

We show that locally favored structural and chemical orders in melt control the glass-forming ability of metallic alloys.
Abstract
The origin of glass formation is one of the most fundamental issues in glass science. The glass-forming ability (GFA) of multicomponent systems, such as metallic glasses and phase-change materials, can be enormously changed by slight modifications of the constituted elements and compositions. However, its physical origin remains mostly unknown. Here, by molecular dynamics simulations, we study three model metallic systems with distinct GFA. We find that they have a similar driving force of crystallization, but a different liquid-crystal interface tension, indicating that the latter dominates the GFA. Furthermore, we show that the interface tension is determined by nontrivial coupling between structural and compositional orderings and affects crystal growth. These facts indicate that the classical theories of crystallization need critical modifications by considering local ordering effects. Our findings provide fresh insight into the physical control of GFA of metallic alloys and the switching speed of phase-change materials without relying on experience.
INTRODUCTION
In principle, any liquid can be solidified upon cooling into either a crystal or a glass. This bifurcation phenomenon can be controlled by the cooling rate, R. The slowest R to bypass crystallization to form glass is called the critical cooling rate, Rc, which characterizes how easily a system is to be vitrified. Glass-forming ability (GFA) is usually quantified by Rc and negatively correlated with it. GFA is a critical issue in the field of metallic glass (MG) (1–3) because it can be controlled over many orders of magnitude by a slight change of its composition or a small addition of a specific element. Thus, MG is the best system to study the physical mechanism of GFA. Among the known MGs, Rc can differ over 16 orders of magnitude (4–6).
Because of the combination of amorphous structures and metallic bonds, MGs have shown many record-breaking properties that make themselves outstanding from traditional glasses and alloys (7, 8). Thus, they have been considered promising alternatives to conventional materials in various applications (9, 10). However, the achievable GFA primarily impedes this possibility. Despite the importance and high demand for the future development of desired MGs, the critical physical factor controlling GFA remains unclear. It is widely known that alloying is crucial for improving GFA. However, the underlying physical principles are mostly unknown. Thus, the fabrication of MGs largely relies on empirical rules. This problem is also crucial for phase-change materials (11–14), which are usually multicomponent mixtures of chalcogenides. For phase-change materials, the rapid switching speed is generally realized by poor GFA, opposite to MGs. To improve this situation, the physical understanding of the mechanism of the GFA is critical, both fundamentally and technologically.
Several empirical rules have been proposed either by treating MGs as hard sphere–like models aiming to maximize disordered packing capability (15–17) or by correlating GFA to various thermodynamic parameters that can be measured after obtaining the amorphous state (18–21). There is also an effort to estimate thermodynamic parameters from the high-temperature liquid (22). More recently, many geometrical structural descriptors from computer simulations have been used to describe the GFA of model MGs (23, 24). However, these phenomenological models do not have general validity. From the most fundamental viewpoint, glass formation is the consequence of the avoidance of crystallization (18, 25, 26). Unveiling the origin of the difference in crystallization kinetics of MGs should provide a physical basis to understand the factors controlling GFA.
A particularly vital question concerning multicomponent alloys, including MGs and phase-change materials, is the specificity of these systems. Similar alloying is possible for hard sphere–like systems such as colloids, but the variety of atoms with different characters and their combination for metallic alloys make the dimensions of the parameter space to explore high GFA vast. So, we require guiding physical principles to design useful materials with the desired GFA.
Here, we aim to unravel the fundamental physical mechanism of distinct GFAs of MGs by investigating their crystallization behaviors by molecular dynamics (MD) simulations. By studying the physical factors controlling crystal nucleation and growth, we find that (i) the most critical factor determining the crystallization rate is the liquid-crystal interface energy and (ii) the interface energy increases by the reduction of crystal-like preordering in a supercooled liquid state, which is caused by the nontrivial coupling between structural and compositional (or “chemical” in the MG terminology) orderings that create frustrations against crystallization. We also reveal that the structural and compositional differences across the liquid-crystal interface are of great significance not only in the crystal nucleation process but also in crystal growth. These findings suggest that the thermodynamic driving force, i.e., the chemical potential difference between the liquid and crystal phases, plays a minor role in determining GFA, contrary to the widespread belief.
RESULTS
Glass-forming ability
To study the origin of GFA, we carefully choose three prototypical metallic systems, Cu50Zr50 (CuZr), Ni50Al50 (NiAl), and Zr, so that they have quite different GFAs in experiments but share similar physical properties (27–29). These similarities are crucial for elucidating the origin of GFA. First, the crystals to be formed are body-centered cubic (bcc)–type for all the systems (for the binary alloys, B2 structure, an ordered bcc structure consisting of two simple cubic interpenetrating sublattices, and for Zr, bcc structure). Second, these binary alloys have a similar atomic size ratio. Here, the atomic size ratio is defined as the radius ratio of the smaller element to the larger one. The atomic size ratios of CuZr and NiAl are around 0.81 and 0.88, respectively. Third, their dynamical properties are similar, characterized by nearly identical liquid fragility (see Materials and Methods and fig. S1). Fragility characterizes how steeply the structural relaxation time τα of glass-forming liquids increases upon cooling (30). A liquid with a steeper increase of τα upon cooling is called a more fragile, or less strong, liquid. Strong and fragile liquids are characterized by nearly Arrhenius and super-Arrhenius behaviors, respectively. Recently, an interesting pathway of crystal nucleation through a chemically ordered intermediate was reported for CuAu alloys (31), but the crystal nucleation paths of our systems are direct and straightforward, which allows us to elucidate the physical origin controlling the GFAs.
To quantify their distinct GFA in computer simulations, we quench them at various cooling rates, R (see Materials and Methods for details). From the R dependence of the crystallinity of the quenched solids, f(Sij) (Fig. 1A), we estimate Rc of NiAl and Zr as 109.9 and 1013.6 K/s, respectively. It is well known experimentally that CuZr is an outstanding bulk MG former among binary alloys (27, 28). Consistently with this fact, we cannot see any crystallization in our simulations for both continuous cooling with R ≥ 109 K/s and isothermal annealing in the supercooled state over microseconds (see fig. S1). Thus, we use the experimental value of Rc of about 102.4 K/s for CuZr in Fig. 1A for comparison. There are 11 orders of magnitude difference in Rc among the three systems.
Fig. 1. GFA and crystallization thermodynamics.
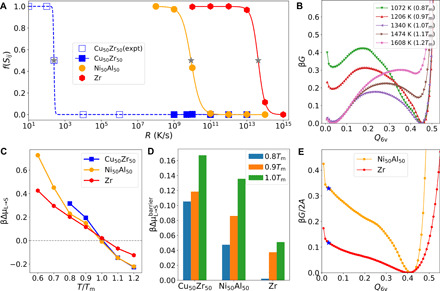
(A) Fraction of the crystalline atoms of the solid phase, f(Sij), obtained upon cooling for various Rs. The filled symbols are the data obtained from MD simulations, whereas the open symbols represent the experimental data for CuZr. The gray stars locate Rc for each material estimated from the criterion of f(Sij) = 0.5. The lines are fits to the data. (B) Free-energy profiles of CuZr liquids with respect to the collective variable Q6v at different temperatures. (C) Temperature dependence of the thermodynamic driving force scaled by the thermal energy for crystallization per particle, βΔμL → S. (D) Thermal energy–scaled free-energy barriers for the liquid-to-crystal transformation, , measured from the corresponding free-energy profiles. (E) Free-energy profiles of NiAl and Zr at Tm with the presence of planar crystal/liquid interfaces (A, the interface area). The blue stars indicate the position of the corresponding free energy of the equilibrium liquid phase.
This enormously different GFA implies that these systems have very different crystallization resistances. From the classical nucleation theory (CNT) (18, 32), the homogeneous nucleation frequency I in a supercooled liquid is given by
| (1) |
where kn is a constant, D is the translational diffusion coefficient, and β = 1/kBT (kB, the Boltzmann constant; T, the temperature). ΔGc is the free-energy barrier for the nucleation of a critical crystal nucleus. This barrier is determined by the balance between the free-energy gain from the chemical potential difference between the liquid (L) and crystal phases (S), ΔμL → S, and the free-energy cost associated with the formation of the interface between L and S, ϒ. More specifically, the barrier scaled by the thermal energy kBT is expressed solely by the dimensionless quantities as βΔGc ∼ (βϒ)3/(βΔμL → S)2 (33). According to CNT, after the nucleation of crystals, their growth rate is given by
| (2) |
where k0 is a constant. The overall crystallization rate Ξ is approximated by (IU3)1/4, which is directly linked to GFA. This relation tells us that three major physical factors govern how easily an MG can form. The first is the translational diffusion constant D, which appears as a prefactor for both I and U, but the difference in D among the three systems is so small that it has little influence on GFA. The second is the driving force of crystallization scaled by the thermal energy, βΔμL → S, and the third is the interface tension scaled by the thermal energy, βϒ. In the following, we will unravel the roles of these factors in controlling crystallization.
