

Nanoscale biomaterial tools reveal impacts of lymphatic mechanisms of transport on CD8+ T cell immunity elicited in melanoma.
Abstract
Revealing the mechanisms that underlie the expansion of antitumor CD8+ T cells that are associated with improved clinical outcomes is critical to improving immunotherapeutic management of melanoma. How the lymphatic system, which orchestrates the complex sensing of antigen by lymphocytes to mount an adaptive immune response, facilitates this response in the context of malignancy is incompletely understood. To delineate the effects of lymphatic transport and tumor-induced lymphatic and lymph node (LN) remodeling on the elicitation of CD8+ T cell immunity within LNs, we designed a suite of nanoscale biomaterial tools enabling the quantification of antigen access and presentation within the LN and resulting influence on T cell functions. The expansion of antigen-specific stem-like and cytotoxic CD8+ T cell pools was revealed to be sensitive to the mechanism of lymphatic transport to LNs, demonstrating the potential for nanoengineering strategies targeting LNs to optimize cancer immunotherapy in eliciting antitumor CD8+ T cell immunity.
INTRODUCTION
Melanoma remains a substantial clinical problem, representing the sixth most common cancer worldwide, with only ~20% of patients surviving at least 5 years following diagnosis of advanced disease and ~41 to 55% of patients demonstrating recurrent disease (1). Immunotherapy, which focuses on reinvigorating antitumor immune responses, particularly on the part of T cells, has emerged as the most promising previously unidentified approach to increase patient survival through its potential to both treat advanced disseminated disease and protect against recurrence. However, response rates in advanced melanoma are disappointingly low: Only ~10 to 33% of patients respond to immune checkpoint blockade (ICB) therapy (2). Enhanced survival is independently associated with several features of a robust antitumor CD8+ T cell immune response including the number of these cells within the tumor (3). More specifically, higher levels of CD8+ T cells expressing granzyme B (GzmB), a marker of degranulation, and interferon-γ (IFN-γ), an inflammatory cytokine, along with immune checkpoint and antigen (Ag) experience marker programmed cell death 1 (PD1) within the tumor are associated with improved survival (4–6). The abundance of CD8+ T cells expressing PD1 in combination with transcription factor Tcf1, which marks a stem-like cell subtype with high proliferative potential and capacity to differentiate into cells with antitumor lytic functions (7, 8), is also a prognostic indicator in melanoma (5, 6). Thus, the generation of both high-quantity and high-quality Ag-primed T cells in the tumor microenvironment (TME) is an important step in inducing effective antitumor immunity, the holy grail to broaden the benefits afforded by cancer immunotherapy.
To this end, the generation of a large and robust pool of tumor-specific CD8+ T cells, the target of ICB and many other immunotherapeutic strategies, relies on priming of T cells by Ag-presenting cells (APCs) (9). The activation status and Ag presentation by APCs, which include dendritic cells (DCs) and macrophages, affect the differentiation of naïve T cells and, thus, their resulting functionality. To facilitate Ag sensing by rare cognate T cells, encounters with APC-presented Ag are tightly orchestrated within lymphocyte-rich secondary lymphoid organs. These include lymph nodes (LNs), whose structure is specialized to facilitate both tight interactions between immune cell subsets (10) and Ag dispersal among cell subtypes that are locally distributed in a spatially prescribed manner. Specifically, Ag derived from peripheral tissues accesses LNs by lymphatic vessel–mediated lymph drainage to the downstream, draining LN (dLN) (10–12). This lymph enters the LN at the subcapsular sinus (SCS), a barrier lined with fibroblastic reticular cells, APCs, and lymphatic endothelial cells (10). From the SCS, lymph-borne solutes diffuse into the LN cortex where B cells, other APCs, and intrafollicular T cells reside (10, 13, 14) or, in a highly size-restricted manner (10, 13, 15), enter conduits to reach the T cell zones of the LN paracortex. APCs also patrol peripheral tissues before migration to dLNs under both steady-state and inflamed conditions (11, 12, 16, 17) to disperse in a chemotactically driven manner among discrete LN zones (16–20). Ag access to strategically distributed APCs colocalized with LN-resident lymphocytes is, thus, dependent on a number of barriers that are sensitive to the mechanism of transport to the LN.
In response to disease, both the tumor and dLN remodel in cancer, with potential impacts on Ag sensing and the resulting magnitude and functions of the antitumor immune response. TME-localized immunosuppression has been widely recognized as a cancer hallmark (21) with consistent and advancing suppression occurring throughout tumor development and progression (22). In particular, T cell activation in the TME is impaired (3, 5, 6) and tumor-associated DCs show high PD1 ligand and B7 family expression, two features associated with impaired prognosis (6). Likewise, immunosuppressive tumor-associated macrophages (23) as well as regulatory T cells and myeloid-derived suppressor cells infiltrate the TME to high extents (24). Melanomas (25, 26), similar to other cancer types (27–30), have also been shown to have high peritumoral lymphatic expansion, a feature associated with increased lymphatic metastasis (27–30). As such, multiple immunosuppressive pathways active within the TME have the potential to be recapitulated within LNs downstream of progressing primary tumors (so-called sentinel LNs or TdLNs). These include suppressed T cell responses by tumor-induced myeloid-derived suppressor cells, inhibition of DC-mediated T cell priming (31), and local persistence of regulatory T cells (32). Moreover, cellular components of LN structures also remodel in cancer. For example, fibroblastic reticular cells, which help to maintain the SCS of the LN (33, 34), exhibit increased permeability and activation in melanoma (35). Similarly, biophysical features of the remodeled TME associated with tumor immune suppression (36, 37) have been observed in TdLNs, including alterations in tissue stiffness and intranodal pressures (38) that parallel changes within LNs in other immune-regulated pathologies (39–41). How these remodeling pathways affect the distribution of tumor-derived Ag among cells within the TdLN to modulate the development of Ag-specific T cell immunity, however, remains undefined.
In this work, we designed a suite of biomaterial tools to enable the analysis of lymphatic transport, cellular association, and Ag presentation to delineate the concerted influence of these interrelated processes on the generation of CD8+ T cell immunity within LNs. This includes a tracer system for the evaluation of macromolecular transport via lymphatic vessels to cells within the dLN and a synthetic Ag system to evaluate Ag presentation and its sensing by T cells. We applied these systems to assess the impacts of lymphatic transport mechanism on Ag accessibility to and presentation by cells of the dLN, the resulting functions of the expanded Ag-specific CD8+ T cell pool, and how each of these processes is modified in the tumor context. CD8+ T cell immunity elicited within TdLNs was found to be compartmentalized by route of lymphatic transport and to remain therapeutically functional in a melanoma mouse model. These results provide previously unknown insight into how immunotherapeutic strategies for melanoma might be improved by leveraging the unique microenvironment and Ag-sensing capabilities of TdLNs.
RESULTS
Constitutive transport barriers and mechanisms regulate access by LN-resident immune cells to skin-derived macromolecules
A panel of fluorescent tracers (table S1 and fig. S1) (11, 42) was co-infused into the skin of naïve animals and, at various times after administration, dLNs were analyzed for both total levels of tracer accumulation as well as tracer distribution profiles among resident leukocytes. Thirty- and 10-nm dextrans (500 and 40 kDa, respectively) versus 500-nm polystyrene spheres were implemented to compare the extents of lymphatic transport to the dLN, resulting from either of two modes of lymphatic transport from the periphery—passive lymph drainage versus active transport mediated by emigrating APCs (fig. S1).
Ten- and 30-nm dextrans accumulated within the dLN at early time points after injection (fig. S1), particularly in the SCS (Fig. 1A and fig. S2, A and B). Correspondingly, an increasing depth of penetration by 10-nm relative to 30-nm dextran was observed in the parenchyma of dLNs (Fig. 1B, top), as would be expected given the size exclusion barrier of quiescent (noninflamed) LN conduits (13–15). Spheres (500 nm) instead appreciably accumulated at detectable levels within dLNs only 1 to 3 days after injection (Fig. 1A and figs. S1 and S2, A and B) and, in contrast to smaller dextran tracers, did so not within the SCS of dLN but instead deeper in the LN parenchyma (Fig. 1B, bottom, and fig. 1C). As a result, small dextrans accumulated within the LN far more capsule-proximally compared to the large spheres (Fig. 1C). This is consistent with the dextrans having been transported by direct uptake into lymph after injection and drainage into dLNs, as is supported by high levels of dLN accumulation at all analyzed times after injection (fig. S1), as well as their restriction to the SCS. Contrastingly, larger spheres were not appreciably detected within dLNs by confocal imaging until after the 4-hour analysis time point (fig. S1). Likewise, the 500-nm tracer appears as punctate points primarily deeper within the parenchyma of the LN, in locations accessed by skin migratory cells (16, 17, 19). This is consistent with their transport to dLNs being mediated by cells migrating from the peripheral tissue of administration, a process that transpires more slowly (16, 17, 19). Resulting levels of total cells within dLNs that associated with tracers were highest for dextrans directly transported via lymph compared to the overall very low level of total 500-nm sphere association with dLN cells (~100 to 1000× difference). The 10-nm dextran associated with a higher number of cells (~5×) compared to 30-nm dextran (Fig. 1D), a result consistent with the 10-nm dextrans’ capacity to more deeply penetrate the LN parenchyma (Fig. 1, A and B, and figs. S1 and S2, A and B) despite overall lower levels of dLN accumulation (fig. S1).
Fig. 1. Hydrodynamic size regulates biomolecular access to different regions and cells of LNs draining site of intradermal injection.
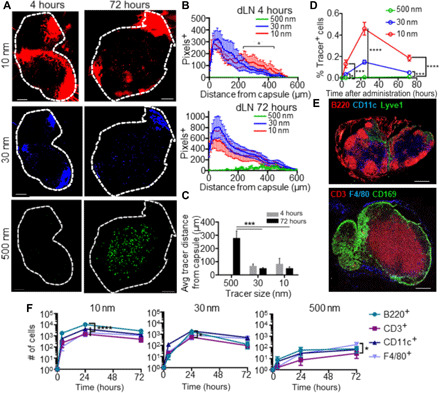
(A to C) Ten-, 30-, and 500-nm tracer distributions within LN draining the intradermal injection site 4 and 72 hours after administration. (A) Representative dLN images. (B) Quantified tracer distances from the dLN capsule. (C) Average tracer-to-LN capsule distance. (D) Total number of tracer+ cells within dLNs 4, 24, and 72 hours after intradermal injection, as measured by flow cytometry. (E) Immunohistochemically measured distributions of cellular subtypes within in LNs. (F) Frequencies of marker-positive cells of total CD45+ cells that are tracer positive within LNs draining the injection site 4, 24, and 72 hours after intradermal administration, assessed flow cytometrically. Scale bars, 200 μm; * indicates significance by two-way analysis of variance (ANOVA) (B to D) or one-way ANOVA (F) with Tukey’s comparison (* indicates P < 0.05, *** indicates P < 0.005, **** indicates P < 0.001). n = 5 to 8 mice; (A) to (D) and (F) are representative of at least two independent experiments.
Within LNs, leukocyte distributions are tightly organized in a spatially defined manner, with B cells (the substantial majority of B220+ dLN-resident cells), DCs (predominantly CD11c+), and SCS macrophages (CD169+) versus T cells (CD3+) being more LN capsule proximal versus distal, respectively (Fig. 1E and fig. S2C). To determine the effects of lymphatic transport mechanism on patterns of leukocyte uptake, tracer uptake was flow cytometrically assessed using a cell extraction protocol that liberated leukocyte but not stromal cell populations of analyzed LNs (43, 44). Consistent with their distinct patterns of spatial distributions within dLNs, tracers of different sizes exhibited differential access to not only differing numbers of dLN-resident cells (Fig. 1D) but also differing leukocyte (CD45+) subtypes (Fig. 1F). In particular, 10-nm tracers were found to accumulate at all measured times most prodigiously in B220+ (B) cells, again consistent with their efficient transit into LN conduits after accumulation within the SCS (Fig. 1F, left) (13–15). In contrast, the 30-nm tracer, which, once transited to the LN, was more restricted to the SCS, was found within more CD11c+ (DC) rather than B220+ cells (Fig. 1F, middle). Further differentiation into more defined cell subtypes [B cells, T cells, plasmacytoid DCs (pDCs), conventional DCs (cDCs), SCS macrophages (SSMs), medullary sinus macrophages (MSMs), medullary cord macrophages (MCMs), dermal DCs (dDCs), and Langerhans cells (LCs) (fig. S3)] revealed the 30- but not the 10-nm tracer to be restricted to LN-resident phagocytic leukocytes positioned at the sinus, while the 500-nm tracer was primarily within skin migratory cells, e.g., dDCs and LCs (fig. S2D and Fig. 3). Constitutive barriers to lymphatic transport and transit into the LN parenchyma via conduits thus substantially influence the resulting distribution of tracers among LN-resident leukocytes.
