

Abstract
Endothelial function declines progressively across stages of the menopause transition; however, the mechanisms contributing to this decline are unknown. We hypothesized that differences in endothelial function among pre-, peri, and postmenopausal women are related to differences in estradiol and oxidative stress. Brachial artery flow-mediated dilation (FMD) was measured in 87 healthy women categorized by menopause stage (24 premenopausal, 17 early and 21 late perimenopausal, and 25 postmenopausal) before and after 3 days of ovarian hormone suppression (gonadotropin releasing hormone antagonist [GnRHant]) alone, and an additional 3 days of GnRHant with concurrent transdermal estradiol or placebo add-back treatment. In 82 women, FMD during acute vitamin C (antioxidant) infusion was measured before and after GnRHant + add-back. Before GnRHant, FMD was different among groups (p < 0.005; reduced across stages of menopause). Vitamin C increased FMD in late peri- and post- (p < 0.005) but not pre- or early perimenopausal women (p > 0.54). After GnRHant alone, FMD decreased in pre- and peri- (p < 0.01), but not postmenopausal women, and was restored to premenopausal levels by estradiol add-back in the pre- and perimenopausal groups. Vitamin C improved FMD in pre-, peri-, and postmenopausal women on GnRHant + placebo. There was no effect of vitamin C on FMD in women on GnRHant + estradiol. These observations support the concept that the decline in endothelial function across the menopause transition is related to the loss of ovarian estradiol. The decline in estradiol may alter redox balance, thereby increasing oxidative stress and impairing endothelial function.
Electronic supplementary material
The online version of this article (10.1007/s11357-020-00236-7) contains supplementary material, which is available to authorized users.
Keywords: Aging, Menopause, Sex hormones, Women, Vascular biology
Introduction
Endothelial dysfunction, characterized by reduced endothelial-dependent vasodilation, is a significant predictor of cardiovascular events in postmenopausal women (Rossi et al. 2008). Because the vascular endothelium plays a key role in the maintenance of vascular health, the loss of normal endothelial function is believed to be a critical step in the initiation and progression of atherosclerosis (Lakatta and Levy 2003). We previously demonstrated that endothelial dysfunction is apparent in the early stages of menopause (perimenopause) and, based on cross-sectional comparisons, worsens with the loss of ovarian function and duration of menopause (Moreau et al. 2012a). Because stage of menopause and chronological age are strongly correlated, it is difficult to isolate the effects of changes in the hormone milieu on the vascular endothelium from the aging process in cross-sectional study designs. Whether the decline in endothelial function during the menopause transition is initiated by changes in ovarian function and sex hormone levels is not clear. Moreover, the mechanisms underlying the impairment in endothelial function across the menopause transition are also unknown. If changes in ovarian function and sex hormone levels trigger the decline in endothelial function, understanding the biological mechanisms would be important for developing interventions to preserve the endothelium and maintain vascular health during the menopause transition.
Oxidative stress, which represents the imbalance between the production and destruction of reactive oxygen species (ROS), contributes to endothelial dysfunction in postmenopausal women (Moreau et al. 2013a). Elevated ROS can impair endothelial function by suppressing nitric oxide (NO) synthesis and scavenging NO, thereby reducing the bioavailability of NO (O’Donnell and Freeman 2001; Kojda and Harrison 1999). Estradiol has direct antioxidant effects and is thought to play an inhibitory role in the production and/or scavenging of ROS (Keaney et al. 1994; Sudoh et al. 2001; Lam et al. 2006). However, whether oxidative stress is mechanistically linked with endothelial dysfunction across the menopause transition and whether this is related to changes in estradiol levels is unknown.
Accordingly, in the present study, we tested the hypothesis that the impairment in endothelial function among women of varying menopausal status is associated with differences in sex hormones and, specifically, estradiol levels. To test this hypothesis, endothelial function was measured via brachial artery flow-mediated dilation (FMD) before and after 3 days of ovarian hormone suppression (gonadotropin-releasing hormone antagonist [GnRHant]) in pre-, peri-, and postmenopausal women. Brachial artery FMD is a well-established measure of conduit artery endothelium-dependent vasodilation. We hypothesized FMD would decrease following GnRHant in premenopausal and perimenopausal but would not change in postmenopausal women. Next, to isolate the role of estradiol, FMD was measured after an additional 3 days of GnRHant in women randomized to also receive placebo or estradiol treatment. We hypothesized that, when compared to placebo, estradiol would reverse the effect of GnRHant on FMD in pre- and perimenopausal women and increase FMD in postmenopausal women.
Additionally, we tested the hypothesis that the impairment in endothelial function in response to the suppression of ovarian estradiol is related to increased oxidative stress. FMD was measured during control (saline) conditions and during acute antioxidant treatment (vitamin C infusion) before and after GnRHant with concurrent placebo or estradiol treatment.
Methods
The study took place at the Colorado Clinical and Translational Sciences Institute Clinical and Translational Research Center (CCTSI CTRC). All procedures were reviewed and approved by the Colorado Multiple Institutional Review Board (COMIRB). All women gave their written informed consent to participate. This study was registered using the clinicaltrials.gov identifier NCT00608062. The authors had full access to all the data in the study and take responsibility for its integrity and the data analysis.
Population
The present study included healthy women aged 18–75 years who were categorized according to the Stages of Reproductive Aging Workshop (STRAW) criteria (Soules et al. 2001) and as described previously (Moreau et al. 2012a). Postmenopausal women had gone through menopause naturally. Inclusion criteria were fasted glucose < 126 mg/dL, resting blood pressure < 140/90 mmHg, sedentary or recreationally active (< 3 days/week vigorous exercise), nonsmokers, and healthy as determined by medical history, physical examination, standard blood chemistries (chemistry panel, CBC, and thyroid stimulating hormone), and ECG at rest and during incremental treadmill exercise. Additionally, participants had not used oral contraceptives or hormone therapy for at least 6 months, were not taking medications that influence cardiovascular function (i.e., antihypertensive, lipid lowering medications), and had not used vitamin supplements or anti-inflammatory medications for at least 4 weeks prior to the vascular visit.
Study design
The participants underwent baseline (i.e., pre-intervention) measurement of endothelial function (described below) during days 7–10 after the onset of menses (i.e., mid-follicular phase) in premenopausal and, when possible, perimenopausal women so that vascular comparisons between premenopausal and perimenopausal would be at a similar cycle stage. Some perimenopausal women were tested regardless of menstrual cycle phase because menses did not occur after 2 months. Participants then underwent a 6-day ovarian hormone suppression intervention that was composed of two phases: phase I—3 days of ovarian suppression alone, and phase II—3 days of ovarian suppression plus randomization to either estradiol or placebo add-back treatment. Vascular assessments were repeated after phase I and phase II. For phase I, the women were categorized into 4 groups: premenopausal, early perimenopausal, late perimenopausal, and postmenopausal. For phase II, the women were categorized into 3 groups: premenopausal, perimenopausal, and postmenopausal.
Phase I: ovarian hormone suppression alone
Following baseline testing, all women were administered a subcutaneous injection of a gonadotropin-releasing hormone antagonist (GnRHant; Ganirelix acetate, Antagon, Organon Pharmaceuticals, USA) dosed at 0.5 mg. This was followed by daily subcutaneous injections of GnRHant dosed at 0.25 mg/day. Ganirelix competes with natural GnRH for binding to membrane receptors on pituitary cells, and induces a rapid, reversible suppression of luteinizing hormone (LH) and follicle-stimulating hormone (FSH). The onset of LH suppression is ~ 1–2 h with the 0.5 mg dose; serum estradiol levels are reduced within 24–48 h and are maintained using the 0.25 mg dose (Oberyé et al. 1999). On day 3, blood samples were obtained and blood pressure and brachial artery FMD were reassessed at the same time of day as the baseline assessment.
Phase II: sex hormone add-back intervention
To isolate the effects of the loss of estradiol from the suppression of all ovarian hormones, women continued GnRHant (0.25 mg/day) treatment for three additional days and were randomly assigned to concurrent transdermal placebo or estradiol dosed at 0.075 mg/day. Blood samples were obtained, and blood pressure, heart rate, and brachial artery FMD were reassessed on the last day, at the same time of day as the baseline and phase I assessments. The dose of estradiol was chosen so that estradiol concentrations would increase to levels typically observed in the mid to late follicular phase of the menstrual cycle (~ 367–551 pmol/L). Participants, as well as the investigators involved in the acquisition and/or analysis of data, were blinded to estradiol and placebo treatment status.
Measurements
Participant characteristics
Screening blood pressure was measured in triplicate in the seated position with a semi-automated device (Dinamap, Johnson & Johnson) over the brachial artery as previously described (Tanaka et al. 2000). Total (percent of total mass) body fat was determined using dual-energy x-ray absorptiometry (Hologic Discovery). Peak oxygen consumption, a measure of peak aerobic power, was determined using an incremental treadmill protocol as described previously (Tanaka et al. 1997). Leisure time physical activity was determined by the Modifiable Activity Questionnaire (Pereira et al. 1997). Dietary composition and caloric intake were determined from 3-day food record and were analyzed by the CCTSI CTRC Bionutrition Core as described previously (Stevenson et al. 1995).
