

Abstract
Objective
The novel morpholino antisense oligonucleotide viltolarsen targets exon 53 of the dystrophin gene, and could be an effective treatment for patients with Duchenne muscular dystrophy (DMD). We investigated viltolarsen’s ability to induce dystrophin expression and examined its safety in DMD patients.
Methods
In this open‐label, multicenter, parallel‐group, phase 1/2, exploratory study, 16 ambulant and nonambulant males aged 5–12 years with DMD received viltolarsen 40 or 80 mg/kg/week via intravenous infusion for 24 weeks. Primary endpoints were dystrophin expression and exon 53 skipping levels.
Results
In western blot analysis, mean changes in dystrophin expression (% normal) from baseline to Weeks 12 and 24 were − 1.21 (P = 0.5136) and 1.46 (P = 0.1636), respectively, in the 40 mg/kg group, and 0.76 (P = 0.2367) and 4.81 (P = 0.0536), respectively, in the 80 mg/kg group. The increase in mean dystrophin level at Weeks 12 and 24 was significant in the 80 mg/kg group (2.78%; P = 0.0364). Patients receiving 80 mg/kg showed a higher mean exon 53 skipping level (42.4%) than those receiving 40 mg/kg (21.8%). All adverse events were judged to be mild or moderate in intensity and none led to study discontinuation.
Interpretation
Treatment with viltolarsen 40 or 80 mg/kg elicited an increasing trend in dystrophin expression and exon 53 skipping levels, and was safe and well tolerated. The decline in motor function appeared less marked in patients with higher dystrophin levels; this may warrant further investigation. This study supports the potential clinical benefit of viltolarsen.
Introduction
Duchenne muscular dystrophy (DMD) is an X‐linked, rapidly progressive, and lethal neuromuscular disease caused by mutations in the dystrophin gene, which results in the absence or deficiency of the dystrophin protein. 1 , 2 Dystrophin is a 427‐ kDa protein with 3685 amino acids that is localized in the sarcolemma of skeletal muscle fibers. 3 The dystrophin‐associated protein complex acts as a cytoskeletal integrator that is critical for the stability of the muscle cell membrane; therefore, the absence of dystrophin results in cell membrane damage and muscle degeneration. 4 , 5 The activation of fibroblasts during tissue repair leads to fibrosis, infiltration of adipocytes, tissue scarring, and reduced myocyte regeneration. 6
The initial symptoms of DMD (e.g., delayed walking, frequent falls, difficulty running, and climbing stairs) are generally observed between the ages of one and three years. 7 Continuous muscle fiber degeneration leads to skeletal muscle wasting and incapacitating weakness, which also affects respiratory and cardiac muscles. Eventually, decreased lower‐limb muscle strength and joint contractures result in the loss of ambulation by eight to 14 years of age, followed by the need for assisted ventilation and, subsequently, death during the thirties or earlier. 7
The global prevalence of DMD is estimated to be 21.2/100,000 school‐aged boys. 8 There is no cure for DMD, and current therapies focus on alleviating symptoms and managing complications. 9 Current treatment guidelines in Japan 9 recommend steroid therapy, which is the only available treatment with a proven effect on improving muscle weakness. 10 Therefore, an effective treatment method that can be used in the early stages of DMD is strongly desired.
Exon skipping therapy has recently gained attention as a potential novel treatment for DMD. 11 , 12 Antisense‐mediated exon skipping results in the skipping of a target exon to restore the reading frame. 13
Viltolarsen is a novel morpholino antisense oligonucleotide designed by the National Center of Neurology and Psychiatry (NCNP) and Nippon Shinyaku Co., Ltd. to target exon 53. 14 It is expected that viltolarsen will be an effective treatment for patients with deletions of exons including, but not limited to, 43–52, 45–52, 47–52, 48–52, 49–52, 50–52 or 52 alone in the dystrophin gene. In a phase 1 investigator‐initiated trial, 14 intravenous treatment with viltolarsen (1.25, 5, or 20 mg/kg once weekly, for 12 weeks) was well tolerated. High exon skipping level and dystrophin expression were confirmed in one patient in the 20 mg/kg group. The purpose of this phase 1/2 dose‐finding study was to investigate the efficacy, safety, and pharmacokinetics of viltolarsen at 40 or 80 mg/kg once a week for 24 weeks in Japanese patients with DMD.
Methods
Study design
This was an open‐label, multicenter, parallel‐group, phase 1/2, dose‐finding, exploratory study (Fig 1A) conducted from April 2016 to November 2017 in five centers in Japan (JAPIC CTI‐163291). At the time of registration, the registration center set four possible combinations of the administration group (40 mg/kg group, 80 mg/kg group) and the muscle biopsy period (12 weeks, 24 weeks). DMD patients were allocated as evenly as possible in terms of ambulant/nonambulant status in each group. Patients with a deletion amenable to exon 53 skipping were randomly assigned to viltolarsen 40 mg/kg or 80 mg/kg groups. The study treatment started at 40 mg/kg weekly. Once the safety of viltolarsen 40 mg/kg was confirmed and was considered appropriate by the safety monitoring committee, enrollment into the viltolarsen 80 mg/kg group was initiated.
Figure 1.
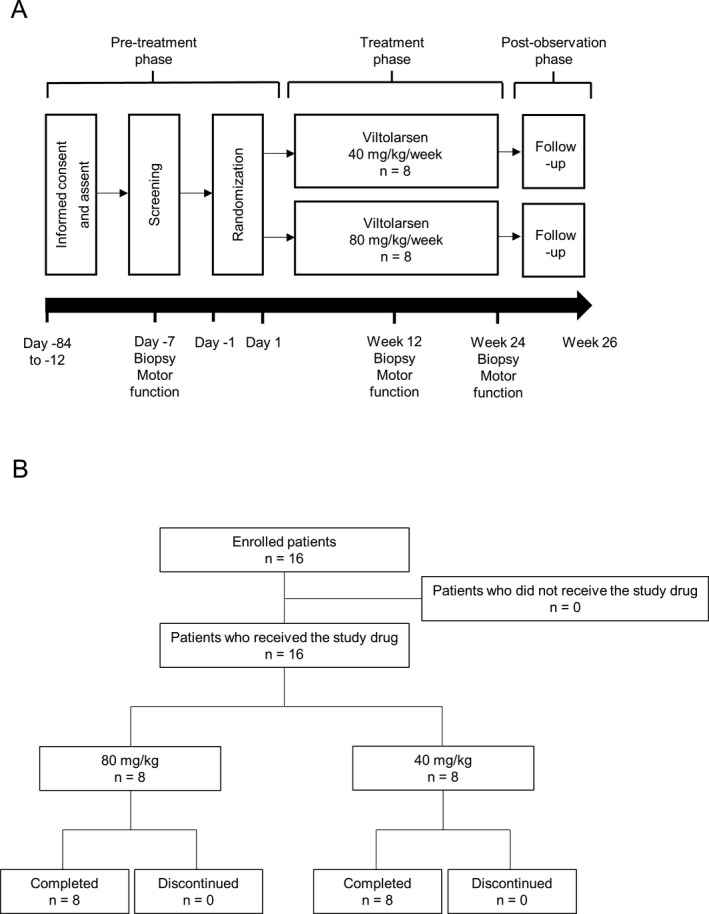
Study design and patient disposition. A, The schematic indicates the three study phases (pre‐treatment, treatment, and post‐observation) and the timing of the assessments conducted during each phase. B, Patients received viltolarsen 40 mg/kg weekly or viltolarsen 80 mg/kg weekly for 24 weeks. All 16 patients enrolled were included in the analyses.
The study was conducted in accordance with the ethical principles of the Declaration of Helsinki, Pharmaceutical Affairs Law, Good Clinical Practice, and associated Japanese regulations. The institutional review board at each participating center reviewed and approved the study protocol and associated documents. All patients and/or their guardians provided written informed consent before study enrollment.