Crystal nucleation thermodynamics
We first characterize the free-energy profiles of crystallization for various degrees of supercooling by using well-tempered metadynamics (see Materials and Methods). Figure 1B shows the free-energy profiles for CuZr as a function of the bond-orientational order parameter Q6v, which is the critical structural order parameter of crystallization, for different temperatures (see also fig. S2). All the free-energy profiles show the simple double-well shape, whose two minima with low and high Q6v correspond to the liquid and crystal phases, respectively. Figure 1C shows the calculated βΔμL → S for the three systems. Notably, βΔμL → S is quite similar to each other for the same degree of supercooling, especially for shallow supercooling. Counterintuitively, CuZr, with the best GFA, has the largest driving force for crystallization. This finding is further corroborated by the empirical estimation of βΔμL → S through (Tm/T − 1)ΔHm/kBTm (18), where ΔHm is the enthalpy of fusion per particle (see fig. S2). Nevertheless, the barriers to prevent crystallization in the free-energy profiles are quite different (Fig. 1D). Consistently with the order of GFA, the barrier height increases in the order of Zr, NiAl, and CuZr. Furthermore, the decrease in the barrier with decreasing T is smaller in the same order. Because the driving force is similar, such a barrier difference must be induced by different interface energy. To verify this point, we directly calculate ϒ at Tm for NiAl and Zr by an enhanced sampling method (see Materials and Methods) (34). We note that the diffusive behavior on their free-energy landscapes is effective enough for efficient sampling (see fig. S3). However, such a diffusive behavior of phase transition for CuZr is very difficult to reach so that it is hard to obtain the converged free-energy profile. This difficulty stems from the strong topological and chemical frustrations against crystallization in CuZr (see below). The nucleation process of the systems we studied follows the two-step scenario (35, 36): The orientational symmetry develops first, followed by the density change (see fig. S3). Here, we note that the calculated interfacial energy (and also the free-energy profiles in bulk) is between the liquid and crystalline phases and not between the liquid phase and the intermediate state with high bond orientational order (BOO) but without translational order. From the free-energy profiles in Fig. 1E, βϒ of NiAl is estimated as 2.81 times of that of Zr. The interfacial energy of our studied systems is only weakly dependent on the crystallographic plane (see fig. S3). This has also been supported by the previous finding (37) that the crystal growth rates along different crystallographic planes are barely different in both CuZr and NiAl (see discussion below). Because βΔGc is proportional to the cubic power of βϒ, this difference should result in a substantial difference in the nucleation rate. Considering the dynamical properties and the driving force, we anticipate that βϒ of CuZr should be larger than that of NiAl.
Topological frustration against crystallization
According to the two-order-parameter model (26, 38), frustration against crystallization, or competing ordering, is crucial to control the fate of a liquid upon cooling. Concerning the local atomic arrangements, there are two types of structural orderings in these metallic liquids. One is local icosahedral ordering (ICO), whose symmetry is incompatible with crystal. Its importance was first pointed out by Frank (39). These locally favored structures are spontaneously formed in the sea of the normal liquid structure. The other is crystal-like bond orientational ordering (CRYO), which has the same local orientational symmetry as the equilibrium crystal. The existence of CRYO is a natural consequence of the lowest free-energy state being the crystalline state. While ICO acts as the source of frustration, or impurities, against crystallization, CRYO tends to promote the formation of long-range density ordering (25, 35). The strength of the competition between ICO and CRYO determines the ease of crystallization. For Zr, for example, local icosahedral and bcc orders were reported to be competing orders by ab initio MD simulations (40). Therefore, both ICO and CRYO and their relationship should play a critical role in determining GFA (25, 41, 42). The existence of ICO in metallic liquids and alloys has been verified by many experimental observations (43–45). The role of ICO has been widely studied for MGs (40, 46–51). However, the role of CRYO in determining GFA and the relationship between ICO and CRYO have long been overlooked.
To rationalize the difference in βϒ, we investigate these structural orderings in the supercooled metallic liquids (see Materials and Methods for details). In Fig. 2A, we compare the change of the fraction of the atoms in the icosahedral environments with decreasing temperature. We can see that the faction of atoms involved in ICO decreases in the order of CuZr, NiAl, and Zr, and the fraction increases more slowly in this order upon cooling. In particular, Zr has very few ICO and already becomes unstable to crystallization below 0.8Tm. The temperature dependence of ICO can be well described by the two-order-parameter model (26, 38, 41, 52) (solid curves in Fig. 2A), which confirms the validity of this model to MGs. The spatial distributions of ICO are also visualized in Fig. 2C. These results demonstrate that ICO in these MGs promotes glass formation and leads to topological frustration against crystallization. Microscopically, the more substantial amount of ICO leads to a more substantial structural contrast between the liquid and crystal, causing larger βϒ. Considering the similar βΔμL → S, the formation of ICO does not necessarily mean the weaker driving force of crystallization. Thus, the prevention of crystallization by ICO is not through the decrease in βΔμL → S, but through the increase in βϒ.
Fig. 2. Topological orderings in supercooled liquids.
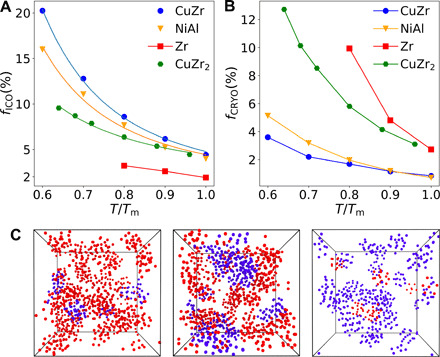
(A) Temperature dependence of the fraction of atoms in the icosahedral environments (ICO) in the four systems: CuZr, NiAl, Zr, and CuZr2. The solid lines for the data of CuZr, NiAl, and CuZr2 are fits to the two-order-parameter model. (B) Temperature dependence of the fraction of atoms involved in crystal-like environments (CRYO). The order of the quantity and growth speed of fCRYO is opposite to that of fICO. (C) Snapshots of supercooled states with red atoms in ICO and blue atoms in CRYO environments at 0.6Tm for CuZr (left) and NiAl (middle), and at 0.8Tm for Zr (right).
Next, we turn our attention to CRYO. Figure 2B illustrates how the fraction of the atoms involved in CRYO changes with lowering the temperature. The spatial distributions of CRYO are also shown in Fig. 2C together with those of ICO for the three systems. The order of the degree of CRYO is the opposite of that of ICO. We can see that the faction of atoms involved in CRYO increases in the order of CuZr, NiAl, and Zr, and the fraction increases much faster in this order upon cooling. Zr has the highest fraction of CRYO, and its growth upon cooling is the fastest. In contrast, CuZr has only a tiny fraction of CRYO. Moreover, with decreasing T/Tm, the spatial correlation of CRYO in Zr grows quickly, whereas it almost does not change in CuZr (see fig. S4). It is worth emphasizing that the appearance of CRYO in MGs reflects the formation of CRYO (i.e., crystal precursors) but does not mean the formation of crystals (26, 35, 36, 52, 53). Because CRYO has the same local orientational symmetry as the equilibrium crystal, its formation reduces βϒ and results in the reduction of the crystallization resistance, contrary to ICO. Thus, suppressing CRYO should help with glass formation.
Therefore, the competition between these geometrical structures is of great importance in determining the GFA by affecting βϒ. However, the topological orderings alone are not enough to understand the GFA of metallic systems, unlike the other systems previously studied (33, 35, 36), as will be shown below.
Chemical frustration against crystallization
It is widely known that besides topological ordering, chemical ordering (54, 55) plays a substantial role in GFA: For example, a large negative heat of mixing is one of the empirical criteria for the high GFA of MGs (2, 3). The direct evidence of the atomic-scale heterogeneity caused by chemical ordering was recently reported (56). However, the microscopic understanding of the effect of chemical ordering on crystallization and its link to topological ordering has remained elusive. The difference in the character between the two types of orders makes the physical understanding of their precise roles difficult: Topological order, mainly driven entropically, grows rapidly upon cooling, but chemical order has only a weak temperature dependence because attractions far beyond the thermal energy stabilize it. Here, we focus on this issue.