Fig. 3. Passive lymphatic drainage in the naïve and melanoma contexts.
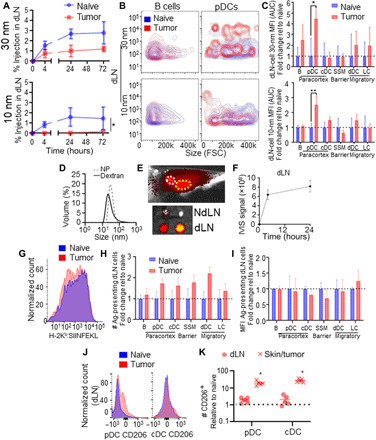
(A) Thirty- and 10-nm dextran bulk accumulation in LNs draining skin or day 7 B16F10 melanomas over time. (B) Representative flow cytometry plots of 30- and 10-nm dextran accumulation within B cells and pDCs of the dLN. FSC, forward scatter. (C) MFI of 30- and 10-nm tracer signal in LNs draining the skin or B16F10 melanomas over 72 hours after injection (AUC). (D) Dynamic light scattering measurements of 500-kDa (30 nm) dextrans and Ag-NPs. (E) NPs (30 nm) injected intradermally accumulate within axillary (white circle) and brachial (yellow circle) dLNs 24 hours after intradermal injection (positive pixels as collected by IVIS imaging are red-yellow, negative background is gray scale), quantified in (F). Flow cytometry histograms of Ag presentation (25D1.16 staining for H-2Kb:SIINFEKL) among CD45+ cells within dLNs (G), number of dLN cells presenting Ag relative to the naïve condition (H), and as MFI of H-2Kb:SIINFEKL signal of positive dLN cells (I) 24 hours after intradermal injection of Ag-NP. CD206 signal (J) among pDCs and cDCs in the skin/tumor and dLN of naïve and day 10 B16F10-bearing animals, quantified in (K), normalized to values in naïve skin and dLN. * indicates significance by one-way ANOVA with Tukey’s post hoc comparison (* indicates P < 0.05, ** indicates P < 0.01); n = 5 to 6 animals; (A) to (F) are representative of at least two independent experiments.
Melanomas mature peripheral DCs and alter the dynamics of cell-trafficked Ag to dLNs, resulting in modified Ag presentation
Tumors locally modulate the functions of APCs, which are necessary for the effective generation of antitumor immunity (fig. S4). As such, tumor effects on APCs within TdLNs were evaluated. DCs in both skin/tumor and dLNs of day 10 B16F10 melanoma–bearing mice were found to exhibit both higher CD86 and lower CD206 levels [by both number of cells and mean fluorescence index (MFI) of positive cells] among DEC205+ DCs (both dDCs and LCs) compared to naïve animals (Fig. 2, A and B). Thus, melanomas induce maturation and decreased tolerogenicity among skin-derived DCs in both the tumor and the dLN, with potential effects on DC trafficking, phagocytosis, and Ag presentation.
Fig. 2. APC migration from the skin to the dLN in the naïve and melanoma contexts.
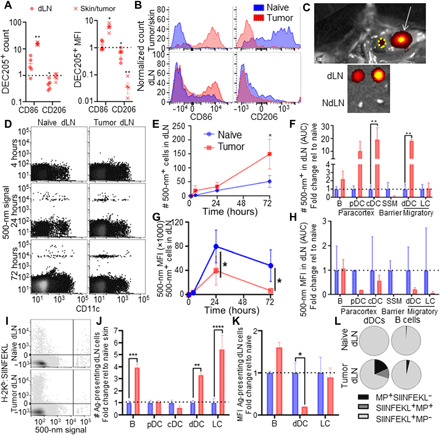
CD86 and CD206 signal (A and B) in CD11c+DEC205+ cells of the skin or tumor and dLN of naïve and day 10 B16F10-bearing animals, with the MFI and number of cells expressing CD86 and CD206 quantified, both normalized to values in the naïve skin/LNs. (C) IVIS (in vivo imaging system) imaging showing a site of 500-nm injection (white arrow) and LN drainage (yellow circle), demonstrating specific accumulation in dLNs after 72 hours (positive pixels as collected by IVIS imaging are red-yellow, negative background is gray scale). (D) Accumulation of 500-nm tracer+ cells in the dLN over time, analyzed flow cytometrically. Five hundred–nanometer accumulation among CD45+ cells in LNs draining skin or day 7 B16F10 melanomas over time, assessed flow cytometrically (E). Fold change in the area under the curve (AUC) (0 to 72 hours) of number of cells containing MPs within the dLN relative to naïve accumulation (F). MFI of MP fluorescent signal (G) in LNs draining the MP-injected skin or B16F10 melanomas over time of all MP-containing cells, and as fold change of AUC (0 to 72 hours) within individual cell subtypes relative to naïve tissues at 4, 24, and 72 hours (H). Representative flow cytometry plots of 500-nm tracer uptake and Ag presentation, as assessed by 25D1.16 staining for H-2Kb:SIINFEKL 72 hours after Ag-MP injection into naïve skin or day 7 B16F10 melanomas (I). Number (J) and MFI (K) of cells presenting SIINFEKL Ag in the TdLN relative to the naïve skin dLN at 72 hours after Ag-MP administration as determined by 25D1.16 staining for H-2Kb:SIINFEKL. (L) MP and H-2Kb:SIINFEKL (SIINFEKL) positivity among dLN B cells and dDCs in naïve and tumor contexts 72 hours after Ag-MP administration. * indicates significance by one-way ANOVA; n = 5 to 6 animals (* indicates P < 0.05, ** indicates P < 0.01, *** indicates P < 0.005, **** indicates P < 0.0001); (D) to (H) are representative of at least two independent studies.
In addition to the immunological microenvironment, tumors also induce local remodeling of the tissue interstitium and vasculature, hallmarks of the disease (21) likely to influence lymphatic transport and resulting Ag access within the TdLN (fig. S4). B16F10 melanomas, for example, exhibit increased lymphatic vascularization and T cell infiltration compared to the naïve skin (fig. S5), recapitulating spontaneous melanoma models (35). As such, the tracer panel (Fig. 1 and fig. S1) was implemented to probe lymphatic transport effects on Ag access and how this is affected by disease (fig. S4). After injection into day 7 B16F10 tumors, 500-nm tracers were transported to TdLNs (Fig. 2C) and were associated with dLN-resident cells at extents greater than those seen in LNs draining the naïve skin, as determined by flow cytometric measurement of tracer fluorescence (Fig. 2, D and E). Of the migratory cells assessed, numbers of 500-nm+ dDCs but not LCs were increased (Fig. 2, D and F). Numbers of 500-nm+ cDCs, an LN-resident cell subtype, were also increased (Fig. 2, D and F), presumably due to tracer handoff from immigrating 500-nm+ cells (18, 45). As mature APCs tend to be more migratory (20, 46), this is in line with changes in APC maturation induced by the tumor (Fig. 2, A and B). When evaluated on a per-cell basis, more cells within dLNs were 500 nm+ overall (Fig. 2E), although among all CD45+ 500-nm+ cells, 500-nm tracer MFI was decreased at both 24 and 72 hours after injection in tumor-bearing compared to naïve animals (Fig. 2G). Likewise, the 500-nm MFI trended downward within individual cell subtypes (Fig. 2H). These data can be interpreted in two ways. As tumors induce APCs to mature and become more migratory, they may also become less phagocytic, bringing less Ag per cell to the dLN. It is also possible that decreases in measured per-cell tracer MFIs result from tracer dilution within TME because of the rapidly growing tissue mass. Nevertheless, these data demonstrate that, overall, APC trafficking from the skin is sustained within melanomas.
As alterations in APC maturation, total migrating cell numbers, and their carried payload are likely to affect Ag presentation within dLNs, animals were subjected to antigenic challenge via an engineered synthetic Ag system (fig. S4). In this configuration, 500-nm particles [microparticles (MPs)] were covalently attached via a disulfide linkage that is reversible within the reducing intracellular environment of APCs to the H-2Kb peptide of chicken albumin (OVA257-264) with an N-terminal cysteine (fig. S6) (47, 48). These COVA257-264-MPs were injected into the skin of naïve or tumor of B16F10 melanoma–bearing animals, and Ag presentation was assessed 72 hours later through measurement of H-2Kb:OVA257-264 staining (Fig. 2I) (49). Consistent with the known transfer of lymphatic-transported Ag between LN-resident cells (18, 45), most cells presenting Ag within dLNs were found to not contain the 500-nm sphere (Fig. 2I). Examining all H-2Kb:OVA257-264+ cells, numbers of lymph-migrating dDCs and LCs along with LN-resident B cells presenting Ag were found to be increased by the tumor (Fig. 2J). However, Ag presentation by dLN-resident cDCs or pDCs was not modified by the presence of the tumor (Fig. 2J). Simultaneously, the amount of Ag presented by each H-2Kb:OVA257-264+ dDC (flow cytometrically assessed per cell MFI) was lower in LNs draining Ag-MP–injected tumors compared to LNs draining Ag-MP–injected skin (Fig. 2K). LCs and B cells, on the other hand, exhibited Ag presentation MFI that was unchanged in the tumor compared to the naïve context (Fig. 2K). The increase in the number of Ag-presenting LCs, which interact primarily with CD8+ T cells (16, 19, 50), thus suggests an increase in presentation of tumor-derived Ag to LN-resident CD8+ T cells in melanoma-bearing animals. dDCs, on the other hand, are reported to prime/interact with CD4+ T cells and B cells (16, 19, 50). The diminished ratio of Ag-presenting MP+ cells (Fig. 2L) is suggestive of dDCs transferring Ag for presentation by B cells (Fig. 2, I and L), thus providing a mechanism for enhanced presentation of peripheral tissue–derived Ag to B cells of the dLN, a hypothesis that requires further experimentation to confirm. These results overall suggest that changes in APC maturation and migration induced by the tumor result in enhanced access of 500-nm spheres to TdLN cells as well as increased Ag presentation by dDCs, LCs, and B cells within TdLNs.
Melanomas alter access of lymph-derived nanoscale solutes and DC phenotype within LNs
Disease influences on lymph-draining Ag accumulation and uptake by cells within dLNs were next interrogated. Tracers 30 and 10 nm in hydrodynamic diameter simultaneously administered as one injection into the day 7 B16F10 melanomas accumulated within TdLNs over 72 hours at reduced levels compared to that seen in dLNs after injection in naïve skin (Fig. 3A). Nevertheless, levels of both 30- and 10-nm tracer association with B cells within TdLNs remained high with respect to per-tracer positive cell MFI over 72 hours (Fig. 3B, left, and Fig. 3C). With respect to pDCs, which reside more distal from the LN SCS within the LN parenchyma and are thus more restricted from lymph access (51), tracer association was increased within tumor compared to naïve skin dLN (Fig. 3B, right, and Fig. 3C). This is consistent with the increased permeability of the LN fibroblastic reticular cell–lined conduits within LNs draining melanomas (11, 35), suggesting that 30-nm tracers in TdLNs are available to be sampled by previously inaccessible cell types within LNs draining the naïve (tumor-free) skin. These results suggest that despite disease-induced alterations in lymphatic drainage, TdLN remodeling sustains access of lymph-draining solutes to resident leukocytes.
