTD conformational rearrangement. This conformational change represents a notable difference, which may contribute to the unique IO and AO gating properties of KCNQ1. Although KCNQ1 is known to conduct predominantly at the AO state in the heart with KCNE1 association, it instead conducts primarily at the IO state in epithelial cells with KCNE3 association (18, 19). Modulation of KCNQ1 occupancy of the AO state thus represents a critical aspect of normal physiology. Here, we provide a mechanism for how CaM interacts with unexpected channel locations, such as the S2-S3 linker and the CTD hub, to control the AO state. In KCNQ1, LQTS-associated mutations such as Q1-R360G (11) and CaM-F142L (46) may cause channel dysfunction by disrupting the CaM-VSD switching mechanism. Arrhythmia-associated CaM mutants are known to affect other key cardiac channels, such as CaV1.2 (47), suggesting that some CaM mutations may lead to complex multichannel arrhythmogenic mechanism.
MATERIALS AND METHODS
Site-directed mutagenesis
Point mutations were made in KCNQ1 channel and CaM using overlap extension and high-fidelity polymerase chain reaction. DNA sequencing confirmed the presence of all mutants made. Mutant complementary RNA (cRNA) was made with the mMessage T7 polymerase kit (Applied Biosystems–Thermo Fisher Scientific).
Channel expression in Xenopus oocytes
Stage V or VI oocytes were obtained from Xenopus laevis by laparotomy in accordance with the protocol approved by the Washington University Animal Studies Committee (protocol #20190030). Oocytes were digested by collagenase (0.5 to 0.7 mg/ml, Sigma-Aldrich, St. Louis, MO) and injected with channel cRNAs (Drummond Nanoject, Broomall). Each oocyte was injected with cRNAs (9.2 ng) of WT or mutant KCNQ1. For experiments with KCNE1 or CaM coexpression, KCNE1 and CaM cRNAs were coinjected at 3:1 (KCNQ1:KCNE1) and 1:1 (KCNQ1:CaM) mass ratio, respectively. All VCF and some KCNQ1 constructs were injected with double the mass of cRNA to boost channel expression. Injected oocytes were incubated in ND96 solution [96 mM NaCl, 2 mM KCl, 1.8 mM CaCl2, 1 mM MgCl2, 5 mM Hepes, 2.5 mM CH3COCO2Na, and 1:100 penicillin-streptomycin (pH 7.6)] at 18°C for 2 to 6 days before recording.
TEVC and VCF
Microelectrodes were made with thin wall borosilicate glass (B150-117-10, Sutter Instrument, Novato, CA) by a micropipette puller (P-97 or P-1000, Sutter Instrument, Novato, CA). The pipette resistance was 0.5 to 3 megohm when filled with 3 M KCl solution and submerged in ND96 solution. TEVC and VCF experiments were recorded in ND96 solutions at room temperature. Whole-cell currents were recorded with a CA-1B amplifier (Dagan, Minneapolis, MN) driven by Patchmaster (HEKA, Holliston, MA) software. Current recordings were sampled at 1 kHz and low-pass–filtered at 2 kHz. For VCF experiments, the KCNQ1 channels were labeled by incubating oocytes expressing KCNQ1 channels for 1 hour on ice in 10 μM Alexa Fluor 488 C5-maleimide (Molecular Probes, Eugene, OR) in high K+ solution [98 mM KCl, 1.8 mM CaCl2, and 5 mM Hepes (pH 7.6)]. The oocytes were washed and incubated in ND96 after labeling. VCF was performed by recording currents with the same instruments as TEVC. The fluorescence signals were measured by a Pin20A photodiode (OSI Optoelectronics, Hawthorne, CA) and an EPC10 patch clamp amplifier (HEKA, Holliston, MA) sampled at 1 kHz and filtered at 200 Hz. The EPC10 amplifier was controlled by the CA-1B amplifier to ensure simultaneous current and fluorescence measurements. All chemicals were obtained from Sigma-Aldrich (St. Louis, MO).
