

Abstract
Guillain‐Barré syndrome (GBS) is widely regarded as a “pure” peripheral nervous system disorder. However, this simplistic interpretation belies the fact that central nervous system involvement, often manifesting as derangements in mental status can occur as a complication of the “pure” form of the disorder, as part of GBS variants, as well as in a number of mimic disorders. Despite being common in clinical practice, there is no guidance in the literature as to how to approach such scenarios. Herein, we detail our approach to these cases.
Introduction
Guillain‐Barré syndrome (GBS) is the commonest cause of acute flaccid quadriparesis in the developed world. 1 The classic syndrome is one of acute inflammatory demyelinating polyradiculoneuropathy characterized by the rapid onset of areflexic weakness, usually affecting the limbs both proximally and distally, and by definition reaching nadir within 4 weeks. 2 In a quarter of cases, respiratory function becomes compromised, requiring intubation and ventilation. 3 Other characteristic features include autonomic instability, back pain secondary to radicular inflammation, and sensory symptoms without striking sensory signs. 1 Diagnosis remains clinical, though features such as cerebrospinal fluid albuminocytologic dissociation and neurophysiologic evidence of demyelinating polyradiculoneuropathy (both often absent early in the disease) can be helpful. Numerous forme frustes of GBS have also been defined, including axonal variants, pharyngeal‐brachial variant, isolated bilateral facial nerve palsy, and the anti‐Gq1b antibody spectrum of disorders. 2
Central nervous system dysfunction is classically considered not to be a part of GBS. However, in our experience, it is not uncommon for alterations in mental status to be present in patients who appear to otherwise have GBS. This has often left us perplexed, questioning our original diagnosis and uncomfortably adrift in our approach. After many hours of scouring the literature in search of guidance, we noted a dearth of information on this topic, prompting us to write this review.
General Approach
An altered sensorium in a patient who appears to have GBS deserves more than just flippant acknowledgement. Indeed, while clouding of consciousness or neuropsychiatric symptoms can be intrinsic features of GBS, they may also portend significant complications of the disease, treatment‐related adverse events, or importantly, may suggest an alternative diagnosis. This review presents clinicians with a pragmatic, practical approach to assessing patients with suspected GBS who exhibit unanticipated CNS signs, herein termed “GBS + CNS” (see Figure 1). As this is intended as a clinical review, the discussion has purposefully been structured in terms of the questions which clinicians should ask themselves as they approach such cases. We also present a decision tree (Figure 2), to which clinicians may refer in their diagnostic process.
Figure 1.
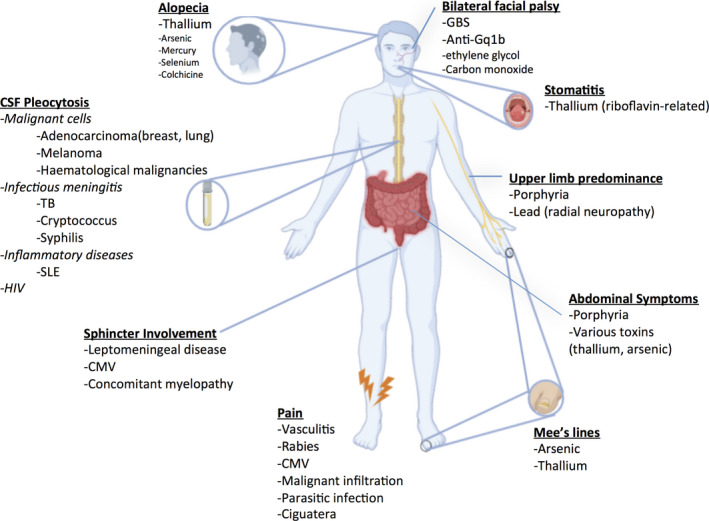
Clinical clues suggesting GBS mimics with altered mental status (GBS + CNS). CSF, cerebrospinal fluid; CMV, cytomegalovirus; HIV, human immunodeficiency virus; SLE, systemic lupus erythematosus; TB, Tuberculosis.
Figure 2.
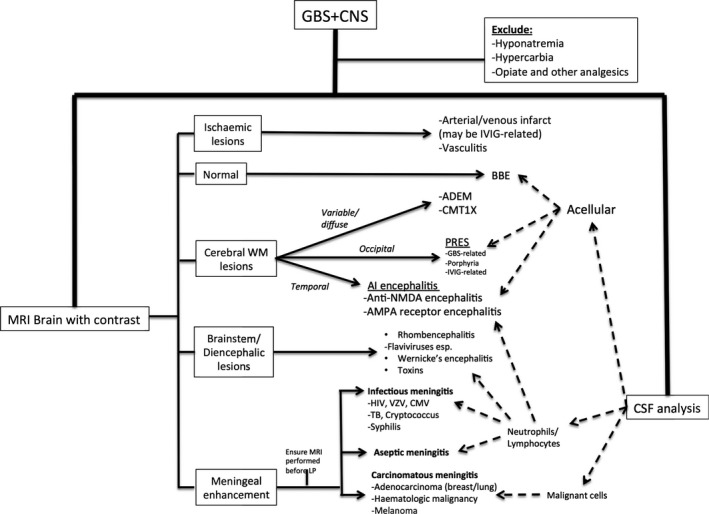
Clinical algorithm for approaching cases of suspected GBS + CNS. ADEM, acute disseminated encephalomyelitis; AI, autoimmune; AMPA, α‐amino‐3‐hydroxy‐5‐methyl‐4‐isoxazolepropionic acid; BBE, Bickerstaff brainstem encephalitis; CMT 1X, Charcot‐Marie‐Tooth type 1X; CSF, cerebrospinal fluid; CMV, cytomegalovirus; HIV, human immunodeficiency virus; IVIG, intravenous immunoglobulin; LP, lumbar puncture; MRI, magnetic resonance imaging; NMDA, N‐Methyl‐ d‐aspartate; PRES, posterior reversible encephalopathy syndrome; TB, Tuberculosis; VZV, varicella zoster virus; WM, white matter.
Could the Observed Mental Status Changes be Part of the Guillain‐Barré Syndrome?
Broadly speaking, mental status changes as part of a “true” GBS illness can comprise either:
Intrinsic neuropsychiatric manifestations of the disorder, or
CNS dysfunction resulting from disease‐related autonomic or metabolic derangements.
Intrinsic neuropsychiatric manifestations of GBS
Clinical descriptions of GBS primarily focus on peripheral nervous involvement, and rightly so. Nevertheless, it must be borne in mind that CNS dysfunction, ranging from mild neuropsychiatric symptoms (depression, anxiety) 4 to deep unconsciousness (mainly in children, where GBS may coexist with acute disseminated encephalomyelitis‐ADEM) is recognized as part of the GBS spectrum. 5 , 6 , 7 In fact, a systematic study of critically ill adult patients with GBS found that almost one third had mental status abnormalities. 8 Encephalopathy was disproportionately common in patients with higher CSF protein levels and autonomic dysfunction, had peak incidence at one week following disease onset, and tended to persist for a further week. Hypnagogic or hypnopompic vivid dreams charged in emotional content, illusions (visual, tactile, auditory, and illusions of body tilt), visual and tactile hallucinations as well as delusions (usually paranoid) were all examples of mental status abnormalities reported in this study as intrinsic GBS‐related features. 8
Though GBS‐related neuropsychiatric alterations remain a diagnosis of exclusion coming at the end of any decision tree once every reasonable effort has be made to rule out other causes, we thought it important to highlight its existence right from the outset in this review.
Metabolic Disturbances associated with GBS
Metabolic disturbances are common among hospitalized patients regardless of underlying diagnosis. Specific to GBS however, symptomatic hyponatremia secondary to the syndrome of inappropriate antidiuretic hormone secretion (SIADH), CO2 narcosis (as mentioned before, 25% of patients require ventilation as a result of weakness‐related hypoventilation) and decubitus‐related infections should all be actively searched for.
SIADH is particularly common, affecting roughly 50% of GBS patients. Its pathogenesis is incompletely understood, but multiple theories have been proposed. These include increased ADH release secondary to hypothalamic damage or immune mechanisms, 9 , 10 resetting of hypothalamic osmoreceptors 11 and altered renal tubular sensitivity to ADH. 12 It is more common in older patients and those with bulbar dysfunction and/or ventilator dependency, 12 and manifests clinically with altered mental status, seizures and occasionally coma (depending on the rapidity and severity of hyponatremia, and subsequent fluid shifts) and biochemically with euvolemic, hypo‐osmolar hyponatremia. In those receiving IVIG, it should not be confused with IVIG‐related pseudohyponatremia, 13 an analytic artifact which is easily differentiated from true hyponatremia by the absence of hypo‐osmolality. 14 Hyponatremia must be managed with care, as overly rapid correction can precipitate osmotic demyelination syndromes, producing permanent, rather than temporary alterations of consciousnesss. 15
Autonomic dysfunction
Autonomic dysfunction secondary to demyelination of autonomic nerve fibers is considered a hallmark feature of GBS, being present in at least two thirds of cases. 16 Blood pressure variability is particularly characteristic, occasionally culminating in either sustained severe systolic hypertension or cardiovascular collapse. 17
While malignant hypertension may in and of itself produce mental status changes, one condition to be particularly mindful of in GBS + CNS is posterior reversible encephalopathy syndrome (PRES). PRES is a clinico‐radiologic entity manifesting classically with headache, nausea, vomiting, visual changes, seizures, encephalopathy, and focal neurologic deficits alongside radiologic evidence of vasogenic edema predominating in posterior brain regions. 18 Its pathophysiologic basis is incompletely understood but rapidly developing hypertension with failure of cerebral autoregulation is thought to be critical. The relative lack of sympathetic innervation of the posterior circulation potentially accounts for preferential involvement of the posterior brain regions. Most cases of GBS‐related PRES have occurred in women aged> 55 years. 19 Both GBS‐associated dysautonomia and IVIG treatment offer fertile territory for its development.