The influence of these changes in lymph-borne nanoscale solute access by TdLN cells on Ag presentation was evaluated using the synthetic Ag system instead composed of COVA257-264 disulfide tethered to lymphatic-draining nanoparticles (NPs) composed of Pluronic-stabilized poly(propylene sulfide) NPs (figs. S4 and S6) (12, 47, 48, 52–54). By virtue of their size (Fig. 3D), these NPs transit the interstitium, drain into lymphatic vessels, and accumulate rapidly within the dLN (Fig. 3E) for prolonged times after injection (Fig. 3F) in a manner similar to that of the fluorescent dextran tracers (Fig. 3D and fig. S1). Administration of these lymph-draining Ag-NPs thus allows cross-presentation by APCs of lymph-accessed Ag to be assessed through measurement of H-2Kb:OVA257-264 staining. Lymph drainage of Ag tethered to the NPs administered intratumorally did not result in increased numbers of cross-presenting total (Fig. 3G) or individual cell subtypes within TdLNs (Fig. 3H), nor was the mean per-cell MFI of H-2Kb:OVA257-264 staining altered relative to naïve dLNs (Fig. 3I). Consistent with this, CD206 expression, which is associated with DC tolerogenicity (55), was enhanced among pDCs within both the tumor and the TdLN and among cDCs of the tumor (Fig. 3, J and K). This suggests that despite increased tracer access (Fig. 3, B and C), cross-presentation of lymph-borne Ag by LN-resident APCs, which overall exhibit higher tolerogenicity, is unaffected by the tumor. This is in opposition to trends in Ag presentation resulting from Ag-MP treatment phenotype (Fig. 2, A and B), further motivating the elucidation of the ramifications of these lymphatic transport mechanism–specific changes on T cell Ag sensing in the tumor context.
Melanoma dLNs are niches that support survival and Ag experience by tumor Ag–specific T cells
Changes in Ag presentation and phenotype of APCs induced by melanomas have the potential to substantially affect Ag sensing and responses by T cells. As such, CD8+ T cell immunity that develops against endogenous tumor Ag in various immune microenvironments of melanoma-bearing animals was interrogated. Both the tumor and TdLNs were found to be highly enriched in presented tumor Ag in animals bearing day 7 B16F10 melanomas that expressed OVA (Fig. 4, A and B, and fig. S7). Accordingly, proliferation 72 hours after transfer of tumor Ag–specific (OT-I) CD45.2 cells into recipient CD45.1 mice bearing B16F10-OVA tumors 7 days after implantation (figs. S8 and S9) was extensive in both the tumor and its TdLNs but not the NdLN or spleen (Fig. 4C). With respect to the density of live tumor Ag–specific donor cells per total live cells, proliferating OT-I donor cells were highly concentrated in the TdLN but not other analyzed tissues (Fig. 4D). Notably, however, proliferating donor CD8+ T cells within the tumor exhibited low or poor viability, in contrast to cells within TdLNs (and, to a lesser extent, NdLNs and spleens) that were largely viable (Fig. 4, E and F). Frequencies of PD1 but not CD39 expressing donor CD8+ T cells, which have presumably experienced Ag but did not become exhausted (56, 57), within TdLNs were also high, in contrast to donor cells measured within tumors, spleens, and non-draining lymph nodes (NdLNs) (Fig. 4, G and H). Densities of a stem-like subtype of PD1-expressing donor CD8+ T cells that are CXCR5+CD39−, which provide the proliferative burst in response to αPD1 ICB therapy (7, 8), were greater in TdLN compared to the tumor, spleen, and NdLN (Fig. 4I). Thus, the TdLN supports the enhanced proliferation and survival of tumor Ag–specific T cells, a subset of which is stem-like (7, 8), in contrast to the tumor and other lymphoid tissues, and could act as a source of tumor Ag–specific T cells to regulate antitumor immunity and response to immunotherapy.
Fig. 4. The TdLN is a niche that supports Ag-specific CD8+ T cell priming and survival.
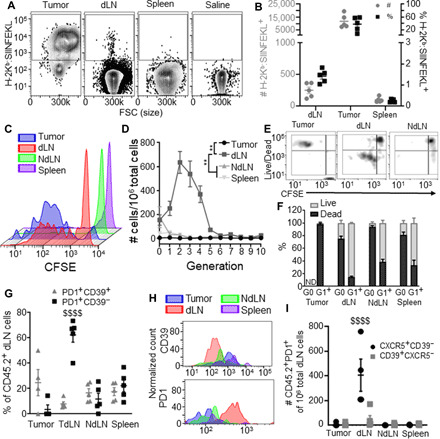
SIINFEKL presentation (measured by 25D1.16 staining for H-2Kb:SIINFEKL) by CD45+ cells (A) and cDCs (B) of the tumor, dLN, and spleen in B16F10-OVA tumor–bearing animals. Representative carboxyfluorescein diacetate succinimidyl ester (CFSE) dilution histograms (C) showing proliferation by tumor Ag–specific CD45.2+ donor cells in the tumor, dLN, NdLN, and spleen 72 hours after transfer into day 7 tumor-bearing CD45.1 animals, as quantified in (D). Representative flow cytometry plots (E) and quantification (F) of proliferative generation (CFSE signal) and viability tumor Ag–specific donor cells in the tumor, dLN, and NdLN. (G to I) Phenotype of tumor Ag–specific donor cells in the tumor, dLN, NdLN, and spleen 72 hours after transfer into day 7 tumor-bearing CD45.1 mice. (G and H) CD44, CD39, and PD1 expression quantification (G) and representative histograms (H). (I) CXCR5 and CD39 expression quantification. * indicates significance by two-way ANOVA with Tukey’s comparison (** indicates P < 0.01); $ indicates significance relative to all other groups by two-way ANOVA with Tukey’s comparison ($$$$ indicates P < 0.0001); n = 5 to 8 animals; (A) to (I) are representative of two experiments.
Mechanism of lymphatic transport regulates Ag presentation and T cell sensing within the LN
To assess how the mechanism of lymphatic transport of Ag from peripheral tissues to dLNs influences responses by Ag-specific CD8+ T cells localized to within dLNs (Fig. 5A), the synthetic Ag system that leverages either an MP or NP carrier to restrict lymphatic transit to dLN via cell-mediated versus passive lymph drainage, respectively (fig. S6), was administered into naïve animals. Responses 3 days after administration by both recipient CD8+ T cells and donor CD45.2 OT-I CD8+ cells (fig. S8) adoptively transferred 1 day before Ag challenge (Fig. 5B, i) or endogenous CD8+ T cells 7 days after administration (Fig. 5B, ii) were subsequently monitored (figs. S6, S8, and S9). As would be expected given programmed proliferation by CD8+ T cells in response to Ag experience (58), the extent of proliferation by responding T cells within dLNs 3 days after challenge was equivalent between animals challenged with NPs versus MPs for both responding donor and recipient cells (Fig. 5C and fig. S10). However, NP-mediated Ag challenge resulted in sustained high levels of CD8+ dLN cells, whereas the number of CD8+ cells after MP challenge was reduced at 7 days after challenge (Fig. 5D). This suggests that CD8+ T cells primed via NPs versus MPs exhibit differences in their dLN recruitment, retention, and/or viability during response to Ag priming. Consistent with the substantially higher levels of dLN accumulation, NP-primed cells resulted in higher numbers of PD1+ cells within dLNs compared to MP-primed cells both 3 and 7 days after challenge (Fig. 5, E and F). More PD1+ donor cells 3 days and endogenous cells 7 days after Ag challenge were also Tcf1+Tim3− (Fig. 5F), indicating that antigenic priming via drained NPs results in greater expansion of the stem-like CD8+ T cell pool within dLNs compared to cell-trafficked MP-tethered Ag. The number of recipient/endogenous CD25+ T cells was somewhat higher 3 but not 7 days after MP challenge, a difference not seen in donor cells (Fig. 5, E and F). Equivalent levels of CD39+ CD8+ T cells (both endogenous and donor) within dLNs resulted from treatment with both Ag-conjugated NPs and MPs (fig. S10). Functional responses assessed by ex vivo restimulation with OVA257-264 revealed more GzmB+- and IFN-γ–producing donor cells 3 days after challenge in response to MP- as opposed to NP-mediated Ag challenge (Fig. 5, G and H). Numbers of GzmB-expressing cells within dLNs were also higher in response to Ag challenge using MPs compared to NPs both 3 and 7 days after treatment, and IFN-γ–producing cells were higher in OT-I cells responding to MP challenge at day 3 (Fig. 5, G and H). Numbers of tumor necrosis factor–α (TNFα)– and interleukin-2 (IL-2)–producing cells remained constant (fig. S10).
Fig. 5. Extent of expansion and quality of Ag-specific CD8+ T cell pool in response to Ag is dependent on the mechanism of lymphatic transport to the dLN.
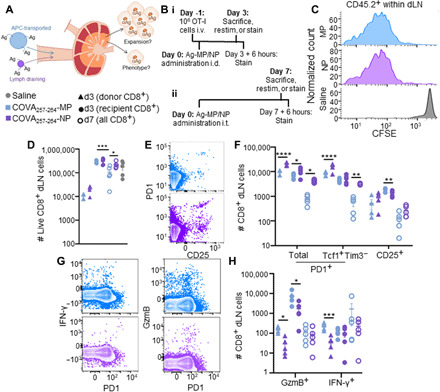
Schematic outlining the approach (A) and experimental design (B) for synthetic Ag system to assess the influence of lymphatic transport mechanism on CD8+ T cell responses elicited in dLNs. (C) Representative CFSE plots of viable Ag-specific, CD45.2+CD8+ dLN donor cells 72 hours after treatment with synthetic Ag systems. (D) Number of live CD8+ donor and recipient cells in dLNs 3 or 7 days after treatment with synthetic Ag systems. Saline includes pooled data from T cells collected both 3 and 7 days after treatment with synthetic Ag systems. (E) Representative flow cytometry plot of PD1 and CD25 expression by dLN CD8+ T cells 7 days after treatment with synthetic Ag systems. (F) Number of marker-expressing cells in the dLN 3 or 7 days after treatment with synthetic Ag systems. (G) Representative flow cytometry plot of IFN-γ, GzmB, and PD1 expression by CD8+ T cells in the dLN 3 days after treatment with synthetic Ag systems. (H) Number of cytokine-producing Ag-specific donor cells 3 or 7 days after treatment with synthetic Ag systems. * indicates significance by one-way ANOVA (* indicates P < 0.05, ** indicates P < 0.01, *** indicates P < 0.005, **** indicates P < 0.0001); n = 5 to 7 mice.
Together, these results reveal that Ag sensing and elicitation of functional cytotoxic CD8+ T cells are compartmentalized between the mechanism of Ag transport to dLNs. Specifically, lymphatic transport to dLNs via trafficking APCs leads to the induction of T cells with more cytotoxic functions. However, lymph drainage of Ag results in higher overall levels of Ag experience with dLNs, effects reflected by higher numbers of induced PD1+ cells and the generation of more stem-like CD8+ T cells. Both of these cell subsets have the potential to induce protective benefits against skin Ag and tumor development.