Electrophysiology data analysis
Data were analyzed with MATLAB (MathWorks, MA) software. The G-V relationship calculation: The instantaneous tail currents following test pulses were normalized to the maximum current. The G-V relationship was fitted with a single Boltzmann equation in the form of
where V is the test pulse voltage, V1/2 is the half-activation voltage, and Vs controls the steepness of the Boltzmann equation. Vs is related to RT/zF, where R is the gas constant, T is the temperature, z is the equivalent valence, and F is the Faraday constant. The F-V relationship calculation: Linear photobleaching correction was applied to each fluorescence trace by subtracting the line obtained by linear fitting of the 2-s fluorescence signal before the start of the test pulse. The F-V relationship was calculated using fluorescence signals at the end of the test pulses and normalized to the maximum fluorescence signal change. The F-V relationship was then fitted with a double Boltzmann distribution in the form of
where Vhalf,1 and Vhalf,2 correspond to the half-activation voltages for F1 and F2, respectively. Other variables are similarly defined as in the G-V relationship fits. One-way analysis of variance (ANOVA) followed by Tukey’s post hoc test was used to compare V1/2 values from multiple mutants. All other statistical significance calculations were performed with Student’s t test.
Current activation kinetics were quantified by fitting the raw 4-s test-pulse currents to a biexponential function in the form of
Each current trace was baseline-corrected using the mean current at the holding potential for each trace. The fast current component was first estimated by fitting the first 0.5 s of the current. A second overall fit was then applied to the entire 4-s test pulse current, with the fast time constant constrained within ±25% of the first fit. All normalized currents shown were normalized by the fitted steady-state amplitude (Afast + Aslow + Coffset). The fraction of total current carried by the fast component was calculated with the equation Afast/(Afast + Aslow).
MD simulations
The human CTD-bent structure [Protein Data Bank (PDB) ID 6UZZ] and the CTD-straight without KCNE3 (PDB ID 6V01) (13) were simulated in equilibrium with three replicas amounting up to ~1 μs per system. Similar to the 1-μs trajectory of the Xenopus structure from (48), we used a 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) membrane, as well as explicit solvent neutralized with 0.1 M KCl. The bound PIP2 molecules were kept in the CTD-straight system. Because the PIP2 binding site is unknown in the CTD-bent conformation, this system was simulated without PIP2, similar to the prior 1-μs trajectory from (48). Missing loops were modeled with MODELLER and the discrete optimized protein energy (DOPE) score (49). The systems were built with CHARMM-GUI, and equilibration was done according to the standard six-step CHARMM-GUI procedure (50), extending the sixth step to 3 ns.
The three simulations of the Xenopus structure (12) were launched from the minimized and equilibrated system, i.e., the first frame, of the already published 1-μs production trajectory (48), by generating new velocities according to a Maxwell distribution. Each new simulation was equilibrated for 375 ps while reducing restraints on the protein atoms, according to the standard six-step CHARMM-GUI procedure (50). The total simulation time of the three trajectories was 1.2 μs.
Each system was simulated in an NPT ensemble (constant number of particles, pressure and temperature) with a 2-fs time step. Temperature was held constant at 303.15 K by the Nosé-Hoover thermostat (51) during production and the V-rescale thermostat (52) during equilibration. Pressure was maintained at 1 bar with the semi-isotropic Parrinello-Rahman barostat (53) during production and the Berendsen barostat (54) during equilibration. Short-ranged electrostatics were computed with a 1.2-nm cutoff, such that the switching function started at 1.0 nm, while long-ranged electrostatics were modeled with particle mesh Ewald (PME) (55). Moreover, CHARMM36 force field (56) and GROMACS 2018 (57) were used, water was modeled with the 3-point rigid water model TIP3P, and bonds involving hydrogens were constrained with LINCS (58). The simulation parameters of the 1-μs-long trajectory of the Xenopus structure (12) are previously described in (48). This was simulated in equilibrium with GROMACS 2016 (57).
The root mean square deviation along each trajectory was calculated with MDTraj 1.9.3 (59). Structural visualization was done with Visual Molecular Dynamics (VMD) (60).
Creating a residue-residue network
Semibinary contact map
Because continuous contact maps yield more reliable networks than binary (32), we used a continuous contact map, such that distances below c = 0.45 nm were weighted by 1, and larger distances were down-weighted continuously until a cutoff, dcut = 0.8 nm, where the interactions were deemed insignificant. This was done with a truncated Gaussian kernel which is K(d) = 1 if d ≤ c and if d > c. The denominator, , makes sure that the contact kernel is continuous at d = c. To determine σ, we used K(dcut = 0.8) = 10−5 so that .
