

Abstract
As human lifespan increases and the population ages, diseases of aging such as Alzheimer’s disease (AD) are a major cause for concern. Although calorie restriction (CR) as an intervention has been shown to increase healthspan in many species, few studies have examined the effects of CR on brain aging in primates. Using postmortem tissue from a cohort of extremely aged rhesus monkeys (22–44 years old, average age 31.8 years) from a longitudinal CR study, we measured immunohistochemically labeled amyloid beta plaques in Brodmann areas 32 and 46 of the prefrontal cortex, areas that play key roles in cognitive processing, are sensitive to aging and, in humans, are also susceptible to AD pathogenesis. We also evaluated these areas for cortical neuron loss, which has not been observed in younger cohorts of aged monkeys. We found a significant increase in plaque density with age, but this was unaffected by diet. Moreover, there was no change in neuron density with age or treatment. These data suggest that even in the oldest-old rhesus macaques, amyloid beta plaques do not lead to overt neuron loss. Hence, the rhesus macaque serves as a pragmatic animal model for normative human aging but is not a complete model of the neurodegeneration of AD. This model of aging may instead prove most useful for determining how even the oldest monkeys are protected from AD, and this information may therefore yield valuable information for clinical AD treatments.
Electronic supplementary material
The online version of this article (10.1007/s11357-020-00259-0) contains supplementary material, which is available to authorized users.
Keywords: Aging, Alzheimer’s disease, Amyloid plaques, Diet, Neurodegeneration, Neuron number
Introduction
Aging and neurodegeneration
The US population is becoming increasingly old, with one in five Americans predicted to be 65 years old or older by 2030 [1, 2]. Associated with this shift in population distribution are concerns about the socioeconomic costs of chronic poor health and fragility. Cognitively, older adults can show mild changes including reductions in processing speed, attention, and memory retrieval [3, 4]. Critically, older adults are also at risk for developing age-related neurodegenerative diseases, such as Alzheimer’s disease (AD; [2]). In fact, the chance of developing AD and other dementias continues to increase markedly with age: after the age of 65, risk for developing AD is predicted to double every 5 years ([5]; Alzheimer’s Association 2019). An additional burden is unpaid care for AD patients, estimated by the year 2050 to bring the societal cost of AD to over $1 trillion annually [6–8]. For these reasons, characterizing relevant animal models of cognitive aging and AD is of major value in order to identify pathological mechanisms as well as to develop and test novel treatments.
Mechanisms and therapeutic interventions
Normative aging of the brain does not result in overt neuron loss, even though mild brain atrophy can occur; this may reflect deterioration of dendrites, shrinkage of neurons, or reduction in white matter [9, 10]. Other physical changes in the brain, such as microgliosis and astrogliosis, the accumulation of amyloid beta (Aβ) plaques, and tauopathy also occur with normal aging and can alter cognitive function [4, 11]. Amyloid beta, a primary focus of this study, is a protein derived from the aptly named amyloid precursor protein (APP). While soluble forms of Aβ exist, insoluble Aβ has the capacity to form extracellular plaques which accumulate across age, and are considered to range anywhere from broadly pathological, to neurotoxic; for a more complete discussion of Aβ across aging as well as AD, see Duyckaerts et al. [12].
Patients with AD show these aforementioned changes, but on an accelerated time course, and with much higher levels of Aβ and phosphorylated tau in association with pronounced neuron death [12]. While there has been limited short-term treatment management success using the few FDA-approved drugs, there are currently no available medications that effectively treat the underlying causes to slow or arrest the disease itself. Approaches that target reductions in Aβ have thus far been unsuccessful in slowing or reversing the progression of AD, with these failures necessitating the exploration of alternative therapeutic approaches [13–17].
Calorie restriction (CR) in various animal models has long been studied as an intervention to attenuate aging processes to prolong healthspan and lifespan [18–21]. For example, in rodents, CR has been shown to benefit brain aging by extending life span by 25%, mitigating hippocampal gene methylation, decreasing apoptosis, increasing neurogenesis, and improving cognitive function ([19, 21–23].
Assessing the effects of CR in humans is understandably difficult as it is impossible to completely control the environment, adherence to diet regimens is poor, and participant retention is low. Yet, the Comprehensive Assessment of Long-term Effects of Reducing Caloric Intake of Energy (CALERIE) trial, a multi-center clinical study of CR, has provided valuable insight on the effects of health parameters [24]. Improved cardiometabolic health was the primary outcome of this trial, but enhanced mood and sleep in the CR group indicate that there may be central effects as well [25]. However, in order to more easily identify the mechanisms by which CR affects the brain and body, it is necessary to have available a preclinical animal model for which environmental factors such as diet, temperature, photoperiod, and medication can be strictly controlled. This also allows for optimal tissue sample collection and processing due to a minimized postmortem interval.
Preclinical rodent models
Compared to humans and nonhuman primates (NHP), rodents have relatively short lifespans allowing for more rapid experimental turnaround in longitudinal studies. However, rodents do not naturally develop measurable cognitive impairments or pathological features of AD such as Aβ plaques [26]. To compensate for these phylogenetic differences, transgenic approaches have been partially successful in replicating AD neuropathology and have contributed significantly to our understanding of basic mechanisms. For instance, Halagappa et al. [27] have shown that CR is capable of reducing levels of both Aβ and phosphorylated tau in hippocampi of a triple-transgenic mouse model of AD. These findings are promising insights into possible mechanisms but are not always successful in translation ([28]; for a review of mouse AD models, see [29]). For example, neurocognitive testing, which is widely regarded as the gold standard in assessment of AD risk and progression [3, 6, 29, 30], lacks the appropriate cognitive test batteries for complex tasks, such as episodic memory or robust executive function for mouse models of MCI and AD. Many mouse models of AD are indeed impaired on one or more neurocognitive measures, but tasks must be simple for effective acquisition and testing [29]. Thus, rodent models are insufficient for evaluating the complexities of AD in areas such as temporal development, cognitive deficits, complete brain pathology, and non-cognitive neuropsychiatric symptoms.
The rhesus macaque model
To bridge the translational gap between humans and lower vertebrates, nonhuman primates (NHPs) have been used as a preclinical model to study human aging. The rhesus macaque (Macaca mulatta) is a well-studied NHP that shows close genetic homology to humans [31], an extended lifespan with similar developmental phases and physiology [32], and a capacity for learning and performing complex cognitive tasks, which decline naturally with age [33–36]. In contrast to rodents, rhesus monkeys develop cortical Aβ plaques during aging, without any experimental manipulations [37, 38]. As Aβ is a hallmark of AD and does not need to be induced in this species, it has been proposed that naturally aging rhesus macaques may be a suitable AD model (i.e., [39]). In addition, there is some evidence that early stages of pathological tauopathy, another hallmark of the disease, may be present in middle-aged and old monkeys, though these reports have been mixed [40, 41]. However, another major pathological hallmark of AD—that is, neuron loss—has not been observed in studies of macaque monkeys that have used modern stereology [42–48]. A notable exception from Smith et al. [49] reported significant neuron loss in the PFC BA 8A of aged macaques compared to younger controls, with no neuronal loss in adjacent BA 46. However, as a prelude to the present study, a larger cohort of aging monkeys at Boston University was evaluated using the same ROI and methods as Smith et al. [49], and failed to find any cell loss (see Online Resource Fig. S1). Importantly, this finding calls into question whether neuron loss occurs anywhere in the aging rhesus monkey brain, at least up to the age of 35, the oldest subject in these stereological studies.
Aims of the current study
The rhesus macaque is the most widely studied NHP model of human aging with an average lifespan of 26 years [30], and a maximum reported lifespan of 40 years [50]. In 1987, the NIA’s intramural research program initiated a longitudinal study of CR in NHPs and includes some of the oldest rhesus macaques in recorded history, at well over 40 years of age [50–52]. Although it remains unclear why some monkeys lived well beyond the reported average lifespan, postmortem tissue from these oldest-old animals may provide valuable insights into very advanced NHP aging. For example, because aging is the primary risk factor for AD diagnosis [53], there is increased likelihood of detecting clinical AD pathology such as neuron loss in the brains of these very old animals. In the current study, postmortem brain tissue was obtained from a subset of these aged NIA animals and examined for differences between diet groups (CR and control) and the effects of age [32].
We initiated our analysis with the PFC, which has been shown to contain a very high number of Aβ plaques in aged rhesus monkeys [37]. Within the PFC, we focused on BA 46, a key cognitive area with well-documented sensitivity to aging in both humans and monkeys [54–57], and BA 32, a region with many implications in clinical aging [58–61], and which has not yet been fully characterized in aging monkeys. Importantly, because BA 32 is located within the same coronal sections as BA 46, it can serve as an internal comparison for each subject. We reasoned that if neuron death does occur in NHPs, it is likely to be revealed in the current cohort of very old monkeys, as it is exceedingly rare for macaques to live beyond the age of 40 years. However, three animals in this cohort lived beyond this age, with one other nearly reaching 40, at 39.4 years. This cohort has an average age of 31.8 years and contains some of the oldest known rhesus monkeys; therefore, brain changes that occur due to aging are likely to become evident within this population. To assess neuron numbers, PFC sections were immunostained for the neuronal marker NeuN, with parallel sections immunostained with the Aβ marker 4G8. Both markers were quantified using unbiased stereology. We hypothesized that Aβ plaque load would increase with age in BA 46 and 32, and might possibly be attenuated by CR, while neuronal cell number, similar to previous studies, would be stable across age, even in the oldest-old animals. If neuronal loss occurs, we hypothesized we would observe it only in the oldest monkeys, and/or those with the highest plaque load.