Could the GBS Treatment be Responsible?
One should always consider that GBS + CNS could be an iatrogenic phenomenon resulting from either disease‐modifying or symptomatic treatment administered for GBS.
Intravenous immunoglobulin (IVIG) has become a standard of GBS care worldwide, in an attempt to hasten recovery. 20 Though often perceived as a benign treatment, it is certainly not devoid of adverse effects. The incidence of thrombotic events is variable (1–16% in the literature), generally occurring within 24 hr of administration of IVIG. 21 Patients with a history of prior thrombosis, advanced age, high total daily dose, and underlying prothrombotic tendencies or atherosclerosis are at heightened risk. Such thromboses can involve both cerebral arterial and venous systems, and should be considered in GBS + CNS patients who are receiving IVIG, especially if other suggestive signs (either focal neurological deficits or –especially in intracranial venous thromboses‐ papilloedema, headache or seizures) are present. 21 Aseptic meningitis affects 0.5–1% of IVIG‐treated patients, generally begins within 48 h of starting the infusion and can rarely lead to mental status alterations, generally alongside fever, headache and meningismus. 22 , 23 CSF examination in such cases reveals an aseptic (often neutrophilic) pleocytosis, elevated CSF protein and high opening pressure. 22 The symptoms are often indistinguishable from viral or bacterial meningitides in the early stages; the condition generally settles spontaneously in 5–7 days. 23 As mentioned before, IVIG administration can also trigger PRES.
Opiate and nonopiate analgesics (e.g., pregabalin, gabapentin), commonly administered for neuropathic pain in this population, should also make the shortlist of potential culprits for mental status alteration. Additionally, in ventilated patients, one should always consider the effects of sedative medications (or withdrawal therefrom) on mental status.
Am I Dealing with a GBS Variant (esp. Anti‐Gq1b Ssease)?
The next point to consider is whether the patient could be suffering from a related, but distinct disorder, particularly anti‐Gq1b antibody‐related disease. 24 Anti‐Gq1b syndromes manifest along a spectrum, ranging from the peripheral nervous system predominant Miller‐Fisher syndrome (typified by the clinical triad of ataxia, areflexia and ophthalmoplegia) through to central involvement in the form of Bickerstaff’s brainstem encephalitis, defined by a combination of external ophthalmoplegia AND either altered level of consciousness –ranging from mild drowsiness to coma‐ OR hyper‐reflexia 25 , 26 , 27 with a multitude of forme frustes including acute ophthalmoparesis and acute ataxic neuropathy in between. 24 , 28
Up to 50 percent of patients with Bickerstaff’s brainstem encephalitis have coexisting GBS, but limb weakness generally follows the development of ophthalmoplegia/ataxia rather than being present at the onset. 27 Nerve conduction studies in these patients frequently demonstrate axonal involvement. Other common symptoms include facial weakness, bulbar dysfunction, blepharoptosis, mydriasis, nystagmus, and proprioceptive deficits; 25–30% may display nonspecific MRI abnormalities, consisting of T2‐weighted hyperintensities in the brainstem, thalamus, cerebellum, or cerebral white matter. 27
Am I Wrong, and This Actually is Not GBS?
Having considered disease‐related CNS involvement/complications, side‐effects of therapy and disease variants, one should ponder whether mental status changes actually suggest the presence of a GBS mimic. Indeed, numerous disorders, some of which are eminently treatable, manifest with the combination of acute paresis alongside CNS dysfunction.
While an extensive list of such disorders is given in Tables 1, 2, 3, 4, 5, these inventories are often unhelpful in the clinical setting. We find the mnemonic MIND (Metabolic + Malignancy, Infectious/Immune/Inherited, Nutritional, Drugs/toxins) useful to classify these remaining diseases, the most pertinent of which we discuss below.
Table 1.
Causes of GBS + CNS in the presence of an initial “correct” GBS diagnosis.
| Condition | Clinical Clues | Additional Investigations |
|---|---|---|
| 1. Initial Diagnosis Correct | ||
| Intrinsic feature of GBS illness |
|
MRI Brain normal EMG/NCS may be suggestive CSF albuminocytologic dissociation |
| Intrinsic feature of GBS variant | ||
| Anti‐Gq1b disease |
|
Serum anti‐Gq1b antibody positivity Brain MRI usually normal |
| Complications of disease | ||
| Metabolic derangement | ||
| Hyponatremia (SIADH) |
|
Serum sodium, Serum Osmolality, Urine Osmolality. |
| Hypercarbia (respiratory muscle weakness) |
|
Arterial blood gas ‐ CO2 levels |
| Infections | Signs of pulmonary, urinary or skin infection especially | Elevated WCC, CRP; Abnormal CXR or urinalysis |
| Autonomic instability | ||
| Posterior reversible encephalopathy syndrome (PRES) |
|
T2 and FLAIR hyperintensities predominantly in posterior brain regions |
| Complication of treatment | ||
| IVIG‐relatesd | ||
| Hypercoagulability(also rarely encountered with plasmapheresis) | Focal neurological deficits(arterial/venous)+/‐seizures, headache, papilloedema(venous) | MRI Brain: Diffusion restriction; vascular imaging help define cause and extent of thrombosis |
| PRES | as above | as above |
| Aseptic meningitis |
|
Culture‐negative CSF pleocytosis |
|
Medications
|
|
Medication administration review |
CNS, Central Nervous System; CRP, c‐reactive protein; CO2, carbon dioxide; CSF, cerebrospinal fluid; CXR, chest X‐ray; EMG, Electromyography; FLAIR, fluid‐attenuated inversion recovery; GBS, Guillain‐Barré Syndrome; IVIG, intravenous immunoglobulin; MRI, magnetic resonance imaging; NCS, nerve conduction studies; PRES, posterior reversible encephalopathy syndrome; SIADH, syndrome of inappropriate ADH secretion; WCC, white cell count
Table 2.
Initial diagnosis incorrect‐ consider “MIND” disorders: M = Metabolic/Malignancy.
| Condition | Clinical Clues | Additional Investigations |
|---|---|---|
| Metabolic/Malignant | ||
| Metabolic | ||
| Porphyria |
+/‐ triggers: Surgery, infection, enzyme‐inducing medications |
Elevated urinary aminolevulinic acid and porphobilinogen during attack |
| Thyrotoxicosis |
|
Low serum TSH Elevated serum T3 and free T4 |
| Mitochondrial disorders | PDH complex deficiency especially can present recurrent peripheral weakness in childhood. GBS + CNS occasionally encountered in other mitochondrial disorders |
‐Bloods: Lactic acidosis, hyperalaninemia ‐MRI: Symmetrical basal ganglia/brainstem lesions ‐Muscle biopsy ‐Genetic analysis |
| Malignant | ||
| Carcinomatous meningitis |
History of cancer, esp:
|
CSF: Malignant cells MRI with contrast: Meningeal contrast uptake |
| Lymphoma (Non‐Hodgkin>>>Hodgkin) |
GBS‐like illnesses due to either: 1. Direct neural invasion (neurolymphomatosis, lymphomatous meningitis) or, 2.Immune mechanisms |
MRI Brain: generally abnormal‐enhancing mass lesion(s) CSF: cytology, flow cytometry Systemic staging: PET‐CT Biopsy of affected tissue(brain, vitreous) necessary for diagnosis. |
|
Paraneoplastic syndromes 1
|
CNS involvement:
PNS involvement:
|
Onco‐neuronal antibody screen Search for primary malignancy |
CSF: cerebrospinal fluid; CNS: central nervous system; CT: computed tomography; GBS: Guillain‐Barré Syndrome; MRI: magnetic resonance imaging; PDH: pyruvate dehydrogenase; PET: positron emission tomography; PNS: peripheral nervous system; TSH: thyroid‐stimulating hormone.
Presentation usually largely dominated by central nervous system involvement. PNS signs rare, but occasionally reported
Table 3.
