

Abstract
Anemia of chronic inflammation (ACI) is a frequently diagnosed anemia and portends an independently increased morbidity and poor outcome associated with multiple underlying diseases. The pathophysiology of ACI is multifactorial, resulting from the effects of inflammatory cytokines which both directly and indirectly suppress erythropoiesis. Recent advances in molecular understanding of iron metabolism provide strong evidence that immune mediators, such as IL-6, lead to hepcidin-induced hypoferremia, iron sequestration, and decreased iron availability for erythropoiesis. The role of hepcidin-ferroportin axis in the pathophysiology of ACI is stimulating the development of new diagnostics and targeted therapies. In this review, we present an overview of and rationale for inflammation-, iron-, and erythropoiesis-related strategies currently in development.
Keywords: Anemia, inflammation, iron metabolism
INTRODUCTION
Anemia of chronic inflammation (ACI) was previously termed anemia of chronic disease and is considered the second most frequent anemia in the world after iron deficiency anemia (IDA).1,2 ACI is found in conditions associated with activation of the immune response, including chronic infections, autoimmune diseases, and malignancy. The presence of anemia in these conditions is considered evolutionarily advantageous in light of the iron dependence of pathogens and rapidly replicating cells. Thus, restricting iron availability serves to limit growth of pathogens and malignant cells at the expense of hemoglobin synthesis. Considered more broadly, the relative fatigue associated with illness and anemia serves to limit energy expenditure, better used to support the immune system, and prevents rapid spread of infectious pathogens, protecting species from ubiquitous exposure.
Anemia associated with chronic renal disease, congestive heart failure, and solid organ transplant is also categorized as ACI due to an inflammatory component in these conditions. In contrast, idiopathic anemia of the elderly often presents with typical features of ACI although inflammation in this anemia remains presumptive. For the purposes of this manuscript, we offer a perspective on ACI in its broadest definition although the specific details may not apply identically to all diseases on this list (e.g., in chronic renal insufficiency, anemia is in large part driven by inadequate erythropoietin or “erythropoietin resistance” although therapeutic benefit in anemia management may be gained by suppressing hepcidin).
ACI is independently associated with poor outcome and a decrease in quality of life measures. Standard approaches, such as parenteral iron and erythropoiesis stimulating agents (ESAs), are not consistently effective.3 Thus, despite its frequency and significant impact both on morbidity and mortality, the pathophysiology of ACI is incompletely understood and effective therapies are lacking. The goals of this manuscript are to
describe the clinical presentation and diagnosis of ACI,
elucidate our current understanding of iron metabolism and its mechanistic relationship with erythropoiesis, and
discuss potential therapeutic modalities.
DEFINITION AND CLINICAL OVERVIEW OF ACI
ACI is characterized as a mild, normocytic normochromic anemia with hemoglobin ranging from 8 to 10 g/dL, with elevated inflammatory markers (e.g., CRP, ESR, IL-6) and decreased circulating serum iron concentration and transferrin saturation despite ample iron stores (e.g., serum ferritin > 100 ng/mL). In the setting of inflammation, ACI may be difficult to differentiate from IDA, and IDA may coexist with ACI. In the majority of cases, IDA in the setting of ACI is a consequence of blood loss, either gastrointestinal or genitourinary, and diagnosing site of and halting the source of bleeding are both required. Furthermore, iron absorption may be impaired (e.g., celiac disease). Thus, understanding when administration of iron is expected to at least in part improve anemia drives the need for identify IDA as a possible component of ACI.
The traditional gold standard approach to identify IDA in ACI is Prussian-blue staining for iron within macrophages on a bone marrow biopsy specimen. However, this approach has been challenged in light of its invasiveness, high inter-observer variability, and poor correlation between iron-loaded macrophages and iron bioavailability in high hepcidin states, e.g., ACI. Furthermore, because serum ferritin functions as an acute phase reactant, it alone is insufficiently specific to identify IDA in ACI, unless very low (i.e., serum ferritin < 10 ng/mL). Serum ferritin is in equilibrium with cellular ferritin, largely derived from macrophages where its expression is increased when cellular iron increases, as in ACI.4 Thus, elevated ferritin concentration does not exclude a possible response to parenteral iron therapy.5
Additional parameters have been proposed to provide evidence of IDA in ACI. Soluble transferrin receptor 1 (sTfR1) concentration and sTfR1/log ferritin have been proposed as estimates of iron deficient erythropoiesis since circulating sTfR1 concentration is proportional to cellular expression of the membrane-associated TfR1, increases with increased cellular iron requirements and cellular proliferation,6–8 and is unaffected by inflammation. However, sTfR1 assays have not been standardized or widely adopted in light of the lack of convincing data of their utility.9
PATHOPHYSIOLOGY OF ACI
The pathophysiology of ACI is multifactorial, resulting from the effects of inflammatory cytokines such as TNF-α, interferon-γ, and IL-1 which suppress erythroid precursor differentiation and erythropoietin responsiveness,10–12 impeding the bone marrow’s ability to compensate for enhanced erythrophagocytosis by cytokine-activated splenic macrophages. In addition, inflammation may also influence erythropoietin production and renal excretion of hepcidin.13–15 However, there is now some consensus that ACI is in large part a consequence of altered iron metabolism resulting from the immune modulation of an underlying chronic disease in which immune mediators, such as IL-6 and possibly other cytokines involved in host defense, lead to hepcidin-induced hypoferremia, resulting in iron sequestration within the reticuloendothelial system, decreasing iron availability for erythropoiesis (Table 1). Although treatment of the underlying disease remains at the center of therapy for ACI, recent advances in molecular understanding of the hepcidin-ferroportin axis are stimulating the development of new targeted therapies.
Table 1.
Pathophysiology of anemia of chronic inflammation
| Increased inflammatory cytokines effect | Consequences on erythropoiesis |
|---|---|
| Increased macrophage activation |
|
| Suppressed erythroid precursor differentiation | Fewer RBC |
| Decreased erythropoietin responsiveness | Fewer erythroid precursors |
| Decreased erythropoietin production | Fewer erythroid precursors |
| Increased circulating hepcidin concentration |
|
Cytokines, IL-6 in particular, stimulate the hepatic hormone hepcidin. IL-6 induction of hepcidin expression is mediated through STAT3 signaling in hepatocytes. More recent data suggests that IL-6 may have a secondary suppressive effect on erythroid precursors.16 In addition, the loss of IL-6 and hepcidin in mice results in a milder anemia and more rapid recovery of hemoglobin in a well-established mouse model of ACI.17 Furthermore, IL-6 knockout animals exhibited faster bone marrow recovery relative to hepcidin knockout animals.