Materials and methods
Animals
All procedures were conducted in accordance with the Guide for the Care and Use of Laboratory Animals and approved by the NIA Intramural Research Program Animal Care and Use Committee. Subjects included 18 rhesus macaques (5 females; 13 males) housed at the NIH Animal Center (Table S2, Online Resource). These animals represented a subpopulation of the NIA longitudinal study of CR in NHPs, initiated in the late-1980s with approximately 120 subjects. Notably, this subset included all subjects with PFC brain tissue that was not over-fixed (and therefore unusable for the IHC protocol detailed below), that were available at the time of the current study. Age at death (ranging from 22 to 44 years) was not significantly different between the control (n = 10, mean = 31.29 years, SEM = ± 1.33 years) and CR (n = 8, mean = 32.49 years, SEM = ± 2.82 years) groups (t = 0.414, p > 0.6). Of note, blood pressure, which is associated with some clinical neuropathological conditions, did not change statistically over time in either control animals or those on CR (data not shown). As previously described, animals were housed in standard primate caging with controlled temperature and humidity and a 12-h light-dark cycle [52, 62].
The diet was naturally sourced, with the potential advantage of having micronutrients such as phytochemicals and ultra-trace minerals included. However, we note that there was the possibility for batch-to-batch variation [32, 63]. CR and control monkeys received the same food, supplemented with additional vitamins and minerals to ensure a 100% daily level for CR monkeys. Thus, with the larger food intake, these levels were exceeded in control monkeys. Control animals were maintained on a diet to approximate natural ad libitum levels: a stable ration based on age and body size in order to avoid confounding obesity effects. Animals in the CR group were fed 30% less than individual sex-, age-, and weight-matched controls, and these diet conditions were maintained for the duration of the study from age of CR onset through death, which ranged from 1 to 15 years in duration. See supplementary Table S1 (Online Resource) for additional demographics.
Specially formulated monkey chow was distributed twice per day at 0630 and 1300, which was supplemented with daily food enrichment. Drinking water was provided ad libitum. Approximately 3 h after the 0630 meal, any unfinished food was removed and a low calorie treat was provided. For the second meal, at 1300, any left-over food was not removed, so animals had continued access during the dark cycle (1800). However, in general, the monkeys tended to eat before lights-out. Because intermittent daily fasting was not a variable that was under consideration when the study began 30 years ago, if and when the afternoon ration was totally consumed was not monitored.
At necropsy, brains were collected, rinsed with 0.9% saline, and hemisected along the medial longitudinal plane. Immediately following collection, right hemispheres were immersed in 0.1 M phosphate buffer (pH 7.4) with 4% paraformaldehyde fixative while the left hemispheres were flash frozen for biochemical assay. The right hemispheres were maintained in fixative at 4 °C for 5–9 days (notably, three animals in this study had longer fixation times, from 52 to 116 days, but have retained antigenicity). After fixation, brains were rinsed in 0.1 M phosphate buffer and transferred to cryoprotectant consisting of 0.1 M phosphate buffer with 10% glycerol and 2% DMSO. After equilibration (1–2 days), they were transferred to 20% glycerol with 2% DMSO for 3–4 days after which they were blocked in the coronal plane posterior to the corpus callosum and flash frozen in isopentane at − 75 °C, then stored at − 80 °C. This cryoprotection method almost totally eliminates freezing artifact and avoids the shrinkage inherent in sucrose cryoprotection [64]. For processing, the right hemisphere was removed from − 80 °C storage and cut into parallel sets of frozen sections, eight at 30 μm thick and one at 60 μm so that adjacent sections in each series were spaced 300 μm apart. After cutting, each series of sections was transferred to cryovials containing 0.1 M phosphate buffer plus 15% glycerol, equilibrated overnight at 4 °C, and then stored at − 80 °C until processed. This method of storing frozen sections has been shown to preserve antigenicity for at least a decade [65].
One eighth of one series through the frontal block of each animal was used to process each antibody (i.e., 4G8 and NeuN). From this series, there was a median of 7 equally spaced sections per animal (~ 2.4-mm intervals) spanning the entire brain region containing BA 46. We were able to use the same PFC brain sections to evaluate both BA 46 and BA 32. BA 32 is shorter in the rostral-caudal axis, so only 4–5 sections per animal contained BA 32.
Immunohistochemistry
Vials containing 1/8th of a set in the series (i.e., sections every 2.4 mm) for all animals were removed simultaneously from − 80 °C and thawed at room temperature. Immunohistochemistry was performed on cryoprotected free-floating PFC sections transferred into 6-well culture plates and stored overnight in 7.6 pH Tris-buffered saline (TBS) at 4 °C. Between each step of the process outlined below, sections were rinsed with TBS three times for 10 min. All steps were performed on a slowly rotating agitation plate at room temperature unless otherwise noted.
To identify neurons, a well-validated primary antibody against NeuN was used to stain cortical neurons (1:2000, MilliporeSigma, Darmstadt, Germany). Aβ was visualized using 4G8 primary antibody (1:5000, Biolegend, San Diego, CA, USA), which identifies both the amyloid precursor protein (APP) and native species of Aβ cleaved from APP, including but not limited to Aβ40 and Aβ42. On day 1 of immunostaining, endogenous peroxidase activity was blocked using a 1% H202 solution for 45 min after which nonspecific binding was blocked using 5% normal horse serum for 30 min. Sections were then incubated with the primary antibody overnight at 4 °C. On day 2, they were transferred to secondary antibody (Vector biotinylated horse anti-mouse, Burlingame, CA, USA, 1:500) for 60 min. Then, the signal was amplified with Avidin-Biotin Complex (manufacturer’s instructions; Vector; Burlingame, CA, USA) for 90 min and immune-positive labeling visualized by exposing the sections to nickel chloride-enhanced 3,3′-diaminobenzadine tetrahydrochloride (manufacturer’s instructions; Sigma-Aldrich, St. Louis, MO, USA) for 5 min. After this step, sections were rinsed, stored overnight at 4 °C, then mounted on glass microscope slides, dehydrated using increasing concentrations of ethanol baths followed by xylenes, and finally sealed with DPX mounting medium (Electron Microscopy Services, Hatfield, PA, USA).
Stereology
All analyses were done with the investigator blind to subject characteristic (age, sex, group, etc.). Blind, coded slides were scanned at × 20 objective in one z-plane using a Leica Aperio AT2 slide scanner (Wetzlar, Germany). Aβ plaques were manually quantified from these images using the area fraction fractionator (AFF) probe in Stereo Investigator 2018 (MBF Bioscience, Williston, VT, USA), and neuronal cells were quantified using the optical fractionator procedure found in the same program. AFF is a form of Cavalieri point counting based on a systematic random sampling grid, which uses manually determined areas of positive staining to estimate a fraction of the entire region. The same blinded investigator manually completed stereological counts on all sections of all animals, in order to eliminate inter-rater variability and to ensure consistent criteria were used to differentiate intracellular staining from amyloid plaques. In brief, the digitized histologically stained images were imported into Stereo Investigator, and counting frames systematically randomly placed across manually delineated ROIs in each brain section (determined with Scalable Brain Atlas Markov Cortical Regions Atlas 2014, regions 46d, 46v, 9-46d, and 9-46v for BA 46, and region 32 for BA 32). After counting was complete, we verified that sampling for each subject had Gunderson’s coefficient of error (an estimate of the accuracy of the counting scheme) scores of < 0.10. Aβ plaques were determined to be positive if they were larger than a pyramidal neuron (i.e., to exclude intraneuronal Aβ) and ellipsoid (as opposed to linear vasculature staining), and only plaque area inside the counting frame was marked positive.
Neurons were counted using the optical fractionator probe in the same program if the nucleus was visible or if the cell was morphologically deemed to be a neuron (Bhattacherjee et al. [66]) and was contained either within the counting frame, or if the cell body or processes contacted the green bottom or right edges of the frame. Neurons were not counted if cell bodies or processes were in contact with the red top and left edges of the counting frame. See Fig. 1 for representative examples of high and low plaque burdens.
Fig. 1.

Age-related changes in the density of amyloid beta plaques in the rhesus macaque PFC. a, b Low-power representative micrographs depicting PFC sections from a 22.4-year-old and a 40.3-year-old animal, respectively. High-power representative photomicrographs emphasizing the difference in PFC plaque density between the same 22.4-year-old (c) and 40.3-year-old animal (d). Scale bars, 2 mm and 200 μm for the upper and lower panels, respectively
Statistical analysis
Two animals of the initial twenty subjects were excluded from all analyses as statistical outliers in high Aβ percent area coverage (i.e., area fractions of these animals were higher than 1.5x the determined interquartile range added to the third quartile). Both animals were CR females in the early-30’s age range. Future studies with more animals will determine whether these animals represent true outliers or are indicative of a phenotype for which we do not yet have the power to detect.
Linear regressions and correlations were performed in GraphPad Prism (San Diego, CA), with alpha level set at 0.05. To calculate neurons per mm2, the estimated total number of neurons per animal was divided by the sum of the area of all ROIs for that animal, as calculated by Stereo Investigator software. Neurons per mm2 was used instead of neuron count estimations to normalize data, as animals varied in number of available PFC sections due to varying brain size. Estimated area fraction of Aβ (i.e., fraction of the area covered by Aβ plaques, or percentage) was calculated by Stereo Investigator.
Results
Qualitative staining
PFC neurons positively stained with anti-NeuN antibody occurred densely through the cortex but were absent in the underlying white matter. In the prefrontal cortex, NeuN-positive neurons displayed three different patterns of staining: nuclear staining, cytoplasmic staining, or both. Since this pattern has not been previously reported, we ruled out a cohort effect using PFC sections from two additional male middle-aged (i.e., mid-20’s) rhesus monkeys that were part of a separate aging study at Boston University, and were processed identically to the NIA samples. They showed the same three NeuN phenotypes (data not shown). In contrast, the occipital cortex in both cohorts of animals showed a more traditional cellular staining pattern with more nuclear and less cytoplasmic NeuN expression (Fig. S2, Online Resource).