The semibinary contact between residues si and sj from all frames were averaged to form the final semibinary contact map
Mutual information
Mutual information (MI) was used to estimate correlation of residue movements. The MI, I, between residue si and sj was estimated on the basis of distances from their respective equilibrium positions. The residue position was taken as the centroid of the residue, thus accounting for both side chain and backbone motions. MI is calculated as
where Hi is the entropy of residue si,
The density was estimated with Gaussian mixture models (GMM) (61–63) with the GMM implementation in Scikit-learn 0.21.2 (64). The number of Gaussian components was chosen with the Bayesian information criterion (65), varying between one and five components.
The number of frames affects not only the density estimate but also the approximated entropy. To increase accuracy, parametric bootstrapping was done by repeatedly drawing Nframes new samples from the estimated density. Using the new samples, new MI matrices were estimated. This was repeated 10 times. The final MI matrix was taken as the average of all estimated matrices.
Information flow analysis
The MI matrix and semibinary contact map make up the full residue network with adjacency matrix elements Aij = CijIij.
Definition of current flow betweenness
Assuming that information spreads from source residues S0 to sink residues S1 on the network according to diffusion, or equally a random walk, where the sink nodes are absorbing, we can use current flow betweenness (here referred to as information flow) to describe the allosteric pathways.
First, the network Laplacian is computed as, L = D − A, where is the diagonal degree matrix. The inverse reduced Laplacian, , is obtained by removing absorbing nodes (i.e., sink nodes) from the Laplacian, inverting this matrix, and then again including zero rows and columns at the absorbing nodes.
Given a supply vector, , which represented an injection of one unit information (or current) in a single source node that would exit in the sink nodes, the information flow through an inner node (not source or sink), or residue, si was calculated as
where . The information flow through the source and sink nodes were set to zero to only consider inner states in the analysis. The final information flow was averaged over the source nodes of one subunit
Symmetrization over subunits
The information flow through each residue was calculated for sources in each subunit, one at a time. The resulting information flow profiles were centered on the first subunit. The information flow was then replicated over the structure, and the total information flow through one residue was taken as the average contribution from each subunit. The average and SD over the structure were calculated from the replicated information flows.
Identifying important residues via hubs
To reveal the communication between the VSD and the pore and highlight hubs with important residues, two different pairs of sources and sinks were studied:
1) Gating charges on S4 (R1, R2, Q3, and R4)–S349
2) CCaM (residues 82-148 in CaM)–S349
This analysis was done on the CTD-bent structure with source/sink pairs 2 versus 1 and on CTD-straight versus CTD-bent using source/sink pair 1. For the CTD-bent structure, the same analysis was also performed by defining the gating charges as the sink and the pore-gate residue as the source. We found that our conclusions were conserved regardless of information flow direction (fig. S8F). To highlight differences, we compared the information flow profiles with simple subtraction of information flow at mutual residues (i.e., residues that are present in both structures). The SD of the delta information flow was calculated with the square root of the added variance of each information flow profile.
Correlation of information flow profiles
The results from the Xenopus KCNQ1 simulations were compared to human KCNQ1 simulations by first converting the Xenopus residue numbering to the human structure. Linear regression was performed, and the Pearson correlation coefficient was computed. This was done on the information flow of mutual residues. The linear regression was done with Scikit-learn 0.21.2 (64).
Residue interaction analysis
Interactions were quantified by the minimum distance between all heavy atoms (nonhydrogen atoms) of the two residues along the second half of each trajectory. The interaction distances were calculated using the python package MDTraj 1.9.3 (59). The distance densities over the trajectory were estimated with GMM, with the number of components chosen by Bayesian information criterion by varying between one and six components. The average density was scaled between 0 and 1. The SE was calculated over the four subunits.
