

Abstract
Alzheimer’s disease (AD) is the most common neurodegenerative disease, and its main hallmark is the deposition of amyloid beta (Aβ) peptides. However, several clinical trials focusing on Aβ-targeting agents have failed recently, and thus new therapeutic leads are focusing on alternate targets such as tau protein pathology, Aβ-metal induced oxidative stress, and neuroinflammation. To address these different pathological aspects of AD, we have employed a multifunctional compound, L1 [4-(benzo[d]thiazol-2-yl)-2-((4,7-dimethyl-1,4,7-triazonan-1-yl)-methyl)-6-methoxyphenol], that integrates Aβ-interacting and metal-binding fragments in a single molecular framework, exhibits significant antioxidant activity and metal chelating ability, and also rescues neuroblastoma N2A cells from Cu2+-induced Aβ neurotoxicity. Along with demonstrating in vivo Aβ-binding and favorable brain uptake properties, L1 treatment of transgenic 5×FAD mice significantly reduces the amount of both amyloid plaques and associated phosphorylated tau (p-tau) aggregates in the brain by 40–50% versus the vehicle-treated 5×FAD mice. Moreover, L1 mitigates the neuroinflammatory response of the activated microglia during the Aβ-induced inflammation process. Overall, these multifunctional properties of L1 to attenuate the formation of amyloid plaques and associated p-tau aggregates while also reducing the microglia-mediated neuroinflammatory response are quite uncommon among the previously reported amyloid-targeting chemical agents, and thus L1 could be envisioned as a lead compound for the development of novel AD therapeutics.
Keywords: Alzheimer’s disease, amyloid beta peptide, Aβ, amyloid plaques, oxidative stress, phosphorylated tau aggregation, p-tau, neuroinflammatio, microglia activation
Graphical Abstract
INTRODUCTION
Alzheimer’s disease (AD) is an irreversible neurodegenerative disorder that gradually destroys memory and thinking skills, thereby devastating the quality of life for patients and their families, and is a huge burden not only on patients but especially on caregivers.1,2 The main neuropathological hallmark of the brains of AD patients is the deposition of amyloid plaques comprising mainly the amyloid beta (Aβ) peptide, and in light of the amyloid cascade hypothesis the reduction of amyloid deposition in the brain has been the main target for the development of AD therapeutics.3 More recently, soluble Aβ oligomers were found to be the most neurotoxic species and are directly implicated in synapse loss and neuronal injury4,5 as the soluble Aβ oligomers can perturb signal transduction via the interaction with lipid membranes or membrane-bound receptors leading to a loss of function and cell death.6,7
However, currently there are only five FDA-approved drugs for the symptomatic relief related to AD, and only a few of the ongoing clinical trials targeting the various Aβ aggregates have shown significant results toward the approval of new AD drugs.8–10 In addition, several clinical trials focusing on Aβ-targeting drugs have failed recently,11,12 and thus the amyloid cascade hypothesis is being reconsidered and new therapeutic leads are focusing on alternate targets such as tau protein pathology or neuroinflammation.13–15 Thus, it has been proposed that therapeutic agents targeting the tau pathology and neuroinflammation could be more effective than Aβ-targeting agents, since tau hyperphosphorylation and phosphorylated tau (p-tau) aggregation is more closely correlated to the cognitive and clinical symptoms of AD than the amyloid plaque formation,13–15 although the amyloid plaques have been shown to facilitate the initial p-tau aggregation surrounding the amyloid plaques and the spread and formation of intracellular neurofibrillary tangles (NFTs).16,17 Therefore, it is important to develop chemical agents that can control both amyloid plaque formation and the associated p-tau aggregation as more efficient AD therapeutics.
Another pathological aspect of AD is that transition metal ions (Cu, Zn, and Fe) are found in abnormally high concentration within the amyloid plaque deposits, and these metal ions were also shown to stabilize neurotoxic Aβ oligomers,18–22 while Aβ-Cu and Aβ-Fe species can catalyze the formation of reactive oxygen species (ROS) and lead to oxidative stress and neuroinflammation.23–26 Due to the unwanted interactions of the metal ions with the Aβ peptides and the abnormal accumulation of metal ions in the amyloid plaques, bifunctional chelators (BFCs) such as clioquinol (CQ) and 8-hydroxyquinoline analog (PBT2) containing both Aβ-interacting and metal-binding sites have been developed to address the disruption of metal homeostasis.27,28 These BFCs were shown to restore cognitive function in an AD mouse model29–31 and to modulate the interaction of metal ions with the Aβ peptides via the formation of a ternary complex (Aβ-BFC-metal) that reduces the toxicity of metal-induced Aβ or ROS production.28,30–35 In spite of these promising in vitro results, however, there are very few examples of BFCs that have shown to control the Aβ aggregation process in vivo in transgenic AD mice.33–35
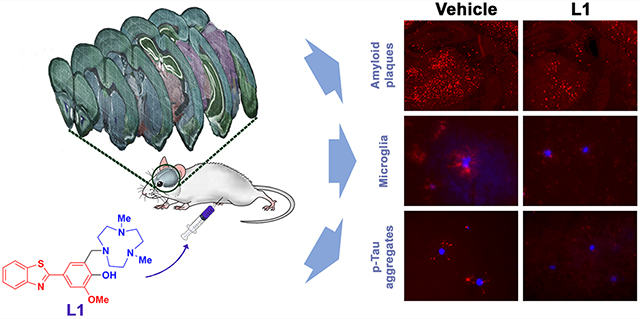
We have also designed and developed BFCs that integrate Aβ-interacting and metal-binding groups in a single molecular framework (Scheme 1).36–39 These bifunctional systems were shown to control metal-mediated Aβ aggregation in vitro via strong interactions with Aβ aggregates and metal ions.36–38 Furthermore, the high affinity of the BFCs toward metal ions as well as Aβ species allowed the use of the 64Cu complexes of these BFCs as positron emission tomography (PET) imaging agents for the detection of Aβ aggregates in vivo.39 Herein we report the in vivo therapeutic efficacy of a multifunctional compound (MFC), 4-(benzo[d]thiazol-2-yl)-2-((4,7-dimethyl-1,4,7-triazonan-1-yl)methyl)-6-methoxyphenol (L1), that contains a 2-phenylbenzothiazole fragment for Aβ binding, a triazacyclononane (TACN) macrocycle as a high-affinity metal chelator, and a vanillin moiety with antioxidant and antiamyloidogenic properties.40–43 While the coordination chemistry, in vitro affinity for Aβ aggregates, in vitro Aβ modulation, and in vivo 64Cu-PET imaging application of L1 have been reported recently,36–39 herein we are reporting totally new results for L1, focusing on its ability to attenuate several pathological hallmarks of AD in transgenic 5×FAD mice. The effect of L1 on metal-mediated Aβ toxicity was evaluated via in vitro cytotoxicity, antioxidant activity, and Aβ aggregation kinetic assays, and the blood-brain barrier (BBB) permeability of L1 and its ability to bind Aβ aggregates in vivo was confirmed through fluorescence microscopy and two-photon microscopy. We have also demonstrated that upon treatment of transgenic 5×FAD mice with L1 for 30 days, the level of amyloid plaques in the treated AD mouse brains was shown to decrease by up to 50% vs the vehicle-treated AD mouse brains. More importantly, we have found that L1 suppresses the p-tau aggregation near the amyloid plaques, as well as limits the activation of microglia near amyloid plaques by 30–44% vs the vehicle-treated AD mouse brains, suggesting that L1 can truly act as a multifunctional chemical agent that can target several pathological aspects of AD and thus could be envisioned as a lead compound for the development of novel AD therapeutics.
