

Abstract
The spatial distribution of 29 per- and polyfluoroalkyl substances (PFASs) in seawater was investigated along a sampling transect from Europe to the Arctic and two transects within Fram Strait, located between Greenland and Svalbard, in the summer of 2018. Hexafluoropropylene oxide-dimer acid (HFPO-DA), a replacement compound for perfluorooctanoic acid (PFOA), was detected in Arctic seawater for the first time. This provides evidence for its long-range transport to remote areas. The total PFAS concentration was significantly enriched in the cold, low-salinity surface water exiting the Arctic compared to warmer, higher-salinity water from the North Atlantic entering the Arctic (260±20 pg/L versus 190±10 pg/L). The higher ratio of perfluoroheptanoic acid (PFHpA) to perfluorononanoic acid (PFNA) in outflowing water from the Arctic suggests a higher contribution of atmospheric sources compared to ocean circulation. An east-west cross section of the Fram Strait, which included seven depth profiles, revealed higher PFAS concentrations in the surface water layer than in intermediate waters and a negligible intrusion into deep waters (>1000 m). Mass transport estimates indicated a net inflow of PFASs with ≥8 perfluorinated carbons via the boundary currents and a net outflow of shorter-chain homologues. We hypothesize that this reflects higher contributions from atmospheric sources to the Arctic outflow and a higher retention of the long-chain compounds in melting snow and ice.
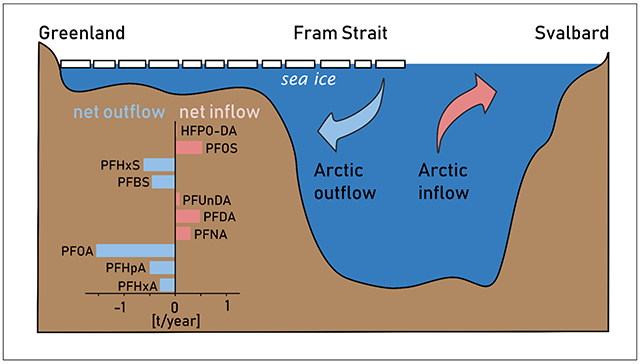
GRAPHICAL ABSTRACT
INTRODUCTION
Long-chain perfluoroalkyl carboxylic acids (PFCAs, CnF2n+1COOH, n ≥ 7), perfluoroalkyl sulfonic acids (PFSAs, CnF2n+1SO3H, n ≥ 6), and their precursors are recognized as global contaminants of high concern. Due to their persistence, bioaccumulation potential, toxicity and long-range transport potential, perfluorooctane sulfonic acid (PFOS) and perfluorooctanoic acid (PFOA) are regulated globally under the Stockholm Convention on Persistent Organic Pollutants (POPs).1-2 The evaluation of perfluorohexane sulfonic acid (PFHxS) is currently ongoing.3 In addition to regulated per- and polyfluoroalkyl substances (PFASs), an OECD study identified more than 4700 structurally similar compounds on the worldwide market.4 They include replacements for long-chain PFCAs and PFSAs, such as shorter-chain homologues and per- and polyfluoroether carboxylic and sulfonic acids (PFECAs and PFESAs), but also legacy PFASs that have already been in use for several years.
An ether-based replacement compound for PFOA, which has been increasingly studied over the last years, is the dimer acid of hexafluoropropylene oxide (HFPO-DA), marketed as its ammonium salt under the trade name GenX.5 It was added to the candidate list of substances of very high concern under REACH, the European chemicals’ regulation, in July 2019.6 In a large-scale study, the compound was detected in 153 of 160 surface water samples taken in China, Europe, the United States and Korea with a median concentration of 0.95 ng/L.7 However, field studies investigating the potential long-range transport of HFPO-DA are still lacking.
An important region to investigate oceanic long-range transport of organic pollutants to remote areas is the Fram Strait, located between Svalbard and Greenland. In the eastern Fram Strait, warm, saline Atlantic water enters the Arctic Ocean in the West Spitsbergen Current (WSC), whereas in western Fram Strait, cold, low-saline water and sea ice, as well as Atlantic Water that has spent significant time in the Arctic, exit the Arctic Ocean via the East Greenland Current (EGC). Fram Strait is the only deep connection between the Arctic Ocean and the remainder of global oceans and has the highest water volume fluxes both pole- and equatorward.8
Prior studies have investigated the distribution of PFASs in oceanic surface waters,9-14 whereas data on deeper ocean waters is limited.15-16 Strong differences between the available vertical profiles, taken in oceanic regions other than Fram Strait, indicate that intrusion of PFASs into deep waters is influenced by stratification, mixing and deep water formation in the respective sampling area.17 In the Labrador Sea, an arm of the North Atlantic Ocean with young deep water (< 10 years18), PFASs were detected down to 3500 m depth.16 In contrast, PFASs were only detectable above 150 m depth in the Nansen and Amundsen Basins of the Arctic Ocean,15 where deep water is much older (up to 450 years19) and does not reflect a period of substantial PFAS production. More information on the vertical distribution of PFASs in the oceans is essential to reduce uncertainties in global PFAS mass balances and assess the role of the oceans as a sink of PFASs.
In this study, surface water samples were taken along a transect from Europe to the Arctic and along two transects within Fram Strait. Additionally, water was collected from different depths of the water column (5 to 3,117 m) at seven stations in Fram Strait. The aims of the study were (i) to investigate the occurrence and distribution of 29 PFASs, especially HFPO-DA and other emerging PFASs, along the oceanic transport pathway from Europe to the Arctic; ii) to provide knowledge on the transport and sources of PFASs in water masses entering and exiting the Arctic Ocean at different depths and iii) to estimate mass flows of PFASs through the deep water passage of Fram Strait.
MATERIALS AND METHODS
Sample Collection
Seawater samples were collected during expedition PS114 of the research icebreaker Polarstern between 10 July and 3 August 2018.20 Forty surface water samples were taken at approximately 11 m depth using the ship’s seawater intake system (stainless steel pipe). Sampling locations included a latitudinal transect from the European continent to the Arctic (57°N to 79°N at ~5°E, n=21) and a longitudinal and latitudinal transect within Fram Strait (9°E to 18°W at ~79°N, n=14, and 78°N to 80°N at ~0°EW, n=5). Additionally, a CTD/rosette sampler was used at seven stations in Fram Strait to collect water samples (n=58) at five to twelve depths from bottom (up to 3,117 m) to surface (5 or 10 m). The samples were stored at 5 °C in 1 L polypropylene (PP) bottles and extracted on board within one week after sampling. A map showing the sampling locations is provided in Figure 1. An overview of the sampling coordinates, sampling depths and physicochemical parameters is given in Tables S1 and S2 and Figure S1.