In the equilibrium B2 crystal, the nearest neighbors of each atom consist of eight other-type atoms as the closest, and another six same-type atoms located a bit farther. A substantial deviation from this crystalline chemical order should frustrate crystallization. To see this effect, we introduce a variable cα, which is the fraction of the atoms of type α in the first coordination shell of a center of type α. Figure 3 (A and C) shows the distributions of cα for perfectly coordinated ICO and CRYO, respectively, around Cu and Ni (see fig. S4 for typical examples of atomic configurations of ICO with chemical inhomogeneity seen in CuZr). The corresponding distributions for all types of such orderings, including the imperfect ones, are depicted in Fig. 3 (B and D). Obviously, both CRYO and ICO in both systems have a strong chemical preference, but which is quite different between them. In CuZr, both CRYO and ICO tend to prefer the composition CuZr2, which significantly deviates from the targeted crystal composition (Cu:Zr = 1:1). In contrast, for NiAl, they tend to prefer the local composition close to the crystal one. This fact indicates that the transformation of CRYO and ICO to the crystal should be much easier for NiAl than CuZr from the aspect of chemical order. For CuZr, CRYO looks like a crystalline precursor topologically, but it is not chemically. Adjustment of local composition delays crystal nucleation. Furthermore, because of the composition difference between ICO and CRYO, as shown in Fig. 3B, the transformation of ICO to CRYO should not be easy. Therefore, ICO in CuZr is unique and generates a stronger hindrance to crystallization. Although with almost the same geometry, the properties of ICO in different MGs can be significantly different.
Fig. 3. Chemical orderings on top of topological orderings in supercooled liquids.
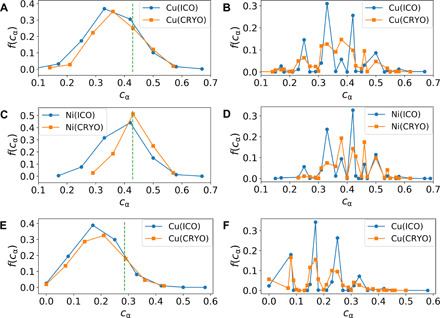
(A and C) Distributions of the local compositions cα of perfectly coordinated ICO (with 12 nearest neighbors) and CRYO (with 14 nearest neighbors) for CuZr (A) and NiAl (C) at 0.6Tm, respectively. The dashed line marks the equilibrium B2 crystal composition. α represents the smaller atom (i.e., Cu or Ni). In CuZr and NiAl, both ICO and CRYO prefer to have smaller species as their centers. (B and D) Corresponding distributions of cα of all ICO and CRYO (including both perfect and imperfect ones) for CuZr (B) and NiAl (D), respectively. ICO and CRYO show distinct cα distributions in CuZr, but similar distributions in NiAl. (E) Distributions of cα of perfectly coordinated ICO (with 12 nearest neighbors) and CRYO (with 14 nearest neighbors) centered at Cu in CuZr2 at ∼0.6Tm. The dashed line indicates the composition of the equilibrium crystal (body-centered tetragonal lattice) of CuZr2. (F) Corresponding distributions of cα of all ICO and CRYO (including both perfect and imperfect ones) in CuZr2. As in the case of NiAl, ICO and CRYO in CuZr2 prefer to have similar local compositions. We note that the basic trend for all composition distributions is rather independent of the temperature.
To further highlight the importance of chemical frustration in determining the GFA, we additionally study the CuZr2 system, which has a GFA lower than CuZr but better than NiAl in experiments (57–59). The better GFA of CuZr2 than NiAl is further confirmed directly by our simulations (see fig. S5). However, the topological orderings in Fig. 2 (A and B) tell us that CuZr2 has less ICO and more CRYO than NiAl. This topological feature would suggest CuZr2 as a poorer glass former than NiAl, but which is against the truth. Now, we turn our attention to the chemical compositions of these structural orders. Figure 3 (E and F) shows that ICO and CRYO in CuZr2 have the chemical compositions similar to those of NiAl. However, the critical fact is that these preferred local compositions strongly deviate from the target crystal one, which is different from NiAl. Therefore, we can conclude that it is the chemical frustration rather than topological frustration that leads to the better GFA of CuZr2 than NiAl: The effect of chemical ordering overwhelms that of the topological one in preventing crystallization of CuZr2.
The results in Fig. 3 represent three possible relationships between chemical and topological orderings: (i) ICO and CRYO have different chemical preferences, and both deviate from the equilibrium crystal (the case of CuZr); (ii) ICO and CRYO have similar chemical preferences, and both deviate from the crystal (the case of CuZr2); (iii) ICO and CRYO have similar chemical preferences and both close to the crystal (the case of NiAl). These findings demonstrate the critical role of the nontrivial coupling between topological and chemical orderings in governing the GFA of metallic alloys, which has not been recognized before. This chemical effect may be unique to atomic glasses such as bulk MGs and chalcogenide glasses because they are usually made of multiple elements with distinct pair interactions.
We also calculate the SD of the local composition from the crystal, δcα, for all atoms in CuZr and NiAl. As shown in Fig. 4A, the compositional deviation is smaller for NiAl than CuZr. The deviation tends to decrease upon cooling for Ni, Al, and Zr, but increase for Cu. This tendency indicates that ICO in CuZr more strongly acts against crystallization at large supercooling. The presence of chemical ordering is further validated from the partial structure factors, which of the Cu-Cu and Ni-Ni pairs are shown in Fig. 4B (see others in fig. S6). Unlike topological orderings, chemical ordering is weakly dependent on temperature (see fig. S6). This fact indicates that the appearance of chemical ordering is mainly determined by the interatomic potentials among different pairs (see fig. S7). For example, in CuZr, there is a substantial difference in the interaction strength and range among different pairs: The Cu-Cu pair has the weakest interaction and shortest range. Thus, Cu has a strong preference to have Zr as its nearest neighbors. It explains why there is always excess Zr in the first coordination shell of Cu in both CuZr and CuZr2. In contrast, the pair interactions in NiAl are very similar and induce only weak chemical preferences, making the arrangements of different species toward a specific crystal lattice easy to take place. In the context of CNT, the fact that CuZr suffers from much stronger chemical frustration than NiAl indicates that the compositional gradient at the liquid-crystal interface is much larger in CuZr than in NiAl, which results in the larger βϒ in the former than in the latter. Our study demonstrates that chemical frustration plays a substantial role in driving glass formation in a previously unknown manner, i.e., through its nontrivial coupling to topological order.
Fig. 4. Chemical frustration during cooling.
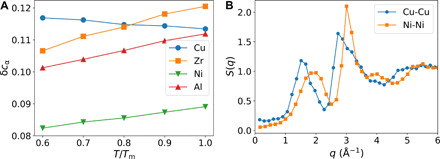
(A) Temperature dependence of the overall SD of cα from the ideal B2 crystal, δcα, for CuZr and NiAl. α represents both species in this analysis. (B) Structure factors of the Cu-Cu pair of CuZr and the Ni-Ni pair of NiAl at 0.6Tm. Similar results are obtained at other temperatures. The presence of a pre-peak at low q manifests chemical ordering in the supercooled liquid. The length scale corresponding to the pre-peak position of CuZr is longer than that of NiAl.
The free energy of a liquid can be reduced by local orientational and chemical orderings. How these orderings grow upon cooling is determined by the balance between the interaction energy and entropy. Our study shows that the degree of crystal-like ordering formed under the frustration effects of noncrystalline orientational (e.g., ICO) and chemical orderings determines the thermal energy–scaled interface energy and thus the GFA. We note that the difference in the interaction potential among various atom pairs and the non-additivity of the potential common in metallic alloys play a critical role in chemical ordering. We further speculate that the chemical ordering effect may become even more prominent in multicomponent alloys of very high GFA.
The kinetics of crystal growth
In principle, the growth kinetics is also important in crystallization (20, 32). Equation 2 suggests that there may be little difference among these materials because the values of D and βΔμL → S are quite similar. Nevertheless, we check this possibility below. To do so, we used the seeding method (see Materials and Methods for details) because spontaneous crystallization is hard to see in simulations, especially for CuZr. First, we identify the critical nucleus size, for which the chance of the seed to grow or dissolve becomes equal (fig. S8). Table 1 shows the number of atoms in the critical nucleus, Nc. For the same degree of supercooling, Nc is larger in the order of GFA, consistently with the higher critical nucleation barrier. The ratio of the corresponding critical nucleus size rc at 0.8Tm between NiAl and Zr is 1.60, which is close to the ratio of 1.29 estimated from the CNT prediction of rc = 2βϒ/(ρSβΔμL → S) (ρS is the number density of the solid phase) and the results of metadynamics simulations. Nc of Zr becomes very small below 0.8Tm, and thus, the liquid becomes unstable against crystallization, which is consistent with our MD simulation results. Considering the similar βΔμL → S for the three systems, the difference in Nc should originate from βϒ, as we discussed above.
