

Abstract
Background:
Posterior cortical atrophy (PCA) and logopenic progressive aphasia (LPA) are two of the most common variants of atypical Alzheimer’s disease (AD). Both PCA and LPA are associated with relative sparing of hippocampus compared to neocortex, although hippocampal atrophy is observed. It is unclear whether regional patterns of hippocampal subfield involvement differ between PCA and LPA, and whether they differ from typical AD.
Objective:
To assess volume of specific subfields of the hippocampus in PCA, LPA and typical AD.
Methods:
Fifty-nine patients with PCA and 77 patients with LPA were recruited and underwent T1-weighted MRI and Pittsburgh Compound B (PiB) PET at Mayo Clinic. Thirty-six probable AD patients and 100 controls were identified from the Alzheimer’s Disease Neuroimaging Initiative. Hippocampal subfield volumes were calculated using Freesurfer, and volumes were compared between PCA, LPA, AD and controls using Kruskal-Wallis and Dunn tests.
Results:
The LPA and PCA groups both showed the most striking abnormalities in CA4, presubiculum, molecular layer of the hippocampus, molecular and granule cell layers of the dentate gyrus and the hippocampal-amygdala transition area, although atrophy was left-sided in LPA. PCA showed smaller volume of right presubiculum compared to LPA, with trends for smaller volumes of right parasubiculum and fimbria. LPA showed a trend for smaller volumes of left CA1 compared to PCA. The AD group showed smaller volumes of the right subiculum, CA1 and presubiculum compared to LPA.
Conclusion:
Patterns of hippocampal subfield atrophy differ across the different syndromic variants of AD.
Keywords: Hippocampus, hippocampal subfields, Magnetic Resonance Imaging, Alzheimer’s disease, posterior cortical atrophy, logopenic progressive aphasia
INTRODUCTION
Alzheimer’s disease (AD) is a major and increasing global burden to the economy and quality of life, with 16-20 million people affected world-wide [1, 2]. Alzheimer’s disease can be classified clinically into typical and atypical AD [3]. The typical clinical pattern of AD starts with episodic memory loss which is related to hippocampal degeneration, which then progresses to affect other cognitive domains. In contrast, in atypical AD, episodic memory impairment is not the first symptom [4], with patients instead presenting with symptoms related to neocortical abnormalities. Two common forms of atypical AD are posterior cortical atrophy (PCA) and logopenic progressive aphasia (LPA). PCA develops when there is damage to the parietal and occipital lobes of the brain [5]. Its core features include visuospatial and perceptual deficits, as well as features of Gerstmann syndrome, Balint syndrome, alexia and apraxia [6, 7]. In contrast, LPA reflects abnormalities to left temporoparietal regions that are important to language, and is characterized by anomia, word retrieval problems, poor sentence repetition, phonological impairments, and working memory deficits [8–11].
The hippocampus plays an important role in learning and memory [12]. Although it is not severely affected early in atypical AD, it often becomes atrophic over time. For instance, a whole hippocampus volumetric analysis using voxel-based morphometry (VBM) found that there was a reduction in gray matter volume in the right hippocampus in PCA compared to controls [5]. Other VBM studies have shown reduced volume of the hippocampus in PCA [5, 13–16] and LPA [15] patients compared to controls. Less, however, is known about patterns of involvement of specific subfields of the hippocampus in PCA and LPA. Volumetric analysis of hippocampal subfields involves approximating the volume of each sub-region of the hippocampus, namely the cornu ammonis (CA1, CA2, CA3, CA4), dentate gyrus (DG) subiculum, presubiculum, fimbria and hippocampal fissure. The subfields CA2, CA3 and DG are input structures responsible for encoding, while CA1 and subiculum are output structures and play a role in retrieval [17, 18]. The subiculum plays a role in spatial information processing, memory and temporal control of behavior [19]. CA2 plays a role in cognition, especially in social memory and object recognition [20]. One study found that PCA was associated with reduced volume of the left presubiculum, right subiculum, right molecular and granule cell layers of the DG (GC-ML-DG), right molecular layer and the right hippocampal amygdala transition area (HATA) [21]. That study also found that PCA showed less involvement of CA1 compared to typical AD. Little is known about hippocampal subfield abnormalities in LPA, although one study did identify subfield abnormalities in a cohort of non-semantic primary progressive aphasia patients, with greater changes observed in the left hemisphere[22].
In this study, we aimed to investigate differences in volume of the hippocampal subfields between PCA and LPA, and compare both groups to typical AD. We hypothesized that PCA and LPA will show similar patterns of abnormalities in hippocampal subfields, although with more left-sided patterns of involvement in LPA, and that both groups will show different patterns of abnormalities compared to typical AD. A better understanding of hippocampal abnormalities will improve understanding of the neurobiology of atypical AD.
METHODS
Participants
Patients who fulfilled clinical criteria for PCA [7] and LPA [23] were recruited from the Mayo Clinic Department of Neurology, Rochester MN, by the Neurodegenerative Research Group (NRG) between 7th July 2010 and 28th February 2020. The diagnosis of PCA was based on clinical consensus criteria [7]. Briefly, patients were included if the chief complaint was a progressive visuospatial or perceptual problem, the visuospatial/perceptual deficits were corroborated on neuropsychological testing and the visuospatial/perceptual deficits were more severe than deficits in all other cognitive domains. Ophthalmologic examination must have been normal within 3 months of presentation. The diagnosis of LPA was based on clinical consensus criteria which requires impaired word retrieval, naming, and repetition of sentences and phrases, and at least three of either phonological errors, spared single-word comprehension and object knowledge, spared motor speech and absence of agrammatism [23]. Both LPA and PCA patients underwent neurological and neuropsychological evaluations, Pittsburgh Compound B (PiB) PET imaging and a volumetric head MRI. Patients were included in the study if they showed evidence of beta-amyloid positivity with a PiB global standardized uptake ratio (SUVR) of greater than 1.48 [24]. One hundred and forty one patients, 82 with LPA and 59 with PCA were included in the study. All patients gave informed consent to participate in the project and the project protocol was approved by Institutional Review Board (IRB) of Mayo Clinic.