Initial diagnosis incorrect‐ consider “MIND” disorders: I = Infections/Immune/Inherited.
| Condition | Clinical Clues | Additional Investigations |
|---|---|---|
| Infections | ||
|
Enteroviruses
|
Anterior horn cell involvement ‐> paralysis +/‐Brainstem encephalitis |
RT‐PCR of cerebrospinal fluid |
|
Flaviviruses
|
‐Anterior horn cell involvement ‐> asymmetric paralysis (except Zika virus, which produces classic GBS) ‐Encephalitis ‐Travel history important ++ |
Virus‐specific IgM and IgG antibodies |
|
Lyssavirus
|
‐Prodromal pain in bitten limb ‐Early fasciculations ‐Rapidly progressive course |
RT‐PCR of saliva Rabies antibodies in CSF or serum |
| HIV |
‐GBS occurs early ‐CNS signs occur late |
HIV p24 antigen, PCR viral load, ELISA or Western blot |
|
Herpesviruses ‐CMV ‐VZV |
CMV:
VZV:
|
Positive CSF PCR for CMV/VZV |
| Lyme |
|
CSF and serum Lyme ELISA +/‐ confirmatory Western blot |
|
Other infections
|
|
Serum +/‐ CSF testing for individual pathogens |
|
Parasites:
|
|
Serum and CSF eosinophilia Specific diagnosis requires ELISA or immunoblot |
| Immune | ||
| Vasculitis |
SLE:
EGPA:
|
SLE:
EGPA
|
| Neurosarcoidosis |
+/‐ Lung, joint, eye, skin, and lymph node involvement. |
CSF pleocytosis CSF ACE levels may be elevated Definite diagnosis: biopsy |
| Sjogren syndrome |
‐F>>M(9:1 ratio) ‐Ocular and oral dryness ‐Musculoskeletal pain and fatigue +/‐pulmonary, renal or biliary tract involvement. Neuro: ‐Sensory neuropathy/neuronopathy |
Anti‐SSA and/or anti‐SSB antibodies Schirmer’s test ‐ ocular dryness Minor salivary gland biopsy |
|
Autoimmune encephalitides 1 ‐AMPA ‐anti‐NMDA ‐Anti‐CASPR2 |
+/‐ Neuropathy |
Autoimmune encephalitis antibody panel T2/FLAIR hyperintensities in medial temporal lobes CSF pleocytosis |
| Inherited | ||
| CMT 1X |
Triggers: infections, exertion, trauma, altitude. |
Genetics: GJB1 pathogenic variants |
ACE, angiotensin‐converting enzyme; AMPA, α‐amino‐3‐hydroxy‐5‐methyl‐4‐isoxazolepropionic acid; ANCA, Anti‐neutrophil cytoplasmic antibodies; CASPR2, Contactin‐associated protein‐like 2;CMT 1X, Charcot‐Marie‐Tooth type 1X; CMV, cytomegalovirus; CNS, central nervous system; CSF, cerebrospinal fluid; CXR, chest X‐ray; dsDNA, double‐stranded DNA; EGPA, eosinophilic granulomatosis with polyangiitis; ELISA, enzyme‐linked immuosorbent assay; EV, enterovirus; F, female; FLAIR, fluid‐attenuated inversion recovery; GBS, Guillain‐Barré Syndrome; HIV, human immunodeficiency virus; M, male; MPO, myeloperoxidase; NMDA, N‐Methyl‐ d‐aspartate; RT‐PCR, reverse transcription polymerase chain reaction; SLE, systemic lupus erythematosus; VZV, varicella zoster virus
Presentation usually largely dominated by central nervous system involvement. PNS signs rare, but occasionally reported
Table 4.
Initial diagnosis incorrect‐ consider “MIND” disorders: N = nutritional.
| Condition | Clinical Clues | Additional Investigations |
|---|---|---|
| Nutritional | ||
| Thiamine (Vitamin B1) |
+/‐Thiamine Neuropathy +/‐Cardiac involvement |
Serum thiamine levels (rarely useful‐long turnaround time)
|
| Vitamin B121 |
+/‐ neuropathy, cognitive changes(rare) |
Low serum vitamin B12 Elevated homocysteine and methylmalonic acid Macrocytic anemia with Hypersegmented neutrophils |
| Copper 1 |
|
Low serum copper Low serum ceruloplasmin |
Presentation usually largely dominated by peripheral nervous system involvement. CNS signs rare, but occasionally reported
Table 5.
Initial diagnosis incorrect‐ consider “MIND” disorders: D = drugs/toxins.
| Condition | Clinical Clues | Additional Investigations |
|---|---|---|
| Drugs/toxins | ||
| Arsenic |
|
Elevated urine arsenic levels |
| Thallium |
|
Elevated urinary and blood thallium levels |
| Lead 1 |
|
FBC: microcytic anemia with basophilic stippling of RBCs Blood lead levels: increased |
| Selenium |
+/‐Peripheral neuropathy |
serum selenium: elevated 24 hr urine selenium excretion: elevated |
| Botulism 1 |
|
Toxin identification: serum, stool, or food sample ‐Mouse bioassays, ELISA, PCR |
| Snake envenomation 1 |
+/‐ autonomic instability
|
Clinical diagnosis‐ descriptions of the causative snake may help identification |
|
Ethylene glycol Diethylene Glycol 2 |
+/−CNS signs esp. parkinsonism |
‐Increased anion gap metabolic acidosis ‐Increased osmolal gap ‐Calcium oxalate crystalluria ‐Elevated serum glycol levels |
| Toluene 2 |
|
Elevated serum toluene levels |
| Colchicine |
|
|
| Nitrous oxide |
|
Vitamin B12 levels usually low‐normal, or low (but can be normal) Serum MMA usually elevated |
| Heroin and other opiates |
|
|
| Acrylamide |
|
Usually intentional poisoning |
| Dioxins |
+/‐Encephalopathy: irritability, restlessness, insomnia and stupor |
Exposures: occupational or intentional poisoning Serum dioxin levels are diagnostic |
|
Hexane/ Hexacarbons 2 |
|
|
| Methyl bromide |
‐Sensorimotor neuropathy +/‐encephalopathy |
Exposures: fumigants, refrigeration materials, fire extinguishers |
| Tin 2 |
|
Exposure: electronics, plastics and soldering industries Urine tin levels peak day 4‐10 |
| Trichlorethylene 2 |
|
Exposure(occupational): degreasing solvents, flame retardants, cleaning solutions |
| Carbon disulfide 2 |
|
Exposure: occupational; disulfiram overdose |
| Carbon monoxide 2 |
+/‐ polyradiculoneuritis and bilateral facial palsy |
Elevated blood carboxyhemoglobin levels |
| Organophosphates |
|
Decreased red blood cell and plasma cholinesterase levels |
| Scorpion envenomation |
+/‐Encephalopathy; flaccid quadriparesis |
|
| Mercury |
|
Exposure: occupational, contaminated seafood, dental amalgam Elevated blood and urine mercury levels |
| Ciguatoxin 1 |
|
|
| Tetrodotoxin 1 |
|
Exposure: Inadequately prepared puffer fish (Japan) |
CNS, Central Nervous System; CO2, carbon dioxide; CSF, cerebrospinal fluid; CT, computed tomography; ELISA, enzyme‐linked immunosorbent assay; EMG, Electromyography; FBC, full blood count; GBS, Guillain‐Barre Syndrome; MMA, Methylmalonic acid; MRI, Magnetic resonance imaging; NCS, nerve conduction studies; PCR, polymerase chain reaction; PET, Positron emission tomography; PNS, Peripheral Nervous System; RBC, red blood cells; RT‐PCR, reverse transcriptase PCR.
Presentation usually largely dominated by peripheral nervous system involvement. CNS signs rare, but occasionally reported;
Presentation usually largely dominated by central nervous system involvement. PNS signs rare, but occasionally reported
Metabolic and Malignancy
Metabolic
The principal metabolic conditions to rule out in the setting of GBS + CNS are the porphyrias. These inherited disorders, caused by partial enzyme deficiencies in the haem biosynthetic pathway, are extremely rare and most neurologists will go a whole career without encountering a case.
Porphyrias are usually dominantly inherited, though only 10‐20% of gene carriers will ever suffer an acute attack. 29 Attacks are often precipitated by increased flux through the haem synthesis pathway, such as by pathway enzyme induction (generally by drugs ‐hormonal contraceptives, certain antibiotics and anticonvulsants‐ but also by other factors including low carbohydrate intake and alcohol binging), or increased metabolic demand through infection, or surgery. 29 Under these circumstances, insufficient enzyme function results in accumulation of toxic haem precursors, and clinical manifestations. Though no sex differences in gene carriage exist, women of reproductive age are five times more likely to experience an acute porphyric attack, probably due to hormonal fluctuations. 29 , 30
Porphyric attacks often begin with abdominal pain, at times so severe as to result in exploratory laparotomies (ironically aggravating the attack), followed by the development of neuropsychiatric symptoms. These may be mild (insomnia, agitation) but if unchecked progress to confusion, delirium and seizures. 31 Up to 40% of affected individuals will develop a combined autonomic and motor‐predominant axonal porphyric neuropathy, starting within 1 month of symptom onset in 80% of cases and sometimes as early as 3 days. 31 In contrast to GBS, porphyric neuropathy often begins in the arms, an important clinical clue 32 . Autonomic dysfunction is often profound; sinus tachycardia is almost universal, while extreme blood pressure lability is not uncommon. PRES is in fact the most common neuroimaging finding during an attack. Cerebrospinal fluid albuminocytologic dissociation and SIADH‐related hyponatremia (detected in 40% of patients) may further confuse the diagnostic process 31 , 33 . Urine often darkens upon standing, a fortuitous observation which has provided the clue to diagnosis in some cases.
