

Abstract
Tumors are characterized by extracellular matrix (ECM) deposition, remodeling, and cross-linking that drive fibrosis to stiffen the stroma and promote malignancy. The stiffened stroma enhances tumor cell growth, survival and migration and drives a mesenchymal transition. A stiff ECM also induces angiogenesis, hypoxia and compromises anti-tumor immunity. Not surprisingly, tumor aggression and poor patient prognosis correlate with degree of tissue fibrosis and level of stromal stiffness. In this review, we discuss the reciprocal interplay between tumor cells, cancer associated fibroblasts (CAF), immune cells and ECM stiffness in malignant transformation and cancer aggression. We discuss CAF heterogeneity and describe its impact on tumor development and aggression focusing on the role of CAFs in engineering the fibrotic tumor stroma and tuning tumor cell tension and modulating the immune response. To illustrate the role of mechanoreciprocity in tumor evolution we summarize data from breast cancer and pancreatic ductal carcinoma (PDAC) studies, and finish by discussing emerging anti-fibrotic strategies aimed at treating cancer.
Keywords: Mechanoreciprocity, Cancer, ECM, CAF, Fibrosis
1. Introduction
Tissue homeostasis requires maintenance of its structure and function. Extracellular matrix (ECM) composition, organization and stiffness influence cell adhesion-dependent cytoskeletal tension to modulate tissue structure and thus is a key regulator of tissue homeostasis. Not surprisingly, pathological conditions linked to ECM stiffening that abnormally elevates cytoskeletal tension, including liver fibrosis and chronic pancreatitis are accompanied by perturbations in tissue structure and function and associate with increased risk of malignancy [1–4]. Indeed, the physical properties or stiffness of the ECM, has increasingly been recognized as an important component of the tumor microenvironment (TME), and is now appreciated as a key factor that not only fosters malignant transformation but also regulates tumor aggression. Consistently, solid tumors are stiffer than healthy tissue and this feature has been exploited to detect cancer either by physical palpation or using imaging modalities such as magnetic resonance imaging, computerized tomography and elastography (Fig. 1) [5,6]. Moreover, breast cancer and pancreatic ductal adenocarcinoma (PDAC) aggression both associate with a stiffer ECM, and experimental models attest to causal links between tissue mechanics and malignancy.
Fig. 1.
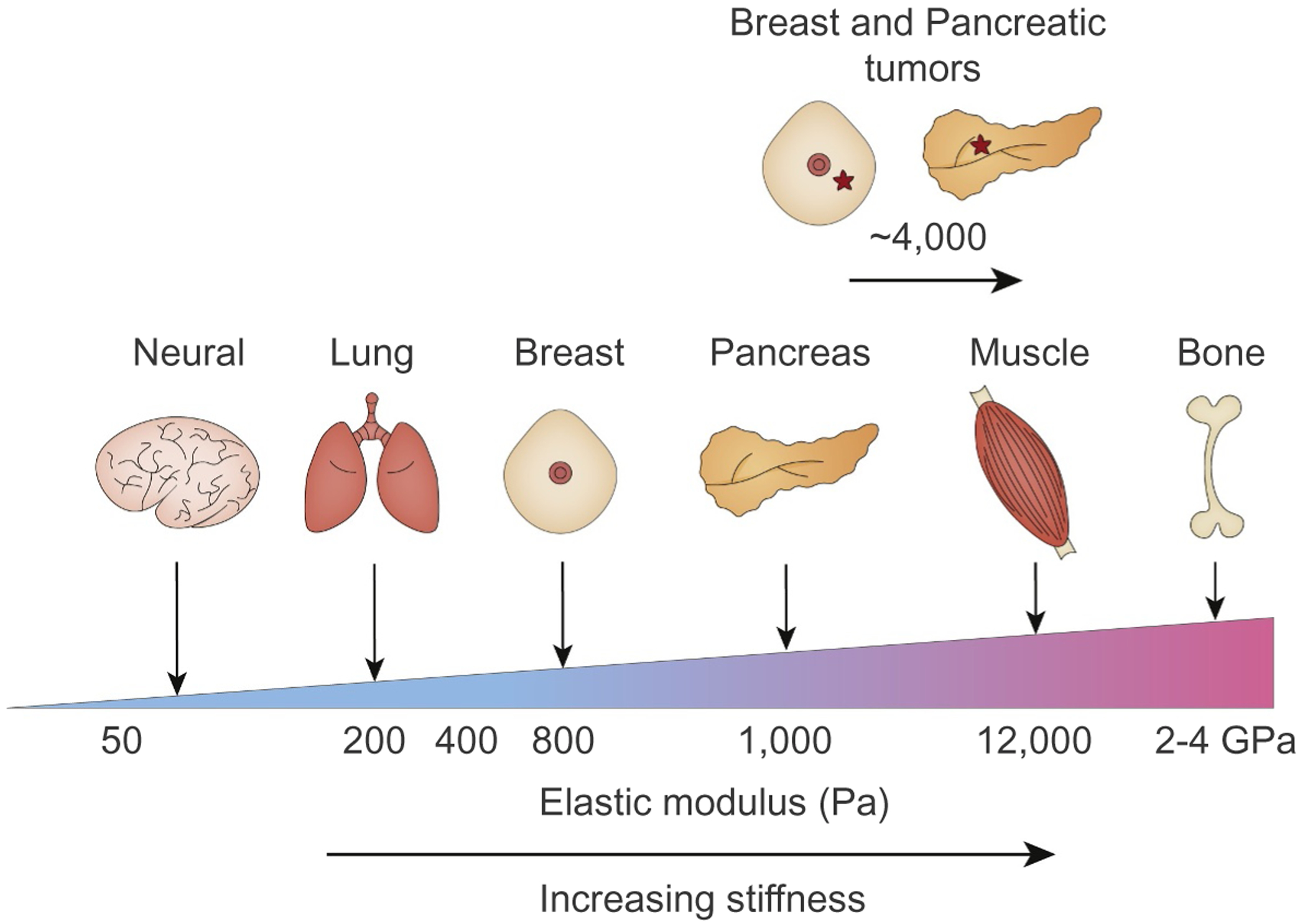
Elastic moduli in healthy human tissues and tumors. Cells within a tissue interact with their ECM which is tuned to a specific elastic modulus (measured in Pa) that dictate the function of the tissue. The ECM in the brain, lung or breast is relatively soft (compliant; <100 Pa), whereas the ECM in tissues that are exposed to high mechanical loading such as skeletal muscle and bone are by comparison stiff (>100 kPa). Soft ECMs promote neural cell growth, survival and intercellular connections, and critically permits the expansion of lung alveoli and mammary epithelial cells associated with breathing and milk delivery. By contrast, stiff ECMs favor osteoblast cell differentiation and cardiac contractility function. Tumors are often fibrotic, and the ECM is stiffer than that found in a healthy tissue (~4–10 kPa), and this ECM stiffness induces cytoskeletal tension that perturbs tissue organization and function. Critically reducing cytoskeletal tension reverts the malignant phenotype of tumor cells and inhibiting ECM stiffening prevents malignant transformation.