For comparison with our patients, we identified patients diagnosed with probable AD and cognitively unimpaired controls from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) (www.loni.ucla.edu/ADNI). The ADNI was launched in 2003 as a public-private partnership, led by Principal Investigator Michael W. Weiner, MD. The primary goal of ADNI has been to test whether serial magnetic resonance imaging, positron emission tomography, other biological markers, and clinical and neuropsychological assessment can be combined to measure the progression of mild cognitive impairment and early AD. The AD patients in ADNI all met NINCDS/ADRDA criteria for probable AD[25]. We selected only individuals who had undergone a 3T MRI on a GE scanner (to match our cohort) and were aged between 60 and 75 years at the time of scan, which resulted in 37 AD patients and 100 cognitively unimpaired controls. We selected this age range in order to identify a relatively pure and representative typical AD group. We have previously shown that typical AD patients under age 60 have atypical patterns of atrophy and older patients have a large contribution from other pathologies [3]. The AD group was median (inter-quartile range) 71 (66, 73) years old at the time of scan, with 49% females and Mini-Mental State Examination scores of 24 (23, 25). The control group was 70 (67, 73) years old, with 61% female and Mini-Mental State Examination scores of 29 (29, 30).
Neurological and neuropsychological evaluations
All neurological assessments for the PCA and LPA patients were performed by one of two Behavioral Neurologists (KAJ, JGR). The neurological evaluation included the Montreal Cognitive Assessment (MoCA) [26] to assess general cognitive function, the Clinical Dementia Rating (CDR) Scale [27] to assess functional ability, the Boston Diagnostic Aphasia Examination (BDAE) repetition subtest [28] to assess sentence repetition, the brief questionnaire version of the Neuropsychiatric Inventory (NPI-Q) [29] to assess psychiatric and behavioral features, and the Movement Disorder Society-sponsored revision of the Unified Parkinson’s Disease Rating Scale (MDS-UPDRS) Part III [30] to assess Parkinsonism. All neuropsychological tests were administered by a trained psychometrist and overseen by a board-certified Neuropsychologist (MMM). These tests included the Visual Object and Space Perception (VOSP) Battery incomplete letters [31] test to assess visuoperceptual function, the Rey-Osterrieth (Rey-O) complex figure test [32] to assess visuoconstructional abilities, the Wechsler Memory Scale III (WMS-III) visual reproduction I/II [33] to assess visual memory, the Auditory Verbal Learning Test (AVLT) to assess verbal memory[34], and the Boston Naming Test (BNT) to assess confrontational word retrieval [35]. The Rey-O, WMS-III% retention and AVLT % recall scores were expressed as Mayo Older American Normative (MOANS) age-adjusted scale scores which are constructed to have a mean of 10 and a standard deviation of 3 among cognitively healthy people.
MRI acquisition
The LPA and PCA patients were scanned with a 3.0 Tesla MRI (GE) scanner and the protocol included a 3D magnetization prepared rapid acquisition gradient echo (MPRAGE). The MRI scanning parameters used are as follows: sagittal plane, TR/TE/TI = 2300/3/900 ms, pixel spacing of 1.0156 × 1.0156, flip angle of 8°, slice thickness = 1.2 mm, repetition time of 7.032 msec, in-plane matrix = 256 × 256 and field of view (FOV) = 26 cm. This sequence was developed to be consistent with the ADNI protocol (published on http://adni.loni.usc.edu/methods/documents/mri-protocols/). All images were corrected for gradient non-linearity and intensity inhomogeneity.
Hippocampal subfield volume analysis
Hippocampal subfields were segmented using FreeSurfer version 6.0 [36, 37]. Each scan undergoes the standard volumetric FreeSurfer pipeline using a probabilistic atlas of the hippocampal formation, and then hippocampal subfield segmentation is performed. Briefly, the hippocampal subfield segmentation uses a Bayesian modelling approach, which generates a model on the MRI image around the hippocampal area and this model is optimized using the Generalized Expectation Maximization (GEM) algorithm [38]. This optimized model is then used to obtain an optimal segmentation, which is given below:
where Ŝ is obtained by assigning each voxel to the label with the highest posterior probability and is the estimate of the model parameters for approximating anatomical labeling [37]. Each hippocampus was segmented into the following subfields: CA1, CA2, CA3, CA4, GC-ML-DG, subiculum, HATA, presubiculum, parasubiculum, fimbria, molecular layer of the hippocampus (HP), hippocampal tail and hippocampal fissure. Whole hippocampal volume was also calculated, as well as total intracranial volume (TIV) to allow a correction for head size. Hippocampal subfield segmentations and corresponding MR images were visually inspected for quality using Freesurfer’s Freeview tool. A total of eight cases were removed due to failed segmentations (five LPA, one AD, and two controls), resulting in final group sizes of 77 LPA, 59 PCA, 36 AD and 98 controls. Example hippocampal segmentations are shown in Figure 1.
Fig. 1.
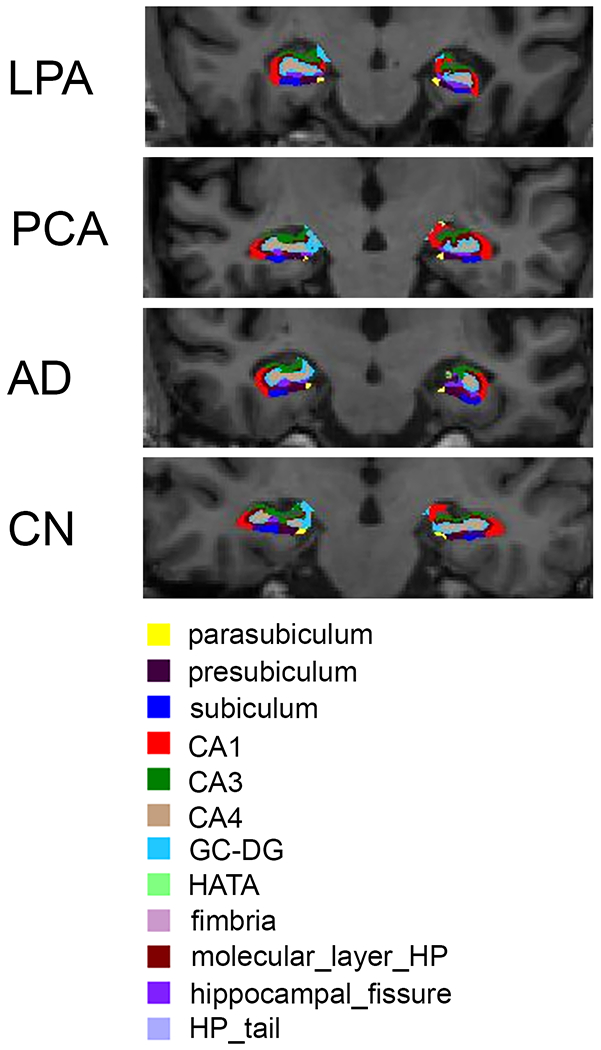
Representative examples of hippocampal subfield segmentations from a patient with PCA, LPA, AD, and a control. The left side of the brain is shown on the right of each image.