Supplementary Material
Acknowledgments
Funding: This work was supported by grants from NIH R01HL126774 (J.C.), R01NS092570 (J.R.S.), F30HL151042 (P.W.K.), grants from the Science for Life Laboratory, and the Göran Gustafsson Foundation (L.D.). The simulations were performed on resources provided by the Swedish National Infrastructure for Computing (SNIC) at the PDC Centre for High Performance Computing (PDC-HPC). Author contributions: P.W.K. and J.C. initiated this work. J.C. and L.D. directed the approaches used. The experimental work was conducted by P.W.K., J.S., K.M.W., A.K.D., and A.H.C. The computational work was conducted by A.M.W. All authors participated in data analysis. P.W.K., A.M.W., J.C., and L.D. wrote the paper with input from all authors. Competing interests: J.S. and J.C. are cofounders of a startup company VivoCor LLC, which is targeting IKs for the treatment of cardiac arrhythmia. The authors declare that they have no other competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Data to reproduce the simulation results is available at: https://osf.io.jt568/. The code used for network analysis is available at https://github.com/delemottelab/allosteric-pathways.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/50/eabd6798/DC1
REFERENCES AND NOTES
- 1.Barhanin J., Lesage F., Guillemare E., Fink M., Lazdunski M., Romey G., K(V)LQT1 and lsK (minK) proteins associate to form the I(Ks) cardiac potassium current. Nature 384, 78–80 (1996). [DOI] [PubMed] [Google Scholar]
- 2.Sanguinetti M. C., Curran M. E., Zou A., Shen J., Spector P. S., Atkinson D. L., Keating M. T., Coassembly of K(V)LQT1 and minK (IsK) proteins to form cardiac I(Ks) potassium channel. Nature 384, 80–83 (1996). [DOI] [PubMed] [Google Scholar]
- 3.Wang H. S., Pan Z., Shi W., Brown B. S., Wymore R. S., Cohen I. S., Dixon J. E., McKinnon D., KCNQ2 and KCNQ3 potassium channel subunits: Molecular correlates of the M-channel. Science 282, 1890–1893 (1998). [DOI] [PubMed] [Google Scholar]
- 4.Schroeder B. C., Waldegger S., Fehr S., Bleich M., Warth R., Greger R., Jentsch T. J., A constitutively open potassium channel formed by KCNQ1 and KCNE3. Nature 403, 196–199 (2000). [DOI] [PubMed] [Google Scholar]
- 5.Ghosh S., Nunziato D. A., Pitt G. S., KCNQ1 assembly and function is blocked by long-QT syndrome mutations that disrupt interaction with calmodulin. Circ. Res. 98, 1048–1054 (2006). [DOI] [PubMed] [Google Scholar]
- 6.Shamgar L., Ma L., Schmitt N., Haitin Y., Peretz A., Wiener R., Hirsch J., Pongs O., Attali B., Calmodulin is essential for cardiac IKS channel gating and assembly: Impaired function in long-QT mutations. Circ. Res. 98, 1055–1063 (2006). [DOI] [PubMed] [Google Scholar]
- 7.Zhang H., Craciun L. C., Mirshahi T., Rohacs T., Lopes C. M., Jin T., Logothetis D. E., PIP(2) activates KCNQ channels, and its hydrolysis underlies receptor-mediated inhibition of M currents. Neuron 37, 963–975 (2003). [DOI] [PubMed] [Google Scholar]
- 8.Zaydman M. A., Silva J. R., Delaloye K., Li Y., Liang H., Larsson H. P., Shi J., Cui J., Kv7.1 ion channels require a lipid to couple voltage sensing to pore opening. Proc. Natl. Acad. Sci. U.S.A. 110, 13180–13185 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shimizu W., Antzelevitch C., Cellular basis for the ECG features of the LQT1 form of the long-QT syndrome: Effects of beta-adrenergic agonists and antagonists and sodium channel blockers on transmural dispersion of repolarization and torsade de pointes. Circulation 98, 2314–2322 (1998). [DOI] [PubMed] [Google Scholar]
- 10.Chen Y. H., Xu S. J., Bendahhou S., Wang X. L., Wang Y., Xu W. Y., Jin H. W., Sun H., Su X. Y., Zhuang Q. N., Yang Y. Q., Li Y. B., Liu Y., Xu H. J., Li X. F., Ma N., Mou C. P., Chen Z., Barhanin J., Huang W., KCNQ1 gain-of-function mutation in familial atrial fibrillation. Science 299, 251–254 (2003). [DOI] [PubMed] [Google Scholar]
- 11.Hedley P. L., Jorgensen P., Schlamowitz S., Wangari R., Moolman-Smook J., Brink P. A., Kanters J. K., Corfield V. A., Christiansen M., The genetic basis of long QT and short QT syndromes: A mutation update. Hum. Mutat. 30, 1486–1511 (2009). [DOI] [PubMed] [Google Scholar]
