

Abstract
Antemortem tau positron emission tomography imaging suggests elevated tau pathology in autosomal dominant versus late‐onset Alzheimer’s disease at equivalent clinical stages, but does not implicate the specific tau pathologies responsible. Here we made stereological measurements of tau neurofibrillary tangles, neuritic plaques, and neuropil threads and found compared to late‐onset Alzheimer’s disease, autosomal dominant Alzheimer’s disease showed even greater tangle and thread burdens. Regional tau burden resembled that observed in tau imaging of a separate cohort at earlier clinical stages. Finally, our results suggest tau imaging measures total tau burden in Alzheimer’s disease, composed predominantly of tangle and thread pathology.
Introduction
Antemortem tau positron emission tomography (PET) imaging suggests elevated tau pathology in autosomal dominant (ADAD) versus late‐onset Alzheimer’s disease (LOAD) at equivalent clinical stages. Compared to LOAD, ADAD has shown elevated 18F‐flortaucipir 1 radioligand binding in prefrontal, premotor, and inferior parietal cortices, 2 as well as precuneus and lateral parietal cortices. 3 However, PET imaging does not implicate specific tau pathologies responsible. Previous work quantitatively comparing AD tau pathology with PET imaging has typically been performed in a single individual, 4 , 5 and it is not known whether these results generalize, given the disease heterogeneity of both ADAD 6 and LOAD 7 . To investigate which tau pathologies contribute to elevated 18F‐flortaucipir binding in ADAD versus LOAD cohorts, we made stereological measurements of three major features of AD tau pathology: neurofibrillary tangles, neuritic plaques, and neuropil threads.
Methods
Protocols for the study have received prior approval by the local Institutional Review Board of each Dominantly Inherited Alzheimer Network site. Informed consent was obtained from each participant.
Cases selected for postmortem study were participants in the Dominantly Inherited Alzheimer Network (n = 7) or in studies directed by the Knight Alzheimer Disease Research Center (n = 10) (Table 1). These individuals met the inclusion criteria of high AD neuropathological change 8 without comorbid neurodegenerative or vascular disease.
Table 1.
Cohort demographics.
| Neuropathology cohort | Imaging cohort | |||
|---|---|---|---|---|
| LOAD | ADAD | LOAD | ADAD | |
| Number | 10 | 7 | 35 | 14 |
| Age at visit, years (SD) | 74.9 (6.75) | 50 (12.5) | ||
| Age at onset, years (SD) | 63.1 (9.83) | 38.4 (4.65) | 48.3 (0.83) 1 | |
| Age at death, years (SD) | 73.4 (8.29) | 44.9 (7.47) | ||
| Female (%) | 6 (60%) | 4 (57.1%) | 19 (54.3%) | 8 (57.1%) |
| MMSE at visit, score (SD) | 25.3 (3.88) | 21.9 (6.40) | ||
| CDR at visit, score (0/0.5/1/2/3) | 0.657 (0/26/8/1/0) | 0.714 (0/12/1/0/1) | ||
| CDR at death, score (0/0.5/1/2/3) | 2.75 (0/0/1/0/7) | 3 (0/0/0/0/6) | ||
| APOE ε4 (%) | 7/9 (77.8%) | 1/7 (14.3%) | 22/34 (64.7%) | 4/14 (28.6%) |
| Family Mutation APP/PSEN1/PSEN2 | 0/7/0 | 1/12/1 | ||
| Aβ plaque score (A0/1/2/3) | 3 (0/0/0/10) | 3 (0/0/0/7) | ||
| NFT stage (B0/1/2/3) | 3 (0/0/0/10) | 3 (0/0/0/7) | ||
| Neuritic plaque score (C0/1/2/3) | 2.9 (0/0/1/9) | 3 (0/0/0/7) | ||
Includes estimated age at onset using expected years to symptom onset (EYO).