Statistical analyses
To allow comparison between patients with different TIV, we used a proportion method which expresses the brain volume (v) as a proportion of TIV. The normalized volume (ṽ) is computed as ṽ = v/TIV.
We compared continuous demographic and clinical features using Pearson’s Chi-square test for association on the categorical variables (gender and handedness) and unpaired t-tests for the remaining continuous clinical features. All p-values were reported after correction for false discovery rate (FDR) using the Benjamini-Hochberg method [39]. Whole hippocampal volume and subfield volumes were compared across all four groups (LPA, PCA, AD, controls) using the Kruskal Wallis test coupled with the Dunn test for paired comparisons. The p-values for the Dunn test were adjusted using the Benjamini-Hochberg FDR approach [39]. In addition, we computed Cohen d for paired group comparisons. Cohen d can be interpreted in terms of the amount of non-overlap of the clinical group in relation to the control group. Cohen’s d is large if 2.0 ≤ d ≤ 0.8; medium: 0.5 ≤ d < 0.8; small: 0.0 ≤ d < 0.5. Note that the larger the value of d, the larger the percentage of non-overlap. The Cohen d (version 0.8.0), Kruskal-Wallis (version 0.99.35) and Dunn test (version 1.3.5) were performed using R package (version 3.6.2).
RESULTS
Comparison of PCA with LPA using demographics and clinical characteristics
Demographic and clinical features are shown in Table 1. The LPA patients had significantly shorter diseases duration and were older than the PCA patients. Consistent with their clinical syndromes, LPA patients performed significantly worse than PCA on the BNT and the BDAE repetition, while PCA patients performed more poorly on the VOSP Letters and Rey-O Complex Figure Test. The PCA patients also performed more poorly on CDR and NPI-Q.
Table 1:
Demographic and clinical characteristics of patients with LPA and PCA
| Features | LPA (N = 77) | PCA (N = 59) | FDR p-values |
|---|---|---|---|
| Gender (female) | 46 (60%) | 36 (61%) | 0.88 |
| Handedness (right) | 68 (88%) | 54 (92%) | 0.68 |
| Age Onset (years) | 64 (57, 70) | 58 (54, 63) | 0.0002 |
| Age MRI (years) | 68 (61, 73) | 62 (58.5, 69.5) | 0.02 |
| Education | 16 (14, 16) | 16 (13, 18) | 0.66 |
| Disease duration (years) | 3.6 (2.7, 4.9) | 4.7 (3.9, 6.8) | 0.0004 |
| Global PiB SUVR | 2.4 (2.2, 2.6) | 2.5 (2.3, 2.6) | 0.29 |
| BNT | 9 (6.5, 12) | 12 (9, 14) | 0.02 |
| BDAE repetition | 6 (4, 8) | 8 (6, 10) | 0.02 |
| NPI-Q | 2 (1, 4) | 3.5 (2, 6) | 0.03 |
| CDR sum of boxes | 3 (1.5, 4.5) | 4 (2, 7.5) | 0.02 |
| WMS-III VR% retention MOANS | 8 (6, 10.5) | 7.5 (4, 9.8) | 0.29 |
| AVLT long term recall MOANS | 6 (4, 7) | 6 (4, 8) | 0.68 |
| VOSP letters | 19.5 (18, 20) | 11.5 (5.25, 16) | <0.0001 |
| MOCA | 16 (10, 19) | 18 (10, 20) | 0.62 |
| MSD-UPDRS III | 3 (1.375, 6) | 2 (0, 8) | 0.29 |
| Rey-O MOANS | 6 (2, 9.25) | 2 (2, 2) | 0.0002 |
Data are shown as Q1 (Q1, Q3) or N (%). Significant adjusted p-values are bolded. AVLT: Auditory Verbal Learning Test, Rey-O-MOANS: Rey Osterrieth Mayo Older American Normative Scale, MOCA: Montreal Cognitive Assessment Battery; CDR: Clinical Dementia Rating, VOSP: visual object and space perception, WMS-III VR% retention MOANS: Wechsler Memory Scale III Visual Reproduction MOANS, BNT: Boston Naming Test, BDAE: Boston Diagnostic Aphasia Examination, NPI-Q: Neuropsychiatric Inventory Brief Questionnaire, MSD-UPDRS III: Movement Disorder Society Unified Parkinson’s Disease Rating Scale Part III.
Comparison of whole and subfield Hippocampal volumes
The comparison of whole and subfield hippocampal volumes between groups is shown in Tables 2 and 3. All hippocampal subfields, except for the right hippocampal fissure, showed significant differences among LPA, PCA, AD and controls based on the Kruskal-Wallis test (Table 2). All remaining hippocampal subfields were smaller in LPA, PCA and AD compared to controls, except for the left hippocampal fissure which did not differ between AD and controls.