IRON METABOLISM AND HEPCIDIN REGULATION
Iron must be tightly regulated, because iron deficiency results in anemia and when iron is in excess, free iron promotes generation of reactive oxygen species, causing tissue injury and organ failure. Dietary absorption, storage, and recycling of iron maintain its stable concentration in circulation. The majority of systemic iron is contained within hemoglobin in circulating red blood cells. Iron storage, in the form of cellular ferritin, is located within hepatocytes and splenic macrophages. Muscle contains iron in the form of myoglobin for oxygen storage, and all cells require small amounts of iron for synthesis of iron-containing proteins for cell metabolism. Iron movement between sites of absorption, storage, and utilization through circulation occurs bound to transferrin; of the 20 to 25 mg of iron delivered daily, only 2 to 4 mg exist in circulation as transferrin-bound iron at any one time. Most of the iron found in circulation is recycled after erythrophagocytosing red blood cells by splenic macrophages. As iron loss is not modulated during systemic iron deficiency or overload, total body iron control is mainly regulated during dietary iron absorption and recycling.
The peptide hormone hepcidin is the principal regulator of iron homeostasis,18–20 including dietary iron absorption, iron recycling by macrophages, and the release of iron from hepatic stores. Hepcidin down-regulates iron release into plasma by binding to and functionally down-regulating ferroportin 1, the sole iron exporter.21,22 Hepcidin expression is induced by circulating iron and tissue iron stores,23,24 as well as inflammatory cytokines and suppressed by hypoxia,25 irondeficiency, and ineffective erythropoiesis. The bone morphogenic protein (BMP) pathway is critical in iron regulation of hepcidin expression with a specific role for hemojuvelin as an iron-specific BMP co-receptor and stimulant of the BMP receptor in iron overload states.26,27 HFE/TfR2 interacts with hemojuvelin and potentiates the BMP signaling pathway (e.g., BMP6 as the essential and specific BMP receptor ligand for iron-related signaling) and hepcidin transcription.27,28
Iron absorption is greatly increased in response to hemorrhage or erythropoietin in conditions of ample iron stores. As a result, the effect of erythropoiesis on intestinal iron absorption is dominant over iron in regulating hepcidin. The mechanisms of this suppressive effect have not been fully elucidated but likely include factor(s) secreted by erythroid precursors.29–33 Growth differentiation factor 15 and twisted gastrulation 1 are candidates identified in β-thalassemic humans and mice, respectively, but their roles have not been confirmed.34,35 More recently, erythroferrone (ERFE) has been identified as a physiological erythroid-derived hepcidin suppresor, increased in bone marrow from β-thalassemic and phlebotomized wild type mice, suppressing hepcidin expression in vitro (Figure 1).36 Furthermore, circulating ERFE concentrations are increased after erythropoietin administration and in β-thalassemic mice.37 However, ERFE appears to play a minimal role in baseline erythropoiesis and iron regulation with ERFE knockout mice exhibiting only mild anemia during the postnatal period36; its main function is likely to facilitate iron mobilization during recovery from anemia.
Figure 1.
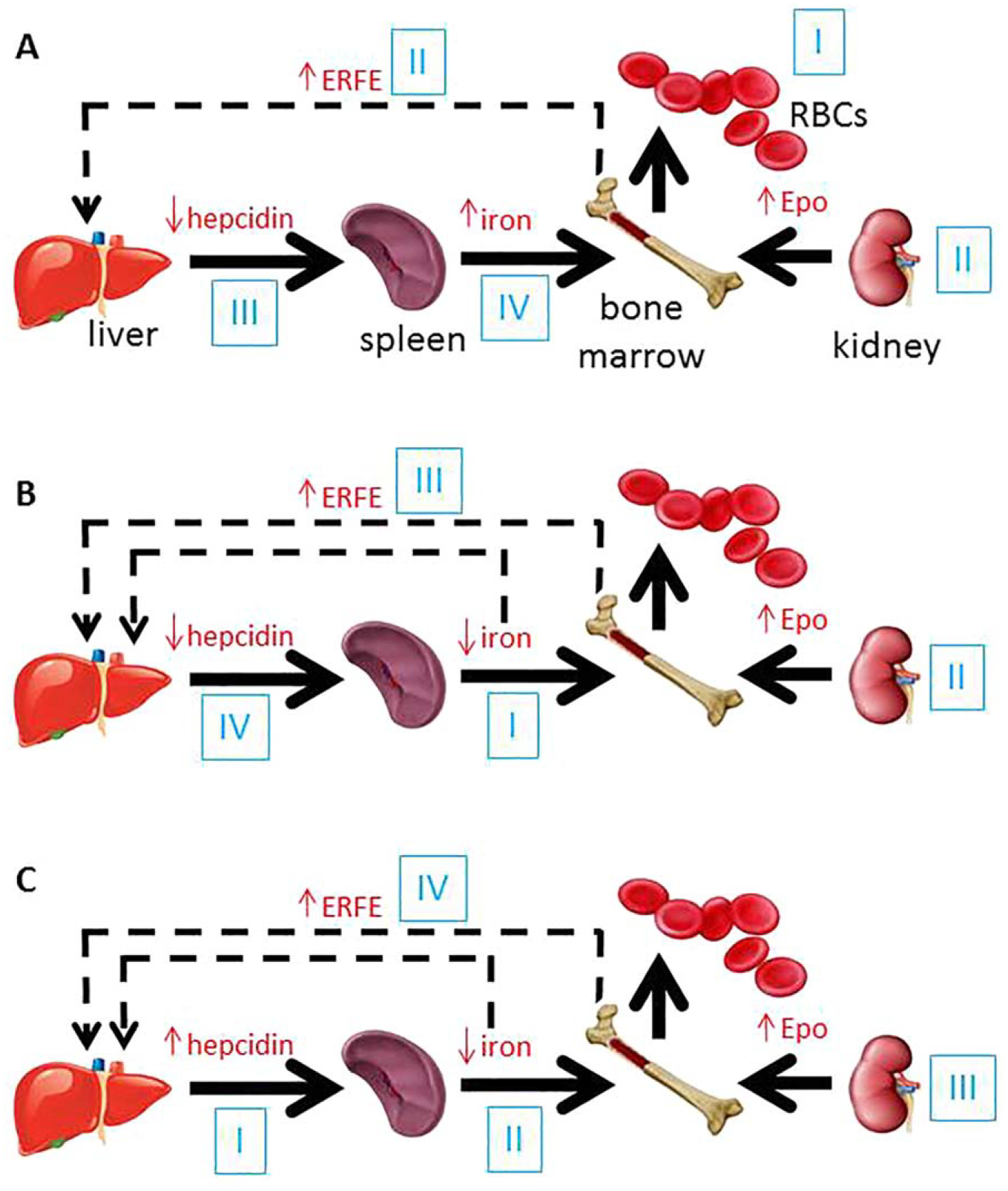
Proposed steps in the cross-talk between erythropoiesis and iron metabolism in conditions associated with iron dysregulation. (A) In acute blood loss (I), resultant hypoxia leads to increased erythropoietin (II) and consequent increase in erythroferrone (II) which suppresses hepcidin (III) and enables an increase in iron absorption and iron recycling, resulting in increased circulating iron (IV) available for erythropoiesis. (B) In iron deficiency anemia, decreased circulation iron concentration (I) leads to anemia and resultant hypoxia which results in increased erythropoietin (II) and subsequently increased erythroferrone (III), suppressing hepcidin (IV) to increase iron absorption and recycling. (C) In anemia of chronic inflammation, IL-6 driven increase in hepcidin (I) results in the sequestration of iron in macrophages, enterocytes, and hepatocytes, decreasing iron release into the circulation (II). As a consequence, iron restricted erythropoiesis and anemia result in compensatory increase in erythropoietin (III) and erythroferrone (IV), the later providing negative feedback to suppress hepcidin (RBCs = red blood cells; Epo = erythropoietin; ERFE = erythroferrone).