“Mechanoreciprocity” is a term that has been coined to describe how a cell “tunes” its actomyosin cytoskeletal tension in response to the stiffness of its ECM, and thereafter how a cells intrinsic contractile phenotype will in turn remodel and stiffen its local ECM until cells reach a state of “tensional homeostasis” or “mechano equilibrium” [7]. In this article, we review the reciprocal physical interactions between cells and the ECM in the context of malignancy. We begin by describing the ECM and discussing basic concepts of mechanotransduction. We then describe the role of cancer-associated fibroblasts (CAFs) in generating the fibrotic, stiffened stroma and how this influences tumor progression. We thereafter summarize data from breast cancer and PDAC studies that illustrate the role of mechanoreciprocity in tumor evolution, and discuss emerging therapeutic strategies aimed at targeting fibrosis to improve cancer treatment.
2. Mechanical properties of the ECM
2.1. Structural organization of the ECM
The ECM is the non-cellular component present within all tissues. The ECM not only provides structural support for resident cells but also critical biochemical and biomechanical cues that drive morphogenesis and tissue-specific differentiation and maintain tissue homeostasis [8]. Although the basic building blocks of the ECM are water, proteins and polysaccharides, the ECM is a highly dynamic structure that is constantly being remodeled through enzymatic and non-enzymatic post-translational modifications that alter its instructive capacity [9]. Functionally discrete tissues are thus defined by unique ECM compositions and topology that are achieved through dynamic and reciprocal biochemical and biomechanical dialogues between the various cellular constituents of the tissue.
The ECM is broadly classified as either basement membrane (BM) or interstitial matrix. The BM which surrounds cells such as epithelial, endothelial and hepatocytes, is composed of a laminin and collagen IV network that is linked by a perlecan and nidogen network [8]. The BM not only provides structural support but also orchestrates the establishment of cell polarity and binds critical growth factors and cytokines that regulates cell differentiation and maintains tissue homeostasis [10,11]. Although the basic building blocks are conserved, the BM in each tissue has a specific composition and structure that is specifically tuned to the functional requirement of the organ system.
Interstitial ECMs are composed of proteoglycans and fibrous proteins that maintain tissue hydration and mechanical strength. The proteoglycans in the interstitial ECM (e.g. hyaluronic acid (HA)) bind water through their glycosaminoglycan (GAG) chains. GAGs are un-branched polysaccharide chains composed of repeating disaccharide units that are quite hydrophilic and they adopt highly extended formations that bind water to provide hydration and permit compression resistance in the tissue. Fibrillar collagens are the main structural component of the interstitial ECM that contribute to the tensile strength of the tissue. Heterotypic fibrils of collagens I, III, and V are the main fibrillar collagens that are assembled into large mechanically resilient fibers. Collagen I fibril assembly involves a number of enzymatic post-translational modifications, including the hydroxylation of lysine residues by lysyl hydroxylase 2 (LH2), glycosylation of hydroxylysine residues, and covalent cross-linking between lysine or hydroxylysine residues by lysyl oxidases (LOX) and LOX-like (LOX-L) members; all of which function to strength collagen fibrils [12]. Fibronectin is another fibrous protein, which is intimately involved in collagen fibrillogenesis [13,14]. Fibronectin is secreted as a dimer, and following integrin receptor engagement and actomyosin-dependent cell contraction their cryptic binding sites are exposed which allows them to bind to one another to induce fibronectin fibril assembly and confers a stretching phenotype to the fibers [15]. Nascent collagen molecules preferentially co-localize with relaxed fibronectin fibers, and in turn, dominate as the load-bearing structure and prevent further stretching of fibronectin [13,14]. The mechanical dynamic that exists between collagen and fibronectin fibrillogenesis is just one example of the mechanical regulation of ECM assembly and homeostasis. Many other interactions contribute to collagen organization including small leucine-rich repeat proteoglycans (SLRPs) such as decorin, and FACIT (Fibril Associated Collagens with Interrupted Triple helices) collagens. These interactions and modifications provide the collagenous, interstitial ECM with its unique physical properties. In order to understand the complex processes that underlie tumor evolution, we need to understand how the tumor ECM is altered from its healthy state, and how the chemical and physical composition and topography of the ECM are sensed and interpreted, and thereby influence cancer cell behavior.
2.2. The fibrotic tumor stroma
Tumors are “fibrotic wounds that do not heal” and chronic fibrosis is a risk factor for cancer [16,17]. For example, idiopathic lung fibrosis is an independent risk factor for lung cancer, and the fibrosis induced by epidermolysis bullosa correlates with increased risk for metastatic melanoma [18,19]. Wound repair is initiated by the infiltration of inflammatory cells that secrete growth factors, cytokines and matrix metalloproteinases (MMPs) that recruit fibroblasts and remodel the fibrin clot. The recruited fibroblasts synthesize and deposit ECM proteins that generate a provisional matrix and induce mechanical stresses that promote the differentiation of fibroblasts into myofibroblasts and stimulate keratinocyte migration. The high contractility of myofibroblasts facilitates wound closure and secreted MMPs remodel the collagenous matrix to permit wound resolution [20]. Tumor fibrosis by contrast is characterized by chronic inflammation, elevated numbers of contractile myofibroblasts that secrete abundant ECM proteins and remodeling enzymes that reorganize, cross-link and stiffen the matrix, and cytokines and growth factors that stimulate tumor cell proliferation and invasion yielding a markedly different stroma (Fig. 2) [21–26].
Fig. 2.
