

Abstract
Nucleoside monophosphate prodrugs that are eventually bioconverted to the active nucleoside triphosphate (NTP) offer the potential to deliver increased intracellular NTP levels and/or organ-specific NTP enhancement. There are several classes of monophosphate prodrugs that have been applied to HCV drug discovery, and some of these approaches are currently being evaluated in humans. This review discusses recent advances in monophosphate prodrug approaches to improve oral absorption, stability and pharmacokinetic profile, including their advantages and potential pitfalls.
Introduction
HCV is a positive-stranded RNA virus, which was identified in 1989 as a member of the Flaviviridae family [1]. The resultant infection is a serious problem affecting approximately 3% of the worldwide population, according to the World Health Organization (WHO). The WHO also estimates that 4 million people contract HCV each year. Although the early stages of HCV are usually asymptomatic, and approximately 20% of infected individuals naturally clear the virus, the majority of infections progress to chronic infection. In addition, >20% of chronically infected individuals will eventually develop more serious liver problems, such as cirrhosis, hepatocellular carcinoma or liver failure requiring liver transplantation [2–4]. Indeed, in industrialized nations, HCV infection is the leading cause for liver transplants [5,6]. Unfortunately, reinfection often occurs post-transplantation [5]. The virus is transmitted parenterally by contaminated blood, usually from sharing contaminated needles, but also from improper sterilization of medical, dental, body piercing or tattoo equipment. Heterosexual transmission and vertical transmission (infected mother-to-child transmission during the birth process) of HCV can also occur, but are rare [7,8]
Presently, there is no vaccine for HCV, although early attempts have shown encouraging results [9]. Chimpanzees are currently the most useful animal model to study antiviral agents for HCV infections; however, ethical concerns, increased costs and other restrictions make large studies difficult. The current treatments that have been approved by the US Food and Drug Administration for chronic HCV are limited to pegylated interferon-α alone or in combination with ribavirin [10]. Unfortunately, these treatments have limited efficacy with response rates of only 40–50% for the genotype-1-infected population, which is the most prevalent genotype in the US and China [11,12]. This treatment also has major side effects, including depression, anaemia, central nervous system toxicity, fatigue, flu-like symptoms, mild alopecia, thyroid dysfunction and teratogenic effects [13–19]. The combination of pegylated interferon-α (given by injection) and ribavirin probably does not directly act on the virus and its mechanism is not well-characterized [20], but is thought to aid the immune system in the clearance of the virus from infected individuals. In addition, the drug combination is not well-tolerated in individuals coinfected with other viruses such as HIV [21].
There is an unmet medical need for safer and more effective treatments for HCV, and two major drug targets have been identified: the NS3/4A serine protease and NS5B RNA-dependent RNA polymerase (RdRp) [22,23]. Peptidomimetics comprise the majority of compounds studied for the NS3/4A serine protease target. Non-nucleoside and nucleoside analogue inhibitors are the two main categories of RdRp inhibitors, which differ based on their chemical structure and mode of action. There have been several modified nucleosides reported that exhibit anti-HCV activity, most with modifications at the 2′ or 4′ positions of the sugar. However, potential problems for the use of these nucleoside analogues are poor cell permeability or unsuitability of the modified nucleoside as a substrate for cellular kinases, for which compatibility is needed in order to transform the nucleoside into a biologically active nucleoside triphosphate (NTP). Because HCV lacks virus-encoded nucleoside kinase expression, conversion to the biologically active NTP needs to be accomplished by cellular kinases [24]; however, poor turnover to the biologically active species is routinely seen. In many cases, it is the conversion to the nucleoside monophosphate (NMP) that is the rate-limiting step [20], but there are exceptions [25]. The anabolism and pharmacokinetic (PK) requirements of nucleoside and nucleotide analogue inhibitors of viral RNA are likely more stringent relative to DNA for polymerization in light of the one to two orders of magnitude higher intracellular levels of ribonucleoside triphosphates (TPs) that typically are in the μM range in the RNA [26]. Furthermore, the challenge of delivering sufficient concentrations of anti-HCV NTP analogues to the HCV polymerase provides an additional barrier to drug development. The NTP itself cannot be considered as a drug candidate because of degradation by intracellular phosphodiesterases and/or alkaline phosphatases and, in particular, because of poor cell permeability.
Prodrug strategies wherein a pharmacologically inactive compound must be converted to the biologically active species in vivo have been widely employed in the nucleoside area in an attempt to circumvent these problems. The development of monophosphorylated nucleoside prodrugs, thereby, bypassing the rate-limiting step of the initial phosphorylation, has been extensively explored [27–30].
Once the target compound is identified and tested in a cell-based assay, a dose–response curve (50% effective concentration [EC50] and EC90) is determined for promising compounds (Figure 1). Prodrugs that target the liver potentially offer an additional challenge of requiring liver enzymes for unmasking of the monophosphate (MP), which might not be present in the cell line used for antiviral activity evaluation [31]. The chemical stability of the prodrug in cell culture media, buffer and plasma will help eliminate prodrugs with no potential for oral delivery or with a short half-life (t1/2). The NTP is synthesized and tested against the HCV RdRp to confirm chain termination. Metabolism studies in human hepatocytes follow, but can be done much earlier for prodrugs that are not unmasked in various cell-based assays. After in vitro selection of resistant HCV and further testing against these mutants, tests in simulated gastric fluid (SGF), simulated intestinal fluid (SIF) and human liver microsomes provide additional stability data prior to in vivo toxicity studies (there are instances where these steps can be skipped and compounds can be tested directly in vivo). Finally, in vivo studies are performed in various animal models to determine PK profiles [32].
Figure 1.
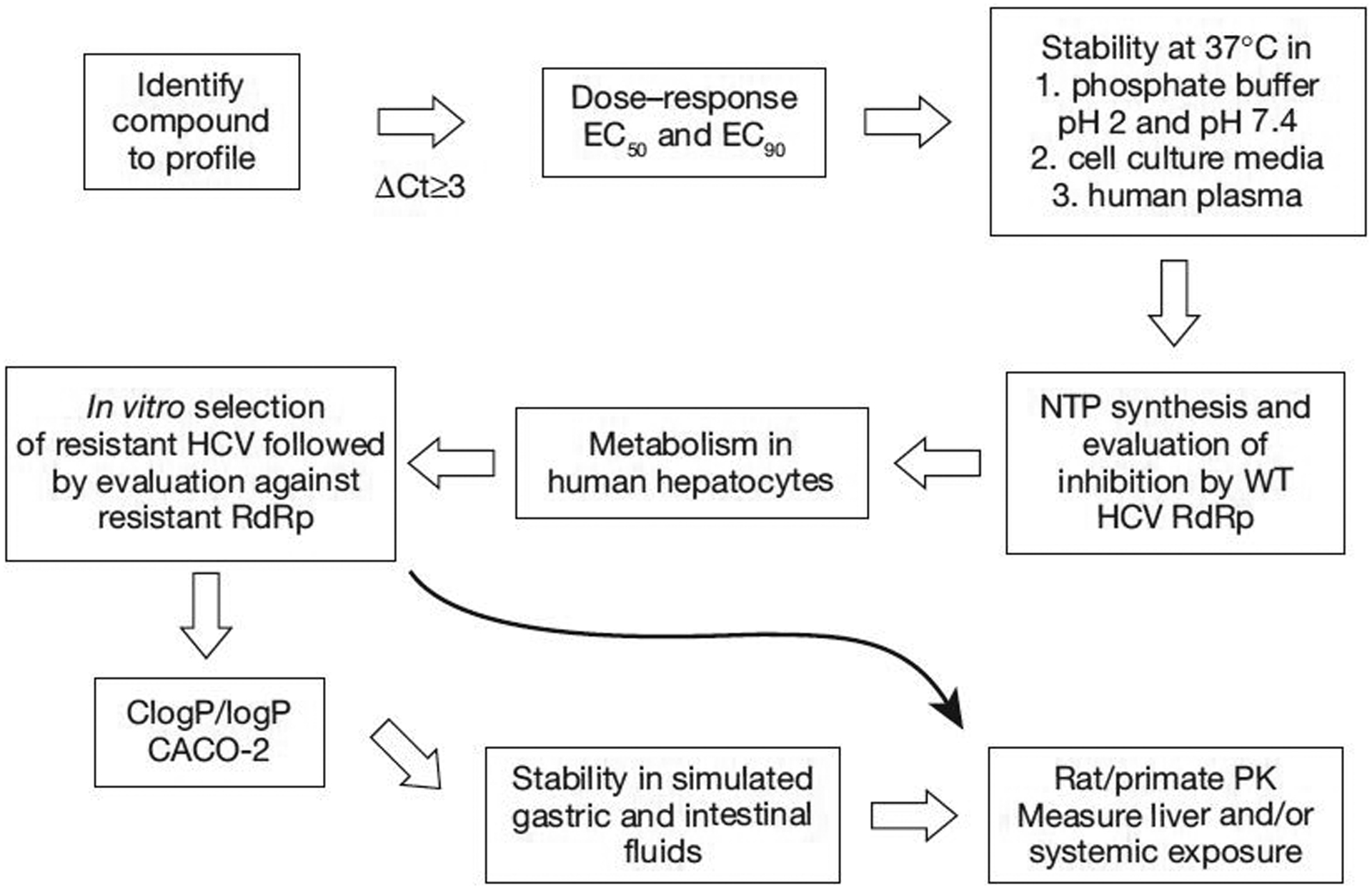
HCV prodrug evaluation strategy
CACO-2, human epithelial colorectal adenocarcinoma cells; ClogP/logP, partition coefficient; EC50, 50% effective concentration; EC90, 90% effective concentration; NTP, nucleoside triphosphate; PK, pharmacokinetics; RdRp, RNA-dependent RNA polymerase; WT, wild-type; ΔCt, change in cycle threshold.
Currently, the most advanced MP prodrugs might bestow several problems, which include intracellular delivery of toxic agents, a racemic phosphorous centre, a weak nucleoside–MP phosphorous–oxygen bond that is prone to cleavage, and the preparation of many MP prodrugs that occur in low to very low yields. There are several classes of NMP prodrugs that have been successfully applied to HCV therapeutics; only the most significant from mid-2007 forward will be the subject of this review.
Aryloxy phosphoramidate
As discussed earlier, some nucleoside analogues have poor cell permeability and/or are not suitable substrates for cellular kinases, which transform it to the biologically active NTP. This problem can be alleviated by the use of a substituted MP prodrug strategy that would be converted, initially, to the NMP and then further to the NTP. It was suggested during the study of potential prodrugs for HIV in the 1990s that nucleosides with a 5′-phosphoramidate group might be able to bypass the problematic initial kinase-promoted phosphorylation step and eventually lead to the active triphosphorylated species [33]. Indeed, this was the case and these aryloxy phosphoramidate prodrugs, termed ProTides [34], have been incorporated in the study of many viral diseases, including recently to HCV. There have been many reports of modified nucleoside Pro-Tides that show biological activity against HCV in vitro (subgenomic HCV replicon RNA) as well as in vivo. It has been shown that the TP of 2′-C-methylcytidine is quite potent against the isolated NS5B enzyme (50% inhibitory concentration [IC50]=0.025 μM), but the parent nucleoside, 2′-C-methylcytidine, shows only modest activity (EC50=2–7 μM) in a subgenomic cell-based replicon assay [35]. Recent studies indicate that conversion of 2′-C-methylcytidine to the biologically active NTP is a problematic pathway and, in particular, the conversion to the NMP is a rate-limiting step [36].
Merck & Co., Inc. [37] has recently reported a thorough evaluation of different phosphoramidate prodrugs of 2′-C-methylcytidine, which included NM-107 (Figure 2). The introduction of the ProTide group at the 5′-position shows promising activity. Structure–activity relationship studies were performed by varying both the alanine ester portion and the aryloxy portion of the phosphoramidate and representative examples are shown in Figure 2. Starting with the ethyl ester and 4-chlorophenyl phosphoramidate (1), an EC50 value of 1 μM was obtained with no apparent cytotoxicity (50% cell cytotoxicity [CC50]>20 μM). Because the toxicity of 4-chlorophenol was less understood and less studied, the unsubstituted phenol substituent was also tested and a twofold increase in activity without any cytotoxicity was observed. Further substitution to a more lipophilic 1-napthyl (1-Nap) group resulted in comparable activity (EC50=0.2 μM) and a slight increase in cytotoxicity (CC50 ranging from >20 μM for 2 to 15 μM for 3). Using a more lipophilic ester moiety (butyl versus ethyl) resulted in a twofold increase in activity with an EC50 of 0.09 μM and use of the 2-propylpentyl ester gave even more potent activity with an EC50 of 0.04 μM, but with an increased cytotoxicity (CC50=2 μM).