PATHOPHYSIOLOGY OF IRON SEQUESTRATION
The resultant increase in circulating hepcidin concentration leads to increased binding, internalization, and degradation of ferroportin, expressed on cells involved in iron metabolism (e.g., duodenal enterocytes, macrophages, hepatocytes, and placental cells). As ferroportin exports iron into the circulation, its degradation traps iron within cells, leading to increased cellular ferritin and ultimately serum ferritin concentration (in equilibrium with ferritin inside cells). As a consequence, inflammation negatively affects erythropoiesis by sequestering iron away from erythroid precursors (i.e., within bone marrow macrophages), inducing iron restricted erythropoiesis (Figure 2).
Figure 2.
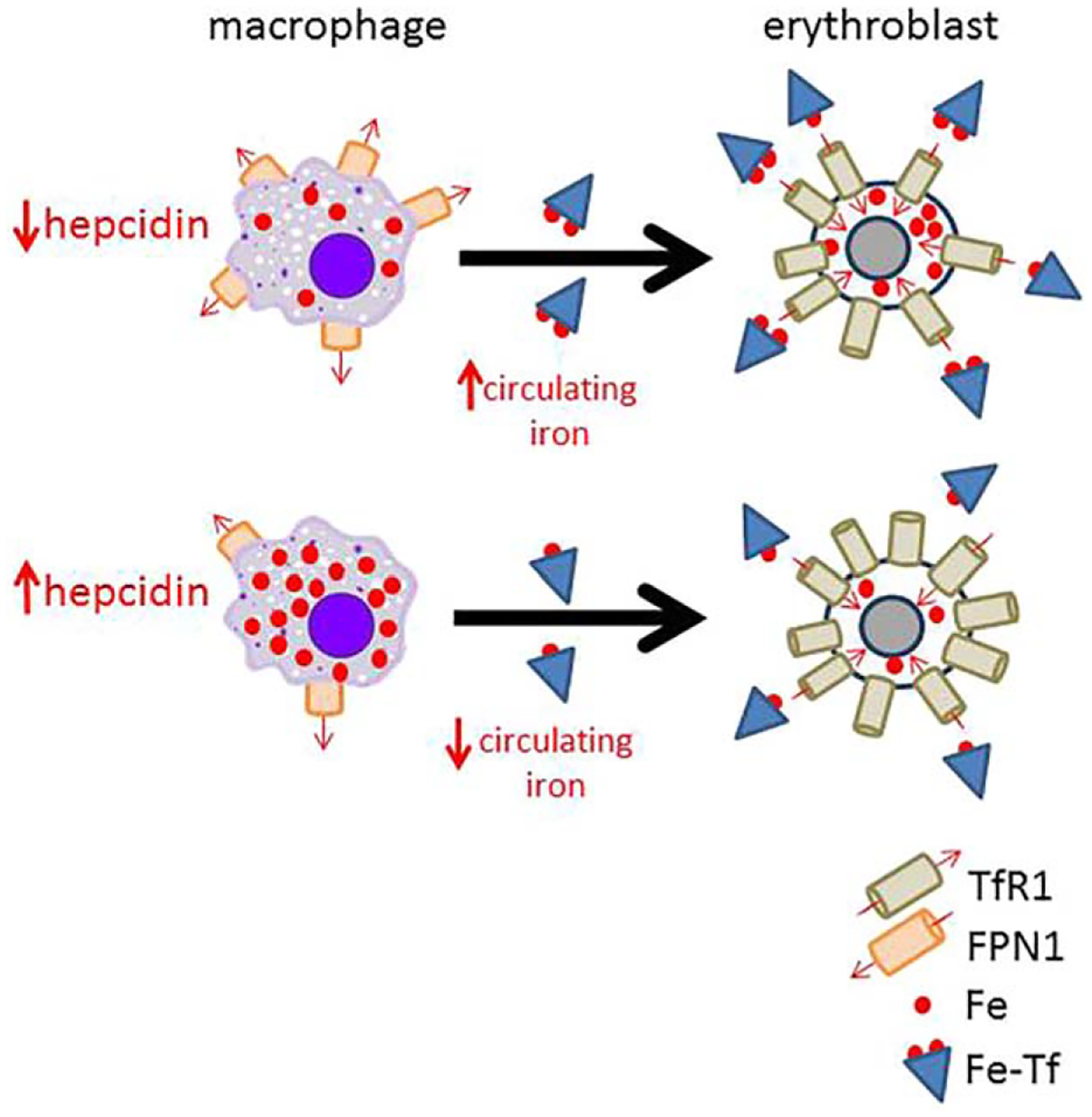
Model of hepcidin’s effect on iron availability for erythropoiesis. Because hepcidin functions post-translationally to influence iron efflux from cells by binding and internalizing ferroportin 1, the only known iron exporter, present on cells involved in iron metabolism (e.g., macrophages), decreased hepcidin leads to more ferroportin 1 present on cell membranes and consequently more iron efflux into circulation where it is available for uptake by erythroblasts via iron loaded transferrin binding to transferrin receptor 1. This happens in iron deficiency anemia. Alternatively, when hepcidin is high, its binding of ferroportin 1 prevents iron egress from cells and results in iron sequestration within macrophages (e.g., splenic macrophages, involved in iron recycling from senescent red blood cells), leading to iron restricted erythropoiesis. This happens in anemia of chronic inflammation (TfR1 = transferrin receptor 1; FPN1 = ferroportin 1; Fe = iron; Fe-Tf = iron loaded transferrin).