Neuropathological assessment of cases involved expert evaluation of histology slides representing 16 brain areas from the left side of each brain. 9 Stereology focused on tissues sampled in the coronal plane, including the frontal lobe (middle frontal gyrus), temporal lobe (superior and middle temporal gyri), parietal lobe (inferior parietal lobe including angular gyrus), occipital lobe (calcarine sulcus and peristriate cortex), parahippocampal gyrus, and hippocampal subfield CA1 (both sampled at the level of the lateral geniculate nucleus). From these regions, stereological measurements of PHF‐1 (a gift from Dr. Peter Davies) immunostained tangles, plaques, and threads were made using the Area Fraction Fractionator probe in Stereo Investigator 10 (MBF Bioscience, Williston, VT, USA).
In a separate cohort (ADAD n = 14, LOAD n = 35), antemortem 18F‐flortaucipir PET was quantified using regional standardized uptake value ratios (SUVRs). 10 These individuals met the inclusion criteria of having a Clinical Dementia Rating (CDR®) 11 greater than 0; individuals with LOAD additionally had positive amyloid PET imaging. 12 Regional SUVRs of interest were defined by FreeSurfer 13 regions best corresponding to neuropathology regions: caudal middle frontal cortex, middle temporal cortex, inferior parietal cortex, pericalcarine cortex, parahippocampal cortex, and hippocampus.
Regional differences across and within ADAD and LOAD in neuropathology and imaging were assessed using the Kruskal–Wallis test. Post hoc Wilcoxon rank‐sum tests were performed with Bonferroni–Holm multiple comparisons correction to assess which regions showed elevated tau burden in ADAD versus LOAD.
Results
Tangle, plaque, thread burden, and SUVR showed statistically significant regional differences across ADAD and LOAD. Only tangle burden and SUVR showed significant regional differences within ADAD and LOAD as well (Fig. 1).
Figure 1.
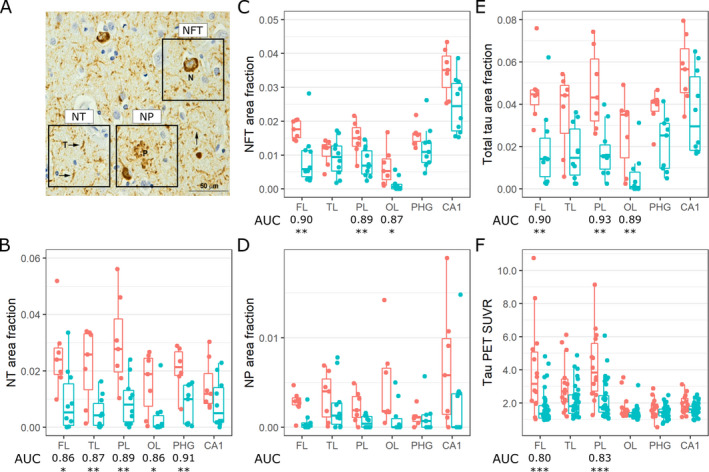
(A) Exemplar PHF‐1 immunostained neuropil threads (NT), neurofibrillary tangles (NFT), and neuritic plaques (NP). (B) Regional NT burden in the frontal lobe (FL), temporal lobe (TL), parietal lobe (PL), occipital lobe (OL), parahippocampal gyrus (PHG), and hippocampal subfield CA1 in ADAD and LOAD. (C) Regional NFT burden. (D) Regional NP burden. (E) Regional total tau (NT + NFT+NP) burden. (F) Regional 18F‐flortaucipir PET imaging SUVRs. Asterisks denote P‐values < 0.05 (*), 0.01 (**), and 0.001 (***) for regionwise Wilcoxon rank‐sum tests between ADAD and LOAD. Only area under the curves (AUCs, the probability that a randomly selected ADAD individual has a higher regional tau burden than a randomly selected LOAD individual) that are statistically significant after Bonferroni–Holm multiple comparisons correction are displayed.
Compared to LOAD, tangle burden in ADAD was significantly elevated outside the temporal lobe. Patterns of regional tangle burden resembled those of regional SUVRs. However, while CA1 is highest in median tangle burden, the hippocampus has only the third and fourth highest median SUVR in ADAD and LOAD, respectively (Fig. 1C&F).
Neuritic plaque burden was elevated in ADAD, but no post hoc test reached statistical significance after multiple comparisons correction. Thread burden was elevated outside CA1 (Fig. 1B&D).