Figure 1:

A) Surface water sampling locations (red dots; samples taken at 11 m depth) along a latitudinal transect from the European continent to the Arctic Ocean (N1-N21), and a longitudinal (F1-F15) and latitudinal (P1-P5) transect within Fram Strait. Arrows represent ocean currents (NCC: Norwegian Coastal Current; NwAC: Norwegian Atlantic Current; WSC: West Spitsbergen Current; EGC: East Greenland Current).8,31 B) Surface water concentrations [pg/L] of detected PFASs along the sampling transect from Europe to the Arctic in connection with water depth [m] at the respective sampling sites.
Target Analytes and Chemicals
Twenty-nine PFASs were analyzed: 11 PFCAs (C4 to C14), five PFSAs (C4, C6, C7, C8, C10), including the linear and branched isomers of PFOS (L-PFOS and Br-PFOS), the cyclic PFSA perfluoro-4-ethylcyclohexanesulfonate (PFECHS), six PFECAs and PFESAs (HFPO-DA, hexafluoropropylene oxide-trimer and tetramer acid (HFPO-TrA, HFPO-TeA); 4,8-dioxa-3H-perfluorononanoic acid (DONA); 6:2 and 8:2 Cl-PFESA), two perfluoroalkyl phosphinic acids (PFPiAs) (6:6 PFPiA, 6:8 PFPiA) and four precursors (perfluorooctane sulfonamide (FOSA) (branched and linear) and three fluorotelomer sulfonic acids (4:2 FTSA, 6:2 FTSA, 8:2 FTSA)). Ten mass-labelled PFASs were used as internal standards (IS), namely seven isotopically labelled PFCAs (perfluoro-[13C4]-butanoic acid (13C4-PFBA), perfluoro-[1,2-13C2]-hexanoic acid (13C2-PFHxA), 13C4-PFOA, perfluoro-[1,2,3,4,5-13C5]-nonanoic acid (13C5-PFNA), perfluoro-[1,2-13C2]-decanoic acid (13C2-PFDA), perfluoro-[1,2-13C2]-undecanoic acid (13C2-PFUnDA), perfluoro-[1,2-13C2]-dodecanoic acid (13C2-PFDoDA), two PFSAs (perfluorohexane-[18O2]-sulfonic acid (18O2-PFHxS), 13C4-PFOS) and one PFECA (13C3-HFPO-DA). 13C8-PFOA served as the injection standard. Detailed information on analytical standards and on chemicals used for sample extraction and cleaning is presented in Tables S3 and S4.
Sample Extraction and Instrumental Analysis
To avoid PFAS losses, water samples were not filtered before analysis.21 Solid phase extraction (SPE) for one liter of the samples was performed on board as described previously.22 Briefly, an IS mix was added to the samples (400 pg of each standard) before they were loaded onto preconditioned SPE cartridges (Oasis mixed-mode weak anion-exchange/reversed-phase (WAX), 6 cc, 500 mg sorbent, 60 μm particle size, Waters, USA). The cartridges were dried under vacuum and stored at −20 °C. Further processing of the samples was performed after the cruise at Helmholtz-Zentrum Geesthacht (Section S1). PFASs were analyzed by liquid chromatography coupled to tandem mass spectrometry (LC-MS/MS) (Tables S5 and S6). The quantification procedure is explained in Section S2.
Quality Assurance and Quality Control
Before the cruise, all reagents and materials were tested for the presence of target PFASs. Only SPE cartridges packed with sorbent lots for which tests showed no detectable levels of PFOA and other target PFASs were taken on board (Figure S2). Throughout the analysis, all containers and equipment were rinsed three times with methanol prior to usage.
Different types of blanks were analyzed to be able to trace back possible contamination sources (Figure S3). In the instrumental blanks, no PFASs were detected, whereas particular PFASs were detected in the laboratory blanks (n=10) and field blanks (n=6) in the low pg/L range (C5 to C8 PFCAs, L-PFOS and Br-PFOS, 6:2 FTSA) (Table S7). As concentrations in the field blanks were marginally higher than in the laboratory blanks, all results were corrected by subtracting the average concentration of the respective compound in the field blanks from the concentration in the sample. For PFASs present in field blanks, method detection limits (MDLs) and method quantification limits (MQLs) were calculated as the mean field blank concentration plus 3 or 10 times the standard deviation (SD), respectively. For PFASs other than those, MDLs and MQLs were derived from a signal-to-noise ratio of 3 or 10, observed in low-level samples. MDLs ranged from 3.3 pg/L (PFDoDA) to 75 pg/L (HFPO-TeA) (Table S7). PFBA was an exception as comparatively high concentrations of the compound were determined in both the laboratory and the field blanks, strongly varying between different SPE batches (mean±SD 350±460 pg/L). Consequently, no results are reported for PFBA. Statistical treatment of results <MDL is explained in Section S3.
To compare the two sampling techniques, a sample was taken with the stainless steel pipe (seawater intake at 11 m depth) when surface water samples were taken with the CTD/rosette sampler (10 or 5 m depth) (n=7). The results showed no significant differences between sampling techniques (two-sample t-test, α=0.05; Table S8). For the CTD/rosette sampler, the three deep water samples V1/1, V3/1 and V7/1 served as additional field blanks because all target analytes were <MDL.
Matrix-spike recovery tests at a spiking level of 400 pg/L resulted in relative recoveries from 73±1% to 111±5%, except for the long-chain compounds PFTrDA, PFTeDA, 6:6 PFPiA (64, 63 and 69%) and 6:8 PFPiA (39±2%) (Table S9). Recoveries of the internal standards are given in Table S10. To verify the trueness of the method, the certified reference material IRMM-428 was analyzed. ∣z-scores∣ were satisfactory with values <2 (Table S11 and Figure S4).
Calculation of PFAS Mass Transport Estimates
To calculate PFAS mass transport estimates through Fram Strait (79°N), reported in [kg/year], the mean PFAS concentration of different water masses, measured in summer 2018 [pg/L], was multiplied by the average water volume transport [m3/s] from the MIT general circulation model (MITgcm) ECCO v4.17 The MITgcm has a horizontal resolution of 1°×1° in most regions and higher resolution in the Arctic (40 km×40 km). Advection fields are from Estimating the Circulation & Climate of the Ocean (ECCO-v4) climatology, optimized in ECCO-v4 to produce a best fit to in situ and satellite observations of the physical ocean state.23-25 Boundary currents used to calculate PFAS mass flows were defined based on prior work (Section S4).26 PFAS mass transport estimates based on water transport data derived from MITgcm were compared to that derived from a mooring array.26
RESULTS AND DISCUSSION
PFAS Concentrations and Composition Patterns in Surface Water
Eleven PFASs were detected in surface water samples from the cruise: HFPO-DA, C6 to C12 PFCAs and three PFSAs (PFBS, PFHxS and L-PFOS/Br-PFOS) (Tables S12 to S16). Eighteen compounds analyzed were excluded from the results due to background contamination (PFBA), matrix interferences (PFPeA), poor spike accuracy (PFTrDA, PFTeDA; 6:6 PFPiA and 6:8 PFPiA) and concentrations below the method detection limits, which were <20 pg/L for DONA, 6:2 Cl-PFESA, 8:2 Cl-PFESA and L-/Br-FOSA, and >20 pg/L for PFECHS, PFHpS, PFDS, HFPO-TrA and HFPO-TeA and the three FTSAs (Table S7).