Malignancy
One should be particularly cautious about making a diagnosis of GBS in patients with known or suspected malignant disease. Not only are these patients prone to developing weakness through direct nerve infiltration in leptomeningeal metastatic disease, but they are often exposed to vast arrays of potentially neurotoxic chemotherapies as well as frequently suffering nutritional deficiencies.
Leptomeningeal metastases in particular can mimic GBS. These affects 5‐8% of patients with solid tumors (especially adenocarcinomas of the breast and lung, and melanoma), and up to 15% of those with hematologic malignancies. 34 , 35 Most affected individuals have a known malignancy, though occasionally it can be the presenting symptom. 35 Clinical manifestations are protean. Involvement of the spinal nerve roots can produce rapidly progressive weakness, dermatomal sensory loss, radicular pain, and sphincter involvement, while intracranial disease can compromise cranial nerve function. 35 Symptomatic intracranial hypertension, resulting from impaired CSF resorption often ensues, manifesting as headache, vomiting and altered consciousness. 34 MRI with contrast, which should be performed prior to lumbar puncture, shows meningeal contrast uptake in the majority of cases though the gold standard for diagnosis remains the detection of malignant cells in spinal fluid, which may require repeated examination of large volumes of CSF or occasionally meningeal biopsy.
Differentiation between leptomeningeal metastases and GBS can be challenging in early disease. Clues to the diagnosis of leptomeningeal metastases include asymmetry of weakness and reflexes, and sphincter involvement. Additionally, though repeated CSF examinations may be required to identify malignant cells, other CSF parameters (opening pressure, protein, cell count, glucose) are almost always abnormal. 34
Paraneoplastic neurologic syndromes must also be borne in mind. These complex, multifaceted disorders often combine both central and peripheral nervous system (PNS) involvement. 36 In contrast to leptomeningeal disease, paraneoplastic syndromes usually precede the detection of the primary malignancy. 36 GBS‐like paraneoplastic syndromes occur mostly in the presence of anti‐Hu antibodies, which is often secondary to small cell lung cancer. The anti‐Hu syndrome generally produces a severe sensory neuronopathy which may mimic sensory variants of GBS, but in 5% mixed motor (often due to a motor neuronopathy) and sensory involvement is the predominant feature; 37 weakness may be so profound as to require mechanical ventilation. Additionally, patients frequently develop brainstem or limbic encephalitis. 37 Patients with anti‐CRMP5 antibodies, either alone or in combination with anti‐Hu, may display a similar syndrome. 38
Infectious/Immune/Inherited
Infectious
Several neurotropic (particularly viral) infections have the potential to produce acute flaccid weakness and encephalitis, and are thus important differential diagnoses of GBS + CNS. These agents vary in their mode of transmission, target population, geographic distribution and endemic versus epidemic nature. Important clues to a potential infectious etiology include: (1) recent travel to endemic areas (pathogen specific), (2) fever, rash, meningism, (3) CSF pleocytosis (cell type may give a clue to the etiology) and (4) associated neurological features e.g., extrapyramidal signs in Japanese encephalitis. A brief account of the most notable offenders is given below.
Enteroviruses
Though historically poliovirus was the principal infectious agent responsible for acute flaccid myelitis, occasionally combined with polioencephalitis, successful vaccination programs have eradicated this disease from much of the developed world. 39 Nevertheless, sporadic outbreaks are still occasionally encountered in the developing world and in unvaccinated populations. Outbreaks of other “polio‐like” enteroviruses, predominantly affecting children, and displaying similar tropism for anterior horn cells have emerged in recent years. Examples include enterovirus 71 and enterovirus D68, both causing severe acute flaccid paralysis and brainstem‐predominant encephalitis, sometimes associated with classic hand foot and mouth skin lesions and bulbar or cranial nerve involvement, respectively. 40 , 41 , 42
Flaviviruses
A number of arthropod‐borne neuroinvasive flaviviruses, in particular Japanese encephalitis (JEV), West Nile (WNV) and tick‐borne encephalitis (TBE) viruses are well recognized to cause acute flaccid paralysis accompanied by encephalitis. 43 Generally transmitted to humans through the bite of a mosquito (JEV and WNV) or tick, most infections remain clinically inapparent or produce only mild viral symptoms. In a small proportion however, progression to invasive neurological disease occurs, often with devastating consequences. 43 , 44 , 45 , 46 , 47 Flavivirus‐related acute motor syndromes are generally asymmetric and the result of a severe, destructive motor neuronopathy. 46 Encephalitis characteristically involves brainstem and diencephalic regions, occasionally producing marked thalamic signal abnormalities described as the “double doughnut” sign. This regional predilection for deep gray matter likely explains the frequent extrapyramidal manifestations encountered with several of these disorders.
West Nile virus (WNV) is a mosquito‐borne virus encountered predominantly in the Americas. 46 West Nile virus encephalitis, predominantly a disease of the elderly or immunocompromised, 46 , 48 affects three quarters of symptomatic cases, 46 and in 20% of individuals is accompanied within 3–7 days of onset by an acute motor neuronopathy, manifesting as acute flaccid paralysis. 49 A viral exanthem, present in 25% of cases, may be an important clinical clue. Extrapyramidal complications not uncommonly accompany the encephalopathy. 50
TBE is endemic in Eastern Europe and central Asia. Different subtypes of the virus exist, with varying propensity for producing brainstem‐predominant meningoencephalitis and acute flaccid paralysis. 51 Both the likelihood of symptomatic infection and of encephalitis increases with age, being most prevalent in those aged over 50 years. 52
In contrast, Japanese encephalitis, the most common cause of viral encephalitis in Asia, is primarily a disease of the young, with three quarters of those affected being aged under 14 years. 44 However, immunologically naive travellers of all ages are susceptible to infection, and since most tourists are adults, infections in returning travellers reflect this demographic. 53 , 54 JE predominantly manifests with fever, headache and altered level of consciousness; in older individuals, behavioral change, sometimes mistaken for psychiatric disease, may be the sole presenting feature. 44 Movement disorders, particularly dystonia/parkinsonism frequently develop, as do seizures. Acute flaccid paralysis coexists with encephalitis in 30% of cases, and though generally lower‐limb predominant and asymmetric, can mimic GBS. 44
Lyssaviruses
Rabies is a viral zoonosis acquired through the bite of an infected animal, though 25% of cases have no recollection of animal bites. Rabies is primarily known for its furious form manifesting with hydrophobia, aerophobia, muscle spasms and limbic encephalitis, but 20‐30% of affected individuals present with the paralytic variant, which is thought to result from a combination of anterior horn cell involvement and mixed axonal/demyelinating neuropathy. The neuropathy is possibly immune‐mediated. In its initial stages, paralytic rabies is clinically and electrodiagnostically indistinguishable from GBS. 55 , 56 Indeed, GBS is the most common misdiagnosis at onset.
Following inoculation, the virus travels to the central nervous system through retrograde axoplasmic flow. It begins replicating in the dorsal root ganglion, producing viral ganglionitis, often characterized by excruciating pain in the affected limb. 55 , 56 Further centripetal spread leads to involvement of the brainstem, hippocampus and hypothalamus, with progressive deterioration in level of consciousness. 55 Most people seek medical attention within 2 weeks of symptom onset; most will be dead within another 2 weeks. Important clinical clues such as prodromal fever in a patient from a known endemic region, urinary incontinence, percussion myoedema, monomelic onset or single limb predominance, fasciculation in the affected limb, and severe pain may be present, though many exhibit “typical” GBS syndromes. 57 Cranial nerve and autonomic involvement is common, just as in GBS. 55
Retroviruses
Though the most common peripheral nerve manifestation of HIV infection is a length‐dependent sensory‐predominant neuropathy, GBS‐like presentations can occur, generally as a seroconversion illness or early in the course of infection, before significant immunosuppression has occurred. That being said, HIV‐associated GBS (and its variants such as Miller‐Fisher syndrome) are also reported in late‐stage AIDS patients, as well as during immune reconstitution inflammatory syndromes. 58 , 59 HIV‐associated GBS is clinically indistinguishable from “classic” disease, and though conventional teaching dictates that CSF pleocytosis is a common finding, in fact, studies show that the CSF is not infrequently acellular. 60
Coexisting encephalopathy can occur for multiple reasons, including opportunistic infection (cerebral toxoplasmosis, cryptococcal meningitis, tuberculosis, cytomegalovirus encephalitis), malignancy (CNS lymphoma), progressive multifocal leukoencephalopathy, and HIV‐associated dementia. 61 In contrast to GBS‐like manifestations however, CNS involvement generally develops in the setting of systemic immunosuppression, meaning that the combination of the two as GBS + CNS is uncommon, but nevertheless important to outrule.