Figure 2.
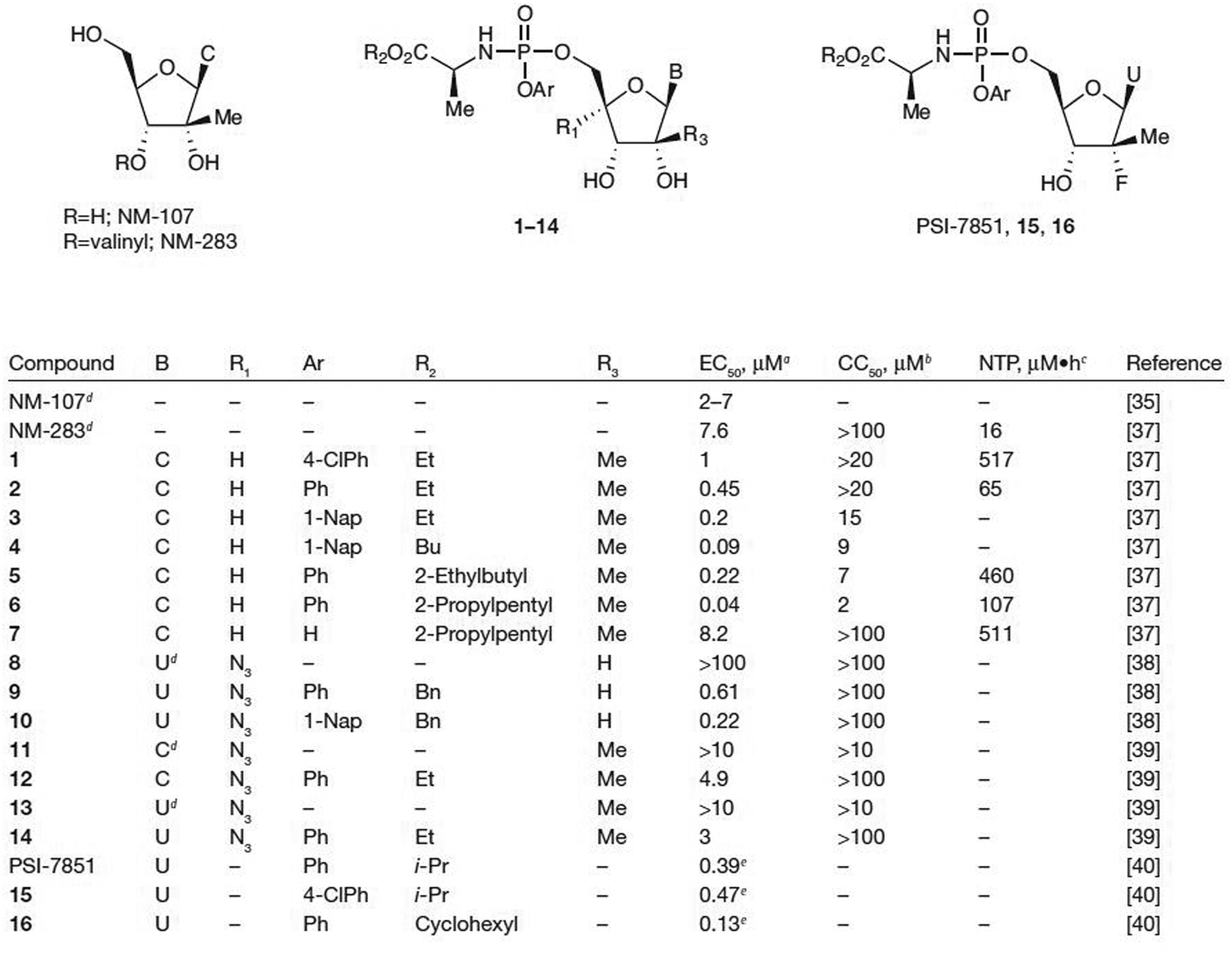
Antiviral activity and cytotoxicity of ProTide prodrugs of pyrimidine analogues
aEffective concentration of compound required to reduce HCV replication by 50% (EC50). bConcentration of inhibitor reducing cell viability by 50% (CC50). cIntracellular nucleoside triphosphate (NTP) concentration after incubation at 10 μM with human hepatocytes. dParent nucleoside. eEffective concentration of compound required to reduce HCV replication by 90%. Bn, benzyl; Bu, butyl; C, cytosine; Et, ethyl; i-Pr, isopropyl; Me, methyl; Ph, phenyl; ProTide, aryloxy phosphoramidate; U, uracil; 1-Nap, 1-napthyl; 4-CIPh, 4-chlorophenyl.
On the basis of anti-HCV data obtained in cell culture for the ProTides of 2′-C-methylcytidine, additional studies were performed with 1 in human hepatocytes [37]. High intracellular concentrations of the 2′-C-methylcytidine-TP were obtained after incubation at 10 μM for 4 h. When expressed as the area under the curve between 0 and 4 h, 517 μM•h of 2′-C-methylcytidine-TP was found: a 32-fold increase over NM-283 (valopicitabine, the valine prodrug of NM-107) under the same conditions. These promising results indicate successful intracellular delivery of the 2′-C-methylcytidine-MP and apparent benefit to bypassing the initial kinase-driven phosphorylation step. The stability of prodrugs 1 and 5 were tested in the plasma of various species. No significant degradation was seen in either dog or human plasma after 5 min for either prodrug; however, significant degradation in both rat and mouse plasma was observed. Further examination of prodrug 5 showed that it was moderately stable in hamster plasma after 5 min. Because of this stability in hamster plasma and in vitro potency, the PK profile of 5 was evaluated. Oral administration of 5 at 30 μmol/kg to hamsters resulted in low (1.1 μM) liver NTP levels at 6 h, indicating low bioavailability, but was twofold higher than NM-283. The stability was also tested in SGF and the compound was found to be stable with no degradation up to 3 h; thus, the low bioavailability could be caused by either poor absorption or degradation in the intestine. Intramuscular injection at a much lower dose of 1.5 μmol/kg resulted in liver NTP levels of 3.6 and 2.2 μM at 3 h and 6 h, respectively. Subcutaneous injection at the same dose resulted in even higher liver NTP levels of 5.6 and 10.1 μM at 3 h and 6 h, respectively. By contrast, oral dosing of NM-283 resulted in liver NTP levels of 0.48 μM and subcutaneous injection at a dose 5.5-fold higher than the dose used for 5 gave liver NTP levels below the lower limit of detection. Hence, MP prodrug 5 showed improved in vivo liver exposure when compared with the prodrug NM-283.
Merck & Co., Inc. [37] explored the removal of the aryloxy portion of the ProTide to eliminate the release of potentially toxic phenol resulting in hydroxy phosphoramidate prodrug 7 with the more lipophilic 2-propylpentyl ester. Although 7 was less active in the cell-based replicon assay (EC50=8.2 μM), high NTP levels in human hepatocytes (511 μM•h) were observed. The hydroxy phosphoramidate prodrug 7 showed good stability in plasma from various species and a favourable metabolic stability profile in rat and human liver fractions. High NTP levels of 39 μM were obtained in hamster liver after subcutaneous injection of 1.5 μmol/kg; however, low bioavailability was seen when dosed orally at 30 μmol/kg.
McGuigan and colleagues [38] have also synthesized a series of ProTide prodrugs of 4′-azidouridine (8). The prodrugs that contained the more lipophilic Ar=1-Nap proved to be the most potent. Most of the prodrugs showed greater anti-HCV potency than the inactive parent nucleoside 8. For example, an EC50 value of 0.61 μM for 9 (Ar=phenyl and R2=benzyl) and 0.22 μM for 10 (Ar=1-Nap and R2=benzyl; Figure 2) was observed. All of the prodrugs as well as the parent nucleoside 8 were non-toxic in the replicon assay (CC50>100 μM); however, additional in vivo studies, such as oral bioavailability, were not reported. This study reinforces their earlier conclusion that a separate ProTide motif optimization process is required for each nucleoside analogue versus a given target.
Other recent phosphoramidate prodrug studies include the prodrugs of 2′-C-methyl-4′-azidocytidine (11) and 2′-C-methyl-4′-azidouridine (13), which were studied by Schinazi and colleagues [39]. Both parent nucleosides 11 and 13 had an EC50 value >10 μM, whereas the corresponding prodrugs 12 and 14 had EC50 values of 4.9 and 3 μM, respectively. Again, neither prodrug displayed any toxicity, even up to 100 μM; however, in vivo studies were not reported.
Pharmasset, Inc. [40] has also recently reported 2′-deoxy-2′-fluoro-2′-C-methyl-uridine ProTide prodrugs, PSI-7851, 15 and 16 (Figure 2). Submicromolar activity in the replicon assay was reported with impressive EC90 values as low as 0.13 μM for prodrug 16. A monotherapy trial for PSI-7851 (40 treatment-naive HCV-infected patients allocated to 4 arms with 10 patients per arm, each arm with 8 active and 2 placebo patients receiving either 50, 100, 200 or 400 mg) showed a dose-dependent decrease in HCV RNA up to −1.95 log10 IU/ml from baseline at day 3 with 400 mg/day oral dosing [41]. However, the S282T mutation, common among 2′-C-methyl-substituted nucleoside analogues, was selected by PSI-7851 in an HCV subgenomic replicon in Huh7 cells [42]. Earlier work in mutated HCV replicons had shown that the S282T substitution in NS5B severely debilitates replication and no compensatory residues were encoded to increase the replication fitness [43]. The intracellular metabolism of PSI-7851 was assessed using both cellular and biochemical assays to determine the mechanism of PSI-7851 activation. The study indicated that five steps are required to reach PSI-7851-TP (PSI-7409): four enzyme-dependent steps and one non-enzymatic step [44]. Similar to the results found in earlier studies [34], the first step involves ester hydrolysis catalysed by either cathepsin A or carboxylesterase 1. The resulting carboxylic acid attacks the P-centre and displaces phenol. Subsequently, the histidine triad nucleotide binding protein 1 catalyses the removal of the amino acid to form the NMP. Final conversion to the NTP is accomplished first by uridine MP-cytidine MP kinase-1 catalysed conversion to the nucleoside diphosphate followed by final catalysis by nucleoside diphosphate kinase to form the NTP.
The ProTide strategy is not limited to pyrimidine nucleosides. Several purines and substituted purines have been converted to their corresponding 5′-phosphoramidate prodrugs and tested for antiviral activity against HCV (Figure 3). Interestingly, 2′-C-methylguanosine (17) and 2′-C-methyladenine (18) have rather different activity profiles. Although the EC50 for nucleoside 17 is 10.1 μM against HCV in Huh 5–2 cells, the EC50 for nucleoside 18 is 0.25 μM, suggesting that guanosine analogue 17 might not be a suitable substrate for the cellular kinases responsible for the initial phosphorylation, whereas 18 is a viable substrate [6]. However, most phosphoramidate prodrugs of 2′-C-methylguanosine (19–23 with the exception of the tert-butyl [t-Bu] ester 23) show rather good antiviral activity in the cell-based replicon assay with an EC50 as low as 0.12 μM and no cytotoxicity for prodrug 22 (all CC50>50 μM), suggesting that the 5′-MP prodrug is indeed delivered intracellularly and efficiently converted to the biologically active NTP by cellular kinases, at least at the in vitro level. Phosphoramidate prodrugs of 2′-C-methyladenine (18) showed no significant increase in antiviral activity in the replicon assay compared with the parent nucleoside (18 versus 24 or 25; Figure 3). These data demonstrate that the prodrugs 24 and 25 offer no advantage to intracellular transport versus parent nucleoside 18, and that the initial kinase-driven phosphorylation of 18 is not a rate-limiting step towards the formation of 2′-C-methyladenine-TP. The t-Bu esters 23 and 26 were inactive in replicon assays, which might be related to the stability of tertiary esters to enzyme-mediated hydrolysis. Inhibitex, Inc. [45] recently reported preliminary results for INX-189, a ProTide of a 2′-C-methylguanosine analogue of undisclosed structure. In a cell-based HCV replicon assay, an EC50 value of 0.01 μM and an EC90 value of 0.04 μM were reported with no apparent mitochondrial toxicity up to 1 μM. PK studies in rats and non-human primates indicated that NTP levels exceeding the EC90 value were achieved for ≥24 h following 14-day oral dosing of a human equivalent dose of 100 mg. As seen with PSI-7851, the S282T mutant was also selected in vitro and was moderately resistant (≤10-fold increase in EC50) to INX-189 [46].