- 12.Sun J., MacKinnon R., Cryo-EM structure of a KCNQ1/CaM complex reveals insights into congenital long QT syndrome. Cell 169, 1042–1050.e9 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sun J., MacKinnon R., Structural basis of human KCNQ1 modulation and gating. Cell 180, 340–347.e9 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cui J., Voltage-dependent gating: Novel insights from KCNQ1 channels. Biophys. J. 110, 14–25 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Barro-Soria R., Rebolledo S., Liin S. I., Perez M. E., Sampson K. J., Kass R. S., Larsson H. P., KCNE1 divides the voltage sensor movement in KCNQ1/KCNE1 channels into two steps. Nat. Commun. 5, 3750 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hou P., Kang P. W., Kongmeneck A. D., Yang N.-D., Liu Y., Shi J., Xu X., White K. M., Zaydman M. A., Kasimova M. A., Seebohm G., Zhong L., Zou X., Tarek M., Cui J., Two-stage electro–mechanical coupling of a KV channel in voltage-dependent activation. Nat. Commun. 11, 676 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Osteen J. D., Gonzalez C., Sampson K. J., Iyer V., Rebolledo S., Larsson H. P., Kass R. S., KCNE1 alters the voltage sensor movements necessary to open the KCNQ1 channel gate. Proc. Natl. Acad. Sci. U.S.A. 107, 22710–22715 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Taylor K. C., Kang P. W., Hou P., Yang N. D., Kuenze G., Smith J. A., Shi J., Huang H., White K. M., Peng D., George A. L., Meiler J., McFeeters R. L., Cui J., Sanders C. R., Structure and physiological function of the human KCNQ1 channel voltage sensor intermediate state. eLife 9, e53901 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zaydman M. A., Kasimova M. A., McFarland K., Beller Z., Hou P., Kinser H. E., Liang H., Zhang G., Shi J., Tarek M., Cui J., Domain-domain interactions determine the gating, permeation, pharmacology, and subunit modulation of the IKs ion channel. eLife 3, e03606 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hou P., Eldstrom J., Shi J., Zhong L., McFarland K., Gao Y., Fedida D., Cui J., Inactivation of KCNQ1 potassium channels reveals dynamic coupling between voltage sensing and pore opening. Nat. Commun. 8, 1730 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hou P., Shi J., White K. M., Gao Y., Cui J., ML277 specifically enhances the fully activated open state of KCNQ1 by modulating VSD-pore coupling. eLife 8, e48576 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu D., Delaloye K., Zaydman M. A., Nekouzadeh A., Rudy Y., Cui J., State-dependent electrostatic interactions of S4 arginines with E1 in S2 during Kv7.1 activation. J. Gen. Physiol. 135, 595–606 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Babu Y. S., Bugg C. E., Cook W. J., Structure of calmodulin refined at 2.2 A resolution. J. Mol. Biol. 204, 191–204 (1988). [DOI] [PubMed] [Google Scholar]
- 24.Alaimo A., Villarroel A., Calmodulin: A multitasking protein in Kv7.2 potassium channel functions. Biomolecules 8, 57 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bartos D. C., Morotti S., Ginsburg K. S., Grandi E., Bers D. M., Quantitative analysis of the Ca2+-dependent regulation of delayed rectifier K+ current IKs in rabbit ventricular myocytes. J. Physiol. 595, 2253–2268 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chang A., Abderemane-Ali F., Hura G. L., Rossen N. D., Gate R. E., Minor D. L. Jr., A calmodulin C-lobe Ca2+-dependent switch governs Kv7 channel function. Neuron 97, 836–852.e6 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gamper N., Shapiro M. S., Calmodulin mediates Ca2+−dependent modulation of M-type K+ channels. J. Gen. Physiol. 122, 17–31 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Haitin Y., Attali B., The C-terminus of Kv7 channels: A multifunctional module. J. Physiol. 586, 1803–1810 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sihn C.-R., Kim H. J., Woltz R. L., Yarov-Yarovoy V., Yang P.-C., Xu J., Clancy C. E., Zhang X.-D., Chiamvimonvat N., Yamoah E. N., Mechanisms of calmodulin regulation of different isoforms of Kv7.4 K+ channels. J. Biol. Chem. 291, 2499–2509 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sachyani D., Dvir M., Strulovich R., Tria G., Tobelaim W., Peretz A., Pongs O., Svergun D., Attali B., Hirsch J. A., Structural basis of a Kv7.1 potassium channel gating module: Studies of the intracellular c-terminal domain in complex with calmodulin. Structure 22, 1582–1594 (2014). [DOI] [PubMed] [Google Scholar]