The sum of the detected PFASs ranged from 140 pg/L in surface water from the Greenland Sea to 850 pg/L in the North Sea (mean 340 pg/L). The replacement compound HFPO-DA was detected in 90% of the samples with a mean concentration of 30 pg/L. The substance with the highest mean concentration was the legacy compound PFOA (66 pg/L), having a detection frequency of 100%. HFPO-DA and PFOA contributed on average 8±4%, or respectively 20±3% to the total PFAS concentration (ΣPFASs). In addition, the short-chain perfluoroalkyl acids (PFAAs) PFHxA, PFHpA and PFBS and the long-chain homologues PFNA and L-/Br-PFOS had high detection frequencies (93-100%), contributing to ΣPFASs with 11% to 17% per compound.
Transport and Sources of PFASs along a Latitudinal Transect from Europe to the Arctic
ΣPFASs in surface water samples decreased from the North Sea continental shelf, to the continental slope, to deep ocean regions of the Norwegian Sea and Greenland Sea (Figure 1, Table S14). This underlines the influence of ocean currents and fronts on PFAS concentrations in surface water. The Norwegian Coastal Current (NCC) affects sampling locations N1 to N5 in the northeastern part of the North Sea. In this region, PFAS concentrations (mean±SD 770±73 pg/L, this study) were about a factor eight lower than in coastal areas (6,000±790 pg/L)22, reflecting mixing and dilution effects with increasing distance from continental sources. From N5, still in the North Sea on the continental shelf (water depth: 148 m), to N6, in the Norwegian Sea on the continental slope (water depth: 900 m), ΣPFASs dropped from 780 pg/L to 430 pg/L. Here, the NCC continues in northeast direction on the continental shelf, whereas samples N6 to N11 were collected on the continental slope, along which the Norwegian Atlantic Current (NwAC) flows, extending the Gulf Stream and the North Atlantic Current in northeast direction.27 The change in water masses between N5 and N6 is demonstrated by a salinity increase (32.59 psu to 34.69 psu), reflecting less freshwater influences in the Norwegian Sea compared to the North Sea. Both North and Baltic Sea are known to be influenced by PFAS sources on the European continent,28 explaining the significantly higher ΣPFASs in the samples from the continental shelf than those from the continental slope which are mainly influenced by Atlantic waters.
The main branch of the NwAC continues along the Norwegian shelf edge in northeast direction,27 whereas further surface water samples of the transect were taken in deep ocean regions in a northward direction (N12 to N23, water depth 2,450 m-3,200 m). Consequently, these samples were less influenced by currents carrying water from the North Atlantic and Europe, explaining the significantly lower ΣPFASs in surface water samples taken north of 68°N (230±51 pg/L) than in samples from the continental slope and shelf (370±54 pg/L and 770±73 pg/L).
Looking at individual PFASs, the compounds differed in the extent of the concentration decrease crossing the European continental shelf front. HFPO-DA showed the highest decrease from the NCC to the NwAC (70%, 91±26 pg/L to 25±5 pg/L). Concentrations of C6 to C8 PFCAs, PFHxS and L-PFOS dropped between 40-60%, whereas C9 to C12 PFCAs showed a smaller (PFNA and PFDA, 20% and 10%) or no decrease (PFUnDA and PFDoDA) (Figure S5).
These differences reflect combined effects of differing primary releases in source regions in the past, transport lag times, and mixing of water masses of different origin. Transit times derived from anthropogenic radionuclide tracers are one to two years from North Sea coastal waters to 60°N in the NCC and another year to eastern Fram Strait.29 In addition to North Sea coastal waters, the NCC carries waters from the Baltic Sea, which has a water turnover time of about 30 years,30 and recirculating Atlantic waters.31 The NwAC carries waters from the North Atlantic that can be affected by multiple PFAS sources, among them the fluoropolymer production sites at the North American east coast. A time lag on the scale of decades is modeled between PFAS release into North American coastal waters and input to the Arctic because of slow oceanic transport and mixing processes.17 Consequently, it can be assumed that historic emissions are still of relevance in the NwAC, whereas the NCC is influenced by both, recent PFAS emissions in North Sea coastal areas and historic emissions in the Baltic Sea and NwAC source regions.
The comparatively strong decrease of HFPO-DA from the NCC to the NwAC underlines that European coastal waters are currently a more important source for oceanic inputs of this compound into the Arctic than North Atlantic waters (Section S5). In contrast, the negligible drop of C9 to C12 PFCAs suggests that these compounds are predominantly the products of semivolatile precursor oxidation, in which case one would not expect to see the gradient from NCC to NwAC waters, and/or play a minor role in recent European emissions. The latter is consistent with the phase-out of long-chain PFCAs in Western Europe and the transition to the production and use of replacement compounds such as HFPO-DA.32 However, historic emissions of C9 to C12 PFCAs still play a relevant role with regard to Arctic inflow, having a proportion of approximately 25% of ΣPFASs in waters from the NwAC.
Potential Long-Range Transport of the Replacement Compound HFPO-DA
Although the concentration of HFPO-DA decreased stepwise along the latitudinal transect from the European continent to Fram Strait, the compound was detected in 19 of 21 samples. In deep ocean regions of the Norwegian Sea and Greenland Sea (69°N-79°N), its surface water concentration ranged from <MDL to 26 pg/L (mean±SD 16±9 pg/L), approximately a factor three lower than that of PFOA (range 38-56 pg/L, mean 47±7 pg/L) (Table S12).
These are the first findings of HFPO-DA in seawater from a remote region without known local sources, indicating that the compound undergoes long-range transport. This is consistent with a modeling study predicting similar overall persistence POV (≈1039 days) and characteristic travel distance CTD in water (≈1745 km) of HFPO-DA and PFOA.33
According to Annex D of the Stockholm Convention, field data are accepted as evidence for the long-range transport of a chemical if 1) measured levels are available in locations distant from the sources of its release that are of potential concern; 2) monitoring data show that long-range environmental transport of the chemical may have occurred via air, water, or migratory species.34 The mere detection of a chemical in a remote region cannot necessarily be understood as evidence of long-range transport, as the potential influence of local sources has to be considered.35 In the investigated region, this includes potential sources on Svalbard,36 in northern Scandinavia and Greenland. The most distant sampling point from land where HFPO-DA was detected in this study is N15, approximately 600 km away from Svalbard and northern Scandinavia and approximately 800 km away from Greenland. This provides a minimum estimate for long-range transport of HFPO-DA.