Outcomes
The primary endpoint was the efficacy of viltolarsen at 40 or 80 mg/kg once‐weekly for 24 weeks, as measured by dystrophin protein expression and exon 53 skipping efficiency in muscle biopsy samples at Weeks 12 and 24. The secondary endpoints were motor function (changes from baseline to Week 24 as measured by the 6‐Minute Walk Test [6MWT], Timed Up and Go Test [TUG], Time to Stand Test [TTSTAND], Time to 10 Meter Run/Walk Test [TTRW], and quantitative muscle strength testing on hip flexion/extension, knee joint flexion/extension, and ankle flexion/extension), safety (vital signs, clinical laboratory tests, immunological tests, heart and lung function, and the occurrence of adverse events [AEs] and adverse drug reactions [ADRs]), and the pharmacokinetics of viltolarsen (by assessments at Weeks 1 and 24). Additionally, we evaluated the effects of viltolarsen on serum creatine kinase (CK) levels.
Patients
Eligible patients were ambulant or nonambulant Japanese males with DMD and were aged ≥ 5 years and < 18 years. Inclusion criteria for this study were a genetic deletion amenable to exon 53 skipping based on the multiplex ligation‐dependent probe amplification method; no DNA polymorphism at exon 53 that prevented duplex formation of viltolarsen; suitable muscle tissue (anterior tibial muscle or biceps) for efficacy measurement; life expectancy of at least one year after informed consent; and QTc < 450 ms before administration (Fridericia’s correction) (if the patient had bundle branch block, a QTc < 480 ms was allowed). Patients had to be able to be hospitalized at least from the day of the initial administration of viltolarsen to the day after the second administration, and on the days of the third and fourth administration, to secure their safety.
The main exclusion criteria were participation in clinical studies of read‐through therapy, exon skipping therapy, utrophin overexpression therapy, or gene therapy to restore dystrophin expression; forced vital capacity estimation < 50%; ejection fraction < 40% or fractional shortening < 25% by echocardiography; severe cardiomyopathy; severe hepatic or kidney disorder; positivity for hepatitis B surface antigen, hepatitis C virus antibody, or human immunodeficiency virus antibody; immune deficiency or autoimmune disease; severe cognitive defect; treatment with other investigational drugs within 90 days of treatment period start; not using acceptable contraception methods; previous medical history of serious drug hypersensitivity; or participation in previous clinical studies of viltolarsen.
Prohibited concomitant drugs included other investigational drugs as well as phosphodiesterase type 5 inhibitors, idebenone, coenzyme Q10 preparations, resveratrol preparations, muscle enhancing steroids (oxandrolone), and adenosine triphosphate disodium salt preparations. From 90 days before treatment to the end of the postobservation period, the use of systemic corticosteroids was permitted only if their dosage and administration were not changed, physical therapy or exercise therapy that could influence the test drug’s efficacy assessment was permitted if the content and frequency were not changed, and any surgical procedure that could influence the efficacy assessment of the study drug was prohibited.
Study procedure
During the pre‐treatment period of 3–12 weeks, patients attended three visits and underwent a muscle biopsy and baseline motor function evaluation at Day − 7. During the 24‐week treatment period (Weeks 1 to 24), patients received either viltolarsen 40 or 80 mg/kg weekly by intravenous infusion over 1 hour.
The doses used in the study (40 and 80 mg/kg) were based on exposure data from the investigator‐initiated trial 14 and a nonclinical toxicity study in cynomolgus monkeys and mice. In terms of safety, doses were set such that the expected maximum plasma concentration (Cmax) and area under the plasma concentration‐time curve (AUC0‐t) in humans calculated from linear regression analysis using pharmacokinetics data from the investigator‐initiated trial did not exceed the Cmax and AUC0‐t at the tolerated dose in monkeys and mice (200 mg/kg and 240 mg/kg). In terms of efficacy, the dose was set to reach the Cmax and AUC0‐t of one patient in the investigator‐initiated clinical trial who had marked dystrophin protein levels.
The treatment duration was set to 24 weeks because the 12‐week treatment in the previous investigator‐initiated clinical trial led to marked increases in dystrophin protein levels only in one patient and clinical trials of similar drugs administered for 24 weeks or longer also demonstrated marked increases in dystrophin protein levels. 15 , 16
During the one‐week postobservation period, patients attended two follow‐up visits. No changes in the dosage and administration of systemic corticosteroids were permitted within 90 days prior to treatment until the end of the observation period.
Assessment of outcomes
Dystrophin protein levels were evaluated by western blotting and quantified using double immunofluorescent staining (immunohistochemistry) to determine dystrophin/spectrin intensity ratios and the number of dystrophin‐positive fibers in muscle biopsy samples collected before administration and at Week 12 and Week 24. Muscle tissue biopsy was performed on the tibialis anterior muscles or biceps brachii muscle, according to methods described previously. 14 For dystrophin and exon skipping evaluations, 50% of patients (n = 4) in each treatment group underwent a muscle biopsy after 12 weeks of treatment whilst the remaining 50% underwent a muscle biopsy after 24 weeks of treatment.
Western blot assessment
Dystrophin protein was extracted from 10 × 10 µm frozen muscle tissue biopsies using 125 mM Tris‐HCl pH 6.4, 4% w/v SDS, 4 M urea, and protease inhibitors. Protein concentrations were measured using bicinchoninic acid. Lysates were denatured (98°C, 3 min), and 12 µg of total protein were subjected to electrophoresis (150 V, 75 min; NuPAGE Novex 3%–8% tris‐acetate gel; Thermo Fisher Scientific). Separated proteins were transferred onto a polyvinylidene fluoride membrane using a semidry blotting system (4 mA/cm2, 30 min). The primary antibody was ab15277 dystrophin rabbit polyclonal antibody (Abcam, Cambridge, UK) diluted 1:500, and the secondary antibody was Histofine Simple Stain MAX‐PO (Multi), diluted 1:100 (Nichirei, Tokyo, Japan). Alpha‐actinin was probed with A7811 α‐actinin mouse monoclonal antibody (Sigma‐Aldrich, MO, USA) diluted 1:10,000. Bands were detected (ECL Prime western blotting system; GE Healthcare) and imaged (LuminoGraph I chemiluminescent system; Atto, Tokyo, Japan). Calibration samples were prepared by diluting normal control (with 100% dystrophin) with negative control (with 0% dystrophin), and generation of a 5‐point calibration curve (1%, 2%, 3.3%, 10%, and 33%). The dystrophin percentage per sample was calculated based on the calibration curve using a log‐transformed linear regression model. Assessments were conducted by blinded assessors. The lower limit of quantitation of the validated method used in the study was 1%. When a measured value was below this limit, the value was estimated using standard curve extrapolation.
Immunofluorescence assessment
We used a modified version of the method reported previously. 14 , 17 Frozen muscle sections (10 µm) from normal control and pre‐ and post‐treatment samples were placed on a microscope slide and probed with NCL‐SPEC1 spectrin mouse monoclonal antibody (Leica Biosystems, Wetzlar, Germany) diluted 1:100 and ab15277 dystrophin rabbit polyclonal antibody (Abcam, Cambridge, UK) diluted 1:200, then incubated (1 h, room temperature). The secondary antibodies were Alexa Fluor 488 F(ab’)2 fragment goat anti‐mouse IgG (H + L) (A11017, Life Technologies) and Alexa Fluor 647 goat anti‐rabbit IgG (H + h) (A21245, Life Technologies), both diluted 1:500 for incubation (30 min, room temperature). After mounting slides in ProLong Gold antifade reagent (P36934, Invitrogen, Carlsbad, CA, USA), images were obtained on a TCS‐SP5 confocal microscope (Leica Microsystems). All microscopic fields of patient sections were captured under the same settings as normal controls to avoid arbitrary assessment. MetaMorph (Molecular Devices, San José, CA, USA) with customized scripts was used for image processing. Based on the spectrin image, MetaMorph recognized a sarcolemmal region. Fluorescence signal intensities of spectrin and dystrophin were quantified, and the dystrophin/spectrin intensity ratio was calculated from both image intensities. The number and proportion of dystrophin‐positive fibers were also calculated. Based on the distribution of the dystrophin/spectrin intensity ratio of each fiber in the normal control muscle biopsy sections, muscle fibers with a ratio higher than the first percentile were considered to be dystrophin‐positive.