ROLE OF IRON-, ERYTHROPOIESIS-, AND HYPOXIA-INDUCED REGULATION OF HEPCIDIN IN THE INFLAMMATORY RESPONSE
Some crosstalk between IL-6 induced STAT3 signaling and BMP/SMAD signaling to hepcidin has been documented. For example, the SMAD binding site on the hepcidin promoter remains essential for IL-6 mediated hepcidin expression,38–40 and liver-specific Smad4 knockout mice demonstrate diminished hepcidin responsiveness to IL-6.41 In vitro experiments reveal that methods blocking BMP receptor signaling inhibit IL-6-mediated hepcidin expression,27,42 providing a rationale for modulating the BMP pathway to alter hepcidin expression in the context of ACI.
Increased erythropoietic demand, e.g., after phlebotomy or ESA administration, results in the mobilization of iron for erythropoiesis. The term “functional iron deficiency” refers to insufficient iron availability at the site of erythroblast production, despite adequate body iron stores and typically applies to the high hepcidin state in patients with renal insufficiency. However, broadly speaking, ACI, another high hepcidin condition, can also conceptually be referred to as functionally iron deficient. As a consequence of this iron restricted erythropoiesis, ERFE increases and possibly provides some counter-regulation of elevated hepcidin. In fact, ERFE knockout mice treated with heat-killed Brucella abortus, a well-accepted model of ACI, exhibited delayed hemoglobin recovery as a consequence of inadequate hepcidin suppression.43 Thus, suppressed erythropoiesis in ACI may enable further increase of hepcidin expression.
Iron homeostasis is also regulated by the hypoxia-mediated pathway, specifically, the involvement of hypoxia-inducible factors (HIFs) mediating hepcidin expression. Under normoxic conditions HIF1α is hydroxylated and marked for degradation by a complex of proteins containing von Hippel Landau (VHL). Hypoxia or mutations in VHL lead to HIF stabilization and transport to the nucleus where it functions as a transcription factor, affecting the expression of multiple genes in the erythropoietic cascade.44 Vhl knockout mice exhibit persistent HIF1α hydroxylation with compensatory increases in serum erythropoietin, splenomegaly, and hepcidin suppression.45 However, a concurrent Vhl and erythropoietin knockout results in hepcidin de-repression, suggesting that hepcidin suppression by hypoxia is mediated by erythropoiesis.46
USE OF HEPCIDIN AS A DIAGNOSTIC TOOL
In light of the central role hepcidin plays in the pathophysiology of ACI, the diagnostic potential of hepcidin has been studied extensively. Mass spectrometry, MALDITOF, and ELISA testing have all been used to determine hepcidin concentration in various clinical settings. This method has been useful in distinguishing an IDA component within ACI. Although hepcidin is elevated in patients with ACI relative to healthy controls, the coexistence of IDA and ACI results in a significant decrease in hepcidin relative to patients solely with ACI. This finding suggests that suppressive signaling as a consequence of iron restriction dominates stimulatory inflammatory signaling. However, standard cut-offs to enable clinical application of hepcidin in this clinical setting are not yet available. Despite this, relatively high hepcidin has been associated with lower mean corpuscular hemoglobin in anemic patients and impaired response to ESAs in an animal model of ACI.47,48
THE RATIONALE OF HEPCIDIN-LOWERING AGENTS
The goal of anemia management is to reverse patient symptoms attributable to decreased hemoglobin. Clinically, anemia has a broad and varied presentation including shortness of breath, lethargy, fatigue, skin pallor, palpitations, tachycardia, systolic flow murmurs, and angina, which can be difficult to distinguish from symptoms of the underlying disease, complicating the decision regarding treating anemia in addition to treating the cause of inflammation. Overall, resolution of anemia in ACI parallels therapy and improvement of the underlying inflammatory condition although treatment of anemia is associated with improved quality of life. Many translational studies focus on the reversing hepcidin overexpression to mitigate anemia in ACI. In addition to ACI, other diseases in which hepcidin concentration is increased include iron refractory iron deficiency anemia due to an autosomal recessive mutation in TMPRSS6, malignancy-associated anemia, and chronic renal insufficiency. Thus, when treatment of the underlying disease is difficult or impractical, severe anemia is typically treated with ESAs and parenteral iron, but their limited efficacy has dampened enthusiasm even in the face of even mild risk. Hepcidin antagonists are being developed targeting either the hepcidin molecule of its regulators (e.g., BMP, TfR2) in the hope that the potential therapeutic efficacy of hepcidin antagonists may extend to even mild anemias.
METHODS FOR PHARMACOLOGICAL REDUCTION OF HEPCIDIN
Via the inflammatory signal
Several diseases in the ACI category (e.g., Castleman disease and rheumatoid arthritis) have benefited from Phase I and II clinical trials with hepcidin lowering agents (Table 2). Therapeutic monoclonal antibodies against IL-6 receptor (tocilizumab), IL-6 (siltuximab), and TNFα (golimumab or infliximab) resulted in decreased hepcidin and improved anemia.49–53 In addition, in light of the STAT3-dependent signaling by IL-6, downstream inhibition of STAT3 signaling (e.g., curcumin, AG490, and PpYLKTK) is also expected to decrease hepcidin in humans, already demonstrated in vitro and in preclinical studies in vivo.54–56 However, this class of agents is unlikely to come to market as a consequence of their non-specific effects (all), competing iron chelating properties (curcumin), and suboptimal pharmacokinetics (AG490 and PpYLKTK). Last, multiple approaches aimed at manipulating the BMP/SMAD signaling pathway to hepcidin have also been employed to reduce hepcidin expression.
Table 2.
Hepcidin antagonists in development for ACI
| Mechanism | Agents | Stage |
|---|---|---|
| Cytokine inhibition | Antibodies to IL-6 receptor, IL-6, or TNFα | Phase I and II trials; FDA approved for other indications |
| Intracellular inflammatory cascade | STAT3 inhibitors | Development halted |
| BMP inhibition | Heparins; antibodies to ALK subtypes, HJV or BMP6 | Translational |
| EPO upregulation | PDH2 inhibitors | Translational |
| Direct hepcidin inhibition | Antibodies to hepcidin; hepcidin inhibitors | Phase I trials complete; further trials ongoing |
ACI = anemia of chronic inflammation; IL-6 = interleukin 6; TNFα = tumor necrosis factor alpha; FDA = Food and Drug Administration; STAT3 = signal transducer and activator of transcription 3; BMP = bone morphogenic protein; ALK = activin-like kinase; HJV = hemojuvulin; EPO = erythropoietin; PDH2 = pyruvate dehydrogenase 2.