Blood sampling
Fasted plasma concentrations of glucose, insulin, total (Roche Diagnostic Systems, Indianapolis, IN) and high-density-lipoprotein (HDL-C, Diagnostic Chemicals, Ltd, Oxford CT) cholesterol were determined using enzymatic/colorimetric methods, and low-density-lipoprotein cholesterol (LDL-C) was determined using the Friedewald equation (Friedewald et al. 1972). Serum concentrations of FSH, estradiol, progesterone, and sex hormone-binding globulin were measured using chemiluminescense (Beckman Coulter). Estrone was measured by radioimmunoassay and total testosterone by a 1-step competitive assay (Beckman Coulter). Norepinephrine was measured using HPLC from serum with EGTA/glutathione preservative added, and plasma endothelin-1 and interleukin (IL)-6 were measured using an enzyme-linked immunoassay (Davy et al. 1995; Goddard and Webb 2000). Oxidized LDL, an indirect measure of oxidative stress, was determined with ELISA plate assays (Alpco Diagnostics, Windham, NH). Total antioxidant status (TAS), a measure of the overall antioxidant defenses, was determined on serum samples using the Randox Laboratories enzymatic kit (Oceanside, CA), and plasma total glutathione was quantified using highly specific liquid chromatography tandem mass spectrometry (LC-MS/MS) assay (Beehler et al. 1989). High-sensitivity C-reactive protein (hs-CRP) was measured using immunoturbidimetric method. All assays were performed by the CCTSI-CTRC Core laboratory, with the exception of total glutathione, which was performed by the University of Colorado Clinical Research & Development Service Center, Quantitative Mass Spectrometry Laboratory.
Endothelial function
The women were studied in the supine position following an overnight fast with proper hydration (water only) and abstinence from exercise for at least 20 h on the days of the main experimental vascular measurements. To control for potential diet effects on vascular function, participants consumed standardized meals for 2 days before each vascular visit. The diets were constructed by the CTRC Bionutrition Core based on dietary composition and caloric intake from 3-day food records. Prior to starting the vascular measures, an intravenous catheter was placed into an antecubital vein for saline and vitamin C infusion and blood sampling (described below).
Brachial artery FMD was determined following 5 min of forearm cuff occlusion with duplex ultrasonography (GE Vivid I) using a multi-frequency linear-array transducer as previously described (Celermajer et al. 1992; Eskurza et al. 2004a; Moreau et al. 2012b). The dilation of the brachial artery in response to the stimulus of forearm ischemia is dependent on the release of vasodilators, predominantly NO, from the vascular endothelium (Doshi et al. 2001). Vascular endothelium-independent dilation was determined in a subsample of women (N = 45) by measuring brachial artery dilation in response to sublingual nitroglycerine (glyceryl trinitrate [GTN], 0.4 mg) continuously for 10 min. All images were coded by number and masked for group assignment and condition. Images were considered valid if clear vascular boundaries were able to be identified; those without clear boundaries were not included in the analyses. All procedures conformed strictly with published guidelines for assessing FMD in human subjects (Corretti et al. 2002).
Oxidative stress-mediated suppression of endothelial function
The contribution of oxidative stress to endothelial dysfunction was determined using a well-described experimental model using vitamin C to temporarily and reversibly reduce ROS (Hornig et al. 1998; Eskurza et al. 2004b; Moreau et al. 2005, 2006, 2007). Briefly, after 20 min of intravenous normal saline infusion (control) and after 20 min of intravenous vitamin C (ascorbic acid) infusion, supine blood pressure and heart rate were measured in triplicate, followed by brachial artery FMD and GTN-mediated vasodilation. The concentration of the vitamin C solution was 0.06 g ascorbic acid per kg fat-free mass per 100 mL normal saline. A bolus of 100 mL ascorbic acid solution was given at 5 mL/min over 20 min followed by a constant infusion at 1.7 mL/min until vascular testing was completed. Total ascorbic acid dose did not exceed 7.5 g. This dose of vitamin C improves carotid artery compliance, femoral artery blood flow, and brachial artery FMD in estrogen-deficient postmenopausal women (Moreau et al. 2005, 2007, 2013a), and restores brachial artery FMD in healthy older men (Eskurza et al. 2004a). The difference in FMD following vitamin C vs. saline was taken as a measure of tonic suppression of FMD by ROS. Blood sampling for sex hormones, metabolic risk factors, and other blood markers occurred after the 20-min bolus saline infusion. Only blood sampling for sex hormones, TAS, IL-6, CRP, ET-1, and norepinephrine was repeated after the GnRHant intervention.
Statistical analysis
All data elements were examined using descriptive statistics and graphical summaries; skewed distributions were improved by transformation. Analyses were conducted using either SPSS software version 26 (IBM Corp.) or SAS software version 9.4 M5 (SAS Institute Inc., Cary, NC, USA). An ANOVA was used to assess the main effect of menopause stage in baseline participant characteristics. To determine the effects of GnRHant alone (i.e., phase I), a mixed ANOVA with menopause stage (premenopausal, early and late perimenopausal, and postmenopausal) as a between-subject factor, and before vs. after GnRHant alone as the within-subject factor, was used to determine the effects of GnRHant on brachial artery FMD measured during the control condition (saline). Blood and hemodynamic parameters were examined in a similar fashion. When the overall F statistic was significant (p < 0.05), paired Student’s t tests for within-group contrasts and independent Student’s t tests for between-group contrasts were performed to identify significant differences among the mean values. In secondary analyses, the change in FMD with GnRHant was adjusted for age using ANCOVA.
To determine the effects of GnRHant plus add-back treatment of either placebo or estradiol. (i.e., phase II), repeated measurements of FMD were modeled using a general linear mixed model with maximum likelihood estimation. This approach is conceptually identical to repeated measures analysis of variance but has the advantage of using all available data; estimates are unbiased under the assumption that missing data are missing at random. An unstructured covariance structure allowing for heterogeneity in the parameter estimates for each level of menopause stage (premenopausal, perimenopausal, and postmenopausal) was chosen for the model based on using Akaike’s information criterion (AIC) to compare various covariance structures. The study was not powered to detect differences among the two perimenopausal groups; therefore, the early and late perimenopausal groups were collapsed into one group (i.e., perimenopausal). A cell means model was used to estimate the mean FMD level for each combination of treatment, menopause stage, and study phase (baseline, phase I, and phase II). Linear contrasts were then constructed to estimate all effects of interest, such as the within-group effects of differences in treatment or the between-group differences. This approach was used when analyzing differences between the placebo and estradiol add-back groups and for analyzing group differences between the vitamin C and saline conditions. Standard statistical conventions of performing 2-sided tests and p values < 0.05 designating statistically significant results were used. Data are reported as means ± SD unless otherwise specified.
Results
A total of 155 women consented to participate. During screening, 33 women did not qualify (12 premenopausal, 11 early- and 3 late- perimenopausal, and 7 postmenopausal). Thirty-one withdrew from participation: 23 before baseline testing and randomization; 7 after baseline testing, but before ovarian suppression intervention and randomization; and one after beginning the ovarian suppression intervention; 91 completed phases I and II. Four participants did not have usable FMD data due to poor image quality or technical issues with image acquisition; results for the remaining 87 women are presented below.
The clinical characteristics of the women who participated in phase I are presented in Table 1. Clinical characteristics of participants by menopause stage and group randomization (i.e., placebo or estradiol) are presented in the on-line data supplement in Table S1. There was a significant effect of menopause stage on total body fat content, total cholesterol, peak aerobic power, oxidized LDL, and glutathione (all p < 0.05); these parameters were not different at baseline in women randomized to placebo vs. estradiol treatment. In the subset of women who had dietary records (N = 64), there were no differences in caloric intake, macronutrients, or selected antioxidants (i.e., vitamins C and E) among the groups (all p > 0.22; data not shown). There were significant main effects of menopause stage on baseline FSH, estradiol, estrone, progesterone, and testosterone (Table 2, all p < 0.05); these effects were not different at baseline in women randomized to placebo or estradiol treatment (see on-line data supplement Table S2).
Table 1.