Therapeutically functional antitumor T cell responses in the TME result from Ag priming of CD8+ T cells within TdLNs
The ramifications of altered APC Ag access, maturation, and Ag presentation within TdLNs as a result of melanoma lymphatic drainage with respect to elicited functional T cell responses were assessed using the synthetic Ag system (figs. S4 and S6). Specifically, Ag-MPs or -NPs were administered intratumorally on day 0 into animals bearing parental (non–OVA-expressing) B16F10 melanomas 7 days after implantation. End point analyses of both donor CD45.2 OT-I CD8+ cells (fig. S8) adoptively transferred 1 day before Ag challenge and recipient CD8+ T cells 3 days after administration (i) or endogenous CD8+ T cells 7 days after administration (ii) were performed. Similar to responses observed in tumor-free animals (Fig. 5), proliferation of donor Ag-specific T cells within TdLNs 3 days after treatment was roughly equivalent regardless of the mechanism of lymphatic transport of Ag and irrespective of the presence of the day 7 B16F10 melanoma (fig. S10). In contrast to responses seen in naïve animals (Fig. 5C), CD8+ T cell numbers within TdLNs in response to MP- and NP-tethered Ag were also roughly equivalent (Fig. 6A). However, when evaluating the quality of responses elicited, subtle differences were noted. Ag-MP treatment resulted in an increase in PD1+ and PD1+Tcf1+Tim3− CD8+ T cell numbers within TdLNs 7 days but not 3 days after administration (Fig. 6B). However, with Ag-NP treatment, the levels of both PD1+ and PD1+Tcf1+Tim3− Ag–specific donor and PD1+ recipient CD8+ T cells were decreased within TdLNs relative to LNs draining tumor-free skin 3 days after administration (Fig. 6B). CD25+ and CD39+ cell numbers showed less consistent trends across time in Ag-NP–treated tumor-bearing animals, with a decrease in CD25+ cells 3 days and endogenous CD39+ cells 7 days after treatment. However, Ag-MP treatment resulted in diminished levels of CD25+ cells 3 days after treatment but increases in both CD25+ and CD39+ cells 7 days after treatment (fig. S10). With respect to cytotoxic CD8+ T cell immunity, levels of cytokine-producing cells (both donor and endogenous) within TdLNs elicited by Ag-MP treatment, on the other hand, were largely unchanged, save a reduction in IL-2–expressing cells 3 days after treatment (fig. S10). Ag-NP treatment resulted in diminished IFN-γ– and IL-2–expressing CD8+ T cells within TdLNs 7 days after administration (fig. S10). However, numbers of GzmB-expressing CD8+ T cells, which are degranulated and can directly kill tumor cells, elicited by Ag-MP, but not Ag-NP, treatment were diminished within TdLNs at both 3 and 7 days after administration compared to LNs draining tumor-free skin (Fig. 6C). This decrease within the TdLN coincided with a substantial increase in GzmB+ CD8+ T cells in the TME 7 days after treatment compared to saline-treated tumors (Fig. 6D). In contrast, although stem-like (PD1+Tcf1+Tim3−) CD8+ T cells were expanded within TdLNs by both synthetic Ag systems, their numbers within the TME remained relatively unchanged irrespective of treatment type (Fig. 6E). However, numbers of PD1+Tcf1−Tim3+ cells, which are derived from stem-like PD1+Tcf1+Tim3− cells, as they transition into an effector phenotype (7, 8), were substantially increased within the TME day 7 after administration of either Ag-MP or -NP, although more so with Ag-NP treatment (Fig. 6E). Moreover, ~20 to 30% of tumor-infiltrating CD8+ donor cells were found to be viable in response to either Ag-MP or -NP treatment (Fig. 6F), in sharp contrast to endogenous, tumor Ag–specific CD8+ cells infiltrating the tumor that are largely (>90%) nonviable (Fig. 4, E and F) (59). Overall, Ag-NP/MP treatment resulted in higher total CD8+ T cell infiltration into tumors (Fig. 6G), a characteristic that has been independently associated with increased survival in patients with melanoma (3). These results suggest that priming of CD8+ T cell immunity within TdLNs results in improved quality and quantity of TME-infiltrating CD8+ T cells.
Fig. 6. Expansion of CD8+ T cell TdLNs in response to administration of synthetic Ag systems affects tumor-infiltrating T cells and has antitumor functions.
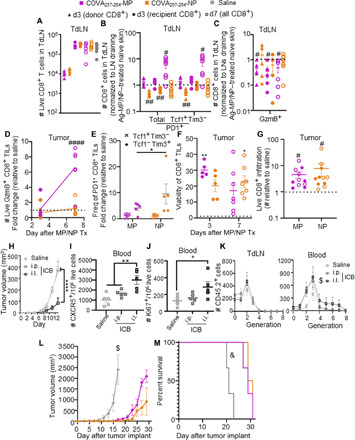
Number (A) and phenotype (B) of viable CD8+ cells in TdLNs 3 and 7 days after Ag-MP or -NP treatment. Saline (A) includes cells collected both 3 and 7 days after treatment with synthetic Ag systems, with no difference between analysis day. (C) Quantification of GzmB-producing Ag-specific donor cells as in (B). (D) Number of TME-localized GzmB+ CD8+ cells in Ag-MP– or Ag-NP–treated B16F10 tumors relative to saline-treated tumors. TILs, tumor-infiltrating lymphocytes. (E) Frequency of Tcf1 and Tim3 expressing PD1+ Ag-specific donor CD8+ cells in Ag-MP– or Ag-NP–treated B16F10 tumors relative to saline-treated tumors. (F) Viability (as %) of CD8+ T cells within the TME. The dashed line indicates viability of CD8+ T cells in B16F10-OVA tumors (Fig. 4), statistics relative to the dashed line. (G) Number of live CD8+ T cells infiltrating Ag-MP/NP–treated tumors relative to saline-treated tumors. (H) Tumor growth of B16F10 tumor after αPD1 treatment in the ipsilateral (i.l.) forearm, intraperitoneal (i.p.), or saline treatment days 5, 7, and 9 after tumor implantation. Number of Ag-specific CXCR5+Tim3−CD45.2+ (I) and Ki67+CD45.2+ (J) donor cells in the blood of tumor-bearing mice. (K) Proliferative generations of tumor Ag–specific donor CD8+ T cells in the TdLN (left) and blood (right). B16F10-OVA tumor growth (L) and animal survival (M) after intratumor treatment in contralateral B16F10 tumor resected 3 days after treatment with saline or Ag-MP/NP. # indicates significance against normalized value of 1.0 by one-sample t test (# indicates P < 0.05, ## indicates P < 0.01, #### indicates P < 0.0001); * indicates significance by one-way ANOVA with Tukey’s post hoc test (* indicates P < 0.05, ** indicates P < 0.01, **** indicates P < 0.0001); $ indicates significance relative to all other groups by RM ANOVA (repeated-measures ANOVA) with Tukey’s post hoc test; & indicates significance relative to all other groups by log-rank test; n = 5 to 10 animals; (H), (L), and (M) are representative of two independent experiments.
To assess the impacts of enhanced tumor infiltration of effector-like PD1+Tcf1−Tim3+ and effector GzmB+ CD8+ T cells on therapeutic responses, two classes of tumor immunotherapy were evaluated. First, we evaluated the effects of monoclonal antibody (mAb)–mediated blockade of PD1 signaling, which results in a proliferative burst by stem-like PD1+Tcf1+Tim3− CD8+ T cells (7). We recently reported (60) the improved immunotherapeutic effects of this therapy when directed to TdLNs using locoregional administration, e.g., administration in the skin ipsilateral to the tumor, resulting in co-drainage by mAb to the TdLN, compared to administration using either a conventional systemic route of mAb administration (intraperitoneal). When the effects of TdLN-directed versus systemic ICB using αPD1 mAb on tumor Ag–specific donor OT-I (CD45.2) CD8+ T cells 3 days after transfer into CD45.1 mice bearing B16F10-OVA melanomas were assessed, the former was associated with higher densities of both circulating CXCR5+Tim3− [stem-like (7); Fig. 6I] and Ki67+ (proliferating; Fig. 6J) CD8+ T cells. With respect to proliferative generation traced via analyses of fluorescently labeled cell dilution, TdLN-directed ICB (ipsilateral) also resulted in higher numbers of circulating donor cells in later (G4 to 8) generations compared to saline or intraperitoneally ICB-treated animals (Fig. 6K). In contrast, earlier generations of donor cells were generally found within TdLNs compared to tumors irrespective of treatment (Fig. 6K). Together with observations of increased tumor infiltration by PD1+Tcf1−Tim3+ CD8+ cells resulting from Ag-MP/NP treatment (Fig. 6E), this is consistent with stem-like PD1+Tcf1+Tim3− cells expanded within TdLNs being mobilized and accumulating in their differentiated state as effector-like cells within the TME. This is also consistent with reports of T cells disseminating from the dLN subsequently losing Tcf1 expression (61). Augmented immunotherapeutic effects from TdLN-directed ICB thus appear to be associated with mobilization of stem-like and responding CD8+ T cells from TdLNs into the circulation to result in TME infiltration and antitumor effects.
Second, the potential for the synthetic Ag-MP/NP system that expands the effector CD8+ T cell pool to protect against tumor growth was evaluated. To assess the effectiveness of the different subpopulations of T cells primed by each Ag system at controlling tumor growth, Ag covalently tethered to either MPs or NPs was administered intratumorally into a day 7 B16F10 (non-OVA expressing) melanoma. Three days after synthetic Ag administration, the primary tumor was excised. In so doing, animal survival was determined not by the treated primary tumor but instead by the growth of a secondary, OVA-expressing B16F10 tumor that was implanted in the contralateral dorsal skin. Irrespective of conjugation to either MPs or NPs, treatment with the synthetic Ag resulted in retardation of growth of the contralateral B16F10-OVA melanoma (Fig. 6L), effects that prolonged animal survival (Fig. 6M). In these studies, the excision of the primary tumor (receiving the Ag-MP/NP injection) left only primed cells within TdLNs in addition to those that had already disseminated from the treated TME. As cells expanded within the TME exhibit low viability (Fig. 4, E and F), therapeutic effects can, thus, be interpreted to be dLN-mediated. This implicates tumor lymphatic transport of passively drained or cell-trafficked Ag as inducing CD8+ T cell immunity that has antitumor functionality. While both the MP and NP treatment resulted in decreased tumor growth and enhanced survival, both synthetic Ag systems provided similar survival benefits, indicating that expansion of either stem-like or effector cells within the dLN can enhance antitumor responses to a similar degree. Together, these data support the concept that the TdLN represents a niche for expansion of therapeutically relevant CD8+ T cell subsets implicated in the efficacy of tumor immunotherapy.
DISCUSSION
Long overlooked for other than their prognostic value, LNs represent increasingly attractive therapeutic targets in cancer therapy applications (12, 54, 60, 62). LNs function as “transit hubs” of the adaptive immune system that facilitate co-mingling of APCs and lymphocytes within microenvironments conducive to the generation of adaptive immune responses. We show here two engineered material systems inspired by the way Ag is transported to LNs via the lymphatic vasculature to delineate in a quantitative fashion how lymphatic transport mechanism influences LN-localized signaling to CD8+ T cells and the impact of malignancy on these processes. By applying these biomaterials tools to the integrated analysis of lymphatic physiology effects on CD8+ T cell immunity relevant to tumor immunotherapy, this work expands upon the currently limited understanding of how Ag transport and interactions between the tumor and LN-resident cells, as mediated by the lymphatic system occurring in melanoma. Our findings elaborate the potential for nanoengineering approaches to harness various lymphatic transport mechanisms to optimize melanoma immunotherapy in eliciting and mobilizing various classes of antitumor CD8+ T cell immunity.
A fluorescent tracer panel designed to quantify lymphatic transport as mediated by migrating APCs versus lymph drainage revealed that alterations in cellular uptake and LN remodeling compensate to sustain access of tumor-derived solutes by LN-resident cells. Specifically, skin-resident DCs become more migratory, reaching the dLN in higher numbers. This is in line with reports of enhanced migratory and decreased phagocytic capacity of activated DCs (20, 46). Others have shown that CD11b+ dDCs that express major histocompatibility complex II (MHC-II) access the melanoma dLN at similar rates to the naïve skin dLN (63); however, by examining all dDCs and LCs, we demonstrate enhanced mobility of the skin DC population as a whole. Evaluating this entire cell population facilitates examination of CD8+ T cell responses in this context, instead of CD4+ T cell responses elicited by MHC-II+ DCs. In addition, lymph-derived, nanoscale solutes are also accessible to cells normally restricted from sampling lymph by the SCS and other structural barriers within the LN. The melanoma dLN has been shown to remodel (35), enhancing intra-LN access to tracers of this size range. Thus, these findings are in line with current understandings of melanoma-induced remodeling locally and in the TdLN. Despite its simplicity, limitations in the tracer system used herein include the comparisons between dextrans and spheres, which differ in physical and chemical properties (42).