Via the iron signal
Heparin, a glycosaminoglycan and frequently used agent in the treatment and prophylaxis of patients with existing or propensity toward blood clots, respectively, has a high affinity for BMP. Thus, heparin inhibits hepcidin expression by binding to and sequestering BMPs.57 Furthermore, heparins without anticoagulating effects have been developed, demonstrating reversal of anemia in mouse models of ACI58 and heparins with high sulfation grade and low anticoagulant activity are specifically able to block hepcidin in vitro and in preclinical studies in vivo.59,60 These agents are poised for randomized clinical trials. Hemojuvulin (HJV), also known as HFE2, is a BMP co-receptor, enhancing hepcidin expression and soluble HJV,42 resulting from furin-mediated cleavage of cellular HJV,61 competes for binding of BMP to BMP receptor.27,62 Administration of either neutralizing anti-HJV antibodies (AbbVie Pharmaceuticals) or sHJV fused with IgG Fc fragment result in hepcidin suppression63 and correct anemia in murine ACI models.64,65 Clinical studies using such agents in renal failure patients are underway.
Antibodies and small molecule inhibitors of BMP/SMAD have demonstrated preclinical efficacy in decreasing hepcidin expression. For example, anti-BMP6 antibodies reduce hepcidin expression and increase serum iron concentration in mice.66,67 In addition, dorsomorphin, an inhibitor of BMP/SMAD signaling, prevents hepcidin induction by IL-6 by blocking type I BMP receptors ALK2, ALK3, and ALK6, inducing hyperferremia in mice.68 Furthermore, dorsomorphin derivatives, e.g., LDN-193189, prevent hepcidin induction and anemia in murine ACI and renal failure models.65,69–71 However, because dorsomorphin and its derivatives do not specifically target BMP/SMAD signaling to hepcidin, they are considered suboptimal candidates for therapeutic development. TP-0184, a specific ALK2 inhibitor (Toledo Pharmaceuticals), has recently been shown to suppress hepcidin in mouse models of ACI and cancer,72 but further studies are required to evaluate its full translational potential.
Via erythropoietic signal
Novel therapeutics targeting stabilization of HIFs provide promise in the treatment of ACI, enabling hepcidin blockade mediated via erythropoiesis, simultaneously stimulating erythropoiesis and iron availability. For example, propyl hydroxylase domain-2 (PDH2) inhibitors stabilize HIF-1 and HIF-2, stimulating endogenous erythropoietin production to suppress hepcidin and enable greater iron availability for erythropoiesis.46,73,74 Other hypoxia inducible hormones such as platelet-derived growth factor BB interfere with endoplasmic reticulum stress-inducible signaling pathway involving CREB-H to suppress hepcidin.75,76 In light of the recent discovery of erythroferrone and its negative regulation of hepcidin, modulation of this pathway is a clear area for future investigation.
Direct hepcidin inhibition
Direct hepcidin inhibitors, e.g., neutralizing antibodies, have demonstrated efficacy in increasing circulating and available iron for erythropoiesis, reversing anemia and increasing responsiveness to ESAs in mouse models of ACI77 and increasing serum iron concentration and erythropoiesis in monkeys.78 LY2787106, a monoclonal anti-hepcidin antibody (Eli Lily), has been developed and proposed as potential therapy in conditions of excess hepcidin,79 recently demonstrating high tolerability, transient iron mobilization, and increased reticulocytosis in a Phase I study of cancer-related anemia patients.80 PRS-080, a PEGylated anticalin with specific binding and neutralization of hepcidin (Pieris Pharmaceuticals), demonstrated suppression of hepcidin activity in vitro and in mice81 with decreased hepcidin and transiently increased transferrin saturation at doses above 0.4 mg/kg in a Phase I study in healthy volunteers,82 warranting further investigations in anemic patients. Last, NOX-H94, a spiegelmer (PEGylated nonnatural occurring mirror-image L-oligoribonucleotide) hepcidin antagonist (NOXXON Pharma AG), binds hepcidin with high affinity and specificity. NOX-H94 has been demonstrated to improve ACI in monkeys83 and prevent hypoferremia in LSP-injected humans volunteers.84 As a consequence, additional clinical trials in renal insufficiency, ACI, and cancer-related anemia are in the planning phases.
CONCLUSION
Anemia in cancer and inflammatory and autoimmune diseases is a common clinical condition often neglected by treating physicians. This challenge is a consequence of inadequate easy and standardized diagnostic tools, incomplete appreciation of variability in therapeutic targets for hemoglobin, insufficient guidelines to estimate the degree of functional iron deficit, and how to tailor therapy with parenteral iron, ESAs, and RBC transfusion for individual patients. Furthermore, it is only recently that we have enhanced our understanding of the complexity of regulation of iron metabolism and cross-talk with erythropoiesis. Based on this, novel therapeutics aiming to modulate hepcidin and increase iron availability for erythropoiesis are being explored from multiple directions. It is noteworthy to consider different therapeutic strategies in ACI associated with different underlying diseases and the potential of hepcidin-targeted therapeutics to enable some degree of physiological self-regulation to prevent inadequate or excessing dosing in individual patients. Many of these therapeutic approaches have been validated in preclinical and clinical settings, with some currently being further explored in randomized clinical trials and are poised to improve management of iron maldistribution and anemia in ACI.
Acknowledgments
Disclosure of grants or other funding: No disclosure of grants or other funding.
Footnotes
Conflict of Interest: The authors declare that they have no conflict of interest.