Staining of Aβ with 4G8 also revealed multiple immunostaining patterns. As expected, Aβ plaques were detected only in cortical regions, but many animals also showed varying degrees of vasculature amyloidosis as well as presumed intracellular 4G8 labeling (Fig. 2 and Fig. 3), neither of which were counted as positive plaque labeling. Of note, no major phenotypic differences were seen between BA 46 and BA 32.
Fig. 2.
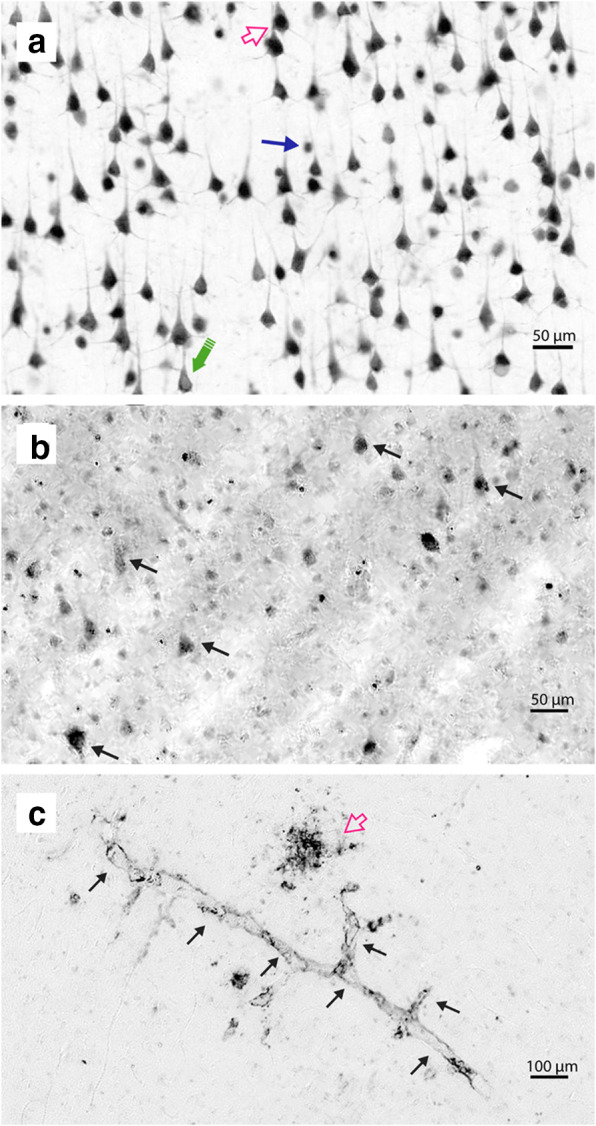
NeuN and 4G8 immunostaining phenotypes in the rhesus macaque PFC. a Phenotypes of NeuN staining. Open pink arrow indicates a neuron with both nuclear and cytoplasmic staining, closed blue arrow points to a cell with only nuclear staining, and the large green arrow emphasizes a neuron with only cytoplasmic staining. All animals showed all three phenotypes in the PFC. b 4G8 staining, which is presumed to be intracellular. Contrast has been enhanced to more clearly show the size and shape of staining. Arrows indicate examples of staining, which can be morphologically identified as pyramidal neurons. c 4G8 vasculature staining. Black arrows indicate the immunostained capillary, while the pink open arrow indicates a diffuse plaque. Scale bar in (a) applies to panel (b) as well: 50 μm. Scale bar (c) 100 μm
Fig. 3.

4G8 immunostaining phenotypes in the rhesus macaque PFC. The top row shows full cortical sections from BA 46 in three animals, representing various staining phenotypes. All three images are shown at the same magnification, encapsulating all cortical layers, and with the pial surface at the top of the image. a 4G8 staining in a 30.7-year-old animal is nearly exclusively intracellular. b Only plaques and vasculature are stained in a 34.1-year-old animal. c 4G8-labeled plaques and vasculature as well as intracellular staining in a 41.2-year-old animal. The middle row contains some representative Aβ plaque phenotypes. d Multiple very diffuse plaques cluster together with light intracellular staining. e A larger plaque with distinctive intracellular 4G8 staining. f A singular plaque with very little intracellular staining surrounding it. The bottom panels are representative images of vasculature staining phenotypes. g, h Larger 4G8-positive blood vessels. i Smaller stained vasculature, with intracellular staining. j A 4G8-positive capillary in cross-section. Note the difference in scale between panel (j) and other panels
Stereological analysis of plaques and neuron numbers
After plaque density was quantified via Stereo Investigator’s optical fraction fractionator, linear regression was applied to calculate whether trajectories of plaque accumulation and neuron number varied based on dietary treatment group. Pearson’s correlations were used to evaluate relationships between measured outcomes (i.e., relationships between neuron density and plaque area fraction). Results showed that there were no differences in plaque accumulation due to sex or genetic origin (Chinese or Indian) from which rhesus monkeys in this study were derived, as the slopes of these linear regressions were not significantly different (Fig. S3 and S4, Online Resource). Therefore, both sexes and both genetic origins were combined across subsequent analyses.
BA 46
In BA 46 (Fig. 4), Aβ plaque load increased significantly as a factor of age (F[1,16] = 5.640, p = 0.030), consistent with previous observations. Additionally, there was no difference in plaque density across age in controls versus animals on CR (slope: F[1,14] = 0.013, p > 0.9; intercept: F[1,14] = 0.200, p > 0.6; pooled regression equation: y = 0.040x–0.711). Also in BA 46, even in these extremely aged animals, neuron number was not significantly changed with age (F[1,16] = 0.139, p > 0.7), and the number was similar irrespective of dietary treatment (slope: F[1,14] = 0.365, p > 0.3; intercept: F[1,15] = 0.302, p > 0.5). Additionally, density of plaques and density of neurons were not significantly correlated in BA 46 (r = 0.196, p > 0.4).
Fig. 4.
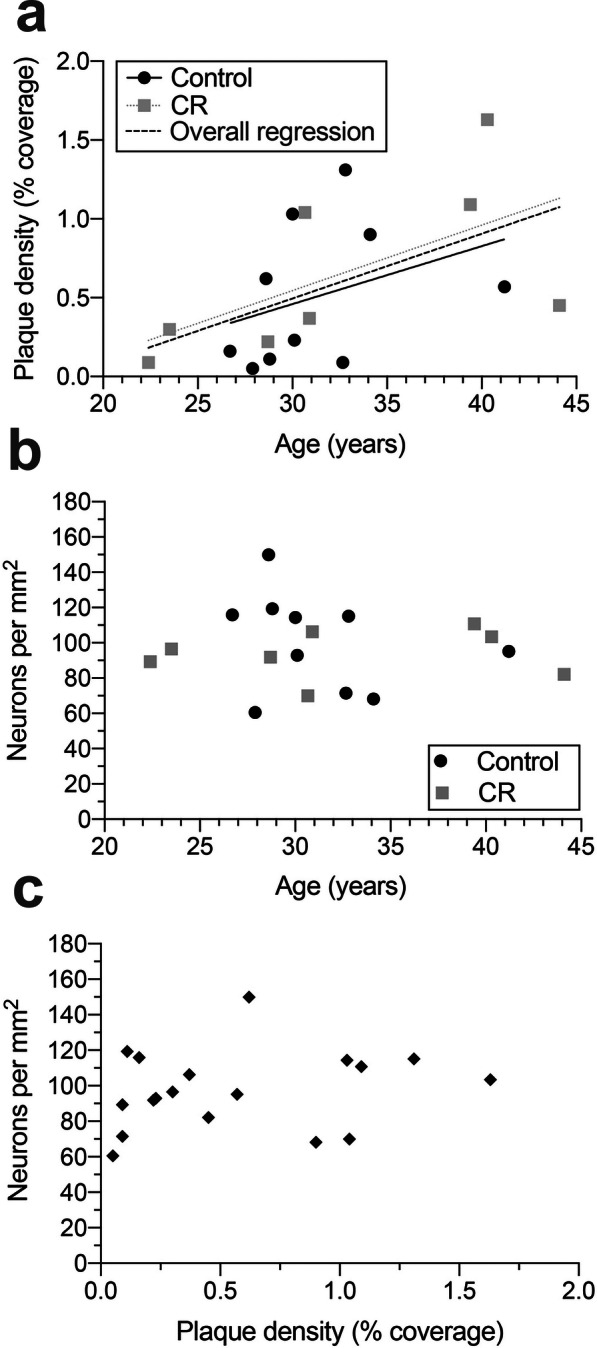
Aβ plaque and neuron density in BA 46 of the PFC. a There was a significant overall increase in plaque density with age (dashed line; F[1,16] = 5.640, p = 0.030; pooled regression equation: y = 0.040x–0.711), but no effect of dietary treatment (slope: F[1, 14] = 0.013, p > 0.9; intercept: F[1,15] = 0.198, p > 0.6). Solid black line indicates control group, while dotted gray line shows regression for the CR animals, in both (a) and (b). b There was no significant age-related change in neuron number in BA 46 (F[1,16] = 0.139, p > 0.7), nor significant differences between treatment groups (slope: F[1,14] = 0.365, p > 0.3; intercept: F[1,15] = 0.302, p > 0.5). c There was no significant correlation between Aβ plaque density and neuron number in BA 46 (r = 0.196, p > 0.4)
BA 32
Using the same tissue sections and same stereological methodology, we noted a significant increase in Aβ plaque density in BA 32 across age (F[1,16] = 7.797, p = 0.013; Fig. 5). As in BA 46, neuronal number did not change with age and was similar irrespective of dietary treatment (slope: F[1,14] = 0.003, p > 0.9; intercept: F[1,15] = 0.164, p > 0.6; pooled regression equation: y = 0.144x–3.391). Despite relatively high plaque density in BA 32, there was no significant decrease in neuron density with age (F[1,16] = 0.080, p > 0.7), nor were Aβ plaque density and neuron density significantly correlated (r = − 0.018, p > 0.9). We did not observe any dietary treatment effect for either measure in BA 32 (Aβ plaque density: slope: F[1,14] = 0.003, p > 0.9; intercept: F[1,15] = 0.164, p > 0.6 and neuron density: slope: F[1,14] = 0.176, p > 0.6; intercept: F[1,15] = 0.006, p > 0.9).