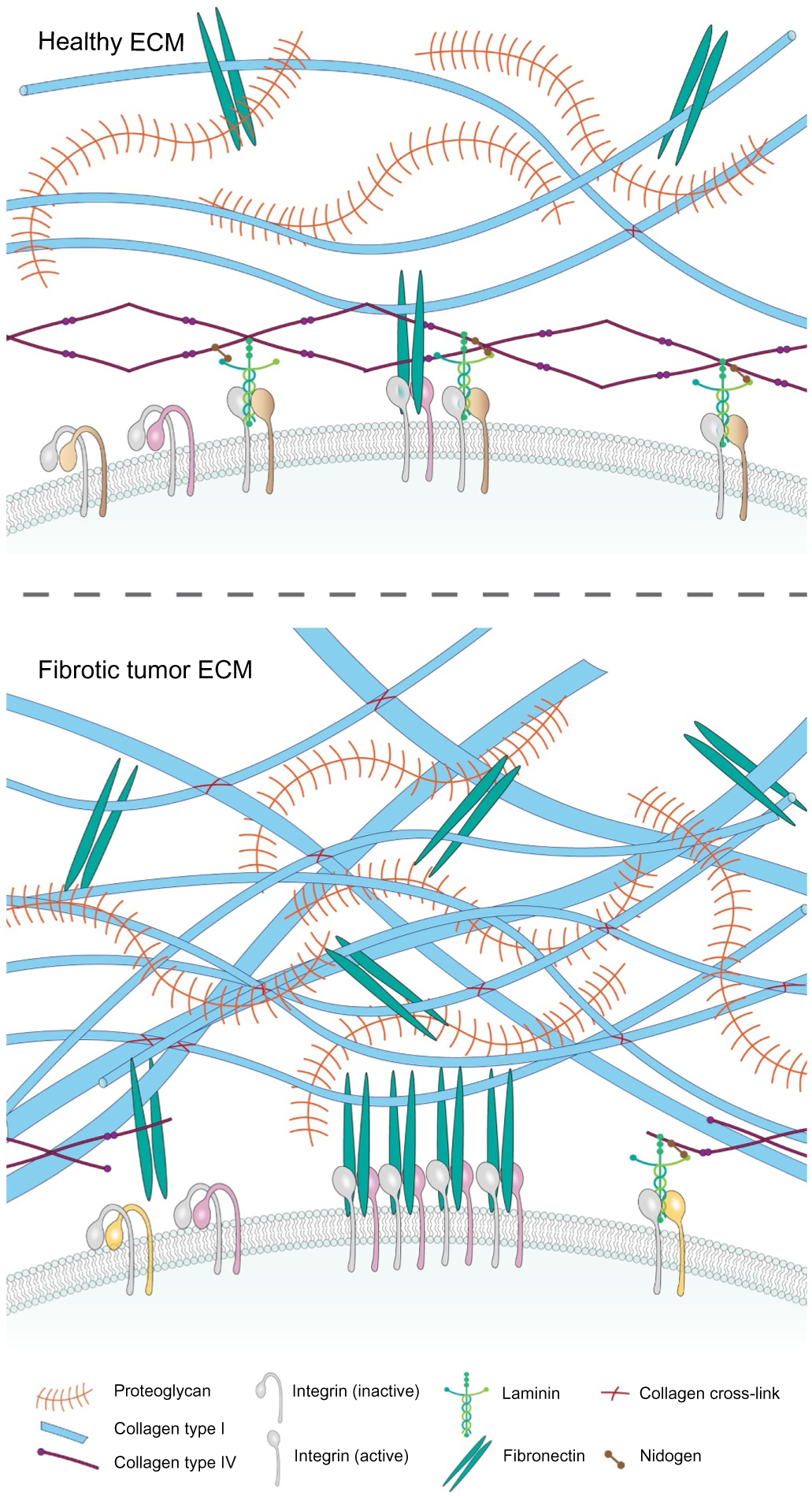
ECM homeostasis in healthy tissues and tumors. The interstitial ECM is composed of collagens, glycoproteins including fibronectin and proteoglycans, and water. Cells ligate to the ECM through transmembrane bound receptors such as integrins, which activate intracellular signaling and induce cytoskeletal reorganization to modify cell growth, survival and motility. The process by which cells sense mechanical signals from their microenvironment and translate these into biochemical signals is termed mechanotransduction. Activated integrins in cells ligating a soft ECM, assemble nascent, dynamic adhesions. By contrast, cells ligating a stiff ECM assemble stable focal adhesions as the resistance here favors the unfolding of tension sensitive adhesion plaque proteins such as talin and vinculin, which interact and nucleate multiple proteins that either stimulate downstream biochemical signaling cascades or activate GTPases that induce actin cytoskeletal reorganization. In a fibrotic tumor, the production and remodeling of the ECM is disturbed. CAFs produce increased amounts of ECM, as well as growth factors and enzymes that induce its remodeling and post-translational cross-linking that stiffens and aligns its fibrils to increases its tensile properties, enhance its density and elevate the compressive force experienced by cells within the tissue.
In healthy tissues, interstitial collagen is isotropically oriented, whereas the collagen in tumors is often aligned and anisotropic. Aligned collagen fibrils in tumors not only reflect differences in ECM composition including an increased ratio between collagen I/III but tracks with tumor aggression and has been exploited to predict breast cancer patient outcome using a tumor-associated collagen signature (TACS3) [22,27–30]. Similarly, highly aligned stromal collagen correlates with reduced post-surgical survival in patients with PDAC [31].
The filamentous components of the ECM undergo internal rearrangements in response to applied stresses conferring ECM networks with mechanical properties of both liquids and solids; defined as viscoelastic [32]. Accordingly, ECM fibers exhibit a non-linear elasticity: fibers are strain-stiffening materials (or increase the elasticity as the strain increases). ECM fibers also present a viscoelastic response [32–34]. This means that as a result of local ECM contraction, the stiffness sensed by neighboring cells hundreds of microns away can be magnitudes higher [35–37]. However, whether the increased ECM stiffness caused by strain-stiffening is responsible for long range communication between cells remains inconclusive. Another proposed mechanism that could explain how cells can propagate signals multiple cell diameters through the ECM, is described by the cell tension-driven formation of aligned fibers of e.g. collagen or fibrin [35–38]. Regardless, these findings suggest that in fibrotic tumors, cell-mediated fiber organization can exert profound effects, not only on neighboring adjacent cells, but also on cells hundreds of microns away.
The normal healthy tissue stroma transitions into a dense fibrotic tumor stroma through the progressive accumulation, alignment and post-translational cross-linking of fibrillar ECMs including collagens type I and III. Consistently, breast, pancreas, lung and colon cancer aggression are associated with levels and extent of dense, linearized and cross-linked ECM in the tissue [21,22,27,28,39,40]. Collagen cross-linking is a multi-step process initiated by LHs, which catalyze lysine (Lys) hydroxylation (Hyl). LOX and LOX-L family members then catalyze the oxidative deamination Lys and Hyl to generate reactive aldehydes (Lysald and Hylald, respectively) that in turn form spontaneous cross-links with opposing Lys or Hyl residues [41,42]. In healthy soft connective tissues such as skin, Lysald-derived collagen cross-links (LCCs) are abundant, while Hylald-derived cross-links (HLCCs) are abundant in load-bearing tissues such as bone [21]. In tumors LH2 may stabilize, organize and stiffen the collagen matrix by switching collagen cross-links from LCCs to HLCCs [21,43]. LH2 is upregulated in breast and lung cancers, and elevated levels correlate with ECM stiffness, tumor aggression and reduced survival [21,44]. These cross-links prevent the sliding of fibers relative to one another when the ECM is subject to external load, leading to changes in the plasticity (irreversible deformations) of the matrix. Collagen cross-linking also increases the resistance of the ECM to applied force, which manifests as an increased elastic modulus of the ECM [45]. Moreover, while healthy tissues demonstrate a balance between ECM synthesis and enzymatic degradation, in a highly cross-linked diseased tissue, this balance is tipped in favor of ECM synthesis primarily due to the inability of MMPs to digest collagen [46].