REFERENCES
- 1.Weiss G, Goodnough LT. Anemia of chronic disease. N Engl J Med. 2005; 352:1011–1023. [DOI] [PubMed] [Google Scholar]
- 2.Matzner Y, Levy S, Grossowicz N, Izak G, Hershko C. Prevalence and causes of anemia in elderly hospitalized patients. Gerontology. 1979; 25:113–119. [DOI] [PubMed] [Google Scholar]
- 3.Sun CC, Vaja V, Babitt JL, Lin HY. Targeting the hepcidin-ferroportin axis to develop new treatment strategies for anemia of chronic disease and anemia of inflammation. Am J Hematol. 2012; 87:392–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cohen LA, Gutierrez L, Weiss A, et al. Serum ferritin is derived primarily from macrophages through a nonclassical secretory pathway. Blood. 2010; 116: 1574–1584. [DOI] [PubMed] [Google Scholar]
- 5.Cazzola M, Ponchio L, de Benedetti F, et al. Defective iron supply for erythropoiesis and adequate endogenous erythropoietin production in the anemia associated with systemic-onset juvenile chronic arthritis. Blood. 1996; 87:4824–4830. [PubMed] [Google Scholar]
- 6.Cazzola M, Beguin Y. New tools for clinical evaluation of erythron function in man. Br J Haematol. 1992; 80: 278–284. [DOI] [PubMed] [Google Scholar]
- 7.Skikne BS, Flowers CH, Cook JD. Serum transferrin receptor: A quantitative measure of tissue iron deficiency. Blood. 1990; 75:1870–1876. [PubMed] [Google Scholar]
- 8.R’Zik S, Beguin Y. Serum soluble transferrin receptor concentration is an accurate estimate of the mass of tissue receptors. Exp Hematol. 2001; 29:677–685. [DOI] [PubMed] [Google Scholar]
- 9.Infusino I, Braga F, Dolci A, Panteghini M. Soluble transferrin receptor (sTfR) and sTfR/log ferritin index for the diagnosis of iron-deficiency anemia. A meta-analysis. Am J Clin Pathol. 2012; 138:642–649. [DOI] [PubMed] [Google Scholar]
- 10.Wang CQ, Udupa KB, Lipschitz DA. Interferon-gamma exerts its negative regulatory effect primarily on the earliest stages of murine erythroid progenitor cell development. J Cell Physiol. 1995; 162:134–138. [DOI] [PubMed] [Google Scholar]
- 11.Grigorakaki C, Morceau F, Chateauvieux S, Dicato M, Diederich M. Tumor necrosis factor alpha-mediated inhibition of erythropoiesis involves GATA-1/GATA-2 balance impairment and PU.1 over-expression. Biochem Pharmacol. 2011; 82:156–166. [DOI] [PubMed] [Google Scholar]
- 12.Means RT Jr, Dessypris EN, Krantz SB. Inhibition of human erythroid colony-forming units by interleukin-1 is mediated by gamma interferon. J Cell Physiol. 1992; 150:59–64. [DOI] [PubMed] [Google Scholar]
- 13.Krajewski J, Batmunkh C, Jelkmann W, Hellwig-Burgel T. Interleukin-1beta inhibits the hypoxic inducibility of the erythropoietin enhancer by suppressing hepatocyte nuclear factor-4alpha. Cell Mol Life Sci. 2007; 64: 989–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jelkmann W Proinflammatory cytokines lowering erythropoietin production. J Interferon Cytokine Res. 1998; 18:555–559. [DOI] [PubMed] [Google Scholar]
- 15.Vannucchi AM, Grossi A, Rafanelli D, Statello M, Cinotti S, Rossi-Ferrini P. Inhibition of erythropoietin production in vitro by human interferon gamma. Br J Haematol. 1994; 87:18–23. [DOI] [PubMed] [Google Scholar]
- 16.McCranor BJ, Kim MJ, Cruz NM, et al. Interleukin-6 directly impairs the erythroid development of human TF-1 erythroleukemic cells. Blood Cells Mol Dis. 2014; 52:126–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gardenghi S, Renaud TM, Meloni A, et al. Distinct roles for hepcidin and interleukin-6 in the recovery from anemia in mice injected with heat-killed Brucella abortus. Blood. 2014; 123:1137–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Krause A, Neitz S, Magert HJ, et al. LEAP-1, a novel highly disulfide-bonded human peptide, exhibits antimicrobial activity. FEBS Lett. 2000; 480:147–150. [DOI] [PubMed] [Google Scholar]
- 19.Park CH, Valore EV, Waring AJ, Ganz T. Hepcidin, a urinary antimicrobial peptide synthesized in the liver. J Biol Chem. 2001; 276:7806–7810. [DOI] [PubMed] [Google Scholar]
- 20.Ganz T Hepcidin–a regulator of intestinal iron absorption and iron recycling by macrophages. Best Pract Res Clin Haematol. 2005; 18:171–182. [DOI] [PubMed] [Google Scholar]
- 21.Nemeth E, Tuttle MS, Powelson J, et al. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science. 2004; 306:2090–2093. [DOI] [PubMed] [Google Scholar]
- 22.Donovan A, Lima CA, Pinkus JL, et al. The iron exporter ferroportin/Slc40a1 is essential for iron homeostasis. Cell Metab. 2005; 1:191–200. [DOI] [PubMed] [Google Scholar]
- 23.Pigeon C, Ilyin G, Courselaud B, et al. A new mouse liver-specific gene, encoding a protein homologous to human antimicrobial peptide hepcidin, is overex-pressed during iron overload. J Biol Chem. 2001; 276: 7811–7819. [DOI] [PubMed] [Google Scholar]
- 24.Nemeth E, Rivera S, Gabayan V, et al. IL-6 mediates hypoferremia of inflammation by inducing the synthesis of the iron regulatory hormone hepcidin. J Clin Invest. 2004; 113:1271–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nicolas G, Chauvet C, Viatte L, et al. The gene encoding the iron regulatory peptide hepcidin is regulated by anemia, hypoxia, and inflammation. J Clin Invest. 2002; 110:1037–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Truksa J, Peng H, Lee P, Beutler E. Bone morphogenetic proteins 2, 4, and 9 stimulate murine hepcidin 1 expression independently of Hfe, transferrin receptor 2 (Tfr2), and IL-6. Proc Natl Acad Sci USA. 2006; 103: 10289–10293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Babitt JL, Huang FW, Xia Y, Sidis Y, Andrews NC, Lin HY. Modulation of bone morphogenetic protein signaling in vivo regulates systemic iron balance. J Clin Invest. 2007; 117:1933–1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schmidt PJ, Toran PT, Giannetti AM, Bjorkman PJ, Andrews NC. The transferrin receptor modulates Hfe-dependent regulation of hepcidin expression. Cell Metab. 2008; 7:205–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Weizer-Stern O, Adamsky K, Amariglio N, et al. Downregulation of hepcidin and haemojuvelin expression in the hepatocyte cell-line HepG2 induced by thalassaemic sera. Br J Haematol. 2006; 135:129–138. [DOI] [PubMed] [Google Scholar]