Fig. 5.
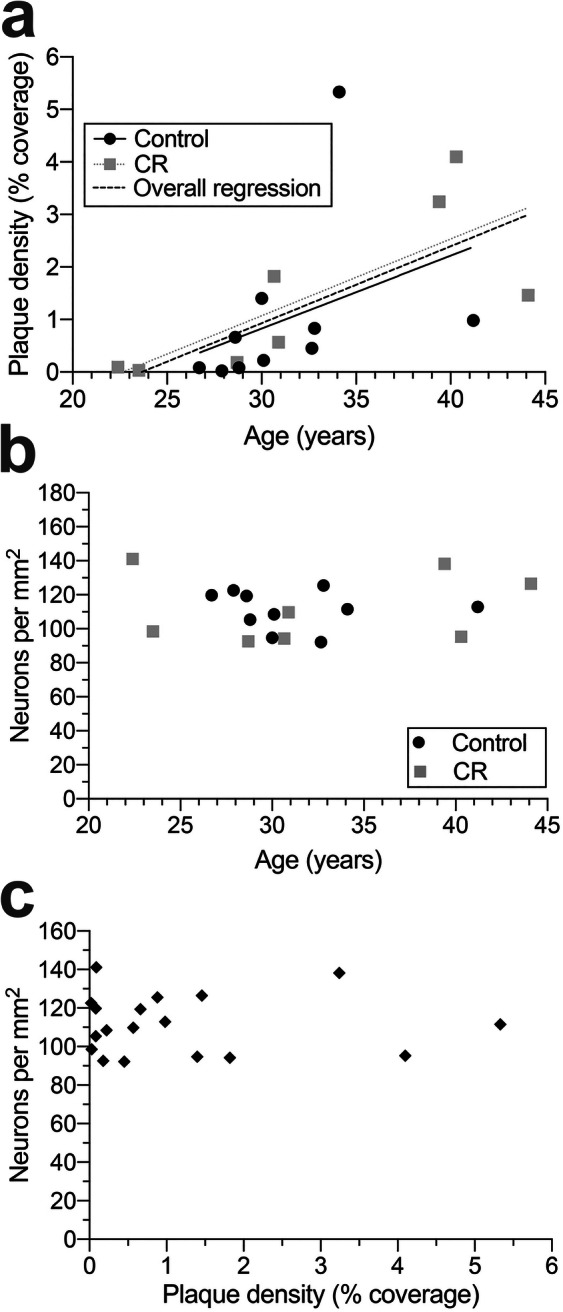
Aβ plaque and neuron density in BA 32 of the cingulate. a There was a significant overall increase in plaque density with age (dashed black line; F[1,16] = 7.797, p = 0.013; pooled regression equation: y = 0.144x–3.391), but no significant effect of dietary treatment (slope: F[1,14] = 0.003, p > 0.9; intercept: F[1,15] = 0.164, p > 0.6). Dashed black line shows significant overall increase in Aβ plaques across age, solid black line represents the control group, while gray dotted line indicates CR group regression, in both (a) and (b). b There was no significant age-related change in neuron density in BA 32(F[1,16] = 0.080, p > 0.7), nor significant differences between treatment groups (slope: F[1,14] = 0.176, p > 0.6; intercept: F[1,15] = 0.006, p > 0.9). c There was no significant correlation between Aβ plaque density and neuron number in BA 32 (r = − 0.018, p > 0.9)
Interestingly, plaque densities in BA 46 and 32 were significantly correlated (r = 0.727, p < 0.001; line of best fit: y = 0.229x + 0.296; Fig. 6a). However, when comparing density of neurons between BA 46 and BA 32, there was no significant correlation between areas (r = 0.147, p > 0.5; Fig. 6b).
Fig. 6.
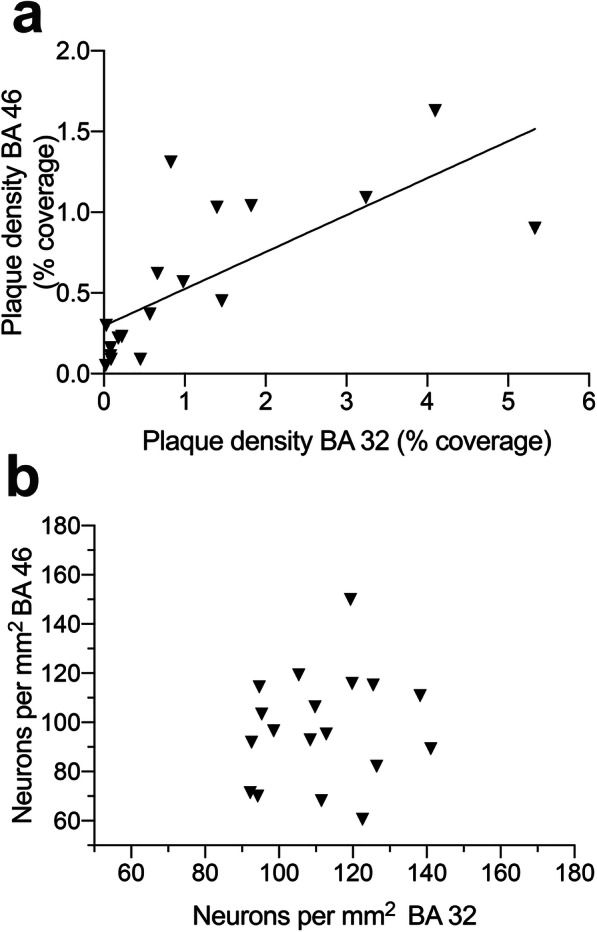
Correlations between BA 46 and BA 32. a Aβ plaque density is significantly correlated between BA 46 and BA 42 (r = 0.727, p < 0.001). b No significant correlation exists between neuron number in BA 46 and BA 32 (r = 0.147, p>0.5). This is thought to be due to cytoarchitechtonic differences between regions, as all layers of cortex were quantified
Discussion
Clinical significance
Rhesus macaques are considered to have a lifespan ratio of about 3:1 to that of humans [40, 67]. The oldest animal in the current cohort lived for more than 44 years, well beyond the average lifespan in captivity of 26 years [30] and exceeding an earlier reported maximum lifespan of 40 years [50]. By analyzing tissue from a cohort of the oldest recorded rhesus macaques, the present data extend current knowledge about the aging rhesus macaque brain. Importantly, the data provide insights into the similarities and differences between the brain of aging rhesus macaques and that of humans undergoing normative and pathological aging.
Although the aging macaque brain has an age-related increase in amyloid plaques that is suggestive of human AD pathology, it is not a definitive model of naturally occurring AD for numerous reasons. For example, both humans and rhesus macaques have Aβ peptides that are generally cleaved at a length of 40 or 42 [68, 69]. In humans, Aβ40 is found in ten-fold higher concentrations than Aβ42 [69], even though the Aβ plaques are predominantly comprised of Aβ42. In contrast, in rhesus macaques, Aβ40 is the primary component of the Aβ plaques [68]. Thus, the relative levels of these peptides seem to be an important differentiator between plaques in monkeys and in AD patients. On a higher level, this study supports others which posit that Aβ plaques are not necessarily an early sign of AD, and may, in fact, be unrelated to more blatantly pathological aspects of the disease, such as loss of cortical neurons and dementia. Clinical data repeatedly shows that many aged humans accumulate Aβ plaques with no signs of mild or overt dementia [70]. In a similar fashion, amyloid levels were not correlated with cognitive ability in aged monkeys [71].
While Aβ plaques are a classical hallmark of AD, some theories suggest that soluble forms of Aβ may be more important for pathological aging than insoluble, plaque-forming confirmations (reviewed in [72]). While plaque load (i.e., insoluble Aβ) increased across age in this study, there was no evidence of effect on neuron number. Therefore, future studies of soluble Aβ may help elucidate the importance of peptide length and solubility in the toxicity of Aβ in humans, and the lack of neurotoxicity in macaques.
Finally, human data shows that prefrontal cortical areas like BA 46 and the less well-studied BA 32 are vulnerable to neurodegeneration in AD [3, 55, 60, 73]. Hence, this first analysis of the cortex of extremely old rhesus monkeys began with BA 46 [55], and BA 32. The extreme age of the subjects along with identification of brain areas that are typically affected by aging was designed to increase the probability that neuronal loss might be identified in relationship to Aβ. Furthermore, we hypothesized that while Aβ plaque load would increase with age in both dietary groups, and might be associated with neuron loss, these effects of aging might be attenuated by CR. While Aβ plaque increase was quantitatively confirmed, there was no effect of CR nor was any neuron loss observed. Nevertheless, it remains unclear as to whether CR confers any benefit to brain aging in primates as many other factors such as dendritic and synaptic effects remain to be explored.
Staining patterns
NeuN
In the rhesus monkey PFC, we saw three distinct NeuN immunostaining phenotypes: nuclear staining, cytoplasmic staining, or both, in the same neuron. NeuN (neuronal nuclear antigen) has been identified as the splicing protein Fox-3, which can be found in varying levels in cytoplasm as well as in neuronal nuclei [74, 75], suggesting that the differential NeuN staining observed here may be due to differential expression of Fox-3 splicing protein. While the exact function of Fox-3 is unknown, it is specific to post-mitotic neurons in the brain [75]. NeuN is traditionally considered a nuclear marker; however, in the current study, animals of both sexes, different ages, and different genetic backgrounds all showed these three phenotypes of NeuN throughout the PFC regions examined. Of note, occipital cortices from the same animals showed more traditional nuclear NeuN staining patterns suggesting this may be a PFC-specific phenomenon ([74, 76]; Online Resource Fig. S2). Further information on NeuN staining can be found in Duan et al. [77].