2.3. Mechanosensing and mechanotransduction
Cells within a tissue are constantly subjected to force (compression, tensile and sheer stress; Box 1). All cells actively sense and respond to these forces via a process termed mechanotransduction. Cells sense externally applied force through conformational changes in transmembrane proteins such as stretch-activated ion channels, cadherins and integrins (mechanosensing). Mechanical cues are then translated into biochemical signals within the cell to modify cell behavior (mechanotransduction). Thus, when cellular integrins experience shear flow their ectodomain undergoes a conformational change that induces a high-affinity state that fosters ligand binding (integrin activation) (Fig. 3). Force can also influence the lifetime of ECM-integrin adhesions as is illustrated by integrin αIIbβ3 which exhibits a slip-bond behavior in which the ligand bond lifetime is shortened, or with integrin α5β1 which demonstrates a catch-bond behavior that prolongs the bond lifetime [47,48]. Force can also reinforce integrin adhesions by promoting the unfolding of protein domains in ECM ligands including fibronectin fibrils where force exposes cryptic bindings sites that promote α5β1 integrin engagement (mechanical reinforcement; [49]), that promotes fibronectin assembly [50].
Box 1. Biomechanical concepts in tumor biology.
Stress:
describes the internal resistance produced when an object is deformed by an external force, measured as force per unit area and expressed in pascals (Pa), where 1 Pa = 1 N/m2. There are three types of stress: tensile (pulling) and compression stress (pushing) are due to a force applied perpendicular to the object surface; shear stress results from a force that acts parallel to the object surface.
Strain:
describes the deformation of an object relative to its original length, measured in percent.
Stiffness:
describes the elasticity of a material or the property of restoration to its original shape after deformation, measured in pascals (Pa). Stiffness is related to elasticity however; stiffness may change as the force increases and is not a characteristic property of the object.
Elasticity:
describes the ability of an object to return to its original shape after removal of a force. Elasticity is described by the modulus of elasticity, which is defined as the ratio of stress to strain. Youngs modulus (E) describes the elasticity of a material subjected to tensile or compression loading. Shear modulus (G) describes the shear elasticity of a material subjected to shear loading.
Viscoelasticity:
is the property of materials that exhibit both elastic and viscous properties when undergoing deformation. The strain of viscous materials is time-dependent, whereas elastic materials is time-independent.
Mechanoreciprocity:
describes the bi-directional mechanical interaction between a cell’s response to external force by reciprocally exerting force.
Fig. 3.
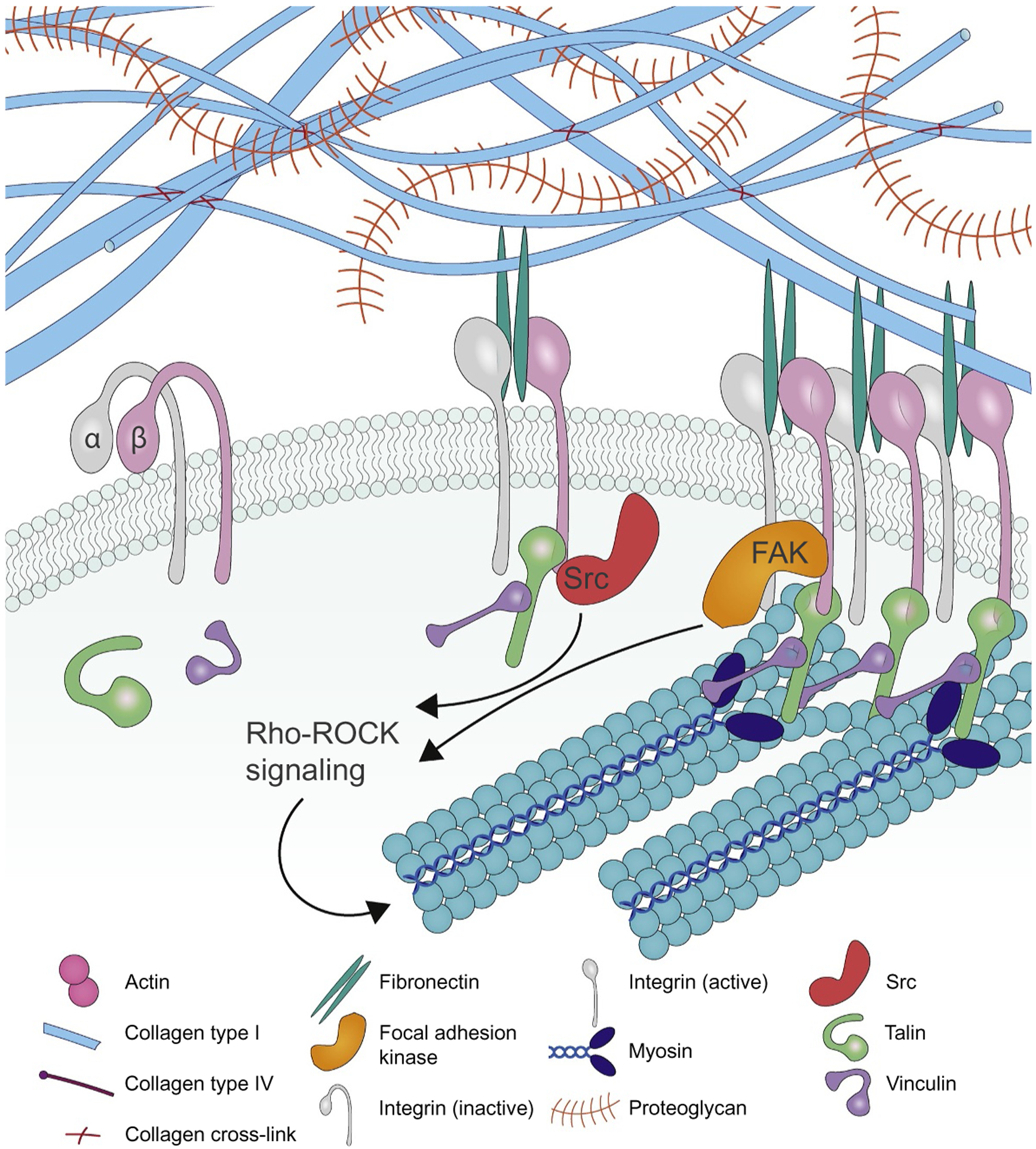
Integrin-dependent mechanotransduction. Integrins exist in a resting, inactive state and can be activated by internal (inside-out activation) or external (outside-in activation) cues including extracellular force (outside-in) or actomyosin contractility or tension (inside-out). Integrin activation is mediated by conformational changes in the integrin ectodomain that shifts the integrin from a low- to a high-affinity ligand binding state. A sufficient force upon integrin engagement will trigger the force-dependent unfolding of talin to expose vinculin binding sites. Vinculin binding to talin promotes its unfolding and recruits a suite of adhesion plaque proteins including Src, paxillin, α-actinin, and FAK that trigger downstream signaling and initiate actin reorganization and RhoGTPase-mediated actomyosin contractility to drive focal adhesion maturation.