- 30.Vokurka M, Krijt J, Sulc K, Necas E. Hepcidin mRNA levels in mouse liver respond to inhibition of erythropoiesis. Physiol Res. 2006; 55:667–674. [DOI] [PubMed] [Google Scholar]
- 31.Kearney SL, Nemeth E, Neufeld EJ, et al. Urinary hepcidin in congenital chronic anemias. Pediatr Blood Cancer. 2007; 48:57–63. [DOI] [PubMed] [Google Scholar]
- 32.Origa R, Galanello R, Ganz T, et al. Liver iron concentrations and urinary hepcidin in beta-thalassemia. Haematologica. 2007; 92:583–588. [DOI] [PubMed] [Google Scholar]
- 33.Kemna EH, Kartikasari AE, van Tits LJ, Pickkers P, Tjalsma H, Swinkels DW. Regulation of hepcidin: Insights from biochemical analyses on human serum samples. Blood Cells Mol Dis. 2008; 40:339–346. [DOI] [PubMed] [Google Scholar]
- 34.Tanno T, Bhanu NV, Oneal PA, et al. High levels of GDF15 in thalassemia suppress expression of the iron regulatory protein hepcidin. Nat Med. 2007; 13: 1096–1101. [DOI] [PubMed] [Google Scholar]
- 35.Tanno T, Porayette P, Sripichai O, et al. Identification of TWSG1 as a second novel erythroid regulator of hepcidin expression in murine and human cells. Blood. 2009; 114:181–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kautz L, Jung G, Valore EV, Rivella S, Nemeth E, Ganz T. Identification of erythroferrone as an erythroid regulator of iron metabolism. Nat Genet. 2014; 46:678–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kautz L, Jung G, Du X, et al. Erythroferrone contributes to hepcidin suppression and iron overload in a mouse model of beta-thalassemia. Blood. 2015; 126: 2031–2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Verga Falzacappa MV, Casanovas G, Hentze MW, Muckenthaler MU. A bone morphogenetic protein (BMP)-responsive element in the hepcidin promoter controls HFE2-mediated hepatic hepcidin expression and its response to IL-6 in cultured cells. J Mol Med. 2008; 86:531–540. [DOI] [PubMed] [Google Scholar]
- 39.Fleming RE. Hepcidin activation during inflammation: Make it STAT. Gastroenterology. 2007; 132:447–449. [DOI] [PubMed] [Google Scholar]
- 40.Huang H, Constante M, Layoun A, Santos MM. Contribution of STAT3 and SMAD4 pathways to the regulation of hepcidin by opposing stimuli. Blood. 2009; 113:3593–3599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang RH, Li C, Xu X, et al. A role of SMAD4 in iron metabolism through the positive regulation of hepcidin expression. Cell Metab. 2005; 2:399–409. [DOI] [PubMed] [Google Scholar]
- 42.Babitt JL, Huang FW, Wrighting DM, et al. Bone morphogenetic protein signaling by hemojuvelin regulates hepcidin expression. Nat Genet. 2006; 38:531–539. [DOI] [PubMed] [Google Scholar]
- 43.Kautz L, Jung G, Nemeth E, Ganz T. Erythroferrone contributes to recovery from anemia of inflammation. Blood. 2014; 124:2569–2574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.van Rooijen E, Santhakumar K, Logister I, et al. A zebrafish model for VHL and hypoxia signaling. Methods Cell Biol. 2011; 105:163–190. [DOI] [PubMed] [Google Scholar]
- 45.Liu Q, Davidoff O, Niss K, Haase VH. Hypoxiainducible factor regulates hepcidin via erythropoietin-induced erythropoiesis. J Clin Invest. 2012; 122: 4635–4644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mastrogiannaki M, Matak P, Mathieu JR, et al. Hepatic hypoxia-inducible factor-2 down-regulates hepcidin expression in mice through an erythropoietin-mediated increase in erythropoiesis. Haematologica. 2012; 97: 827–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Prentice AM, Doherty CP, Abrams SA, et al. Hepcidin is the major predictor of erythrocyte iron incorporation in anemic African children. Blood. 2012; 119: 1922–1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Theurl M, Nairz M, Schroll A, et al. Hepcidin as a predictive factor and therapeutic target in erythropoiesis-stimulating agent treatment for anemia of chronic disease in rats. Haematologica. 2014; 99:1516–1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Casper C, Chaturvedi S, Munshi N, et al. Analysis of inflammatory and anemia-related biomarkers in a randomized, double-blind, placebo-controlled study of siltuximab (anti-IL6 monoclonal antibody) in patients with multicentric castleman disease. Clin Cancer Res. 2015; 21:4294–4304. [DOI] [PubMed] [Google Scholar]
- 50.Isaacs JD, Harari O, Kobold U, Lee JS, Bernasconi C. Effect of tocilizumab on haematological markers implicates interleukin-6 signalling in the anaemia of rheumatoid arthritis. Arthritis Res Ther. 2013; 15:R204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Song SN, Tomosugi N, Kawabata H, Ishikawa T, Nishikawa T, Yoshizaki K. Down-regulation of hepcidin resulting from long-term treatment with an anti-IL-6 receptor antibody (tocilizumab) improves anemia of inflammation in multicentric Castleman disease. Blood. 2010; 116:3627–3634. [DOI] [PubMed] [Google Scholar]
- 52.Song SN, Iwahashi M, Tomosugi N, et al. Comparative evaluation of the effects of treatment with tocilizumab and TNF-alpha inhibitors on serum hepcidin, anemia response and disease activity in rheumatoid arthritis patients. Arthritis Res Ther. 2013; 15:R141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Doyle MK, Rahman MU, Frederick B, et al. Effects of subcutaneous and intravenous golimumab on inflammatory biomarkers in patients with rheumatoid arthritis: Results of a phase 1, randomized, open-label trial. Rheumatology (Oxford). 2013; 52:1214–1219. [DOI] [PubMed] [Google Scholar]
- 54.Jiao Y, Wilkinson J, Di X, et al. Curcumin, a cancer chemopreventive and chemotherapeutic agent, is a biologically active iron chelator. Blood. 2009; 113:462–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fatih N, Camberlein E, Island ML, et al. Natural and synthetic STAT3 inhibitors reduce hepcidin expression in differentiated mouse hepatocytes expressing the active phosphorylated STAT3 form. J Mol Med. 2010; 88:477–486. [DOI] [PubMed] [Google Scholar]
- 56.Zhang SP, Wang Z, Wang LX, Liu SJ. AG490: An inhibitor of hepcidin expression in vivo. World J Gastroenterol. 2011; 17:5032–5034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Poli M, Girelli D, Campostrini N, et al. Heparin: A potent inhibitor of hepcidin expression in vitro and in vivo. Blood. 2011; 117:997–1004. [DOI] [PubMed] [Google Scholar]