Figure 3.
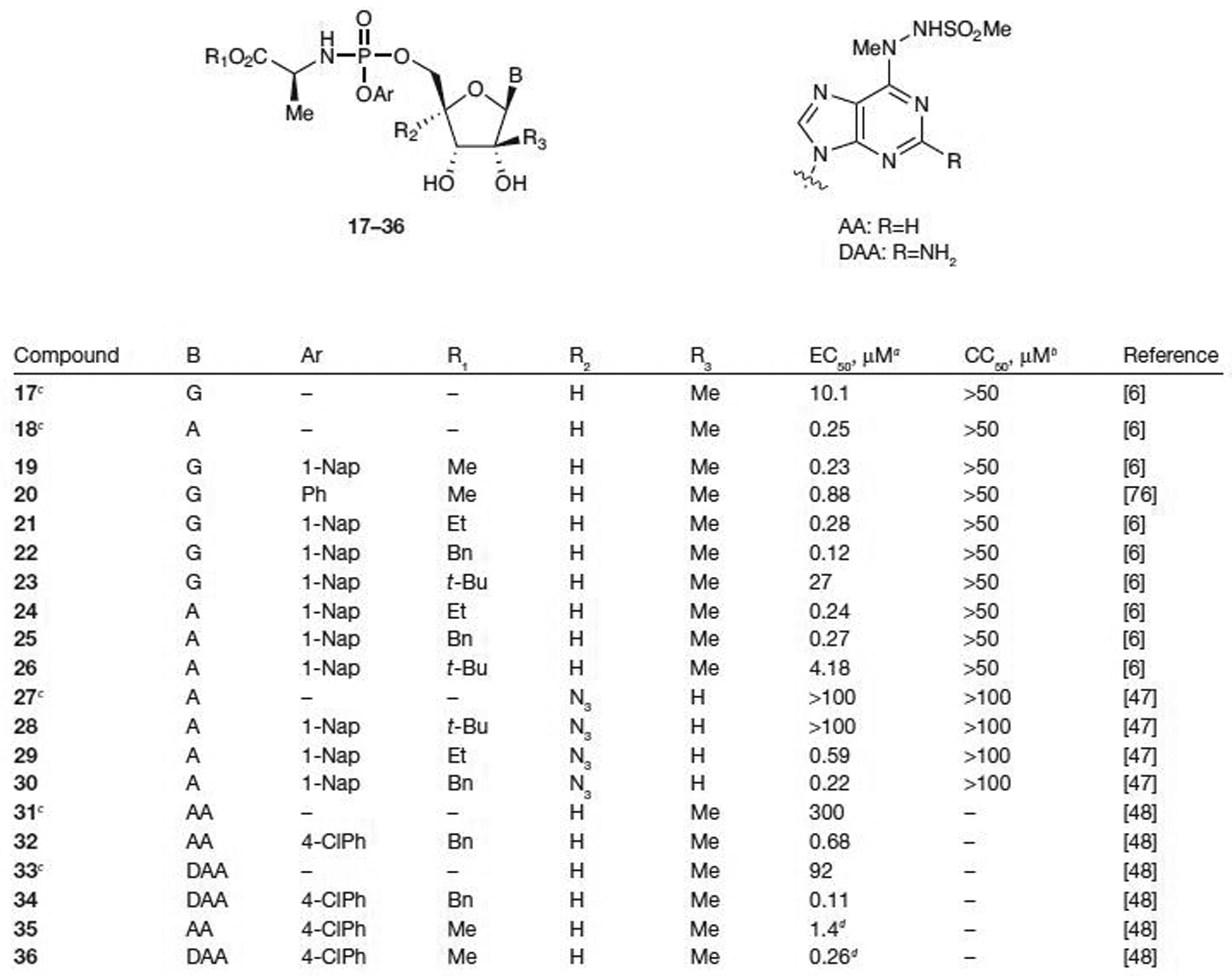
Antiviral activity and cytotoxicity of ProTide prodrugs of purine analogues
aEffective concentration of compound required to reduce HCV replication by 50% (EC50). bConcentration of inhibitor reducing cell viability by 50% (CC50). cParent nucleoside. dα-Methyl alanine methyl ester used as an amino acid. A, adenine; Bn, benzyl; Et, ethyl; G, guanine; Me, methyl; Ph, phenyl; ProTide, aryloxy phosphoramidate; t-Bu, tert-butyl; 1-Nap, 1-napthyl; 4-CIPh, 4-chlorophenyl.
McGuigan and colleagues [47] have shown that ProTide prodrugs of 4′-azidoadenosine (27) also provide an increase in antiviral potency compared with the parent nucleoside while showing no signs of cytotoxicity (CC50>100 μM). When Ar=1-Nap, both the ethyl (29) and benzyl (30) esters show much improved activity with EC50 values of 0.59 μM and 0.22 μM, respectively, over the parent nucleoside (27), which was inactive in the replicon assay (EC50>100 μM). However, as seen in previous examples with other nucleoside analogues (23 and 26), the t-Bu ester (28) was inactive in the replicon assay, which might be related to the stability of tertiary esters to enzyme-mediated hydrolysis [47].
In 2007, Valeant Pharmaceuticals International [48] reported that 6-hydrizinopurine-2′-C-methylnucleosides had poor stability and/or a poor selectivity index. This eventually led to the discovery of the corresponding ProTide prodrugs. Neither of the parent nucleosides displayed significant activity (31 with EC50=300 μM and 33 with EC50=92 μM; Figure 3); however, prodrugs 32 and 34 showed antiviral activity with EC50 values of 0.68 μM (32; Ar=4-chlorophenyl, R1=benzyl and R2=H) and 0.11 μM (34; Ar=4-chlorophenyl, R1=benzyl and R2=H), respectively. The stability of these prodrugs was also tested in human plasma, human SGF and human SIF. Both prodrugs were stable in human plasma and SGF up to 1 h, but unstable in SIF. Utilizing α-methylalanine methyl ester as the amino acid portion of the phosphoramidate gave favourable antiviral activity (EC50=1.4 μM for 35 and 0.26 μM for 36), but more importantly, no degradation was observed in SIF up to 3 h. Additional in vivo studies are currently underway for these prodrugs. Acyloxyethylamino phosphoramidates have also been studied, but poor NTP levels in rat liver after oral dosing suggests low bioavailability [49].
To avoid the release of potentially toxic phenol and other potentially toxic aryloxy derivatives, cyclic phosphoramidate prodrugs have been developed by Meppen et al. [24] and Merck & Co., Inc. [50]. Only cyclic prodrug 37 was more active than NM-283, with an EC50 value of 4 μM. All of the other cyclic prodrugs (38–41; Figure 4) were less active than NM-283 (all EC50 values ≥10 μM), regardless of the ester substituent, in the cell-based replicon assay. However, conversion to the NTP was observed in human hepatocytes for all prodrugs with up to a 10-fold increase compared with the parent NM-283 (NTP concentration ranging from 6–186 μM•h compared with 16 μM•h for NM-107). Subcutaneous injection of prodrug 40 (1.5 μM) resulted in improved NTP formation as high as 2.6 nmol/g in hamster liver after 6 h. The process responsible for the intracellular conversion of the cyclic prodrugs to their corresponding MPs is not well understood.
Figure 4.
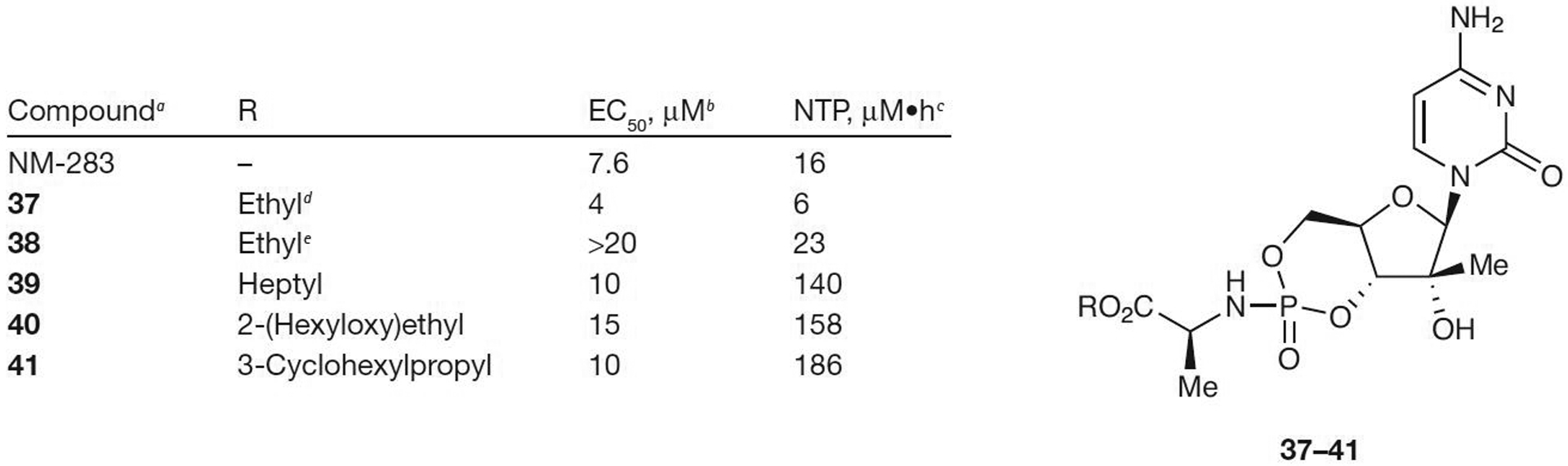
Antiviral activity of cyclic phosphoramidate prodrugs of 2′-C-methylcytosine
aFrom [24] and [50]. bEffective concentration of compound required to reduce HCV replication by 50% (EC50). cIntracellular nucleoside triphosphate (NTP) after incubation at 10 μM with human hepatocytes. dSlow eluting phosphorous diastereomer by reverse phase (RP)-HPLC. eFast eluting phosphorous diastereomer by RP-HPLC.
Both the ProTide prodrugs and the cyclic phosphoramidate prodrugs have shown promising activity in cell-based replicon assays and, in many cases, have produced a significant boost in NTP formation in vivo. Overall as a class, ProTides provide limited systemic exposure upon oral dosing and pose significant potential for toxicity associated with the liberated aryl alcohol, which appears problematic for clinical applications, especially if the drug is to be given at high doses (>400 mg daily). However, the ProTide prodrugs have proven quite valuable in elucidating the anabolic pathway for a number of nucleoside analogues. Their utility in liver-directed clinical applications remains to be determined.
S-acyl-2-thioethyl prodrugs
Another prodrug strategy developed in an effort to increase the cell permeability characteristics of nucleo sides for potential therapeutic use is the incorporation of the S-acyl-2-thioethyl (SATE) group into the 5′-MP. The MP is exposed by a multistep process involving an esterase-mediated cleavage of the thioester to reveal a thioethyl ether, which has been shown to decompose with release of ethylene sulfide (Figure 5) [51,52]. Although the toxicity risk associated with ethylene sulfide has limited the development of SATE-type prodrugs, many groups continue to explore modified SATE-type prodrugs of nucleoside analogues as potential anti-HCV agents.
Figure 5.