Discussion
Antemortem tau PET SUVRs and postmortem tangle burden in frontal and parietal regions were elevated in ADAD versus LOAD. This concordance appears robust at the group level, even though antemortem imaging and postmortem neuropathology were assessed in different cohorts, roughly representing early and late clinical stages of AD, respectively (Table 1). Additionally, the ADAD neuropathology cohort demonstrates an earlier age of onset than the ADAD imaging cohort, suggesting more aggressive forms of AD pathology in the former.
Given differences in clinical stage and age of onset, it is not surprising there are also discordant findings. First, in ADAD, tangle burden is elevated in the occipital lobe relative to LOAD, but SUVR is not. Second, medial temporal lobe regions show some of the highest regional tangle burden in the neuropathology, but not imaging, cohort. There are several potential explanations. First, tau burden may be particularly modest in the medial temporal lobe at early symptomatic stages of AD, but increase substantially by end stage. Second, PET imaging may have difficulty resolving the tau burden of small brain structures compared to histopathological assessment. Finally, some individuals in the imaging cohort may have subtle neuropathological comorbidities that contribute to cognitive impairment, qualifying an amyloid PET positive case with low AD neuropathological change (transentorhinal versus limbic stages of tau pathology) for inclusion in this study.
More discordances between imaging and neuropathology come from patterns of regional neuritic plaque and thread burden. Plaque burden was elevated in ADAD, though no post hoc test reached statistical significance, and thread burden was elevated outside CA1. Ringman et al. 14 found statistically significant elevation of plaque burden in a larger cohort (ADAD n = 60, LOAD n = 120), but used a semi‐quantitative global score for each individual. We could not find any published studies comparing levels of neuropil threads between ADAD and LOAD. We also attribute these discordant findings to differences in clinical stage between neuropathology and imaging cohorts.
Comparing frontal, temporal, and parietal lobe values suggests tau PET SUVRs may correspond best to total tau burden (summed contributions from tangles, plaques, and threads). Similarly, Smith et al. 4 found regional SUVRs correlated best with regional total tau burdens in a single individual with ADAD. However, that study also found threads outnumbered tangles in every studied brain region; we found no significant statistical dominance of thread over tangle burden in any region in either ADAD or LOAD.
Given our findings, we can make two conservative claims. First, although tau PET did not assess individuals in late stages of AD, and our neuropathologic assessments focused on very late stages, the regional pattern of elevated tau radioligand binding is largely concordant with the regional pattern of elevated postmortem total tau burden in ADAD versus LOAD. This suggests regional differences in tau pathology between ADAD and LOAD are consistent throughout their symptomatic stages. We propose that tau PET did not identify the relatively high tau burdens seen in the neuropathological assessment of CA1 and the parahippocampal gyrus because, at the level of the lateral geniculate nucleus, these areas develop far more robust tauopathy only in late stages of AD neuropathological change, 15 , 16 that are more likely to be associated with a terminal Clinical Dementia Rating of 3 than 0.5 or 1. In contrast, the entire hippocampus was assessed in tau PET imaging, which may explain some of the discordance in this comparison.
Second, like tangle burden, thread burden is elevated in ADAD versus LOAD, and across more brain regions, while plaque burden is elevated to a lesser extent. A possible explanation for greater tangle and thread burden in ADAD than LOAD might be that LOAD is often a multifactorial process, with cerebral small vessel disease, TDP‐43, and other co‐pathologies contributing to the clinicopathological phenotype such that less AD neuropathologic change is needed to reach similar states of dementia severity. That said, enhancement of tangle and thread burden in ADAD compared to LOAD without an equally strong enhancement of plaque burden may seem unusual. One explanation suggests tangles and threads are pathophysiologically closely linked, with tangles appearing first, and threads reflecting more severe saturation of neuronal processes by abnormal tau, whereas neuritic plaques develop later, 16 and reflect more focal disturbances that leave remaining neuronal cytosol unperturbed. Another explanation: on sections immunostained for tau, within areas of very dense threads, plaques are occasionally difficult to discern, and might be undercounted.