A synthetic Ag system was used to elaborate the influence of disease on Ag presentation by LN-localized cells resulting from Ag delivered to dLNs via different lymphatic transport mechanisms. Despite sustained access by LN-resident cells to lymph-draining Ag, the extent of Ag presentation by dLN-resident cells was unchanged in the melanoma context, suggesting that Ag presentation is locally suppressed. In contrast, presentation of cell-transported Ag, which is taken up and whose presentation occurs first within the periphery or during intralymphatic transit (17, 64), was increased within TdLNs. We hypothesize that differences in tumor effects between these mechanisms are due to the phenotypes of DCs differentially accessed by each lymphatic transport mechanism. Passively draining Ag was presented primarily by pDCs and cDCs, which exhibit a tolerogenic phenotype, while actively transported Ag was presented by dDCs and LCs, which are more activated in the tumor context. These results are consistent with previous reports demonstrating that both pDCs and cDCs express immunosuppressive molecules within the melanoma dLN (65, 66), while tumor-resident DCs are mature (63). Furthermore, mature DCs have decreased phagocytic capacity but maintain Ag presentation functions (20, 46). The induction of tolerogenic phenotypes in LN-resident DCs by the tumor results in levels of presentation by lymph-draining Ag that are relatively unchanged compared to naïve animals, while activation of skin-resident DCs by the tumor enhances Ag presentation within the TdLN. We note that these studies omitted the analysis of stromal populations, despite their robust Ag presentation and immunomodulatory properties, particularly in the context of melanoma (67, 68).
The combined effects of lymphatic transport mechanism and microenvironment on resulting responses by CD8+ T cells were interrogated using the synthetic Ag system. Lymph-draining Ag was found to increase the expansion of the stem-like CD8+ T cell reservoir within the dLN over cell-trafficked Ag, an effect largely unaltered by the presence of a melanoma. These cells, which provide the proliferative burst in response to αPD1 ICB therapy (7, 8) and whose presence in patients is associated with improved survival (5, 8), can recirculate and differentiate, reaching the TME at higher numbers to improve immunotherapeutic effects. Conversely, CD8+ T cells with effector functions associated with improved survival in melanoma (3, 4) were more robustly induced within dLNs by cell-trafficked Ag and were mobilized to the TME in the tumor context. Each CD8+ T cell subtype that expanded locally within the TdLN in response to lymphatic-transported Ag was furthermore functional and associated with reductions in tumor growth and enhanced survival of melanoma-bearing animals in response to ICB or in situ vaccine immunotherapy. We note that this investigation was limited to the relatively lymphatic-rich B16F10 model (fig. S5) as human melanomas tend to be lymphatic infiltrated (25, 26). However, recent seminal work has demonstrated the correlation between tumor lymphangiogenesis and tumor immune microenvironment and responsiveness to immunotherapy (69, 70), effects attributed directly to the TME. Future studies elaborating immunological responses within TdLNs as they relate to the extent of lymphatic infiltration within the melanoma, in combination with the work presented here, have the potential to benefit the treatment of disease that is resistant to current immunotherapy treatment regimens through LN-directed immunomodulation. Examples include tumors that are poorly immune infiltrated (71), lack niches that support stem-like CD8+ T cell survival and functions (72), and are stroma dense and into which therapeutic delivery is largely restricted (73).
The finding that different subsets of T cells with antitumor functions expand within LNs as a result of Ag transport mechanism has the potential to inform improved immunotherapeutic management of melanoma, particularly the examples described above. ICB therapies have been most effective in patients with expression of immune checkpoints and higher T cell infiltration (74). We show here that lymph-draining vaccines have the potential to further enhance the application of these therapies by robustly expanding the pool of CD8+ T cells that provide the proliferative burst of tumor Ag–specific CD8+ T cells in response to ICB therapy. Immunotherapies have also been found to be more effective in patients with immunologically “hot” tumors that exhibit higher immune infiltration (75). MP-based or depot vaccines that locally recruit and activate APCs within peripheral tissues before T cell expansion within dLNs represent a potential strategy to make tumors more hot by increasing infiltration of functional, effector T cells into the TME, which has been independently associated with survival (3, 4). With the advent of bioengineering approaches in cancer immunotherapy and the emerging appreciation of LNs as druggable, therapeutically relevant tissues, lymphatic-directed drug delivery innovations therefore represent novel approaches to improve translational management of melanoma.
MATERIALS AND METHODS
Experimental design
The objectives of these studies were to understand how cells of the LN access skin- and melanoma-derived Ag on the basis of size and mechanism of transport, how this Ag is presented, how T cells sense Ag in both the tumor and naïve context, and how this influences immunotherapeutic outcomes in mouse models. Experiments were designed with appropriate controls (e.g., saline injected intradermally and intratumorally) and appropriate timelines to assess these outcomes. Sample size was determined using power analysis. End points were predetermined as time points for all nonsurvival studies. End point for survival studies was set at the humane end point (tumors reaching 1.5 cm in any direction or ulcerating and actively bleeding). Animals were randomized to receive different treatments using a random number generator and mixed among cages, and researchers were blinded for survival studies. Survival studies were replicated for a total of two separate studies to confirm results.
Cell culture
B16F10 and B16F10-OVA mouse melanoma cells (obtained from Swartz Laboratory, EPFL, Lausanne, Switzerland) were cultured in Dulbecco’s modified Eagle’s medium (Gibco, Thermo Fisher Scientific Inc., Waltham, MA) with 10% heat-inactivated fetal bovine serum (Gibco, Thermo Fisher Scientific Inc.) and 1% penicillin/streptomycin/amphotericin B from Life Technologies (Carlsbad, CA, USA). Cells were passaged at ~80% confluency and maintained at 37°C with 5% CO2 in a standard incubator.
Animal tumor models
C57Bl/6 or B6 CD45.1 mice were purchased at 6 weeks of age from the Jackson Laboratory (Bar Harbor, ME, USA). All protocols were approved by the Institutional Animal Care and Use Committee (IACUC). For tumor-bearing cohorts, 0.5 × 106 melanoma cells were intradermally implanted into the left dorsal skin of 6- to 8-week-old mice. Tumor dimensions were measured with calipers in three dimensions and reported as an ellipsoidal volume.
Fluorescent tracers
Five hundred–nanometer yellow-green and red fluorescent (505/515 and 580/605 excitation/emission, respectively) carboxylate-modified microspheres were purchased from Thermo Fisher Scientific Inc. Tetramethylrhodamine isothiocyanate (TRITC) dextran (40,000 Da) was purchased from Sigma-Aldrich. Amino-dextran (500,000 Da) (Thermo Fisher Scientific Inc.) was covalently labeled by incubation with Alexa Fluor 647 or 700 NHS (N-hydroxysuccinimide) ester dyes (Thermo Fisher Scientific Inc.) in 0.1 M NaHCO3 at pH 8.4 for 4 hours on a tube rocker. AF647 and AF700 dextran-dye conjugates were purified from unreacted free dye by Sepharose CL-6B gravity column chromatography after conjugation. Purified dextran-fluorophore conjugates were further confirmed free of unconjugated dye by a second Sepharose CL-6B column analysis (11). All reagents were used and maintained under sterile conditions. Hydrodynamic sizes were confirmed before injection by dynamic light scattering using a Zetasizer Nano ZS (Malvern Instruments Ltd., Malvern, UK).
Tracer injections
Fluorescent tracers suspended in saline were co-infused by a syringe pump at a rate of ~300 nl/s directly into the center of the tumors for tumor-bearing groups or into the dermal layer of the skin for naïve groups, using a 27- to 31-gauge needle (Becton Dickinson, Franklin Lakes, NJ) while mice were under anesthesia via isoflurane. For biodistribution experiments, 500-nm red or yellow-green fluorescent microspheres (19 pM), 30-nm AF700 or AF647 dextran (4.8 μM), and 10-nm TRITC dextran (4.8 μM) were co-infused in 10 μl of saline total. Mice were euthanized via CO2 asphyxiation in accordance with American Veterinary Medical Association (AVMA) and local IACUC guidelines at the prescribed times after tracer injection for each experiment.
IVIS imaging
Animals were injected intradermally in the left dorsal skin with AF647-conjugated NPs or 500-nm yellow-green microspheres (MP) as described above. Twenty-four hours after NP injection or 72 hours after MP injection, animals were euthanized via CO2 asphyxiation in accordance with AVMA and local IACUC guidelines. Animals were then dissected to expose axillary and brachial LNs and imaged using a PerkinElmer IVIS (in vivo imaging system) Spectrum CT (Waltham, MA). LNs were then dissected and placed on a black plastic and imaged using a PerkinElmer IVIS Spectrum CT.
Tumor and LN immunohistochemistry and imaging
Tumor, skin, and dLNs were frozen in optimum cutting temperature compound (Sakura Finetek USA Inc., Torrance, CA, USA) in 2-methylbutane (Sigma-Aldrich) chilled by liquid nitrogen and frozen tissue blocks immediately stored at −80°C. For imaging of tracer dLNs, coverslips were mounted onto LN slices (10-μm thickness) using Vectashield mounting medium (Vector Laboratories, Burlingame, CA, USA) with no prior wash steps and within 30 min of imaging. Otherwise, LN, skin, and tumor tissue sections were fixed with prechilled acetone for 10 min at 4°C and subjected to standard immunofluorescence protocols using the following antibodies, which were obtained from Thermo Fisher Scientific Inc. unless otherwise specified: fluorescein isothiocyanate (FITC)–conjugated rat anti-mouse CD31 (1:50), rabbit anti-mouse Lyve-1 (1:250), Alexa Fluor 633 goat anti-rabbit (1:300), Armenian hamster anti-mouse CD3Ɛ (1:50), AF647 goat anti-hamster (1:300; Abcam plc., Cambridge, MA, USA), AF488-conjugated rat anti-mouse CD169 (1:100; BioLegend Inc., San Diego, CA, USA), biotinylated rat anti-mouse F4/80 (1:200; Life Technologies), streptavidin-AF555 (1:400; Life Technologies), biotinylated rat anti-mouse B220 (1:250), and AF488-conjugated Armenian hamster anti-mouse CD11c (1:50; BioLegend Inc.). Blocking and antibody dilutions were performed with 10% donkey serum (Sigma-Aldrich) in Dulbecco’s phosphate-buffered saline (D-PBS). Slides were washed with 0.1% Tween 20 (Sigma-Aldrich) in D-PBS for washing steps, counterstained with DAPI (4′,6-diamidino-2-phenylindole) (VWR International Inc.), and imaged using a 710 NLO confocal microscope (Carl Zeiss Microscopy Ltd., Jena, Germany) with a 20× magnification objective.
CD8+ T cell isolation
OT-I animals were purchased from Charles River Laboratories (Lyon, France) and bred in-house. OT-I animals were euthanized, and the spleens were harvested and disrupted with 18G needles (Becton Dickinson) followed by washing with D-PBS. Cells were passed through a sterile 70-μm cell strainer (Greiner Bio-One, Monroe, NC, USA), washed, and incubated with ACK Lysing Buffer (Lonza Group AG, Basel, Switzerland) for 60 s at room temperature, quenched with D-PBS, washed, and resuspended for counting. Cells were resuspended at 108 cells/ml buffer (2% bovine serum albumin in D-PBS), blocked with normal rat serum, and mixed with CD8+ T cell isolation antibody cocktail (STEMCELL Technologies, Vancouver, Canada), followed by streptavidin-coated magnetic beads (STEMCELL Technologies). Buffer was added to the mixture and placed in a magnet (STEMCELL Technologies, Vancouver, Canada), and the supernatant was collected. Cells were then counted and resuspended in carboxyfluorescein diacetate succinimidyl ester (CFSE; Life Technologies, Carlsbad, CA, USA) and then quenched with ice-cold RPMI medium containing >10% heat-inactivated fetal bovine serum (Life Technologies, Carlsbad, CA, USA). Purity, viability, and CFSE loading were confirmed via flow cytometry on a customized BD LSRFortessa flow cytometer before adoptive transfer (fig. S1). Cells were maintained in sterile conditions before adoptive transfer.
Adoptive transfer
Isolated CD8+ T cells were suspended in sterile saline at a concentration of 2 × 106 cells per 200 μl of sterile saline. After induction of isoflurane anesthesia, the hair over the neck of mice was removed using depilatory cream and cleaned using warm water and ethanol wipes, and suspended cells were injected intravenously via the jugular vein.