Samples N16 and N17 in the southern Greenland Sea, the only samples along the latitudinal transect in which HFPO-DA was not detected, also had a lower ΣPFASs (160 pg/L and 140 pg/L) than the other samples taken in deep water regions (260±25 pg/L). This coincided with a temperature decrease from 8.2 °C (N15) to 5.5°C (N17), indicating that the sampling transect crossed the Arctic Front in this area, which separates waters in the Norwegian Sea, mainly directly originating from the North Atlantic, from apparently less HFPO-DA-contaminated waters in the Greenland Sea, also containing water that had spent significant time in the Arctic.37 The influence of the WSC carrying waters from the south is an explanation for the detection of HFPO-DA in samples N18 to N21, taken north of N16 and N17.
Sources of PFASs to Surface Water Entering and Exiting the Arctic Ocean through Fram Strait
The longitudinal sampling transect within Fram Strait (~79°N) cuts across the ice edge near the prime meridian (0°EW) (Figure S8). East of it, warm, saline Atlantic Water (AW) enters the Arctic Ocean, whereas west of it, low-saline Polar Surface Water (PSW) and sea ice is transported out of the Arctic. The sum of PFASs with a detection frequency >50% (C6 to C9 PFCAs, PFBS and HFPO-DA) was significantly higher in the samples taken west of 0°EW (F6 to F15, ice-covered, 260±20 pg/L) than east of it (F1 to F5, ice-free, 190±10 pg/L) (two-sample t-test, α=0.05, p<0.001) (Figure 2). Additionally, ΣPFASs was higher in samples taken north of the ice edge (P3, 300 pg/L, ice-covered) than south of it (P1, 190 pg/L, ice-free) along the latitudinal transect within Fram Strait (~0°EW) (Table S16).
Figure 2:

PFHpA/PFNA ratio and sum of PFASs with a detection frequency >50% (C6 to C9 PFCAs, PFBS and HFPO-DA) versus salinity [psu] and temperature [°C] in samples taken along a longitudinal transect across Fram Strait (~79°N; surface water samples F1 to F15). Temperature and salinity data was taken from von Appen and Rohardt (2019).44
For individual PFASs, the relative difference was highest for PFHpA (46±7 pg/L west of 0°EW versus 24±3 pg/L east of 0°EW) and lowest for PFNA (33±2 pg/L versus 25±2 pg/L). An exception was HFPO-DA, having a higher mean concentration in eastern Fram Strait (Table S13). Moreover, HFPO-DA was the only compound not showing a significant negative correlation with temperature and salinity (Table S17). Part of the AW entering the Arctic Ocean via the WSC in eastern Fram Strait spends significant time in the Arctic before exiting the Arctic Ocean via the EGC in western Fram Strait. Consequently, tracers transported by ocean currents reach western Fram Strait after longer lag times than eastern Fram Strait.29, 38 Thus, the later start of HFPO-DA production compared to other PFASs may be an explanation for its lower mean concentration west of 0°EW than east of it and the non-negative correlation with salinity and temperature.
In addition to oceanic transport times, the Arctic in- and outflow differs in the amount of freshwater. Reflected in the lower salinity and temperature, freshwater inputs, such as sea ice and glacier melt water, precipitation and river runoff, are of higher relevance in the outflowing EGC than in the inflowing WSC.39 Atmospheric long-range transport and degradation of volatile precursors can be expected to be the most important PFAS source to Arctic freshwater.15, 40-42 The relative importance of atmospheric sources is influenced by the amount of direct emissions of a compound in relation to those of its precursors.43 For example, global emission estimates of individual PFCAs from 1951-2030 show that >74% of PFHpA was released to the environment as degradation products of precursors, whereas >75% of PFNA was emitted from direct sources.32 Consequently, a higher PFHpA/PFNA ratio can be indicative of atmospheric sources. In our study, the PFHpA/PFNA ratio increased from 0.95±0.12 east of 0°EW to 1.40±0.22 west of it and showed a significant negative correlation with salinity (r=−0.79, p<0.001) (Figure 2). This indicates that the contribution of atmospheric PFAS sources is higher to Arctic outflow than to Arctic inflow and may account for the higher ΣPFASs west of 0°EW than east of it.
However, PFHpA/PFNA ratios in both Arctic in- and outflow were still considerably lower than in recent samples from North Sea coastal areas with a PFHpA/PFNA ratio of 5.4±1.1.22 Influenced by recent emissions from point sources, this value reflects global emission estimates with a predicted PFHpA/PFNA ratio of 5.8 from 2016 to 2030 (higher scenario), which is higher than 1951-2002 (range 0.2-1.5) and 2003-2015 (range 0.5-1.7).32 Consequently, the PFHpA/PFNA ratio in both Arctic in- and outflow can be expected to increase within the next years, with a rate that depends on the relative contributions from slow oceanic transport and fast atmospheric transport.
Depth Profiles of PFASs in Fram Strait
PFAS concentrations in different water depths varied among water masses. In general, the sum of PFASs with a detection frequency >50% (C6, C7, C9, C11 PFCAs and PFBS) was higher in the surface water layer, including PSW (170±30 pg/L, n=16) and AW (120±20 pg/L, n=18), than in Recirculating Atlantic Water/Arctic Atlantic Water (RAW/AAW) (90±35 pg/L, n=10) and in Intermediate Water (IW) (73±30 pg/L, n=6). In Deep Water (DW) (n=8), most of the results were <MDL, except for PFHxA, PFHpA, PFNA and PFUnDA, which were detected in one to three samples each (Tables S18 to S20). To show PFAS concentrations in relation to the properties of the respective water masses, temperature versus salinity plots are given in Figure S9.
The distribution of PFASs along the zonal section across Fram Strait is provided in Figure 3. Depth profiles V1 to V3 were taken in the eastern part of Fram Strait, where the warm, high-saline WSC enters the Arctic Ocean. In this area, the AW layer extends down to approximately 600 m, characterized by a potential temperature Θ of >0°C and a potential density isopycnal, referenced to 0 dbar, of σ0≤27.97 kg/m3. During winter, this is the depth to which the water column tends to be mixed, diluting tracers such as PFASs. This is reflected by comparatively stable PFAS concentrations from the surface to the sampling point at 500 m depth and significantly lower concentrations in IW and DW (>1000 m) (Figure 3B). Depth profiles V4 and V5 were taken in the western part of Fram Strait, where the surface layer is dominated by cold, low-density PSW from the Arctic (Θ≤0°C, σ0≤27.70 kg/m3) which reaches down to about 100 m. Below PSW is RAW/AAW (Θ>0°C, σ0>27.70 kg/m3), flowing southwards as well. This water mass comprises recirculating Atlantic Water (a part of the WSC that does not enter the Arctic Ocean but flows westward in Fram Strait before joining the EGC), and Arctic Atlantic Water (transported into the Arctic Ocean by the WSC and flowing around the Arctic Ocean in a cyclonic boundary current before exiting via the EGC).45 Passing the water mass boundary from PSW to RAW/AAW corresponded with a decrease of ΣPFASs from 120 pg/L to 77 pg/L in V4 and 150 pg/L to 74 pg/L in V5, respectively (Figure 3B). As discussed for the surface water samples before, concentrations of specific PFASs were higher in PSW than in AW, especially of PFHpA (42±9 pg/L vs. 25±5 pg/L). In contrast, the long-chain compound PFNA showed comparable concentrations in PSW and AW (31±6 pg/L vs. 31±9 pg/L). Consequently, the gradient between PSW and RAW/AAW was also higher for PFHpA (42±9 pg/L to 21±12 pg/L) than for PFNA (31±9 pg/L to 22±6 pg/L (Figures 4C and 4D). These observations suggest that atmospheric sources playing an important role for the outflowing surface water (PSW) are less relevant for RAW/AAW outflowing below.