Table 2:
Hippocampal volume estimates for LPA, PCA, AD and controls
| Hippocampal subfields | TIV normalized hippocampal volumes | Kruskal-Wallis p-value | |||
|---|---|---|---|---|---|
| LPA | PCA | AD | Control | ||
| Left Hippocampal tail | 0.306 (0.064) | 0.313 (0.052) | 0.290 (0.044) | 0.368 (0.056) | <0.0001 |
| Left subiculum | 0.243 (0.047) | 0.242 (0.057) | 0.236 (0.040) | 0.293 (0.044) | <0.0001 |
| Left CA1 | 0.345 (0.060) | 0.369 (0.063) | 0.365 (0.068) | 0.428 (0.069) | <0.0001 |
| Left hippocampal fissure | 0.109 (0.022) | 0.107 (0.024) | 0.116 (0.024) | 0.121 (0.026) | 0.003 |
| Left presubiculum | 0.173 (0.030) | 0.177 (0.037) | 0.169 (0.037) | 0.211 (0.033) | <0.0001 |
| Left parasubiculum | 0.037 (0.009) | 0.039 (0.009) | 0.037 (0.012) | 0.044 (0.010) | <0.0001 |
| Left molecular layer HP | 0.301 (0.053) | 0.320 (0.054) | 0.313 (0.060) | 0.380 (0.055) | <0.0001 |
| Left GC.ML.DG | 0.157 (0.025) | 0.165 (0.030) | 0.166 (0.029) | 0.197 (0.030) | <0.0001 |
| Left CA3 | 0.112 (0.020) | 0.119 (0.023) | 0.118 (0.025) | 0.144 (0.025) | <0.0001 |
| Left CA4 | 0.139 (0.022) | 0.144 (0.029) | 0.140 (0.030) | 0.174 (0.024) | <0.0001 |
| Left fimbria | 0.038 (0.016) | 0.037 (0.017) | 0.035 (0.015) | 0.046 (0.015) | <0.0001 |
| Left HATA | 0.031 (0.007) | 0.034 (0.007) | 0.034 (0.008) | 0.041 (0.008) | <0.0001 |
| Left whole hippocampus | 1.875 (0.323) | 1.969 (0.350) | 1.936 (0.326) | 2.319 (0.306) | <0.0001 |
| Right hippocampal tail | 0.336 (0.058) | 0.342 (0.067) | 0.316 (0.060) | 0.382 (0.064) | <0.0001 |
| Right subiculum | 0.253 (0.044) | 0.249 (0.047) | 0.229 (0.047) | 0.294 (0.045) | <0.0001 |
| Right CA1 | 0.393 (0.071) | 0.375 (0.071) | 0.360 (0.061) | 0.448 (0.074) | <0.0001 |
| Right hippocampal fissure | 0.120 (0.024) | 0.121 (0.022) | 0.125 (0.028) | 0.126 (0.025) | 0.514 |
| Right presubiculum | 0.174 (0.033) | 0.160 (0.030) | 0.156 (0.029) | 0.198 (0.032) | <0.0001 |
| Right parasubiculum | 0.037 (0.010) | 0.034 (0.008) | 0.035 (0.010) | 0.042 (0.009) | <0.0001 |
| Right molecular layer HP | 0.336 (0.048) | 0.327 (0.059) | 0.311 (0.064) | 0.390 (0.052) | <0.0001 |
| Right GC.ML.DG | 0.182 (0.030) | 0.182 (0.034) | 0.172 (0.037) | 0.208 (0.032) | <0.0001 |
| Right CA3 | 0.136 (0.023) | 0.138 (0.025) | 0.129 (0.024) | 0.155 (0.028) | <0.0001 |
| Right CA4 | 0.160 (0.024) | 0.155 (0.027) | 0.149 (0.027) | 0.180 (0.026) | <0.0001 |
| Right fimbria | 0.037 (0.016) | 0.032 (0.014) | 0.031 (0.012) | 0.046 (0.015) | <0.0001 |
| Right HATA | 0.036 (0.006) | 0.034 (0.008) | 0.036 (0.007) | 0.044 (0.008) | <0.0001 |
| Right whole hippocampus | 2.068 (0.322) | 2.064 (0.319) | 1.926 (0.366) | 2.387 (0.325) | <0.0001 |
GC-ML-DG: Molecular and Granule Cell Layers of the Dentate, HATA: Hippocampal Amygdala Transition Area, PCA: Posterior Cortical Atrophy, LPA: Logopenic Progressive Aphasia, AD: probable Alzheimer’s Disease.
Table 3:
Pair-wise statistics for all hippocampal regions
| Hippocampal subfields | Cohen d and Dunn test Adjusted p-values | |||||
|---|---|---|---|---|---|---|
| LPA~Control | PCA~Control | AD~Control | LPA~PCA | LPA~AD | PCA~AD | |
| Left Hippocampal tail | −1.04 *** | −1.00 *** | −1.46 *** | 0.12 | 0.27 | 0.46† |
| Left subiculum | −1.10 *** | −1.03 *** | −1.34 *** | −0.02 | 0.16 | 0.12 |
| Left CA1 | −1.27 *** | −0.88 *** | −0.92 *** | 0.39† | −0.31 | 0.07 |
| Left hippocampal fissure | −0.50 * | −0.56 ** | −0.23 | −0.08 | −0.28 | −0.35 |
| Left presubiculum | −1.20 *** | −0.98 *** | −1.25 *** | 0.13 | 0.15 | 0.24 |
| Left parasubiculum | −0.69 *** | −0.48 * | −0.68 *** | 0.20 | 0.07 | 0.24 |
| Left molecular layer HP | −1.46 *** | −1.11 *** | −1.20 *** | 0.35 | −0.21 | 0.12 |
| Left GC.ML.DG | −1.46 *** | −1.10 *** | −1.06 *** | 0.29 | −0.35 | −0.04 |
| Left CA3 | −1.38 *** | −1.02 *** | −1.03 *** | 0.34 | −0.28 | 0.05 |
| Left CA4 | −1.49 *** | −1.18 *** | −1.30 *** | 0.17 | −0.04 | 0.11 |
| Left fimbria | −0.54 ** | −0.62 *** | −0.74 *** | −0.09 | 0.18 | 0.08 |
| Left HATA | −1.39 *** | −1.05 *** | −0.93 *** | 0.35 | −0.39† | −0.06 |
| Left whole hippocampus | −1.42 *** | −1.08 *** | −1.23 *** | 0.28 | −0.19 | 0.10 |
| Right hippocampal tail | −0.76 *** | −0.62 *** | −1.05 *** | 0.10 | 0.33 | 0.40 |
| Right subiculum | −0.91 *** | −0.98 *** | −1.43 *** | −0.10 | 0.55 * | 0.42 † |
| Right CA1 | −0.75 *** | −1.01 *** | −1.25 *** | −0.27 | 0.49 * | 0.22 |
| Right hippocampal fissure | −0.22 | −0.21 | −0.02 | 0.02 | −0.19 | −0.18 |
| Right presubiculum | −0.73 *** | −1.22 *** | −1.32 *** | −0.46 * | 0.57 * | 0.12 |
| Right parasubiculum | −0.45 ** | −0.94 *** | −0.70 ** | −0.41† | 0.21 | −0.19 |
| Right molecular layer HP | −1.07 *** | −1.14 *** | −1.41 *** | −0.17 | 0.46† | 0.25 |
| Right GC.ML.DG | −0.85 *** | −0.79 *** | −1.09 *** | 0.01 | 0.32 | 0.30 |
| Right CA3 | −0.74 *** | −0.64 *** | −0.97 *** | 0.08 | 0.29 | 0.36 |
| Right CA4 | −0.79 *** | −0.96 *** | −1.18 *** | −0.21 | 0.44† | 0.22 |
| Right fimbria | −0.58 *** | −0.99 *** | −1.05 *** | −0.37† | 0.41† | 0.04 |
| Right HATA | −1.11 *** | −1.26 *** | −1.14 *** | −0.28 | 0.12 | −0.16 |
| Right whole hippocampus | −0.99 *** | −1.00 *** | −1.37 | −0.01 | 0.42 | 0.41 |
GC-ML-DG: Molecular and Granule Cell Layers of the Dentate, HATA: Hippocampal Amygdala Transition Area, PCA: Posterior Cortical Atrophy, LPA: Logopenic Progressive Aphasia, AD: probable Alzheimer’s Disease.