We note the limitations to this study. First, no individuals in our neuropathology cohort had undergone antemortem tau PET, precluding direct imaging‐neuropathology comparisons. Second, regions included in the neuropathology portion of this study were limited in number and not perfectly correspondent to those from tau PET. Third, most ADAD individuals who came to autopsy were at the end stage of disease. Finally, there is a difference in age of AD symptom onset between the imaging and neuropathology cohorts. Earlier ages of onset appear to be correlated with higher cortical tau PET signal 17 , 18 , 19 and thus there may be a mixed contribution of mutation and early age of onset to the tau PET imaging of the ADAD cohort. From the current study, the tau pathologies responsible for differences observed in tau PET between ADAD versus LOAD were revealed to be predominantly neurofibrillary tangles and neuropil threads. However, our current findings cannot address how temporal progression of tau pathology in ADAD differs from that in LOAD (hypothesized to begin in the brain stem 20 , 21 and suspected to share early‐stage distribution in the medial temporal lobe with primary age‐related tauopathy 22 ). Future work can investigate the temporal progression of AD tau pathology more broadly by studying the relationship between earlier/later ages of AD symptom onset and tau pathology. One possibility is to introduce an early‐onset sporadic AD cohort to help disentangle the contributions of an earlier age of onset form the specific mutations that define the ADAD cohort.
Author Contributions
B.A.G., E.E.F., H.L., R.J.B., B.M.A., R.J.P., T.L.S.B., N.J.C, and J.C.M. were involved in the conception and design of the study. T.R.H. was involved in the acquisition of the data. T.R.H., Y.L., and D.W.C. were involved in the preliminary analysis of the data. C.D.C. was involved in performing the final analysis of the data, writing the manuscript, and making the figures.
The composition of the DIAN study group is listed below.
| Carlos Cruchaga, PhD | Department of Psychiatry, Washington University in St. Louis, St. Louis, MO, USA |
|---|---|
| Anne Fagan, PhD | Department of Neurology, Washington University in St. Louis, St. Louis, MO, USA |
| Alison Goate, DPhil | Department of Genetics and Genomic Sciences, Ichan School of Medicine at Mount Sinai, New York, NY, USA |
| Jason Hassenstab, PhD | Department of Neurology, Washington University in St. Louis, St. Louis, MO, USA |
| Celeste Karch, PhD | Department of Psychiatry, Washington University in St. Louis, St. Louis, MO, USA |
| Eric McDade, DO | Department of Neurology, Washington University in St. Louis, St. Louis, MO, USA |
| Chengjie Xiong, PhD | Department of Neurology, Washington University in St. Louis, St. Louis, MO, USA |
The composition of the DIAN‐TU study group is listed below.
| James J. Lah, MD, PhD | Department of Neurology, Emory University, Atlanta, GA, USA |
|---|---|
| Sarah B. Berman, MD, PhD | Department of Neurology, University of Pittsburgh, Pittsburgh, PA, USA |
| Jared R. Brosch, MD | Department of Neurology, Indiana University, Indianapolis, IN, USA |
| Ghulam Surti, MD | Department of Psychiatry and Human Behavior, Warren Alpert Medical School of Brown University, Providence, USA |
| Christopher H. van Dyck, MD | Alzheimer’s Disease Research Unit, Yale University School of Medicine, New Haven, CT, USA |
| Serge Gauthier, MD | McGill Center for Studies in Aging, Douglas Hospital, Montreal, Canada |
| Mario Masellis, MD, PhD | Division of Neurology, Department of Medicine, Sunnybrook Health Sciences Centre, Toronto, Canada |
Conflicts of Interest
R.J.B. is the principal investigator of the DIAN‐TU, which is supported in part by the DIAN‐TU Pharma Consortium. Eli Lilly and Company and Hoffman‐LaRoche, two members of the DIAN‐TU Pharma Consortium, provided funding for the DIAN‐TU‐001 trial, the former additionally providing technology transfer and precursor for 18F‐flortaucipir, the tau PET radioligand used in this study, and the latter additionally providing payment and reimbursement for speaking fees, advisory boards, and travel expenses of R.J.B. Additionally, B.A.G., Y.L., D.W.C., R.J.P., and T.L.S.B. are also members of the DIAN‐TU, and T.L.S.B. additionally participates as a site investigator in clinical trials sponsored by Eli Lilly and Company.