Scheme 1. Chemical Structures of BFCs 1 and 2,36 3 and 4,37 and L1 [4-(Benzo[d]thiazol-2-yl)-2-((4,7-dimethyl-1,4,7-triazonan-1-yl)methyl)-6-methoxyphenol] and 5–838,39 Developed by Our Groupa.

aThe metal-binding and Aβ-interaction fragments are shown in blue and red, respectively.
RESULTS AND DISCUSSION
Chemical and Biochemical Properties of L1
In order to control the Aβ aggregation mediated by metal ions, we have developed several BFCs that integrate both Aβ-interacting and metal-binding functions within a small molecular framework (Scheme 1).36–39 Among them, we chose L1 to be evaluated as an in vivo therapeutic agent since L1 efficiently inhibited the aggregation of Aβ42 and reduced the neurotoxicity of soluble Aβ42 oligomers in the presence of Cu ions (see below). Furthermore, L1 has nanomolar affinity for Aβ aggregates and exhibits a very high affinity for Cu2+ ions (pCu = 13.3 at pH 7.4, Table 1),38,39 which outcompetes the Cu affinity of the Aβ peptide (estimated to be around 10−10 M),20,21 and it also satisfies the Lipinski’s criteria for central nervous system (CNS) drugs (MW < 450, clogP < 5, number of H-bond donors (HBD) < 3, and number of H-bond acceptors (HBA) < 7) for blood-brain barrier (BBB) penetration (Scheme 1 and Table 1).44 Overall, these data suggest that L1 exhibits strong affinity for Aβ aggregates and Cu2+ ions and displays favorable BBB permeability, thus being suitable for in vivo use as a potential AD drug.
Table 1.
| MW | log Poct | Ki (L1)a | log K ([L1Cu]+) | pCu (−log [Cu]free) |
|---|---|---|---|---|
| 426.6 g/mol | 0.97 ± 0.12 | 170 ± 50 nM | 32.0 ± 0.1 | 12.8 (pH 6.6), 13.3 (pH 7.4) |
Inhibiton constant (Ki) obtained from competitive binding studies to Aβ fibrils vs ThT.40
The Antioxidant Properties of L1
In addition to the Aβ-interacting and metal-binding properties, L1 also exhibits antioxidant properties, likely due to the presence of the guaiacyl (methoxyphenol) group that has been shown previously to act as an oxidation indicator.45 The antioxidant activity of L1 was evaluated by the Trolox equivalent antioxidant capacity (TEAC) assay, which measures the ability to scavenge the radical cation (ABTS•+), as compared to the standard (Trolox).46 Interestingly, the radical scavenging activity of L1 is >1.6 times higher than that of Trolox or glutathione, a known in vivo antioxidant (Figure 1a), strongly suggesting that L1 can efficiently quench free radicals and thus prevent oxidative damage.
Figure 1.

(a) Antioxidant activity of Trolox, glutathione, and L1 at 1, 3, 6, and 15 min assessed by the TEAC assay. The TEAC values of glutathione and L1 are normalized to the antioxidant activity of Trolox (shown by the dashed line). Conditions: [ABTS•+] = 750 μM; [compound] =25–100 μM. (b) Inhibitory activity of L1 toward Cu-mediated hydroxyl radical generation, evaluated by monitoring the fluorescence intensity of CCA-OH over time. Conditions: [CCA] = 100 μM; [CuSO4]=40μM; [ascorbic acid] = 400 μM; [L1] = 0–80 μM; λem = 395 nm; λem = 450 nm. The error bars represent the standard deviation from three independent experiments.
We have also investigated the antioxidant activity of L1 toward hydroxyl radicals generated by Cu2+ ions in the presence of reducing agents (a Fenton-type reaction), which is closely relevant to metal-Aβ-induced radical generation.47 Coumarin-3-carboxylic acid (CCA), which is converted to the highly fluorescent derivative 7-hydroxycoumarin-3-carboxylic acid (CCA-OH) in the presence of hydroxyl radicals generated by the Cu2+ ions and ascorbic acid, was used as an indicator.48 It was observed that the amount of fluorescent CCA-OH generated after 30 min decreased as the concentration of L1 increased (Figure 1b), and almost no CCA-OH was formed when 1 equiv of L1 was added relative to the amount of Cu2+ ions, indicating that the production of the hydroxyl radicals was suppressed by the formation of the 1:1 L1-Cu complex.38 Taken together, these results suggest that the potent antioxidant properties of L1 lead to the alleviation of metal-Aβ-induced oxidative stress by scavenging free radicals and also controlling the direct metal-mediated radical production. Since L1 has Aβ-interacting, metal-binding, and antioxidant properties, it can thus act as a multifunctional compound (MFC) with several desirable properties related to AD.
Pulsed Hydrogen-Deuterium Exchange (HDX) Stud- ies of Cu-Mediated Aβ42 Aggregation
Metal ions are closely involved in Aβ oligomerization, the stabilization of Aβ oligomers, and prevention of degradation of Aβ, thus resulting in Aβ oligomer buildup in the brain.20,49 Furthermore, we have previously confirmed that Cu2+ sped up the formation of soluble Aβ42 oligomers and increased their stability, which led to a significant increase in their neurotoxicity.50 In particular, we have used pulsed hydrogen-deuterium exchange (HDX) mass spectrometry to validate that the presence of Cu2+ retarded the Aβ42 aggregation process by stabilizing the soluble Aβ42 aggregates. This mass spectrometry technique has allowed us previously to directly monitor the time-dependent Aβ aggregation process by hydrogen-deuterium exchange, without addition any fluorescence dyes or additional modification of the Aβ peptides, in which cases such additives could potentially affect the Aβ aggregation process.51 Using this developed HDX platform, we have investigated the effect of L1 on the Aβ42 aggregation process in the presence of Cu2+. For an increased structural resolution of Aβ42 aggregation, we have applied pepsin digestion following the pulsed HDX labeling and analyzed the three peptic peptide fragments (Aβ1–19, Aβ20–35, and Aβ36–42) that cover the entire Aβ42 peptide sequence. As shown previously, during the Aβ42 aggregation process in the presence of Cu2+ a longer lag phase at the ∼60% protection level was observed, which was proposed to correspond to soluble Aβ42 oligomers, followed by a gradual growth to large Aβ42 aggregates.51 In contrast, Aβ42 aggregation in the presence of Cu2+ and L1 showed a shorter lag phase with a lower level of protection (∼40%) followed by a sharper growth, suggesting that L1 limits the formation of oligomeric species during the Aβ42 aggregation process (Figure 2a). When the t1/2 values obtained from the fitting of the pulsed HDX data to a modified Finke-Wetsky (F-W) model,51,52 all three peptic peptides of Aβ42 showed that both the lag and growth phases were significantly shortened in the presence of L1 and Cu2+ (Figure 2b). Overall, these results support that L1 accelerates the structural reorganization to form large Aβ42 aggregates with a high protection level of ∼80%, and these species were found to correspond to nonfibrillar amorphous aggregates as observed by TEM.38 Furthermore, the acceleration of Aβ42 structural reorganization process is attributable to the destabilization of the Cu-Aβ42 oligomers by L1, which outcompetes the Aβ42 species for Cu2+ binding. This conclusion is further supported by the previously reported native gel/Western blotting and TEM data on the effect of L1 on metal-mediated Aβ42 aggregation.38
Figure 2.