- 31.Adams P. J., Ben-Johny M., Dick I. E., Inoue T., Yue D. T., Apocalmodulin itself promotes ion channel opening and Ca2+ regulation. Cell 159, 608–622 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Botello-Smith W. M., Luo Y., Robust determination of protein allosteric signaling Pathways. J. Chem. Theory Comput. 15, 2116–2126 (2019). [DOI] [PubMed] [Google Scholar]
- 33.Duclert-Savatier N., Bouvier G., Nilges M., Malliavin T. E., Building graphs to describe dynamics, kinetics, and energetics in the d-ALa:D-Lac ligase VanA. J. Chem. Inf. Model. 56, 1762–1775 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fernandez-Marino A. I., Harpole T. J., Oelstrom K., Delemotte L., Chanda B., Gating interaction maps reveal a noncanonical electromechanical coupling mode in the Shaker K+channel. Nat. Struct. Mol. Biol. 25, 320–326 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Park K., Kim D., Modeling allosteric signal propagation using protein structure networks. BMC Bioinformatics 12 (Suppl 1), S23 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.U. Brandes, D. Fleischer, in Annual Symposium on Theoretical Aspects of Computer Science (Springer, 2005), pp. 533–544. [Google Scholar]
- 37.Newman M. E., A measure of betweenness centrality based on random walks. Soc. Networks 27, 39–54 (2005). [Google Scholar]
- 38.Lu Z., Klem A. M., Ramu Y., Coupling between voltage sensors and activation gate in voltage-gated K+ channels. J. Gen. Physiol. 120, 663–676 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lehman A., Thouta S., Mancini G. M. S., Naidu S., van Slegtenhorst M., McWalter K., Person R., Mwenifumbo J., Salvarinova R., Guella I., McKenzie M. B., Datta A., Connolly M. B., Kalkhoran S. M., Poburko D., Friedman J. M., Farrer M. J., Demos M., Desai S., Claydon T., Loss-of-function and gain-of-function mutations in KCNQ5 cause intellectual disability or epileptic encephalopathy. Am. J. Hum. Genet. 101, 65–74 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kasimova M. A., Zaydman M. A., Cui J., Tarek M., PIP2-dependent coupling is prominent in Kv7.1 due to weakened interactions between S4-S5 and S6. Sci. Rep. 5, 7474 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang Q., Zhou P., Chen Z., Li M., Jiang H., Gao Z., Yang H., Dynamic PIP2 interactions with voltage sensor elements contribute to KCNQ2 channel gating. Proc. Natl. Acad. Sci. U.S.A. 110, 20093–20098 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Choveau F. S., De la Rosa V., Bierbower S. M., Hernandez C. C., Shapiro M. S., Phosphatidylinositol 4,5-bisphosphate (PIP2) regulates KCNQ3 K+ channels by interacting with four cytoplasmic channel domains. J. Biol. Chem. 293, 19411–19428 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu Y., Xu X., Gao J., Naffaa M. M., Liang H., Shi J., Wang H. Z., Yang N. D., Hou P., Zhao W., White K. M., Kong W., Dou A., Cui A., Zhang G., Cohen I. S., Zou X., Cui J., A PIP2 substitute mediates voltage sensor-pore coupling in KCNQ activation. Commun. Biol. 3, 385 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li Y., Gao J., Lu Z., McFarland K., Shi J., Bock K., Cohen I. S., Cui J., Intracellular ATP binding is required to activate the slowly activating K+ channel IKs. Proc. Natl. Acad. Sci. U.S.A. 110, 18922–18927 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ben-Johny M., Yue D. T., Calmodulin regulation (calmodulation) of voltage-gated calcium channels. J. Gen. Physiol. 