Acknowledgments
Data collection and sharing for this project was supported by the Knight Alzheimer Disease Research Center (Knight ADRC, NIH grants P50AG005681, P01AG026276, P01AG03991), the Dominantly Inherited Alzheimer Network (DIAN, NIH U19AG032438), and the DIAN Trials Unit (DIAN‐TU). This manuscript has been reviewed by DIAN Study investigators for scientific content and consistency of data interpretation with previous DIAN Study publications. The authors thank the altruism of the participants and their families and contributions of the Knight ADRC and DIAN research and support staff at each of the participating sites for their contributions to this study. For the provision of brain tissue and staining, we acknowledge the Knight ADRC Neuropathology Core and the DIAN Neuropathology Core (NIH P01AG003991). The authors also thank support for tau PET imaging in DIAN (NIH R01AG052550‐01A1), as well as the Neuroimaging Informatics and Analysis Center (NIH P30NS098577). C.D.C. thanks the support from the NSF GRFP (DGE‐1745038). B.A.G. thanks the support from the NIH (K01AG053474). J.C.M. thanks the support from the NIH (UF1AG032438). The DIAN‐TU is supported by the Alzheimer’s Association, GHR Foundation, an anonymous organization, and the DIAN‐TU Pharma Consortium (https://dian.wustl.edu/our‐research/the‐pharma‐consortium/). This research for the DIAN‐TU‐001 Trial has received support from the Alzheimer’s Association, Eli Lilly and Company, Roche, NIH grants U01AG042791, U01AG042791‐S1 (FNIH and Accelerating Medicines Partnership), R01AG046179, R01AG53267‐S1, Avid Radiopharmaceuticals, GHR Foundation, and an anonymous organization. In‐kind support has been received from CogState and Bracket. The authors thank the altruism of the participants and their families and contributions of the DIAN and DIAN‐TU research and support staff at each of the participating sites (https://dian.wustl.edu/wp‐content/uploads/2019/04/DIAN‐TU‐Publications_Acknowledgement_V26.pdf) for their contributions to this study. Avid Radiopharmaceuticals (a wholly owned subsidiary of Eli Lilly) provided technology transfer and precursor for 18F‐flortaucipir.
Funding Information
This study was supported by the Knight Alzheimer Disease Research Center (Knight ADRC, NIH grants P50AG005681, P01AG026276, P01AG03991), the Dominantly Inherited Alzheimer Network (DIAN, NIH grants U19AG032438, R01AG052550‐01A1), the DIAN Trials Unit (DIAN‐TU, NIH grants U01AG042791, U01AG042791‐S1, R01AG046179, R01AG53267‐S1, as well as support from the Alzheimer's Association, GHR Foundation, an anonymous organization, the DIAN‐TU Pharma Consortium, Eli Lilly and Company, Roche, Avid Radiopharmaceuticals, CogState and Bracket), and the Neuroimaging Informatics and Analysis Center (P30NS098577). The authors were supported by grants from the NSF (DGE‐1745038) and NIH (K01AG053474, UF1AG032438).
Funding Statement
This work was funded by Eli Lilly and Company grant ; Alzheimer's Association grant ; NSF grant DGE‐1745038; Avid Radiopharmaceuticals grant ; GHR Foundation grant ; Bracket grant ; NIH grants K01AG053474, P01AG003991, P01AG026276, P50AG005681, R01AG046179, R01AG052550‐01A1, R01AG53267‐S1, U01AG042791, U01AG042791‐S1, U19AG032438, and UF01AG032438; Roche grant ; CogState grant .