(a) Pulsed HDX results for three peptic peptide fragments from Aβ42 in the presence of Cu2+ (green) and Cu2+ and L1 (blue). Peptide fragments are Aβ1–19, Aβ20–35, and Aβ36–42. All lines are fits using a modified F-W model in MathCAD. (b) Comparison of t1/2 values obtained from the fits to the modified F-W model. (c) Cell viability of N2A cells (normalized to a 1% DMSO control) after 40 h treatment with Aβ42 and Cu2+ in the absence or presence of L1, assessed by the Alamar Blue assay. Conditions: [Aβ42] = 20μM; [Cu2+] = 20 μM; [L1] = 2 μM. The error bars represent the standard deviation from five independent experiments, and the statistical analysis was evaluated according to one-way ANOVA (***p < 0.001).
Inhibition of Metal-Induced Aβ42 Neurotoxicity
To confirm the inhibition activity of L1 against metal-induced Aβ neurotoxicity, we have performed cell viability assays using mouse neuroblastoma Neuro-2a (N2A) cells that differentiate into neurons under neural induction media.53 Since L1 was shown to be toxic at concentrations higher than 5 μM (Figure S1), we used 2 μM of L1, which is nontoxic to N2A cells, corresponding to a 1:10 L1/Aβ-Cu ratio. After 40 h incubation of N2A cells treated with Aβ42 and Cu2+ in the absence or the presence of L1, the cell viability was measured using the Alamar Blue assay. Whereas the N2A cells treated with Aβ42 and Cu2+ showed low viability due to Cu2+-mediated stabilization of soluble Aβ42 oligomers,50 addition of L1 in the presence of Aβ42 and Cu2+ had a dramatic effect in restoring the cell viability to control levels, even at substoichiometric levels (Figure 2c). We propose that the beneficial effect of L1 against Cu2+-induced Aβ42 neurotoxicity can be attributed to three features of L1: (1) its antioxidant activity, (2) the inhibition of Cu2+-mediated ROS formation through strong metal chelation,38,39 and (3) the inhibition of metal-mediated Aβ42 oligomerization. The antioxidant assays described above showed that L1 had powerful radical scavenging activity and efficiently repressed the Cu2+-mediated ROS generation. Moreover, the Aβ42 aggregation pulsed HDX kinetic data suggested that L1 reduced the stability of soluble Cu2+-Aβ42 oligomers and promoted the formation of nontoxic amorphous Aβ42 aggregates. Therefore, we can conclude that the various properties of L1 are operating cooperatively to provide a significant beneficial effect against the neurotoxicity of Cu2+-stabilized soluble Aβ42 oligomers.
It is also important to contrast the beneficial antineurotoxic properties of L1 with the previously reported BFCs 1 and 2 (Scheme 1), which have the opposite effect of exacerbating the neurotoxicity of Aβ42 species by promoting the formation of Cu2+-stabilized soluble Aβ42 oligomers and also the disaggregation of Aβ42 fibrils.36 This stark difference in properties is likely due to the different metal-chelating fragments of these compounds and their different metal binding affinity, since all three compounds contain the same Aβ-interacting fragment, yet L1 contains a triazacyclononane (TACN) macrocycle, while 1 and 2 contain a bis(2-pyridylmethyl)amine or (2-pyridylmethyl)methylamine metal-binding fragment, respectively. Thus, this suggests the interaction of metal ions with different Aβ42 aggregates, including soluble Aβ42 oligomers, might play an important role in Aβ42 neurotoxicity in vivo and that carefully designed compounds could have a pronounced antineurotoxic activity.
Two-Photon Fluorescence Imaging of Amyloid Plaques in Live AD Mice
Given that L1 successfully stained and visualized amyloid plaques ex vivo, as evidenced by the observed strong fluorescence signal in AD mouse brain sections,39 we have employed 5×FAD transgenic mice to assess the in vivo availability of L1 for Aβ binding. To circumvent the potential limitation of BBB permeability, L1 was intracranially administrated to the brains of 5×FAD or WT mice, and then a thinned-skull cranial window was installed. After intravenous injection of dextran-Texas Red (70 kDa) to visualize the blood vessels, the amyloid plaques were imaged by two-photon microscopy (Figures 3 and S2). While a negligible fluorescence intensity was observed for L1 in the WT mouse brain, a significant fluorescence intensity was clearly visible in the 5×FAD mouse brains, which was assigned to the binding of L1 to the parenchymal amyloid plaques. The observed regions stained with L1 were 10–20 μm, similar in size and morphology to the amyloid plaques observed in the brain sections of age-matched 5×FAD mice stained ex vivo with thioflavin S (ThS), Congo Red, and L1 (see below).54,55 Overall, these results reveal that L1 vividly binds amyloid plaques in live AD mice without any significant nonspecific binding observed in WT mice, further validating its potential utility for in vivo modulation of Aμ aggregation and its neurotoxicity.
Figure 3. In vivo two-photon fluorescence brain images of 5×FAD.

(a) or WT (b) mice after intracranial injection of L1. The fluorescence intensity from L1 was monitored at 435–485 nm under two-photon excitation at 770 nm. Dextran-Texas Red (70 kDa) was intravenously injected for blood vessel imaging prior to two-photon microscopy, and its fluorescence was monitored at 565–615 nm under excitation at 543 nm. The regions within the white rectangles in the left panels are shown magnified in the right panels (scale bar 50 μm).
In Vivo Regulation of Amyloid Plaques in 5xFAD Mice
To evaluate the in vivo therapeutic efficacy of L1, we have administered daily the MFC L1 to 5×FAD mice (1 mg/kg of body weight) via intraperitoneal injection for a period of 30 days. Since the amyloid plaques start to deposit in the deep cortex and subiculum of 5×FAD mice at ∼2 months of age,55 we chose 3-month-old 5×FAD mice that are assumed to exhibit multiple forms of Aβ in their brains, including monomeric Aβ, soluble Aβ oligomers, intraneuronal Aβ aggregates, and amyloid plaques.56 As a control group, other 5×FAD mice were treated with the vehicle (1% DMSO in PBS). After 30 days, the brains were harvested, and 50 μm thick brain sections were obtained. The brain sections were immunostained with the AF594-conjugated HJ3.4 (AF594-HJ3.4) antibody, which binds to a wide range of amyloid aggregates.57 Excitingly, while the vehicle-treated brain sections showed only the fluorescence signal corresponding to the immunostained amyloid plaques (Figure S3), the 5×FAD brain sections from the treated mice exhibited an intrinsic fluorescence intensity corresponding to L1 that accumulated in the brain during the treatment, and appreciable colocalization between L1 and the amyloid plaques immunostained with AF594-HJ3.4 was observed (Figure 4). These staining studies thus strongly suggest that when intraperitoneally administered into the 5×FAD mouse, L1 can pass through the BBB and accumulate within the amyloid deposits to a detectable amount, further confirming that L1 can bind to native amyloid aggregates of various types, similar to an amyloid antibody.