Flow cytometry analysis
Axillary and brachial dLNs were pooled and incubated with collagenase D (1 mg/ml) (Sigma-Aldrich) in D-PBS with calcium and magnesium for 1 hour at 37°C, passed through a 70-μm cell strainer (Greiner Bio-One, Monroe, NC, USA), washed, and resuspended in a 96-well plate (VWR International Inc.) for staining. Lung tissues were treated with the same procedure as LNs. Spleen capsules were disrupted using 18G needles, and the cell suspension was passed through a 70-μm strainer, pelleted and then incubated with blood cell lysis buffer (Sigma-Aldrich) for 7 min at room temperature, diluted with D-PBS, washed, and resuspended. Liver tissues were disrupted with 18G needles, passed through a 70-μm cell strainer, and centrifuged at 60g for 1 min to remove large debris, and the supernatant was collected into a different tube and centrifuged at 300g for 5 min. The pellet was resuspended, layered onto lymphocyte separation medium (Thermo Fisher Scientific Inc.), and centrifuged for 20 min at 400g. The mononuclear cell layer was recovered, incubated with red blood cell lysis buffer as before, washed, and resuspended for staining. All antibodies for flow cytometry were from BioLegend Inc. unless otherwise stated. Cells were blocked with anti-mouse CD16/CD32 (clone, 2.4G2) (Tonbo Biosciences, San Diego, CA, USA) for 5 min on ice, washed, and then stained with a fixable viability dye eFluor 455UV (1:1000; eBioscience, San Diego, CA, USA) for 15 min on ice or fixable viability dye Zombie Aqua (1:100; BioLegend Inc., San Diego, CA, USA) for 30 min at room temperature, before quenching with 0.1% bovine serum albumin in D-PBS (flow cytometry buffer). Antibodies were prepared in flow cytometry buffer at the following dilutions on the basis of preliminary titrations: phycoerythrin (PE) anti-mouse CD45.2 (1.25:100), AF700 anti-mouse CD25 (0.5:100), BV785 anti-mouse PD1 (1.25:100), AF647 anti-mouse CXCR5 (0.5:100), PerCP anti-mouse CD3 (2.5:100), APC-Cy7 anti-mouse CD8 (2.5:100), PE-Cy7 anti-mouse CD39 (5:100), and BV421 anti-mouse CD44 (5:100) for T cell distribution analyses (Fig. 1); BV711 anti-mouse CD45.1 (1.25:100), BV605 anti-mouse CD3 (2.5:100), APC-Cy7 anti-mouse CD8 (2.5:100), and BV786 anti-mouse PD1 (1.25:100), PerCP anti-mouse CD69 (1.25:100), or PerCP anti-mouse CD45 (0.5:100) for T cell restimulation analysis (Figs. 3 and 6); PerCP anti-mouse CD45 (0.625:100), BV711 anti-mouse CD3 (1.25:100), FITC anti-CD8 (0.3125:100), BV786 anti-mouse PD1 (1.25:100), PE-Cy7 anti-mouse CD39 (2.5:100), and anti-mouse CD25 (1:100) for T cell vaccination response studies (Figs. 3 and 6); PE-Cy7 anti-mouse CD11b (0.625:100), BV421 anti-mouse CD11c (5:100), BV605 anti-mouse CD169 (5:100), BV650 anti-mouse B220 (2:100), BV711 anti-mouse CD3 (1.25:100), BV785 anti-mouse F4/80 (2.5:100), and PE anti-mouse H-2Kb:SIINFEKL (5:100; clone 25-D1.16) for Ag presentation experiments; or PerCP anti-mouse CD45 (1.25:100), AF700 anti-CD11b (1.25:100), PE-Cy7 anti-CD11c (1.25:100), APC-Cy7 anti-mouse MHC-II (2.5:100), BV605 anti-mouse CD206 (5:100), PE anti-mouse CD86 (5:100), APC anti-mouse DEC205 (1:100), and BV711 anti-mouse B220 (1.25:100) for DC phenotyping experiments. Cells were fixed with 4% paraformaldehyde (VWR International Inc.) for Ag presentation and biodistribution experiments. For intracellular staining, cells were incubated with FoxP3/Transcription Factor Fixation/Permeabilization solution (eBioscience, Thermo Fisher Scientific Inc.) for 60 min on ice in the dark. Cells were then incubated with PE anti-mouse Tcf1 (1.25:100; Becton Dickinson), APC anti-GzmB (2.5:100), and BV605 anti-Ki67 (1.25:100) in FoxP3/Transcription Factor Fixation/Permeabilization buffer (eBioscience, Thermo Fisher Scientific Inc.) for 75 min on ice in the dark. For ex vivo cytokine staining, cells were suspended in IC Fixation Buffer (eBioscience, Thermo Fisher Scientific Inc.) for 60 min at room temperature in the dark. Cells were then incubated with APC anti-mouse GzmB (2.5:100), PE anti-mouse IFN-γ (1.25:100), AF700 anti-mouse IL-2 (0.5:100), and PE-Cy7 anti-mouse TNF-α (1.25:100) in IC Permeabilization Buffer (eBioscence, Thermo Fisher Scientific Inc.) for 60 min at room temperature in the dark. Cells were then resuspended in FACS (fluorescence-activated cell sorting) buffer and kept at 4°C until analyzed with a customized BD LSRFortessa flow cytometer (BD Biosciences, Franklin Lakes, NJ, USA). Compensation was performed using AbC, ArC, or UltraComp compensation beads (Thermo Fisher Scientific Inc.), and data were analyzed using FlowJo software version 10 (FlowJo LLC, Ashland, OR).
Peptide-conjugated nanoparticle synthesis and administration
Pyridyl disulfide–functionalized NPs (PDS-NPs) were prepared as previously described (76). Cysteine-modified OVA257-264 (CSIINFEKL) was dissolved in Milli-Q water at 1 mg/ml and added 1:1 to PDS-NPs (40 mg/ml). The disulfide displacement reaction proceeded overnight at room temperature with stirring. After reacting, COVA257-264-NP was separated from unreacted peptide by size exclusion chromatography using a CL-6B column. Fractions containing peptide were identified by reacting with fluorescamine, and PEG (polyethylene glycol)–containing fractions (NPs) were determined using an iodine assay. Fractions containing COVA257-264-NP were combined and concentrated to the appropriate dose using 30-kDa MWCO (molecular weight cutoff) spin filters and sterilized by filtration through a 0.22-μm syringe filter. COVA257-264-conjugated NP or MP in sterile saline was injected intradermally in the center of the tumor or into the dermal layer of the skin (naïve animals) of C57/Bl6 mice.
Tracer diffusion analyses
Capillary tubes were loaded with rat tail collage type I solution (5.6 mg/ml) (Corning Inc., Corning, NY, USA) in D-PBS and allowed to gel overnight at 37°C. Yellow-green spheres (500 nm), FITC-conjugated 30-nm dextran, and TRITC 10-nm dextran were loaded in separate tubes, and fluorescence imaging was performed every 4 min for 2 hours at 37°C with a Zeiss AxioObserver Z1 inverted microscope (Carl Zeiss) with a 4× magnification objective. Image analysis was performed using ImageJ software (77).
Tracer biodistribution analyses
At 4, 24, and 72 hours after tracer injection, mice were euthanized, and the tumor-draining axillary and brachial LNs were harvested and homogenized in D-PBS using 1.4-mm acid-washed zirconium grinding beads with a FastPrep-24 automated homogenizer. Whole-tissue homogenate fluorescence was measured with a Synergy H4 BioTek plate reader (BioTek Instruments Inc., Winooski, VT, USA), compensation was applied, and fluorescent tracer amounts and concentrations were calculated from standard curves made by spiking individual naïve tissue homogenates with tracer solution. Tracer concentration within tissues was calculated by application of standard curves generated in tissue homogenates to the fluorescent readouts. To determine percent injection, this concentration was multiplied by a dilution factor to account for the PBS added to the tissue after excision and the portion of the tissue measured on the plate reader (tubes were weighed before tissue was added, after homogenization, and after plating to determine dilution factor). Percent injection was then defined as the amount of tracer divided by the known amount of tracer injected into the animal. We note that in these experiments, only total fluorescence is measured via end point analyses. As such, this methodology cannot distinguish between rates of lymphatic drainage versus LN accumulation.
Ex vivo restimulation
After cell isolation (as above, in flow cytometry), 30% of LN samples, 5% of spleen samples, or 5% of tumor cells were plated in a sterile 96-well U-bottom plate. SIINFEKL peptide (1 μg/ml) in 200 μl of IMDM (Iscove’s modified Dulbecco’s medium) with 10% heat-inactivated fetal bovine serum and 0.05 mM β-mercaptoethanol (Sigma-Aldrich) was added to each sample and then incubated for a total of 6 hours at 37°C with 5% CO2. Three hours into the incubation period, brefeldin A (50 μg/ml) (Sigma-Aldrich) was added to each sample. Cells were then stained for flow cytometry as above.
Tumor resection surgery
Animals are anesthetized using isoflurane in oxygen and then given sustained-release buprenorphine (1 mg/kg) and ketoprofen (5 mg/kg) via intraperitoneal injection as analgesics. The animals are then placed on a warming bed, and a sterile drape is placed to expose only the tumor and surrounding skin. Povidone-iodine is applied to the skin three times to sterilize the surgical area. Sterile scissors are then used to excise and remove the tumor. Sterile wound clips are used to close the wound. The animal is monitored throughout recovery and returned to its cage. Wound clips are removed 10 days after surgery, and animals are monitored every other day to ensure well-being and examined for infection or irritation surrounding the surgical site. All procedures are approved by the IACUC.
ICB therapeutic analysis
On day 7 of B16F10-OVA tumor growth in CD45.1 mice, isolated, CFSE-labeled CD8+ OT-I T cells (CD45.2+) were administered intravenously into the jugular vein. On day 9 of B16F10-OVA tumor growth, 150 μg of each of anti-PD1 mAb and anti-cytotoxic T lymphocyte Ag 4 mAb was administered either intraperitoneally or intradermally in the forelimb, which results in mAb accumulation within the TdLN but not NdLNs (60). Saline administered intradermally in the forelimb served as control. Animals were euthanized on day 11, and lymphocyte responses were analyzed as described above.
Statistical analysis
Data are represented as the means accompanied by SEM, and statistics were calculated using Prism 6, 7, and 8 software (GraphPad Software Inc., La Jolla, CA, USA). Statistical significance was defined as P < 0.05, 0.01, and 0.001, respectively, unless otherwise specified. Area under the curve was calculated using the built-in Prism analysis tool.