4G8
Although the focus of the present study was on extracellular Aβ plaques, it was interesting that many animals showed apparent intracellular labeling with the 4G8 antibody. As double-labeling was not performed with NeuN and 4G8, we cannot be completely certain that the observed staining was entirely intraneuronal. However, intraneuronal Aβ has been implicated as a possible precursor to extracellular plaques [78] and has been reported in macaques and humans of varying ages [79, 80]. Therefore, it is plausible that light cytoplasmic 4G8 staining indicates labeling of either intraneuronal APP or Aβ [75, 81].
In addition to plaques and presumed intracellular 4G8 labeling, rhesus monkeys showed 4G8 blood vessel staining in both small and large vessels. Aβ angiopathy seemed to be present to some extent in all of the animals in the current study. In humans, vascular Aβ is not uncommon, nor is it specifically related to AD; roughly a quarter of aged adults show this phenotype, regardless of AD status. In the human population, Aβ angiopathy has been linked to several different mutations in the APP gene, and occurs most commonly in larger vessels [12]. As shown in Fig. 2, both large and small diameter vessels are Aβ-positive within this cohort, and a more in-depth histological exploration of specific phenotypes may reveal more clinically relevant information.
In regard to Aβ plaques, human amyloidosis generally consists of both diffuse (present in macaques) and dense core (not typically observed in macaques) plaques. This is also of interest as dense core plaques are more strongly correlated with cognitive decline in humans [29], providing additional support to the idea that the rhesus macaque likely mimics normative aging rather than AD. Aβ levels in this study (~ 1% area coverage in area 46, and ~ 2–3% coverage in area 32) also do not reach the 7 + % Aβ area coverage seen in the PFC in late stages of diagnosed AD in the clinic [73]. The existence of a very high plaque load may be necessary before cell death or pathological cognitive decline occurs, so the relatively low levels seen here in both PFC regions might indicate that the density is simply insufficient to induce an AD phenotype, especially if the plaques are mainly Aβ40 rather than Aβ42.
Lack of diet effects
Animals in the current study represent a subset from a larger study which showed a significant effect of CR on healthspan [32], leading to the hypothesis that these benefits may translate to healthier brain aging in the CR group as well. However, no CR effect on the age-related increase in amyloid load emerged in analyses of BA 46 or 32. This could be due to the fact that the overall CR effect on other measures of health span is small, or, rather, that CR has effects on other factors such as dendritic atrophy [82], synapse loss [35] or inflammation [83], as opposed to Aβ plaques.
There was large heterogeneity amongst the subjects in this study, as the cohort included both sexes, animals of differing genetic backgrounds (i.e., both Chinese and Indian ancestry), and began CR diets at different stages of life (see Table S2 in the Online Resource for individual information). Small effects were more difficult to detect due to this diversity, in combination with a relatively small sample size compared to human clinical studies, yet this variability is not unlike the heterogeneity found in clinical populations. As ~ 20 additional monkeys from the original study are still alive, more definitive conclusions about the effects of aging and diet on markers of brain pathology will hopefully become more apparent as these subjects reach the end of life, and their brains are added to the current analysis as well as to future analytic assays.
It also needs to be emphasized that unlike dietary controls in many other calorie restriction studies [32, 84–86], the control animals in the current study were not fed ad libitum with continuous access to food, but instead receive a standardized caloric intake that was designed to prevent obesity. It has been hypothesized that the benefits of CR are primarily driven by a lack of overeating, i.e., a return to a more evolutionarily relevant state of fewer calories overall, and longer periods of time between feedings [87, 88]. If this is the case, we may simply be seeing a “floor” effect, as both the CR and the control group animals escape the deleterious effects of overeating and obesity. This theory is supported by Bodkin et al. [50], who showed that when caloric intake was only mildly restricted (i.e., restricted enough to prevent obesity), it resulted in reduced morbidity and improved mortality compared to ad libitum control animals. With the assumption that CR is simply a return to evolutionarily relevant dietary standards, restriction beyond recommended caloric intake may not be advantageous in Aβ plaque reduction or overall brain health in primates.
Similar patterns—that is, a significant increase in Aβ plaques across age, but no differences due to CR—have also emerged in another cohort of CR macaques, albeit in the hippocampus. Sridharan et al. [86] report an age-related increase in Aβ plaques in hippocampi of monkeys in the University of Wisconsin Dietary Restriction and Aging Study without a CR effect on Aβ plaque load. However, the level of hippocampal astrogliosis was indeed attenuated by CR, similarly to what has been previously shown in rodents [86, 89]. Given similar Aβ findings in both studies, it is likely that CR is not effective at reducing Aβ plaque burden in NHPs, even in the presence of other brain health benefits. This contrasts with transgenic rodent AD models which have shown attenuated plaque formation as well as improved cognitive scores using CR interventions [21, 27, 85]. Moreover, like some clinical studies [90], prior NHP studies showed no correlation between cognitive scores and Aβ plaques [71], but did show other qualitative and quantitative benefits of CR on brain aging such as reduced iron deposition and beneficial lowering of proinflammatory cytokines [83, 91]. Therefore, it may be pertinent to shift focus in this model to identifying other potential biomarkers of aging and neurodegeneration, which may be more relevant to normative aging. It is also important to distinguish the rhesus model from genetically manipulated rodent models of overt, AD-like damage.
Absence of neuron loss in extreme old age
Finally, it is worth noting that even in this cohort of extremely aged subjects, there was no loss of neurons. To put this into perspective, other studies of neuron number in BA 46 did not include any animals over 32 years of age, while all animals in this cohort survived past the age of 22, and six of which were older than the oldest previously studied animal (i.e., older than 32 years old). For example, in Smith et al. [49], aged subjects had an average age of 25.4 years, while Peters et al., (1994), had a group of five aged subjects ranging from 25 to 32. In both of these cases, even when compared to young adult groups, there was no evidence of neuron loss in BA 46. Our results extend these findings to reveal that even the oldest-old rhesus macaques do not show any significant loss of neurons in either of the PFC regions examined. Overall, this comparison leaves little doubt that in normal aging rhesus macaques, despite age-related amyloid accumulation, there is no age-related loss of neurons in BA 46 of the PFC.
Conclusions
Both BA 46 and BA 32 are brain regions that show age-related increases in Aβ plaque load in rhesus macaques, but neither region displays neuron loss, even in the oldest-old rhesus monkeys. Similarly, stability in neuron number across age is shown in PFC BA 8A (Supplemental data). As more brains become available for analysis, nuances in sex differences, genetic backgrounds, age at CR onset, and more parameters will be explored.
Differential staining patterns of NeuN were observed in the PFC, whereas, the occipital cortices of the same animals contained commonly observed phenotypes. To our knowledge, we are the first group to identify these three immunostaining phenotypes within the rhesus PFC. These patterns are presumed to be due to varying levels of Fox-3; however, for what function is currently unknown.
The percent coverage of Aβ plaques in BA 46 and BA 32 increased significantly with age, but not at a cost to neuron density in the oldest-old individuals. The question is why? Perhaps chronic clinical health issues (obesity, high blood pressure, diabetes etc.) which are suppressed in animal husbandry programs, retard age-related neuropathology. Moreover, regulation of amyloid metabolism in aged monkeys shows a lack of 2-mer amyloid isoforms [92], whereas the addition of amyloid oligomers induces pathology [93, 94]. Continued efforts to study the monkey model will provide insights into mechanisms that underlie these differences and could lead to novel preventions and treatments for AD.
Electronic supplementary material
(DOCX 2118 kb)
Acknowledgments
The authors would like to acknowledge the contributions of Dr. Sathya Srinivasan in the Imaging and Histology core at the Oregon National Primate Research Center for his expertise in stereological methodology.
Code availability
Not applicable.
Abbreviations
- Aβ
Amyloid beta
- AD
Alzheimer’s disease
- APOE
Apolipoprotein E
- APP
Amyloid precursor protein
- BA 32
Brodmann Area 32 of the cingulate cortex
- BA 46
Brodmann Area 46 of the prefrontal cortex
- CDC
Centers for Disease Control and Prevention
- CR
Calorie restriction
- MCI
Mild cognitive impairment
- NeuN
Neuronal nucleic protein
- NHP
Nonhuman primate
- NIA
National Institute on Aging
- PFC
Prefrontal cortex
- ROI
Region of interest
- TBS
7.6 pH Tris-buffered saline
- US
United States of America
Funding
This work was supported by the National Institutes of Health (NIH) grants AG043640, AG055378, AG062220, OD011092 and the Intramural Research Program, National Institute on Aging, NIH.
Data availability
Contact corresponding author for data.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflicts of interest.
Ethics approval
This study makes use of animal tissue from a study approved by the National Institute on Aging Intramural Research Program Animal Care and Use Committee.
Consent to participate
Not applicable.