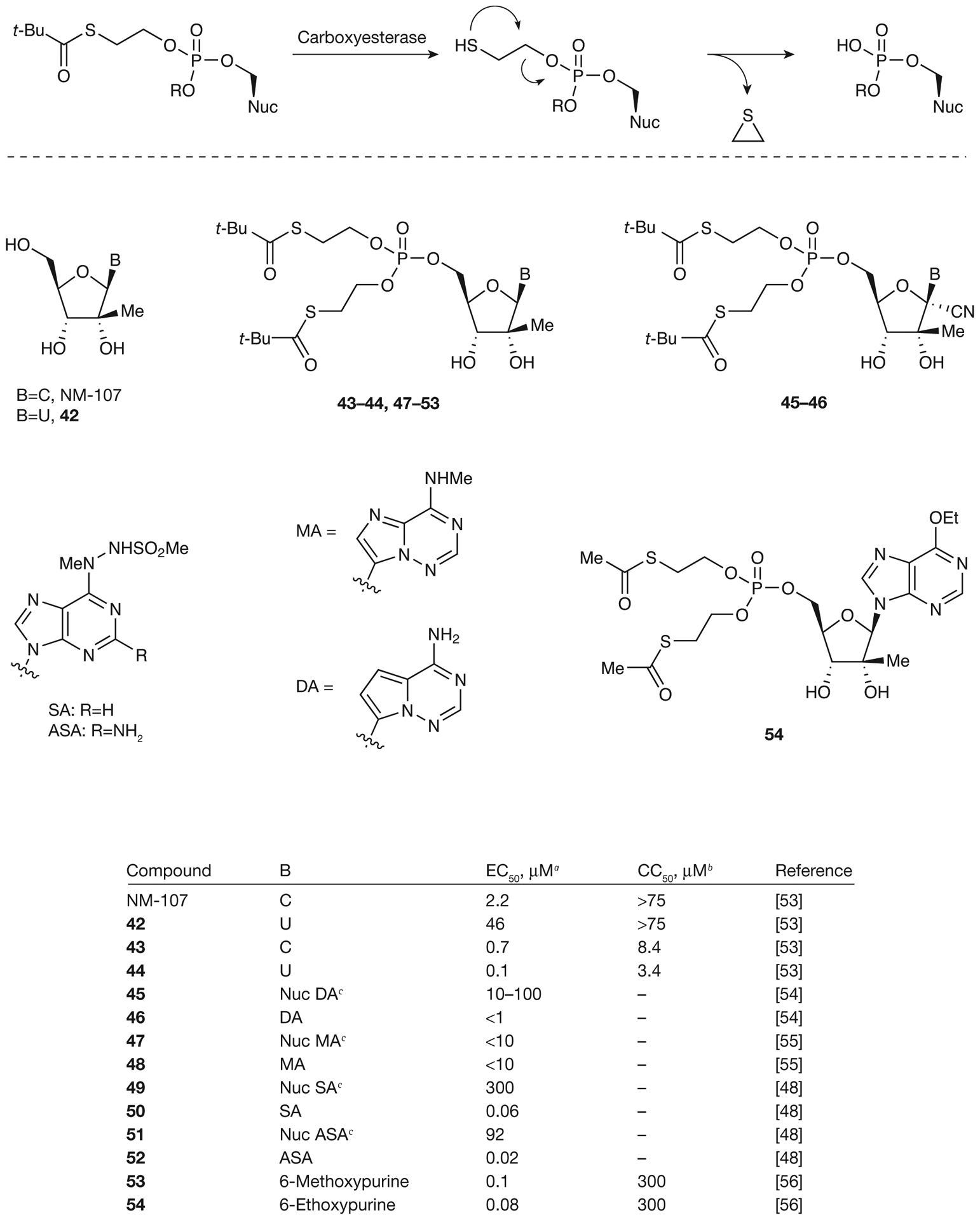
Metabolic activation and antiviral activity of SATE monophosphate prodrugs
aEffective concentration of compound required to reduce HCV replication by 50% (EC50). bConcentration of inhibitor reducing cell viability by 50% (CC50). cParent nucleoside (Nuc). C, cytosine; SATE, S-acyl-2-thioethyl; t-Bu, tert-butyl; U, uracil.
Benzaria et al. [53] from Idenix Pharmaceuticals, Inc. have shown that bis(t-BuSATE) MP prodrugs of NM-107 and 2′-C-methyluridine (42) exhibit a large improvement in the antiviral activity: from EC50=2.2 μM for NM-107 to 0.7 μM for prodrug 43 and from 46 μM for 2′-C-methyluridine (42) to 0.1 μM for prodrug 44. However, an increase in cytotoxicity was also observed with both prodrugs exhibiting CC50 values ≤8.4 μM. Several SATE prodrugs of modified adenine derivatives (46, 48, 50 and 52–54; Figure 5) have also been synthesized by Gilead Sciences [54,55] and Valeant Pharmaceuticals International [48,56], and have been tested for anti-HCV activity. In all cases, the increased lipophilicity of the SATE prodrugs led to improved cell penetration capabilities and coincided with greater potent activity with EC50 values as low as 0.02 μM for prodrug 52. However, these prodrugs showed poor stability in human plasma with t1/2 in the order of min. Further investigation into cyclic SATE prodrugs was also attempted by Valeant Pharmaceuticals International [57] in order to obtain a better PK profile (Figure 6). Again, in all cases, the cyclic SATE prodrugs (55–58) displayed an enhancement in antiviral activity, with EC50 values as low as 0.01 μM for prodrug 58 and no significant cytotoxicity observed for any of the prodrugs (CC50>50 μM). The stability of cyclic SATE prodrugs 55–58 was also observed in SIF and SGF with no significant degradation detected in 2 h. The ultimate goal was to deliver these cyclic MP prodrugs to hepatocytes; therefore, prodrug 58 was further tested for stability in rat plasma. After incubation in rat plasma for 1 h, only 4% of 58 remained unchanged with various metabolites observed (59% of cyclic MP, 19% of 5′-MP and 18% of 51). However, prodrug 58 was stable in human and monkey plasma for 1 h suggesting that this class of prodrugs might be able to be administered by an intravenous route.
Figure 6.
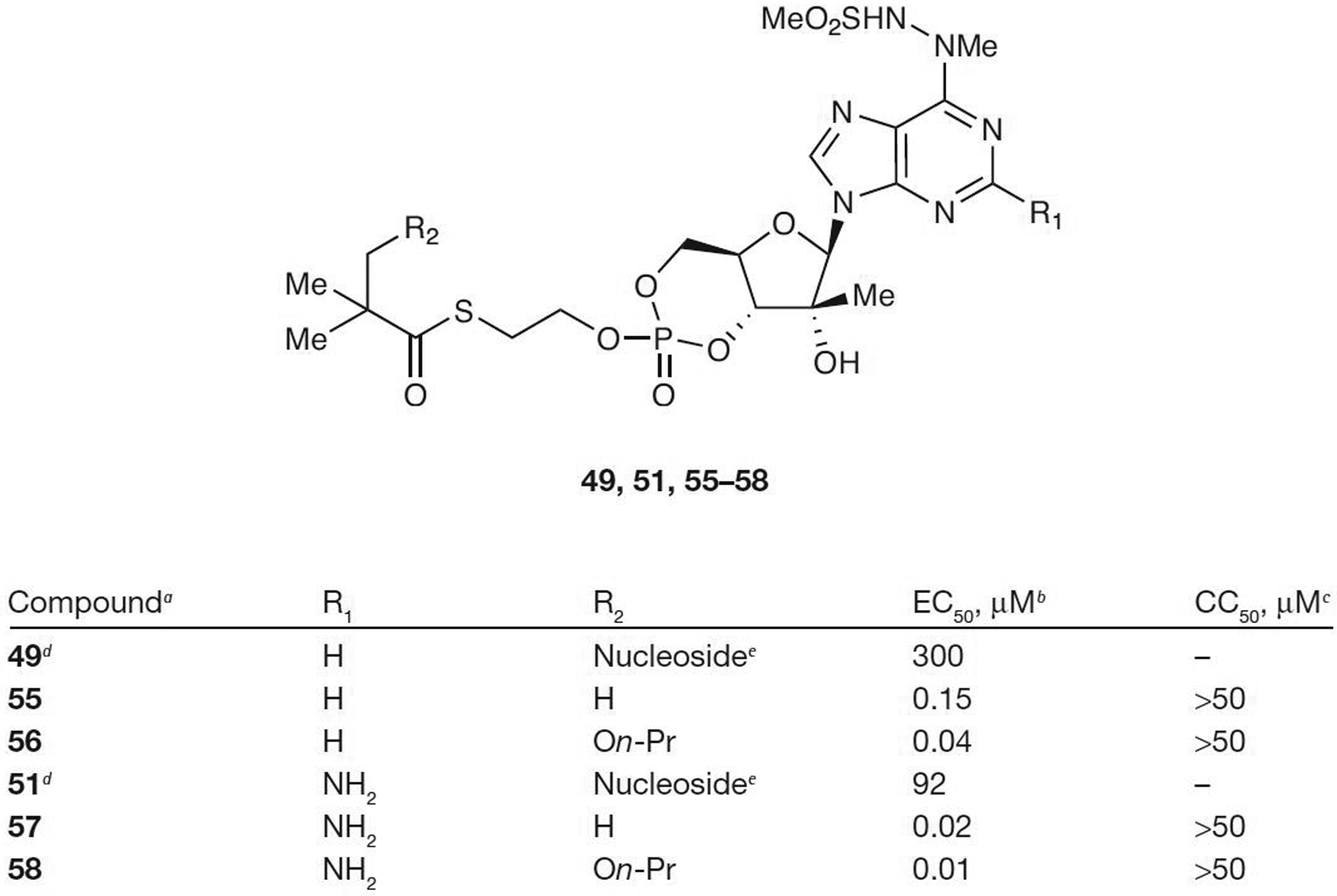
Antiviral activity and cytotoxicity of cyclic mono-SATE prodrugs
aFrom reference [57]. bEffective concentration of compound required to reduce HCV replication by 50% (EC50). cConcentration of inhibitor reducing cell viability by 50% (CC50). dFrom reference [48]. eParent nucleoside. On-Pr, On-propyl; SATE, S-acyl-2-thioethyl.
A hybrid prodrug that combined the lipophilicity of the SATE prodrugs with the stability of the ProTide phosphoramidates was recently disclosed from Idenix Pharmaceuticals, Inc. [58] (Figure 7). The mono-SATE N-benzyl phosphoramidate prodrugs 60 and 61 both displayed improved potency (EC50<1 μM) compared with the parent nucleoside, 2′-C-methylguanosine (17; EC50>10.1 μM). The cytosine prodrug 59 showed similar activity to NM-107 in replicon assays with both resulting in an EC50 value of 1–10 μM. However, intracellular NTP levels of liver extracts were much higher for 59 than for NM-107 (1,838 pmol/106 cells versus 10 pmol/106 cells in monkeys and 991 pmol/106 cells versus 19 pmol/106 cells in humans after 24 h incubation at 10 μM). It was also found that chimpanzee hepatic NTP levels were 10–50-fold higher for 59 than NM-107 after oral dosing of 50 mg/kg/day for 14 days. This mixed prodrug showed promising in vivo stability and activity against HCV. However, formation of the toxic alkylating agent ethylene sulfide might severely limit the clinical potential of this hybrid MP prodrug.
Figure 7.
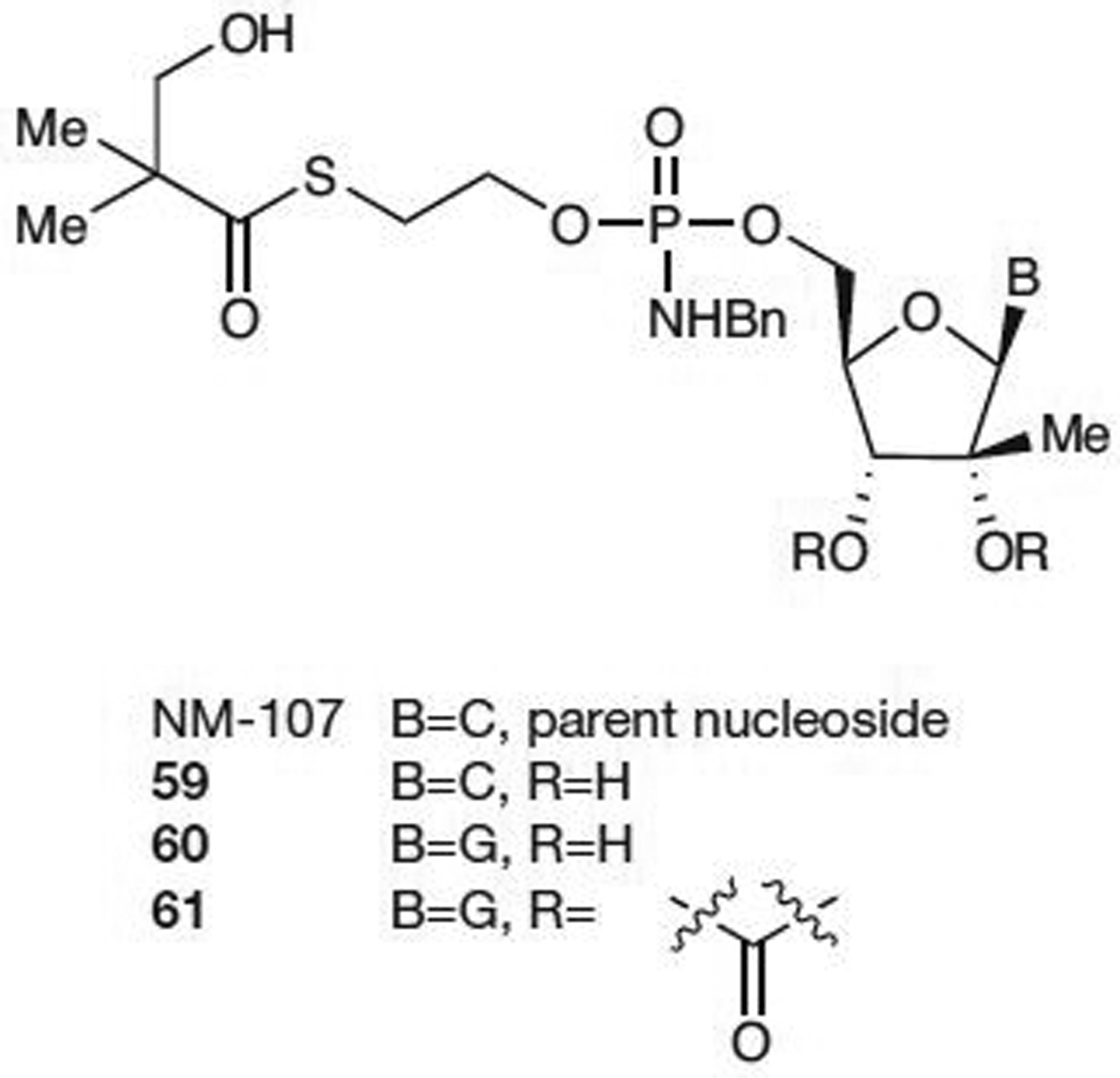
Mixed mono-SATE ProTide prodrugsa
aFrom reference [58]. C, cytosine; G, guanosine; ProTide, aryloxy phosphoramidate; SATE, S-acyl-2-thioethyl.