Figure 3:

Distribution of A) water masses, B) sum of PFASs with a detection frequency >50% (C6, C7, C9, C11 PFCAs and PFBS), C) temperature, D) salinity from bottom to surface and E) PFHpA and F) PFNA from 600 m depth to surface along the east-west cross section at ~79°N (depth profiles V1 to V6, cruise stations PS114_12, 9, 25, 43, 46 and 49). Black dots represent samples analyzed for PFASs. Temperature and salinity data was taken from von Appen et al. (2019).48 For bottom bathymetry, GEBCO_2014 gridded data was used. Data were plotted using Ocean Data View.49
Figure 4:

PFAS mass transport estimates through Fram Strait via the boundary currents, calculated by combining measured PFAS concentrations (mean±SD) in Atlantic Water (AW), Polar Surface Water (PSW) and Recirculating/Arctic Atlantic Water (RAW/AAW) from this study with the average water volume transport [m3/s] for the respective water mass over the years 2010-2015, derived from the MITgcm ECCO v4 model (Section S4). Positive values describe northward fluxes into the Arctic Ocean, whereas negative values describe southward fluxes to the Nordic Seas. The modeled average water volume transport was 3.7·106 m3/s for AW, −0.6·106 m3/s for PSW and −3.8·106 m3/s for RAW/AAW. To calculate the mean value of each water mass in this figure, all values <MDL were replaced by √2/2 times the MDL.
AW and PSW in Fram Strait have an apparent mean age of approximately a decade, whereas intermediate waters have an apparent age of approximately 50 to 70 years, respectively.26 Concentrations of HFPO-DA were below detection limit in older intermediate waters, reflecting production and use that began after 2000.
Depth profiles of PFASs in this study were different from those of more hydrophobic legacy POPs such as polychlorinated biphenyls (PCBs) and dichlorodiphenyltrichloroethanes (DDTs), for which highest concentrations in Fram Strait were not found in the upper water layers, but in intermediate or deep waters.46 This was expected because for hydrophobic POPs, which partition readily to organic carbon and suspended particles, particle settling is suggested to be a dominant transport pathway to deeper water layers (“biological pump”). On the contrary, lateral and vertical transport due to physical mixing is thought to play a more important role for less hydrophobic compounds such as PFASs (“physical pump”).15, 17, 47 In agreement with this, the differences in PFAS concentrations and composition patterns between the upper and the deeper water layers, as well as the negligible intrusion of PFASs into DW indicate that physical mixing processes are more relevant for vertical PFAS transport in Fram Strait than sorption to sinking particles.
In a previous study, PFAS detection was generally limited to the upper water layers (<150 m) in four vertical water column profiles from the central Arctic.15 The authors attributed this to subsequent dilution and mixing of AW after entering the Arctic Ocean. This is consistent if looking at the water mass transport. Starting from V2 and V3 with comparatively constant PFAS concentrations down to 500 m (this study), water masses are further transported northwards and reach the sampling location PS80/227 of the previous study,15 at which PFASs could still be detected down to 250 m, before arriving at the three other sampling locations in the central Arctic, in which PFASs were non-detectable in the AW layer below 150 m.
PFAS Mass Transport Estimates through Fram Strait
For compounds with ≥8 perfluorinated carbon atoms (C9 to C11 PFCAs and L-/Br-PFOS) and HFPO-DA, summing mass flow estimates for the different water masses resulted in a net transport into the Arctic Ocean, whereas C6 to C8 PFCAs as well as C4 and C6 PFSAs showed a net transport out of the Arctic Ocean (Figure 4). A comparable net transport pattern was observed when the calculation was based on water transport data derived from a mooring array instead of the MITgcm (Table S23, Figure S10). Different statistical treatments of values below the MDL (zero, √2/2*MDL and MDL) did not change the observation of a chain-length dependent net transport (Table S22).
Estimated PFAS mass flows through Fram Strait in 2018 corresponded to approximately 2% (PFOA) to 13% (PFHxA) of the annual average of the higher value of estimated global cumulative emissions from 2003 to 2015.32 In modeling studies, PFOA mass flows into the Arctic through direct emissions and oceanic transport were estimated to be 8-23 t/year (1991-2004)50, 9-20 t/year (2000-2005)51 and 8-22 t/year (2005)52. The result of our study, combining measured PFOA concentrations and modeled water volume transport, is in the same order of magnitude (6.4±1.0 t/year) for oceanic input through Fram Strait, the major gateway to the Arctic Ocean.
A possible explanation for the chain-length dependent net transport through Fram Strait is the different contribution of atmospheric sources to each homologue in the Arctic. Atmospheric transport to remote regions is fast (days to weeks) in comparison to oceanic transport (years to decades).43 This suggests a rapid response of atmospheric inputs to changes in production, i.e. the industrial shift from long-chain PFSAs, PFCAs and their precursors to shorter-chain homologues. In snow pits from High Arctic ice caps, which receive inputs solely from atmospheric sources, concentrations of PFOS and its precursor perfluorooctane sulfonamide (FOSA) decreased in the early 2000s.53-54 Rapid declines in FOSA observed in North Atlantic pilot whales were also attributed to fast changes in atmospheric inputs, reflected in seawater concentrations.55 In contrast, the oceanic response is slower, which is demonstrated by a higher contribution of long-chain homologues to ΣPFASs in the Arctic inflow than in water from regions close to emission areas, such as the North Sea. As our study indicates that atmospheric inputs are of higher relevance for outflowing Arctic surface waters, the higher share of short-chain homologues and their precursors in recent emissions is a possible explanation for their higher concentrations in outflowing waters, resulting in a net southward transport.
In addition, fractionation of the compounds in the melting snowpack and sea ice due to different physicochemical properties of PFASs with varying chain length may be important. In a laboratory study, short-chain PFCAs and PFSAs were released early during a snowmelt period, whereas the more hydrophobic long-chain PFAAs were enriched in the late meltwater and particle fractions.56 PFCAs showed a similar behavior to PFSAs with one perfluoroalkyl moiety less, indicating a higher affinity of the sulfonic group to the snow grain surface than the carboxylate group, as also observed for sediment.57 Consequently, the higher retention of the long-chain compounds in snow, sea ice and terrestrial environments may serve as an additional explanation for the observation that for compounds with ≥8 perfluorinated carbons, the Arctic output is lower than the input.