RNA assessment
Total RNA was extracted from 2 × 10‐ mm diameter, 50‐µm thick frozen muscle sections using the RNeasy Micro Kit (Qiagen, Venlo, The Netherlands) and analyzed by reverse transcription‐polymerase chain reaction (RT‐PCR) using a Qiagen OneStep RT‐PCR Kit. The RT‐PCR program (GeneAmp PCR System 9700, Thermo Fisher Scientific) was: RT (50°C, 30 min) and heat denaturation (95°C, 15 min); 35 cycles of denaturation (94°C, 1 minute), annealing (60°C, 1 minute), extension (72°C, 1 minute); and final extension (72°C, 7 minutes). Reaction products were electrophoresed on agarose gels (Experion Automated Electrophoresis Station; Bio‐Rad, Hercules, CA, USA). Bands with and without exon 53 were excised, purified, and sequenced using a BigDye Terminator v3.1 Cycle Sequencing Kit (Thermo Fisher Scientific). Percent skipping was calculated as [molarity of skipped band/(sum of the molarities of the skipped + unskipped bands)] x 100.
The forward primer was one of the following according to the individual patient deletion (the number in the primer name indicates the exon targeted):
44F: 5'‐CCTGAGAATTGGGAACATGC‐3'
46F: 5'‐AACCTGGAAAAGAGCAGCAA‐3'
48F: 5'‐CCAAGAAGGACCATTTGACG‐3'
The reverse primer for RT‐PCR was 54/55R, while 54R was used for sequencing:
54/55R: 5'‐TCTCGCTCACTCACCCTTTT‐3'
54R: 5'‐GTGGACTTTTCTGGTATCAT‐3'
Motor function evaluations
The exercise function evaluation procedures (6MWT, TUG, TTSTAND, and TTRW) were conducted in a standardized manner by trained clinical evaluators during the last two visits in the pre‐treatment period (with the visit closest to study drug administration defined as baseline), then again at Week 12 and at Week 24 after the administration of viltolarsen, and at treatment discontinuation. Authorized physical therapists or doctors who were trained by a Master Physical therapist performed the motor function evaluations at each site, following standardized procedures.
Quantitative muscle strength testing was conducted using a microFET® (Hogan Industries, Draper, UT, USA) handheld dynamometer measuring flexion/extension of the hip, knee joint, and ankle. The maximum value of either the left or right muscle strength was used for each target muscle at each time point. Serum CK was measured at Day − 14 during the preobservation period; at Weeks 1, 5, 9, 13, 17, and 21 during the treatment period; at Week 25 during the postobservation period; and at treatment discontinuation.
Pharmacokinetics
Blood samples for pharmacokinetics were collected at pretreatment; 30 minutes after infusion start; end of infusion; and 15 minutes and 1, 2, 4, 8, and 23 hours after the end of infusion on Day 1 of Week 1 and Week 24 and end of infusion of Week 2. Urine samples were collected from infusion start to 24 hours after the infusion start on Day 1 and Week 24.
Pharmacokinetic parameters included plasma viltolarsen concentration, Cmax, time to reach Cmax, AUC0–t, elimination half‐life, apparent volume of distribution at steady state, apparent total body clearance of the drug from plasma, and urinary excretion rate. Liquid chromatography‐mass spectrometry/mass spectrometry was used to measure the concentration of viltolarsen in plasma and urine.
Safety assessment
AEs and ADRs were recorded using the Preferred Term according to the Medical Dictionary for Regulatory Activities/J (ver. 20.1). The incidence of events (number of events in the safety analysis population) was calculated for each treatment group.
Statistical methods
Sample size was not calculated statistically. Owing to the rarity of the disease and study feasibility, we planned to enroll a total of 16 patients to evaluate the efficacy of viltolarsen using dystrophin expression as an index.
Preplanned analytical populations were the full analysis set (FAS), the safety analysis set, and the pharmacokinetic analysis set. The full analysis set included all enrolled patients who had at least one efficacy evaluation result and were treated with the study drug without any violation of the study protocol. The safety analysis set included all enrolled patients who had at least one safety evaluation result and received the study drug at least once without any Good Clinical Practice violation. The pharmacokinetic analysis set included all patients who received the study drug and had enough plasma and/or urine samples collected and without any protocol violation that would significantly influence pharmacokinetic parameters.
For primary and secondary endpoints, descriptive statistics were calculated. For differences before and after administration, descriptive statistics were calculated for each treatment group at each time point.
A correlation analysis was performed post hoc, with Pearson’s correlation coefficient and regression line assessed with the following parameters: correlation of dystrophin expression with plasma drug concentration, 6MWT, TUG, TTSTAND, TTRW at Week 24; exon deletion site and age at baseline; and correlation of exon 53 skipping level with plasma drug concentration, exon deletion site, and age at baseline.
The assessment parameters were compared before and after study drug administration via an unpaired t‐test for each patient and a paired t‐test for each treatment group. For comparison between treatment groups, unpaired t‐tests and two‐way analysis of variance were conducted. For the motor function evaluation, time (s) was converted to velocity (1/s) for analysis. Missing data owing to disease progression were handled as 0 for TUG, TTSTAND, and TTRW at the first visit for each applicable assessment. Pearson’s correlation coefficient was used for correlation analysis, and logarithmic Cmax and AUC0‐t were used to obtain plasma drug concentrations. Changes from baseline were calculated for dystrophin, exon 53 skipping level, 6MWT, TUG, TTSTAND, and TTRW. Statistical tests were two‐sided with a 95% confidence interval (CI), and the significance level was set at 5%. SAS® software version 9.4 (SAS Institute Inc., Cary, NC, USA) was used to conduct statistical analyses.
Results
Patients
Patients received viltolarsen 40 mg/kg weekly (n = 8) or viltolarsen 80 mg/kg weekly (n = 8) for 24 weeks (Fig. 1B). All 16 patients enrolled were included in the FAS, safety, and pharmacokinetic analysis populations.
The main patient characteristics and clinical features in both dose groups are shown in Table 1. Overall, patients’ mean age was 8.4 ± 2.0 years, 81.3% were ambulant, and 87.5% had previously received steroid treatment. The most common dystrophin mutations were deletions of exons 45–52 in 37.5%, 48–52 in 18.8%, and 52 alone in 18.8%. In general, both dose groups were numerically similar in terms of baseline characteristics.
Table 1.
Patients’ characteristics. In general, both dose groups were numerically similar in terms of baseline characteristics.
| Viltolarsen | |||
|---|---|---|---|
|
40 mg/kg (n = 8) |
80 mg/kg (n = 8) |
Total (n = 16) |
|
| Age (years), mean± SD | 8.4 ± 1.9 | 8.4 ± 2.3 | 8.4 ± 2.0 |
| Range | 5–11 | 5–12 | 5–12 |
| Height (cm), mean ± SD | 119.7 ± 14.5 | 120.8 ± 10.9 | 120.3 ± 12.4 |
| Range | 98.0–140.7 | 100.3–134 | 98.0–140.7 |
| Weight (kg), mean ± SD | 26.8 ± 10.1 | 29.8 ± 11.3 | 28.3 ± 10.5 |
| Range | 16.0–41.7 | 16.0–52.1 | 16.0–52.1 |
| Body mass index (kg/m2), mean ± SD | 18.0 ± 2.8 | 19.9 ± 5.0 | 19.0 ± 4.0 |
| Range | 15.6–23.6 | 15.3–30.2 | 15.3–30.2 |
| Mutation (deleted exon), n (%) | |||
| 43–52 | 0 | 0 | 0 |
| 45–52 | 3 (37.5) | 3 (37.5) | 6 (37.5) |
| 47–52 | 0 | 0 | 0 |
| 48–52 | 2 (25.0) | 1 (12.5) | 3 (18.8) |
| 49–52 | 2 (25.0) | 0 | 2 (12.5) |
| 50–52 | 1 (12.5) | 1 (12.5) | 2 (12.5) |
| 52 | 0 | 3 (37.5) | 3 (18.8) |
| Steroid treatment, n (%) | |||
| Yes | 7 (87.5) | 7 (87.5) | 14 (87.5) |
| No | 1 (12.5) | 1 (12.5) | 2 (12.5) |
| Independent ambulation, n (%) | |||
| Ambulant | 6 (75.0) | 7 (87.5) | 13 (81.3) |
| Nonambulant | 2 (25.0) | 1 (12.5) | 3 (18.8) |
SD = standard deviation.