In December 2009, Idenix Pharmaceuticals, Inc. [59] disclosed results for IDX-184 (presumed structure is compound 60 based on their patent filing; Figure 7) from a 3-day Phase I proof-of-concept study. This double-blinded, placebo-controlled, monotherapy, dose-escalation study enrolled 41 treatment-naive HCV genotype-1-infected patients into four dosing cohorts (25 mg, 50 mg, 75 mg and 100 mg) [60,61]. Mean viral load decreases ranged from 0.47 log10 in the 25 mg group to 0.74 log10 in the 100 mg group after 3 days of treatment.
The use of either the bisSATE MP prodrug at the 5′-position or the 5′,3′-cyclic SATE MP prodrug has increased the lipophilicity of the nucleotide enabling the intracellular delivery of MPs resulting in potent anti-HCV activity in the cell-based replicon assays. There still remains a stability issue with these compounds that needs to be resolved if oral administration is to be realized. Improved oral bioavailability and a favourable pharmacokinetic profile were found with the mixed mono-SATE ProTide prodrug. However, formation of the toxic ethylene sulfide, an alkylating agent, as a by-product could severely limit the clinical potential of this group of MP prodrugs. Hence, the dose selected for human studies should be low enough to avoid releasing large quantities of ethylene sulfide.
Phosphate/phosphonate prodrugs
Simple alkyl esters of nucleoside MP are metabolically stable and not suitable for use as prodrugs [27]; however, modified phosphate esters of nucleosides have been evaluated for the treatment of HCV. Both acyloxyalkyl esters and aryl-substituted cyclic phosphate esters (HepDirect prodrugs [62]) as well as acyclic phosphonates have led to some promising in vitro and in vivo results.
Acyloxyalkyl ester
One of the most commonly used prodrug for the phosphate group is the acyloxyalkyl ester. The carboxylate or carbonate ester is cleaved enzymatically by esterases generating a transient hydroxymethyl intermediate (62; Figure 8), which rapidly loses formaldehyde to generate the deprotected phosphate group (63) [27]. However, oral bioavailability of this type of prodrug can be limited as the esterases responsible for this cleavage are found in numerous tissues and at high levels [63]. Also, the intracellular generation of formaldehyde raises some toxicity concerns for prolonged treatment; however, there are antiviral phosphonate prodrugs of this type that have been approved by the US Food and Drug Administration currently on the market [27].
Figure 8.
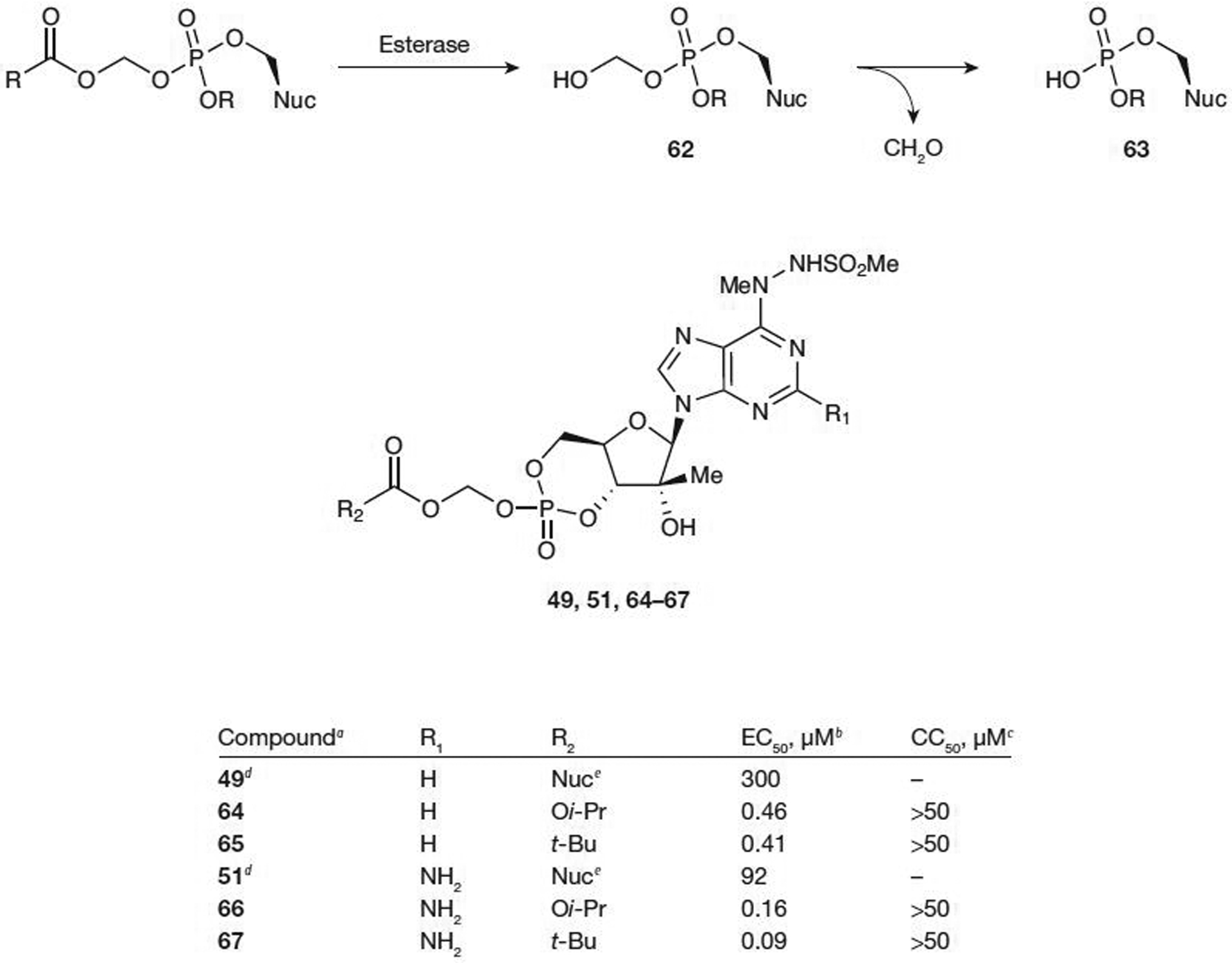
Metabolic activation and antiviral activity of cyclic monophosphate prodrugs of modified adenosines
aFrom reference [57]. bEffective concentration of compound required to reduce HCV replication by 50% (EC50). cConcentration of inhibitor reducing cell viability by 50% (CC50). dFrom reference [48]. eParent nucleoside (Nuc). Oi-Pr, O-isopropyl; t-Bu, tert-butyl.
Valeant Pharmaceuticals International [57] recently disclosed a cyclic acyloxyalkyl ester prodrug of a modified adenine nucleoside that shows activity towards HCV in a cell-based replicon assay. Significant increases of activity were observed in comparison with the parent nucleoside (49; EC50=300 μM) with both the isopropylcarbonate 64 prodrug (EC50=0.46 μM) and the pivaloyloxymethyl prodrug (POM; 65; EC50=0.41 μM; Figure 8). There was no cytotoxicity observed up to 50 μM. Slightly improved potency was noted when R1=NH2 (66 and 67; Figure 8) with observed EC50 values as low as 0.09 μM and with no apparent cytotoxicity (CC50>50 μM). Both the carbonate prodrug and the POM prodrug were found to be stable in SIF and SGF with no significant degradation detected after 2 h. The stability in SIF is in contrast to the 5′-phosphoramidate prodrug of the same nucleoside and could be further evaluated in advanced studies as a potential drug candidate.
HepDirect prodrugs
Because esterases are expressed throughout the gut, small intestine, plasma, muscle and various other tissues, activation of prodrugs by this route can often be problematic. After ester cleavage, the resulting ionic phosphate species have little to no membrane permeability, so the distribution is largely limited to the site of metabolism. This often leads to toxicity in non-target organs throughout the body [63]. A prodrug was designed by Erion et al. [64] from Metabasis Therapeutics, Inc. in 2004 in an effort to deliver NMP analogues in high concentration specifically to the liver for the treatment of HBV and HCV infections, and hepatocellular carcinoma. In order to achieve this goal, rapid activation of the prodrug (specifically in the liver) and good stability in aqueous solutions, such as blood, was necessary. Substituted arylpropanyl esters achieved both of these goals. Benzylic oxidation of the prodrug 68 by cytochrome P450 in the liver gives hydroxylated prodrug 69, which, after rearrangement, undergoes β-elimination to give the activated drug (71) and an aryl enone by-product (Figure 9). The β-elimination is relatively slow, allowing for the hydroxylated prodrug to pass through the liver and be distributed to tissues. In this pathway, 69 would be in equilibrium with the ring-opened ketone 70 and the equilibrium should favour the ring closed 69. Two anticancer drugs on the market (cyclophosphamide and ifosfamide) are activated in a similar manner.
Figure 9.