Figure 4.

Fluorescence microscopy images of brain sections from 5×FAD mice treated with L1 for 30 days. The brain sections were immunostained with the AF594-HJ3.4 antibody. The fluorescence signals from AF594-HJ3.4 antibody and L1 were monitored at 600–660 nm and 435–485 nm under excitation at 340–380 nm and 540–580 nm, respectively (AF594-HJ3.4 antibody, red; L1, blue; scale bar 100 μm). The in vivo L1 was appreciably colocalized with the amyloid plaques immunostained with AF594-HJ3.4 (Pearson’s correlation coefficient 0.41).
For the quantitative analysis of the amyloid plaques, immunostaining with the AF594-HJ3.4 antibody and staining with ThS and Congo Red were carried out for the brain sections of the treated 5×FAD mice. These staining results can provide quantitative information on the amyloid deposits in brain sections54,58 and can show a similar trend in Aβ levels as that obtained from analysis of brain homogenates.59 Prior to quantitative analysis, both the L1- and the vehicle-treated brain sections were costained with ThS and the AF594-HJ3.4 antibody, and as expected ThS stained the amyloid plaques in the 5×FAD mice brain sections in a significantly colocalized pattern to that observed for AF594-HJ3.4 (Pearson’s correlation coefficients of 0.59 and 0.52, respectively, Figure S4). We have quantified the amyloid plaques in the brain sections by analyzing the fluorescence intensity of the stained plaques of 10 brain sections per mouse that were sliced from uniformly distributed locations between the frontal lobe and the occipital lobe, thus covering evenly the entire mouse brain. The brain section images with three different staining methods (AF594-HJ3.4 antibody, ThS, and Congo Red, respectively) consistently displayed that the number and the fluorescence intensity of the amyloid plaques were significantly diminished in the brains from the L1-treated 5×FAD mice, as compared to those from the vehicle-treated 5×FAD mice (Figures 5a,b and S5–S7). Since the fluorescence intensity of the staining reagent is proportional to the concentration of amyloid plaques, it was determined that the amount of Aβ plaques in the L1-treated mice were sharply reduced by ∼45% when compared to those of control group (Figures 5b, S6b, and S7b). To support the quantification of amyloid plaques based on the fluorescence intensity analysis of the stained brain sections, the total amounts of Aβ42 (and Aβ40) species found in soluble (i.e., PBS-soluble) and insoluble (i.e., guanidine-soluble) brain lysates were quantified by the enzyme-linked immunosorbent assay (ELISA). Importantly, a significant reduction in the amount of soluble and insoluble Aβ42 species was found in the L1-treated brain lysate vs the vehicle-treated control group (a 38% and 50% reduction for soluble and insoluble Aβ42 species, respectively, Figure 5c) and a 49% reduction for the insoluble Aβ40 species (Figure S8). This reduction of the in vivo amount of various Aβ species is well correlated with the imaging analysis of brain sections stained with ThS, Congo Red, and the AF594-HJ3.4 antibody.
Figure 5.

(a) Representative fluorescence microscopy images of the AF594-HJ3.4 antibody-stained brain sections from 5×FAD mice treated with L1 or vehicle. The fluorescence of AF594 was monitored with a standard TRITC filter set. Scale bar: 1 mm. (b) Total number and fluorescence intensity of amyloid plaques in the AF594-HJ3.4 immunostained brain sections from 5×FAD mouse treated with L1 or vehicle. When compared to vehicle-treated mice, the amyloid burden in the L1-treated mice was reduced by 49% and 43% based on the number of amyloid plaques and fluorescence intensity, respectively. The fluorescence intensity and number of amyloid plaques were obtained as the sum obtained from seven brain sections per mouse. Error bars represent the standard deviation (n = 3 mice), and the statistical analysis was evaluated according to one-way ANOVA (*p < 0.05). (c) PBS-soluble (left) and guanidine-soluble (right) Aβ42 levels from brain tissues, quantified using ELISA. Compared to vehicle-treated mice brains, total amount of the Aβ42 species in the L1-treated mice brains were reduced by 38% and 50% in PBS- and guanidine-soluble brain homogenates, respectively. Error bars represent standard deviation (n = 3 mice), and the statistical analysis was evaluated according to one-way ANOVA (**p < 0.01).
Attenuation of Aβ-Induced Neuroinflammation and p-tau Aggregation in 5xFAD Mice
Since L1 has important antioxidant properties and also modulates the neurotoxicity of Aβ aggregates, we have also investigated the effect of L1 on other important pathological aspects of AD, such as the activation of microglia as a neuroinflammatory response and the aggregation of the phosphorylated tau (p-tau) protein. First, a fluorescently labeled antibody for the microglia-specific Iba1 protein was employed to visualize the activated microglia, which play a critical role in the neuroinflammatory response during AD progression.60,61 Excitingly, microglia activation was appreciably suppressed near the amyloid plaques observed in the brain sections from the L1-treated 5×FAD mice, compared to those from the vehicle-treated 5×FAD mice (Figures 6a and S9), and the quantification of the fluorescence intensity of the immunostained-Iba1 antibody revealed a significant reduction of 43% of microglia activation in the L1-treated mouse brains versus the control group (Figure 6b). These results nicely complement the observation that L1 alleviates the neurotoxicity of Aβ aggregates and suppresses metal-mediated ROS generation and oxidative stress, which in turn leads to the attenuation of neuroinflammation mediated by activated microglia near the amyloid plaques. While the 5×FAD mouse model does not exhibit a pronounced tau pathology, a considerable amount of extracellular p-tau aggregates was still observed in the cortex and hippocampus regions of the 5×FAD transgenic mice.62 Therefore, we have also employed a fluorescently labeled antibody, AT8, to immunostain and quantify the amount of p-tau aggregates surrounding the amyloid plaques. Importantly, the amount of amyloid plaque-associated p-tau aggregates in the L1-treated mouse brains versus the vehicle-treated controls were dramatically reduced by 44% and 30% based on the fluorescence intensity and number of p-tau aggregates, respectively, both in the cortex and in the hippocampus areas of the brain (Figures 6a, 6c, and S10). This appreciable result is in contrast to the recent amyloid-targeting antibody immunotherapies showing effective reduction of Aβ deposition but no effect on p-tau aggregation in vivo.13,63–65 While the role of Aβ species in the initial p-tau aggregation, their formation into intracellular neurofibrillary tangles, and their neurotoxicity is still unclear,13 these results suggest that L1 decreases the amount of p-tau aggregates in the extracellular space surrounding the amyloid plaques.
Figure 6.