Significant p values shown as
<0.05,
<0.01,
<0.001,
trend-level of p<0.10
For LPA, the regions with the largest Cohen d values (>1.2) in comparison to controls were left CA4, left molecular layer HP, left GC-ML-DG, left whole hippocampus, left HATA, left CA3, left CA1 and left presubiculum (Table 3). For PCA, the only regions with a Cohen d value larger than 1.2 were the right HATA and right presubiculum. For AD, the regions with the largest Cohen d values (>1.2) were left hippocampal tail, right subiculum, right molecular layer HP, right whole hippocampus, left subiculum, right presubiculum, left CA4, left presubiculum, right CA1 and left whole hippocampus.
The PCA group showed smaller volume of the right presubiculum compared to LPA. There were also trends (corrected Dunns p values <0.1) for PCA to have smaller volumes of right parasubiculum, and fimbria compared to LPA, and for LPA to show smaller volumes of left CA1 compared to PCA (Table 3, Figure 2). The AD group showed smaller volumes of the right subiculum, CA1 and presubiculum compared to LPA (Table 3, Figure 2). There were also trends for AD to have smaller volumes of the right molecular layer HP, CA4, and fimbria compared to LPA, and LPA to have smaller volume of the left HATA compared to AD (Table 3, Figure 2). There were trends for AD to have smaller volumes of the right subiculum and left hippocampal tail compared to PCA.
Fig. 2.
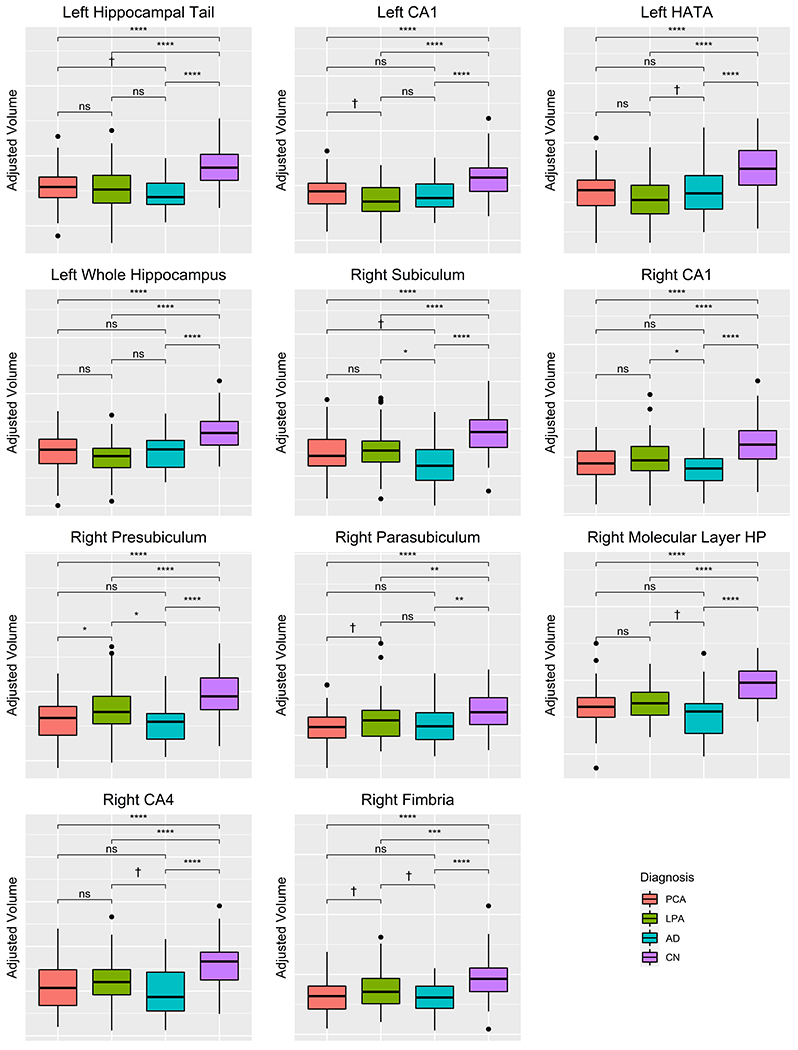
Box-plots across groups for hippocampal subfields that showed differences between LPA, PCA, and AD. Values are adjusted for TIV. *<0.05, **<0.01, ***<0.001, ****<0.0001, †trend-level of p < 0.10.
DISCUSSION
This study utilizing a large number of well characterized LPA and PCA patients demonstrates that both syndromes are associated with volume loss of the majority of the hippocampal subfields, however, LPA showed more asymmetric left-sided involvement and smaller volumes of left CA1 compared to PCA, and PCA showed greater involvement of the right presubiculum, parasubiculum and fimbria. Regional differences were also observed when comparing both variants to typical AD.
There was a large degree of overlap between LPA and PCA, with both syndromes showing volume loss across the hippocampal regions. The subfields that showed the most severe involvement in PCA were the right presubiculum and HATA, followed by right CA4, left and right molecular layer, left GC-ML-DG, left HATA and left subiculum. These regions are very similar to those identified in PCA in a previous study[21]. The CA4, presubiculum, molecular layer, GC-ML-DG and HATA were also amongst the most severely affected structures in LPA, although in LPA we observed a striking asymmetry with greatest involvement of left hemisphere regions. This is not unexpected given that LPA is a language disorder and commonly targets the left hemisphere. The PCA cohort showed a slight right-sided predominance, with the two most affected structures being on the right, but also showed severe involvement of many subfields in the left hemisphere.
Differences were observed between PCA and LPA in specific subfields. The PCA patients showed greater involvement of the right presubiculum, and trends for greater involvement of the right parasubiculum and fimbria compared to LPA. The pre and parasubiculum both receive inputs from the parietal lobe and evidence has suggested that they play a role in creating a visuospatial representation of the world, i.e. a scene, critical in both memory, navigation and spatial processing[40]. It is, therefore, possible that damage to the pre and parasubiculum in PCA could be contributing to the visuospatial deficits characteristic of this syndrome. The fimbria is a white matter tract that begins at the posterior end of the hippocampus and transitions into the fornix which is the main connecting tract of the limbic system [41]. The fimbria-fornix pathway plays a crucial role in spatial memory [42]. The posterior location of the fimbria may make it particularly vulnerable in PCA, since neurodegeneration is observed in posterior temporal and parietooccipital regions[5]. We also observed a trend for LPA to have a smaller volume of the left CA1 compared to PCA. This was somewhat unexpected given that the CA1 subfield plays a crucial role in contextual retrieval [43], incremental value learning [44] and episodic autobiographical memory [45], and is associated with typical amnestic AD[46–49]. Abnormalities in CA1 have previously been identified in patients with primary progressive aphasia and were related to visual memory performance[22]. While the LPA patients in our cohort performed comparably to the PCA patients in both visual and verbal memory performance, both tests can be confounded by non-memory language and visuoperceptual deficits, plus the PCA patients had a longer disease duration.