Table 1. The number of atoms in the critical nucleus, Nc, estimated from the seeding simulations.
The systematic error is determined by the crystal structure and lattice constant.
| T/Tm | CuZr | NiAl | Zr |
| 0.8 | 307 | 229 | 51 |
| 0.7 | 113 | 89 | 9 |
| 0.6 | 89 | 51 | – |
Now, we investigate how the seeded critical nucleus grows with time. The crystal growth rate is determined by the balance between particles attaching to and detaching from the nucleus. To see the net effect, we consider the net number of atoms attached at every moment, Nattach, as the number of particles crystallized from the initial liquid state. The growth of Nattach is shown in Fig. 5A for CuZr, NiAl, and Zr. We can see that Nattach of CuZr increases with time much slower than NiAl, consistently with the previous study (37). CuZr exhibits a linear growth mode, while NiAl shows faster exponential growth. For Zr, the particle attachment is so fast that crystallization finishes almost instantaneously once it starts after the short incubation. This behavior is a consequence of the fact that Nc is so small that the critical nucleus cannot be stable.
Fig. 5. Crystal growth kinetics and its structural mechanism in supercooled liquids.
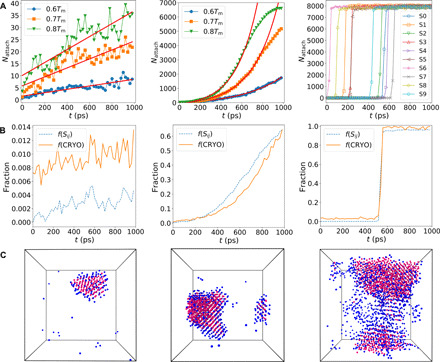
The figures of each row from left to right are for CuZr, NiAl, and Zr, respectively. (A) Temporal change in the net number of particles attached to the crystal nucleus, Nattach, at different undercooling. CuZr exhibits a linear growth mode, as indicated by the solid lines from linear fits. For NiAl, the data at 0.6Tm are multiplied by a factor of 5 for clarity. The solid curves are exponential fits. The deviations of the data from the solid curves are due to the finite sizes of the systems. For Zr, we show the temporal change of Nattach for 10 independent simulation runs at 0.7Tm. The system crystallizes quickly after some incubation, and then, the growth finishes almost instantaneously. (B) Comparisons of the fraction of the crystallized atoms (dashed curve) and that of the precursors (or CRYO) (solid curve) in the remaining liquid phase at 0.7Tm. (C) Snapshots of the spatial distribution of the crystalline phase (magenta atoms) and the precursors (or CRYO) (blue atoms). The degree of wettability of the precursors to the crystal increases with a decrease in GFA.
To account for the different kinetics among the three systems, we study how the precursors defined as CRYO in the liquid state influence the rate of particle attachment to the crystal. We show in Fig. 5B the temporal changes in the fraction of the crystallized atoms, f(Sij), and that of the precursors, f(CRYO), during isothermal annealing at 0.7Tm for CuZr, NiAl, and Zr. This result indicates that the precursors always mediate the crystal growth, but the degree and nature of preordering (both topological and chemical) are significantly different among the three systems, as discussed above. For CuZr, the amount of CRYO around the nucleus is so low that it cannot wet the crystal nucleus, as visualized in the left panel of Fig. 5C. Furthermore, the chemical composition of the preorder is different from that of the crystal. Thus, the crystal phase cannot quickly grow into the liquid because of the substantial structural and compositional differences between the two phases across the interfaces. In contrast, there is much more CRYO for NiAl (the middle panel of Fig. 5C). In the early stage before the crystal starts to grow, crystal precursors form around the critical nucleus. The fraction of the particles in CRYO becomes higher than that of crystalline particles. In the very late stage, they approach each other due to the finite-size effect. The amount of CRYO in Zr is even higher, and the surrounding liquid has a strong tendency to form crystal-like preordering. The chemical frustration is also absent in the monoatomic system. This fact indicates that the nucleus is thoroughly wet by CRYO during its growth (the right panel of Fig. 5C). These results indicate that CRYO also plays a critical role in determining the crystal growth kinetics by tuning the properties of the liquid-crystal interfaces.
Because the factors D and βΔμL → S in Eq. 2 are so similar in these systems, CNT predicts that the crystal growth rate should also be similar. In contrast, our study shows that their particle attachment rates are significantly different, and even the underlying growth mechanisms are different. This result shows the severe failure of CNT in predicting the crystallization kinetics of metallic liquids at large supercooling. This failure may originate from ignorance of the preordering in the liquid phase near the growth front of the crystal in CNT. More specifically, enhancement of the liquid phase’s wettability to the crystal through preordering has been overlooked in the previous theories of crystal growth, including CNT. We note that the structural ordering in the liquid phase becomes more substantial at larger supercooling. Our finding suggests that classical theory needs fundamental modifications to take structural and chemical orderings into account. It may be of substantial importance for phase-change materials (11–13, 20).
DISCUSSION
We have studied all the physical factors controlling the crystallization kinetics for the three typical metallic systems, with the critical cooling rates (i.e., GFAs) differing over 10 orders of magnitude. Previously, Tang and Harrowell (37) found that the maximum U in NiAl is about 20 times that in CuZr, which is consistent with our estimation from the particle attachment rates we measured. This difference leads to the 9.46 times difference in Ξ, but which is far smaller than the actual eight orders of magnitude difference. Thus, I should be dominant in the difference of Ξ. Furthermore, βΔμL → S is similar among the three systems, and its weak difference is compensated by the difference of U in determining Ξ. Thus, we conclude that the substantial difference of I, and thus the GFA, i.e., Ξ, is primarily determined by the interface tension scaled by the thermal energy, βϒ.
Our conclusion that the interface energy is the dominant factor controlling GFA should hold general validity in various glass-forming systems, although its source may differ (33). Thus, we infer that it can be used as a guiding physical principle to control the GFA of any materials. The scenario based on the competing orderings between CRYO and ICO supports the validity of the two-order-parameter model (26, 52) for crystallization and vitrification. Our study reveals that the unique feature of metallic alloys stems from the importance of chemical ordering in these systems on top of topological ordering. Both substantial structural and compositional differences across the liquid-crystal interface contribute to the increase of βϒ, which favors glass formation. It should be stressed that the impact of chemical ordering is not only the source of the thermodynamic frustration but also the kinetic constraint. The crucial point is that the adjustment of the chemical composition requires a long time that scales as , where is the characteristic length of the chemical heterogeneity that is larger than the interatomic distance (see, for instance, Fig. 4B).
Concerning the topological frustration, enhancing ICO and suppressing CRYO would promote glass formation if they are incompatible. Thus, both ICO and CRYO, and their relationship, should be considered to determine the GFA. This point has been overlooked so far. If a metastable icosahedral phase tends to nucleate or the Laves phase or quasicrystal is the primary crystalline phase, the competing ordering effects disappear because ICO becomes compatible with CRYO (42). Then, βϒ would be largely reduced so that (quasi-)crystallization proceeds efficiently if the chemical frustration effects are weak. The nucleation barrier reduction for the metastable quasicrystal phase by ICO in melts has been experimentally observed in metallic systems such as TiZrNi and MgZnYb (43, 60). Devitrification toward quasicrystals triggered by extremely small interface tension has also been observed by experiments in some Zr-based bulk MG with rich ICO (48). This also solves the controversy in simulations (23, 61, 62), concerning how ICO affects the GFA in some CuZr-based alloys. Therefore, it is not just the amount of ICO, but its competition with CRYO is critical in glass formation.