Two patients in the 40 mg/kg group had treatment interruptions: one because of an AE (influenza) and the other because of an ADR (eczema). However, each of these patients missed only a single dose; therefore, treatment compliance was considered to be high over the course of the study.
Efficacy
Primary efficacy endpoint
From the western blot analysis, the changes in dystrophin expression from baseline to Weeks 12 and 24, expressed as a mean percentage of normal control tissue (standard deviation), were − 1.21 (3.26), P = 0.5136 and 1.46 (1.59), P = 0.1636 in the 40 mg/kg group, and 0.76 (1.02), P = 0.2367 and 4.81 (3.11) P = 0.0536 in the 80 mg/kg group, respectively (Fig. 2A). Fourteen out of 16 patients showed a dystrophin level increase (Table 2). For one patient (Patient 04) in the 40 mg/kg group, the baseline dystrophin level was 6.39%. Subsequent remeasurement using a different muscle block showed the baseline dystrophin level to be 0.51%. The initial band detected was of a lower molecular weight than expected and was considered nonspecific. Overall, the dystrophin expression level tended to increase depending on the dose and treatment duration. In particular, three of four patients in the 80 mg/kg group who underwent muscle biopsy at 24 weeks showed large increases in dystrophin expression from baseline (7.85%, 6.42%, and 4.30%). When the changes in dystrophin level from baseline to Weeks 12 and 24 were combined, the increase in the 80 mg/kg group was statistically significant (2.78% ± 3.05; P = 0.0364).
Figure 2.
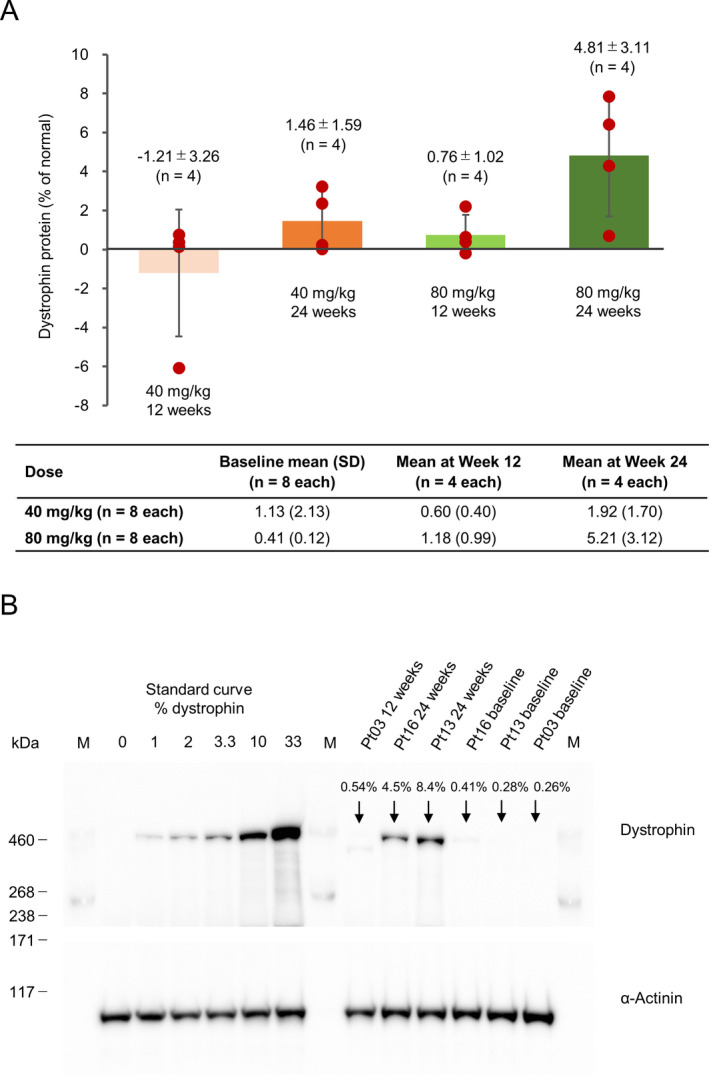
Dystrophin protein level by western blot (mean ± SD). A, Changes from baseline to 12 or 24 weeks. The figure shows the difference in the amount of dystrophin protein from baseline to 12 or 24 weeks (red circle: value for each case, bar: average value) ± standard deviation, and the table below the figure shows the amount of dystrophin protein before administration of viltolarsen (baseline) and the mean value (standard deviation) of the measured values of dystrophin protein at 12 and 24 weeks. “% of normal” = Data from Duchenne muscular dystrophy patients/data from healthy people, with % of normal calculated at baseline and after administration. The y‐axis indicates the changes (% normal 0 to Week 12 and % normal 0 to Week 24). After treatment, mean data include each of the four patients who had a biopsy performed, and dystrophin protein levels were evaluated at Week 12 and Week 24 in each group. Therefore, the mean change includes changes from baseline to Week 12 and Week 24. B, Western Blot images of patients 03, 13, and 16 after once‐weekly treatment with 80 mg/kg viltolarsen. The image depicts the data from one of the triplicate experiments conducted for this measurement. At baseline, there were no visible bands indicating the presence of dystrophin for these patients. However, after 24 weeks of treatment, clear bands for dystrophin appeared for both patients 13 and 16. SD, standard deviation.
Table 2.
Dystrophin and exon 53 skipping quantification results in each patient.