Baseline characteristics of premenopausal (pre), early and late perimenopausal (peri), and postmenopausal (post) women
| Parameter | Pre | Early peri | Late peri | Post | p value |
|---|---|---|---|---|---|
| n | 24 | 17 | 21 | 25 | |
| Ethnicity | 0.709 | ||||
|
Hispanic or Latino Not Hispanic or Latino Unknown |
4 (16.7) 19 (79.2) 1 (4.2) |
2 (11.1) 15 (83.3) 0 (0.0) |
2 (9.5) 19 (90.5) 0 (0.0) |
4 (16.0) 20 (80.0) 1 (4.0) |
|
| Race | 0.524 | ||||
|
Asian African American White More than one race |
3 (12.5) 1 (4.2) 17 (70.8) 3 (12.5) |
1 (5.6) 1 (5.6) 15 (83.3) 0 (0.0) |
0 (0.0) 1 (4.8) 18 (85.7) 2 (9.5) |
1 (4.0) 0 (0.0) 22 (88.0) 2 (8.0) |
|
| Age, years | 33 ± 7 | 49 ± 3 | 50 ± 4 | 58 ± 6 | < 0.001 |
| Body mass, kg | 65.5 ± 13.3 | 66.5 ± 6.4 | 68.0 ± 12.8 | 70.1 ± 12.0 | 0.59 |
| BMI | 24.1 ± 5.3 | 25.0 ± 2.7 | 24.4 ± 4.3 | 26.6 ± 4.4 | 0.21 |
| Total body fat, % | 30 ± 7 | 35 ± 5 | 36 ± 7 | 39 ± 4 | < 0.001 |
| Systolic BP, mmHg | 109 ± 7 | 113 ± 12 | 115 ± 13 | 119 ± 14 | 0.063 |
| Diastolic BP, mmHg | 71 ± 6 | 72 ± 7 | 71 ± 8 | 74 ± 9 | 0.53 |
| Total cholesterol, mmol/L | 3.9 ± 0.8 | 4.3 ± 0.7 | 4.3 ± 0.9 | 4.7 ± 0.9 | 0.006 |
| LDL cholesterol, mmol/L | 2.3 ± 0.6 | 2.5 ± 0.7 | 2.6 ± 0.8 | 2.9 ± 0.9 | 0.058 |
| Glucose, mmol/L | 4.7 ± 0.5 | 4.8 ± 0.4 | 4.5 ± 0.5 | 4.8 ± 0.7 | 0.21 |
| Insulin, pmol/La | 36 (18–60) | 28(21–56) | 30 (18–60) | 30 (24–66) | 0.71 |
| Oxidized LDL, U/L | 43.7 ± 15.7 | 41.8 ± 11.4 | 50.5 ± 17.0 | 53.4 ± 17.3 | 0.033 |
| Glutathione, μM | 5.1 ± 2.4 | 5.6 ± 2.4 | 5.8 ± 1.0 | 4.6 ± 2.1 | 0.029 |
| TAS, mmol/L | 1.40 ± 0.19 | 1.34 ± 0.15 | 1.31 ± 0.13 | 1.30 ± 0.15 | 0.16 |
| CRP, mg/dLa | 0.7 (0.3–0.7) | 0.9 (0.5–1.9) | 0.9 (0.5–1.7) | 1.1 (0.5–1.4) | 0.54 |
| IL-6, pg/mLa | 0.70 (0.56–1.29) | 0.70 (0.58–1.23) | 0.93 (0.62–1.40) | 0.92 (0.62–1.24) | 0.80 |
| Endothelin-1, pg/mLc | 5.2 ± 1.2 | 6.0 ± 1.2 | 6.2 ± 1.7 | 6.3 ± 1.2 | 0.15 |
| Norepinephrine, pg/mLa | 168 (106–225) | 245 (211–291) | 227 (159–272) | 231 (147–317) | 0.21 |
| LTPAb | 17.2 ± 11.8 | 17.3 ± 12.8 | 15.1 ± 11.7 | 14.9 ± 18.1 | 0.91 |
| VO2peak, ml/kg/min | 33.1 ± 6.0 | 27.6 ± 4.4 | 27.3 ± 5.4 | 24.4 ± 3.4 | < 0.001 |
Values are mean ± SD, unless otherwise indicated. aValues are median (interquartile range). bn = 86; cn = 84. BMI, body mass index; BP, blood pressure; LDL, low-density lipoprotein; TAS, total antioxidant status; CRP, C-reactive protein; IL-6, interleukin-6; LTPA, leisure time physical activity; VO2peak, peak aerobic power
Table 2.
Serum reproductive hormone concentrations at baseline and after gonadotropin-releasing hormone antagonist alone and with concurrent placebo or estradiol add-back treatment in premenopausal (pre), perimenopausal (peri), and postmenopausal (post) women
| Placebo | Estradiol | |||||
|---|---|---|---|---|---|---|
| Pre | Peri | Post | Pre | Peri | Post | |
| n | 12 | 21 | 13 | 12 | 17 | 12 |
| FSH, μIU/mL | ||||||
|
Baseline# GnRHant GnRHant + add-back |
7.2 ± 3.1 7.0 ± 2.4 6.5 ± 1.8 |
39.0 ± 32.0*‡ 26.1 ± 21.5*‡ 29.9 ± 24.6*‡ |
85.1 ± 29.9*†‡ 54.7 ± 19.5*†‡ 56.2 ± 20.8*†‡ |
5.5 ± 1.1 5.8 ± 2.0 5.0 ± 1.7 |
41.3 ± 39.1*‡ 26.9 ± 21.0*‡ 23.1 ± 16.3*‡ |
73.9 ± 33.5*†‡ 51.7 ± 30.7*†‡ 39.2 ± 24.7*§‡|| |
| Estradiol, pmol/La | ||||||
|
Baseline# GnRHant GnRHant + add-back |
270 (128–484) 127 (96–243)‡ 134 (117–183)‡ |
147 (62–316) 99 (37–321) 106 (37–231) |
37 (37–42) 37 (37–42) 37 (37–39) |
229 (117–332) 147 (87–183)‡ 202 (140–481)§|| |
360 (46–508) 92 (37–468) 187 (81–569)§|| |
39 (37–49) 37 (37–42) 165 (128–223)§||‡ |
| Progesterone, nmol/La | ||||||
|
Baseline# GnRHant GnRHant + add-back |
2.1 (1.0–3.1) 1.7 (0.7–3.7) 1.0 (0.6–2.1) |
1.3 (1.0–1.9) 1.0 (0.6–2.5)‡ 1.6 (0.7–4.9) |
0.9 (0.6–1.3) 0.6 (0.3–1.1) 0.6 (0.6–1.1) |
1.8 (0.7–2.5) 1.3 (0.4–2.9) 1.0 (0.5–2.7) |
1.1 (0.6–2.9) 1.0 (0.6–1.9) 0.8 (0.6–3.6) |
1.1 (0.6–1.3) 1.0 (0.3–1.3) 1.0 (0.6–1.0) |
| Estrone, pmol/La | ||||||
|
Baseline# GnRHant GnRHant + add-back |
226 (147–278) 130 (106–232)‡ 191 (126–242) |
137 (102–265) 141 (9–220) 118 (83–213)‡ |
96 (89–124) 104 (72–120) 107 (80–113) |
191 (130–257) 143 (130–167)‡ 211 (144–377)§|| |
159 (115–303) 118 (81–194) 192 (130–364)§|| |
93 (76–148) 96 (75–145) 150 (108–226)§||‡ |
| SHBG, nmol/L | ||||||
|
Baseline GnRHant GnRHant + add-back |
58 (36–81) 56 (36–81) 55 (38–71) |
66 (37–98) 70 (41–111) 69 (44–112) |
47 (37–76) 52 (41–80) 52 (39–89) |
55 (36–86) 51 (36–67) 52 (39–69) |
61 (40–103) 62 (42–83) 64 (43–88) |
67 (39–104) 70 (39–106) 74 (41–107) |
| Testosterone, nmol/La | ||||||
|
Baseline# GnRHant GnRHant + add-back |
1.0 (0.7–1.5) 1.0 (0.6–1.6) 0.9 (0.6–1.4)|| |
0.7 (0.6–0.9) 0.6 (0.6–0.8) 0.6 (0.6–0.7) |
0.6 (0.6–0.8) 0.6 (0.6–0.7) 0.6 (0.6–0.6)‡ |
1.1 (0.7–1.5) 0.8 (0.7–1.2)‡ 0.9 (0.6–1.2) |
0.7 (0.6–1.2) 0.6 (0.6–1.1) 0.6 (0.6–0.9) |
0.7 (0.6–1.0) 0.6 (0.6–0.9) 0.6 (0.6–0.7) |
Values are mean ± SD, unless otherwise indicated. aValues are median (interquartile range). #p < 0.05 main effect of menopause stage; *p < 0.016, compared to pre; †p < 0.016 vs. peri; ‡p < 0.016 vs. baseline of the same menopause stage; §p < 0.016 vs. placebo of same menopause stage; ||p < 0.016 vs. GnRHant of same menopause stage; GnRHant, gonadotropin-releasing hormone antagonist; FSH, follicle-stimulating hormone; SHBG, sex hormone-binding globulin
Effects of GnRHant alone and with concurrent placebo or estradiol add-back treatment on reproductive hormones and other blood parameters
Following GnRHant alone, serum FSH decreased in early and late perimenopausal and postmenopausal women; estradiol and estrone decreased in premenopausal and late perimenopausal women, and progesterone decreased in late perimenopausal and postmenopausal women (see on-line data supplement Table S2, all p < 0.025).
Following the GnRHant plus add-back the decrease in FSH persisted in perimenopausal and postmenopausal women, regardless of add-back group, and decreased further in the postmenopausal treated with GnRHant with concurrent estradiol (all p < 0.005; Table 2). Similarly, the decrease in estradiol persisted, and testosterone also decreased in premenopausal women treated with placebo add-back (p < 0.05), whereas only estrone decreased in perimenopausal women. Estradiol add-back treatment increased estradiol and estrone in premenopausal and perimenopausal women (p < 0.01) to where it was no longer different from baseline levels (p > 0.40); estradiol levels were increased above baseline levels in postmenopausal women (p < 0.005).
There were no significant effects of GnRHant alone or with add-back treatment on TAS, inflammatory markers ET-1, or norepinephrine (Table 3).
Table 3.