Cells translate force by activating intracellular signaling pathways. For example, force applied to ECM-integrin adhesions promotes the formation of focal adhesions that recruit adhesion plaque proteins to trigger signaling cascades and cytoskeletal reorganization. Many of the proteins within focal adhesions also undergo force-induced conformational changes. Force exposes cryptic binding sites in talin and vinculin. Talin directly interacts with the cytoplasmic domain of the integrin beta chain and tension-stabilized interactions between talin and vinculin acts as a molecular clutch to bind the actin cytoskeleton [51]. The slow loading rates induced in cells interacting with a soft ECM fail to induce talin unfolding and vinculin recruitment before the slip-bond between the integrin and its ligand ruptures. By contrast, in cells interacting with a stiff ECM the high loading rate induces vinculin-dependent clutch reinforcement and facilitates catch-bond formation. Consequently, both catch-bond–dependent adhesion strengthening, and vinculin-dependent clutch reinforcement are essential for triggering downstream signaling by integrins including the activation of FAK and the nuclear translocation of the transcription factor YAP [48,52]. Force can also alter the conformation of intracellular signaling molecules such as the tension induced exposure of tyrosine motifs in p130Cas (also known as BCAR1) that are phosphorylated by Src kinases and that serve as a docking hub for signaling molecules [53,54]. Force can additionally remodel protein-protein interactions in focal adhesions, as was demonstrated for adhesion-associated LIM domain-containing proteins [55]. Force can also promote integrin clustering to drive focal adhesion assembly. Theoretical analysis suggests that lateral cross-linking of adjacent integrins by adaptor proteins allows the redistribution of the tensile load between adjacent bonds within the cluster, increases integrin rebinding rates, and extends the duration of mechanotransduction [56]. The functional significance of integrin clustering has been demonstrated in a tumor model, where enhanced β1 integrin clustering induced by the V737N point mutation in the β1 integrin transmembrane domain, but not constitutive β1 integrin activation induced by G429N point mutation in the β1 integrin ectodomain, can bypass the requirement of ECM stiffness for inducing FAK activation and malignant phenotypes in tumor cells cultured on soft substrates [57,58]. Integrin clustering may be further modified by regulation of cell surface glycans on the protein and lipid backbones that form the glycocalyx [59]. A bulky glycocalyx and in particular rigid glycoproteins such as mucin 1, create a kinetic trap that both applies tension on the integrin to activate it (outside-in activation) but also promotes integrin clustering and focal adhesion assembly [60,61].
Cells then modify the composition, organization and elasticity of their microenvironment, and reciprocally adjust their behavior in response to this tensile resistance (mechanoreciprocity). ECM tensile resistance increases actomyosin-mediated tension and induces cytoskeletal remodeling that promote ECM remodeling and stiffening. For example, activation of Rho-associated protein kinase (ROCK) phosphorylates myosin light chain (MLC) to enable cells to pull on and remodel the ECM through actomyosin-mediated contractility [62,63]. ROCK can also inhibit the phosphatase that acts on MLC, further enhancing its phosphorylation and activity. Force can also trigger sustained cellular responses by altering gene expression. For instance, force-mediated biochemical signaling can induce the expression of ECM proteins (fibronectin and collagen) and ECM-modifying proteins (MMPs and LOX) which remodel and stiffen the surrounding microenvironment and reinforce mechanosignaling. In this manner, cells can alter the composition, organization and elasticity of their tissue microenvironment and alter their adhesions and cell shape and orientation to tune their behavior according to the magnitude, direction and nature of applied mechanical stress. Cells maintain a state of tensional homeostasis by adjusting to balance forces in order to maintain function and integrity within a heterogeneous tissue [64].
3. Cancer-associated fibroblasts in the tumor stroma
3.1. CAF function
Fibroblasts synthesize and remodel the interstitial matrix and are therefore aptly named the engineers of the ECM. Not surprisingly, fibroblasts are essential for tissue repair and homeostasis [65]. In wound healing, a variety of growth factors and cytokines stimulate fibroblast recruitment. These recruited fibroblasts deposit ECM proteins, which elevates mechanical stress in the wound, that in turn, induces the transdifferentiation of fibroblasts into myofibroblasts, through a process characterized by the de novo expression of α smooth muscle actin (αSMA) [66]. In response to transforming growth factor (TGF) β1, fibroblasts produce the EDA splice variant of fibronectin and this in turn enhances cellular tension through fibronexus adhesions [67,68]. The elevated cellular tension promotes the assembly of focal adhesions and the recruitment of αSMA to the actomyosin fibers [69]. Notably, while smooth muscle cell contraction is rapid and short in duration, αSMA-positive myofibroblasts contract the ECM over longer time periods, to permit permanent tissue contraction [70]. This chronic tissue contraction is mediated by both calcium-calmodulin-MLC kinase-dependent contraction and Rho-ROCK-myosin light chain phosphatase-mediated contraction [71]. Chronic tissue contraction also stiffens the ECM, and via EDA-fibronectin, promotes the tension-induced release of TGFβ1 from the latent complex thereby activating it and further amplifying fibroblast activation through a feedforward reinforcement circuit. Actomyosin-mediated contraction in fibroblasts also activates YAP/TAZ and MRTF and enhances ECM remodeling by transcriptionally increasing ECM protein expression, thereby linking physical stress with gene transcription in myofibroblasts [72–74]. In a healthy tissue, wound resolution either induces myofibroblast apoptosis or reverts their phenotype towards a quiescent fibroblastic state [75–77]. In this manner mechanical and biochemical signaling control myofibroblast differentiation and phenotype.
Fibroblasts within the TME are collectively termed CAFs, and in many solid tumor types, correlate with tumor aggression and reduced survival [78–80]. Similar to myofibroblasts, CAFs were originally identified as αSMA-positive fibroblast-like cells [81,82]. However, CAFs are fundamentally different from normal, and even wound-associated fibroblasts. Key distinctions include the fact that fibroblasts are in a resting, non-proliferative and low metabolic state. Wound-associated fibroblasts and CAFs alike, are characterized by loss of certain fibroblast markers (fibroblast activation protein; FAP) and acquisition of muscle-like markers (αSMA), as well as increased ECM synthesis and remodeling, and a contractile phenotype. The distinction between wound-associated fibroblasts and CAFs is the sensitivity of fibroblasts to undergo apoptosis, and/or dedifferentiate to a resting state following resolution of the wound. Moreover, CAFs, and not normal fibroblasts, promote tumor cell proliferation and migration [83–86]. Through direct contact, CAFs can also pull and lead tumor cells away from the primary tumor, and thereby support metastasis [87,88]. Like normal fibroblasts, CAFs are recruited to the tumor stroma by growth factors secreted by tumor cells and they are likely converted to myofibroblasts by similar signals to those found in wounds. Initially, CAFs are likely recruited to the tumor to repair the injured tissue where they may initially restrain tumor cell invasion. However, as the tumor evolves the CAFs continue to deposit ECM proteins, secrete growth factors and contract and remodel the ECM. As a consequence, CAFs re-organize and cross-link collagen to induce stiff and oriented collagen fibers along which tumor cells can migrate [89]. Thereafter, the MMPs secreted and activated by the tumor cells and CAFs facilitate BM degradation and promote tumor cell migration to foster malignant transformation and tumor cell dissemination. In this manner, a mechanical feedback loop exists between CAF activation status and ECM stiffening [15,90].