(a) Representative fluorescence microscopy images of the AF594-Iba1 and AF594-AT8 immunostained brain sections (cortex and hippocampus areas) from 5×FAD mice treated with L1 or vehicle. All fluorescence images are the maximum intensity projection images obtained from 30 Z-sections collected at 1 μm intervals. Color: red, AF594-Iba1 or AF594-AT8 antibody; blue, ThS. Scale bar 50 μm. (b) Quantification of the fluorescence intensity of AF594-Iba1 immunostained brain sections from 5×FAD mice, showing that the extent of microglia activation in the L1-treated mice was reduced by 43% versus that in the vehicle-treated mice. The fluorescence intensity was the sum obtained from eight fluorescence images randomly chosen in four brain sections per mouse. Error bars represent standard deviations (n = 3 mice), and the statistical analysis was evaluated according to one-way ANOVA (**p < 0.01). (c) Quantification of the number and fluorescence intensity of AF594-AT8 immunostained brain sections from 5×FAD mice, showing that the p-tau aggregates in the L1-treated mice were reduced by 44% and 30% based on the fluorescence intensity and number of p-tau aggregates, respectively. The fluorescence intensity and number of p-tau aggregates were obtained as the sum obtained from eight fluorescence images randomly chosen in four brain sections per mouse. Error bars represent standard deviations (n =3 mice), and the statistical analysis was evaluated according to one-way ANOVA (*p < 0.05, ***p < 0.001).
Overall, these results are quite exciting, since they show that L1 can be taken up in the brain of 5×FAD mice and is not degraded rapidly (as confirmed by the detection of its fluorescence in the brain sections of the L1-treated mice) and that L1 binds in vivo to the amyloid plaques, leading to an appreciable reduction of the amyloid plaques upon treatment of the 5×FAD mice with L1. Moreover, L1 mitigates the activated microglia-mediated neuroinflammatory response and also reduces the amount of extracellular p-tau aggregates surrounding the amyloid plaques, which is quite uncommon among the many amyloid-targeting agents that have been employed in recent clinical trials. Therefore, by virtue of the multifunctional properties of L1 described above, we conclude that the MFC L1 reduced considerably the development of several pathological hallmarks of AD and thus could exhibit therapeutic efficacy for delaying the progression of this disorder.
CONCLUSIONS
Herein we report that the MFC L1, which incorporates Aβ-interacting and metal-binding groups into a small molecule framework, exhibits in vivo properties that suggest a potential therapeutic efficacy against AD. Due to the high affinity of L1 for both Aβ species and Cu ions, L1 outcompetes Aβ species for binding of Cu2+ ions and thus can prevent the formation of Cu-stabilized soluble Aβ oligomers and promotes the formation nontoxic, amorphous aggregates. In addition, L1 exhibits appreciable antioxidant properties, including radical scavenging activity and suppression of metal-mediated radical generation, and contributes to the recovery of neuroblastoma N2A cells from Cu2+-induced Aβ neurotoxicity. Along with its favorable BBB permeability, L1 showed efficient Aβ-binding in the 5×FAD mouse brains, which allowed for in vivo modulation of Aβ aggregation and protection against metal-induced Aβ neurotoxicity. Interestingly, the quantification of amyloid plaques in brain sections based onficationquant immunostaining and ELISA analysis revealed up to 50% reduction of amyloid plaques in the L1-treated 5×FAD mice versus the vehicle-treated mice. Importantly, L1 also alleviated the neuroinflammatory response in vivo by reducing the activation of microglia and also reduces the amount of extracellular p-tau aggregates surrounding the amyloid plaques, thus holding potential for delaying the progression of AD. These properties are quite uncommon among the previously reported amyloid-targeting chemical agents, and in the near future we plan to evaluate the ability of L1 to have a beneficial effect on the learning and memory performance of AD mice.
METHODS
The general experimental methods, Aβ peptide preparation, Trolox equivalent antioxidant capacity (TEAC) assays, coumarin-3-carboxylic acid (CCA) assays, pulsed hydrogen-deuterium exchange (HDX) studies, and HDX data analysis, cell viability, and multiphoton in vivo imaging studies are described in the Supporting Information.
Treatment of Mice with L1
All animal studies have been approved by the Washington University Animal Studies Committee, and the handling of mice was performed in accordance with institutional regulations. Three-month old 5×FAD mice were divided into two groups. The two groups of 3 mice each were treated daily with a freshly prepared solution of L1 (1 mg/kg of body weight in 200 μL of PBS, pH 7.4, 1% DMSO) or vehicle (200 μL of PBS, pH 7.4, 1% DMSO) via intraperitoneal injection. After 30 days, all mice were sacrificed under deep anesthesia for perfusion and brain harvesting.
Mouse Brain Section Preparation.
All mice treated with L1 and vehicle were anesthetized with ketamine/xylazine (5 μL/g) via intraperitoneal injection and perfused with PBS containing 0.3% heparin. After being harvested, brains were divided into two hemispheres for immunohistochemistry and Aβ quantification. The right hemisphere was fixed in 4% paraformaldehyde in PBS (pH 7.4) at 4 °C overnight and preserved in 30% sucrose (w/v) in PBS (pH 7.4) at 4 °C for 3 days. The serial brains were coronally sectioned (thickness 50 μm) on a freezing sliding microtome. The brain sections were kept in a cryoprotectant solution of 30% ethylene glycol (v/v) and 15% sucrose (w/v) in 0.1 M phosphate buffer (pH 7.4) at −20 °C. The left hemisphere was frozen with dry ice and kept at −80 °C.
Mouse Brain Section Staining
Brain sections were permeabilized in PBS with 0.25% Triton-X100 for 30 min at room temperature and washed with PBS (3 × 5 min). To quantify the Aβ aggregates, the brain sections were treated with ThS solution (0.005% in PBS) or with Congo Red solution (5 μM, in PBS) for 30 min. After sequential washes with PBS (1 × 5 min), 50% EtOH/PBS (1 × 5 min), and PBS (3 × 5 min), the brain sections were mounted with mounting media. For immunostaining, the brain sections were treated with blocking solution (0.1% Triton-X100, 0.2% dry milk, and 1% BSA in PBS) at room temperature for 1 h and then incubated with AF594-conjugated anti-Aβ antibody (AF594-HJ3.4 antibody) solution (1:1000 dilution in blocking solution) at room temperature for 1 h.57 After staining, the brain sections were washed with PBS (3 × 5 min) and mounted with mounting media. For double staining with ThS and an antibody, the brain sections were stained with ThS, followed by the same incubation and washing steps used for ThS staining alone. After incubation with the blocking solution, the brain sections were treated with antibody solution (AF594-HJ3.4, AF594-Iba1, or AF594-AT8) diluted in the blocking solution (1:1000) at room temperature for 1 h and mounted with mounting media. For AF594-Iba1 staining, antigen retrieval was performed by pretreating the brain sections with citrate buffer (10 mM sodium citrate, 0.05% Tween 20, pH 6.0) for 30 min at 80 °C. Fluorescence images were visualized using either a Nikon Eclipse 80i, EVOS FL Auto 2, or Hamamatsu NanoZoomer fluorescence microscope. The maximum intensity projection images of the AF594-Iba1- and AF594-AT8-stained brain sections were obtained from 30 Z-sections collected at 1 μm intervals. The number and fluorescence intensity of amyloid plaques, microglia cells, and aggregated p-tau in each brain section were quantitatively analyzed by the ImageJ program under the same minimum and maximum values of fluorescence intensity. Colocalization analysis and determination of the Pearson’s correlation coefficient was performed with the imaging software Fiji (ImageJ 1.52p).