Blood parameters at baseline and after gonadotropin releasing hormone antagonist alone and with concurrent placebo or estradiol add-back treatment in premenopausal (Pre), perimenopausal (Peri), and postmenopausal (Post) women
| Placebo | Estradiol | |||||
|---|---|---|---|---|---|---|
| Parameters | Pre | Peri | Post | Pre | Peri | Post |
| n | 12 | 21 | 13 | 12 | 17 | 12 |
| TAS, nmol/L | ||||||
| Baseline | 1.4±0.2 | 1.3±0.1 | 1.3±0.2 | 1.4±0.2 | 1.3±0.2 | 1.3±0.2 |
| GnRHant | 1.5±0.1 | 1.3±0.1 | 1.4±0.3 | 1.4±0.2 | 1.3±0.2 | 1.3±0.1 |
| GnRHant+Add-back | 1.4±0.1 | 1.3±0.1 | 1.3±0.2 | 1.4±0.1 | 1.3±0.1 | 1.3±0.2 |
| IL-6, pg/mLa | ||||||
| Baseline | 0.7(0.6-1.3) | 0.8(0.6-1.3) | 0.8(0.6-1.2) | 0.8(0.5-1.3) | 0.8(0.5-1.3) | 0.9(0.6-1.8) |
| GnRHant | 0.9(0.6-1.1) | 1.1(0.6-1.3) | 1.0(0.8-1.6) | 0.7(0.5-1.1) | 1.1(0.8-1.5) | 0.9(0.7-1.8) |
| GnRHant+Add-back | 0.8(0.6-1.4) | 1.0(0.7-1.1) | 1.1(0.8-1.5) | 0.9(0.7-1.9) | 1.1(0.7-1.4) | 1.2(0.8-2.1) |
| CRP, mg/La | ||||||
| Baseline | 0.7(0.3-3.7) | 0.9(0.5-1.9) | 1.1(0.8-1.4) | 0.7(0.3-2.3) | 0.9(0.5-1.5) | 1.1(0.3-3.9) |
| GnRHant | 0.8(0.4-4.2) | 0.9(0.5-1.6) | 1.3(0.7-2.1) | 1.2(0.5-2.2) | 0.9(0.6-2.2) | 1.4(0.3-3.8) |
| GnRHant+Add-back | 0.7(0.3-3.3) | 1.0(0.5-1.4) | 1.1(0.7-2.2) | 1.1(0.3-2.6) | 0.9(0.6-1.8) | 1.5(0.5-4.0) |
| ET-1, pg/mL b | ||||||
| Baseline # | 5.2±0.9 | 6.2±1.6 | 6.4±1.1 | 5.2±1.5 | 5.9±1.3 | 6.2±1.4 |
| GnRHant | 5.5±0.9 | 5.9±0.9 | 6.5±0.9 | 5.5±1.7 | 6.0±1.8 | 6.5±2.1 |
| GnRHant+Add-back | 5.0±0.7 | 5.7±1.9 | 6.4±0.9 | 6.0±1.1 | 5.9±1.6 | 6.0±1.8 |
| Norepinephrine, pg/mLa | ||||||
| Baseline # | 184(101-217) | 247(164-295) | 242(228-331) | 161(108-230) | 227(168-280) | 231(127-349) |
| GnRHant | 156(103-252) | 233(159-341) | 217(180-282) | 144(116-164) | 235(183-282) | 380(156-362) |
| GnRHant+Add-back | 158(111-229) | 217(152-282) | 221(162-303) | 146(118-214) | 231(168-295) | 204(168-280) |
Values are mean ± SD, unless otherwise indicated. a Values are median (interquartile range). b n=84 # P < 0.05 main effect of menopause stage. GnRHant gonadotropin releasing hormone antagonist; TAS total antioxidant status; IL-6 interluekin-6; CRPC-reactive protein; ET-1 endothelin-1
Effects of GnRHant alone and with concurrent placebo or estradiol add-back treatment on brachial artery FMD
At baseline, brachial artery FMD was different across the stages of the menopause transition (Fig. 1a, p < 0.005), and GTN-mediated vasodilation tended to be different across menopause stages (Fig. 1b, p = 0.08). Following GnRHant alone, brachial artery FMD decreased from baseline by ~ 24% in both premenopausal and early perimenopausal women (Fig. 1a, p < 0.005), ~ 11% in late perimenopausal women (p = 0.001), and ~ 9% in postmenopausal women (p = 0.015). The change in FMD in premenopausal women was significantly different from late perimenopausal and postmenopausal women (Fig. 2, p < 0.01), and significantly different between early perimenopausal and postmenopausal women (p < 0.05). The effect of menopause stage remained after adjusting for age (p = 0.012). After 3 days of GnRHant, FMD was no longer different between early perimenopausal women and postmenopausal women. There was a significant main effect of GnRHant on GTN-mediated vasodilation (N = 45, Fig. 1b; p < 0.005), but the effect was not different by menopause stage.
Fig. 1.

Ovarian hormone suppression decreased endothelial function in premenopausal and perimenopausal women. a Brachial artery FMD and b nitroglycerine (GTN)-mediated vasodilation before (black bars) and following GnRH antagonist (GnRHant) treatment (gray bars). Data are means ± SE. *p < 0.01 vs. premenopausal women under the same condition; †p < 0.05 vs. baseline condition early perimenopausal; ‡p < 0.01 vs. baseline condition of the same group. In the subsample of women (N = 45; Pre, premenopausal, N = 15; Early Peri, early perimenopausal, N = 8; Late Peri, late perimenopausal, N = 9; Post, postmenopausal, N = 23) who had GTN-mediated dilation performed, there was a significant main effect of time with all groups decreasing after GnRHant; p < 0.005)
Fig. 2.
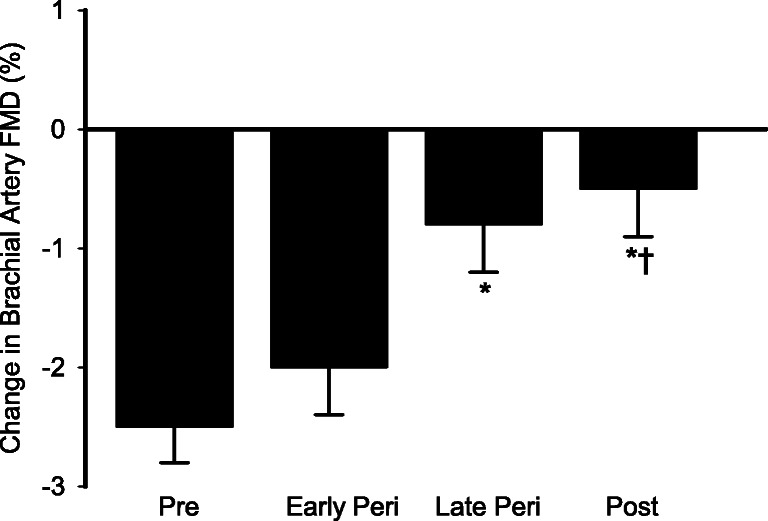
The change in endothelial function (brachial artery FMD) following GnRH antagonist treatment was dependent on menopausal state. Data are means ± SE. *p < 0.01 vs. premenopausal women; †p < 0.05 vs. early perimenopausal women
The decrease in brachial artery FMD following GnRHant alone was restored to baseline (pre-GnRHant) following GnRHant with add-back of estradiol in premenopausal women (Fig. 3). In perimenopausal and postmenopausal women, FMD was increased above baseline levels (both p < 0.001) with estradiol add-back, such that they were no longer different from premenopausal baseline levels (both p > 0.10). Brachial artery FMD was decreased from baseline in GnRHant with placebo add-back in all menopausal stages. There were no effects of GnRHant alone or with add-back treatments on brachial artery diameter, blood pressure, or heart rate (see on-line data supplement in Tables S3 and S4). There was no effect of estradiol add-back treatment on GTN-mediated vasodilation (data not shown), possibly due to small sample sizes in (5–7/group).
Fig. 3.
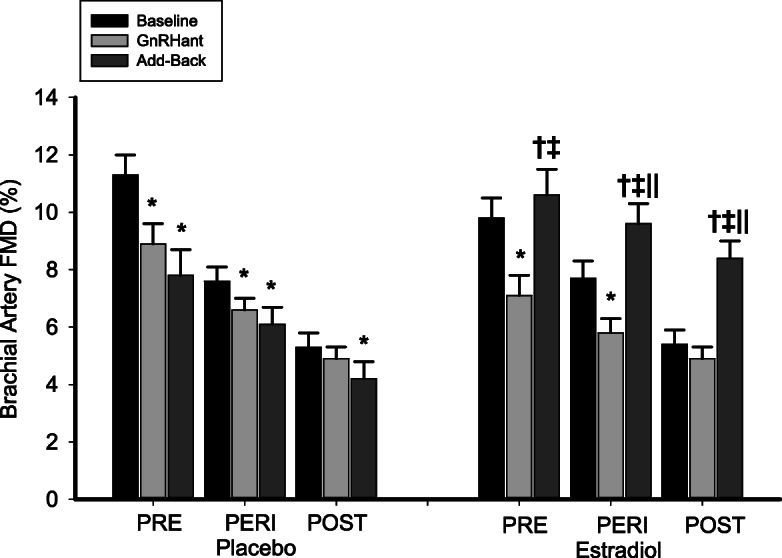
Estradiol add-back treatment reversed the impairment in endothelial function with ovarian suppression alone in premenopausal women and increased endothelial function in perimenopausal and postmenopausal women to normal premenopausal levels. Brachial artery FMD before and following GnRH antagonist (GnRHant) alone and with placebo or estradiol add-back treatment. Data are means ± SE. *p < 0.01 vs. baseline of the same group; †p < 0.05 vs. GnRH antagonist alone condition of the same group; ‡p < 0.01 vs. baseline condition of the same group; ‖p < 0.001 vs. placebo add-back condition of the same group
Vitamin C effects on brachial artery FMD at baseline, following GnRHant alone and with concurrent placebo or estradiol add-back treatment
At baseline, FMD during vitamin C infusion was completed in 82 women (22 premenopausal, 16 early perimenopausal, 21 late perimenopausal, and 23 postmenopausal). Vitamin C infusion increased brachial artery FMD in late perimenopausal and postmenopausal women (both p < 0.01), but had no effect in premenopausal or early perimenopausal women (both p > 0.50; Fig. 4). Brachial artery FMD during the vitamin C infusion remained lower in postmenopausal compared to premenopausal women but group differences between premenopausal and late perimenopausal women and among the perimenopausal and postmenopausal groups were no longer significant. Basal brachial artery diameter increased during the vitamin C infusion in all groups (data not shown), but there were no differences in diameters among the groups. There was a significant effect of vitamin C on brachial artery vasodilation to GTN (p = 0.004) that did not differ by menopause stage.