CAFs reside in almost all solid tumors; however, their contribution to the stromal population varies between different types of cancers. For example, breast and pancreatic cancers display high CAF density, whereas brain, kidney and ovarian cancers display lower CAF density [91]. While CAF density has been associated with poor prognosis, the CAF population remains poorly defined in terms of origin, subtype and biology in part due to their extreme phenotypic heterogeneity coupled with the lack of specific, definitive markers.
3.2. CAF heterogeneity
Historically, CAFs were identified by αSMA expression, and studies underestimated the complexity of CAF heterogeneity, with adoption of the misconception that CAFs represent a homogenous and static population of stromal cells [82]. It is now appreciated that not all CAFs express the classical marker αSMA. Recent advances in studies of CAF heterogeneity indicate multiple CAF subtypes coexist in the TME, each influencing the tumor in a unique manner with tumor-promoting or tumor-restrictive roles [79,92]. Currently, there is no consensus to the molecular definition of CAFs [93]. In principle, determining specific pro-tumorigenic CAF subtypes would provide direction for the development of CAF-targeted anti-cancer therapies. Studies in breast cancer and PDAC have since compared the expression profiles of multiple molecular markers associated with CAFs and demonstrated functionally distinct CAF subsets [79,94–96]. In breast cancer, CAF subtypes have been associated with certain tumor subtypes. For example, CAFs with low expression of β1 integrin, αSMA, FAP, and PDGFRβ associate primarily with luminal subtypes (less aggressive), whereas CAFs positive for these CAF-associated markers were predominantly found in human epidermal growth factor receptor 2 (HER2) and triple negative breast cancers (TNBC) (more aggressive) [79]. Moreover, TNBC-associated CAFs demonstrate immune-suppressive functions by recruiting and activating FoxP3+ regulatory T-cells (Treg), whereas HER2-associated CAFs do not. Although these CAF subsets did not correlate with patient survival, the authors linked CAF subsets to histological grade and tumors from patients that received chemotherapy treatment. Similarly, two CAF subtypes were identified in PDAC as CD10-negative and CD10-positive, with the latter having a more pro-tumorigenic role. This finding was supported in both breast and lung cancers, with CD10-positive CAFs (CD10+ GPR77+) promoting tumorigenesis and chemoresistance [97]. In PDAC, one study identified two distinct CAF subsets; αSMAHighFAP+ myofibroblastic CAFs (MyCAF), which were responsive to TGFβ1, and αSMALow inflammatory CAFs (iCAF), which secreted inflammatory mediators such as IL-6 [95]. Moreover, MyCAFs were located adjacent to the tumor, while iCAFs were located in the dense stroma. These studies allude to the notion that tumors influence CAF heterogeneity. Such that, PDAC-derived IL-1 pushes CAFs towards an iCAF phenotype, while TGFβ1 pushes towards a MyCAF phenotype. Moreover, TGFβ1 can push iCAFs towards a MyCAF phenotype by antagonism of the pro-inflammatory IL-1/JAK/STAT pathway [92]. These studies suggest that fibroblasts can adopt multiple cell states, ranging from a pro-inflammatory to an anti-inflammatory, myofibroblast-like phenotype [92,95]. Together, these reports indicate diversity of CAFs, in regard to marker expression, spatial distribution and tumor subtype. The resolution of CAF heterogeneity is an essential to enable the development of CAF-based therapeutic strategies.
3.3. CAF immune cell interactions
Chronic inflammation has been implicated in malignant transformation and metastasis. The infiltrating macrophages in a chronically inflamed tissue secrete factors such as TGFβ that stimulate the resident stromal fibroblasts to synthesize and secrete ECM proteins, MMPs and increase the levels and activity of collagen cross-linking enzymes that remodel and stiffen the tissue stroma [22,98,99]. Indeed, early during cancer development immune cell-derived IL-1β activates nuclear factor-κB (NF-κB) signaling in CAFs to instruct their production of a tumor-promoting inflammatory response [100]. Accordingly, over time chronic inflammation will induce tissue fibrosis. Whether chronic inflammation increases risk to malignancy and promotes malignancy and tumor aggression by inducing tissue fibrosis remains an open question. What is clear is that the stromal CAF secretome can modify tumor immunity by influencing innate immune cell recruitment and activation and polarizing the adoptive immune response towards a pro-tumor phenotype. CAF secreted IL-6 recruits tumor-associated macrophages and promotes their transition to an immunosuppressive phenotype (M2). Moreover, the expression of CAFs and M2 markers is associated with poor clinical outcome in colorectal cancer and oral squamous cell carcinoma patients [101–103]. CAFs also recruit and induce differentiation of Tregs to repress anti-tumor immunity [104]. The secretome of CAFs can also enhance the tumor promoting function of CD4+ Helper T (Th) lymphocytes, favoring the tumor-promoting Th2 and Th17 phenotype over the tumor protective Th1 response. Indeed, CAFs can directly reduce the activation of CD8+ cytotoxic T cells and natural killer (NK) cells by expressing inhibitory immune checkpoint signals, programmed death-ligand (PD-L)1 and PDL-2 [105], or they can secrete immune suppressive factors, such as prostaglandin E2 (PGE2) [106]. In this way, CAFs have the capacity to recruit and stimulate immunosuppressive cells as well as inhibit various immune effector cells.
The dense, stiffened fibrotic ECM induced through CAF activity can also physically prevent immune cells from efficiently infiltrating the tumor to limit T cell contact with cancer cells [107]. Indeed, immune exclusion has been linked to a lack of anti-PDL1 therapy response, particularly in some tumors with a gene signature that reflects high TGFβ1 signaling in the tissue CAFs [108]. Consistently, co-treating mouse breast and colorectal tumors with inhibitors against TGFβ and PD-L1 reduced CAF expression of ECM-modifying enzymes to enable T cell penetration which reduced the primary and metastatic tumor burden presumably by overcoming their immune exclusion phenotype [109]. A note of caution is needed however, since ECM remodeling can release growth factors and cytokines that promote tumor cell growth and recruit immune suppressive myeloid cells, as well as unmask cryptic binding sites in the ECM that could deleteriously alter immune cell interactions, and may also promote the unrestrained dissemination and expansion of tumor cells that express high levels of immune suppressive glycoproteins such as sialic acid that can severely compromise checkpoint inhibitor response [110].