- 58.Poli M, Asperti M, Naggi A, et al. Glycol-split nonanticoagulant heparins are inhibitors of hepcidin expression in vitro and in vivo. Blood. 2014; 123:1564–1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Poli M, Asperti M, Ruzzenenti P, et al. Oversulfated heparins with low anticoagulant activity are strong and fast inhibitors of hepcidin expression in vitro and in vivo. Biochem Pharmacol. 2014; 92:467–475. [DOI] [PubMed] [Google Scholar]
- 60.Asperti M, Naggi A, Esposito E, et al. High sulfation and a high molecular weight are important for anti-hepcidin activity of heparin. Front Pharmacol. 2015; 6: 316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Silvestri L, Pagani A, Camaschella C. Furin-mediated release of soluble hemojuvelin: A new link between hypoxia and iron homeostasis. Blood. 2008; 111:924–931. [DOI] [PubMed] [Google Scholar]
- 62.Lin L, Goldberg YP, Ganz T. Competitive regulation of hepcidin mRNA by soluble and cell-associated hemojuvelin. Blood. 2005; 106:2884–2889. [DOI] [PubMed] [Google Scholar]
- 63.Boser P, Seemann D, Liguori MJ, et al. Anti-repulsive guidance molecule C (RGMc) antibodies increases serum iron in rats and cynomolgus monkeys by hepcidin downregulation. AAPS J. 2015; 17:930–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kovac S, Boser P, Cui Y, et al. Anti-hemojuvelin antibody corrects anemia caused by inappropriately high hepcidin levels. Haematologica. 2016; 101:e173–e176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Theurl I, Schroll A, Sonnweber T, et al. Pharmacologic inhibition of hepcidin expression reverses anemia of chronic inflammation in rats. Blood. 2011; 118:4977–4984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Meynard D, Kautz L, Darnaud V, Canonne-Hergaux F, Coppin H, Roth MP. Lack of the bone morphogenetic protein BMP6 induces massive iron overload. Nat Genet. 2009; 41:478–481. [DOI] [PubMed] [Google Scholar]
- 67.Andriopoulos B Jr, Corradini E, Xia Y, et al. BMP6 is a key endogenous regulator of hepcidin expression and iron metabolism. Nat Genet. 2009; 41:482–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yu PB, Hong CC, Sachidanandan C, et al. Dorsomorphin inhibits BMP signals required for embryogenesis and iron metabolism. Nat Chem Biol. 2008; 4:33–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Steinbicker AU, Sachidanandan C, Vonner AJ, et al. Inhibition of bone morphogenetic protein signaling attenuates anemia associated with inflammation. Blood. 2011; 117:4915–4923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mayeur C, Kolodziej SA, Wang A, et al. Oral administration of a bone morphogenetic protein type I receptor inhibitor prevents the development of anemia of inflammation. Haematologica. 2015; 100:e68–e71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sun CC, Vaja V, Chen S, et al. A hepcidin lowering agent mobilizes iron for incorporation into red blood cells in an adenine-induced kidney disease model of anemia in rats. Nephrol Dial Transplant. 2013; 28: 1733–1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Peterson P, Soh KK, Sol Lee Y, et al. ALK2 inhibition via TP-0184 abrogates inflammation-induced hepcidin expression and is a potential therapeutic for anemia of chronic disease. Annual meeting of the American Society of Hematology, 2015, Orlando, FL. [Google Scholar]
- 73.Wilkinson N, Pantopoulos K. IRP1 regulates erythropoiesis and systemic iron homeostasis by controlling HIF2alpha mRNA translation. Blood. 2013; 122:1658–1668. [DOI] [PubMed] [Google Scholar]
- 74.Soni H Prolyl hydroxylase domain-2 (PHD2) inhibition may be a better therapeutic strategy in renal anemia. Med Hypotheses. 2014; 82:547–550. [DOI] [PubMed] [Google Scholar]
- 75.Vecchi C, Montosi G, Zhang K, et al. ER stress controls iron metabolism through induction of hepcidin. Science. 2009; 325:877–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sonnweber T, Nachbaur D, Schroll A, et al. Hypoxia induced downregulation of hepcidin is mediated by platelet derived growth factor BB. Gut. 2014; 63:1951–1959. [DOI] [PubMed] [Google Scholar]
- 77.Sasu BJ, Cooke KS, Arvedson TL, et al. Antihepcidin antibody treatment modulates iron metabolism and is effective in a mouse model of inflammation-induced anemia. Blood. 2010; 115:3616–3624. [DOI] [PubMed] [Google Scholar]
- 78.Cooke KS, Hinkle B, Salimi-Moosavi H, et al. A fully human anti-hepcidin antibody modulates iron metabolism in both mice and nonhuman primates. Blood. 2013; 122:3054–3061. [DOI] [PubMed] [Google Scholar]
- 79.Grebenchtchikov N, Geurts-Moespot AJ, Trentmann S, et al. Engineered human lipocalin as an antibody mimetic: Application to analysis of the small peptide hormone hepcidin. Clin Chem. 2014; 60:897–899. [DOI] [PubMed] [Google Scholar]
- 80.Vadhan-Raj S, Abonour R, Goldman JW, et al. Phase 1 Study of a hepcidin antagonist, LY2787106, in cancer-associated anemia. Annual meeting of the American Society of Hematology, 2015, Orlando, FL. [Google Scholar]
- 81.Hohlbaum AM, Trentman S, Gille H, et al. Discovery and preclinical characterization of a novel hepcidin antagonist with tunable PK/PD properties for the treatment of anemia in different patient populations. Annual meeting of the American Society of Hematology, 2011, San Diego, CA. [Google Scholar]
- 82.Moebius U, Feuerer W, Fenzl E, van Swelm R, Swinkels DW, Hohlbaum A. A Phase I Study investigating the safety, tolerability, pharmacokinetics and pharmacodynamic activity of the hepcidin antagonist PRS-080#022. Results from a randomized, placebo controlled, double-blind study following single administration to healthy subjects. Annual meeting of the American Society of Hematology, 2015, Orlando, FL. [Google Scholar]
- 83.Schwoebel F, van Eijk LT, Zboralski D, et al. The effects of the anti-hepcidin Spiegelmer NOX-H94 on inflammation-induced anemia in cynomolgus monkeys. Blood. 2013; 121:2311–2315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.van Eijk LT, John AS, Schwoebel F, et al. Effect of the antihepcidin Spiegelmer lexaptepid on inflammation-induced decrease in serum iron in humans. Blood. 2014; 124:2643–2646. [DOI] [PMC free article] [PubMed] [Google Scholar]