Implications
This study adds empirical evidence to modelling assessments33 indicating that HFPO-DA, similar to PFOA, fulfills the criterion of long-range transport. Consequently, it is a compound of global environmental concern and it should be evaluated with regard to future regulations. The recent detection of another ether-based PFAS (6:2 Cl-PFESA) in East Greenland marine mammals58 highlights the potential role of long-range oceanic transport for delivering emerging PFASs to the Arctic food web.
The detection of the replacement compound HFPO-DA in a remote area also underlines the importance of complementing targeted methods with non-target approaches such as database screening or analytical approaches using high-resolution mass spectrometry to identify novel PFASs of relevance. Earlier identification of compounds of emerging concern in source regions could trigger mitigation measures before the substances become ubiquitously distributed.59-60
The differences in Arctic in- and outflow highlight that the interplay of changes in source patterns, oceanic and atmospheric long-range transport as well as ice and snow as links between the atmosphere and the ocean has to be considered to assess the fate of PFASs in the Arctic. The results of this study indicate that atmospheric inputs are an important factor when assessing if the Arctic is a sink or source of PFASs. Future research on the relative importance of direct and indirect sources in remote areas can improve the understanding on the ultimate fate of PFASs.
The PFAS depth profiles in Fram Strait demonstrate that knowledge of the ocean circulation, vertical and lateral stratification as well as physical mixing processes is crucial for understanding the large-scale distribution and fate of PFASs. Using PFOS as an example, prior modeling results showed that weakened Atlantic Meridonial Overturning Circulation (AMOC) due to climate change is likely to increase the magnitude of POPs entering the Arctic Ocean.17 Additionally, the loss of perennial glacial mass and snow due to climate change could release long-chain PFASs deposited during earlier periods of high use, reinforcing the role of the Arctic as a secondary source.42, 61-62 Consequently, future PFAS depth profiles in Fram Strait and other key regions of the global oceanic circulation, as well as mechanistic studies on the post-deposition characteristics of PFASs in snow and ice interacting with ocean surface water, can help to elucidate how PFAS cycling is linked to climate change.
Supplementary Material
ACKNOWLEDGMENTS
Ship time was provided under grant AWI_PS114_03. The authors gratefully thank the captain and the crew of the research vessel Polarstern for assistance during the sampling.
This work was supported in kind by the EISPAC project (NE/R012857/1), part of the Changing Arctic Ocean programme, jointly funded by the UKRI Natural Environment Research Council (NERC) and the German Federal Ministry of Education and Research (BMBF). EMS and CW acknowledge support from the National Institute for Environmental Health Sciences (NIEHS) Superfund Research Program (P42ES027706).
Footnotes
Supporting Information. Details on sample collection, PFAS analysis, QA/QC measures and data analysis (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).BRS Secretariat; Secretariat of the Basel, Rotterdam and Stockholm Conventions. Listing of perfluorooctane sulfonic acid, its salts and perfluorooctane sulfonyl fluoride. UNEP/POPS/COP.4-SC-4/17 2009. http://chm.pops.int/TheConvention/ConferenceoftheParties/Meetings/COP4/COP4Documents/tabid/531/Default.aspx (accessed 01 November 2019). [Google Scholar]
- (2).BRS Secretariat; Secretariat of the Basel, Rotterdam and Stockholm Conventions. Listing of perfluorooctanoic acid (PFOA), its salts and PFOA-related compounds. UNEP/POPS/COP.9-SC-9/12 2019. http://chm.pops.int/TheConvention/ConferenceoftheParties/Meetings/COP9/tabid/7521/Default.aspx (accessed 01 November 2019). [Google Scholar]
- (3).BRS Secretariat; Secretariat of the Basel, Rotterdam and Stockholm Conventions. POPRC Recommendations for listing chemicals – Chemicals under review. 2019. http://chm.pops.int/Convention/POPsReviewCommittee/Chemicals/tabid/243/Default.aspx (accessed 05 November 2019).
- (4).OECD; Organisation for Economic Co-operation and Development. Toward a new comprehensive global database of per- and polyfluoroalkyl substances (PFASs): Summary report on updating the OECD 2007 list of per- and polyfluoroalkyl substances (PFASs). Series on Risk Management 2018, 39, 1–24. [Google Scholar]
- (5).Wang Z; Cousins IT; Scheringer M; Hungerbühler K Fluorinated alternatives to long-chain perfluoroalkyl carboxylic acids (PFCAs), perfluoroalkane sulfonic acids (PFSAs) and their potential precursors. Environ. Int 2013, 60, 242–248. [DOI] [PubMed] [Google Scholar]
- (6).ECHA; European Chemicals Agency. Candidate list of substances of very high concern for authorisation. 2019. https://echa.europa.eu/candidate-list-table (accessed 05 January 2020).
- (7).Pan Y; Zhang H; Cui Q; Sheng N; Yeung LWY; Sun Y; Guo Y; Dai J Worldwide distribution of novel perfluoroether carboxylic and sulfonic acids in surface water. Environ. Sci. Technol 2018, 52 (14), 7621–7629. [DOI] [PubMed] [Google Scholar]
- (8).Beszczynska-Möller A; Fahrbach E; Schauer U; Hansen E Variability in Atlantic water temperature and transport at the entrance to the Arctic Ocean, 1997–2010. ICES Journal of Marine Science 2012, 69 (5), 852–863. [Google Scholar]
- (9).Yamashita N; Kannan K; Taniyasu S; Horii Y; Petrick G; Gamo T A global survey of perfluorinated acids in oceans. Mar. Pollut. Bull 2005, 51 (8-12), 658–668. [DOI] [PubMed] [Google Scholar]