3.4. CAF phenotype regulation
The biophysical and biochemical properties of the tumor ECM are largely dictated by the subtype and phenotype of the resident CAFs, and their location with the tissue [63]. CAF heterogeneity has been highlighted by deep-sequencing studies conducted using human tumor biopsies [88,111,112]. Functional studies have revealed the relevance of this heterogeneity by showing how some CAFs regulate ECM synthesis, remodeling and stiffening, whereas others primarily function as immune cell modulators [96]. Indeed, recent data suggest that distinct CAF subtypes are located within specific regions of the tumor and indicate that each cancer type may harbor different CAF subtypes [96,113]. This division of CAF location, subtype and function can have profound implications on tumor evolution. For example, in lung fibrosis, fibroblasts are primarily activated by synergistic interactions between tissue tension and TGFβ, and these “activated fibroblasts” produce and remodel the ECM to progressively stiffen the stroma adjacent to the CAFs that compromises alveolar function [72,114]. Clearly, elucidating the spatio-mechanical heterogeneity of CAFs and their surrounding ECM will clarify how specific CAF subsets arise, whether and how they switch from one phenotype to another, and which subsets needed to be targeted to achieve the best therapeutic outcome, while minimizing detrimental effects of broadly targeting all CAFs.
4. Mechanoreciprocity: push me, pull you
In all tissues, the dynamic and reciprocal interplay between cellular contractility and ECM stiffness is key for the maintenance of tissue homeostasis. In tumors, a stiffened ECM and elevated tumor cell contractility due to increased oncogene activity [115] severely disrupts tensional homeostasis [7]. CAFs synthesize, remodel and cross-link the ECM to increase stiffness, and ECM stiffness is feedback to further stiffen, remodel and reorient the ECM [85]. A stiff ECM promotes focal adhesion assembly and enhances cytoskeletal tension to increase growth factor receptor signaling-dependent activation of ERK and PI3K in tumor cells. In the transformed epithelium in particular, the increased cellular tension stimulates cell growth, disrupts cell-cell adhesions, compromises tissue polarity and promotes tumor cell invasion into the stroma [57,58,116]. The stiffened ECM also promotes the release of the transcription factor Twist1 from its cytoplasmic binding partner G3BP2, so that its nuclear translocation can activate genes that promote an epithelial to mesenchymal transition (EMT), which is a tumor state linked to tumor aggression and metastasis [117]. Interestingly, in addition to their effects on the ECM and secretion of soluble factors, CAFs can also modulate tumor cell behavior through direct cell-cell contact. Thus, CAFs can exert tensile force on tumor cells through heterophilic E-cadherin/N-cadherin junctions that recruit the actin associated molecules α-actinin, vinculin, nectin-1 and −2, and afadin to prevent polarity reversal and promote tumor cell detachment to drive their invasion [88,118].
As tumor cells in solid cancers such as the breast and pancreatic proliferate, their increased volume imposes tensile forces on the surrounding fibrotic ECM which in turn constrains the tumor mass [97,119]. The resultant stored stress deforms compliant structures within the tumor including blood and lymphatic vessels, and this deformation compromises vessel structure and function to impair fluid flow that induces hypoxia and prevents lymphatic drainage. This reciprocal interaction between a proliferating and growing tumor and the tense surrounding ECM also creates a feedback loop where surrounding stromal cells are continuously stimulated to further potentiate tumor progression [68]. This growth-induced mechanical tissue stress can also activate the Ret-β/catenin pathway in the tumor cells themselves to potentiate tumor cell growth and invasion and when/if chronic can induce an EMT that can promote metastasis [120].
A stiffness gradient in the ECM, such as that at the invasive front of tumors, promotes the migratory behavior of single cells up the stiffness gradient through a process termed durotaxis, and this phenotype has been implicated in tumor cell dissemination and metastasis [121,122]. Indeed, an ECM stiffness gradient may also foster the migration of tumor aggregates, through a process mediated by the collective contraction of the actomyosin cytoskeleton and retrograde flow of polymerizing actin in the cell cluster [123]. These migrating tumor clusters preferably migrate along stiff EDA-fibronectin fibers using integrin α9β1 [124]. In this regard, CAFs can tunnel through the ECM to create collagen, fibronectin and tenascin C enriched tracks along which tumor cells can migrate; the relevance of which was clinically validated in head and neck squamous cell carcinoma specimens [87]. Indeed, CAFs can deposit and create fibronectin fiber tracks on their surface along which tumor cells can migrate using α5β1 integrins [125]. CAFs can also exert actomyosin-dependent forces on the BM to generate MMP-mediated thinning and stromal heterogeneity previously implicated in tumor progression [126]. Tumor cells and presumably CAFs can also strain-stiffen the ECM through increased actomyosin contraction and cytoskeletal tension [127]. This strain-stiffened ECM can thereafter create stress fields that can be sensed hundreds of microns away where it can distally stimulate single cell and collective cell migration to foster tumor cell dissemination [38].
Many oncogenes implicated in cancer such as mutant Ras stimulate Rho GTPases and ROCK to increase actomyosin contractility in the transformed cells. Increased tumor cell tension can activate integrins via inside-out signaling and stimulate the remodeling of the tumor adjacent stroma. The importance of this phenotype was illustrated by studies in Ras-induced squamous cell carcinoma, whereby activated Ras stimulated ROCK activity in the tumor cells to increase their actomyosin contractility. The high contractility of the Ras pre-transformed cells induced the remodeling, cross-linking and stiffening of the tumor adjacent stroma. The stiffened tumor adjacent ECM and increased tumor cell contractility activated the tumor cell integrins which in turn promoted β-catenin activation and drove tumor cell growth and ultimately malignant transformation [115]. Tumor cells can also synthesize ECM components that are distinct from those generated by CAFs and these ECM proteins have been implicated in tumor cell metastasis [25,128]. Critically, over time and in response to chronic stiffness some cancer cells soften, and the degree of their softening correlates with their metastatic potential [129–131]. The stiffness of a cell depends primarily on the mechanical properties of the nucleus and cytoskeleton with some contribution from the cellular organelles [132].
5. Mechanoreciprocity in solid tumors
Mammographic density (MD) associates with an overall increase in lifetime risk for malignancy [133,134]. MD not only reflects a higher epithelial density in the tissue but detects the increased levels of fibrillar collagen that stiffens the breast tissue [135]. Stromal stiffness also associates with breast cancer aggression, in which the more aggressive subtypes (HER2 and TNBC) contain more linearized collagen and have a stiffer stroma than the less aggressive subtypes (luminal A and luminal B) [22]. The abundance of fibrillar collagen in a primary breast tumor is a significant risk factor for patient survival [136]. Tumor collagen abundance also associates with distant metastasis in TNBC [137], and was shown to promote tumor aggression and metastasis in experimental mouse models [138] (Fig. 4).
Fig. 4.