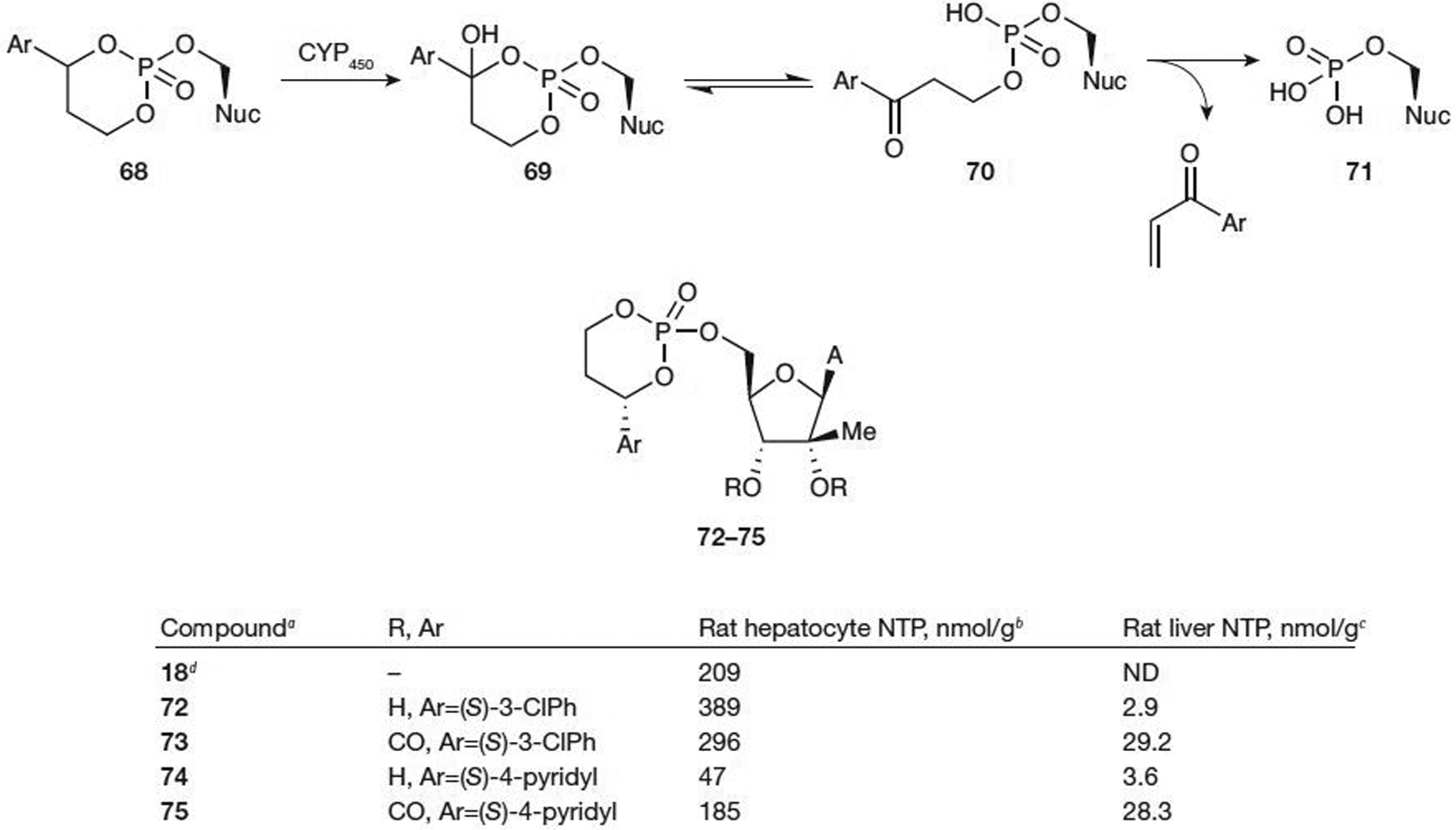
Metabolic activation and antiviral activity of HepDirect prodrugs of modified adenosines
aFrom reference [65]. bConcentration of nucleoside triphosphate (NTP) at 2 h after incubation at 25 μM. cConcentration of NTP at 3 h after a 50 mg/kg oral dose. dParent nucleoside (Nuc). CYP450, cytochrome P450; ND, not determined; (S)-3-CIPh, (S)-3-chlorophenyl.
Various HepDirect prodrugs of substituted nucleosides have been tested for NTP formation [65]. Activation of prodrugs 72–75 (Figure 9) to the NTP would require cleavage of the arylpropanyl group as well as two subsequent phosphorylations by cellular kinases. In some cases, NTP in rat hepatocytes was increased for the HepDirect prodrugs compared with the parent nucleoside (72–75; Figure 9); increased NTP was observed in only two out of four rats. Testing the oral bioavailability of prodrugs 72 and 74 led to disappointing low NTP levels in rat liver of approximately 3 nmol/g. However, decreasing the hydrogen bond donating capability of the nucleoside by the incorporation of a 2′,3′-carbonate protecting group resulted in good formation of the NTP at a dosing level of 50 mg/kg (NTP values were 29.2 nmol/g for 73 and 28.3 nmol/g for 75). Prodrug 75 was stable with a t1/2 of 13 h in pH 7 buffer at 25°C and was efficiently converted to 74 in rat plasma at 37°C without any further degradation.
Modifications to both the base and the sugar of the HepDirect prodrugs have led to some promising results. Prodrugs 77, 79, 81 and 83 all showed increased levels of NTP in rat hepatocytes compared with the parent nucleoside (Figure 10) [66–68]. Administration of the prodrugs intraperitoneally by injection at 5 mg/kg resulted in high NTP for prodrugs 79 and 83 compared with the parent nucleoside (NTP from 0.73 nmol/g for 78 to 78.4 nmol/g for 79 and from 0.1 nmol/g for 82 to 24.5 nmol/g for 83). Unfortunately, oral dosing only resulted in good NTP levels in rat liver for prodrug 79 (NTP=23.3 nmol/g), but at a lower dose of only 10 mg/kg compared with the previous study of 50 mg/kg.
Figure 10.
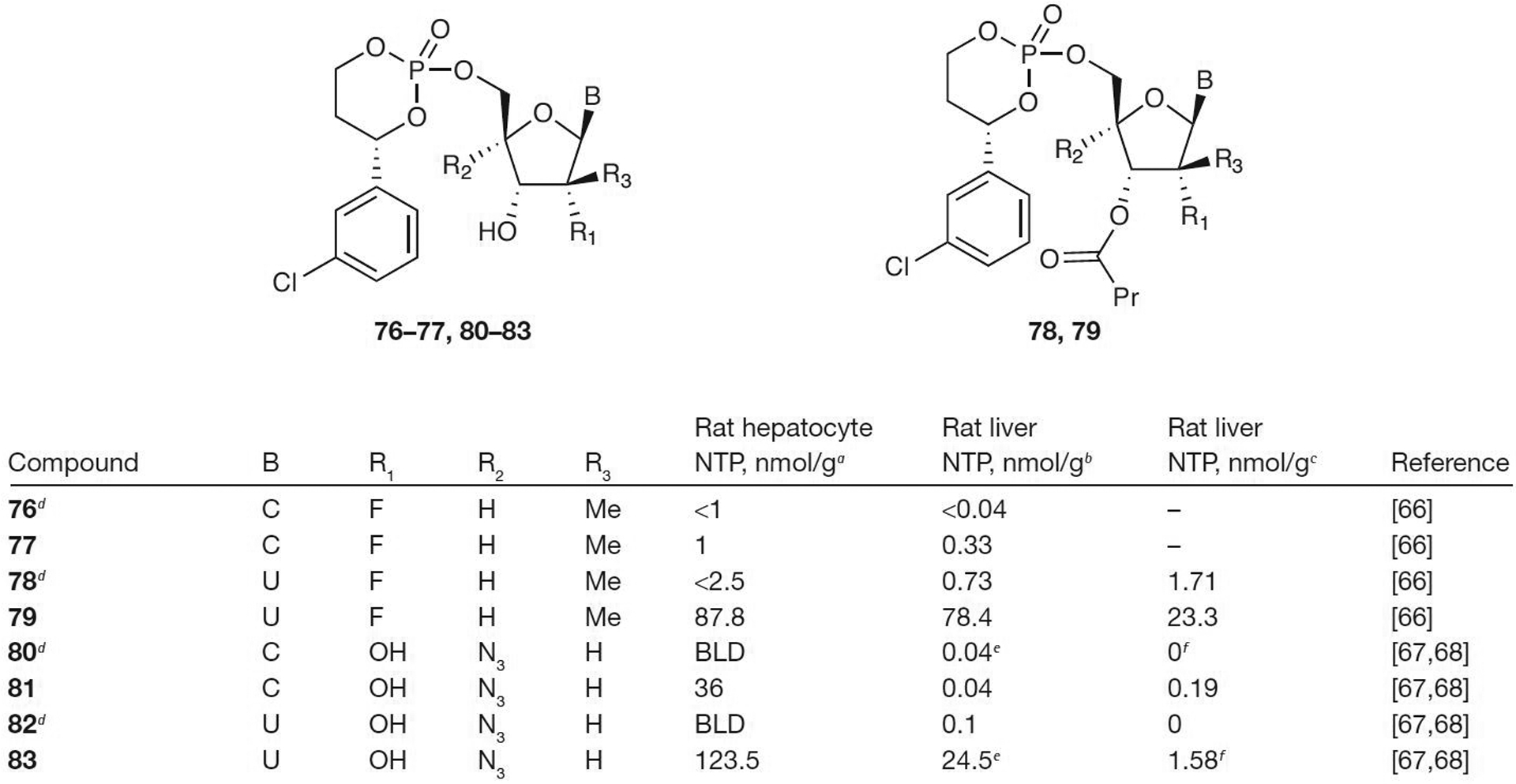
Antiviral activity of HepDirect prodrugs of pyrimidines
aConcentration of nucleoside triphosphate (NTP) at 2 h after incubation at 25 μM. bConcentration of NTP at 3 h after 5 mg/kg intraperitoneal injection. cConcentration of NTP at 3 h after an oral dose of 10 mg/kg. dParent nucleoside. e4 mg/kg. f8 mg/kg. BLD, below limit of detection; C, cytosine; Me, methyl; Pr, propyl; U, uracil.
HepDirect prodrugs show promise by delivering the monophosphorylated nucleoside prodrug to the liver. Once in the liver, metabolic activation by cytochrome P450 releases the active compound. High levels of NTP can be observed via intraperatonial injection or even with oral administration of the prodrug. Despite this promise, to date, no HepDirect prodrugs have been approved for human use. Furthermore, formation of potentially reactive by-products, such as aryl vinyl ketone, will require further evaluation.
Phosphonate prodrugs
Another way to bypass the initial phosphorylation of nucleosides is with the phosphonate group. Acyclic nucleoside phosphonates have been used to treat a variety of viral diseases, including HIV and HBV. Hostetler and colleagues [69] have developed alkoxyalkyl esters of acyclic adenines that show activity in an HCV replicon system. Both the R- and S-enantiomers of the octadecyloxyethyl ester and hexadecyloxy ester of 9-[3-hydroxyl-2-(phosphonomethoxy)-propyl]adenine (84–87; Figure 11) show activity for both genotypes 1B and 2A. The S-enantiomers were more active than the R-enantiomers with 85 being the most potent (EC50=1.3 μM for genotype 1B and 0.7 μM for genotype 2A). The octadecyloxyethyl esters were more potent than the hexadecyloxy esters (85 and 87 versus 84 and 86; Figure 11), but the hexadecyloxy esters were less cytotoxic. The inclusion of the ester increased the bioavailability of 84 and it has been estimated at 74% compared with intraperitoneal injection [70]. These nucleotide analogues were also found to be active against a number of double-stranded DNA viruses, including vaccinia virus, cowpox virus, human and murine cytomegalovirus and adenovirus [70]. Further studies on these compounds are currently underway.
Figure 11.
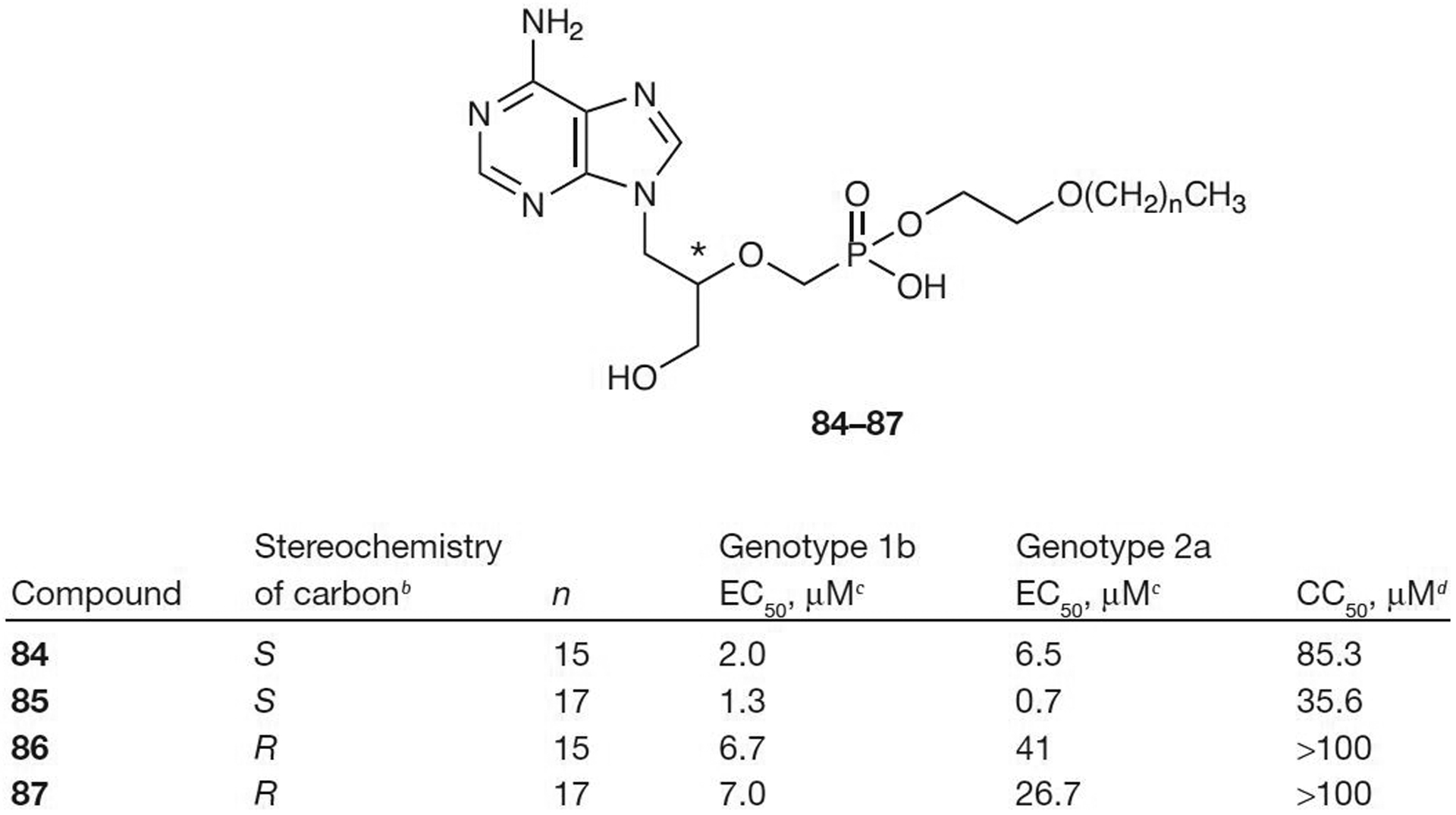
Acyclic nucleoside phosphonatesa
aFrom reference [69]. bAt the position indicated with an asterisk. cEffective concentration of compound required to reduce HCV replication by 50% (EC50) dConcentration of inhibitor reducing cell viability by 50% (CC50).