143, 679–692 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Crotti L., Johnson C. N., Graf E., De Ferrari G. M., Cuneo B. F., Ovadia M., Papagiannis J., Feldkamp M. D., Rathi S. G., Kunic J. D., Pedrazzini M., Wieland T., Lichtner P., Beckmann B. M., Clark T., Shaffer C., Benson D. W., Kaab S., Meitinger T., Strom T. M., Chazin W. J., Schwartz P. J., George A. L. Jr., Calmodulin mutations associated with recurrent cardiac arrest in infants. Circulation 127, 1009–1017 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dick I. E., Joshi-Mukherjee R., Yang W., Yue D. T., Arrhythmogenesis in Timothy Syndrome is associated with defects in Ca2+-dependent inactivation. Nat. Commun. 7, 10370 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liin S. I., Yazdi S., Ramentol R., Barro-Soria R., Larsson H. P., Mechanisms underlying the dual effect of polyunsaturated fatty acid analogs on Kv7.1. Cell Rep. 24, 2908–2918 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fiser A., Do R. K. G., Modeling of loops in protein structures. Protein Sci. 9, 1753–1773 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jo S., Kim T., Iyer V. G., Im W., CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 29, 1859–1865 (2008). [DOI] [PubMed] [Google Scholar]
- 51.Nosé S., A unified formulation of the constant temperature molecular dynamics methods. J. Chem. Phys. 81, 511–519 (1984). [Google Scholar]
- 52.Bussi G., Donadio D., Parrinello M., Canonical sampling through velocity rescaling. J. Chem. Phys. 126, 014101 (2007). [DOI] [PubMed] [Google Scholar]
- 53.Parrinello M., Rahman A., Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 52, 7182–7190 (1981). [Google Scholar]
- 54.Berendsen H. J., Postma J. P. M., van Gunsteren W. F., DiNola A., Haak J. R., Molecular dynamics with coupling to an external bath. J. Chem. Phys. 81, 3684–3690 (1984). [Google Scholar]
- 55.Darden T., York D., Pedersen L., Particle mesh Ewald: An N· log (N) method for Ewald sums in large systems. J. Chem. Phys. 98, 10089–10092 (1993). [Google Scholar]
- 56.Huang J., MacKerell A. D. Jr., CHARMM36 all-atom additive protein force field: Validation based on comparison to NMR data. J. Comput. Chem. 34, 2135–2145 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Abraham M. J., Murtola T., Schulz R., Páll S., Smith J. C., Hess B., Lindahl E., GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1, 19–25 (2015). [Google Scholar]
- 58.Hess B., Bekker H., Berendsen H. J., Fraaije J. G., LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 18, 1463–1472 (1997). [Google Scholar]
- 59.McGibbon R. T., Beauchamp K. A., Harrigan M. P., Klein C., Swails J. M., Hernandez C. X., Schwantes C. R., Wang L. P., Lane T. J., Pande V. S., MDTraj: A modern open library for the analysis of molecular dynamics trajectories. Biophys. J. 109, 1528–1532 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Humphrey W., Dalke A., Schulten K., VMD: Visual molecular dynamics. J. Mol. Graph. 14, 33–38 (1996). [DOI] [PubMed] [Google Scholar]
- 61.Dempster A. P., Laird N. M., Rubin D. B., Maximum likelihood from incomplete data via the EM algorithm. J. R. Stat. Soc. B. Methodol. 39, 1–22 (1977). [Google Scholar]
- 62.Westerlund A. M., Harpole T. J., Blau C., Delemotte L., Inference of calmodulin’s Ca2+-dependent free energy landscapes via Gaussian Mixture Model Validation. J. Chem. Theory Comput. 14, 63–71 (2018). [DOI] [PubMed] [Google Scholar]
- 63.Westerlund A. M., Delemotte L., InfleCS: Clustering Free Energy Landscapes with Gaussian Mixtures. J. Chem. Theory Comput. 15, 6752–6759 (2019). [DOI] [PubMed] [Google Scholar]
- 64.Pedregosa F., Varoquaux G., Gramfort A., Michel V., Thirion B., Grisel O., Blondel M., Prettenhofer P., Weiss R., Dubourg V., Vanderplas J., Passos A., Cournapeau D., Scikit-learn: Machine learning in Python. J. Mach. Learn. Res. 12, 2825–2830 (2011). [Google Scholar]
- 65.Schwarz G., Estimating the dimension of a model. Ann. Stat. 6, 461–464 (1978). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/50/eabd6798/DC1