References
- 1. Chien DT, Szardenings AK, Bahri S, et al. Early clinical PET imaging results with the novel PHF‐tau radioligand [F18]‐T808. J Alzheimer’s Dis 2014;38:171–184. [DOI] [PubMed] [Google Scholar]
- 2. Schöll M, Ossenkoppele R, Strandberg O, et al. Distinct 18F‐AV‐1451 tau PET retention patterns in early‐ and late‐onset Alzheimer’s disease. Brain 2017;140:2286–2294. [DOI] [PubMed] [Google Scholar]
- 3. Gordon BA, Blazey TM, Christensen J, et al. Tau PET in autosomal dominant Alzheimer’s disease: Relationship with cognition, dementia and other biomarkers. Brain 2019;142:1063–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Smith R, Wibom M, Pawlik D, et al. Correlation of in Vivo [18 F]Flortaucipir with postmortem Alzheimer disease tau pathology. JAMA Neurol 2019;76:310–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ringman JM, Gylys KH, Medina LD, et al. Biochemical, neuropathological, and neuroimaging characteristics of early‐onset Alzheimer’s disease due to a novel PSEN1 mutation. Neurosci Lett 2011;487:287–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ringman JM, Goate A, Masters CL, et al. Genetic heterogeneity in alzheimer disease and implications for treatment strategies. Curr Neurol Neurosci Rep 2014;14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Badhwar AP, McFall GP, Sapkota S, et al. A multiomics approach to heterogeneity in Alzheimer’s disease: focused review and roadmap. Brain 2020;143:1315–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Montine TJ, Phelps CH, Beach TG, et al. National institute on aging‐Alzheimer’s association guidelines for the neuropathologic assessment of Alzheimer’s disease: a practical approach. Acta Neuropathol 2012;123:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cairns NJ, Perrin RJ, Franklin EE, et al. Neuropathologic assessment of participants in two multi‐center longitudinal observational studies: The Alzheimer Disease Neuroimaging Initiative (ADNI) and the Dominantly Inherited Alzheimer Network (DIAN). Neuropathology 2015;35:390–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Su Y, D’Angelo GM, Vlassenko AG, et al. Quantitative analysis of PiB‐PET with freesurfer ROIs. PLoS One 2013;8:e73377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Morris JC. The clinical dementia rating (cdr): Current version and scoring rules. Neurology 1993;43:2412–2414. [DOI] [PubMed] [Google Scholar]
- 12. Su Y, Flores S, Wang G, et al. Comparison of Pittsburgh compound B and florbetapir in cross‐sectional and longitudinal studies. Alzheimer’s Dement (Amst) 2019;11:180–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fischl B, Salat DH, Busa E, et al. Whole brain segmentation: Automated labeling of neuroanatomical structures in the human brain. Neuron 2002;33:341–355. [DOI] [PubMed] [Google Scholar]
- 14. Ringman JM, Monsell S, Ng DW, et al. Neuropathology of autosomal dominant Alzheimer disease in the National Alzheimer coordinating center database. J Neuropathol Exp Neurol 2016;75(3):284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lace G, Savva GM, Forster G, et al. Hippocampal tau pathology is related to neuroanatomical connections: an ageing population‐based study. Brain 2009;132:1324–1334. [DOI] [PubMed] [Google Scholar]
- 16. Braak H, Braak E. Neuropathological stageing of Alzheimer‐related changes. Acta Neuropathol 1991;82:239–259. [DOI] [PubMed] [Google Scholar]
- 17. Pontecorvo MJ, Devous MD, Navitsky M, et al. Relationships between flortaucipir PET tau binding and amyloid burden, clinical diagnosis, age and cognition. Brain 2017;140:748–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ossenkoppele R, Schonhaut DR, Schöll M, et al. Tau PET patterns mirror clinical and neuroanatomical variability in Alzheimer’s disease. Brain 2016;139:1551–1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. La JR, Visani AV, Baker SL, et al. Prospective longitudinal atrophy in Alzheimer’s disease correlates with the intensity and topography of baseline tau‐PET. Sci Transl Med 2020;12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Busch C, Bohl J, Ohm TG. Spatial, temporal and numeric analysis of Alzheimer changes in the nucleus coeruleus. Neurobiol Aging 1997;18:401–406. [DOI] [PubMed] [Google Scholar]
- 21. Braak H, Thal DR, Ghebremedhin E, Del Tredici K. Stages of the pathologic process in Alzheimer Disease: Age categories from 1 to 100 years. J Neuropathol Exp Neurol 2011;70:960–969. [DOI] [PubMed] [Google Scholar]
- 22. Crary JF, Trojanowski JQ, Schneider JA, et al. Primary age‐related tauopathy (PART): a common pathology associated with human aging. Acta Neuropathol 2014;128:755–766. [DOI] [PMC free article] [PubMed] [Google Scholar]