Supplementary Material
Acknowledgments
We thank A. Schudel for technical assistance. Funding: This work was supported by U.S. National Institutes of Health grants R01CA207619 (S.N.T.), U01CA214354 (S.N.T.), T32GM008433 (M.J.O. and N.A.R.), and T32EB021962 (D.M.F.). M.P.M. was a National Science Foundation Graduate Research Fellow. Author contributions: M.J.O. performed T cell and Ag presentation analysis experiments. N.A.R. performed Ag access experiments. M.J.O. analyzed all data. M.P.M. and D.M.F. prepared NPs and MPs and assisted in technical experimentation. M.J.O. and S.N.T. designed experiments and wrote the manuscript. H.T.K. provided clinical knowledge and directional advice throughout project development and manuscript preparation. Competing interests: The authors declare that they have no competing interests. Data and materials availability: The cell lines can be provided by S.N.T. pending scientific review and a completed material transfer agreement. Requests for the cell lines should be submitted to susan.thomas@gatech.edu. All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/50/eabd7134/DC1
REFERENCES AND NOTES
- 1.Koolen P. G. L., Matos T. R., Ibrahim A. M. S., Sun J., Lee B. T., Frankenthaler R. A., Lin S. J., Recurrence rates over 20 years in the treatment of malignant melanoma: Immediate versus delayed reconstruction. Plast. Reconstr. Surg. Glob. Open. 5, e1378 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Larkin J., Chiarion-Sileni V., Gonzalez R., Grob J. J., Cowey C. L., Lao C. D., Schadendorf D., Dummer R., Smylie M., Rutkowski P., Ferrucci P. F., Hill A., Wagstaff J., Carlino M. S., Haanen J. B., Maio M., Marquez-Rodas I., McArthur G. A., Ascierto P., Long G. V., Callahan M. K., Postow M. A., Grossman K., Sznol M., Dreno B., Bastholt L., Yang A., Rollin L. M., Horak C., Hodi S., Wolchock J. D., Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N. Engl. J. Med. 373, 23–34 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Erdag G., Schaefer J. T., Smolkin M. E., Deacon D. H., Shea S. M., Dengel L. T., Patterson J. W., Slingluff C. L. Jr., Immunotype and immunohistologic characteristics of tumor-infiltrating immune cells are associated with clinical outcome in metastatic melanoma. Cancer Res. 72, 1070–1080 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nirschl C. J., Suárez-Fariñas M., Izar B., Prakadan S., Dannenfelser R., Tirosh I., Liu Y., Zhu Q., Devi K. S. P., Carroll S. L., Chau D., Rezaee M., Kim T.-G., Huang R., Fuentes-Duculan J., Song-Zhao G. X., Gulati N., Lowes M. A., King S. L., Quintana F. J., Lee Y.-S., Krueger J. G., Sarin K. Y., Yoon C. H., Garraway L., Regev A., Shalek A. K., Troyanskaya O., Anandasabapathy N., IFNγ-dependent tissue-immune homeostasis is co-opted in the tumor microenvironment. Cell 170, 127–141.e15 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sade-Feldman M., Yizhak K., Bjorgaard S. L., Ray J. P., de Boer C. G., Jenkins R. W., Lieb D. J., Chen J. H., Frederick D. T., Barzily-Rokni M., Freeman S. S., Reuben A., Hoover P. J., Villani A.-C., Ivanova E., Portell A., Lizotte P. H., Aref A. R., Eliane J.-P., Hammond M. R., Vitzthum H., Blackmon S. M., Li B., Gopalakrishnan V., Reddy S. M., Cooper Z. A., Paweletz C. P., Barbie D. A., Stemmer-Rachamimov A., Flaherty K. T., Wargo J. A., Boland G. M., Sullivan R. J., Getz G., Hacohen N., Defining T cell states associated with response to checkpoint immunotherapy in melanoma. Cell 175, 998–1013.e20 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Obeid J. M., Erdag G., Smolkin M. E., Deacon D. H., Patterson J. W., Chen L., Bullock T. N., Slingluff C. L., PD-L1, PD-L2 and PD-1 expression in metastatic melanoma: Correlation with tumor-infiltrating immune cells and clinical outcome. Onco. Targets. Ther. 5, e123107 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Im S. J., Hashimoto M., Gerner M. Y., Lee J., Kissick H. T., Burger M. C., Shan Q., Hale J. S., Lee J., Nasti T. H., Sharpe A. H., Freeman G. J., Germain R. N., Nakaya H. I., Xue H.-H., Ahmed R., Defining CD8+T cells that provide the proliferative burst after PD-1 therapy. Nature 537, 417–421 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Miller B. C., Sen D. R., Al Abosy R., Bi K., Virkud Y. V., LaFleur M. W., Yates K. B., Lako A., Felt K., Naik G. S., Manos M., Gjini E., Kuchroo J. R., Ishizuka J. J., Collier J. L., Griffin G. K., Maleri S., Comstock D. E., Weiss S. A., Brown F. D., Panda A., Zimmer M. D., Manguso R. T., Hodi F. S., Rodig S. J., Sharpe A. H., Haining W. N., Subsets of exhausted CD8+ T cells differentially mediate tumor control and respond to checkpoint blockade. Nat. Immunol. 20, 326–336 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Francis D. M., Thomas S. N., Progress and opportunities for enhancing the delivery and efficacy of checkpoint inhibitors for cancer immunotherapy. Adv. Drug Deliv. Rev. 115, 33–42 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.O’Melia M. J., Lund A. W., Thomas S. N., The biophysics of lymphatic transport: Engineering tools and immunological consequences. iScience 22, 28–43 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rohner N. A., Thomas S. N., Melanoma growth effects on molecular clearance from tumors and biodistribution into systemic tissues versus draining lymph nodes. J. Control. Release 223, 99–108 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Thomas S. N., Vokali E., Lund A. W., Hubbell J. A., Swartz M. A., Targeting the tumor-draining lymph node with adjuvanted nanoparticles reshapes the anti-tumor immune response. Biomaterials 35, 814–824 (2014). [DOI] [PubMed] [Google Scholar]
- 13.Roozendaal R., Mempel T. R., Pitcher L. A., Gonzalez S. F., Verschoor A., Mebius R. E., von Andrian U. H., Carroll M. C., Conduits mediate transport of low-molecular-weight antigen to lymph node follicles. Immunity 30, 264–276 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gretz J. E., Norbury C. C., Anderson A. O., Proudfoot A. E. I., Shaw S., Lymph-borne chemokines and other low molecular weight molecules reach high endothelial venules via specialized conduits while a functional barrier limits access to the lymphocyte microenvironments in lymph node cortex. J. Exp. Med. 192, 1425–1440 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sixt M., Kanazawa N., Selg M., Samson T., Roos G., Reinhardt D. P., Pabst R., Lutz M. B., Sorokin L., The conduit system transports soluble antigens from the afferent lymph to resident dendritic cells in the T cell area of the lymph node. Immunity 22, 19–29 (2005). [DOI] [PubMed] [Google Scholar]
- 16.Kissenpfennig A., Henri S., Dubois B., Laplace-Builhé C., Perrin P., Romani N., Tripp C. H., Douillard P., Leserman L., Kaiserlian D., Saeland S., Davoust J., Malissen B., Dynamics and function of Langerhans cells in vivo: Dermal dendritic cells colonize lymph node areas distinct from slower migrating Langerhans cells. Immunity 22, 643–654 (2005). [DOI] [PubMed] [Google Scholar]
- 17.Kitano M., Yamazaki C., Takumi A., Ikeno T., Hemmi H., Takahashi N., Shimizu K., Fraser S. E., Hoshino K., Kaisho T., Okada T., Imaging of the cross-presenting dendritic cell subsets in the skin-draining lymph node. Proc. Natl. Acad. Sci. U.S.A. 113, 1044–1049 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Allan R. S., Waithman J., Bedoui S., Jones C. M., Villadangos J. A., Zhan Y., Lew A. M., Shortman K., Heath W. R., Carbone F. R., Migratory dendritic cells transfer antigen to a lymph node-resident dendritic cell population for efficient CTL priming. Immunity 25, 153–162 (2006). [DOI] [PubMed] [Google Scholar]
- 19.Klechevsky E., Morita R., Liu M., Cao Y., Coquery S., Thompson-Snipes L., Briere F., Chaussabel D., Zurawski G., Palucka A. K., Reiter Y., Banchereau J., Ueno H., Functional specializations of human epidermal Langerhans cells and CD14+ dermal dendritic cells. Immunity 29, 497–510 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.de Vries I. J. M., Krooshoop D. J. E. B., Scharenborg N. M., Lesterhuis W. J., Diepstra J. H. S., van Muijen G. N. P., Strijk S. P., Ruers T. J., Boerman O. C., Oyen W. J. G., Adema G. J., Punt C. J. A., Figdor C. G., Effective migration of antigen-pulsed dendritic cells to lymph nodes in melanoma patients is determined by their maturation state. Cancer Res. 63, 12–17 (2003). [PubMed] [Google Scholar]
- 21.Hanahan D., Weinberg R. A., Hallmarks of cancer: The next generation. Cell 144, 646–674 (2011). [DOI] [PubMed] [Google Scholar]
- 22.van der Burg S. H., Arens R., Ossendorp F., van Hall T., Melief C. J. M., Vaccines for established cancer: Overcoming the challenges posed by immune evasion. Nat. Rev. Cancer 16, 219–233 (2016). [DOI] [PubMed] [Google Scholar]
- 23.Najafi M., Hashemi Goradel N., Farhood B., Salehi E., Nashtaei M. S., Khanlarkhani N., Khezri Z., Majidpoor J., Abouzaripour M., Habibi M., Kashani I. R., Mortezaee K., Macrophage polarity in cancer: A review. J. Cell. Biochem. 120, 2756–2765 (2019). [DOI] [PubMed] [Google Scholar]
- 24.Marzagalli M., Ebelt N. D., Manuel E. R., Unraveling the crosstalk between melanoma and immune cells in the tumor microenvironment. Semin. Cancer Biol. 59, 236–250 (2019). [DOI] [PubMed] [Google Scholar]
- 25.Shields J. D., Borsetti M., Rigby H., Harper S. J., Mortimer P. S., Levick J. R., Orlando A., Bates D. O., Lymphatic density and metastatic spread in human malignant melanoma. Br. J. Cancer 90, 693–700 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shayan R., Karnezis T., Murali R., Wilmott J. S., Ashton M. W., Taylor G. I., Thompson J. F., Hersey P., Achen M. G., Scolyer R. A., Stacker S. A., Lymphatic vessel density in primary melanomas predicts sentinel lymph node status and risk of metastasis. Histopathology 61, 702–710 (2012). [DOI] [PubMed] [Google Scholar]
- 27.Zhang S., Yi S., Zhong D., Gong M., Cai Y., Zou L., Intratumoral and peritumoral lymphatic vessel density both correlate with lymph node metastasis in breast cancer. Sci. Rep. 7, 40364 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mori D., Yamasaki F., Shibaki M., Tokunaga O., Lateral peritumoral lymphatic vessel invasion can predict lymph node metastasis in esophageal squamous cell carcinoma. Mod. Pathol. 20, 694–700 (2007). [DOI] [PubMed] [Google Scholar]
- 29.Lin M., Ma S.-P., Lin H.-Z., Ji P., Xie D., Yu J.-X., Intratumoral as well as peritumoral lymphatic vessel invasion correlates with lymph node metastasis and unfavourable outcome in colorectal cancer. Clin. Exp. Metastasis 27, 123–132 (2010). [DOI] [PubMed] [Google Scholar]
- 30.Hara T., Makino T., Yamasaki M., Tanaka K., Tamashita K., Nogi Y., Saito T., Takahashi T., Kurokawa Y., Tatsumi M., Nakajima K., Morii E., Eguchi H., Doki Y., Peritumoral lymphatic vessels associated with resistance to neoadjuvant chemotherapy and unfavorable survival in esophageal cancer. Ann. Surg. Oncol. 27, 3762–3769 (2020). [DOI] [PubMed] [Google Scholar]