- (10).Ahrens L; Barber JL; Xie Z; Ebinghaus R Longitudinal and latitudinal distribution of perfluoroalkyl compounds in the surface water of the Atlantic Ocean. Environ. Sci. Technol 2009, 43 (9), 3122–3127. [DOI] [PubMed] [Google Scholar]
- (11).Zhao Z; Xie Z; Moller A; Sturm R; Tang J; Zhang G; Ebinghaus R Distribution and long-range transport of polyfluoroalkyl substances in the Arctic, Atlantic Ocean and Antarctic coast. Environ. Pollut 2012, 170, 71–77. [DOI] [PubMed] [Google Scholar]
- (12).Busch J; Ahrens L; Xie Z; Sturm R; Ebinghaus R Polyfluoroalkyl compounds in the East Greenland Arctic Ocean. J. Environ. Monit 2010, 12 (6), 1242–1246. [DOI] [PubMed] [Google Scholar]
- (13).Cai M; Zhao Z; Yin Z; Ahrens L; Huang P; Cai M; Yang H; He J; Sturm R; Ebinghaus R; Xie Z Occurrence of perfluoroalkyl compounds in surface waters from the North Pacific to the Arctic Ocean. Environ. Sci. Technol 2012, 46 (2), 661–668. [DOI] [PubMed] [Google Scholar]
- (14).Benskin JP; Muir DCG; Scott BF; Spencer C; De Silva AO; Kylin H Perfluoroalkyl acids in the Atlantic and Canadian Arctic Oceans. Environ. Sci. Technol 2012, 46 (11), 5815–5823. [DOI] [PubMed] [Google Scholar]
- (15).Yeung LWY; Dassuncao C; Mabury S; Sunderland EM; Zhang X; Lohmann R Vertical profiles, sources, and transport of PFASs in the Arctic Ocean. Environ. Sci. Technol 2017, 51 (12), 6735–6744. [DOI] [PubMed] [Google Scholar]
- (16).Yamashita N; Taniyasu S; Petrick G; Wei S; Gamo T; Lam PKS Perfluorinated acids as novel chemical tracers of global circulation of ocean waters. Chemosphere 2008, 70 (7), 1247–1255. [DOI] [PubMed] [Google Scholar]
- (17).Zhang X; Zhang Y; Dassuncao C; Lohmann R; Sunderland EM North Atlantic Deep Water formation inhibits high Arctic contamination by continental perfluorooctane sulfonate discharges. Global Biogeochem. Cycles 2017, 31 (8), 1332–1343. [Google Scholar]
- (18).Rhein M; Kieke D; Steinfeldt R Advection of North Atlantic Deep Water from the Labrador Sea to the southern hemisphere. Journal of Geophysical Research: Oceans 2015, 120 (4), 2471–2487. [Google Scholar]
- (19).Schlosser P; Bayer R; Bönisch G; Cooper LW; Ekwurzel B; Jenkins WJ; Khatiwala S; Pfirman S; Smethie WM Pathways and mean residence times of dissolved pollutants in the ocean derived from transient tracers and stable isotopes. Sci. Total Environ 1999, 237-238, 15–30. [Google Scholar]
- (20).von Appen W-J The Expedition PS114 of the Research Vessel POLARSTERN to the Fram Strait in 2018. Reports on polar and marine research, Bremerhaven, Alfred Wegener Institute for Polar and Marine Research: Bremerhaven1, 2018, 723; pp. 84; doi: 10.2312/BzPM_0723_2018. [DOI] [Google Scholar]
- (21).Chandramouli B; Benskin JP; Hamilton MC; Cosgrove JR Sorption of per- and polyfluoroalkyl substances (PFASs) on filter media: Implications for phase partitioning studies. Environ. Toxicol. Chem 2015, 34 (1), 30–36. [DOI] [PubMed] [Google Scholar]
- (22).Joerss H; Apel C; Ebinghaus R Emerging per- and polyfluoroalkyl substances (PFASs) in surface water and sediment of the North and Baltic Seas. Sci. Total Environ 2019, 686, 360–369. [DOI] [PubMed] [Google Scholar]
- (23).Forget G; Campin JM; Heimbach P; Hill CN; Ponte RM; Wunsch C ECCO version 4: an integrated framework for non-linear inverse modeling and global ocean state estimation. Geosci. Model Dev 2015, 8 (10), 3071–3104. [Google Scholar]
- (24).Forget G; Ferreira D; Liang X On the observability of turbulent transport rates by Argo: supporting evidence from an inversion experiment. Ocean Sci. 2015, 11 (5), 839–853. [Google Scholar]
- (25).Forget G; Ponte RM The partition of regional sea level variability. Progress in Oceanography 2015, 137, 173–195. [Google Scholar]
- (26).Stöven T; Tanhua T; Hoppema M; von Appen WJ Transient tracer distributions in the Fram Strait in 2012 and inferred anthropogenic carbon content and transport. Ocean Sci. 2016, 12 (1), 319–333. [Google Scholar]
- (27).Mork KA; Skagseth Ø A quantitative description of the Norwegian Atlantic Current by combining altimetry and hydrography. Ocean Sci. 2010, 6 (4), 901–911. [Google Scholar]
- (28).Ahrens L; Gerwinski W; Theobald N; Ebinghaus R Sources of polyfluoroalkyl compounds in the North Sea, Baltic Sea and Norwegian Sea: Evidence from their spatial distribution in surface water. Mar. Pollut. Bull 2010, 60 (2), 255–260. [DOI] [PubMed] [Google Scholar]
- (29).Smith JN; McLaughlin FA; Smethie WM Jr.; Moran SB; Lepore K Iodine-129, 137Cs, and CFC-11 tracer transit time distributions in the Arctic Ocean. Journal of Geophysical Research: Oceans 2011, 116 (C4). [Google Scholar]
- (30).HELCOM; Baltic Marine Environment Protection Commission. State of the Baltic Sea – Second HELCOM holistic assessment 2011-2016. Baltic Sea Environment Proceedings 2018, 155. [Google Scholar]
- (31).Winther NG; Johannessen JA North Sea circulation: Atlantic inflow and its destination. Journal of Geophysical Research (Oceans) 2006, 111, 1–12.20411040 [Google Scholar]
- (32).Wang Z; Cousins IT; Scheringer M; Buck RC; Hungerbühler K Global emission inventories for C4–C14 perfluoroalkyl carboxylic acid (PFCA) homologues from 1951 to 2030, Part I: production and emissions from quantifiable sources. Environ. Int 2014, 70, 62–75. [DOI] [PubMed] [Google Scholar]
- (33).Gomis MI; Wang Z; Scheringer M; Cousins IT A modeling assessment of the physicochemical properties and environmental fate of emerging and novel per- and polyfluoroalkyl substances. Sci. Total Environ 2015, 505, 981–991. [DOI] [PubMed] [Google Scholar]
- (34).SSC Secretariat, Secretariat of the Stockholm Convention. Stockholm Convention on Persistent Organic Pollutants. Text and Annexes, revised in 2017 http://www.pops.int/TheConvention/Overview/TextoftheConvention/tabid/2232/Default.aspx (accessed: 03 May 2020).