Herpesviruses
The most important herpesviruses to consider in GBS + CNS syndromes are cytomegalovirus (CMV) and varicella zoster virus (VZV). Systemic CMV infection rarely occurs outside of extreme immunosuppression as in AIDS and transplant recipients. Retinitis (producing painless visual loss) is the most common presentation, followed by gastrointestinal involvement 62 . Neurological disease occurs in less than 1% of cases. 62 CMV encephalitis commonly produces lethargy, confusion, cranial nerve palsies, and nystagmus. 62 Neuroimaging may be normal but CMV PCR on CSF is positive in over 90% of patients. CMV polyradiculopathy, which may mimic GBS, is typified by lower‐limb predominant, excruciatingly painful, ascending flaccid areflexic weakness, and is generally only encountered once CD4 counts drop under 50 cells/mm3. In contrast to GBS, sphincter disturbance is common at presentation, and CSF often has a neutrophilic pleocytosis. 62 Early diagnosis is paramount, as this offers the best hope of survival and functional recovery.
Similarly, VZV can trigger a GBS‐like illness as part of a primary infection, or indeed may produce a meningomyeloradiculitis as part of a reactivation illness. 63 In both settings, coexistent meningoencephalitis can compromise mental function. In contrast to CMV, immunosuppression is not a prerequisite to developing invasive neurological disease, though advancing age is a risk factor for reactivation. 64 The typical vesicular rash is a useful clue, but may be absent‐ zoster sine herpete.
Bacterial infections
Bacterial infections, Campylobacter jejuni in particular, mostly either trigger GBS through immune activation and molecular mimicry, or produce infectious meningitis with involvement of spinal and cranial nerve roots. The list of potential bacterial triggers is vast, has been extensively reviewed, 65 and is outside the scope of this review. Neuroborreliosis is however worth mentioning, as it commonly manifests with a painful, potentially GBS‐mimicking spinal polyradiculitis, and cranial polyneuritis. 66 Concomitant Lyme encephalitis is rare (about 5% of cases), and usually a late manifestation, but can resemble psychotic or dementing illnesses. 66 , 67 Scrub typhus has similarly been associated with GBS alongside mental status alterations (scrub meningoencepahlitis). 68 Meningeal infiltration with tuberculosis, cryptococcus and syphilis should always be considered, especially in the immunosuppressed. 69
Immune
Vasculitides, with their multisystemic involvement and propensity to masquerade, are important to rule out in GBS + CNS. Though traditional medical teaching dictates that vasculitic neuropathies manifest as mononeuritis multiplex, in fact, a not infrequent presentation is that of symmetric sensorimotor polyneuropathy, at times mimicking GBS. Neuropathy may be the presenting feature of systemic inflammatory disease prior to systemic involvement, and in some cases vasculitides remain confined to the peripheral nerves. 70 , 71 CNS manifestations of vasculitis are protean, ranging from confusion, cognitive decline and altered mental status through to neuropsychiatric abnormalities (especially in lupus) and focal neurological signs, often the result of ischemic lesions.
In theory, any vasculitis could cause these complications, but in practice most cases of GBS + CNS result from eosinophilic granulomatosis with polyangiitis or systemic lupus erythematosus. 72 , 73 , 74 , 75 Clinicians should therefore have a low threshold for serological testing for these disorders, even with “barn door” GBS. Systemic symptoms and signs such as fever, weight loss, purpuric rash, active urinary sediment as well as axonal involvement on nerve conduction studies should alert physicians to the diagnosis. Early nerve biopsy, or biopsy of other involved tissue, in suspect cases may secure the diagnosis and allow initiation of immunosuppressive treatment.
In patients with a subacute neurocognitive syndrome, the autoimmune encephalitides should also be kept in mind. Their syndromic presentations vary according to the associated antibody and the age of the patient, but for the most part comprise combinations of cognitive decline, psychiatric symptoms, movement disorders, seizures, and autonomic dysfunction. 76 , 77 If present, signal abnormalities on MRI and CSF pleocytosis are further pointers to the diagnosis. 76 , 77 Rarely, autoimmune encephalitides may also have peripheral nerve manifestations mimicking GBS, though this may be overlooked owing to the significant disturbances in mental status. In particular, antibodies targeting glutamate receptors (NMDA and AMPA receptors) have been reported to produce a destructive, generally motor‐predominant neuritis mimicking GBS, 78 which may precede or develop concurrently with the classic CNS manifestations. 79 The underlying pathomechanisms remain unclear. Certainly, peripheral nerves are rich in NMDA and AMPA receptors, 80 opening a number of possibilities, including co‐targeting of central and peripheral epitopes by common antibodies, exposure of peripheral NMDA and AMPA receptors as a result of neuritis with secondary CNS manifestations (akin perhaps to the pathophysiology of post‐HSV anti‐NMDA receptor encephalitis), immune‐mediated motor neuronopathy (glutamate receptors are highly distributed in anterior horn cells) or the presence of other coexisting autoimmune or paraneoplastic antibodies. 78 Patients with CASPR2 antibodies typically present with limbic encephalitis, Morvan’s syndrome, dysautonomia, and peripheral nerve hyperexcitability, but an association with a syndrome resembling GBS has been recorded 81 .
Inherited
X‐linked Charcot Marie Tooth disease (CMT 1X) caused by mutations in the GJB1 gene encoding the gap junction protein, connexin‐32, is the second most common inherited demyelinating neuropathy 82 . CMT 1X is characterized by the insidious onset of a slowly progressive peripheral sensorimotor neuropathy, which generally manifests before the age of 25 years, the phenotype being less severe in women due to X chromosome inactivation. However, superimposed on this chronic neuropathy, patients frequently develop transient neurologic dysfunction triggered by febrile illness, physical exertion or high‐altitude exposure. Episodic symptoms can include hemiparesis, paraparesis or quadriparesis, which are often misdiagnosed as GBS; demyelinating features on electrophysiologic testing can lead to misdiagnosis 83 . Transient CNS dysfunction in CMT 1X can also manifest as ataxia, dysarthria or altered level of consciousness and may be associated with ADEM‐like white matter abnormalities on brain imaging, which can persist 82 .
Nutritional Syndromes
The principal nutritional disorder to consider in the differential diagnosis of GBS + CNS is thiamine deficiency, producing not only thiamine neuropathy, but also sometimes Wernicke’s encephalopathy, or the more irreversible Korsakoff syndrome. 84 The classic Wernicke’s triad of confusion, ataxia and ophthalmoplegia can bear striking resemblance to anti‐Gq1b spectrum disorders. Thiamine neuropathy generally produces a symmetric, length‐dependent axonal sensorimotor neuropathy which begins in the lower limbs and progresses centripetally with time, in contrast to GBS which tends to begin with combined proximal and distal weakness. 84 Dysphagia, vocal cord dysfunction, dependent edema, and elevations in serum lactate are other possible clues to the diagnosis. 85
Thiamine deficiency may result from insufficient dietary consumption (chronic alcoholism, anorexia nervosa, low thiamine diets, hyperemesis), impaired intestinal uptake (gut failure, surgical resection or diversion procedures and bariatric surgery) or increased metabolic demand e.g., pregnancy and lactation. 84 , 86 Approximately 2–4 weeks is required for bodily stores to be depleted and clinical manifestations to arise, though this may be shorter under conditions of physiologic stress e.g., pregnancy. A glucose load in the setting of relative or absolute thiamine deficiency may also precipitate the syndrome. 87 Though symmetric MRI signal alterations in the thalami, mammillary bodies and periaqueductal regions may be a tip‐off, empiric treatment should be started upon any clinical suspicion, prior to confirmatory testing. 88
Drugs and Toxins
A myriad of drugs and toxins can produce rapidly progressive paralysis accompanied by central nervous system dysfunction (Table 5). Patients can be exposed to these through inadvertent environmental exposure, self‐harm attempts, criminal poisoning, or as a known side‐effect of prescribed medications. Most toxic neuropathies produce a length‐dependent sensorimotor axonal neuropathy, 89 and such a finding on neurophysiologic testing should always lead one to consider this diagnosis.
Certain toxindromes, particularly arsenic and thallium intoxication, characteristically produce a GBS + CNS syndrome. Early symptoms are relatively nonspecific, frequently leading to delays in diagnosis. Indeed it can take 1‐2 weeks for the complete clinical picture to emerge, by which stage valuable time for therapeutic intervention has been lost.