We note that an empirical GFA indicator, Trg (=Tg/Tm), is estimated as 0.525 and 0.499 for CuZr and NiAl, respectively, from our MD simulations. The small difference of Trg cannot explain a considerable higher GFA of CuZr than NiAl. This indicates that a larger βϒ can enhance the GFA even for an alloy with low Trg (18). This mechanism explains why bulk MGs with high GFA sometimes have a moderate or even low Trg (21). Elevating the interface energy on the basis of the mechanism found here shall serve as a more effective route to design new MGs. Here, we show an example of two multicomponent bulk MGs from experiments, Pd40Ni40P20 and Mg65Cu25Y10 (21). The situations are quite similar to CuZr versus NiAl that we studied. The GFA is higher for PdNiP than for MgCuY. However, their fragility is very similar, being 48 and 44.5, respectively. The difference of Trg is also small, which is 0.589 for PdNiP and 0.562 for MgCuY. The reduced thermodynamic driving force, ΔHm/kBTm, of PdNiP is 1.42, which is larger than the value 0.89 of MgCuY (63, 64). These demonstrate that the interface energy is the critical factor in differentiating their GFAs. Furthermore, Busch et al. (65) reported that several multicomponent MGs have similar chemical potential differences in the supercooled state, but their critical cooling rates differ by many orders of magnitude. In some cases, better glass formers have a higher chemical potential difference. These results further indicate the primary importance of the interfacial energy in governing the GFA of metallic alloys. However, whether the interface energy is always the more dominant factor than the chemical potential difference in controlling GFA should be studied carefully in the future.
Here, we unravel the fundamental physical mechanism of glass formation from binary metallic alloys. As for the practical implications for glass design, our findings indicate that increasing the liquid-crystal interfacial energy can effectively improve the GFA. Microscopically, this can be realized by increasing the contrast of topological and chemical properties of the liquid structure with respect to those of the crystalline solid to be formed. On the one hand, our finding provides the physical rationalization for the Inoue’s empirical rules of glass formation (3), i.e., multicomponent systems, large atomic size ratio, and negative heats of mixing. In the Inoue’s rules, what was missing is the information concerning the crystal to be nucleated. For example, we propose that increasing the degree of contrast in the cohesive energy of the constituents from the one favored to form the crystal would elevate chemical and topological frustration against crystallization and enhance the GFA. Further refinements taking this point into account are essential to increase the predictive power of the GFA. How to exactly transfer our theoretical findings to practical design rules is a promising topic for future study.
In summary, we have examined the roles of the thermodynamic driving force, the interface energy, and the particle attachment rate in crystallization in determining the GFAs of typical MG formers, based on numerical simulations. We have identified the thermal energy–scaled interface energy as the critical factor controlling GFA. It is determined by both topological and chemical frustrations induced by local structural orderings with symmetry and composition incompatible to the crystal, respectively, and their coupling. Furthermore, we show that it is crucial not only in crystal nucleation but also in crystal growth, indicating the fundamental deficiency of the classical theory of crystal growth, in which the interface tension plays no role (see Eq. 2). Our findings reveal the physical principle behind the GFA of multicomponent systems and provide a guiding rule for future glass design. Our results shed fresh light on the glass formation of not only metallic alloys but also broad classes of materials, including phase-change materials (chalcogenides) (11–14, 20), oxides, molecular and ionic systems, pharmaceuticals, cryoprotectants, and frozen foods. For example, phase-change materials have recently been shown to be incipient metals, governed by a bonding mechanism that is distinctively different from metallic bonding (14). Revealing similarities and potential differences in crystallization and vitrification behaviors for these different material classes is an interesting topic for further study.
MATERIALS AND METHODS
MD simulations
All the computer simulations were performed using the open-source MD simulation engine, large-scale atomic/molecular massively parallel simulator (LAMMPS) (66). To describe the atomic interactions of the materials studied, we used the many-body embedded-atom method (EAM) potentials (67, 68). In simulations, periodic boundary conditions in three directions were applied, and the time step for integration was 0.002 ps. The isobaric-isothermal ensemble (NPT) with zero external pressure was generally used unless otherwise stated. The temperature and pressure were controlled by using the Nose-Hoover thermostat and barostat. To study the structural and dynamical characteristics of these materials in equilibrium, we carried out extensive simulations in their supercooled liquid states. We first equilibrated the initial configuration of N = 4000 atoms with the desired composition at a temperature much higher than the melting temperature Tm for 0.5 ns. Tm is obtained from the literature as 1340, 1535, and 2110 K for CuZr, NiAl, and Zr, respectively (37, 69). The liquid was then instantaneously quenched to a supercooled state and relaxed at a target temperature for ∼100τα (τα: structural relaxation time; see below) under NPT. The ensemble was then switched to the Canonical ensemble (NVT) to relax for another ∼100τα for production.
Supercooled liquid dynamics
As for CuZr and NiAl, we characterized τα of the supercooled liquids by computing the self-part of the intermediate scattering function defined as
| (3) |
where N is the total number of atoms and rj(t) is the position vector of atom j at time t. The wave number q used to estimate τα corresponds to the first-peak position of the structure factor S(q), which was calculated directly from
| (4) |
Then, τα was determined as the time at which Fs(q, t) decays to e−1.
The estimated τα can be well fitted by the Vogel-Fulcher-Tammann equation, τα = τ0 exp (B/(T − T0)), in which B and T0 are fitting parameters (see fig. S1A). We determine the fragility parameter from (30). The glass transition temperature, Tg, is referenced as the temperature at which τα = 10 ns. Here, Tg is 704 and 766 K for CuZr and NiAl, respectively. We found that these two systems have rather similar fragilities: The fragility parameter m is 33.7 and 34.2 for CuZr and NiAl, respectively. In fig. S1B, we show the temperature dependence of τα scaled by Tm. We can easily see that at the same degree of undercooling, the dynamics of CuZr is only slightly slower than NiAl.
GFA characterization
To show the GFA difference of the materials in simulations, we first performed trial MD simulations at different cooling rates, R. Then, the temperature dependence of the potential energy E during cooling was calculated (see fig. S1C). We can see that the first-order phase transition, i.e., crystallization, occurs for Zr and NiAl at R = 1012 K/s and R = 109 K/s, respectively. These cooling rates are then lower than their critical cooling rates Rc. For CuZr at R = 109 K/s, on the other hand, the change of the potential energy has no discontinuous jump, indicating glass formation instead of crystallization. To further prove the high GFA of CuZr in simulations, we isothermally annealed a sample of N = 8192 atoms at 800 K for nearly 2 μs and found that no crystallization happens (see fig. S1D). In our study, the cooling rate was ranged from 109 to 1015 K/s. The slowest rate of 109 K/s is limited by the current computational capability.
To quantitatively measure Rc, MD simulations were performed by cooling melts from high to low temperature at various R. Specifically, an initial configuration of 2000 atoms with a designed composition was created in the form of a bcc lattice in a cubic box. The initial melt state was then equilibrated for 300 ps at 2500, 2500, and 3000 K for CuZr, NiAl, and Zr, respectively. Then, they were cooled to 100 K at a constant R. The final temperature configuration was used to judge whether the system is crystallized or not, with the BOO parameters (see below). The lowest cooling rate that we could access in our simulations was ∼109 K/s, which takes 2.4 μs for a single run. Two to 10 independent simulations were performed for the ensemble average, depending on the computational resource. The total simulation time to characterize the Rc values is over 16 μs. We calculated the crystallinity of the low-temperature solids, f(Sij), by measuring the BOO parameter s(i, j). Obviously, for R > Rc, the melt is to be vitrified, and thus, f(Sij) = 0. In contrast, it is to be fully crystallized [f(Sij) = 1] for R < Rc. Therefore, the R dependence of f(Sij) behaves like a “sigmoid” function. By fitting the function f(Sij) = c − d tanh (a · R + b) to the data, where the variables other than R are fitting parameters, Rc is defined as the R value at which f(Sij) = 0.5.
Free-energy profile calculation by metadynamics simulations
In MD simulations, crystallization is usually a rare event, and thus, it is often hard to observe in the computational time scale. It is especially true for the good glass former CuZr. To accelerate crystallization, an advanced sampling technique, such as metadynamics, is necessary. The free-energy landscape of the phase transition, which is inaccessible with conventional simulation methods, is now accurately measurable with enhanced sampling. To the best of our knowledge, there has so far been no study on the GFA of MGs using such a state-of-the-art technique, and thus, the free-energy landscapes of MGs concerning crystallization remain unknown.
Metadynamics encourages the system to explore a wide range of conformational space by continuously adding an external history-dependent Gaussian bias potential to the selected degrees of freedom (70), i.e., collective variables. The ideal collective variable should capture the slowest degree of freedom of the physical behavior under study. The periodically adding bias potential to the slow degree of freedom makes it possible to reach a time scale much longer than that is achievable by standard MD simulations. Therefore, the high-dimensional free-energy landscape can be projected onto one dimension by the representative collective variable, which is reconstructed by removing the bias through reweighting (71, 72). In the well-tempered variant of metadynamics, the Gaussian height decreases with the bias accumulated over time by rescaling, ensuring the bias to converge more smoothly (73). The choice of a suitable collective variable is also critical to improve the efficiency of enhanced sampling.