- 31.Baitsch L., Baumgaertner P., Devêvre E., Raghav S. K., Legat A., Barba L., Wieckowski S., Bouzourene H., Deplancke B., Romero P., Rufer N., Speiser D. E., Exhaustion of tumor-specific CD8+ T cells in metastases from melanoma patients. J. Clin. Invest. 121, 2350–2360 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu Z., Kim J. H., Falo L. D. J., You Z., Tumor regulatory T cells potently abrogate antitumor immunity. J. Immunol. 182, 6160–6167 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cremasco V., Woodruff M. C., Onder L., Cupovic J., Nieves-Bonilla J. M., Schildberg F. A., Chang J., Cremasco F., Harvey C. J., Wucherpfennig K., Ludewig B., Carroll M. C., Turley S. J., B cell homeostasis and follicle confines are governed by fibroblastic reticular cells. Nat. Immunol. 15, 973–981 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Astarita J. L., Cremasco V., Fu J., Darnell M. C., Peck J. R., Nieves-Bonilla J. M., Song K., Kondo Y., Woodruff M. C., Gogineni A., Onder L., Ludewig B., Weimer R. M., Carroll M. C., Mooney D. J., Xia L., Turley S. J., The CLEC-2–podoplanin axis controls the contractility of fibroblastic reticular cells and lymph node microarchitecture. Nat. Immunol. 16, 75–84 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Riedel A., Shorthouse D., Haas L., Hall B. A., Shields J., Tumor-induced stromal reprogramming drives lymph node transformation. Nat. Immunol. 17, 1118–1127 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lu P., Weaver V. M., Werb Z., The extracellular matrix: A dynamic niche in cancer progression. J. Cell Biol. 196, 395–406 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pickup M. W., Mouw J. K., Weaver V. M., The extracellular matrix modulates the hallmarks of cancer. EMBO Rep. 15, 1243–1253 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rohner N. A., McClain J., Tuell S. L., Warner A., Smith B., Yun Y., Mohan A., Sushnitha M., Thomas S. N., Lymph node biophysical remodeling is associated with melanoma lymphatic drainage. FASEB J. 29, 4512–4522 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Maarouf O. H., Uehara M., Kainath V., Solhjou Z., Banouni N., Bahmani B., Jiang L., Yilman O. A., Guleria I., Lovitch S. B., Grogan J. L., Fiorina P., Sage P. T., Bromberg J. S., McGrath M. M., Abdi R., Repetitive ischemic injuries to the kidneys result in lymph node fibrosis and impaired healing. JCI Insight 3, e120546 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huang L., Deng J., Xu W., Wang H., Shi L., Wu F., Wu D., Nei W., Zhao M., Mao P., Zhou X., CD8+ T cells with high TGF-β1 expression cause lymph node fibrosis following HIV infection. Mol. Med. Rep. 18, 77–86 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kityo C., Makamdop K. N., Rothenberger M., Chipman J. G., Hoskuldsson T., Beilman G. J., Grzywacz B., Mugyenyi P., Ssali F., Akondy R. S., Anderson J., Schmidt T. E., Reimann T., Callisto S. P., Schoephoerster J., Schuster J., Muloma P., Ssengendo P., Moysi E., Petrovas C., Lanciotti R., Zhang L., Arévalo M. T., Rodriquez B., Ross T. M., Trautmann L., Sekaly R.-P., Lederman M. M., Koup R. A., Ahmed R., Reilly C., Douek D. C., Schacker T. W., Lymphoid tissue fibrosis is associated with impaired vaccine responses. J. Clin. Invest. 128, 2763–2773 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rohner N. A., Thomas S. N., Flexible macromolecule versus rigid particle retention in the injected skin and accumulation in draining lymph nodes are differentially influenced by hydrodynamic size. ACS Biomater Sci. Eng. 3, 153–159 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Broggi M. A. S., Schmaler M., Lagarde N., Ross S. W., Isolation of murine lymph node stromal cells. J. Vis. Exp. , e51803 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fletcher A. L., Malhotra D., Acton S. E., Lukacs-Kornek V., Bellemare-Pelletier A., Curry M., Armant M., Turley S. J., Reproducible isolation of lymph node stromal cells reveals site-dependent differences in fibroblastic reticular cells. Front. Immunol. 2, 35 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Inaba K., Turley S., Yamaide F., Iyoda T., Mahnke K., Inaba M., Pack M., Subklewe M., Sauter B., Sheff D., Albert M., Bhardwaj N., Mellman I., Steinman R. M., Efficient presentation of phagocytosed cellular fragments on the major histocompatibility complex class II products of dendritic cells. J. Exp. Med. 188, 2163–2173 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kim M. K., Kim J., Properties of immature and mature dendritic cells: Phenotype, morphology, phagocytosis, and migration. RSC Adv. 9, 11230–11238 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hirosue S., Kourtis I. C., van der Vlies A. J., Hubbell J. A., Swartz M. A., Antigen delivery to dendritic cells by poly(propylene sulfide) nanoparticles with disulfide conjugated peptides: Cross-presentation and T cell activation. Vaccine 28, 7897–7906 (2010). [DOI] [PubMed] [Google Scholar]
- 48.Jeanbart L., Ballester M., de Titta A., Corthesy P., Romero P., Hubbell J. A., Swartz M. A., Enhancing efficacy of anticancer vaccines by targeted delivery to tumor-draining lymph nodes. Cancer Immunol. Res. 2, 436–447 (2014). [DOI] [PubMed] [Google Scholar]
- 49.Porgador A., Yewdell J. W., Deng Y., Bennink J. R., Germain R. N., Localization, quantitation, and in situ detection of specific peptide–MHC class I complexes using a monoclonal antibody. Immunity 6, 715–726 (1997). [DOI] [PubMed] [Google Scholar]
- 50.Stoitzner P., Tripp C. H., Eberhart A., Price K. M., Jung J. Y., Bursch L., Ronchese F., Romani N., Langerhans cells cross-present antigen derived from skin. Proc. Natl. Acad. Sci. U.S.A. 103, 7783–7788 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Colonna M., Trinchieri G., Liu Y.-J., Plasmacytoid dendritic cells in immunity. Nat. Immunol. 5, 1219–1226 (2004). [DOI] [PubMed] [Google Scholar]
- 52.Reddy S. T., Rehor A., Schmoekel H. G., Hubbell J. A., Swartz M. A., In vivo targeting of dendritic cells in lymph nodes with poly(propylene sulfide) nanoparticles. J. Control. Release 112, 26–34 (2006). [DOI] [PubMed] [Google Scholar]
- 53.Schudel A., Sestito L. F., Thomas S. N., S-nitrosated poly(propylene sulfide) nanoparticles for enhanced nitric oxide delivery to lymphatic tiss. J. Biomed. Mater. Res. A. 106, 1463–1475 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schudel A., Chapman A. P., Yau M.-K., Higginson C. J., Francis D. M., Manspeaker M. P., Avecilla A. R. C., Rohner N. A., Finn M. G., Thomas S. N., Programmable multistage drug delivery to lymph nodes. Nat. Nanotechnol. 15, 491–499 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wollenberg A., Mommaas M., Oppel T., Schottdorf E.-M., Günther S., Moderer M., Expression and function of the mannose receptor CD206 on epidermal dendritic cells in inflammatory skin diseases. J. Invest. Dermatol. 118, 327–334 (2002). [DOI] [PubMed] [Google Scholar]
- 56.Gupta P. K., Godec J., Wolski D., Adland E., Yates K., Pauken K. E., Cosgrove C., Ledderose C., Junger W. G., Robson S. C., Wherry E. J., Alter G., Goulder P. J. R., Klenerman P., Sharpe A. H., Lauer G. M., Haining W. N., CD39 expression identifies terminally exhausted CD8+ T cells. PLOS Pathog. 11, e1005177 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ahn E., Araki K., Hashimoto M., Li W., Riley J. L., Cheung J., Sharpe A. H., Freeman G. J., Irving B. A., Ahmed R., Role of PD-1 during effector CD8 T cell differentiation. Proc. Natl. Acad. Sci. U.S.A. 115, 4749–4754 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kaech S. M., Ahmed R., Memory CD8+ T cell differentiation: Initial antigen encounter triggers a developmental program in naïve cells. Nat. Immunol. 2, 415–422 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Horton B. L., Williams J. B., Cabanov A., Spranger S., Gajewski T. F., Intratumoral CD8+ T-cell apoptosis is a major component of T-cell dysfunction and impedes antitumor immunity. Cancer Immunol. Res. 6, 14–24 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Francis D. M., Manspeaker M. P., Schudel A., Sestito L. F., O’Melia M. J., Kissick H. T., Pollack B. P., Waller E. K., Thomas S. N., Blockade of immune checkpoints in lymph nodes through locoregional delivery augments cancer immunotherapy. Sci. Transl. Med. 12, eaay3575 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Davidson S., Efremova M., Riedel A., Mahata B., Pramanik J., Huuhtanen J., Kar G., Vento-Tormo R., Hagai T., Chen X., Haniffa M. A., Shields J. D., Teichmann S. A., Single-cell RNA sequencing reveals a dynamic stromal niche that supports tumor growth. Cell Rep. 31, 107628 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Thomas S. N., Schudel A., Overcoming transport barriers for interstitial-, lymphatic-, and lymph node-targeted drug delivery. Curr. Opin. Chem. Eng. 7, 65–74 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gerner M. Y., Mescher M. F., Antigen processing and MHC-II presentation by dermal and tumor-infiltrating dendritic cells. J. Immunol. 182, 2726–2737 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Clausen B. E., Stoitzner P., Functional specialization of skin dendritic cell subsets in regulating T cell responses. Front. Immunol. 6, 534 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Munn D. H., Sharma M. D., Hou D., Baban B., Lee J. R., Antonia S. J., Messina J. L., Chandler P., Koni P. A., Mellor A. L., Expression of indoleamine 2,3-dioxygenase by plasmacytoid dendritic cells in tumor-draining lymph nodes. J. Clin. Invest. 114, 280–290 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Essner R., Kojima M., Dendritic cell function in sentinel nodes. Oncologia 16, 27–31 (2002). [PubMed] [Google Scholar]
- 67.Malhotra D., Fletcher A. L., Astarita J., Lukacs-Kornek V., Tayalia P., Gonzalez S. F., Elpek K. G., Chang S. K., Knoblich K., Hemler M. E., Brenner M. B., Carroll M. C., Mooney D. J., Turley S. J.; Immunological Genome Project Consortium , Transcriptional profiling of stroma from inflamed and resting lymph nodes defines immunological hallmarks. Nat. Immunol. 13, 499–510 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Krishnamurty A. T., Turley S. J., Lymph node stromal cells: Cartographers of the immune system. Nat. Immunol. 21, 369–380 (2020). [DOI] [PubMed] [Google Scholar]
- 69.Fankhauser M., Broggi M. A. S., Potin L., Bordry N., Jeanbart L., Lund A. W., Da Costa E., Hauert S., Rincon-Restrepo M., Tremblay C., Cabello E., Homicsko K., Michielin O., Hanahan D., Speiser D. E., Swartz M. A., Tumor lymphangiogenesis promotes T cell infiltration and potentiates immunotherapy in melanoma. Sci. Transl. Med. 9, eaal4712 (2017). [DOI] [PubMed] [Google Scholar]
- 70.Lund A. W., Wagner M., Fankhauser M., Steinskog E. S., Broggi M. A., Spranger S., Gajewski T. F., Alitalo K., Eikesdal H. P., Wiig H., Swartz M. A., Lymphatic vessels regulate immune microenvironments in human and murine melanoma. J. Clin. Invest. 126, 3389–3402 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Binnewies M., Roberts E. W., Kersten K., Chan V., Fearon D. F., Merad M., Coussens L. M., Gabrilovich D. I., Ostrand-Rosenberg S., Hedrick C. C., Vonderheide R. H., Pittet M. J., Jain R. K., Zou W., Howcroft T. K., Woodhouse E. C., Weinberg R. A., Krummel M. F., Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 24, 541–550 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Jansen C. S., Prokhnevska N., Master V. A., Sanda M. G., Carlisle J. W., Bilen M. A., Cardenas M., Wilkinson S., Lake R., Sowalsky A. G., Valanparambil R. M., Hudson W. H., McGuire D., Melnick K., Khan A. I., Kim K., Chang Y. M., Kim A., Filson C. P., Alemozaffar M., Osunkoya A. O., Mullane P., Ellis C., Akondy R., Im S. J., Kamphorst A. O., Reyes A., Liu Y., Kissick H., An intra-tumoral niche maintains and differentiates stem-like CD8 T cells. Nature 576, 465–470 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Stylianopoulos T., Economides E.-A., Baish J. W., Fukumura D., Jain R. K., Towards optimal design of cancer nanomedicines: Multi-stage nanoparticles for the treatment of solid tumors. Ann. Biomed. Eng. 43, 2291–2300 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Taube J. M., Klein A., Brahmer J. R., Xu H., Pan X., Kim J. H., Chen L., Pardoll D. M., Topalian S. L., Anders R. A., Association of PD-1, PD-1 ligands, and other features of the tumor immune microenvironment with response to anti–PD-1 therapy. Clin. Cancer Res. 20, 5064–5074 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sharma P., Allison J. P., The future of immune checkpoint therapy. Science 348, 56–61 (2015). [DOI] [PubMed] [Google Scholar]
- 76.van der Vlies A. J., O’Neil C. P., Hasegawa U., Hammond N., Hubbell J. A., Synthesis of pyridyl disulfide-functionalized nanoparticles for conjugating thiol-containing small molecules, peptides, and proteins. Bioconjug. Chem. 21, 653–662 (2010). [DOI] [PubMed] [Google Scholar]
- 77.Schneider C. A., Rasband W. S., Eliceiri K. W., NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/50/eabd7134/DC1