Fig. 4.

Acute antioxidant vitamin C infusion increased endothelial function in late perimenopausal and postmenopausal women. Baseline a brachial artery FMD and b nitroglycerine (GTN)-mediated vasodilation before (black bars) and following acute vitamin C infusion (hatched bars). Data are means ± SE (N = 82; Pre, premenopausal, N = 22; Early Peri, early perimenopausal, N = 16; Late Peri, late perimenopausal, N = 21; Post, postmenopausal, N = 23). *p < 0.01 vs. premenopausal women under the same condition; †p < 0.05 vs. baseline condition early perimenopausal; ‡p < 0.01 vs. baseline condition of the same group. For GTN, there was a significant main effect of the time (N = 39; Pre = 11, Early Peri = 7, Late Peri = 11, Post = 10; p = 0.004)
In women randomized to GnRHant with concurrent placebo treatment, the vitamin C infusion reversed the effect of the GnRHant and increased brachial artery FMD back to baseline (saline control) level in premenopausal women, and increased FMD above baseline level in perimenopausal and postmenopausal women (Fig. 5a–c). There was no effect of the vitamin C infusion on FMD in women treated with GnRHant plus estradiol regardless of menopause stage (all p > 0.12).
Fig. 5.

Acute vitamin C treatment reversed the effect of ovarian suppression in placebo-treated women but had no effect in estradiol-treated women. Brachial artery FMD during saline (solid bars) and vitamin C infusion (hatched bars) before (black bars) and following GnRH antagonist (gray bars) plus placebo or estradiol treatment in a premenopausal, b perimenopausal, and c postmenopausal women. Data are means ± SE. *p < 0.01 vs. baseline saline of the same group; †p < 0.05 vs. GnRH antagonist plus add-back saline of the same group
Discussion
In the present study, we extended our previous cross-sectional work to determine the underlying mechanisms for the progressive reduction in endothelial function in healthy women across the stages of the menopause transition. Using an ovarian hormone suppression model, we showed that brachial artery FMD (macrovascular endothelial function) decreases with short-term GnRHant treatment to suppress ovarian function. Moreover, we showed that the effect of GnRHant treatment was reversed with estradiol add-back, suggesting the decline in endothelial function across the menopause transition is related to changes in estradiol levels. Finally, the decline in endothelial function with the change in estradiol appears to be driven by increased oxidative stress, as indicated by an increase in brachial artery FMD with acute antioxidant (vitamin C) treatment in late perimenopausal and postmenopausal women, and in women treated with GnRHant. Collectively, these findings provide proof-of-concept evidence for the decline in estradiol as a triggering event that leads to increased oxidative stress and subsequent endothelial dysfunction in women.
Ovarian hormone regulation of endothelial function
Our previous cross-sectional work demonstrated that vascular endothelial function measured by brachial artery FMD progressively deteriorated across stages of the menopausal transition (Moreau et al. 2012a). The late perimenopausal transition appeared to be the most critical time period for adverse changes in the vasculature, consistent with previous observations (Wildman et al. 2008; Matthews et al. 2009; Santoro and Sutton-Tyrrell 2011). Indeed, although endothelial function was impaired in early perimenopausal compared to premenopausal women, the level of impairment in late perimenopausal women was twice that of age-matched early perimenopausal women (Moreau et al. 2012a). We speculated that this apparent “acceleration” in vascular aging was related to the reduction in estradiol level in the late perimenopausal transition. However, causality is difficult to determine because it is nearly impossible to uncouple the tight association between menopause and aging in cross-sectional comparisons of women in different stages of menopause, or even in a prospective study design. Thus, in the present study, we employed an innovative pharmacological ovarian suppression intervention that has previously been used to distinguish the independent effects of changes in ovarian hormones from other factors (e.g., increased adiposity and blood pressure) that may influence vascular aging, (Shea et al. 2015; Gavin et al. 2018a, b; Melanson et al. 2018) to better understand how changes in ovarian hormones, and specifically estradiol, influence the decline in endothelial function across stages of the menopause transition.
In the present study, we found that suppressing ovarian hormones for 3 days with a GnRHant decreased endothelial function, and that the effect was independent of age and dependent on menopausal state, with greater decreases in brachial artery FMD in premenopausal and early perimenopausal women than in late perimenopausal and postmenopausal women. This finding suggests that changes in ovarian hormones contribute to the decline in endothelial function with the menopause transition in healthy women. We also observed decreases in GTN-mediated vasodilation, suggesting that ovarian hormones may also regulate vascular smooth muscle cell function.
Our finding that GnRHant with estradiol add-back treatment completely reversed the impairment of GnRHant alone on brachial artery FMD in premenopausal women was consistent with previous observations demonstrating greater brachial artery FMD in premenopausal women treated with GnRHant with estradiol compared with GnRHant alone (Meendering et al. 2008; Miner et al. 2011). Our study extends these observations by demonstrating that estradiol add-back increased FMD in perimenopausal and postmenopausal women to the normal premenopausal level. Collectively, these observations support the idea that estrogen protects vascular endothelial function against vascular aging in women, and that the loss of estrogen during the menopause transition may initiate endothelial dysfunction in women.
Oxidative stress and endothelial dysfunction across stages of the menopause transition
One of the key mechanisms underlying endothelial dysfunction in various populations (e.g., aging, diabetes, hypertension, cardiovascular disease) is oxidative stress (Eskurza et al. 2004a; Donato et al. 2007; Heitzer et al. 2001; Taddei et al. 2001), which is an imbalance between the production of ROS and the ability of antioxidants to detoxify ROS (Harman 2006). Excessive ROS production impairs endothelial function by suppressing NO synthesis and by increasing oxidative breakdown of NO, decreasing its overall bioavailability (O’Donnell and Freeman 2001; Kojda and Harrison 1999). Preclinical studies demonstrate that ovariectomized animals have elevated levels of ROS compared to intact animals, and treating ovariectomized animals with estradiol prevents the development of ROS and preserves endothelial function, presumably by protecting NO from ROS scavenging (Keaney et al. 1994; Sudoh et al. 2001). Thus, oxidative scavenging of NO due to a decline in circulating estrogen concentrations could explain the impaired endothelial function with the menopause transition in women. The findings from the present study are consistent with this idea. First, at baseline (i.e., pre- gonadal suppression intervention), brachial artery FMD increased during the systemic infusion of the antioxidant vitamin C in late perimenopausal and postmenopausal women, but there was no effect in premenopausal and early perimenopausal women, possibly because circulating estrogen levels provided protection against oxidative stress. Additionally, the vitamin C infusion rescued the decrease in FMD in premenopausal and perimenopausal women treated with GnRHant plus placebo add-back, and improved FMD in postmenopausal placebo add-back, but had no effect on FMD in women treated with GnRHant plus estradiol add-back. These findings are consistent with a previous observation by Virdis et al., who demonstrated that a local infusion of vitamin C into the brachial artery reversed the impaired resistance vessel endothelial function following oophorectomy in premenopausal women (Virdis et al. 2000). These authors also found no effect of the vitamin C infusion on resistance vessel endothelial function in the women before oophorectomy, in healthy controls, or in oophorectomized women treated with estradiol for 3 months (Virdis et al. 2000). Collectively, these observations are consistent with the idea that declining estradiol levels with the menopausal transition results in oxidative stress that tonically suppresses endothelial function. Interestingly, the vitamin C infusion decreased GTN-mediated vasodilation in all menopause stage groups for reasons that are unclear. Some ROS play important roles in cell signaling, and vitamin C may stimulate ROS production depending on the cellular state (Podmore et al. 1998; Clempus and Griendling 2006). Thus, the acute decrease in GTN-mediated dilation with vitamin C could reflect a disruption of cell signaling of key ROS important for vasodilation, particularly in vascular smooth cells that appear not to be impaired.