Enzyme-Linked Immunosorbent Assay (ELISA)
The frozen brain tissue (left hemisphere) was weighed and homogenized in cold PBS (100 mg/mL, 10% w/v) with a protease inhibitor cocktail (1:100 dilution, ThermoFisher Scientific). After centrifugation at 16000 × g for 20 min at 4 °C, the supernatant containing the soluble Aβ species was separated. The pellet was resuspended in 5 M guanidine HCl/50 mM Tris-HCl (pH 8, 100 mg/mL, 10% w/v) and incubated at room temperature for 4 h. After centrifugation at 16000 × g for 20 min at 4 °C, the supernatant containing insoluble Aβ species was separated. Both supernatants were diluted 1000–10000-fold, and then the levels of soluble and insoluble Aβ species from both supernatants were measured by human Aβ42/Aβ40 ELISA kits, in accordance with the protocol provided by the manufacturer (Invitrogen).
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by research funding from the NIH (R01GM114588 to L.M.M. and P41GM103422 to M.L.G.), Alzheimer’s Association (NIRG 12-259199 to L.M.M.), Washington University Knight Alzheimer’s Disease Research Center (NIH P50AG05681), and the McDonnell Center for Cellular and Molecular Neurobiology at Washington University School of Medicine. Animal surgery and imaging studies were performed at the Hope Center Animal Surgery Core and the Alafi Neuroimaging Lab at Washington University School of Medicine.
Footnotes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acschemneuro.0c00114.
Additional experimental details for antioxidant assays, pulsed HDX assays, cell viability assays, two-photon imaging studies, quantification of amyloid plaques, and additional fluorescence microscopy images (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/acschemneuro.0c00114
Contributor Information
Hong-Jun Cho, Department of Chemistry, University of Illinois at Urbana-Champaign, Urbana, Illinois 61801, United States.
Anuj K. Sharma, Department of Chemistry, Central University of Rajasthan, Ajmer, Rajasthan 305817, India
Ying Zhang, Department of Chemistry, Washington University, St. Louis, Missouri 63130, United States.
Michael L. Gross, Department of Chemistry, Washington University, St. Louis, Missouri 63130, United States.
Liviu M. Mirica, Department of Chemistry, University of Illinois at Urbana-Champaign, Urbana, Illinois 61801, United States; Hope Center for Neurological Disorders, Washington University School of Medicine, St. Louis, Missouri 63110, United States.
REFERENCES
- (1).Kelley AS, McGarry K, Gorges R, and Skinner JS (2015) The burden of health care costs for patients with dementia in the last 5 years of life. Ann. Intern. Med. 163 (10), 729–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Alzheimer’s Association (2019) 2019 Alzheimer’s disease facts and figures. Alzheimer's Dementia 15, 321–387. [Google Scholar]
- (3).Karran E, Mercken M, and Strooper BD (2011) The amyloid cascade hypothesis for Alzheimer’s disease: an appraisal for the development of therapeutics. Nat. Rev. Drug Discovery 10 (9), 698–712. [DOI] [PubMed] [Google Scholar]
- (4).Haass C, and Selkoe DJ (2007) Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid β-peptide. Nat. Rev. Mol. Cell Biol. 8 (2), 101–112. [DOI] [PubMed] [Google Scholar]
- (5).Benilova I, Karran E, and De Strooper B (2012) The toxic Aβ oligomer and Alzheimer’s disease: an emperor in need of clothes. Nat. Neurosci. 15, 349–357. [DOI] [PubMed] [Google Scholar]
- (6).Lesné S, Koh MT, Kotilinek L, Kayed R, Glabe CG, Yang A, Gallagher M, and Ashe KH (2006) A specific amyloid-β protein assembly in the brain impairs memory. Nature 440, 352–357. [DOI] [PubMed] [Google Scholar]
- (7).Lee SJC, Nam E, Lee HJ, Savelieff MG, and Lim MH (2017) Towards an understanding of amyloid-β oligomers: characterization, toxicity mechanisms, and inhibitors. Chem. Soc. Rev. 46 (2), 310–323. [DOI] [PubMed] [Google Scholar]
- (8).Alzheimer’s Association. FDA-approved treatments for Alzheimer’s. (2017). https://www.alz.org/media/Documents/fda-approved-treatments-alzheimers-ts.pdf (accessed February 12, 2019).
- (9).Eisai and Biogen announce detailed results of phase II clinical study of BAN2401 in early Alzheimer’s disease at Alzheimer’s Association International Conference (AAIC) 2018. https://www.eisai.com/news/2018/news201866.html (accessed February 12, 2019).
- (10).von Schaper E (2018) Everything but amyloid: new thinking prompts FDA revamp. Nat. Biotechnol. 36, 483–484. [DOI] [PubMed] [Google Scholar]
- (11).Morris GP, Clark IA, and Vissel B (2014) Inconsistencies and Controversies Surrounding the Amyloid Hypothesis of Alzheimer’s Disease. Acta. Neuropathol. Commun. 2 (1), 135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Makin S (2018) The amyloid hypothesis on trial. Nature 559 (7715), S4–S4. [DOI] [PubMed] [Google Scholar]
- (13).Giacobini E, and Gold G (2013) Alzheimer disease therapy—moving from amyloid-β to tau. Nat. Rev. Neurol. 9 (12), 677–686. [DOI] [PubMed] [Google Scholar]
- (14).Heneka MT, Carson MJ, Khoury JE, Landreth GE, Brosseron F, Feinstein DL, Jacobs AH, Wyss-Coray T, Vitorica J, Ransohoff RM, Herrup K, Frautschy SA, Finsen B, Brown GC, Verkhratsky A, Yamanaka K, Koistinaho J, Latz E, Halle A, Petzold GC, Town T, Morgan D, Shinohara ML, Perry VH, Holmes C, Bazan NG, Brooks DJ, Hunot S, Joseph B, Deigendesch N, Garaschuk O, Boddeke E, Dinarello CA, Breitner JC, Cole GM, Golenbock DT, and Kummer MP (2015) Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 14 (4), 388–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Kametani F, and Hasegawa M (2018) Reconsideration of Amyloid Hypothesis and Tau Hypothesis in Alzheimer’s Disease. Front. Neurosci. 12, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).He Z, Guo JL, McBride JD, Narasimhan S, Kim H, Changolkar L, Zhang B, Gathagan RJ, Yue C, Dengler C, Stieber A, Nitla M, Coulter DA, Abel T, Brunden KR, Trojanowski JQ, and Lee VMY (2018) Amyloid-β plaques enhance Alzheimer’s brain tau-seeded pathologies by facilitating neuritic plaque tau aggregation. Nat. Med. 24, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Leyns CEG, Gratuze M, Narasimhan S, Jain N, Koscal LJ, Jiang H, Manis M, Colonna M, Lee VMY, Ulrich JD, and Holtzman DM (2019) TREM2 function impedes tau seeding in neuritic plaques. Nat. Neurosci. 22 (8), 1217–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Bush AI, and Tanzi RE (2008) Therapeutics for Alzheimer’s disease based on the metal hypothesis. Neurotherapeutics 5 (3), 421–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Que EL, Domaille DW, and Chang CJ (2008) Metals in neurobiology: probing their chemistry and biology with molecular imaging. Chem. Rev. 108 (5), 1517–1549. [DOI] [PubMed] [Google Scholar]