We chose to do all-atom acceleration simulations by using the plug-in PLUMED 2 patched to LAMMPS (71). We performed well-tempered metadynamics with the BOO parameter Q6v as the collective variable relevant to crystallization. Since we are interested in the liquid-to-crystal phase transition, Q6v should be adequate because it can quantitatively reveal the degree of the ordering and differentiate the disordered-liquid phase and the ordered-solid phase (see fig. S2A). We scaled the studied temperatures by Tm so that the results at the same undercooling can be directly compared. To define the collective variable, we first calculated a complex vector for each atom as
| (5) |
where Y6m is the sixth-order spherical harmonics. s(rij) is a switching function that determines whether atom j is the nearest neighbor of atom i or not based on their distance. Q6v is the norm of the mean vector over all atoms. We set the bias factor of the well-tempered metadynamics ensemble to be 50 to 200, depending on the free-energy barrier to be surmounted. The bias potential was constructed by depositing the Gaussian every 2 ps with a width of 0.008 and an initial height of 10kBT. In simulations, the initial state of N = 432 atoms with the desired composition was equilibrated at a target temperature under the NPT ensemble (P = 0) for 1 ns. The well-tempered metadynamics with optimized parameters (by compromising the efficiency and accuracy) were then switched on to enhance the configuration space sampling. To assure the convergence of the free-energy profile, extremely long time metadynamics simulations were carried out until the diffusive behavior of phase transformation was observed (see fig. S2B); it generally takes 4000 ns at many state points. The total simulation time of metadynamics is ∼62 μs. Because of the high complexity of the collective variable, which needs to be calculated at every time step, a huge amount of computational resources have been consumed in all these simulations. From the measured free-energy profiles G(Q6v), we can calculate the Gibbs free-energy difference per particle between the liquid and the crystal by
| (6) |
The free-energy barrier to crystallization, , was estimated by the barrier preventing the liquid phase from transforming to the crystal in the free-energy profiles. It is thus different from the critical barrier in CNT and should rely on the collective variable (74).
Interface energy calculation by enhanced sampling
We carried out enhanced sampling simulations with Q6v (see above) as the collective variable to calculate the interface energy between the crystal and liquid phases. This simulation was only performed at Tm, at which the liquid phase and crystal phase have the same free energy. A perfect B2/bcc crystal lattice of 2304 atoms (18 × 8 × 8 unit cells) with the desired composition was first constructed and divided into two groups. We pinned one group and melted the other at a high temperature by heating. After equilibration, we released the pinned atoms and relaxed all atoms at Tm to make a configuration of crystal-liquid coexistence. After that, we constructed two collective variables, Q6v, for the liquid and crystal phases. On the one hand, to keep the crystal phase during the simulation, we limited its accessible phase space by adding restraint potential. This bias potential is expressed as k0((Q6v − 0.50)/s0)e0, where k0 is an energy constant, s0 is a rescaling factor, and e0 is the exponent of the power law. We selected 0.50 as a lower limit, below which the restraint potential starts acting on the system to prevent it from exploring the other phase space (71, 75). We chose k0 = 100, s0 = 0.008, and e0 = 2 such that this bias potential can effectively constrain the crystal phase so that the bcc/B2 structure cannot be melted during the simulation (see fig. S3). On the other hand, the liquid phase is free to visit its full phase space, which was accelerated by well-tempered metadynamics. The bias potential was constructed by depositing the Gaussians every 2 ps with a width of 0.008 and an initial height of 10kBT. We chose a large bias factor of 500 because of the large system size. During the simulations, the cell was only allowed to expand or contract along the long axis upon vitrification or crystallization. The free-energy profile of phase transformation of the liquid phase was reconstructed by reweighting. Because the liquid and crystal phases have the same free energy at Tm, the additional free-energy difference between the two phases (βΔG) should originate from the crystal-liquid interface. The interface energy was then computed as βΔG/2A, where A is the interface area. A is for NiAl and for Zr. The above description is explicit for calculating the interface energy along the (100) crystallographic interface, while the process is similar for the (110) crystal plane calculation.
Seeding technique simulations
The seeding method is very efficient in evaluating the critical nucleus size of a material. It is especially true for the good glass former CuZr. To determine the critical nucleus size, we embedded a perfect crystalline seed with various sizes in a liquid, and its probability of growing or dissolving was determined. In our study, the seed’s crystal structure should be B2 and bcc for the alloys and the pure metal, respectively, which are the equilibrium crystal structures. In detail, a perfect crystal of a designed composition and suitable lattice constant was initially constructed in a cubic box. The total number of atoms was 8192 (16 × 16 × 16 unit cells). Then, we grouped the atoms in a spherical volume of a designed radius in the center of the box as a seed. The seed was pinned, whereas the rest was equilibrated at a high temperature as aforementioned for 1 ns. Following that, the liquid was instantaneously quenched to an undercooled state and relaxed for ∼10 ps to adjust the thermal condition. Last, the seed and liquid were relaxed at the undercooled temperature for another 1 ns, during which the configurations were produced for analyses. By tuning the seed size and monitoring whether it grows or dissolves, we could measure the temperature-dependent critical nucleus size rc. At the critical size, the nuclei have an equal chance of growing or dissolving. The seed will disappear quickly below the critical size, whereas it grows very fast above the critical size. Ten independent simulations were carried out at each state point. The BOO parameters (see below) were used to characterize the behavior of the system. We also study the crystal growth kinetics by isothermal annealing of a supercooled liquid with a critical crystal nucleus embedded.
Structure analysis
The atomic-level structure was characterized by the radical Voronoi tessellation and the BOO parameters developed by Steinhardt et al. (76). The nearest neighbors and the face area of each polyhedral of each atom determined by the Voronoi analysis were used for the BOO analysis. To calculate the BOO parameters, first, a 2l + 1 vector of the l-fold symmetry for atom i was computed as
| (7) |
where Ni is the number of the nearest neighbors of atom i, m are integers from −l to l, and Ylm is the spherical harmonics. In our calculations, the contribution from the spherical harmonics of each neighbor was weighted by the fraction of the Voronoi face area separating the two atoms Aj in the total face area of the polyhedral Atot (77). The order parameter q6 was calculated as
| (8) |
To evaluate whether the local environment of each atom is crystalline or not, we calculated the normalized scalar product of qlm with l = 6 as
| (9) |
In this formula, atom j is one of the nearest neighbors of atom i and * means conjugate complex. When the value s(i, j) is larger than 0.7, the bond between i and j was considered as crystalline otherwise disordered. If the crystalline bond of an atom exceeds 10, then it was treated as crystalline. Therefore, the fraction of crystalline atoms, f(Sij), in a sample was obtained. It was used to evaluate Rc in Fig. 1A and to monitor the growth of the critical nucleus in Fig. 5. The parameter w6 is defined as
| (10) |
in which the term in parentheses is the Wigner 3-j symbol. In addition, the complex vector Qlm(i) coarse grained over its nearest neighbors was computed as
| (11) |
where Ni is the coordination number of particle i. The corresponding rotational invariant was then calculated from
| (12) |
W6 was calculated similar to w6 and . The combination of these order parameters is useful to detect structural orderings in the disordered liquid state. We treated atoms with w6 < − 0.023 as the central atoms of icosahedral clusters. To identify the CRYO, we combined Q6 and . We found that the threshold value Q6 = 0.25 can well differentiate ordered configurations from disordered ones. Furthermore, the crystal-like ordering of bcc-type prefers to have a positive . These cutoffs have been verified in (36, 78) and in figs. S9 and S10. One advantage of using the BOO parameters is that both perfect and distorted ones are taken into consideration. Note that they are all important for crystallization and glass formation. Furthermore, because we are investigating the crystallization resistance that resulted from competing structural orderings, the atomic cluster running over the first coordination shell, based on which the local symmetry is built, should be considered to characterize both the icosahedral and crystal-like environments.