| Dose | Patient No. | Visit | Dystrophin protein level (%) 1 | Exon 53 skipping level (%) 2 | ||||
|---|---|---|---|---|---|---|---|---|
| Mean ± SD | Difference | Mean ± SD | Difference | |||||
| Mean |
Unpaired t‐test |
Mean |
Unpaired t‐test |
|||||
| 40 mg/kg | 01 |
1 12–13 |
0.41 ± 0.09 1.17 ± 0.23 |
0.76 |
P = 0.0177 |
0.87 ± 0.14 8.57 ± 0.93 |
7.69 |
P = 0.0041 |
| 02 |
1 12–13 |
0.22 ± 0.10 0.36 ± 0.13 |
0.14 |
P = 0.2201 |
1.94 ± 0.09 27.36 ± 4.40 |
25.42 |
P = 0.0098 |
|
| 03 |
1 12–13 |
0.21 ± 0.04 0.58 ± 0.03 |
0.37 |
P = 0.0005 |
1.15 ± 1.00 15.65 ± 4.15 |
14.50 |
P = 0.0213 |
|
| 04 |
1 12–13 |
6.39 ± 1.59 0.31 ± 0.05 |
−6.08 |
P = 0.0220 |
0.00 14.63 ± 3.18 |
14.63 |
P = 0.0153 |
|
| 05 |
1 24–26 |
0.68 ± 0.22 3.91 ± 0.83 |
3.23 |
P = 0.0163 |
1.04 ± 0.36 24.70 ± 0.46 |
23.66 |
P < 0.0001 |
|
| 06 |
1 24–26 |
0.37 ± 0.09 0.59 ± 0.07 |
0.23 |
P = 0.0258 |
2.08 ± 0.14 28.85 ± 0.85 |
26.76 |
P = 0.0002 |
|
| 07 |
1 24–26 |
0.40 ± 0.11 0.41 ± 0.07 |
0.01 |
P = 0.8607 |
2.62 ± 1.77 46.04 ± 1.88 |
43.41 |
P < 0.0001 |
|
| 08 |
1 24–26 |
0.39 ± 0.04 2.75 ± 0.20 |
2.36 |
P = 0.0017 |
1.21 ± 0.45 19.25 ± 0.94 |
18.04 |
P = 0.0001 |
|
| 80 mg/kg | 09 |
1 12–13 |
0.55 ± 0.23 0.35 ± 0.07 |
−0.20 |
P = 0.2656 |
2.62 ± 0.97 33.44 ± 2.51 |
30.81 |
P = 0.0007 |
| 10 |
1 12–13 |
0.40 ± 0.06 2.60 ± 1.86 |
2.20 |
P = 0.1774 |
5.25 ± 4.18 52.30 ± 2.59 |
47.05 |
P = 0.0003 |
|
| 11 |
1 12–13 |
0.38 ± 0.12 1.02 ± 0.09 |
0.63 |
P = 0.0022 |
2.41 ± 0.30 29.97 ± 2.34 |
27.56 |
P = 0.0021 |
|
| 12 |
1 12–13 |
0.36 ± 0.04 0.75 ± 0.03 |
0.39 |
P = 0.0003 |
1.86 ± 0.65 36.81 ± 1.40 |
34.94 |
P < 0.0001 |
|
| 13 |
1 24–26 |
0.23 ± 0.04 8.08 ± 0.70 |
7.85 |
P = 0.0026 |
1.09 ± 0.30 62.46 ± 1.16 |
61.36 |
P < 0.0001 |
|
| 14 |
1 24–26 |
0.63 ± 0.04 7.05 ± 1.97 |
6.42 |
P = 0.0300 |
4.09 ± 0.77 53.14 ± 2.73 |
49.05 |
P = 0.0005 |
|
| 15 |
1 24–26 |
0.36 ± 0.08 1.05 ± 0.04 |
0.69 |
P = 0.0011 |
3.31 ± 0.56 52.28 ± 0.50 |
48.96 |
P < 0.0001 |
|
| 16 |
1 24–26 |
0.35 ± 0.06 4.65 ± 0.43 |
4.30 |
P = 0.0029 |
2.01 ± 0.87 41.42 ± 2.50 |
39.41 |
P = 0.0005 |
|
SD, standard deviation.
Triplicate measurements were made for each item.
Skipping level (%; as the mean of triplicate measurements) was calculated as [molarity of skipped band/(sum of the molarities of the skipped + unskipped bands)] x 100.
Figure 2B shows western blot images from Patients 03, 13, and 16. At baseline, there were no visible bands indicating the presence of dystrophin for any patient. However, after 24 weeks of once‐weekly 80 mg/kg viltolarsen treatment, clear bands for dystrophin appeared for both patients 13 and 16. When measured by immunohistochemistry, the presence of dystrophin was captured by imaging in several patient samples but was numerically detectable only in patient 13. The muscle biopsy in this patient, who received viltolarsen 80 mg/kg, showed an increase in dystrophin staining in the sarcolemma at Week 24 compared with baseline (Fig. 3). Overall, the increase in the number of dystrophin‐positive fibers was not significant in either dose group.
Figure 3.
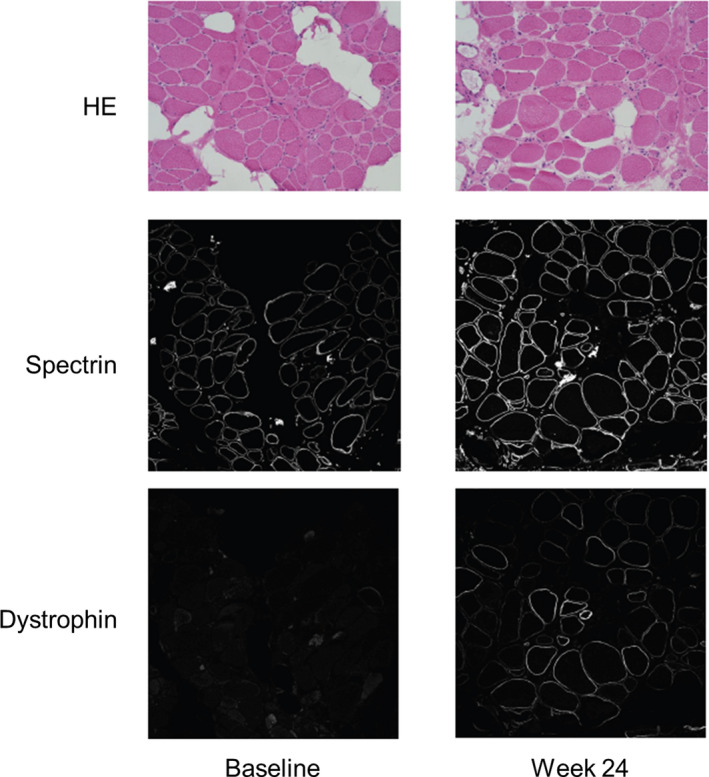
Immunofluorescence images of muscle biopsy samples from patient 13. The muscle biopsy in this patient, who received viltolarsen 80 mg/kg, showed an increase in dystrophin staining in the sarcolemma at Week 24 compared with baseline. HE, hematoxylin and eosin.
Table 2 and Table 3 show the exon skipping levels measured by RT‐PCR. After 12 weeks and 24 weeks of treatment, the exon 53 skipping level increased for all patients, although the increases varied according to the dose and duration of treatment. Overall, patients receiving viltolarsen 80 mg/kg doses showed a higher mean exon 53 skipping level (42.4%) compared with those receiving 40 mg/kg (21.8%) (Table 3). Similarly, those receiving either dose showed a higher mean exon 53 skipping level after 24 weeks of treatment (38.8%) compared with that after 12 weeks of treatment (25.3%).
Table 3.
Exon 53 skipping level by reverse transcription‐polymerase chain reaction.
| Dose | Biopsy week | |||
|---|---|---|---|---|
| 12 | 24 | Total | ||
|
Exon 53 skipping level, mean (%) |
40 mg/kg |
15.6 (n = 4) |
28.0 (n = 4) |
21.8 (n = 8) |
| 80 mg/kg |
35.1 (n = 4) |
49.7 (n = 4) |
42.4 (n = 8) |
|
| Total |
25.3 (n = 8) |
38.8 (n = 8) |
32.1 (n = 16) |
|
Skipping level (%) was calculated as [molarity of the skipped band/(sum of the skipped + unskipped bands)] x 100.
Secondary efficacy endpoints
Performance in all motor function tests declined over time (Fig. 4A). The decline was somewhat smaller at Week 24‐26 among patients receiving viltolarsen 80 mg/kg in relation to the 6MWT and TTRW tests. The correlation coefficients between dystrophin quantity and prevention of motor function decline, measured by TTSTAND and TTRW, were 0.64 and 0.92, respectively, showing a stronger correlation for TTRW (Fig. 4B). In the quantitative muscle tests, all items improved in the viltolarsen 80 mg/kg group after 24 weeks of treatment (Fig. 5). At 24 weeks, the mean change in serum CK was 916.8 U/L (2573.4 U/L) in the 40 mg/kg group and − 417.5 U/L (4006.4 U/L) in the 80 mg/kg group.
Figure 4.