The mechanisms by which estradiol protects the vascular endothelium against oxidative stress are unclear. Estradiol has well-described antioxidant properties and acts through both direct and indirect, and enzymatic and non-enzymatic mechanisms. Estradiol has a phenol-hydroxyl ring that donates hydrogen, enabling estrogen to scavenge major sources of vascular ROS production including nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, oxidized LDL, and hydrogen peroxide (Keaney et al. 1994; Wagner et al. 2001; Song et al. 2009; Stirone et al. 2005; Lu et al. 2007; Xu et al. 2004). Estradiol also increases mitochondrial antioxidant defenses and other intracellular antioxidants (Stirone et al. 2005; Bellanti et al. 2013). Thus, the decline in estradiol with the menopause transition may shift the redox balance to a state of oxidative stress. In this regard, previous observations reported elevated plasma and protein levels of oxidized LDL and other lipoperoxides, and lower levels of the antioxidants superoxide dismutase (SOD) and catalase in perimenopausal and postmenopausal women (Moreau et al. 2005; Bellanti et al. 2013; Sanchez-Rodriguez et al. 2012; Zitnanova et al. 2011). In the present study, oxidized LDL was elevated in late perimenopausal and postmenopausal women, and the antioxidant glutathione was reduced in postmenopausal women. Glutathione is a major intracellular antioxidant that facilitates the detoxification of hydrogen and lipid peroxides, superoxide, and peroxynitrate (Ashfaq et al. 2008). Additionally, TAS, a measure of the cumulative effect of all the chain-breaking antioxidants (e.g., α-tocopheral, ascorbic acid, thiols, uric acid) in plasma, tended to be reduced across menopause stages. Collectively, these observations support the idea that redox balance shifts during the late perimenopausal and early postmenopausal periods, possibly due to changes in estradiol levels.
Other possible contributing mechanisms to the endothelial dysfunction across the stages of the menopause transition in healthy women include inflammation, and/or the modulation by vasoconstrictor hormones (e.g., endothelin-1, norepinephrine, angiotensin II) and local factors (Arenas et al. 2006; Moreau et al. 2013b; Kaplon et al. 2011). It is important to note that all of these factors also contribute to, and are modulated by oxidative stress. In the present study, although plasma norepinephrine and endothelin-1 were elevated across the stages of the menopause transition, there were no significant changes in norepinephrine, endothelin-1, or inflammatory markers with gonadal suppression.
Experimental considerations and limitations
The findings in the present study can only be generalized to healthy, non-smoking sedentary women without evidence of clinical disease. We acknowledge that studying endothelial function after short-term manipulations of ovarian hormones does not recapitulate the chronic changes that occur over the menopause transition. We also recognize that we were not able to separate the long-term effects of menopause from those of chronological aging in this study. However, because many physiological changes (i.e., body composition) concurrent with the menopause transition could exaggerate the effects of declining ovarian hormones on vascular function, a short-term ovarian suppression intervention provides a controlled environment to permit causal inferences about the modulatory influence that ovarian hormones may have on endothelial function in women (Stachenfeld and Taylor 2014). We also did not examine the independent effects of other ovarian hormones (e.g., FSH, progesterone, testosterone) that change with menopause; however, previous studies using gonadal suppression have shown that neither progesterone nor testosterone appear to contribute to endothelial function (Meendering et al. 2008; Miner et al. 2011). In premenopausal women, administration of either medroxyprogesterone acetate or micronized progesterone alone or combined with estradiol abolished the beneficial effect of estradiol on brachial artery FMD (Meendering et al. 2008; Miner et al. 2011), and there were no differences reported in cutaneous microvascular vasodilation between GnRHant with and without testosterone (Wenner et al. 2013). Nonetheless, whether progesterone, testosterone, and/or FSH contribute to the decline in endothelial function during the menopause transition needs further investigation. We also could not address whether estradiol regulated vascular smooth muscle cell function due to small sample sizes (i.e., 5–7/grp) in the placebo and estradiol add-back groups.
Endothelial function was measured using brachial artery FMD, a well-established measure of macrovascular, conduit artery endothelial-dependent dilation, rather than the gold standard of coronary epicardial vasoreactivity, a more invasive and expensive procedure that is limited to individuals undergoing coronary angiography (Flammer et al. 2012). We also recognize that we did not measure nitrites/nitrates in the blood to get a measure of circulating NO to corroborate our FMD findings. Despite the disadvantages of the FMD procedure being that it is technically challenging to perform and requires standardization, the advantages of using brachial artery FMD are that it is non-invasive, easy to access, correlates with coronary endothelial function, and predicts CVD risk (Flammer et al. 2012). Moreover, we standardized our protocol and adhered to evidence-based recommendations for the assessment of FMD in humans (Thijssen et al. 2019). Whether our findings are applicable to other measures of endothelial function, particularly at the microvascular level (e.g., venous occlusion plethysmography, EndoPat), warrants further investigation.
We did not measure the direct effect of the ovarian suppression intervention or vitamin C on ROS. However, vitamin C infusion is a commonly used experimental model to determine the tonic suppression of ROS on endothelial function in human studies (Moreau et al. 2013a; Donato et al. 2007; Taddei et al. 2001; Virdis et al. 2000), and blood concentrations do not necessarily reflect differences at the local vascular level or vascular responsiveness to these factors. We also cannot rule out the possibility that the vasoactive factors mentioned above (i.e., norepinephrine, endothelin-1, inflammatory cytokines) were modulated at the local level with the ovarian suppression. Estradiol concentrations did not significantly decrease in early perimenopausal women with the GnRHant treatment, although we cannot rule out decreases in estrogen production in other tissues (e.g., adipose tissue). Additionally, it is possible that the immunoassay measurement of estradiol used in the present study lacked the sensitivity and precision to accurately measure estradiol and that a more sensitive and specific assay such as LC-MS/MS may have detected significant changes in estradiol. Finally, it is important to note that the findings from this study do not support vitamin C supplementation for the maintenance of endothelial function, but rather to help identify possible mechanistic pathways and potential therapeutic targets for future research and prevention strategies.
Conclusion
The present study provided novel evidence for the independent effect of changes in ovarian hormones, specifically estradiol, as the initiating event for the decline in endothelial function with the menopause transition in healthy women. Additionally, our data suggest that the decline in estradiol causes a shift in redox balance that results in increased vascular oxidative stress, which impairs endothelial function. Future investigations should examine whether maintaining estradiol concentration and/or implementing lifestyle or other preventive strategies that mitigate oxidative stress during the perimenopausal years is effective in preserving or attenuating the decline in endothelial function in women.
Electronic supplementary material
(DOCX 48 kb)
Acknowledgments
We thank the nursing, core laboratory, bionutrition, information systems, and administrative staff of the Clinical and Translational Research Center and the Energy Balance Core of the Nutrition and Obesity Research Center for their support of the study. We also are grateful to the members of our research group who helped with the initiation of the study and carried out day-to-day activities for the project. Finally, we thank the women who volunteered to participate in the study for their time and effort.
Author contributions
K.L.M and W.M.K conceived and designed the research. K.L.H. provided medical oversight of the study participants, evaluated inclusion and exclusion criteria, and reviewed adverse events. J. Klawitter performed and analyzed the glutathione assays. P. Blatchford and K.L.M. performed the statistical analyses. All authors helped in the interpretation of the data, drafting of the manuscript, and approved the final version of the manuscript.
Funding information
This study was supported by the National Institutes of Health awards R01 AG027678, R56 HL114073, U54 AG062319, R01 AG049762, Colorado Clinical and Translational Sciences Institute UL1 TR001082, Colorado Nutrition and Obesity Research Center P30 DK048520, and Eastern Colorado GRECC.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Ethics approval
All procedures were reviewed and approved by the Colorado Multiple Institutional Review Board (COMIRB).