Supplementary Material
Acknowledgments
Y.-C.H. thanks C. Tang and Y. Wang (University of Copenhagen) for helpful discussion. Funding: This work was partially supported by Specially Promoted Research (JP25000002 and JP20H05619) and Scientific Research (A) (JP18H03675) from the Japan Society for the Promotion of Science (JSPS). Y.-C.H. is grateful for the financial support from a JSPS fellowship (JP19F19021). Author contributions: Y.-C.H. and H.T. designed research; H.T. supervised research; Y.-C.H. performed research; Y.-C.H. and H.T. analyzed data and wrote the paper. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the corresponding author.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/50/eabd2928/DC1
REFERENCES AND NOTES
- 1.Egami T., Universal criterion for metallic glass formation. Mater. Sci. Eng. A 226–228, 261–267 (1997). [Google Scholar]
- 2.Johnson W. L., Bulk glass-forming metallic alloys: Science and technology. MRS Bull. 24, 42–56 (1999). [Google Scholar]
- 3.Inoue A., Stabilization of metallic supercooled liquid and bulk amorphous alloys. Acta Mater. 48, 279–306 (2000). [Google Scholar]
- 4.Nishiyama N., Takenaka K., Miura H., Saidoh N., Zeng Y., Inoue A., The world’s biggest glassy alloy ever made. Intermetallics 30, 19–24 (2012). [Google Scholar]
- 5.Klement Jun W., Willens R. H., Duwez P., Non-crystalline structure in solidified gold–silicon alloys. Nature 187, 869–870 (1960). [Google Scholar]
- 6.Zhong L., Wang J., Sheng H., Zhang Z., Mao S. X., Formation of monatomic metallic glasses through ultrafast liquid quenching. Nature 512, 177–180 (2014). [DOI] [PubMed] [Google Scholar]
- 7.Demetriou M. D., Launey M. E., Garrett G., Schramm J. P., Hofmann D. C., Johnson W. L., Ritchie R. O., A damage-tolerant glass. Nat. Mater. 10, 123–128 (2011). [DOI] [PubMed] [Google Scholar]
- 8.Kumar G., Tang H. X., Schroers J., Nanomoulding with amorphous metals. Nature 457, 868–872 (2009). [DOI] [PubMed] [Google Scholar]
- 9.Greer A. L., Metallic glasses on the threshold. Mater. Today 12, 14–22 (2009). [Google Scholar]
- 10.Plummer J., Is metallic glass poised to come of age? Nat. Mater. 14, 553–555 (2015). [DOI] [PubMed] [Google Scholar]
- 11.Wuttig M., Yamada N., Phase-change materials for rewriteable data storage. Nat. Mater. 6, 824–832 (2007). [DOI] [PubMed] [Google Scholar]
- 12.Lencer D., Salinga M., Wuttig M., Design rules for phase-change materials in data storage applications. Adv. Mater. 23, 2030–2058 (2011). [DOI] [PubMed] [Google Scholar]
- 13.Wei S., Lucas P., Angell C. A., Phase-change materials: The view from the liquid phase and the metallicity parameter. MRS Bull. 44, 691–698 (2019). [Google Scholar]
- 14.Kooi B. J., Wuttig M., Chalcogenides by design: Functionality through metavalent bonding and confinement. Adv. Mater. 32, 1908302 (2020). [DOI] [PubMed] [Google Scholar]
- 15.Takeuchi A., Inoue A., Classification of bulk metallic glasses by atomic size difference, heat of mixing and period of constituent elements and its application to characterization of the main alloying element. Mater. Trans. 46, 2817–2829 (2005). [Google Scholar]
- 16.Zhang K., Wang M., Papanikolaou S., Liu Y., Schroers J., Shattuck M. D., O’Hern C. S., Computational studies of the glass-forming ability of model bulk metallic glasses. J. Chem. Phys. 139, 124503 (2013). [DOI] [PubMed] [Google Scholar]
- 17.Laws K. J., Miracle D. B., Ferry M., A predictive structural model for bulk metallic glasses. Nat. Commun. 6, 8123 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Turnbull D., Under what conditions can a glass be formed? Contemp. Phys. 10, 473–488 (1969). [Google Scholar]
- 19.Lu Z. P., Liu C. T., A new glass-forming ability criterion for bulk metallic glasses. Acta Mater. 50, 3501–3512 (2002). [Google Scholar]
- 20.Greer A. L., New horizons for glass formation and stability. Nat. Mater. 14, 542–546 (2015). [DOI] [PubMed] [Google Scholar]
- 21.Johnson W. L., Na J. H., Demetriou M. D., Quantifying the origin of metallic glass formation. Nat. Commun. 7, 10313 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dai R., Ashcraft R., Gangopadhyay A. K., Kelton K. F., Predicting metallic glass formation from properties of the high temperature liquid. J. Non Cryst. Solids 525, 119673 (2019). [Google Scholar]
- 23.Cheng Y. Q., Ma E., Sheng H. W., Atomic level structure in multicomponent bulk metallic glass. Phys. Rev. Lett. 102, 245501 (2009). [DOI] [PubMed] [Google Scholar]
- 24.Yang L., Guo G. Q., Chen L. Y., Huang C. L., Ge T., Chen D., Liaw P. K., Saksl K., Ren Y., Zeng Q. S., La Qua B., Chen F. G., Jiang J. Z., Atomic-scale mechanisms of the glass-forming ability in metallic glasses. Phys. Rev. Lett. 109, 105502 (2012). [DOI] [PubMed] [Google Scholar]
- 25.Shintani H., Tanaka H., Frustration on the way to crystallization in glass. Nat. Phys. 2, 200–206 (2006). [Google Scholar]
- 26.Tanaka H., Bond orientational order in liquids: Towards a unified description of water-like anomalies, liquid-liquid transition, glass transition, and crystallization. Eur. Phys. J. E. 35, 113 (2012). [DOI] [PubMed] [Google Scholar]
- 27.Wang W. H., Lewandowski J. J., Greer A. L., Understanding the glass-forming ability of Cu50Zr50 alloys in terms of a metastable eutectic. J. Mater. Res. 20, 2307–2313 (2005). [Google Scholar]
- 28.Wang D., Li Y., Sun B. B., Sui M. L., Lu K., Ma E., Bulk metallic glass formation in the binary Cu–Zr system. Appl. Phys. Lett. 84, 4029–4031 (2004). [Google Scholar]
- 29.Reutzel S., Hartmann H., Galenko P. K., Schneider S., Herlach D. M., Change of the kinetics of solidification and microstructure formation induced by convection in the Ni–Al system. Appl. Phys. Lett. 91, 041913 (2007). [Google Scholar]
- 30.Angell C. A., Formation of glasses from liquids and biopolymers. Science 267, 1924–1935 (1995). [DOI] [PubMed] [Google Scholar]
- 31.Desgranges C., Delhommelle J., Can ordered precursors promote the nucleation of solid solutions? Phys. Rev. Lett. 123, 195701 (2019). [DOI] [PubMed] [Google Scholar]
- 32.K. F. Kelton, A. L. Greer, Nucleation in Condensed Matter: Applications in Materials and Biology (Pergamon, 2010). [Google Scholar]
- 33.Russo J., Romano F., Tanaka H., Glass forming ability in systems with competing orderings. Phys. Rev. X 8, 021040 (2018). [Google Scholar]
- 34.Angioletti-Uberti S., Ceriotti M., Lee P. D., Finnis M. W., Solid-liquid interface free energy through metadynamics simulations. Phys. Rev. B 81, 125416 (2010). [Google Scholar]
- 35.Kawasaki T., Tanaka H., Formation of a crystal nucleus from liquid. Proc. Natl. Acad. Sci. U.S.A. 107, 14036–14041 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Russo J., Tanaka H., The microscopic pathway to crystallization in supercooled liquids. Sci. Rep. 2, 505 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tang C., Harrowell P., Anomalously slow crystal growth of the glass-forming alloy CuZr. Nat. Mater. 12, 507–511 (2013). [DOI] [PubMed] [Google Scholar]
- 38.Tanaka H., A simple physical model of liquid-glass transition: Intrinsic fluctuating interactions and random fields hidden in glass-forming liquids. J. Phys. Condens. Matter 10, L207–L214 (1998). [Google Scholar]
- 39.Frank F. C., Supercooling of liquids. Proc. R. Soc. Lond. A 215, 43–46 (1952). [Google Scholar]
- 40.Jakse N., Pasturel A., Local order of liquid and supercooled zirconium by Ab Initio molecular dynamics. Phys. Rev. Lett. 91, 195501 (2003). [DOI] [PubMed] [Google Scholar]