Conclusions
HCV continues to be a serious problem throughout the world. With >3% of the worldwide population chronically infected and only two approved drugs for treatment with limited efficacy, improved treatment modalities utilizing drugs with a long intracellular t1/2, such as nucleosides, are needed. In addition, nucleoside analogues are effective against all HCV genotypes and maintain a high genetic barrier to HCV resistance [12,61,71–74]. They are currently the best in any class because of these properties and we anticipate that they will be widely used in the future as a backbone to anti-HCV treatment similar to HIV and lead towards a cure for this disease because HCV does not establish latency [75]. Although modified nucleosides exhibit promising activity against viral diseases such as HCV in cell-based assays, their highly polar nature might complicate their development as an orally available medication. Conversion to the biologically active NTP can be problematic with initial kinase-driven conversion to the MP often rate-limiting. ProTide prodrugs (aryloxy phosphoramidate approach) and cyclic phosphoramidate prodrugs have shown promising activity in vitro; however, toxicity concerns continue to slow development. The increased lipophilicity of the SATE prodrugs has enabled the intracellular delivery of the nucleoside MP in cell-based in vitro assays, but poor stability and potential toxicity remains an issue with this class of compounds. Acyloxyalkyl ester prodrugs of cyclic phosphates show some promise, eliminating the potentially toxic phenolic by-product of the ProTide approach that could be delivered intracellularly. However, formaldehyde is a by-product of this class and thus long-term exposure could be problematic. The HepDirect class of prodrugs is capable of bypassing the first phosphorylation step and selective activation in the liver could reduce systemic toxicity exposure. The presence of cytochrome P450 isoenzymes in the gastrointestinal tract might be problematic and the formation of a potentially toxic aryl vinyl ketone by-product necessitates further studies. Acyclic nucleoside phosphonate prodrugs also show some promising results by bypassing the initial conversion to the NMP and further studies are currently underway.
It is apparent that many of these approaches are quite attractive because of the intracellular increase of NTP levels in the liver and low levels of the parent drug in the plasma. Progress towards this goal has been recently achieved in a clinical trial with PSI-7851, the first ProTide, specifically designed for HCV that produced a 2 log10 decrease in viral load after 3 days of oral treatment at 400 mg once daily. Nevertheless, much more work is warranted to make MP prodrugs a safe and non-toxic clinical reality.
Acknowledgements
This work was supported, in part, by 2P30-AI-050409, R01-AI-076535, 5R37-AI-041980 and 5R37-AI-025899, and by the Center for AIDS Research, Laboratory of Biochemical Pharmacology, Department of Pediatrics, Emory University School of Medicine and Veterans Affairs Medical Center, Decatur, GA, USA (all to RFS).
Disclosure statement
RFS is the principal founder and a Director of RFS Pharma, LLC. He is also a founder and major shareholder of Idenix Pharmaceuticals and Pharmasset, Inc. All his conflicts of interest were reviewed and are managed by Emory University School of Medicine, and Veterans Affairs Medical Center. All other authors declare no competing interests.
References
- 1.Choo QL, Kuo G, Weiner A, Overby LR, Bradley DW, Houghton M. Isolation of a cDNA clone derivied from a blood-borne non-A, non-B viral hepatitis genome. Science 1989; 244:359–362. [DOI] [PubMed] [Google Scholar]
- 2.Tong MJ, El-Farra NS, Reikes AR, Co RL. Clinical outcomes after transfusion-associated hepatitis C. N Engl J Med 1995; 332:1463–1466. [DOI] [PubMed] [Google Scholar]
- 3.Darby SC, Ewart DW, Giangrande PLF, et al. Mortality from liver cancer and liver disease in haemophilic men and boys in UK given blood products contaminated with hepatitis C. Lancet 1997; 350:1425–1431. [DOI] [PubMed] [Google Scholar]
- 4.Poynard T, Ratziu V, Benhamou Y, Opolon P, Cacoub P, Bedossa P. Natural history of HCV infection. Baillieres Best Pract Res Clin Gastroenterol 2000; 14:211–228. [DOI] [PubMed] [Google Scholar]
- 5.Hoofnagle JH, Hepatitis C. The clinical spectrum of disease. Hepatology 1997; 26 Suppl 1:15S–20S. [DOI] [PubMed] [Google Scholar]
- 6.McGuigan C, Perrone P, Madela K, Neyts J. The phosphoramidate ProTide approach greatly enhances the activity of β−2′-C-methylguanosine against hepatitis C virus. Bioorg Med Chem Lett 2009; 19:4316–4320. [DOI] [PubMed] [Google Scholar]
- 7.Ghosn J, Larsen C, Piroth L, et al. Evidence for ongoing epidemic sexual transmission of HCV (2006 to 2007) among HIV-1-infected men who have sex with men: France. 16th Conference on Retroviruses and Opportunistic Infections 8–11 February 2009, Montreal, QC, Canada Abstract 800. [Google Scholar]
- 8.Fishman S, Childs K, Dieterich D, et al. Age and risky behaviors of HIV-infected men with acute HCV infection in New York City are similar, but not identical, to those in a European outbreak. 16th Conference on Retroviruses and Opportunistic Infections 8–11 February 2009, Montreal, QC, Canada Abstract 801. [Google Scholar]
- 9.Manns MP, Foster GR, Rockstroh JK, Zeuzem S, Zoulim F, Houghton M. The way forward in HCV treatment – finding the right path. Nat Rev Drug Discov 2007; 6:991–1000. [DOI] [PubMed] [Google Scholar]
- 10.Hughes CA, Shafran SD. Chronic hepatitis C virus management: 2000–2005 update. Ann Pharmacother 2006; 40:74–82. [DOI] [PubMed] [Google Scholar]
- 11.Manns MP, McHutchison JG, Gordon SC, et al. Peginterferon alfa-2b plus ribavirin compared with interferon alfa-2b plus ribavirin for initial treatment of chronic hepatitis C: a randomised trial. Lancet 2001; 358:958–965. [DOI] [PubMed] [Google Scholar]
- 12.McCown MF, Rajyaguru S, Pogam Sl, et al. The hepatitis C virus replicon presents a higher barier to resistance to nucleoside analogs than to nonnucleoside polymerase or protease inhibitors. Antimicrob Agents Chemother 2008; 52:1604–1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McHutchison JG, Manns M, Patel K, et al. Adherence to combination therapy enhances sustained response in genotype-1-infected patients with chronic hepatitis C. Gastroenterology 2002; 123:1061–1069. [DOI] [PubMed] [Google Scholar]
- 14.Fried MW. Side effects of therapy of hepatitis C and their management. Hepatology 2002; 36 Suppl 1:S237–S244. [DOI] [PubMed] [Google Scholar]
- 15.Pawlotsky JM. Mechanisms of antiviral treatment efficacy and failure in chronic hepatitis C. Antiviral Res 2003; 59:1–11. [DOI] [PubMed] [Google Scholar]
- 16.Gordon CP, Keller PA. Control of hepatitis C: a medicinal chemistry perspective. J Med Chem 2005; 48:1–20. [DOI] [PubMed] [Google Scholar]
- 17.Keicher JD, Roberts CD, Dyatkina NB, inventors; Genelabs Technologies, Inc., assignee. Nucleoside compounds for treating viral infections. United States patent US 0107312 2005. May 19. [Google Scholar]
- 18.Wong T, Lee SS. Hepatitis C: a review for primary care physicians. Can Med Assoc J 2006; 174:649–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Koch U, Narjes F. Recent progress in the development of inhibitors of the hepatitis C virus RNA-dependent RNA polymerase. Curr Top Med Chem 2007; 7:1302–1329. [DOI] [PubMed] [Google Scholar]
- 20.Prakash TP, Prhavc M, Eldrup AB, et al. Synthesisand evaluation of S-acyl-2-thioethyl esters of modified nucleoside 5′-monophosphates as inhibitors of hepatitis C virus RNA replication. J Med Chem 2005; 48:1199–1210. [DOI] [PubMed] [Google Scholar]
- 21.Arends JE, Mudrikova T, Wensing AMJ, et al. High percentage of non-response with peginterferon-alfa-2a monotherapy for the treatment of acute hepatitis C inHIV infected patients. 49th Interscience Conference on Antimicrobial Agents and Chemotherapy 12–15 September 2009, San Francisco, CA, USA Abstract H-222. [Google Scholar]
- 22.De Francesco R, Migliaccia G. Challenges and successes in developing new therapies for hepatitis C. Nature 2005; 436:953–960. [DOI] [PubMed] [Google Scholar]
- 23.Huang Z, Murray MG, Secrist JA III. Recent development of therapeutics for chronic HCV infection. Antiviral Res 2006; 71:351–362. [DOI] [PubMed] [Google Scholar]
- 24.Meppen M, Pacini B, Bazzo R, et al. Cyclic phosphoramidates as prodrugs of 2′-methylcytidine. Eur J Med Chem 2009; 44:3765–3770. [DOI] [PubMed] [Google Scholar]
- 25.Martin LT, Cretton-scott E, Placidi L, et al. In vitro and in vivo metabolism and pharmacokinetics of bis[(t-butyl)-S-acyl-2-thioethyl]-β-l-2′,3′-dideoxy-5-fluorocytidine monophosphate. Nucleosides Nucleotides Nucleic Acids 2000; 19:481–499. [DOI] [PubMed] [Google Scholar]
- 26.Traut TW. Physiological concentrations of purines and pyrimidines. Mol Cell Biochem 1994; 140:1–22. [DOI] [PubMed] [Google Scholar]
- 27.Hecker SJ, Erion MD. Prodrugs of phosphates and phosphonates. J Med Chem 2008; 51:2328–2345. [DOI] [PubMed] [Google Scholar]
- 28.De Clercq E, Field HJ. Antiviral prodrugs – the development of successful prodrug strategies for antiviral chemotherapy. Br J Pharmacol 2006; 147:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Poijärvi-Virta P, Lönnberg H. Prodrug approaches of nucleotides and oligonucleotides. Curr Med Chem 2006; 13:3441–3465. [DOI] [PubMed] [Google Scholar]
- 30.Li F, Maag H, Alfredson T. Prodrugs of nucleoside analogues for improved oral absorption and tissue targeting. J Pharm Sci 2008; 97:1109–1134. [DOI] [PubMed] [Google Scholar]
- 31.Ploss A, Khetani SR, Jones CT, et al. Persistent hepatitisC virus infection in microscale primary human hepatocyte cultures. Proc Natl Acad Sci U S A 2010; 107:3141–3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schinazi RF, Coats SJ, Bassit LC, Lennerstrand J,Nettles JH, Hurwitz SJ. Approaches for the development of antiviral compounds: the case of hepatitis C virus. Handb Exp Pharmacol 2009; 189:25–51. [DOI] [PubMed] [Google Scholar]
- 33.Devine KG, McGuigan C, O’Connor TJ, Nicholls SR, Kinchington D. Novel phosphate derivatives of zidovudine as anti-HIV compounds. AIDS 1990; 4:371–373. [PubMed] [Google Scholar]