- (20).Kepp KP (2012) Bioinorganic chemistry of Alzheimer’s disease. Chem. Rev. 112 (10), 5193–5239. [DOI] [PubMed] [Google Scholar]
- (21).Faller P, Hureau C, and Berthoumieu O (2013) Role of metal ions in the self-assembly of the Alzheimer’s amyloid-beta peptide. Inorg. Chem. 52 (21), 12193–206. [DOI] [PubMed] [Google Scholar]
- (22).Barnham KJ, and Bush AI (2014) Biological metals and metal-targeting compounds in major neurodegenerative diseases. Chem. Soc. Rev. 43 (19), 6727–6749. [DOI] [PubMed] [Google Scholar]
- (23).Barnham KJ, Masters CL, and Bush AI (2004) Neurodegenerative diseases and oxidative stress. Nat. Rev. Drug Discovery 3, 205–214. [DOI] [PubMed] [Google Scholar]
- (24).Smith DG, Cappai R, and Barnham KJ (2007) The redox chemistry of the Alzheimer’s disease amyloid β peptide. Biochim. Biophys. Acta, Biomembr. 1768 (8), 1976–1990. [DOI] [PubMed] [Google Scholar]
- (25).Hureau C, and Faller P (2009) Aβ-mediated ROS production by Cu ions: structural insights, mechanisms and relevance to Alzheimer’s disease. Biochimie 91 (10), 1212–1217. [DOI] [PubMed] [Google Scholar]
- (26).Cheignon C, Tomas M, Bonnefont-Rousselot D, Faller P, Hureau C, and Collin F (2018) Oxidative stress and the amyloid beta peptide in Alzheimer’s disease. Redox Biol. 14, 450–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Bush AI (2003) The metallobiology of Alzheimer’s disease. Trends Neurosci. 26 (4), 207–214. [DOI] [PubMed] [Google Scholar]
- (28).Scott LE, and Orvig C (2009) Medicinal inorganic chemistry approaches to passivation and removal of aberrant metal ions in disease. Chem. Rev. 109 (10), 4885–4910. [DOI] [PubMed] [Google Scholar]
- (29).Adlard PA, Cherny RA, Finkelstein DI, Gautier E, Robb E, Cortes M, Volitakis I, Liu X, Smith JP, Perez K, Laughton K, Li Q-X, Charman SA, Nicolazzo JA, Wilkins S, Deleva K, Lynch T, Kok G, Ritchie CW, Tanzi RE, Cappai R, Masters CL, Barnham KJ, and Bush AI (2008) Rapid restoration of cognition in Alzheimer’s transgenic mice with 8-hydroxy quinoline analogs Is associated with decreased interstitial Aβ. Neuron 59 (1), 43–55. [DOI] [PubMed] [Google Scholar]
- (30).Rodríguez-Rodríguez C, Telpoukhovskaia M, and Orvig C (2012) The art of building multifunctional metal-binding agents from basic molecular scaffolds for the potential application in neurodegenerative diseases. Coord. Chem. Rev. 256 (19), 2308–2332. [Google Scholar]
- (31).Savelieff MG, DeToma AS, Derrick JS, and Lim MH (2014) The ongoing search for small molecules to study metal-associated amyloid-β species in Alzheimer’s disease. Acc. Chem. Res. 47 (8), 2475–2482. [DOI] [PubMed] [Google Scholar]
- (32).Gomes LMF, Mahammed A, Prosser KE, Smith JR, Silverman MA, Walsby CJ, Gross Z, and Storr T (2019) A catalytic antioxidant for limiting amyloid-beta peptide aggregation and reactive oxygen species generation. Chem. Sci. 10 (6), 1634–1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Dedeoglu A, Cormier K, Payton S, Tseitlin KA, Kremsky JN, Lai L, Li X, Moir RD, Tanzi RE, Bush AI, Kowall NW, Rogers JT, and Huang X (2004) Preliminary studies of a novel bifunctional metal chelator targeting Alzheimer’s amyloidogenesis. Exp. Gerontol. 39 (11), 1641–1649. [DOI] [PubMed] [Google Scholar]
- (34).Beck MW, Oh SB, Kerr RA, Lee HJ, Kim SH, Kim S, Jang M, Ruotolo BT, Lee J-Y, and Lim MH (2015) A rationally designed small molecule for identifying an in vivo link between metal-amyloid-β complexes and the pathogenesis of Alzheimer’s disease. Chem. Sci. 6 (3), 1879–1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Beck MW, Derrick JS, Kerr RA, Oh SB, Cho WJ, Lee SJC, Ji Y, Han J, Tehrani ZA, Suh N, Kim S, Larsen SD, Kim KS, Lee JY, Ruotolo BT, and Lim MH (2016) Structure-mechanism-based engineering of chemical regulators targeting distinct pathological factors in Alzheimer’s disease. Nat. Commun. 7, 13115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Sharma AK, Pavlova ST, Kim J, Finkelstein D, Hawco NJ, Rath NP, Kim J, and Mirica LM (2012) Bifunctional compounds for controlling metal-mediated aggregation of the Aβ42 peptide. J. Am. Chem. Soc. 134 (15), 6625–6636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Sharma AK, Kim J, Prior JT, Hawco NJ, Rath NP, Kim J, and Mirica LM (2014) Small bifunctional chelators that do not disaggregate amyloid β fibrils exhibit reduced cellular toxicity. Inorg. Chem. 53 (21), 11367–11376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Sharma AK, Schultz JW, Prior JT, Rath NP, and Mirica LM (2017) Coordination chemistry of bifunctional chemical agents designed for applications in 64Cu PET imaging for Alzheimer’s disease. Inorg. Chem. 56 (22), 13801–13814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Bandara N, Sharma AK, Krieger S, Schultz JW, Han BH, Rogers BE, and Mirica LM (2017) Evaluation of 64Cu-based radiopharmaceuticals that target Aβ peptide aggregates as diagnostic tools for Alzheimer’s disease. J. Am. Chem. Soc. 139 (36), 12550–12558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Klunk WE, Wang Y, Huang G.-f., Debnath ML, Holt DP, and Mathis CA (2001) Uncharged thioflavin-T derivatives bind to amyloid-beta protein with high affinity and readily enter the brain. Life Sci. 69 (13), 1471–1484. [DOI] [PubMed] [Google Scholar]
- (41).Serdons K, Terwinghe C, Vermaelen P, Van Laere K, Kung H, Mortelmans L, Bormans G, and Verbruggen A (2009) Synthesis and evaluation of 18F-labeled 2-phenylbenzothiazoles as positron emission tomography imaging agents for amyloid plaques in Alzheimer’s disease. J. Med. Chem. 52 (5), 1428–1437. [DOI] [PubMed] [Google Scholar]
- (42).Roger M, Lima LMP, Frindel M, Platas-Iglesias C, Gestin J-F, Delgado R, Patinec V, and Tripier R (2013) Monopicolinate-dipicolyl derivative of triazacyclononane for stable complexation of Cu2+ and 64Cu2+. Inorg. Chem. 52 (9), 5246–5259. [DOI] [PubMed] [Google Scholar]