Arsenic
Acute arsenic poisoning generally results from intentional oral administration, often of rodenticides, as suicide or murder attempts. 89 Initial symptoms are generally gastrointestinal in nature, including nausea, vomiting, colicky abdominal pain, and characteristically voluminous ‘bloody rice water’ diarrhea. 90 Vasodilatation, capillary leak and intestinal fluid losses may lead to vasogenic shock. Cardiac arrhythmias and heart failure may develop. 91 Neuropathy generally appears 7–14 days after ingestion. It begins with distal burning dysesthesia, followed by length‐dependent large and small fiber sensory loss, producing ataxia and weakness in a distal‐to‐proximal fashion, which may mimic GBS. 92 Paralysis may be so severe as to require mechanical ventilation. 93 Important clinical clues include the presence of white transverse lines on the nails (Mee’s lines), palmo‐plantar keratosis, hyperpigmentation and desquamation. 90 Acute psychosis and encephalopathy are also described as part of the toxindrome. 90 , 91 , 94 , 95
Thallium
Thallium poisoning, generally encountered following intentional criminal administration, follows a similar pattern to arsenic. Oral ingestion rapidly engenders severe gastrointestinal distress (nausea, vomiting, abdominal pain) followed 2–5 days later by ascending sensory symptoms. 96 , 97 Initial severe dysesthesia of the palms and soles are followed by ascending weakness, hours to days after ingestion. 98 Autonomic involvement is common and cardiovascular compromise may ensue. 98 Central nervous system involvement comprising psychiatric symptoms, movement disorders and encephalopathy are common. 98 Optic neuropathies are also frequently described. 96 , 97 As in arsenic poisoning, patients can develop Mee’s lines. Dermatitis and stomatitis of the variety encountered in pellagra is often seen, as thallium interferes with riboflavin metabolism. Alopecia, beginning two weeks after ingestion, is characteristic and an important clinical clue. 96
Glycol ingestion
Both ethylene and diethylene glycol are found in a number household products including antifreeze, windshield washer fluid and brake fluid. Ingestion produces dose‐dependent toxindromes which develop in phases. 99 The first phase, beginning within hours, is one of inebriation with ataxia, stupor and occasionally seizures. This is followed by a second phase with renal failure, gap metabolic acidosis and cardiopulmonary compromise. 100 Finally, often in a delayed fashion 1–2 weeks after ingestion, patients develop flaccid areflexic limb and cranial nerve weakness (often an inflammatory demyelinating polyradiculoneuropathy) possibly as a reaction to deposition of toxic metabolites. 99 , 100 , 101 Bilateral facial nerve involvement is characteristic, bulbar weakness is common and CSF protein is characteristically elevated; CNS signs, including parkinsonism, may be present. 99 The syndrome is generally only partially reversible. 100 , 101
Environmental toxins
Environmental toxins can also be responsible for GBS + CNS syndromes. One important example is ciguatera poisoning. 102 This condition, mainly encountered in the South Pacific, is caused by ingestion of reef fish contaminated by ciguatoxin. 102 The resulting toxindrome is quite specific, producing severe painful paraesthesia which spread centrifugally, as well as characteristic reversal of thermal sensations, whereby hot objects feel cold, and cold objects burning hot. Hot showers and micturition can be particularly distressing. Weakness, either due to direct involvement of motor nerves or polymyositis, and frequent autonomic involvement may lead to an erroneous diagnosis of GBS. 103 Severe cases may be complicated by delirium, confusion and psychosis.
Iatrogenic causes
Drug‐induced peripheral neuropathies account for < 5% of all neuropathies, apart from in the chemotherapy‐treated population, where the rate may be over 50%. 105 These syndromes generally evolve over weeks to months, and are usually sensory‐predominant with little motor involvement. 105 Mis‐interpretation as GBS is therefore rare.
One notable exception is that of the newer biologic agents, particularly TNF‐alpha antagonists (infliximab, etanercept, adalimumab), which are frequently employed in the treatment of systemic inflammatory/autoimmune disorders. Administration of these agents has been reported to be associated with GBS and its variants. 105 , 106 Interferons have also infrequently been associated with GBS‐like immune neuropathies. 105 CNS involvement in these scenarios could be the result of the primary disease process, opportunistic infection, or concomitant drug‐related CNS demyelination. 107
Conclusion
The above narrative review details a systematic, step‐by‐step approach to clinical cases of apparent GBS complicated by mental status abnormalities, a syndrome which in our opinion is under‐appreciated in the medical literature. Our discussion is not intended as a systematic review of all possible causes, but rather as a logical framework within which to evaluate such patients. While GBS itself can occasionally produce mental status alterations, this should remain a diagnosis of exclusion. Significant depression in level of consciousness, seizures, movement disorders as well as immunosuppression and at‐risk travel histories should prompt exhaustive searches for alternative explanations. Moreover, the medical interview, combined with a high degree of suspicion is often key to identifying various toxindromes, the full clinical pictures of which may take weeks to develop. Many GBS + CNS disorders are treatable, with patient outcomes being critically dependent on early recognition. A structured approach such as that presented here is therefore paramount.
Author Contributions
1) conception and design of the study, 2) acquisition and analysis of data. 3) drafting a significant portion of the manuscript or figures. Eoin Mulroy 1, 2, 3. Neil E Anderson 1, 2, 3
Conflicts of interest
Nothing to report.
Acknowledgments
Eoin Mulroy is supported by the Edmond J Safra Philanthropic Foundation and the National Institute for Health Research University College London Hospitals Biomedical Research Centre.
Funding InformationEoin Mulroy is supported by the Edmond J Safra Philanthropic Foundation and the National Institute for Health Research University College London Hospitals Biomedical Research Centre.
References
- 1. Hughes RAC, Cornblath DR. Guillain‐Barré syndrome. Lancet 2005;366:1653–1666. [DOI] [PubMed] [Google Scholar]
- 2. Willison HJ, Jacobs BC, van Doorn PA. Guillain‐Barré syndrome. Lancet 2016;388:717–727. [DOI] [PubMed] [Google Scholar]
- 3. Fokke C, van den Berg B, Drenthen J, et al. Diagnosis of Guillain‐Barre syndrome and validation of Brighton criteria. Brain 2014;137:33–43. [DOI] [PubMed] [Google Scholar]
- 4. Sangroula D, Durrance R, Bhattarai S, Nandakumar T. Neuropsychiatric debut as a presentation of Guillain‐Barré Syndrome: an atypical clinical case and literature review. J Clin Neurosci 2017;44:245–249. [DOI] [PubMed] [Google Scholar]
- 5. Adamovic T, Riou ÉM, Bernard G, et al. Acute combined central and peripheral nervous system demyelination in children. Pediatr Neurol 2008;39:307–316. [DOI] [PubMed] [Google Scholar]
- 6. Amit R, Glick B, Itzchak Y, et al. Acute severe combined demyelination. Child’s Nerv Syst 1992;8:354–356. [DOI] [PubMed] [Google Scholar]
- 7. Amit R, Shapira Y, Blank A, Aker M. Acute, severe, central and peripheral nervous system combined demyelination. Pediatr Neurol 1986;2:47–50. [DOI] [PubMed] [Google Scholar]
- 8. Cochen V, Arnulf I, Demeret S, et al. Vivid dreams, hallucinations, psychosis and REM sleep in Guillain‐Barré syndrome. Brain 2005;128:2535–2545. [DOI] [PubMed] [Google Scholar]
- 9. Posner JB. Hyponatremia in acute polyneuropathy. Arch Neurol 1967;17:530. [DOI] [PubMed] [Google Scholar]
- 10. Park SJ, Pai KS, Kim JH, Shin JI. The role of interleukin 6 in the pathogenesis of hyponatremia associated with Guillain‐Barré syndrome. Nefrologia 2012;32:114. [DOI] [PubMed] [Google Scholar]
- 11. Penney MD, Murphy D, Walters G. Resetting of osmoreceptor response as cause of hyponatraemia in acute idiopathic polyneuritis. BMJ 1979;2:1474–1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Saifudheen K, Jose J, Gafoor VA, Musthafa M. Guillain‐Barre syndrome and SIADH. Neurology 2011;76:701–704. [DOI] [PubMed] [Google Scholar]