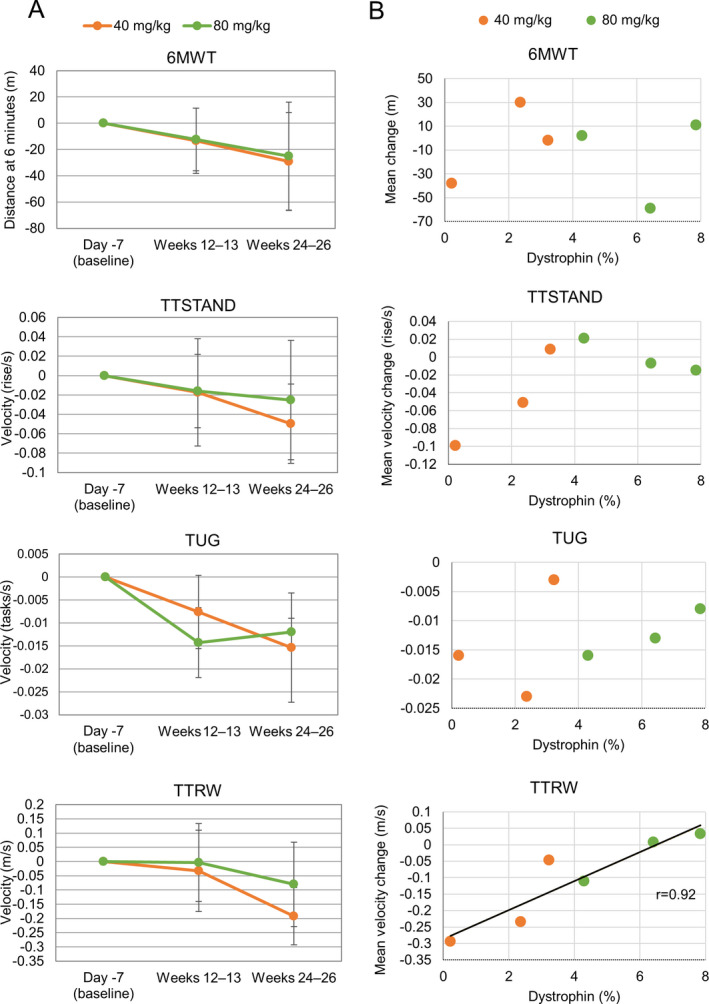
Motor function tests. A, Mean (standard deviation) change from baseline to 24 weeks. Performance in all motor function tests (6‐minute walk test [6MWT], timed up and go [TUG], time to stand [TTSTAND], and time to 10‐meter run/walk [TTRW]) declined over time. B, Correlation between dystrophin protein quantity (western blot analysis) and motor function (at 24 weeks). For each motor function test, a regression line was drawn when the correlation coefficient met a significance level of 5% (one‐sided).
Figure 5.
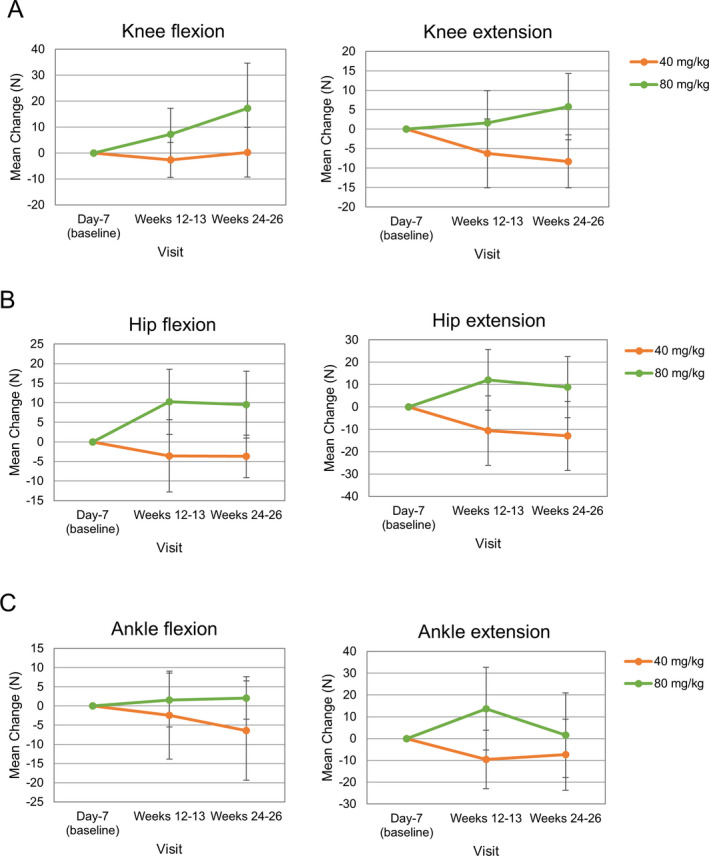
Mean ± standard deviation changes in quantitative muscle tests of the knee (A), hip (B), and ankle (C).
Pharmacokinetics
Figure 6 shows that viltolarsen Cmax and AUC0‐t increased in a dose‐dependent manner. Pharmacokinetic parameters showed that viltolarsen was primarily and immediately excreted in urine and that excretion within 24 h after its administration was similar between the 40 and 80 mg/kg groups.
Figure 6.
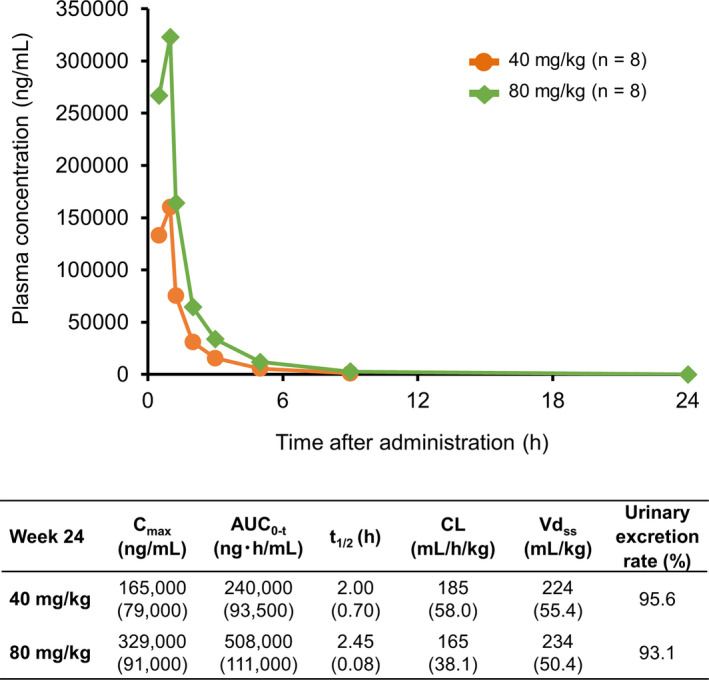
Viltolarsen concentration change in plasma and pharmacokinetic parameters at Week 24. Viltolarsen Cmax and AUC0‐t increased in a dose‐dependent manner. The table shows viltolarsen pharmacokinetic parameters. Data are expressed as mean (standard deviation), except for urinary excretion rate. Cmax = maximum concentration; AUC0–t = area under the plasma concentration‐time curve from time zero to time of last measurable concentration; t1/2 = elimination half‐life; CL = apparent total body clearance of the drug from plasma; Vdss = apparent volume of distribution at steady state.
The correlation coefficients for the change in dystrophin expression levels with Cmax and AUC0‐t were 0.03 and 0.205, respectively, and those for the relationship between exon 53 skipping level and Cmax and AUC0‐t were 0.494 and 0.675, respectively.
Safety
Mean treatment duration was 161.8 days in both groups, and the overall incidences of AEs and ADRs were 87.5% (14/16 patients) and 56.3% (9/16 patients), respectively. The incidences of AEs in the 40 and 80 mg/kg groups were both 87.5% (7/8 patients) and did not increase dose‐dependently. Incidences of ADRs in the 40 and 80 mg/kg groups were 37.5% (3/8 patients) and 75.0% (6/8 patients), respectively.
Table 4 summarizes the treatment‐emergent AEs occurring in two or more patients during the study. The most common treatment‐emergent AEs were contusion in the 40 mg/kg group and nasopharyngitis and upper respiratory tract infection (URTI) in the 80 mg/kg group. No AEs led to treatment discontinuation, and all AEs were judged to be mild or moderate. One case of URTI in the 80 mg/kg group was considered serious (required prolongation of hospitalization); however, it was not judged to be treatment‐related.
Table 4.