- (35).Scheringer M Long-range transport of organic chemicals in the environment. Environ. Toxicol. Chem 2009, 28 (4), 677–690. [DOI] [PubMed] [Google Scholar]
- (36).Skaar JS; Ræder EM; Lyche JL; Ahrens L; Kallenborn R Elucidation of contamination sources for poly- and perfluoroalkyl substances (PFASs) on Svalbard (Norwegian Arctic). Environ. Sci. Pollut. Res 2019, 26 (8), 7356–7363. [DOI] [PubMed] [Google Scholar]
- (37).Walczowski W Frontal structures in the West Spitsbergen Current margins. Ocean Sci. 2013, 9 (6), 957–975. [Google Scholar]
- (38).Dahlgaard H Transfer of European coastal pollution to the arctic: Radioactive tracers. Marine Pollution Bulletin 1995, 31 (1), 3–7. [Google Scholar]
- (39).de Steur L; Peralta-Ferriz C; Pavlova O Freshwater export in the East Greenland Current freshens the North Atlantic. Geophysical Research Letters 2018, 45 (24), 13359–13366. [Google Scholar]
- (40).Pickard HM; Criscitiello AS; Spencer C; Sharp MJ; Muir DCG; De Silva AO; Young CJ Continuous non-marine inputs of per- and polyfluoroalkyl substances to the High Arctic: a multi-decadal temporal record. Atmos. Chem. Phys 2018, 18 (7), 5045–5058. [Google Scholar]
- (41).MacInnis JJ; Lehnherr I; Muir DCG; St. Pierre KA; St. Louis VL; Spencer C; De Silva AO Fate and transport of perfluoroalkyl substances from snowpacks into a lake in the high Arctic of Canada. Environ. Sci. Technol 2019, 53 (18), 10753–10762. [DOI] [PubMed] [Google Scholar]
- (42).Kwok KY; Yamazaki E; Yamashita N; Taniyasu S; Murphy MB; Horii Y Transport of Perfluoroalkyl substances (PFAS) from an arctic glacier to downstream locations: implications for sources. Sci. Total Environ 2013, 447, 46–55. [DOI] [PubMed] [Google Scholar]
- (43).Young CJ; Mabury SA Atmospheric Perfluorinated Acid Precursors: Chemistry, Occurrence, and Impacts In Reviews of Environmental Contamination and Toxicology Volume 208: Perfluorinated alkylated substances; De Voogt P, Ed.; Springer New York: New York, 2010; pp 109. [DOI] [PubMed] [Google Scholar]
- (44).von Appen W-J; Rohardt G Continuous thermosalinograph oceanography along POLARSTERN cruise track PS114. Alfred Wegener Institute, Helmholtz Centre for Polar and Marine Research, Bremerhaven, PANGAEA, 2019. doi: 10.1594/PANGAEA.898225. [DOI] [Google Scholar]
- (45).Rudels B; Björk G; Nilsson J; Winsor P; Lake I; Nohr C The interaction between waters from the Arctic Ocean and the Nordic Seas north of Fram Strait and along the East Greenland Current: results from the Arctic Ocean-02 Oden expedition. Journal of Marine Systems 2005, 55 (1), 1–30. [Google Scholar]
- (46).Ma Y; Adelman DA; Bauerfeind E; Cabrerizo A; McDonough CA; Muir D; Soltwedel T; Sun C; Wagner CC; Sunderland EM; Lohmann R Concentrations and water mass transport of legacy POPs in the Arctic Ocean. Geophysical Research Letters 2018, 45 (23), 12972–12981. [Google Scholar]
- (47).Lohmann R; Jurado E; Dijkstra HA; Dachs J Vertical eddy diffusion as a key mechanism for removing perfluorooctanoic acid (PFOA) from the global surface oceans. Environ. Pollut 2013, 179, 88–94. [DOI] [PubMed] [Google Scholar]
- (48).von Appen W-J; Schaffer J; Rohardt G; Wisotzki A Physical oceanography measured on water bottle samples during POLARSTERN cruise PS114. Alfred Wegener Institute, Helmholtz Centre for Polar and Marine Research, Bremerhaven, PANGAEA, 2019. doi: 10.1594/PANGAEA.898695. [DOI] [Google Scholar]
- (49).Schlitzer R Ocean Data View. https://odv.awi.de, 2018.
- (50).Stemmler I; Lammel G Pathways of PFOA to the Arctic: variabilities and contributions of oceanic currents and atmospheric transport and chemistry sources. Atmos. Chem. Phys 2010, 10 (20), 9965–9980. [Google Scholar]
- (51).Wania F A global mass balance analysis of the source of perfluorocarboxylic acids in the Arctic Ocean. Environ. Sci. Technol 2007, 41 (13), 4529–4535. [DOI] [PubMed] [Google Scholar]
- (52).Armitage J; Cousins IT; Buck RC; Prevedouros K; Russell MH; MacLeod M; Korzeniowski SH Modeling global-scale fate and transport of perfluorooctanoate emitted from direct sources. Environ. Sci. Technol 2006, 43 (24), 9274–9280. [DOI] [PubMed] [Google Scholar]
- (53).Young CJ; Furdui VI; Franklin J; Koerner RM; Muir DCG; Mabury SA Perfluorinated acids in Arctic snow: New evidence for atmospheric formation. Environ. Sci. Technol 2007, 41 (10), 3455–3461. [DOI] [PubMed] [Google Scholar]
- (54).MacInnis JJ; French K; Muir DCG; Spencer C; Criscitiello A; De Silva AO; Young CJ Emerging investigator series: a 14-year depositional ice record of perfluoroalkyl substances in the High Arctic. Environ. Sci.: Processes Impacts 2017, 19 (1), 22–30. [DOI] [PubMed] [Google Scholar]
- (55).Dassuncao C; Hu XC; Zhang X; Bossi R; Dam M; Mikkelsen B; Sunderland EM Temporal shifts in poly- and perfluoroalkyl substances (PFASs) in North Atlantic pilot whales indicate large contribution of atmospheric precursors. Environ. Sci. Technol 2017, 51 (8), 4512–4521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Plassmann MM; Meyer T; Lei YD; Wania F; McLachlan MS; Berger U Laboratory studies on the fate of perfluoroalkyl carboxylates and sulfonates during snowmelt. Environ. Sci. Technol 2011, 45 (16), 6872–6878. [DOI] [PubMed] [Google Scholar]
- (57).Higgins CP; Luthy RG Sorption of perfluorinated surfactants on sediments. Environ. Sci. Technol 2006, 40 (23), 7251–7256. [DOI] [PubMed] [Google Scholar]
- (58).Gebbink WA; Bossi R; Rigét FF; Rosing-Asvid A; Sonne C; Dietz R Observation of emerging per- and polyfluoroalkyl substances (PFASs) in Greenland marine mammals. Chemosphere 2016, 144, 2384–2391. [DOI] [PubMed] [Google Scholar]
- (59).Hollender J; van Bavel B; Dulio V; Farmen E; Furtmann K; Koschorreck J; Kunkel U; Krauss M; Munthe J; Schlabach M; Slobodnik J; Stroomberg G; Ternes T; Thomaidis NS; Togola A; Tornero V High resolution mass spectrometry-based non-target screening can support regulatory environmental monitoring and chemicals management. Environmental Sciences Europe 2019, 31 (1), 42. [Google Scholar]
- (60).Muir D; Zhang X; de Wit CA; Vorkamp K; Wilson S Identifying further chemicals of emerging arctic concern based on ‘in silico’ screening of chemical inventories. Emerging Contaminants 2019, 5, 201–210. [Google Scholar]
- (61).Pućko M; Stern GA; Macdonald RW; Jantunen LM; Bidleman TF; Wong F; Barber DG; Rysgaard S The delivery of organic contaminants to the Arctic food web: Why sea ice matters. Sci. Total Environ 2015, 506-507, 444–452. [DOI] [PubMed] [Google Scholar]
- (62).Chen M; Wang C; Wang X; Fu J; Gong P; Yan J; Yu Z; Yan F; Nawab J Release of perfluoroalkyl substances from melting glacier of the Tibetan Plateau: Insights into the impact of global warming on the cycling of emerging pollutants. Journal of Geophysical Research: Atmospheres 2019, 124 (13), 7442–7456. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.