We also observed differences between both atypical AD variants and typical AD. The typical AD group showed relatively bilateral patterns of atrophy with the most striking loss observed in the hippocampal tail, subiculum, molecular layer, presubiculum, CA1 and CA4. Interestingly, the AD patients did not show increased volume of either hippocampal fissure, in line with a previous study[50]. We found that the right subiculum was smaller in AD compared to both LPA and PCA, suggesting that it may be preferentially affected in amnestic AD. The AD patients also showed smaller volumes of the right CA1 and presubiculum subfields compared to LPA. Previous studies have shown that both CA1 and the subiculum are strongly affected in amnestic AD[49, 51, 52], and one study found smaller CA1 volumes in typical AD compared to PCA[21]. We also found evidence that the left HATA was smaller in LPA compared to AD. In fact, the left HATA showed a larger difference (i.e. AUROC) between LPA and controls than between PCA and controls, suggesting a particular association with LPA. It is unclear whether the HATA contributes to the LPA phenotype, although the left anterior temporal lobe does indeed tend to be involved to a greater degree in LPA than AD and PCA and so it may reflect the anatomy of the disease[16, 53].
A strength of our study was the large number of LPA and PCA patients who all underwent identical MRI as well as amyloid determination with PiB-PET. A limitation, however, was that the PCA group had a longer disease duration at the time of scan and were younger than the LPA group which could have biased the results. The use of ADNI data allowed comparison to a large cohort of controls and to a cohort of probable AD patients. We ensured that the ADNI cases were also scanned on a GE, 3T scanner; however, noise could have been introduced into the analysis since ADNI cases are scanned in multiple sites. We only used T1-weighted MRI scans in order to define hippocampal subfields; a determination which could be improved by including complementary information from T2 weighted MR imaging.
In summary, this study demonstrates that these atypical variants of AD have overlapping neuroanatomical disruptions in the hippocampal subfields, although there are some regional differences which may relate to differences in clinical or neurodegenerative topography between LPA and PCA. There is also evidence that patterns of hippocampal subfield involvement in atypical AD differ somewhat from that observed in typical amnestic AD. Future research is needed to determine the histological correlates of these findings. Given these findings, it may be important to understand disease mechanisms behind neurogenesis in hippocampal subfields, perhaps utilizing techniques such as proteomics and metabolomics that may be important steps towards developing therapeutics.
ACKNOWLEDGMENTS
This study was funded by National Institute of Health (NIH) grants R01AG050603 and R01DC010367 and the Alzheimer’s Association. Data collection and sharing for this project was funded by the Alzheimer’s Disease Neuroimaging Initiative (ADNI) (National Institutes of Health Grant U01 AG024904) and DOD ADNI (Department of Defense award number W81XWH-12-2-0012). ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: AbbVie, Alzheimer’s Association; Alzheimer’s Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc.; Biogen; Bristol-Myers Squibb Company; CereSpir, Inc.; Cogstate; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; EuroImmun; F. Hoffmann-La Roche Ltd and its affiliated company Genentech, Inc.; Fujirebio; GE Healthcare; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC.; Johnson & Johnson Pharmaceutical Research & Development LLC.; Lumosity; Lundbeck; Merck & Co., Inc.; Meso Scale Diagnostics, LLC.; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Takeda Pharmaceutical Company; and Transition Therapeutics. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer’s Therapeutic Research Institute at the University of Southern California. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of Southern California.
Footnotes
CONFLICT OF INTEREST/DISCLOSURE STATEMENT
The authors declare no conflict of interest.
References
- [1].(2020)2020 Alzheimer’s disease facts and figures. Alzheimers Dement 16, 391–460. [DOI] [PubMed] [Google Scholar]
- [2].Prince MJ (2019) World Alzheimer report 2019: the global impact of dementia. [Google Scholar]
- [3].Josephs KA, Tosakulwong N, Graff-Radford J, Weigand SD, Buciuc M, Machulda MM, Jones DT, Schwarz CG, Senjem ML, Ertekin-Taner N, Kantarci K, Boeve BF, Knopman DS, Jack CR Jr., Petersen RC, Lowe VJ, Whitwell JL (2020) MRI and flortaucipir relationships in Alzheimer’s phenotypes are heterogeneous. Ann Clin Transl Neurol 7, 707–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Ghezzi L (2018) Diagnosis of Alzheimer’s Disease Typical and Atypical Forms In Neurodegenerative Diseases: Clinical Aspects, Molecular Genetics and Biomarkers, Galimberti D, Scarpini E, eds. Springer International Publishing, Cham, pp. 21–28. [Google Scholar]