- 34.Cahard D, McGuigan C, Balzarini J. Aryloxy phosphoramidate triesters as Pro-Tides. Mini Rev Med Chem 2004; 4:371–381. [DOI] [PubMed] [Google Scholar]
- 35.Pierra C, Amador A, Benzaria S, et al. Synthesis and pharmacokinetics of valopictabine (NM-283), an efficient prodrug of the potent anti-HCV agent 2′-C-methylcytidine. J Med Chem 2006; 49:6614–6620. [DOI] [PubMed] [Google Scholar]
- 36.Murakami E, Bao H, Ramesh M, et al. Mechanism of activation of β-d-2′-deoxy-2′-fluoro-2′-C-methylcytidine and inhibition of hepatitis C virus NS5B RNA polymerase. Antimicrob Agents Chemother 2007; 51:503–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gardelli C, Attenni B, Donghi M, et al. Phosphoramidate prodrugs of 2′-methylcytidine for therapy of hepatitus C virus infection. J Med Chem 2009; 52:5394–5407. [DOI] [PubMed] [Google Scholar]
- 38.Perrone P, Luoni GM, Kelleher MR, et al. Application of the phosphoramidate protide approach to 4′-azidouridine confers sub-micromolar potency versus hepatitis C virus on an inactive Nucleoside. J Med Chem 2007; 50:1840–1849. [DOI] [PubMed] [Google Scholar]
- 39.Rondla R, Coats SJ, McBrayer TR, et al. Anti-hepatitis C virus activity of novel β-d-2′-C-methyl-4′-azido pyrimidine nucleoside phosphoramidate prodrugs. Antivir Chem Chemother 2009; 20:99–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sofia MJ, Du J, Wang P, Nagarathnam D, inventors; Pharmasset, Inc., assignee. Nucleoside phosphoramidate prodrugs. WO 121634. 2008. October 9. [Google Scholar]
- 41.Lawitz E, Rodriguez-Torres M, Denning J, et al. Potent antiviral activity observed with PSI-7851, a novel nucleotide polymerase inhibitor for HCV, following multiple ascending oral doses for 3 days in patients with chronic HCV infection. Global Antiviral Journal 2009; 5 Suppl 1:96. [Google Scholar]
- 42.Lam AM, Espiritu C, Micolochick Steuer HM. In vitro selection and characterization of hepatitis C virus replicon variants resistant to PSI-7851, a phosphoramidate prodrug of β-d-2′-deoxy-2′-fluoro-2′-C-methyluridine monophosphate. Global Antiviral Journal 2009; 5 Suppl 1:137–138. [Google Scholar]
- 43.Ludmerer SW, Graham DJ, Boots E, et al. Replication fitness and NS5B drug sensitivity of diverse hepatitis C virus isolates characterized by using a transient replication assay. Antimicrob Agents Chemother 2005; 49:2059–2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Murakami E, Tolstykh T, Bao H, Niu C, Furman PA. Mechanism of PSI-7851 activation. Global Antiviral Journal 2009; 5 Suppl 1:96–97. [Google Scholar]
- 45.Patti J, Ames B, Bryant D, et al. Pharmacological properties and in vitro characterization of INX-189, a liver targeted phosphoramidate nucleoside analogue inhibitor of NS5b. 60th Annual Meeting of the American Association for the Study of Liver Diseases 30 October–3 November 2009, Boston, MA, USA Poster 1611. [Google Scholar]
- 46.Kolykhalov A, Chamberlain S, Gorovits E, et al. In vitro characterization of INX-189, a highly potent phosphoramidate nucleoside analogue inhibitor of HCV. Global Antiviral Journal 2009; 5 Suppl 1:95. [Google Scholar]
- 47.Perrone P, Daverio F, Valente R, et al. First example of phosphoramidate approach applied to a 4′-substituted purine nucleoside (4′-azidoadenosine): conversion of an inactive nucleoside to a submicromolar compound versus hepatitis C virus. J Med Chem 2007; 50:5463–5470. [DOI] [PubMed] [Google Scholar]
- 48.Gunic E, Suetying C, Frank R, et al. 6-Hydrazinopurine 2′-methyl ribonucleosides and their 5′-monophsophate prodrugs as potent hepatitis C virus inhibitors. Bioorg Med Chem Lett 2007; 17:2456–2458. [DOI] [PubMed] [Google Scholar]
- 49.Donghi M, Attenni B, Gardelli C, et al. Synthesis and evaluation of novel phosphoramidate prodrugs of 2′-methylcytidine as inhibitors of hepatitis C virus NS5B polymerase. Bioorg Med Chem Lett 2009; 19:1392–1395. [DOI] [PubMed] [Google Scholar]
- 50.Meppen M, Narjes F, Pacini B, Gardelli C, Durette PJ, inventors; Merck & Co., Inc., Instituto Di Richerche Di Biologia Molecolare P. Angeletti S.P.A., assignees. Nucleoside cyclic phosphoramidates for the treatment of RNA-dependent RNA viral infection. WO 079206. 2008. July 3. [Google Scholar]
- 51.Eckstein F, Gish G. Phosphorothioates in molecular biology. Trends Biochem Sci 1989; 14:97–100. [DOI] [PubMed] [Google Scholar]
- 52.Périgaud C, Gosselin G, Lefebvre I, et al. Rational design for cytosolic delivery of nucleoside monphosphates: ‘SATE’ and ‘DTE’ as enzyme-labile transient phosphate protecting groups. Bioorg Med Chem Lett 1993; 3:2521–2526. [Google Scholar]
- 53.Benzaria S, Bardiot D, Bouisset T, et al. 2′-C-Methyl branched pyrimidine ribonucleoside analogues: potent inhibitors of RNA virus replication. Antivir Chem Chemother 2007; 18:225–242. [DOI] [PubMed] [Google Scholar]
- 54.Butler T, Cho A, Kim C, Saunders OL, Zhang L, inventors; Gilead Sciences, Inc., assignee. 1′-Substituted carba-nucleoside analogs for antiviral treatment. WO 132135. 2009. October 29. [Google Scholar]
- 55.Cho A, Kim C, Parrish J, Xu J, inventors; Gilead Sciences, Inc., assignee. Carba-nucleoside analogs for antiviral treatment. WO 132123. 2009. October 29. [Google Scholar]
- 56.Ding Y, Girardet J, Hong Z, et al. Synthesis of 9-(2-β-C-methyl-β-d-ribofuranosyl)-6-substituted purine derivatives as inhibitors of HCV RNA replication. Bioorg Med Chem Lett 2005; 15:709–713. [DOI] [PubMed] [Google Scholar]
- 57.Gunic E, Girardet JL, Ramasamy K, et al. Cyclic monophosphate prodrugs of base-modified 2′-C-methyl ribonucleosides as potent inhibitors of hepatitis C virus RNA replication. Bioorg Med Chem Lett 2007; 17:2452–2455. [DOI] [PubMed] [Google Scholar]
- 58.Sommadossi JP, Gosselin G, Pierra C, Perigaud C, Peyrottes S, inventors; Idenix Pharmaceuticals, Inc., Centre National de la Recherche Scientifique, L’Universite Montpellier Il., assignees. Compounds and pharmaceutical compositions for the treatment of viral infections. WO 082601. 2008. July 10. [Google Scholar]
- 59.Standring DN, Perigaud C, Peyrottes S, et al. Pharmacology of IDX184, a liver-targeting nucleotide prodrug for the treatment of HCV. Global Antiviral Journal 2009; 5 Suppl 1:22. [Google Scholar]
- 60.Lalezari J, Asmuth D, Casir A, et al. Antiviral activity, safety and pharmacokinetics of IDX184, a liver – targeted nucleotide HCV polymerase inhibitor, in patients with chronic hepatitis C. 60th Annual Meeting of the American Association for the Study of Liver Diseases 30 October–2 November 2009, Boston, MA, USA Poster LB18. [Google Scholar]
- 61.Mayers D IDX184 – a novel, liver-targeted, once-a-day nucleoside prodrug for the treatment of chronic HCV infection HEP DART – Frontiers in Drug Development for Viral Hepatitis. Satellite Mini-Symposium: Toward Curative Therapies for Hepatitis C 6–10 December 2009, Kohala Coast, HI, USA. [Google Scholar]
- 62.Erion MD, Bullough DA, Lin C-C, Hong Z. HepDirect PDs for targeting nucleotide-based antiviral drugs to the liver. Curr Opin Investig Drugs 2006; 7:109–117. [PubMed] [Google Scholar]
- 63.Bookser BC, Raffaele NB. High-throughput synthesis of HepDirect prodrugs of nucleoside monophosphates. J Comb Chem 2008; 10:567–572. [DOI] [PubMed] [Google Scholar]
- 64.Erion MD, Reddy KR, Boyer SH, et al. Design, synthesis, and characterization of a series of cytochrome P450 3A-activated prodrugs (HepDirect prodrugs) useful for targeting phosph(on)ate-based prodrugs to the liver. J Am Chem Soc 2004; 126:5154–5163. [DOI] [PubMed] [Google Scholar]
- 65.Hecker SJ, Reddy KR, van Poelje PD, et al. Liver-targeted prodrugs of 2′-methyladenosine for therapy of hepatitus C virus infection. J Med Chem 2007; 50:3891–3896. [DOI] [PubMed] [Google Scholar]
- 66.Hecker SJ, Reddy KR, inventors; Metabasis Therapeutics, Inc. assignee. Nucleoside prodrugs and uses thereof. WO 073506. 2009. June 11. [Google Scholar]
- 67.Bookser BC, Hecker SJ, Reddy RK, Smith DB, Sun Z, inventors; Metabasis Therapeutics Inc., assignee. Antiviral nucleoside compounds. WO 069095. 2009. June 4. [Google Scholar]
- 68.Hecker SJ, Reddy KR, Sun Z, Bookser BC, Smith DB. inventors; Roche Palo Alto LLC, assignee. Nucleoside prodrugs and uses thereof. United States patent US 0209481. 2009. August 20. [Google Scholar]
- 69.Wyles DL, Kaihara KA, Korba BE, Schooley RT, Beadle JR, Hostetler KY. The octadecylonyethyl ester of (S)-9-[3-hydroxy-2-(phosphonomethoxy) propyl]adenine is a potent and selective inhibitor of hepatitis C virus replication in genotype 1A, 1B, and 2A replicons. Antimicrob Agents Chemother 2009; 53:2660–2662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Morrey JD, Korba BE, Beadle JR, Wyles DL,Hostetler KY. Alkoxyalkyl esters of 9-(S)-(3-hydroxy-2-phosphonomethoxypropyl)adenine are potent and selective inhibitors of hepatitis B virus (HBV) replication in vitro and in HBV transgenic mice in vivo. Antimicrob Agents Chemother 2009; 53:2865–2870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Standring D IDX184, a liver-targeted nucleotide prodrug for the treatment of HCV HEP DART – Frontiers in Drug Development for Viral Hepatitis. Satellite Mini-Symposium: Toward Curative Therapies for Hepatitis C 6–10 December 2009, Kohala Coast, HI, USA. [Google Scholar]
- 72.Pharmasset, Inc. Pharmasset: building for success 28th Annual JPMorgan Healthcare Conference 11–14 January 2010, San Francisco, CA, USA (Accessed 2 March 2010) Available from http://files.shareholder.com/downloads/VRUS/733159221x0x344081/b202fb5f-894f-40a0-aa38-abffea446aca/VRUS%20JPM%20Pres%20011410.pdf [Google Scholar]
- 73.Burton JR Jr., Everson GT. HCV NS5B polymerase inhibitors. Clin Liver Dis 2009; 13:453–465. [DOI] [PubMed] [Google Scholar]
- 74.Manns MP, Foster GR, Rockstroh JK, Zeuzem S, Zoulim F, Houghton M. The way forward in HCV treatment-finding the right path. Nat Rev Drug Discov 2007; 6:991–1000. [DOI] [PubMed] [Google Scholar]
- 75.Schinazi RF, Bassit L, Gavegnano C. HCV drug discovery aimed at viral eradication. J Viral Hepat 2010; 17:77–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.McGuigan C, Perrone P, inventors; University College Cardiff Consultants Limited, KU Leuven Research and Development, assignees. Chemical Compounds. WO 062206. 2008. May 29. [Google Scholar]