Consent to participate
All women gave their written informed consent to participate.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- Arenas IA, Armstrong SJ, Xu Y, Davidge ST. Tumor necrosis factor-{alpha} and vascular angiotensin II in estrogen-eeficient rats. Hypertension. 2006;48(3):497–503. doi: 10.1161/01.HYP.0000235865.03528.f1. [DOI] [PubMed] [Google Scholar]
- Ashfaq S, Abramson JL, Jones DP, Rhodes SD, Weintraub WS, Hooper WC, Vaccarino V, Alexander RW, Harrison DG, Quyyumi AA. Endothelial function and aminothiol biomarkers of oxidative stress in healthy adults. Hypertension. 2008;52(1):80–85. doi: 10.1161/hypertensionaha.107.097386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beehler CJ, Simchuk ML, Toth KM, Drake SK, Parker NB, White CW, Berger EM, Sanderson RJ, Repine JE. Blood sulfhydryl level increases during hyperoxia: a marker of oxidant lung injury. J Appl Physiol. 1989;67(3):1070–1075. doi: 10.1152/jappl.1989.67.3.1070. [DOI] [PubMed] [Google Scholar]
- Bellanti F, Matteo M, Rollo T, De Rosario F, Greco P, Vendemiale G, et al. Sex hormones modulate circulating antioxidant enzymes: impact of estrogen therapy. Redox Biol. 2013;1(1):340–346. doi: 10.1016/j.redox.2013.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celermajer DS, Sorensen KE, Gooch VM, Spiegelhalter DJ, Miller OI, Sullivan ID, Lloyd JK, Deanfield JE. Non-invasive detection of endothelial dysfunction in children and adults at risk of atherosclerosis. Lancet. 1992;340(8828):1111–1115. doi: 10.1016/0140-6736(92)93147-F. [DOI] [PubMed] [Google Scholar]
- Clempus RE, Griendling KK. Reactive oxygen species signaling in vascular smooth muscle cells. Cardiovasc Res. 2006;71(2):216–225. doi: 10.1016/j.cardiores.2006.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corretti MC, Anderson TJ, Benjamin EJ, Celermajer D, Charbonneau F, Creager MA, Deanfield J, Drexler H, Gerhard-Herman M, Herrington D, Vallance P, Vita J, Vogel R. International Brachial Artery Reactivity Task Force. Guidelines for the ultrasound assessment of endothelial-dependent flow-mediated vasodilation of the brachial artery: a report of the International Brachial Artery Reactivity Task Force. J Am Coll Cardiol. 2002;39(2):257–265. doi: 10.1016/S0735-1097(01)01746-6. [DOI] [PubMed] [Google Scholar]
- Davy KP, Johnson DG, Seals DR. Cardiovascular, plasma norepinephrine, and thermal adjustments to prolonged exercise in young and older healthy humans. Clin Physiol. 1995;15(2):169–181. doi: 10.1111/j.1475-097X.1995.tb00441.x. [DOI] [PubMed] [Google Scholar]
- Donato AJ, Eskurza I, Silver AE, Levy AS, Pierce GL, Gates PE, Seals DR. Direct evidence of endothelial oxidative stress with aging in humans: relation to impaired endothelium-dependent dilation and upregulation of nuclear factor-kappaB. Circ Res. 2007;100(11):1659–1666. doi: 10.1161/01.RES.0000269183.13937.e8. [DOI] [PubMed] [Google Scholar]
- Doshi SN, Naka KK, Payne N, Jones CJ, Ashton M, Lewis MJ, et al. Flow-mediated dilatation following wrist and upper arm occlusion in humans: the contribution of nitric oxide. Clin Sci (Lond) 2001;101(6):629–635. doi: 10.1042/cs1010629. [DOI] [PubMed] [Google Scholar]
- Eskurza I, Monahan KD, Robinson JA, Seals DR. Effect of acute and chronic ascorbic acid on flow-mediated dilatation with sedentary and physically active human ageing. J Physiol. 2004;556(Pt 1):315–324. doi: 10.1113/jphysiol.2003.057042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eskurza I, Monahan KD, Robinson JA, Seals DR. Ascorbic acid does not affect large elastic artery compliance or central blood pressure in young and older men. Am J Physiol Heart Circ Physiol. 2004;286(4):H1528–H1534. doi: 10.1152/ajpheart.00879.2003. [DOI] [PubMed] [Google Scholar]
- Flammer AJ, Anderson T, Celermajer DS, Creager MA, Deanfield J, Ganz P, Hamburg NM, Lüscher TF, Shechter M, Taddei S, Vita JA, Lerman A. The assessment of endothelial function: from research into clinical practice. Circulation. 2012;126(6):753–767. doi: 10.1161/circulationaha.112.093245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedewald W, Levy R, Fredrickson D. Estimation of the concentration of low-density lipoprotein cholesterol in plasma, without use of the preparative ultracentrifuge. Clin Chem. 1972;18:499–502. doi: 10.1093/clinchem/18.6.499. [DOI] [PubMed] [Google Scholar]
- Gavin KM, Kohrt WM, Klemm DJ, Melanson EL. Modulation of energy expenditure by estrogens and exercise in women. Exerc Sport Sci Rev. 2018;46(4):232–239. doi: 10.1249/JES.0000000000000160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavin KM, Shea KL, Gibbons E, Wolfe P, Schwartz RS, Wierman ME, Kohrt WM. Gonadotropin-releasing hormone agonist in premenopausal women does not alter hypothalamic-pituitary-adrenal axis response to corticotropin-releasing hormone. Am J Physiol Endocrinol Metab. 2018;315(2):E316–EE25. doi: 10.1152/ajpendo.00221.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goddard J, Webb DJ. Plasma endothelin concentrations in hypertension. J Cardiovasc Pharmacol. 2000;35(4):S25–S31. doi: 10.1097/00005344-200000002-00007. [DOI] [PubMed] [Google Scholar]
- Harman D. Free Radical Theory of Aging: an update. Ann N Y Acad Sci. 2006;1067(1):10–21. doi: 10.1196/annals.1354.003. [DOI] [PubMed] [Google Scholar]
- Heitzer T, Schlinzig T, Krohn K, Meinertz T, Munzel T. Endothelial dysfunction, oxidative stress, and risk of cardiovascular events in patients with coronary artery disease. Circulation. 2001;104(22):2673–2678. doi: 10.1161/hc4601.099485. [DOI] [PubMed] [Google Scholar]
- Hornig B, Arakawa N, Kohler C, Drexler H. Vitamin C improves endothelial function of conduit arteries in patients with chronic heart failure. Circulation. 1998;97(4):363–368. doi: 10.1161/01.CIR.97.4.363. [DOI] [PubMed] [Google Scholar]
- Kaplon RE, Walker AE, Seals DR. Plasma norepinephrine is an independent predictor of vascular endothelial function with aging in healthy women. J Appl Physiol. 2011;111(5):1416–1421. doi: 10.1152/japplphysiol.00721.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keaney JF, Jr, Shwaery GT, Xu A, Nicolosi RJ, Loscalzo J, Foxall TL, Vita JA. 17 beta-estradiol preserves endothelial vasodilator function and limits low-density lipoprotein oxidation in hypercholesterolemic swine. Circulation. 1994;89(5):2251–2259. doi: 10.1161/01.CIR.89.5.2251. [DOI] [PubMed] [Google Scholar]
- Kojda G, Harrison D. Interactions between NO and reactive oxygen species: pathophysiological importance in atherosclerosis, hypertension, diabetes and heart failure. Cardiovasc Res. 1999;43(3):562–571. doi: 10.1016/S0008-6363(99)00169-8. [DOI] [PubMed] [Google Scholar]
- Lakatta EG, Levy D. Arterial and cardiac aging: major shareholders in cardiovascular disease enterprises: Part I: aging arteries: a “set up” for vascular disease. Circulation. 2003;107(1):139–146. doi: 10.1161/01.CIR.0000048892.83521.58. [DOI] [PubMed] [Google Scholar]
- Lam KK, Lee YM, Hsiao G, Chen SY, Yen MH. Estrogen therapy replenishes vascular tetrahydrobiopterin and reduces oxidative stress in ovariectomized rats. Menopause. 2006;13(2):294–302. doi: 10.1097/01.gme.0000182806.99137.5e. [DOI] [PubMed] [Google Scholar]
- Lu A, Frink M, Choudhry MA, Hubbard WJ, Rue LW, Bland KI, et al. Mitochondria play an important role in 17β-estradiol attenuation of H2O2-induced rat endothelial cell apoptosis. Am J Physiol Endocrinol Metab. 2007;292(2):E585–EE93. doi: 10.1152/ajpendo.00413.2006. [DOI] [PubMed] [Google Scholar]
- Matthews KA, Crawford SL, Chae CU, Everson-Rose SA, Sowers MF, Sternfeld B, Sutton-Tyrrell K. Are Changes in cardiovascular disease risk factors in midlife women due to chronological aging or to the menopausal transition? J Am Coll Cardiol. 2009;54(25):2366–2373. doi: 10.1016/j.jacc.2009.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meendering JR, Torgrimson BN, Miller NP, Kaplan PF, Minson CT. Estrogen, medroxyprogesterone acetate, endothelial function, and biomarkers of cardiovascular risk in young women. Am J Physiol Heart Circ Physiol. 2008;294(4):H1630–H16H7. doi: 10.1152/ajpheart.01314.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melanson EL, Lyden K, Gibbons E, Gavin KM, Wolfe P, Wierman ME, et al. Influence of estradiol status on physical activity in premenopausal women. Med Sci Sports Exerc. 2018;50(8):1704–1709. doi: 10.1249/MSS.0000000000001598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miner JA, Martini ER, Smith MM, Brunt VE, Kaplan PF, Halliwill JR, Minson CT. Short-term oral progesterone administration antagonizes the effect of transdermal estradiol on endothelium-dependent vasodilation in young healthy women. Am J Physiol Heart Circ Physiol. 2011;301(4):H1716–H1H22. doi: 10.1152/ajpheart.00405.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreau KL, Gavin KM, Plum AE, Seals DR. Ascorbic acid selectively improves large elastic artery compliance in postmenopausal women. Hypertension. 2005;45(6):1107–1112. doi: 10.1161/01.HYP.0000165678.63373.8c. [DOI] [PubMed] [Google Scholar]