- 13. Lawn N, Wijdicks EFM, Burritt MF. Intravenous Immune Globulin and Pseudohyponatremia. N Engl J Med 1998;339:632–632. [DOI] [PubMed] [Google Scholar]
- 14. Lippi G, Aloe R. Hyponatremia and pseudohyponatremia: First, Do No Harm. Am J Med 2010;123:e17. [DOI] [PubMed] [Google Scholar]
- 15. Buffington MA, Abreo K. Hyponatremia. J Intensive Care Med. 2016;31:223–236. [DOI] [PubMed] [Google Scholar]
- 16. Zaeem Z, Siddiqi ZA, Zochodne DW. Autonomic involvement in Guillain‐Barré syndrome: an update. Clin Auton Res 2019;29:289–299. [DOI] [PubMed] [Google Scholar]
- 17. Zochodne DW. Autonomic involvement in Guillain‐Barre syndrome: A review. Muscle Nerve 1994;17:1145–1155. [DOI] [PubMed] [Google Scholar]
- 18. Hinduja A. Posterior reversible encephalopathy syndrome: clinical features and outcome. Front Neurol 2020;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chen A, Kim J, Henderson G, Berkowitz A. Posterior reversible encephalopathy syndrome in Guillain‐Barré syndrome. J Clin Neurosci 2015;22:914–916. [DOI] [PubMed] [Google Scholar]
- 20. Hughes RAC, Swan AV, van Doorn PA. Intravenous immunoglobulin for Guillain‐Barré syndrome. Cochrane Database Syst Rev 2014;2014:CD002063. [DOI] [PubMed] [Google Scholar]
- 21. Guo Y, Tian X, Wang X, Xiao Z. Adverse effects of immunoglobulin therapy. Front Immunol 2018;9:1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Moris G, Garcia‐Monco JC. The challenge of drug‐induced aseptic meningitis. Arch Intern Med 1999;159:1185. [DOI] [PubMed] [Google Scholar]
- 23. Bharath V, Eckert K, Kang M, et al. Incidence and natural history of intravenous immunoglobulin‐induced aseptic meningitis: a retrospective review at a single tertiary care center. Transfusion 2015;55:2597–2605. [DOI] [PubMed] [Google Scholar]
- 24. Shahrizaila N, Yuki N. Bickerstaff brainstem encephalitis and Fisher syndrome: anti‐GQ1b antibody syndrome. J Neurol Neurosurg Psychiatry 2013;84:576–583. [DOI] [PubMed] [Google Scholar]
- 25. Bickerstaff ER. Brain‐stem encephalitis. BMJ 1957;1:1384–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Al‐Din AN, Anderson M, Bickerstaff ER, Harvey I. Brainstem encephalitis and the syndrome of miller fisher a clinical study. Brain 1982;105:481–495. [DOI] [PubMed] [Google Scholar]
- 27. Odaka M, Yuki N, Yamada M, et al. Bickerstaff’s brainstem encephalitis: clinical features of 62 cases and a subgroup associated with Guillain‐Barré syndrome. Brain 2003;126:2279–2290. [DOI] [PubMed] [Google Scholar]
- 28. de Bruyn A, Poesen K, Bossuyt X, et al. Clinical spectrum of the anti‐GQ1b antibody syndrome: a case series of eight patients. Acta Neurol Belg 2019;119:29–36. [DOI] [PubMed] [Google Scholar]
- 29. Stein PE, Badminton MN, Rees DC. Update review of the acute porphyrias. Br J Haematol 2017;176:527–538. [DOI] [PubMed] [Google Scholar]
- 30. Elder G, Harper P, Badminton M, et al. The incidence of inherited porphyrias in Europe. J Inherit Metab Dis 2013;36:849–857. [DOI] [PubMed] [Google Scholar]
- 31. Albers JW, Fink JK. Porphyric neuropathy. Muscle Nerve 2004;30:410–422. [DOI] [PubMed] [Google Scholar]
- 32. Ridley A. The neuropathy of acute intermittent porphyria. QJM An Int J Med. 1969;38(151:307–33. [PubMed] [Google Scholar]
- 33. Puy H, Gouya L, Deybach J‐C. Porphyrias. Lancet 2010;375:924–937. [DOI] [PubMed] [Google Scholar]
- 34. Nayar G, Ejikeme T, Chongsathidkiet P, et al. Leptomeningeal disease: current diagnostic and therapeutic strategies. Oncotarget 2017;8:73312–73328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wang N, Bertalan MS, Brastianos PK. Leptomeningeal metastasis from systemic cancer: review and update on management. Cancer 2018;124:21–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Giometto B. Paraneoplastic neurologic syndrome in the PNS euronetwork database. Arch Neurol 2010;67:330. [DOI] [PubMed] [Google Scholar]
- 37. Graus F. Anti‐Hu‐associated paraneoplastic encephalomyelitis: analysis of 200 patients. Brain 2001. [DOI] [PubMed] [Google Scholar]
- 38. Koike H, Sobue G. Paraneoplastic neuropathy. Handb Clin Neurol 2013;713–726. [DOI] [PubMed] [Google Scholar]
- 39. Kidd D, Williams AJ, Howard RS. Poliomyelitis. Postgrad Med J 1996;72:641–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lee KY. Enterovirus 71 infection and neurological complications. Korean J Pediatr 2016;59:395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Cassidy H, Poelman R, Knoester M, et al. Enterovirus D68 – The New Polio? Front. Microbiol. 2018;13:2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Knoester M, Helfferich J, Poelman R, et al. Twenty‐nine Cases of Enterovirus‐D68–associated acute flaccid myelitis in Europe 2016. Pediatr Infect Dis J 2019;38:16–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sips GJ, Wilschut J, Smit JM. Neuroinvasive flavivirus infections. Rev Med Virol 2012;22:69–87. [DOI] [PubMed] [Google Scholar]
- 44. Griffiths MJ, Turtle L, Solomon T. Japanese encephalitis virus infection. Handb Clin Neurol 2014;561–576. [DOI] [PubMed] [Google Scholar]
- 45. Solomon T, Kneen R, Dung NM, et al. Poliomyelitis‐like illness due to Japanese encephalitis virus. Lancet 1998;351:1094–1097. [DOI] [PubMed] [Google Scholar]
- 46. Jeha LE, Sila CA, Lederman RJ, et al. West Nile virus infection: a new acute paralytic illness. Neurology 2003;61:55–59. [DOI] [PubMed] [Google Scholar]
- 47. Gould E, Solomon T. Pathogenic flaviviruses. Lancet 2008;371:500–509. [DOI] [PubMed] [Google Scholar]
- 48. Solomon T, Fisher AF, Beasley DWC, et al. Natural and nosocomial infection in a patient with west nile encephalitis and extrapyramidal movement disorders. Clin Infect Dis 2003;36:e140–e145. [DOI] [PubMed] [Google Scholar]
- 49. Hehir MK, Logigian EL. Infectious neuropathies. Contin Lifelong Learn Neurol 2014;20:1274–1292. [DOI] [PubMed] [Google Scholar]
- 50. Sejvar J. Clinical manifestations and outcomes of west nile virus infection. Viruses 2014;6:606–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Bender A. Severe tick borne encephalitis with simultaneous brain stem, bithalamic, and spinal cord involvement documented by MRI. J Neurol Neurosurg Psychiatry 2005;76:135–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Steffen R. Epidemiology of tick‐borne encephalitis (TBE) in international travellers to Western/Central Europe and conclusions on vaccination recommendations. J. Travel Med 2016;23(4):taw018. [DOI] [PubMed] [Google Scholar]
- 53. Griggs AC, Fischer M, Hills SL. Japanese encephalitis in travelers from non‐endemic countries, 1973–2008. Am J Trop Med Hyg 2010;82:930–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Buhl MR, Lindquist L. Japanese encephalitis in travelers: review of cases and seasonal risk. J Travel Med 2009;16:217–219. [DOI] [PubMed] [Google Scholar]
- 55. Gadre G, Satishchandra P, Mahadevan A, et al. Rabies viral encephalitis: clinical determinants in diagnosis with special reference to paralytic form. J Neurol Neurosurg Psychiatry 2010;81:812–820. [DOI] [PubMed] [Google Scholar]
- 56. Mitrabhakdi E, Shuangshoti S, Wannakrairot P, et al. Difference in neuropathogenetic mechanisms in human furious and paralytic rabies. J Neurol Sci 2005;238:3–10. [DOI] [PubMed] [Google Scholar]
- 57. Madhusudana S, Sukumaran S. Antemortem diagnosis and prevention of human rabies. Ann Indian Acad Neurol 2008;11:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Hiraga A, Kuwabara S, Nakamura A, et al. Fisher/Gullain–Barré overlap syndrome in advanced AIDS. J Neurol Sci 2007;258:148–150. [DOI] [PubMed] [Google Scholar]
- 59. Piliero PJ, Fish DG, Preston S, et al. Guillain‐barre syndrome associated with immune reconstitution. Clin Infect Dis 2003;36:e111–e114. [DOI] [PubMed] [Google Scholar]
- 60. Brannagan TH, Zhou Y. HIV‐associated Guillain‐Barré syndrome. J Neurol Sci 2003;208:39–42. [DOI] [PubMed] [Google Scholar]
- 61. Manji H. The neurology of HIV infection. J Neurol Neurosurg Psychiatry 2004;75:29i–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Anders H‐J, Goebel FD. Neurological manifestations of cytomegalovirus infection in the acquired immunodeficiency syndrome. Int J STD AIDS 1999;10:151–161. [DOI] [PubMed] [Google Scholar]