- (43).Zhao D, Sun J, Sun B, Zhao M, Zheng F, Huang M, Sun X, and Li H (2017) Intracellular antioxidant effect of vanillin, 4-methylguaiacol and 4-ethylguaiacol: three components in Chinese Baijiu. RSC Adv. 7 (73), 46395–46405. [Google Scholar]
- (44).Pajouhesh H, and Lenz GR (2005) Medicinal chemical properties of successful central nervous system drugs. NeuroRx 2 (4), 541–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Doerge DR, Divi RL, and Churchwell MI (1997) Identification of the colored guaiacol oxidation product produced by peroxidases. Anal. Biochem. 250 (1), 10–17. [DOI] [PubMed] [Google Scholar]
- (46).Jones MR, Mathieu E, Dyrager C, Faissner S, Vaillancourt Z, Korshavn KJ, Lim MH, Ramamoorthy A, Wee Yong V, Tsutsui S, Stys PK, and Storr T (2017) Multi-target-directed phenol-triazole ligands as therapeutic agents for Alzheimer’s disease. Chem. Sci. 8 (8), 5636–5643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Guilloreau L, Combalbert S, Sournia-Saquet A, Mazarguil H, and Faller P (2007) Redox chemistry of copper-amyloid-β: the generation of hydroxyl radical in the presence of ascorbate is linked to redox-potentials and aggregation state. ChemBioChem 8 (11), 1317–1325. [DOI] [PubMed] [Google Scholar]
- (48).Manevich Y, Held KD, and Biaglow JE (1997) Coumarin-3-carboxylic acid as a detector for hydroxyl radicals generated chemically and by gamma radiation. Radiat. Res. 148 (6), 580–591. [PubMed] [Google Scholar]
- (49).Pedersen JT, Teilum K, Heegaard NHH, Østergaard J, Adolph H-W, and Hemmingsen L (2011) Rapid Formation of a Preoligomeric Peptide-Metal-Peptide Complex Following Copper-(II) Binding to Amyloid β Peptides. Angew. Chem., Int. Ed. 50 (11), 2532–2535. [DOI] [PubMed] [Google Scholar]
- (50).Sharma AK, Pavlova ST, Kim J, Kim J, and Mirica LM (2013) The effect of Cu2+ and Zn2+ on the Aβ peptide aggregation 42 and cellular toxicity. Metallomics 5 (11), 1529–1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Zhang Y, Rempel DL, Zhang J, Sharma AK, Mirica LM, and Gross ML (2013) Pulsed hydrogen-deuterium exchange mass spectrometry probes conformational changes in amyloid beta (Aβ) peptide aggregation. Proc. Natl. Acad. Sci. U. S. A. 110, 14604–14609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Morris AM, Watzky MA, Agar JN, and Finke RG (2008) Fitting neurological protein aggregation kinetic data via a 2-step, minimal/"Ockham’s razor" model: the Finke-Watzky mechanism of nucleation followed by autocatalytic surface growth. Biochemistry 47 (8), 2413–2427. [DOI] [PubMed] [Google Scholar]
- (53).Wu G, Fang Y, Lu Z-H, and Ledeen RW (1998) Induction of axon-like and dendrite-like processes in neuroblastoma cells. J. Neurocytol. 27 (1), 1–14. [DOI] [PubMed] [Google Scholar]
- (54).Wengenack TM, Whelan S, Curran GL, Duff KE, and Poduslo JF (2000) Quantitative histological analysis of amyloid deposition in Alzheimer’s double transgenic mouse brain. Neuroscience 101 (4), 939–944. [DOI] [PubMed] [Google Scholar]
- (55).Oakley H, Cole SL, Logan S, Maus E, Shao P, Craft J, Guillozet-Bongaarts A, Ohno M, Disterhoft J, Van Eldik L, Berry R, and Vassar R (2006) Intraneuronal β-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: potential factors in amyloid plaque formation. J. Neurosci. 26 (40), 10129–10140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Ohno M, Chang L, Tseng W, Oakley H, Citron M, Klein WL, Vassar R, and Disterhoft JF (2006) Temporal memory deficits in Alzheimer’s mouse models: rescue by genetic deletion of BACE1. Eur. J. Neurosci. 23 (1), 251–260. [DOI] [PubMed] [Google Scholar]
- (57).Schwetye KE, Cirrito JR, Esparza TJ, Mac Donald CL, Holtzman DM, and Brody DL (2010) Traumatic brain injury reduces soluble extracellular amyloid-β in mice: A methodologically novel combined microdialysis-controlled cortical impact study. Neurobiol. Dis. 40 (3), 555–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Wilcock DM, Gordon MN, and Morgan D (2006) Quantification of cerebral amyloid angiopathy and parenchymal amyloid plaques with Congo red histochemical stain. Nat. Protoc. 1, 1591–1595. [DOI] [PubMed] [Google Scholar]
- (59).Sevigny J, Chiao P, Bussière T, Weinreb PH, Williams L, Maier M, Dunstan R, Salloway S, Chen T, Ling Y, O’Gorman J, Qian F, Arastu M, Li M, Chollate S, Brennan MS, Quintero-Monzon O, Scannevin RH, Arnold HM, Engber T, Rhodes K, Ferrero J, Hang Y, Mikulskis A, Grimm J, Hock C, Nitsch RM, and Sandrock A (2016) The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature 537, 50–56. [DOI] [PubMed] [Google Scholar]
- (60).Meraz-Ríos MA, Toral-Rios D, Franco-Bocanegra D, Villeda-Hernández J, and Campos-Peña V (2013) Inflammatory process in Alzheimer’s Disease. Front. Integr. Neurosci. 7, 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Katsumoto A, Takeuchi H, Takahashi K, and Tanaka F (2018) Microglia in Alzheimer’s disease: risk factors and inflammation. Front. Neurol. 9, 978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Shukla V, Zheng Y-L, Mishra SK, Amin ND, Steiner J, Grant P, Kesavapany S, and Pant HC (2013) A truncated peptide from p35, a Cdk5 activator, prevents Alzheimer’s disease phenotypes in model mice. FASEB J. 27 (1), 174–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Ostrowitzki S, Deptula D, Thurfjell L, Bark Bohrmann B, Brooks DJ, Klunk WE, Ashford E, Yoo K, Xu Z-X, Loetscher H, and Santarelli L (2012) Mechanism of amyloid removal in patients with Alzheimer disease treated with Gantenerumab. Arch. Neurol. 69 (2), 198–207. [DOI] [PubMed] [Google Scholar]
- (64).Doody RS, Thomas RG, Farlow M, Iwatsubo T, Vellas B, Joffe S, Kieburtz K, Raman R, Sun X, Aisen PS, Siemers E, Liu-Seifert H, and Mohs R (2014) Phase 3 trials of Solanezumab for mild-to-moderate Alzheimer’s disease. N. Engl. J. Med. 370 (4), 311–321. [DOI] [PubMed] [Google Scholar]
- (65).Salloway S, Sperling R, Fox NC, Blennow K, Klunk W, Raskind M, Sabbagh M, Honig LS, Porsteinsson AP, Ferris S, Reichert M, Ketter N, Nejadnik B, Guenzler V, Miloslavsky M, Wang D, Lu Y, Lull J, Tudor IC, Liu E, Grundman M, Yuen E, Black R, and Brashear HR (2014) Two phase 3 trials of Bapineuzumab in mild-to-moderate Alzheimer’s disease. N. Engl. J. Med. 370 (4), 322–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.