Consent for publication
Not applicable
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Colby SL, Ortman JM (2015) Projections of the size and composition of the U.S. population: 2014 to 2060. U.S. Census Bureau: Population Estimates and Projections. Retrieved from: https://www.census.gov/content/dam/Census/library/publications/2015/demo/p25-1143.pdf
- 2.Maust D, Langa K, Solway E, Singer D, Kirch M, Kullgren J, et al. Thinking about brain health: University of Michigan National Poll on Healthy Aging; 2019. Available at: http://hdl.handle.net/2027.42/149132
- 3.Harada CN, Natelson Love MC, Triebel KL. Normal cognitive aging. Clin Geriatr Med. 2013;29:737–752. doi: 10.1016/j.cger.2013.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schott JM. The neurology of ageing: what is normal? Pract Neurol. 2017;17:172–182. doi: 10.1136/practneurol-2016-001566. [DOI] [PubMed] [Google Scholar]
- 5.von Strauss E, Viitanen M, De Ronchi D, Winblad B, Fratiglioni L. Aging and the occurrence of dementia. Arch Neurol. 1999;56:587–592. doi: 10.1001/archneur.56.5.587. [DOI] [PubMed] [Google Scholar]
- 6.Deb A, Thornton JD, Sambamoorthi U, Innes K. Direct and indirect cost of managing Alzheimer’s disease and related dementias in the United States. Expert Rev Pharmacoecon Outcomes Res. 2017;2:189–202. doi: 10.1080/14737167.2017.1313118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hurd MD, Martorell P, Delavande A, Mullen KJ, Langa KM. Monetary costs of dementia in the United States. N Engl J Med. 2013;368:1326–1334. doi: 10.1056/NEJMsa1204629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tejada-Vera B. Mortality from Alzheimer’s disease in the United States: data for 2000 and 2010. NCHS data brief. 2013;116:1–8. [PubMed] [Google Scholar]
- 9.Freeman SH, Kandel R, Cruz L, Rozkalne A, Newell K, Frosch MP, Hedley-Whyte ET, Locascio JJ, Lipsitz LA, Hyman BT. Preservation of neuronal number despite age-related cortical brain atrophy in elderly subjects without Alzheimer disease. J Neuropathol Exp Neurol. 2008;67:1205–1212. doi: 10.1097/NEN.0b013e31818fc72f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Marner L, Nyengaard JR, Tang Y, Pakkenberg B. Marked loss of myelinated nerve fibers in the human brain with age. J Comp Neurol. 2003;462:144–152. doi: 10.1002/cne.10714. [DOI] [PubMed] [Google Scholar]
- 11.Furcila D, Defelipe J, Alonso-Nanclares L. A study of amyloid-β and phosphotau in plaques and neurons in the hippocampus of Alzheimer’s disease patients. J Alzheimers Dis. 2018;64:417–435. doi: 10.3233/JAD-180173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Duyckaerts C, Delatour B, Potier MC. Classification and basic pathology of Alzheimer disease. Acta Neuropathol. 2009;118:5–36. doi: 10.1007/s00401-009-0532-1. [DOI] [PubMed] [Google Scholar]
- 13.Cummings JL, Cohen S, Van Dyck CH, Brody M, Curtis C, Cho W, Ward M, Friesenhahn M, Brunstein F, Quartino A, Honigberg LA, Fuji RN, Clayton D, Mortensen D, Ho C, Paul R, Rabe C. A phase 2 randomized trial of crenezumab in mild to moderate Alzheimer disease. Neurology. 2018;90:E1889–E1897. doi: 10.1212/WNL.0000000000005550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Egan MF, Kost J, Voss T, Mukai Y, Aisen PS, Cummings JL, Tariot PN, Vellas B, Van Dyck CH, Boada M, Zhang Y, Li W, Furtek C, Mahoney E, Mozley LH, Mo Y, Sur C, Michelson D. Randomized trial of verubecestat for prodromal Alzheimer’s disease. N Engl J Med. 2019;380:1408–1420. doi: 10.1056/NEJMoa1812840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Farlow M, Arnold SE, Van Dyck CH, Aisen PS, Snider BJ, Porsteinsson AP, Friedrich S, Dean RA, Gonzales C, Sethuraman G, Demattos RB, Mohs R, Paul SM, Siemers ER. Safety and biomarker effects of solanezumab in patients with Alzheimer’s disease. Alzheimers Dement. 2012;8:261–271. doi: 10.1016/j.jalz.2011.09.224. [DOI] [PubMed] [Google Scholar]
- 16.Haass C, Levin J. Hat die Alzheimer-Forschung versagt? : Das Scheitern amyloidbasierter klinischer Studien [Did Alzheimer research fail entirely? : Failure of amyloid-based clinical studies] Nervenarzt. 2019;90:884–890. doi: 10.1007/s00115-019-0751-1. [DOI] [PubMed] [Google Scholar]
- 17.Ostrowitzki S, Lasser RA, Dorflinger E, Scheltens P, Barkhof F, Nikolcheva T, Ashford E, Retout S, Hofmann C, Delmar P, Klein G, Andjelkovic M, Dubois B, Boada M, Blennow K, Santarelli L, Fontoura P. A phase III randomized trial of gantenerumab in prodromal Alzheimer’s disease. Alzheimers Res Ther. 2017;9:1–15. doi: 10.1186/s13195-017-0318-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kishi T, Hirooka Y, Nagayama T, Isegawa K, Katsuki M, Takesue K, Sunagawa K. Calorie restriction improves cognitive decline via up-regulation of brain-derived neurotrophic factor: Tropomyosin-related kinase B in hippocampus of obesity-induced hypertensive rats. Int Heart J. 2015;56:110–115. doi: 10.1536/ihj.14-168. [DOI] [PubMed] [Google Scholar]
- 19.McCay CM, Crowell MF. Prolonging the life span. Science. 1934;39:405–414. [Google Scholar]
- 20.Parrella E, Maxim T, Maialetti F, Zhang L, Wan J, Wei M, Cohen P, Fontana L, Longo VD. Protein restriction cycles reduce IGF-1 and phosphorylated tau, and improve behavioral performance in an Alzheimer’s disease mouse model. Aging Cell. 2013;12:257–268. doi: 10.1111/acel.12049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wahl D, Coogan SCP, Solon-Biet SM, Haran JB, Raubenheimer D, Cogger VC, Mattson MP, Simpson SJ, Le Couteur DG. Cognitive and behavioral evaluation of nutritional interventions in rodent models of brain aging and dementia. Clin Interv Aging. 2017;12:1419–1428. doi: 10.2147/CIA.S145247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gültekin F, Nazıroğlu M, Savaş HB, Çiğ B. Calorie restriction protects against apoptosis, mitochondrial oxidative stress and increased calcium signaling through inhibition of TRPV1 channel in the hippocampus and dorsal root ganglion of rats. Metab Brain Dis. 2018;33:1761–1774. doi: 10.1007/s11011-018-0289-0. [DOI] [PubMed] [Google Scholar]
- 23.Hadad N, Unnikrishnan A, Jackson JA, Masser DR, Otalora L, Stanford DR, Richardson A, Freeman WM. Caloric restriction mitigates age-associated hippocampal differential CG and non-CG methylation. Neurobiol Aging. 2018;67:53–66. doi: 10.1016/j.neurobiolaging.2018.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kraus WE, Bhapkar M, Huffman KM, Pieper CF, Krupa Das S, Redman LM, Villareal DT, Rochon J, Roberts SB, Ravussin E, Holloszy JO, Fontana L. 2 years of calorie restriction and cardiometabolic risk (CALERIE): exploratory outcomes of a multicentre, phase 2, randomised controlled trial. Lancet Diabetes Endocrinol. 2019;7:673–683. doi: 10.1016/S2213-8587(19)30151-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Martin CK, Bhapkar M, Pittas AG, Pieper CF, Das SK, Williamson DA, Scott T, Redman LM, Stein R, Gilhooly CH, Stewart T, Robinson L, Roberts SB. Effect of calorie restriction on mood, quality of life, sleep, and sexual function in healthy nonobese adults the CALERIE 2 randomized clinical trial. JAMA Intern Med. 2016;176:743–752. doi: 10.1001/jamainternmed.2016.1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.LaFerla FM, Green KN. Animal models of Alzheimer’s disease. Cold Spring Harb Perspect Med. 2012;2:1031–1085. doi: 10.1016/B978-0-12-809468-6.00040-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Halagappa VK, Guo Z, Pearson M, Matsuoka Y, Cutler RG, Laferla FM, Mattson MP. Intermittent fasting and caloric restriction ameliorate age-related behavioral deficits in the triple-transgenic mouse model of Alzheimer’s disease. Neurobiol Dis. 2007;26:212–220. doi: 10.1016/j.nbd.2006.12.019. [DOI] [PubMed] [Google Scholar]
- 28.Ingram DK, De Cabo R. Calorie restriction in rodents: caveats to consider. Ageing Res Rev. 2017;39:15–28. doi: 10.1016/j.arr.2017.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Webster SJ, Bachstetter AD, Nelson PT, Schmitt FA, Van Eldik LJ. Using mice to model Alzheimer’s dementia: an overview of the clinical disease and the preclinical behavioral changes in 10 mouse models. Front Genet. 2014;5:1–23. doi: 10.3389/fgene.2014.00088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Colman RJ. Non-human primates as a model for aging. Biochim Biophys Acta - Mol Basis Dis. 2018;1864:2733–2741. doi: 10.1016/j.bbadis.2017.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gibbs RA, Rogers J, Katze MG, Bumgarner R, Weinstock GM, Mardis ER, Remington KA, Strausberg RL, Venter JC, Wilson RK, Batzer MA, Bustamante CD, Eichler EE, Hah MW, Cheng Z, Karro JE, Ma J, Raney B, She X, Cox MJ, Demuth JP, Dumas LJ, Han S, Hopkins J, Karimpour-Fard A, Kim YH, Pollack JR, Vinar T, Addo-Quaye C, Degenhardt J, Alexandr FY, Barber GP, Haussler D, Karolchik D, Kern AD, Kuhn RM, Smith KS, Zwieg AS. Evolutionary and biomedical insights from the rhesus macaque genome. Science. 2007;316:222–234. doi: 10.1126/science.1139247. [DOI] [PubMed] [Google Scholar]