- [5].Whitwell JL, Jack CR Jr., Kantarci K, Weigand SD, Boeve BF, Knopman DS, Drubach DA, Tang-Wai DF, Petersen RC, Josephs KA (2007) Imaging correlates of posterior cortical atrophy. Neurobiol Aging 28, 1051–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Crutch SJ, Lehmann M, Schott JM, Rabinovici GD, Rossor MN, Fox NC (2012) Posterior cortical atrophy. Lancet Neurol 11, 170–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Crutch SJ, Schott JM, Rabinovici GD, Murray M, Snowden JS, van der Flier WM, Dickerson BC, Vandenberghe R, Ahmed S, Bak TH, Boeve BF, Butler C, Cappa SF, Ceccaldi M, de Souza LC, Dubois B, Felician O, Galasko D, Graff-Radford J, Graff-Radford NR, Hof PR, Krolak-Salmon P, Lehmann M, Magnin E, Mendez MF, Nestor PJ, Onyike CU, Pelak VS, Pijnenburg Y, Primativo S, Rossor MN, Ryan NS, Scheltens P, Shakespeare TJ, Suarez Gonzalez A, Tang-Wai DF, Yong KXX, Carrillo M, Fox NC, Alzheimer’s Association IAAsD, Associated Syndromes Professional Interest A (2017) Consensus classification of posterior cortical atrophy. Alzheimers Dement 13, 870–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Gorno-Tempini ML, Dronkers NF, Rankin KP, Ogar JM, Phengrasamy L, Rosen HJ, Johnson JK, Weiner MW, Miller BL (2004) Cognition and anatomy in three variants of primary progressive aphasia. Ann Neurol 55, 335–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Montembeault M, Brambati SM, Gorno-Tempini ML, Migliaccio R (2018) Clinical, Anatomical, and Pathological Features in the Three Variants of Primary Progressive Aphasia: A Review. Front Neurol 9, 692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Marshall CR, Hardy CJD, Volkmer A, Russell LL, Bond RL, Fletcher PD, Clark CN, Mummery CJ, Schott JM, Rossor MN, Fox NC, Crutch SJ, Rohrer JD, Warren JD (2018) Primary progressive aphasia: a clinical approach. J Neurol 265, 1474–1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Botha H, Duffy JR, Whitwell JL, Strand EA, Machulda MM, Schwarz CG, Reid RI, Spychalla AJ, Senjem ML, Jones DT, Lowe V, Jack CR, Josephs KA (2015) Classification and clinicoradiologic features of primary progressive aphasia (PPA) and apraxia of speech. Cortex 69, 220–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Mu Y, Gage FH (2011) Adult hippocampal neurogenesis and its role in Alzheimer’s disease. Mol Neurodegener 6, 85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Firth NC, Primativo S, Marinescu R-V, Shakespeare TJ, Suarez-Gonzalez A, Lehmann M, Carton A, Ocal D, Pavisic I, Paterson RW, Slattery CF, Foulkes AJM, Ridha BH, Gil-Néciga E, Oxtoby NP, Young AL, Modat M, Cardoso MJ, Ourselin S, Ryan NS, Miller BL, Rabinovici GD, Warrington EK, Rossor MN, Fox NC, Warren JD, Alexander DC, Schott JM, Yong KXX, Crutch SJ (2019) Longitudinal neuroanatomical and cognitive progression of posterior cortical atrophy. Brain 142, 2082–2095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Manning EN, Macdonald KE, Leung KK, Young J, Pepple T, Lehmann M, Zuluaga MA, Cardoso MJ, Schott JM, Ourselin S, Crutch S, Fox NC, Barnes J (2015) Differential hippocampal shapes in posterior cortical atrophy patients: A comparison with control and typical AD subjects. Hum Brain Mapp 36, 5123–5136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Migliaccio R, Agosta F, Rascovsky K, Karydas A, Bonasera S, Rabinovici GD, Miller BL, Gorno-Tempini ML (2009) Clinical syndromes associated with posterior atrophy: early age at onset AD spectrum. Neurology 73, 1571–1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Tetzloff KA, Graff-Radford J, Martin PR, Tosakulwong N, Machulda MM, Duffy JR, Clark HM, Senjem ML, Schwarz CG, Spychalla AJ, Drubach DA, Jack CR, Lowe VJ, Josephs KA, Whitwell JL (2018) Regional Distribution, Asymmetry, and Clinical Correlates of Tau Uptake on [18F]AV-1451 PET in Atypical Alzheimer’s Disease. J Alzheimers Dis 62, 1713–1724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Eldridge LL, Engel SA, Zeineh MM, Bookheimer SY, Knowlton BJ (2005) A dissociation of encoding and retrieval processes in the human hippocampus. J Neurosci 25, 3280–3286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Zeineh MM, Engel SA, Thompson PM, Bookheimer SY (2003) Dynamics of the Hippocampus During Encoding and Retrieval of Face-Name Pairs. Science 299, 577–580. [DOI] [PubMed] [Google Scholar]
- [19].O’Mara SM, Sanchez-Vives MV, Brotons-Mas JR, O’Hare E (2009) Roles for the subiculum in spatial information processing, memory, motivation and the temporal control of behaviour. Prog Neuropsychopharmacol Biol Psychiatry 33, 782–790. [DOI] [PubMed] [Google Scholar]
- [20].Pang CC, Kiecker C, O’Brien JT, Noble W, Chang RC (2019) Ammon’s Horn 2 (CA2) of the Hippocampus: A Long-Known Region with a New Potential Role in Neurodegeneration. Neuroscientist 25, 167–180. [DOI] [PubMed] [Google Scholar]
- [21].Parker TD, Slattery CF, Yong KXX, Nicholas JM, Paterson RW, Foulkes AJM, Malone IB, Thomas DL, Cash DM, Crutch SJ, Fox NC, Schott JM (2019) Differences in hippocampal subfield volume are seen in phenotypic variants of early onset Alzheimer’s disease. Neuroimage Clin 21, 101632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Christensen A, Alpert K, Rogalski E, Cobia D, Rao J, Beg MF, Weintraub S, Mesulam MM, Wang L (2015) Hippocampal subfield surface deformity in non-semantic primary progressive aphasia. Alzheimers Dement 1, 14–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Gorno-Tempini ML, Brambati SM, Ginex V, Ogar J, Dronkers NF, Marcone A, Perani D, Garibotto V, Cappa SF, Miller BL (2008) The logopenic/phonological variant of primary progressive aphasia. Neurol 71, 1227–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Whitwell JL, Avula R, Master A, Vemuri P, Senjem ML, Jones DT, Jack CR, Josephs KA (2011) Disrupted thalamocortical connectivity in PSP: a resting state fMRI, DTI, and VBM study. Parkinsonism Relat Disord 17, 599–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].McKhann GM, Knopman DS, Chertkow H, Hyman BT, Jack CR Jr., Kawas CH, Klunk WE, Koroshetz WJ, Manly JJ, Mayeux R, Mohs RC, Morris JC, Rossor MN, Scheltens P, Carrillo MC, Thies B, Weintraub S, Phelps CH (2011) The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 7, 263–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Nasreddine ZS, Phillips NA, Bedirian V, Charbonneau S, Whitehead V, Collin I, Cummings JL, Chertkow H (2005) The Montreal Cognitive Assessment, MoCA: a brief screening tool for mild cognitive impairment. J Am Geriatr Soc 53, 695–699. [DOI] [PubMed] [Google Scholar]
- [27].Hughes CP, Berg L, Danziger WL, Coben LA, Martin RL (1982) A new clinical scale for the staging of dementia. Br J Psychiatry 140, 566–572. [DOI] [PubMed] [Google Scholar]