- 41.Tanaka H., Roles of local icosahedral chemical ordering in glass and quasicrystal formation in metallic glass formers. J. Phys. Condens. Matter 15, L491–L498 (2003). [Google Scholar]
- 42.Tanaka H., Relationship among glass-forming ability, fragility, and short-range bond ordering of liquids. J. Non Cryst. Solids 351, 678–690 (2005). [Google Scholar]
- 43.Kelton K. F., Lee G. W., Gangopadhyay A. K., Hyers R. W., Rathz T. J., Rogers J. R., Robinson M. B., Robinson D. S., First X-ray scattering studies on electrostatically levitated metallic liquids: Demonstrated influence of local icosahedral order on the nucleation barrier. Phys. Rev. Lett. 90, 195504 (2003). [DOI] [PubMed] [Google Scholar]
- 44.Xi X. K., Li L. L., Zhang B., Wang W. H., Wu Y., Correlation of atomic cluster symmetry and glass-forming ability of metallic glass. Phys. Rev. Lett. 99, 095501 (2007). [DOI] [PubMed] [Google Scholar]
- 45.Hirata A., Kang L. J., Fujita T., Klumov B., Matsue K., Kotani M., Yavari A. R., Chen M. W., Geometric frustration of icosahedron in metallic glasses. Science 341, 376–379 (2013). [DOI] [PubMed] [Google Scholar]
- 46.Luo W. K., Sheng H. W., Alamgir F. M., Bai J. M., He J. H., Ma E., Icosahedral short-range order in amorphous alloys. Phys. Rev. Lett. 92, 145502 (2004). [DOI] [PubMed] [Google Scholar]
- 47.Sheng H. W., Luo W. K., Alamgir F. M., Bai J. M., Ma E., Atomic packing and short-to-medium-range order in metallic glasses. Nature 439, 419–425 (2006). [DOI] [PubMed] [Google Scholar]
- 48.Shen Y. T., Kim T. H., Gangopadhyay A. K., Kelton K. F., Icosahedral order, frustration, and the glass transition: Evidence from time-dependent nucleation and supercooled liquid structure studies. Phys. Rev. Lett. 102, 057801 (2009). [DOI] [PubMed] [Google Scholar]
- 49.Cheng Y. Q., Ma E., Atomic-level structure and structure-property relationship in metallic glasses. Prog. Mater. Sci. 56, 379–473 (2011). [Google Scholar]
- 50.Ding J., Cheng Y.-Q., Ma E., Full icosahedra dominate local order in Cu64Zr34 metallic glass and supercooled liquid. Acta Mater. 69, 343–354 (2014). [Google Scholar]
- 51.Gonzalez B., Bechelli S., Essafri I., Piquet V., Desgranges C., Delhommelle J., Competition between crystalline and icosahedral order during crystal growth in bimetallic systems. J. Cryst. Growth 478, 22–27 (2017). [Google Scholar]
- 52.Tanaka H., Role of many-body correlation in slow dynamics of glass-forming liquids: Intrinsic or perturbative. J. Stat. Mech. 2020, 034003 (2020). [Google Scholar]
- 53.Tan P., Xu N., Xu L., Visualizing kinetic pathways of homogeneous nucleation in colloidal crystallization. Nat. Phys. 10, 73–79 (2014). [Google Scholar]
- 54.Gaskell P. H., A new structural model for transition metal–metalloid glasses. Nature 276, 484–485 (1978). [Google Scholar]
- 55.Schwarz R. B., Johnson W. L., Formation of an amorphous alloy by solid-state reaction of the pure polycrystalline metals. Phys. Rev. Lett. 51, 415–418 (1983). [Google Scholar]
- 56.Fujita T., Konno K., Zhang W., Kumar V., Matsuura M., Inoue A., Sakurai T., Chen M. W., Atomic-scale heterogeneity of a multicomponent bulk metallic glass with excellent glass forming ability. Phys. Rev. Lett. 103, 075502 (2009). [DOI] [PubMed] [Google Scholar]
- 57.Altounian Z., Guo-hua T., Strom-Olsen J. O., Crystallization characteristics of Cu-Zr metallic glasses from Cu70Zr30to Cu25Zr75. J. Appl. Phys. 53, 4755–4760 (1982). [Google Scholar]
- 58.Wang Y., Yao J., Li Y., Glass formation adjacent to the intermetallic compounds in Cu-Zr binary system. J. Mater. Sci. Technol. 34, 605–612 (2018). [Google Scholar]
- 59.Levchenko E. V., Evteev A. V., Belova I. V., Murch G. E., Molecular dynamics determination of the time-temperature-transformation diagram for crystallization of an undercooled liquid Ni50Al50 alloy. Acta Mater. 59, 6412–6419 (2011). [Google Scholar]
- 60.Kurtuldu G., Shamlaye K. F., Löffler J. F., Metastable quasicrystal-induced nucleation in a bulk glass-forming liquid. Proc. Natl. Acad. Sci. U.S.A. 115, 6123–6128 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zemp J., Celino M., Schönfeld B., Löffler J. F., Crystal-like rearrangements of icosahedra in simulated copper-zirconium metallic glasses and their effect on mechanical properties. Phys. Rev. Lett. 115, 165501 (2015). [DOI] [PubMed] [Google Scholar]
- 62.Ryltsev R. E., Klumov B. A., Chtchelkatchev N. M., Shunyaev K. Y., Nucleation instability in supercooled Cu-Zr-Al glass-forming liquids. J. Chem. Phys. 149, 164502 (2018). [DOI] [PubMed] [Google Scholar]
- 63.Lu I.-R., Wilde G., Görler G. P., Willnecker R., Thermodynamic properties of Pd-based glass-forming alloys. J. Non Cryst. Solids 250-252, 577–581 (1999). [Google Scholar]
- 64.Busch R., Liu W., Johnson W. L., Thermodynamics and kinetics of the Mg65Cu25Y10 bulk metallic glass forming liquid. J. Appl. Phys. 83, 4134–4141 (1998). [Google Scholar]
- 65.Busch R., Schroers J., Wang W. H., Thermodynamics and kinetics of bulk metallic glass. MRS Bull. 32, 620–623 (2007). [Google Scholar]
- 66.Plimpton S., Fast parallel algorithms for short-range molecular dynamics. J. Comput. Phys. 117, 1–19 (1995). [Google Scholar]
- 67.Mendelev M. I., Kramer M. J., Ott R. T., Sordelet D. J., Yagodin D., Popel P., Development of suitable interatomic potentials for simulation of liquid and amorphous Cu–Zr alloys. Philos. Mag. 89, 967–987 (2009). [Google Scholar]
- 68.Mishin Y., Mehl M. J., Papaconstantopoulos D. A., Embedded-atom potential for B2−NiAl. Phys. Rev. B 65, 224114 (2002). [Google Scholar]
- 69.Gunawardana K. G. S. H., Wilson S. R., Mendelev M. I., Song X., Theoretical calculation of the melting curve of Cu-Zr binary alloys. Phys. Rev. E 90, 052403 (2014). [DOI] [PubMed] [Google Scholar]
- 70.Laio A., Parrinello M., Escaping free-energy minima. Proc. Natl. Acad. Sci. U.S.A. 99, 12562–12566 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tribello G. A., Bonomi M., Branduardi D., Camilloni C., Bussi G., PLUMED 2: New feathers for an old bird. Comput. Phys. Commun. 185, 604–613 (2014). [Google Scholar]
- 72.Tiwary P., Parrinello M., A time-independent free energy estimator for metadynamics. J. Phys. Chem. B 119, 736–742 (2014). [DOI] [PubMed] [Google Scholar]
- 73.Barducci A., Bussi G., Parrinello M., Well-tempered metadynamics: A smoothly converging and tunable free-energy method. Phys. Rev. Lett. 100, 020603 (2008). [DOI] [PubMed] [Google Scholar]
- 74.Niu H., Piaggi P. M., Invernizzi M., Parrinello M., Molecular dynamics simulations of liquid silica crystallization. Proc. Natl. Acad. Sci. U.S.A. 115, 5348–5352 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tribello G. A., Giberti F., Sosso G. C., Salvalaglio M., Parrinello M., Analyzing and driving cluster formation in atomistic simulations. J. Chem. Theory Comput. 13, 1317–1327 (2017). [DOI] [PubMed] [Google Scholar]
- 76.Steinhardt P. J., Nelson D. R., Ronchetti M., Bond-orientational order in liquids and glasses. Phys. Rev. B 28, 784–805 (1983). [Google Scholar]
- 77.Mickel W., Kapfer S. C., Schörder-Turk G. E., Mecke K., Shortcomings of the bond orientational order parameters for the analysis of disordered particulate matter. J. Chem. Phys. 138, 044501 (2013). [DOI] [PubMed] [Google Scholar]
- 78.Leocmach M., Tanaka H., Roles of icosahedral and crystal-like order in the hard spheres glass transition. Nat. Commun. 3, 974 (2012). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/50/eabd2928/DC1