- Moreau KL, Gavin KM, Plum AE, Seals DR. Oxidative stress explains differences in large elastic artery compliance between sedentary and habitually exercising postmenopausal women. Menopause. 2006;13(6):951–958. doi: 10.1097/01.gme.0000243575.09065.48. [DOI] [PubMed] [Google Scholar]
- Moreau KL, Depaulis AR, Gavin KM, Seals DR. Oxidative stress contributes to chronic leg vasoconstriction in estrogen-deficient postmenopausal women. J Appl Physiol. 2007;102(3):890–895. doi: 10.1152/japplphysiol.00877.2006. [DOI] [PubMed] [Google Scholar]
- Moreau KL, Hildreth KL, Meditz AL, Deane KD, Kohrt WM. Endothelial function is impaired across the stages of the menopause transition in healthy women. J Clin Endocrinol Metab. 2012;97(12):4692–4700. doi: 10.1210/jc.2012-2244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreau KL, Meditz A, Deane KD, Kohrt WM. Tetrahydrobiopterin improves endothelial function and decreases arterial stiffness in estrogen-deficient postmenopausal women. Am J Physiol Heart Circ Physiol. 2012;302(5):H1211–H1218. doi: 10.1152/ajpheart.01065.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreau KL, Stauffer BL, Kohrt WM, Seals DR. Essential role of estrogen for improvements in vascular endothelial function dwith endurance exercise in postmenopausal women. J Clin Endocrinol Metab. 2013;98(11):4507–4515. doi: 10.1210/jc.2013-2183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreau KL, Deane KD, Meditz AL, Kohrt WM. Tumor necrosis factor-α inhibition improves endothelial function and decreases arterial stiffness in estrogen-deficient postmenopausal women. Atherosclerosis. 2013;230(2):390–396. doi: 10.1016/j.atherosclerosis.2013.07.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Donnell VB, Freeman BA. Interactions between nitric oxide and lipid oxidation pathways: implications for vascular disease. Circ Res. 2001;88(1):12–21. doi: 10.1161/01.RES.88.1.12. [DOI] [PubMed] [Google Scholar]
- Oberyé JJL, Mannaerts BMJL, Huisman JAM, Timmer CJ. Pharmacokinetic and pharmacodynamic characteristics of ganirelix (Antagon/Orgalutran). part II. dose-proportionality and gonadotropin suppression after multiple doses of ganirelix in healthy female volunteers. Fertil Steril. 1999;72(6):1006–1012. doi: 10.1016/S0015-0282(99)00414-8. [DOI] [PubMed] [Google Scholar]
- Pereira MD, Marques AF, Brenda E, de Castro M. Immediate reconstruction of the central segment of the mandible using the masseter osteomuscular flap. Plast Reconstr Surg. 1997;99(6):1749–1754. doi: 10.1097/00006534-199705010-00045. [DOI] [PubMed] [Google Scholar]
- Podmore ID, Griffiths HR, Herbert KE, Mistry N, Mistry P, Lunec J. Vitamin C exhibits pro-oxidant properties. Nature. 1998;392(6676):559. doi: 10.1038/33308. [DOI] [PubMed] [Google Scholar]
- Rossi R, Nuzzo A, Origliani G, Modena MG. Prognostic role of flow-mediated dilation and cardiac risk factors in post-menopausal women. J Am Coll Cardiol. 2008;51(10):997–1002. doi: 10.1016/j.jacc.2007.11.044. [DOI] [PubMed] [Google Scholar]
- Sanchez-Rodriguez MA, Zacarias-Flores M, Arronte-Rosales A, Correa-Munoz E, Mendoza-Nunez VM. Menopause as risk factor for oxidative stress. Menopause. 2012;19(3):361–367. doi: 10.1097/gme.0b013e318229977d. [DOI] [PubMed] [Google Scholar]
- Santoro N, Sutton-Tyrrell K. The SWAN song: study of women’s health across the nation’s recurring themes. Obstet Gynecol Clin N Am. 2011;38(3):417–423. doi: 10.1016/j.ogc.2011.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shea KL, Gavin KM, Melanson EL, Gibbons E, Stavros A, Wolfe P, Kittelson JM, Vondracek SF, Schwartz RS, Wierman ME, Kohrt WM. Body composition and bone mineral density after ovarian hormone suppression with or without estradiol treatment. Menopause. 2015;22:1045–1052. doi: 10.1097/GME.0000000000000430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song J-Y, Kim M-J, Jo H-H, Hwang S-J, Chae B, Chung J-E, Kwon DJ, Lew YO, Lim YT, Kim JH, Kim JH, Kim MR. Antioxidant effect of estrogen on bovine aortic endothelial cells. J Steroid Biochem Mol Biol. 2009;117(1-3):74–80. doi: 10.1016/j.jsbmb.2009.07.006. [DOI] [PubMed] [Google Scholar]
- Soules MR, Sherman S, Parrott E, Rebar R, Santoro N, Utian W, Woods N. Stages of reproductive aging workshop (STRAW) J Womens Health Gend Based Med. 2001;10(9):843–848. doi: 10.1089/152460901753285732. [DOI] [PubMed] [Google Scholar]
- Stachenfeld NS, Taylor HS. Challenges and methodology for testing young healthy women in physiological studies. Am J Physiol Endocrinol Metab. 2014;306(8):E849–E853. doi: 10.1152/ajpendo.00038.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevenson ET, Davy KP, Seals DR. Hemostatic, metabolic, and androgenic risk factors for coronary heart disease in physically active and less active postmenopausal women. Arterioscler Thromb Vasc Biol. 1995;15(5):669–677. doi: 10.1161/01.ATV.15.5.669. [DOI] [PubMed] [Google Scholar]
- Stirone C, Duckles SP, Krause DN, Procaccio V. Estrogen increases mitochondrial efficiency and reduces oxidative stress in cerebral blood vessels. Mol Pharmacol. 2005;68(4):959–965. doi: 10.1124/mol.105.014662. [DOI] [PubMed] [Google Scholar]
- Sudoh N, Toba K, Akishita M, Ako J, Hashimoto M, Iijima K, Kim S, Liang YQ, Ohike Y, Watanabe T, Yamazaki I, Yoshizumi M, Eto M, Ouchi Y. Estrogen prevents oxidative stress-induced endothelial cell apoptosis in rats. Circulation. 2001;103(5):724–729. doi: 10.1161/01.CIR.103.5.724. [DOI] [PubMed] [Google Scholar]
- Taddei S, Virdis A, Ghiadoni L, Salvetti G, Bernini G, Magagna A, Salvetti A. Age-related reduction of NO availability and oxidative stress in humans. Hypertension. 2001;38(2):274–279. doi: 10.1161/01.HYP.38.2.274. [DOI] [PubMed] [Google Scholar]
- Tanaka H, Desouza CA, Jones PP, Stevenson ET, Davy KP, Seals DR. Greater rate of decline in maximal aerobic capacity with age in physically active vs. sedentary healthy women. J Appl Physiol. 1997;83(6):1947–1953. doi: 10.1152/jappl.1997.83.6.1947. [DOI] [PubMed] [Google Scholar]
- Tanaka H, Dinenno FA, Monahan KD, Clevenger CM, DeSouza CA, Seals DR. Aging, habitual exercise, and dynamic arterial compliance. Circulation. 2000;102(11):1270–1275. doi: 10.1161/01.CIR.102.11.1270. [DOI] [PubMed] [Google Scholar]
- Thijssen DHJ, Bruno RM, van Mil ACCM, Holder SM, Faita F, Greyling A, Zock PL, Taddei S, Deanfield JE, Luscher T, Green DJ, Ghiadoni L. Expert consensus and evidence-based recommendations for the assessment of flow-mediated dilation in humans. Eur Heart J. 2019;40(30):2534–2547. doi: 10.1093/eurheartj/ehz350. [DOI] [PubMed] [Google Scholar]
- Virdis A, Ghiadoni L, Pinto S, Lombardo M, Petraglia F, Gennazzani A, Buralli S, Taddei S, Salvetti A. Mechanisms responsible for endothelial dysfunction associated with acute estrogen deprivation in normotensive women. Circulation. 2000;101(19):2258–2263. doi: 10.1161/01.CIR.101.19.2258. [DOI] [PubMed] [Google Scholar]
- Wagner DC, BonDurant RH, Sischo WM. Reproductive effects of estradiol cypionate in postparturient dairy cows. J Am Vet Med Assoc. 2001;219(2):220–223. doi: 10.2460/javma.2001.219.220. [DOI] [PubMed] [Google Scholar]
- Wenner MM, Taylor HS, Stachenfeld NS. Androgens influence microvascular dilation in PCOS through ET-A and ET-B receptors. vol 7. 2013. [DOI] [PMC free article] [PubMed]
- Wildman RP, Colvin AB, Powell LH, Matthews KA, Everson-Rose SA, Hollenberg S, Johnston JM, Sutton-Tyrrell K. Associations of endogenous sex hormones with the vasculature in menopausal women: the Study of Women's Health Across the Nation (SWAN) Menopause. 2008;15(3):414–421. doi: 10.1097/gme.0b013e318154b6f5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Armstrong SJ, Arenas IA, Pehowich DJ, Davidge ST. Cardioprotection by chronic estrogen or superoxide dismutase mimetic treatment in the aged female rat. Am J Physiol Heart Circ Physiol. 2004;287(1):H165–H171. doi: 10.1152/ajpheart.00037.2004. [DOI] [PubMed] [Google Scholar]
- Zitnanova I, Rakovan M, Paduchova Z, Dvorakova M, Andrezalova L, Muchova J, et al. Oxidative stress in women with perimenopausal symptoms. Menopause. 2011;18(11):1249–1255. doi: 10.1097/gme.0b013e318224fa3d. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(DOCX 48 kb)