- [28].Goodglass H, Kaplan E (1972) Boston Diagnostic Aphasia Examination (BDAE), Lea and Febiger, Philadelphia. [Google Scholar]
- [29].Kaufer DI, Cummings JL, Ketchel P, Smith V, MacMillan A, Shelley T, Lopez OL, DeKosky ST (2000) Validation of the NPI-Q, a brief clinical form of the Neuropsychiatric Inventory. J Neuropsychiatry Clin Neurosci 12, 233–239. [DOI] [PubMed] [Google Scholar]
- [30].Goetz CG (2010) [Movement Disorder Society-Unified Parkinson’s Disease Rating Scale (MDS-UPDRS): a new scale for the evaluation of Parkinson’s disease]. Rev Neurol (Paris) 166, 1–4. [DOI] [PubMed] [Google Scholar]
- [31].Warrington EK, James M (1991) The visual object and space perception battery, Thames Valley Test Company Bury St Edmunds. [Google Scholar]
- [32].Osterrieth PA (1944) Le test de copie d’une figure complexe. Arch Psychol 30, 206–356. [Google Scholar]
- [33].Wechsler D (2008) Wechsler adult intelligence scale–Fourth Edition (WAIS–IV). San Antonio, TX: NCS Pearson; 22, 498. [Google Scholar]
- [34].Rey A (1964) L’examen clinique en psychologie, Presses Universitaires de France, Paris. [Google Scholar]
- [35].Lansing AE, Ivnik RJ, Cullum CM, Randolph C (1999) An empirically derived short form of the Boston naming test. Arch Clin Neuropsychol 14, 481–487. [PubMed] [Google Scholar]
- [36].Iglesias JE, Augustinack JC, Nguyen K, Player CM, Player A, Wright M, Roy N, Frosch MP, McKee AC, Wald LL, Fischl B, Van Leemput K, Alzheimer’s Disease Neuroimaging I (2015) A computational atlas of the hippocampal formation using ex vivo, ultra-high resolution MRI: Application to adaptive segmentation of in vivo MRI. Neuroimage 115, 117–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Van Leemput K, Bakkour A, Benner T, Wiggins G, Wald LL, Augustinack J, Dickerson BC, Golland P, Fischl B (2009) Automated segmentation of hippocampal subfields from ultra-high resolution in vivo MRI. Hippocampus 19, 549–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Dempster AP, Laird NM, Rubin DB (1977) Maximum Likelihood from Incomplete Data Via the EM Algorithm. J Royal Stat Soc 39, 1–22. [Google Scholar]
- [39].Benjamini Y, Hochberg Y (1995) Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J Royal Stat Soc 57, 289–300. [Google Scholar]
- [40].Dalton MA, Maguire EA (2017) The pre/parasubiculum: a hippocampal hub for scene-based cognition? Curr Opin Behav Sci 17, 34–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Powell TP, Guillery RW, Cowan WM (1957) A quantitative study of the fornixmamillo-thalamic system. J Anatomy 91, 419–437. [PMC free article] [PubMed] [Google Scholar]
- [42].Dahmani L, Courcot B, Near J, Patel R, Amaral RSC, Chakravarty MM, Bohbot VD (2020) Fimbria-Fornix Volume Is Associated With Spatial Memory and Olfactory Identification in Humans. Front Sys Neurosci 13, 87–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Ji J, Maren S (2008) Differential roles for hippocampal areas CA1 and CA3 in the contextual encoding and retrieval of extinguished fear. Learn Mem 15, 244–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Jeong Y, Huh N, Lee J, Yun I, Lee JW, Lee I, Jung MW (2018) Role of the hippocampal CA1 region in incremental value learning. Sci Rep 8, 9870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Bartsch T, Dohring J, Rohr A, Jansen O, Deuschl G (2011) CA1 neurons in the human hippocampus are critical for autobiographical memory, mental time travel, and autonoetic consciousness. Proc Natl Acad Sci U S A 108, 17562–17567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Apostolova LG, Mosconi L, Thompson PM, Green AE, Hwang KS, Ramirez A, Mistur R, Tsui WH, de Leon MJ (2010) Subregional hippocampal atrophy predicts Alzheimer’s dementia in the cognitively normal. Neurobiol Aging 31, 1077–1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Mueller SG, Stables L, Du AT, Schuff N, Truran D, Cashdollar N, Weiner MW (2007) Measurement of hippocampal subfields and age-related changes with high resolution MRI at 4T. Neurobiol Aging 28, 719–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].West MJ, Kawas CH, Stewart WF, Rudow GL, Troncoso JC (2004) Hippocampal neurons in pre-clinical Alzheimer’s disease. Neurobiol Aging 25, 1205–1212. [DOI] [PubMed] [Google Scholar]
- [49].Apostolova LG, Dinov ID, Dutton RA, Hayashi KM, Toga AW, Cummings JL, Thompson PM (2006) 3D comparison of hippocampal atrophy in amnestic mild cognitive impairment and Alzheimer’s disease. Brain 129, 2867–2873. [DOI] [PubMed] [Google Scholar]
- [50].Khan W, Westman E, Jones N, Wahlund L- O, Mecocci P, Vellas B, Tsolaki M, Kłoszewska I, Soininen H, Spenger C, Lovestone S, Muehlboeck JS, Simmons A, AddNeuroMed c, for the Alzheimer’s Disease Neuroimaging I (2015) Automated Hippocampal Subfield Measures as Predictors of Conversion from Mild Cognitive Impairment to Alzheimer’s Disease in Two Independent Cohorts. Brain topography 28, 746–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Madusanka N, Choi HK, So JH, Choi BK, Park HG (2019) One-year Follow-up Study of Hippocampal Subfield Atrophy in Alzheimer’s Disease and Normal Aging. Curr Med Imaging Rev 15, 699–709. [DOI] [PubMed] [Google Scholar]
- [52].Mak E, Gabel S, Su L, Williams GB, Arnold R, Passamonti L, Vazquez Rodriguez P, Surendranathan A, Bevan-Jones WR, Rowe JB, O’Brien JT (2017) Multi-modal MRI investigation of volumetric and microstructural changes in the hippocampus and its subfields in mild cognitive impairment, Alzheimer’s disease, and dementia with Lewy bodies. Int Psychogeriatr 29, 545–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Madhavan A, Whitwell JL, Weigand SD, Duffy JR, Strand EA, Machulda MM, Tosakulwong N, Senjem ML, Gunter JL, Lowe VJ, Petersen RC, Jack CR Jr., Josephs KA (2013) FDG PET and MRI in logopenic primary progressive aphasia versus dementia of the Alzheimer’s type. PLoS One 8, e62471. [DOI] [PMC free article] [PubMed] [Google Scholar]