- 32.Mattison JA, Colman RJ, Beasley TM, Allison DB, Kemnitz JW, Roth GS, Ingram DK, Weindruch R, De Cabo R, Anderson RM. Caloric restriction improves health and survival of rhesus monkeys. Nat Commun. 2017;8:1–12. doi: 10.1038/ncomms14063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Moore TL, Killiany RJ, Herndon JG, Rosene DL, Moss MB. Executive system dysfunction occurs as early as middle-age in the rhesus monkey. Neurobiol Aging. 2006;27:1484–1493. doi: 10.1016/j.neurobiolaging.2005.08.004. [DOI] [PubMed] [Google Scholar]
- 34.Nagahara AH, Bernot T, Tuszynski MH. Age-related cognitive deficits in rhesus monkeys mirror human deficits on an automated test battery. Neurobio Aging. 2010;31:1–13. doi: 10.1038/jid.2014.371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Peters A, Rosene DL, Moss MB, Kemper TL, Abraham CR, Tigges J, Albert MS. Neurobiological bases of age-related cognitive decline in the rhesus monkey. J Neuropath Exp Neuro. 1996;55:861–874. doi: 10.1097/00005072-199608000-00001. [DOI] [PubMed] [Google Scholar]
- 36.Rapp P. Neuropsychological analysis of learning and memory in the aged nonhuman primate. Neurobiol Aging. 1993;14:627–629. doi: 10.1016/0197-4580(93)90050-L. [DOI] [PubMed] [Google Scholar]
- 37.Heilbroner PL, Kemper TL. The cytoarchitectonic distribution of senile plaques in three aged monkeys. Acta Neuropathol. 1990;81:60–65. doi: 10.1007/BF00662638. [DOI] [PubMed] [Google Scholar]
- 38.Uno H. The incidence of senile plaques and multiple infarction in aged macaque brain. Neurobiol Aging. 1993;14:673–674. doi: 10.1016/0197-4580(93)90067-L. [DOI] [PubMed] [Google Scholar]
- 39.Gandy S, DeMattos RB, Lemere CA, Heppner FL, Leverone J, Aguzzi A, Ershler WB, Dai J, Fraser P, St George Hyslop P, Holtzman DM, Walker LC, Keller ET. Alzheimer’s Abeta vaccination of rhesus monkeys (Macaca mulatta) Mech Ageing Dev. 2004;125:149–151. doi: 10.1016/j.mad.2003.12.002. [DOI] [PubMed] [Google Scholar]
- 40.Gearing M, Rebeck GW, Hyman BT, Tigges J, Mirra SS. Neuropathology and apolipoprotein E profile of aged chimpanzees: implications for Alzheimer disease. Proc Natl Acad Sci U S A. 1994;91:9382–9386. doi: 10.1073/pnas.91.20.9382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Paspalas CD, Carlyle BC, Leslie S, Preuss TM, Crimins JL, Huttner AJ, van Dyck CH, Rosene DL, Nairn AC, Arnsten AFT. The aged rhesus macaque manifests Braak stage III/IV Alzheimer’s-like pathology. Alzheimers Dement. 2018;14:680–691. doi: 10.1016/j.jalz.2017.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gazzaley AH, Thakker MM, Hof PR, Morrison JH. Preserved number of entorhinal cortex layer II neurons in aged macaque monkeys. Neurobiol Aging. 1997;18:549–553. doi: 10.1016/S0197-4580(97)00112-7. [DOI] [PubMed] [Google Scholar]
- 43.Giannaris EL, Rosene DL. A stereological study of the numbers of neurons and glia in the primary visual cortex across the lifespan of male and female rhesus monkeys. J Comp Neurol. 2012;520:3492–3508. doi: 10.1002/cne.23101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hof PR, Nimchinsky EA, Young WG, Morrison JH. Numbers of Meynert and layer IVB cells in area V1: a stereologic analysis in young and aged macaque monkeys. J Comp Neurol. 2000;420:113–126. doi: 10.1002/(SICI)1096-9861(20000424)420:1<113::AID-CNE8>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 45.Keuker JIH, Luiten PGM, Fuchs E. Preservation of hippocampal neuron numbers in aged rhesus monkeys. Neurobiol Aging. 2003;24:157–165. doi: 10.1016/S0197-4580(02)00062-3. [DOI] [PubMed] [Google Scholar]
- 46.Merrill DA, Roberts JA, Tuszynski MH. Conservation of neuron number and size in entorhinal cortex layers II, III, and V/VI of aged primates. J Comp Neurol. 2000;422:396–401. doi: 10.1002/1096-9861(20000703)422:3<396::AID-CNE6>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 47.Peters A, Morrison JH, Rosene DL, Hyman BT. Feature article are neurons lost from the primate cerebral cortex during normal aging? Cereb Cortex. 1998;8:295–300. doi: 10.1093/cercor/8.4.295. [DOI] [PubMed] [Google Scholar]
- 48.Roberts DE, Killiany RJ, Rosene DL. Neuron numbers in the hypothalamus of the normal aging rhesus monkey: stability across the adult lifespan and between the sexes. J Comp Neurol. 2012;520:1181–1197. doi: 10.1002/cne.22761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Smith DE, Rapp PR, McKay HM, Roberts JA, Tuszynski MH. Memory impairment in aged primates is associated with focal death of cortical neurons and atrophy of subcortical neurons. J Neurosci. 2004;24:4373–4381. doi: 10.1523/JNEUROSCI.4289-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bodkin NL, Alexander TM, Ortmeyer HK, Johnson E, Hansen BC. Mortality and morbidity in laboratory-maintained rhesus monkeys and effects of long-term dietary restriction. J Geron. 2003;58A:212–219. doi: 10.1093/gerona/58.3.B212. [DOI] [PubMed] [Google Scholar]
- 51.Mattison JA, Vaughan KL. An overview of nonhuman primates in aging research. Exp Gerontol. 2017;94:41–45. doi: 10.1016/j.exger.2016.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mattison JA, Black A, Huck J, Moscrip T, Handy A, Tilmont E, Roth GS, Lane MA, Ingram DK. Age-related decline in caloric intake and motivation for food in rhesus monkeys. Neurobiol Aging. 2005;26:1117–1127. doi: 10.1016/j.neurobiolaging.2004.09.013. [DOI] [PubMed] [Google Scholar]
- 53.Matthews KA, Xu W, Gaglioti AH, Holt JB, Croft JB, Mack D, McGuire LC. Racial and ethnic estimates of Alzheimer’s disease and related dementias in the United States (2015–2060) in adults aged ≥65 years. Alzheimers Dement. 2019;15:17–24. doi: 10.1016/j.jalz.2018.06.3063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Peters A, Kemper T. A review of the structural alterations in the cerebral hemispheres of the aging rhesus monkey. Neurobiol Aging. 2012;33:2357–2372. doi: 10.1016/j.neurobiolaging.2011.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Peters A, Leahu D, Moss MB, Mcnally KJ. The effects of aging on area 46 of the frontal cortex of the rhesus monkey. Cereb Cortex. 1994;4:621–635. doi: 10.1093/cercor/4.6.621. [DOI] [PubMed] [Google Scholar]
- 56.Primiani CT, Ryan VH, Rao JS, Cam MC, Ahn K, Modi HR, Rapoport SI. Coordinated gene expression of neuroinflammatory and cell signaling markers in dorsolateral prefrontal cortex during human brain development and aging. PLoS One. 2014;9:e110972. doi: 10.1371/journal.pone.0110972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yuan Y, Chen YPP, Boyd-Kirkup J, Khaitovich P, Somel M. Accelerated aging-related transcriptome changes in the female prefrontal cortex. Aging Cell. 2012;11:894–901. doi: 10.1111/j.1474-9726.2012.00859.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dumas JA, Kutz AM, McDonald BC, Naylor MR, Pfaff AC, Saykin AJ, Newhouse PA. Increased working memory-related brain activity in middle-aged women with cognitive complaints. Neurobiol Aging. 2013;34:1145–1147. doi: 10.1016/j.neurobiolaging.2012.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gigi A, Babai R, Katzav E, Atkins S, Hendler T. Prefrontal and parietal regions are involved in naming of objects seen from unusual viewpoints. Beh Neuro. 2007;121:836–844. doi: 10.1037/0735-7044.121.5.836. [DOI] [PubMed] [Google Scholar]
- 60.Pimontel MA, Kanellopoulos D, Gunning FM. Neuroanatomical abnormalities in older depressed adults with apathy: a systematic review. J Geriatr Psychiatry Neurol. 2019;33:289–303. doi: 10.1177/0891988719882100. [DOI] [PubMed] [Google Scholar]
- 61.Shamy JL, Habeck C, Hof PR, Amaral DG, Fong SG, Buonocore MH, Stern Y, Barnes CA, Rapp PR. Volumetric correlates of spatiotemporal working and recognition memory impairment in aged rhesus monkeys. Cereb Cortex. 2011;21:1559–1573. doi: 10.1093/cercor/bhq210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mattison JA, Roth GS, Beasley TM, Tilmont EM, Handy AM, Herbert RL, Longo DL, Allison DB, Young JE, Bryant M, Barnard D, Ward WF, Qi W, Ingram DK, De Cabo R. Impact of caloric restriction on health and survival in rhesus monkeys from the NIA study. Nature. 2012;489:318–321. doi: 10.1038/nature11432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ingram DK, Cutler RG, Weindruch R, Renquist DM, Knapa JJ, April M, Belcher CT, Clark MA, Hatcherson CD, Marriott BM, et al. Dietary restriction and aging: the initiation of a primate study. J Gerontol. 1990;45:B148–B163. doi: 10.1093/geronj/45.5.b148. [DOI] [PubMed] [Google Scholar]
- 64.Rosene DL, Roy NJ, Davis BJ. A cryoprotection method that facilitates cutting frozen sections of whole monkey brains for histological and histochemical processing without freezing artifact. J Histochem Cytochem. 1986;34:1301–1315. doi: 10.1177/34.10.3745909. [DOI] [PubMed] [Google Scholar]