Adverse events (AEs) occurring in two or more patients. The most common were nasopharyngitis, upper respiratory tract infection, contusion, and beta‐N‐acetyl‐D‐glucosaminidase increased. No AEs led to treatment discontinuation and all AEs were judged to be mild or moderate.
| Treatment‐emergent adverse event, n (%) | Viltolarsen | ||
|---|---|---|---|
|
40 mg/kg n = 8 |
80 mg/kg n = 8 |
Total n = 16 |
|
| Nasopharyngitis | 1 (12.5) | 3 (37.5) | 4 (4.25) |
| Upper respiratory tract infection | 1 (12.5) | 3 (37.5) | 4 (25.0) |
| Contusion | 3 (37.5) | 0 | 3 (18.8) |
| Beta‐N‐acetyl‐D‐glucosaminidase increased | 1 (12.5) | 2 (25.0) | 3 (18.8) |
| Pyrexia | 0 | 2 (25.0) | 2 (12.5) |
| Influenza | 2 (25.0) | 0 | 2 (12.5) |
| Brain natriuretic peptide increased | 1 (12.5) | 1 (12.5) | 2 (12.5) |
| Interleukin level increased | 1 (12.5) | 1 (12.5) | 2 (12.5) |
| Ejection fraction decreased | 0 | 2 (25.0) | 2 (12.5) |
| Pain in extremity | 1 (12.5) | 1 (12.5) | 2 (12.5) |
| Eczema | 1 (12.5) | 1 (12.5) | 2 (12.5) |
| Rash | 1 (12.5) | 1 (12.5) | 2 (12.5) |
| Urticaria | 0 | 2 (25.0) | 2 (12.5) |
ADRs occurring in two or more patients included two cases each (one patient each in the 40 and 80 mg/kg groups) of increased brain natriuretic peptide and increased interleukin, and two cases each (all in the 80 mg/kg group) of fever, increased beta‐N‐acetyl‐D‐glucosaminidase, decreased ejection fraction, and urticaria.
No safety concerns were raised according to the results of clinical laboratory tests, anti‐dystrophin or anti‐viltolarsen antibody levels, 12‐lead electrocardiogram, or lung function tests.
Discussion
After 24 weeks of treatment with viltolarsen 40 or 80 mg/kg once‐weekly, 14 out of 16 patients with DMD with similar baseline characteristics showed a dystrophin level increase by western blot analysis. Exon 53 skipping was detected in all patients. Furthermore, dystrophin and exon skipping levels were shown to increase with increasing dose and duration of treatment. Motor function showed a tendency to decrease over time, although these decreases, measured with the TTSTAND and TTRW tests, were less pronounced in the higher dosage group, while dystrophin levels tended to be higher in the 80 mg/kg group. Additionally, the correlation coefficient for the dystrophin expression level was high in TTRW and TTSTAND.
The most commonly reported AEs were nasopharyngitis and upper respiratory tract infection (25.0% each), and contusion and beta‐N‐acetyl‐D‐glucosaminidase increased (18.8% each). No AEs led to discontinuation, and all AEs were judged to be mild or moderate, including one case of serious upper respiratory tract infection that was considered unrelated to viltolarsen. These findings are consistent with the safety results reported in the viltolarsen phase 1 trial where patients received lower doses (1.25, 5, or 20 mg/kg/weekly) for 12 weeks. 14 Anemia and renal and urinary disorders, observed in the previous phase 1 study, 14 were not observed in this study. In the phase 1 study, anemia may have resulted from a high blood collection volume, and in this study, the absence of anemia may be a result of the smaller blood collection volumes. Regarding renal events, urinary protein was not detected in this study. Coomassie brilliant blue was used in this study to measure urinary protein in 24‐hour pooled urine as it does not cross‐react with viltolarsen, unlike pyrogallol red, which was shown to cross‐react with viltolarsen in the previous phase 1 trial. Overall, no new safety concerns were raised regardless of the viltolarsen dose received.
Our findings are consistent with those of a previous phase 2 study conducted in the US and Canada. 18 Similar to our study, patients treated with viltolarsen 40 and 80 mg/kg per week showed increased drug‐induced dystrophin production and improvements in timed function tests from baseline (TTSTAND, TTRW, and 6MWT); the safety profile was consistent with this study’s findings.
Immunohistochemistry analysis of muscle biopsies showed that dystrophin protein was visible in several patient tissue samples. However, background staining of nonmuscle cell regions meant that we could not detect the change from baseline in dystrophin/spectrin fluorescence intensity ratio or the number of dystrophin‐positive fibers in most patients.
Viltolarsen treatment has the potential to increase dystrophin expression levels in the long term. In a study of the exon 51 skipping drug eteplirsen, the dystrophin level increased by 0.16% and 0.44% before and after 48 weeks of treatment, respectively. 19 Moreover, in a study of the exon 53 skipping drug golodirsen, a significant increase in normal dystrophin to 1.019% at Week 48 from a baseline value of 0.095% (P < 0.001) was quantified using a validated western blot method. 15 In our study, the dose of antisense oligonucleotide used was higher than that used in the eteplirsen and golodirsen studies. In addition, our treatment duration was relatively short, and the conditions of dystrophin measurement differed. However, despite these differences, it is worth noting that higher dystrophin expression levels were demonstrated in our study. Previous reports on the diagnosis of DMD and Becker muscular dystrophy show correlations between clinical data and dystrophin levels. It has been reported that most patients with intermediate dystrophy have >3% dystrophin, 20 , 21 with lower levels of dystrophin associated with milder dystrophinopathy phenotypes. 22 Furthermore, there is evidence of a link between the different genetic mutations and dystrophin levels. 23 , 24 For example, mutations amenable to exon 44 skipping may be associated with dystrophin levels of 0.2%–7.0% of normal, 25 while wheelchair dependence occurs later in DMD patients with such mutations. 26 Therefore, the observed dystrophin level increase in this study (>3% of normal) may have a beneficial effect on motor function in DMD patients; however, longer‐term studies with more patients are needed to confirm improvement and maintenance of motor function in DMD patients.
In contrast to our expectation, an improvement of motor function was not clearly shown; a possible reason is that, while dystrophin expression may change over a few months, a longer evaluation period is necessary to confirm improvements in motor function. 27 Furthermore, the heterogeneous population, which included nonambulant patients, and the lack of appropriate control groups, meant we could not evaluate this accurately.
The main limitations of the study were the small sample size, owing to the rarity of the disease; short treatment duration, particularly for such a slowly progressing disease; and the open‐label design. It is likely that the small sample size contributed to the lack of significant changes in the 40 and 80 mg/kg groups, and no comparisons with a control group were made. Thus, further studies are needed to investigate these nonsignificant trends. The inclusion of both ambulatory and nonambulatory patients may also have affected the outcomes.
In conclusion, patients treated with viltolarsen 40 and 80 mg/kg tended to have increased dystrophin expression and exon 53 skipping level, as evidenced in muscle biopsy samples analyzed by western blot and RT‐PCR. Dystrophin and exon 53 skipping levels increased with dose escalation and duration of treatment. The maximum tolerated dose has not yet been established in humans, and it is feasible that a higher dose of viltolarsen could affect outcomes. The decline in motor function appeared less marked in patients with higher dystrophin levels, and this may warrant further investigation. Viltolarsen was considered safe and well tolerated. DMD may be improved, and progression suppressed, following the expression of dystrophin protein by the administration of viltolarsen. However, further study is needed, involving a double‐blind comparative study using a placebo group as a control, or assessing long‐term efficacy by extrapolating data from a comparative control such as that provided by natural history, possibly with motor function evaluation as the main outcome.
Author Contributions
H.K., Y.I., H.Y., Y.E., T.T., M.T., and S.T. participated in the study design, data collection, data analysis and interpretation. Y.T., T.M., S.O., M.F., and E.T. participated in the data collection. All authors participated in the development of the manuscript, gave final approval of the manuscript for submission, and agree to be accountable for the integrity of the work.