- 63. Steiner I, Kennedy PG, Pachner AR. The neurotropic herpes viruses: herpes simplex and varicella‐zoster. Lancet Neurol 2007;6:1015–1028. [DOI] [PubMed] [Google Scholar]
- 64. Nagel MA, Gilden D. Neurological complications of varicella zoster virus reactivation. Curr Opin Neurol 2014;27:356–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Wakerley BR, Yuki N. Infectious and noninfectious triggers in Guillain‐Barré syndrome. Expert Rev Clin Immunol 2013;9:627–639. [DOI] [PubMed] [Google Scholar]
- 66. Oschmann P, Dorndorf W, Hornig C, et al. Stages and syndromes of neuroborreliosis. J Neurol 1998;245:262–272. [DOI] [PubMed] [Google Scholar]
- 67. Hess A, Buchmann J, Zettl UK, et al. Borrelia burgdorferi central nervous system infection presenting as an organic schizophrenialike disorder. Biol Psychiatry 1999;45:795. [DOI] [PubMed] [Google Scholar]
- 68. Ju IN, Lee JW, Cho SY, et al. Two cases of scrub typhus presenting with Guillain‐Barré syndrome with respiratory failure. Korean J Intern Med 2011;26:474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Corral I, Quereda C, Casado J‐L, et al. Acute polyradiculopathies in HIV‐infected patients. J Neurol 1997;244:499–504. [DOI] [PubMed] [Google Scholar]
- 70. Kissel JT, Slivka AP, Warmolts JR, Mendell JR. The clinical spectrum of necrotizing angiopathy of the peripheral nervous system. Ann. Neurol. 1985;18(2):251–257. [DOI] [PubMed] [Google Scholar]
- 71. Restrepo JF, Rondón F, Matteson EL, et al. Necrotizing Lymphocytic Vasculitis limited to the peripheral nerves: Report of six cases and review. Int J Rheumatol 2009;2009:1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Suggs SP, Thomas TD, Joy JL, et al. Vasculitic neuropathy mimicking Guillain‐barré syndrome. Arthritis Rheum 1992;35:975–978. [DOI] [PubMed] [Google Scholar]
- 73. Ng KK, Yeung HM, Loo KT, et al. Acute fulminant neuropathy in a patient with Churg‐Strauss syndrome. Postgrad Med J 1997;73:236–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. De Toni FL, Amadio S, Scarlato M, et al. A fatal case of Churg‐Strauss syndrome presenting with acute polyneuropathy mimicking Guillain‐Barré syndrome. Neurol Sci 2011;32:937–940. [DOI] [PubMed] [Google Scholar]
- 75. Li X, Wang Y. Systemic lupus erythematosus with acute inflammatory demyelinating polyneuropathy: a case report and review of the literature. J Clin Med Res 2016;8:555–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Graus F, Titulaer MJ, Balu R, et al. A clinical approach to diagnosis of autoimmune encephalitis. Lancet Neurol 2016;15:391–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Cellucci T, Van Mater H, Graus F, et al. Clinical approach to the diagnosis of autoimmune encephalitis in the pediatric patient. Neurol Neuroimmunol Neuroinflammation 2020;7:e663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Wei Y‐C, Huang C‐C, Liu C‐H, et al. Peripheral neuropathy in limbic encephalitis with anti‐glutamate receptor antibodies: case report and systematic literature review. Brain Behav 2017;7:e00779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Prüss H, Hoffmann C, Stenzel W, et al. A case of inflammatory peripheral nerve destruction antedating anti‐NMDA receptor encephalitis. Neurol Neuroimmunol Neuroinflammation 2014;1:e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Coggeshall RE, Carlton SM. Ultrastructural analysis of NMDA, AMPA, and kainate receptors on unmyelinated and myelinated axons in the periphery. J Comp Neurol 1998;391:78–86. [DOI] [PubMed] [Google Scholar]
- 81. Tüzün E, Kinay D, Hacohen Y, et al. Guillain‐Barré‐like syndrome associated with lung adenocarcinoma and CASPR2 antibodies. Muscle Nerve 2013;48:836–837. [DOI] [PubMed] [Google Scholar]
- 82. Abrams C. GJB1 Disorders: Charcot Marie Tooth Neuropathy (CMT1X) and Central Nervous System Phenotypes. Adam MP, Ardinger HH, Pagon RA, al., Ed. GeneReviews® [Internet]. Seattle Univ. Washington, Seattle; 1993–2020. Available from https://www.ncbi.nlm.nih.gov/books/NBK1374/ [date unknown]. [PubMed]
- 83. Parissis D, Ioannidis P, Papadopoulos G, Karacostas D. Charcot‐marie‐tooth disease 1X simulating paraparetic Guillain‐Barre syndrome. Neurologist 2017;22:234–236. [DOI] [PubMed] [Google Scholar]
- 84. Koike H, Ito S, Morozumi S, et al. Rapidly developing weakness mimicking Guillain‐Barré syndrome in beriberi neuropathy: two case reports. Nutrition 2008;24:776–780. [DOI] [PubMed] [Google Scholar]
- 85. Saini M, Lin W, Kang C, Umapathi T. Acute flaccid paralysis: do not forget beriberi neuropathy. J Peripher Nerv Syst 2019;24:145–149. [DOI] [PubMed] [Google Scholar]
- 86. Chandrakumar A, Bhardwaj A, ‘t Jong GW. Review of thiamine deficiency disorders: Wernicke encephalopathy and Korsakoff psychosis. J Basic Clin Physiol Pharmacol 2019;30:153–162. [DOI] [PubMed] [Google Scholar]
- 87. Schabelman E, Kuo D. Glucose before thiamine for Wernicke encephalopathy: A literature review. J Emerg Med 2012. [DOI] [PubMed] [Google Scholar]
- 88. Zuccoli G, Santa Cruz D, Bertolini M, et al. MR imaging findings in 56 patients with wernicke encephalopathy: nonalcoholics may differ from alcoholics. Am J Neuroradiol 2009;30:171–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Misra U, Kalita J. Toxic neuropathies. Neurol. India 2009;57:697. [DOI] [PubMed] [Google Scholar]
- 90. Ratnaike RN. Acute and chronic arsenic toxicity. Postgrad Med J 2003;79:391–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Saha JC, Dikshit AK, Bandyopadhyay M, Saha KC. A review of arsenic poisoning and its effects on human health. Crit Rev Environ Sci Technol 1999;29:281–313. [Google Scholar]
- 92. Goddard MJ, Tanhehco JL, Dau PC. Chronic arsenic poisoning masquerading as Landry‐Guillain‐Barre syndrome. Electromyogr Clin Neurophysiol 1992;32(9):419–423. [PubMed] [Google Scholar]
- 93. Greenberg C, Davies S, McGowan T, et al. Acute respiratory failure following severe arsenic poisoning. Chest 1979;76:596–598. [DOI] [PubMed] [Google Scholar]
- 94. Morton WE, Caron GA. Encephalopathy: an uncommon manifestation of workplace arsenic poisoning? Am J Ind Med 1989;15:1–5. [DOI] [PubMed] [Google Scholar]
- 95. Freeman JW, Couch JR. Prolonged encephalopathy with arsenic poisoning. Neurology 1978;28:853–853. [DOI] [PubMed] [Google Scholar]
- 96. Moore D, House I, Dixon A. Thallium poisoning. Diagnosis may be elusive but alopecia is the clue. BMJ 1993;306:1527–1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Bank WJ, Pleasure DE, Suzuki K, et al. Thallium poisoning. Arch Neurol 1972;26:456–464. [DOI] [PubMed] [Google Scholar]
- 98. Galván‐Arzate S, Santamaría A. Thallium toxicity. Toxicol. Lett. 1998;99(1):1–13. [DOI] [PubMed] [Google Scholar]
- 99. Reddy NJ, Sudini M, Lewis LD. Delayed neurological sequelae from ethylene glycol, diethylene glycol and methanol poisonings. Clin Toxicol 2010;48:967–973. [DOI] [PubMed] [Google Scholar]
- 100. Daubert GP, Katiyar A, Wilson J, et al. Encephalopathy and peripheral neuropathy following diethylene glycol ingestion. Neurology 2006;66:782–783. [DOI] [PubMed] [Google Scholar]
- 101. Hasbani MJ, Sansing LH, Perrone J, et al. Encephalopathy and peripheral neuropathy following diethylene glycol ingestion. Neurology 2005;64:1273–1275. [DOI] [PubMed] [Google Scholar]
- 102. Friedman M, Fleming L, Fernandez M, et al. Ciguatera fish poisoning: treatment, prevention and management. Mar Drugs 2008;6:456–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Gatti C, Oelher E, Legrand AM. Severe seafood poisoning in French Polynesia: A retrospective analysis of 129 medical files. Toxicon 2008;51:746–753. [DOI] [PubMed] [Google Scholar]
- 104. Quod JP, Turquet J. Ciguatera in Réunion Island (SW Indian Ocean): Epidemiology and clinical patterns. Toxicon 1996;34:779–785. [DOI] [PubMed] [Google Scholar]
- 105. Jones MR, Urits I, Wolf J, et al. Drug‐induced peripheral neuropathy: a narrative review. Curr Clin Pharmacol 2020;15:38–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Shin I‐SJ, Baer AN, Kwon HJ, et al. Guillain‐Barré and Miller Fisher syndromes occurring with tumor necrosis factor α antagonist therapy. Arthritis Rheum 2006;54:1429–1434. [DOI] [PubMed] [Google Scholar]
- 107. Kemanetzoglou E, Andreadou E. CNS Demyelination with TNF‐α Blockers. Curr Neurol Neurosci Rep 2017;17:36. [DOI] [PMC free article] [PubMed] [Google Scholar]