- 65.Estrada LI, Robinson AA, Amaral AC, Giannaris EL, Heyworth NC, Mortazavi F, Ngwenya LB, Roberts DE, Cabral HJ, Killiany RJ, Rosene DL (2017) Evaluation of long-term cryostorage of brain tissue sections for quantitative histochemistry. J Histochem Cytochem 65:153–171. PMID: 28080173, PMCID: PMC5298458. [DOI] [PMC free article] [PubMed]
- 66.Bhattacherjee A, Djekidel MN, Chen R, Chen W, Tuesta LM, Zhang Y. Cell type-specific transcriptional programs in mouse prefrontal cortex during adolescence and addiction. Nat Commun. 2019;10:1–18. 10.1038/s41467-019-12054-3. [DOI] [PMC free article] [PubMed]
- 67.Tigges J, Gordon TP, McClure HM, Hall EC, Peters A. Survival rate and life span of rhesus monkeys at the Yerkes regional primate research center. Am J Primatol. 1988;15:263–273. doi: 10.1002/ajp.1350150308. [DOI] [PubMed] [Google Scholar]
- 68.Gearing M, Tigges J, Mori H, Mirra SS. A beta40 is a major form of beta-amyloid in nonhuman primates. Neurobiol Aging. 1996;17:903–908. doi: 10.1016/s0197-4580(96)00164-9. [DOI] [PubMed] [Google Scholar]
- 69.Zheng W, Tsai MY, Wolynes PG. Comparing the aggregation free energy landscapes of amyloid Beta(1-42) and amyloid Beta(1-40) J Am Chem Soc. 2017;139:16666–16676. doi: 10.1021/jacs.7b08089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Edmonds EC, Bangen KJ, Delano-Wood L, Nation DA, Furst AJ, Salmon DP, Bondi MW, for the Alzheimer’s Disease Neuroimaging Initiative Patterns of cortical and subcortical amyloid burden across stages of preclinical Alzheimer’s disease. J Int Neuropsychol Soc. 2016;22:978–990. doi: 10.1017/S1355617716000928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sloane JA, Pietropaolo MF, Rosene DL, Moss MB, Peters A, Kemper T, Abraham CR. Lack of correlation between plaque burden and cognition in the aged monkey. Acta Neuropathol. 1997;94:471–478. doi: 10.1007/s004010050735. [DOI] [PubMed] [Google Scholar]
- 72.Selkoe DJ, Hardy J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol Med. 2016;8:595–608. doi: 10.15252/emmm.201606210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ikonomovic MD, Klunk WE, Abrahamson EE, Mathis CA, Price JC, Tsopelas ND, Lopresti BJ, Ziolko S, Bi W, Paljug WR, Debnath ML, Hope CE, Isanski BA, Hamilton RL, DeKosky ST. Post-mortem correlates of in vivo PiB-PET amyloid imaging in a typical case of Alzheimer’s disease. Brain. 2008;131:1630–1645. doi: 10.1093/brain/awn016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gusel’nikova VV, Korzhevskiy DE. NeuN as a neuronal nuclear antigen and neuron differentiation marker. Acta Naturae. 2015;7:42–47. doi: 10.32607/20758251-2015-7-2-42-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kim KK, Adelstein RS, Kawamoto S. Identification of neuronal nuclei (NeuN) as Fox-3, a new member of the Fox-1 gene family of splicing factors. J Biol Chem. 2009;284:31052–31061. doi: 10.1074/jbc.M109.052969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gittins R, Harrison PJ. Neuronal density, size and shape in the human anterior cingulate cortex: a comparison of Nissl and NeuN staining. Brain Res Bull. 2004;63:155–160. doi: 10.1016/j.brainresbull.2004.02.005. [DOI] [PubMed] [Google Scholar]
- 77.Duan W, Zhang YP, Hou Z, Huang C, Zhu H, Zhang CQ, Yin Q. Novel insights into NeuN: from neuronal marker to splicing regulator. Mol Neurobiol. 2016;53:1637–1647. doi: 10.1007/s12035-015-9122-5. [DOI] [PubMed] [Google Scholar]
- 78.Cruz E, Kumar S, Yuan L, Arikkath J, Batra SK. Intracellular amyloid beta expression leads to dysregulation of the mitogen-activated protein kinase and bone morphogenetic protein-2 signaling axis. PLoS One. 2018;13:1–21. doi: 10.1371/journal.pone.0191696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kimura N, Yanagisawa K, Terao K, Ono F, Sakakibara I, Ishii Y, Kyuwa S, Yoshikawa Y. Age-related changes of intracellular Aβ in cynomolgus monkey brains. Neuropathol Appl Neurobiol. 2005;31:170–180. doi: 10.1111/j.1365-2990.2004.00624.x. [DOI] [PubMed] [Google Scholar]
- 80.LaFerla FM, Green KN, Oddo S. Intracellular amyloid-β in Alzheimer’s disease. Nat Rev Neurosci. 2007;8:499–509. doi: 10.1038/nrn2168. [DOI] [PubMed] [Google Scholar]
- 81.Martin LJ, Sisodia SS, Koo EH, Cork LC, Dellovade TL, Weidemann A, Beyreuther K, Masters C, Price DL. Amyloid precursor protein in aged nonhuman primates. Proc Natl Acad Sci U S A. 1991;88:1461–1465. doi: 10.1073/pnas.88.4.1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Luebke J, Barbas H, Peters A. Effects of normal aging on prefrontal area 46 in the rhesus monkey. Brain Res Rev. 2010;62:212–232. doi: 10.1016/j.brainresrev.2009.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Willette AA, Coe CL, Birdsill AC, Bendlin BB, Colman RJ, Alexander AL, Allison DB, Weindruch RH, Johnson SC. Interleukin-8 and interleukin-10, brain volume and microstructure, and the influence of calorie restriction in old rhesus macaques. Age (Omaha) 2013;35:2215–2227. doi: 10.1007/s11357-013-9518-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Harper JM, Leathers CW, Austad SN. Does caloric restriction extend life in wild mice? Aging Cell. 2006;5:441–449. doi: 10.1111/j.1474-9726.2006.00236.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Schafer MJ, Alldred MJ, Lee SH, Calhoun ME, Petkova E, Mathews PM, Ginsberg SD. Reduction of β-amyloid and γ-secretase by calorie restriction in female Tg2576 mice. Neurobiol Aging. 2015;36:1293–1302. doi: 10.1016/j.neurobiolaging.2014.10.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Sridharan A, Pehar M, Salamat MS, Pugh TD, Bendlin BB, Willette AA, Anderson RM, Kemnitz JW, Colman RJ, Weindruch RH, Puglielli L, Johnson SC. Calorie restriction attenuates astrogliosis but not amyloid plaque load in aged rhesus macaques: a preliminary quantitative imaging study. Brain Res. 2013;1508:1–8. doi: 10.1038/jid.2014.371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Austad S N (2001) Does caloric restriction in the laboratory simply prevent overfeeding and return house mice to their natural level of food intake? Science 2001:pe3. 10.1126/sageke.2001.6.pe3. [DOI] [PubMed]
- 88.Le Bourg E. Does calorie restriction in primates increase lifespan? Revisiting studies on macaques (Macaca mulatta) and mouse lemurs (Microcebus murinus) BioEssays. 2018;40:e1800111. doi: 10.1002/bies.201800111. [DOI] [PubMed] [Google Scholar]
- 89.Major DE, Kesslak JP, Cotman CW, Finch CE, Day JR. Life-long dietary restriction attenuates age-related increases in hippocampal glial fibrillary acidic protein mRNA. Neurobiol Aging. 1997;18:523–526. doi: 10.1016/s0197-4580(97)00102-4. [DOI] [PubMed] [Google Scholar]
- 90.Nelson PT, Alafuzoff I, Bigio E, Bouras C, Braak H, Cairns NJ, Castellani R, Crain B, Davies P, Del Tredic K, Duyckaerts C, Frosch MP, Haroutunian V, Hof PR, Hulette CM, Hyman BT, Iwatsubo T, Jellinger K, Jicha GA, Kövari E, Kukull W, Leverenz J, Love S, Mackenzie IR, Mann DM, Masliah E, McKee A, Montine TJ, Morris JC, Schneider JA, Sonnen JA, Thal DR, Trojanowski JQ, Troncoso JC, Wisniewski T, Woltjer RL, Beach TG. Correlation of Alzheimer disease neuropathologic changes with cognitive status: a review of the literature. J Neuropathol Exp Neurol. 2012;71:362–381. doi: 10.1097/NEN.0b013e31825018f7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kastman EK, Willette AA, Coe CL, Bendlin BB, Kosmatka KJ, McLaren DG, Xu G, Canu E, Field AS, Alexander AL, Voytko ML, Beasley MT, Colman RJ, Weindruch RH, Johnson SC. A calorie-restricted diet decreases brain Iron accumulation and preserves motor performance in old rhesus monkeys. J Neurosci. 2012;32:11897–11904. doi: 10.1523/JNEUROSCI.2553-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zhang J, Chen B, Lu J, Wu Y, Wang S, Yao Z, Zhu L, Qiao Y, Sun Q, Qin W, Zhao Q, Jia J, Wie C. Brains of rhesus monkeys display Aβ deposits and glial pathology while lacking dimers and other Alzheimer’s pathologies. Aging Cell. 2018;18:e12978. doi: 10.1111/acel.12978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Beckman D, Ott S, Donis-Cox K, Janssen WG, Bliss-Moreau E, Rudebeck PH, Baxter MG, Morrison JH. Oligomeric Aβ in the monkey brain impacts synaptic integrity and unduces accelerated cortical aging. PNAS. 2019;116:26239–26246. doi: 10.1073/pnas.1902301116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Forny-Germano L, Lyra eSilva NM, Batista AF, Brito-Moreira J, Gralle M, Boehnke SE, Coe BC, Lablans A, Marques SA, AMB M, Klein WL, Houzel J-C, Ferreira ST, Munoz DP, De Felice FG. Alzheimer’s disease-like pathology induced by amyloid-β oligomer in nonhuman primates. J Neurosci. 2014;34:13629–13643. doi: 10.1523/JNEUROSCI.1353-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(DOCX 2118 kb)
Data Availability Statement
Contact corresponding author for data.