Conflicts of Interest
H.K. has received grants from Taiho, Pfizer Japan, Nippon Shinyaku, Daiichi Sankyo, Chugai, PTC Therapeutics; and personal fees from Sarepta Therapeutics. Y.T. has received grants from Nippon Shinyaku; and personal fees from Daiichi Sankyo and Biogen. T.M. has received grants from Nippon Shinyaku. S.O. has received grants from Nippon Shinyaku, Biogen, and PTC Therapeutics. M.F. has received grants from Nippon Shinyaku and Taiho. E.T. has received grants from Taiho, Nippon Shinyaku, Daiichi Sankyo, and Takeda; and personal fees from Pfizer Japan. Y.I. has received grants from Taiho, Nippon Shinyaku, and Daiichi Sankyo; and personal fees from Astellas Pharma. H.Y. has received grants from Taiho, Nippon Shinyaku, and Daiichi Sankyo; and personal fees from Biogen. S.T. is an officer and board member of the National Center of Neurology and Psychiatry; and has received grants from Nippon Shinyaku, Daiichi Sankyo and The Noguchi Institute. Y.E., M.T., and T.T. are employees of Nippon Shinyaku Co., Ltd.
Acknowledgment
This study was funded by Nippon Shinyaku Co., Ltd. We thank the patients and their families for participating in this study as well as the study team members at each study site. Medical writing and editorial support were provided by Keyra Martinez Dunn, MD, of Edanz Evidence Generation and was funded by Nippon Shinyaku Co., Ltd. The National Center of Neurology and Psychiatry and Nippon Shinyaku Co., Ltd are coinventors of viltolarsen.
Funding Information
This study was funded by Nippon Shinyaku Co., Ltd. Medical writing and editorial support were provided by Keyra Martinez Dunn, MD, of Edanz Evidence Generation and was funded by Nippon Shinyaku Co., Ltd. The National Center of Neurology and Psychiatry and Nippon Shinyaku Co., Ltd are coinventors of viltolarsen.
Funding Statement
This work was funded by Nippon Shinyaku Co., Ltd. grant .
References
- 1. Guiraud S, Chen H, Burns DT, Davies KE. Advances in genetic therapeutic strategies for Duchenne muscular dystrophy. Exp Physiol 2015;100:1458–1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hoffman EP, Brown RH Jr, Kunkel LM. Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell 1987;51:919–928. [DOI] [PubMed] [Google Scholar]
- 3. Koenig M, Hoffman EP, Bertelson CJ, et al. Complete cloning of the Duchenne muscular dystrophy (DMD) cDNA and preliminary genomic organization of the DMD gene in normal and affected individuals. Cell 1987;50:509–517. [DOI] [PubMed] [Google Scholar]
- 4. Stone MR, O'Neill A, Catino D, Bloch RJ. Specific interaction of the actin‐binding domain of dystrophin with intermediate filaments containing keratin 19. Mol Biol Cell 2005;16:4280–4293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gao QQ, McNally EM. The dystrophin complex: Structure, function, and implications for therapy. Compr Physiol 2015;5:1223–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kharraz Y, Guerra J, Pessina P, Serrano AL, Muñoz‐Cánoves P. Understanding the Process of Fibrosis in Duchenne Muscular Dystrophy. BioMed Res Int 2014;2014:1–11. 10.1155/2014/965631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ryder S, Leadley RM, Armstrong N, et al. The burden, epidemiology, costs and treatment for Duchenne muscular dystrophy: an evidence review. Orphanet J Rare Dis 2017;12:79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mah JK, Korngut L, Dykeman J, et al. A systematic review and meta‐analysis on the epidemiology of Duchenne and Becker muscular dystrophy. Neuromuscul Disord 2014;24:482–491. [DOI] [PubMed] [Google Scholar]
- 9. Japanese Society of Neurology , Japanese Society of Child Neurology, National Center of Neurology and Psychiatry, Nankodo: Clinical care and treatment guideline for Duchenne Muscular Dystrophy, 2014. Available at https://www.neurology‐jp.org/guidelinem/dmd.html.
- 10. Takeuchi F, Komaki H, Nakamura H, et al. Trends in steroid therapy for Duchenne muscular dystrophy in Japan. Muscle Nerve 2016;54:673–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Aartsma‐Rus A, Straub V, Hemmings R, et al. Development of exon skipping therapies for Duchenne muscular dystrophy: a critical review and a perspective on the outstanding issues. Nucleic Acid Ther 2017;27:251–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nakamura A. Moving towards successful exon‐skipping therapy for Duchenne muscular dystrophy. J Hum Genet 2017;62:871–876. [DOI] [PubMed] [Google Scholar]
- 13. Aartsma‐Rus A, Fokkema I, Verschuuren J, et al. Theoretic applicability of antisense‐mediated exon skipping for Duchenne muscular dystrophy mutations. Hum Mutat 2009;30:293–299. [DOI] [PubMed] [Google Scholar]
- 14. Komaki H, Nagata T, Saito T, et al. Systemic administration of the antisense oligonucleotide NS‐065/NCNP‐01 for skipping of exon 53 in patients with Duchenne muscular dystrophy. Sci Transl Med 2018;10:eaan0713. [DOI] [PubMed] [Google Scholar]
- 15. Frank DE, Schnell FJ, Akana C, et al. Increased dystrophin production with golodirsen in patients with Duchenne muscular dystrophy [published online ahead of print, 2020 Mar 5]. Neurology 2020;00:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mendell JR, Rodino‐Klapac LR, Sahenk Z, et al. Eteplirsen for the treatment of Duchenne muscular dystrophy. Ann Neurol 2013;74:637–647. [DOI] [PubMed] [Google Scholar]
- 17. Taylor LE, Kaminoh YJ, Rodesch CK, Flanigan KM. Quantification of dystrophin immunofluorescence in dystrophinopathy muscle specimens. Neuropathol Appl Neurobiol 2013;38:591–601. [DOI] [PubMed] [Google Scholar]
- 18. Clemens PR, Rao VK, Connolly AM, et al. Safety, tolerability, and efficacy of viltolarsen in boys with duchenne muscular dystrophy amenable to exon 53 skipping: a phase 2 randomized clinical trial. JAMA Neurol 2020. 10.1001/jamaneurol.2020.1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. EXONDYS 51TM . Highlights of prescribing information, revised September 2016. Available at: https://www.accessdata.fda.gov/drugsatfda_docs/label/2016/206488lbl.pdf.
- 20. Hoffman EP, Fischbeck KH, Brown RH, et al. Characterization of dystrophin in muscle‐biopsy specimens from patients with Duchenne’s or Becker’s muscular dystrophy. N Engl J Med 1998;318:1363–1368. [DOI] [PubMed] [Google Scholar]
- 21. van den Bergen J, Wokke B, Janson A, et al. Dystrophin levels and clinical severity in Becker muscular dystrophy patients. J Neurol Neurosurg Psychiatry 2014;85:747–753. [DOI] [PubMed] [Google Scholar]
- 22. de Feraudy Y, Ben Yaou R, Wahabi K, et al. Very low residual dystrophin levels mitigate dystrophinopathy towards Becker muscular dystrophy. Neuropediatrics 2019;50(Supplement 01):S1–S10. [Google Scholar]
- 23. Bello L, Morgenroth LP, Gordish‐Dressman H, et al. DMD genotypes and loss of ambulation in the CINRG Duchenne Natural History Study. Neurology 2016;87:401–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wang RT, Barthelemy F, Martin AS, et al. DMD genotype correlations from the Duchenne Registry: Endogenous exon skipping is a factor in prolonged ambulation for individuals with a defined mutation subtype. Hum Mutat 2018;39:1193–1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Beekman C, Janson AA, Baghat A, et al. Use of capillary Western immunoassay (Wes) for quantification of dystrophin levels in skeletal muscle of healthy controls and individuals with Becker and Duchenne muscular dystrophy. PLoS One 2018;13:e0195850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. van den Bergen JC, Ginjaar HB, Niks EH, et al. Prolonged ambulation in Duchenne patients with a mutation amenable to exon 44 skipping. J Neuromuscul Dis 2014;1:91–94. [PubMed] [Google Scholar]
- 27. Merlini L, Sabatelli P. Improving clinical trial design for DMD. BMC Neurol 2015;15:153. [DOI] [PMC free article] [PubMed] [Google Scholar]