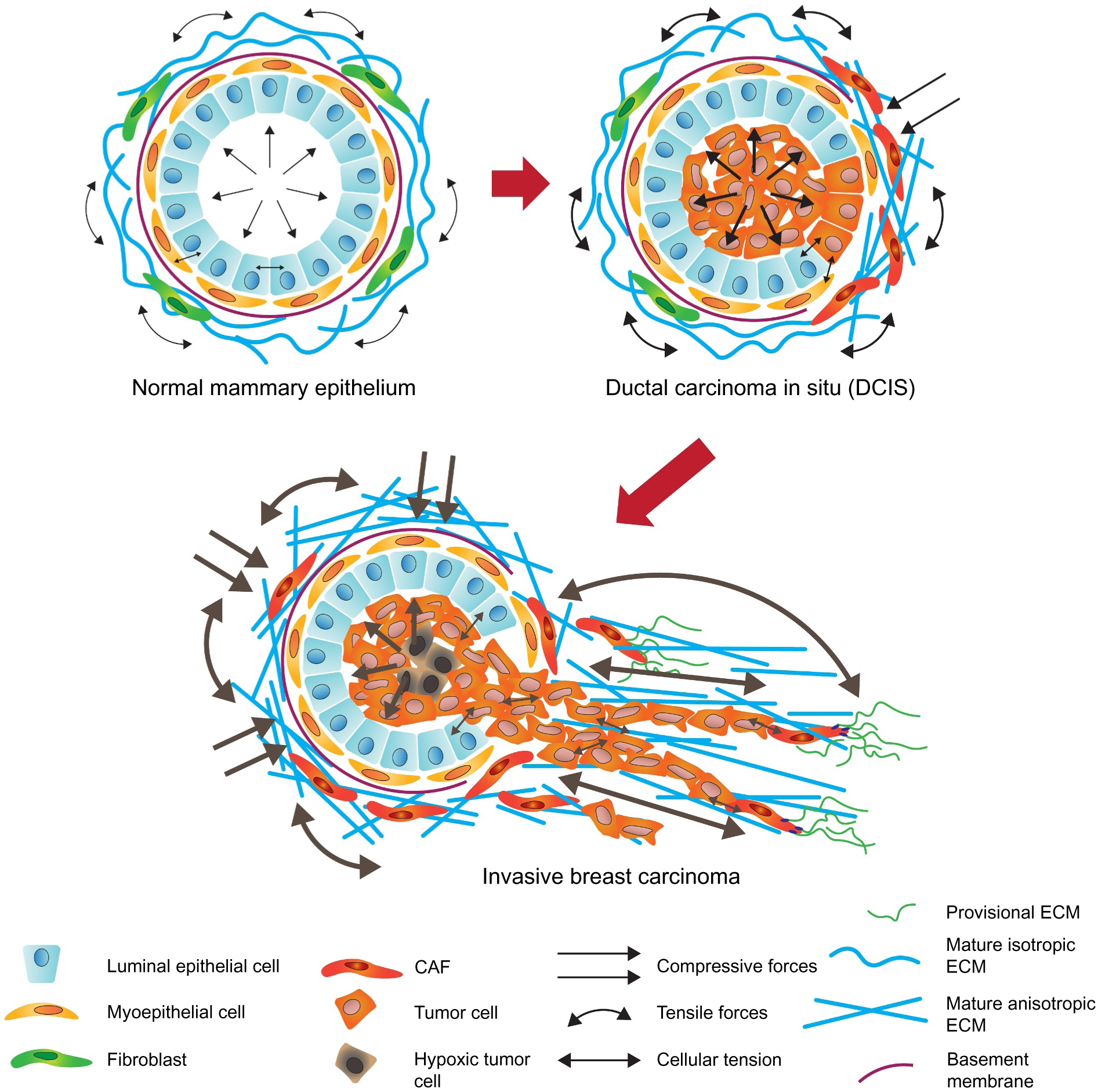
Tumor-associated mechanoreciprocity. A simplified schematic of mechanoreciprocity in breast cancer evolution. Breast ducts are composed of two epithelial linings: inner luminal epithelial cells and outer myoepithelial cells. Breast ducts reside in an ECM populated by fibroblasts, endothelial cells, pericytes, leukocytes, and adipocytes. In a healthy breast, the tension exerted between the epithelium and stroma maintains tensional homeostasis. In ductal carcinoma in situ (DCIS), neoplastic epithelial cells proliferate and fill the lumen of the duct, thereby increasing solid stress. Neoplastic cells secrete factors that activate CAFs in the stroma to synthesize, remodel and stiffen the interstitial stroma, which mechanically resists the expansion of the DCIS lesion. The neoplastic epithelium in DCIS lesions responds to these forces by increasing their actomyosin tension that drives the assembly of focal adhesions to potentiate growth factor-dependent PI3K and ERK signaling and increases tumor cell contractility. Through the combined activity of the contractile tumor epithelium and activated CAFs, the BM surrounding DCIS lesions is compromised, and the collagenous-rich interstitial stroma becomes aligned and perpendicularly reorganized to support the invasion of the transformed breast epithelium into the interstitial stroma.
Transcriptome-wide analyses demonstrated that dramatic and consistent changes in gene expression occur within the breast cancer associated fibroblast and myoepithelial population, and that it is possible to derive a prognostic gene signature (26-gene) that can predict relapse-free survival in breast cancer patients [139–141]. Indeed, one ‘wound-healing’ gene signature, identified using microarray analysis of serum stimulated cultured fibroblasts predicted breast cancer patient survival [142], whereas another identified a predictive association between a stromal gene expression signature and resistance to neoadjuvant chemotherapy [143]. These data clearly implicate CAFs in breast cancer progression and imply the phenotype/genotype of the tumor stroma has potential predictive value.
PDAC which is an aggressive cancer with an overall 5-year survival rate between 6 and 7% [96]. PDAC is characterized by an intense fibrotic stroma, with high abundance of CAFs, immune cells and excessive ECM accumulation, that accounts for most of the tumors volume (50–80%) [96,144–146]. The dense and poorly vascularized stroma compromises the tumor vasculature to inhibit drug penetration and induce hypoxia which promotes therapeutic resistance and tumor aggression [31,147,148]. Not surprisingly, a major challenge in PDAC treatment is overcoming the profound fibrotic response.
In PDAC, tumor cells at both the primary site and metastatic tissues, secrete factors such as TGFβ1 that activate stromal fibroblasts and pancreatic stellate cells (PSCs) to stimulate the synthesis, deposition and cross-linking of the stromal ECM and this ability to activate the stroma is dictated by the tumor cell genotype [23,149–151]. In turn, changes in the ECM may also drive the early stages of tumor formation. Furthermore, pancreatic tumor cells themselves can produce ECM proteins including collagen type IV [152]. Importantly, PDAC fibrosis is most evident in the periductal regions, consistent with the idea that tumor cell tension and paracrine signaling are potent drivers of the unique fibrotic response found in this disease. Clearly PDAC progression and aggression hinge on the complex interplay between the genotype/phenotype of the tumor cells, the nature and abundance of the CAFs or PSCs in the tumor and their respective impact on the ECM and tissue tension. As such clarifying this tumor-stromal dynamic should help improve patient treatment.
6. Anti-fibrotic therapies in cancer treatment
6.1. Targeting the ECM and ECM modulators
The stiff, dense ECM is an attractive anti-tumor cancer target. Not surprisingly, strategies have been developed to target ECM deposition and collagen-modifying enzymes to reduce ECM stiffness. The pharmacological inhibitor of lysyl oxidase (LOX), BAPN and a LOX-specific function blocking antibody both prevented LOX-dependent collagen cross-linking and reduced tissue fibrosis to delay breast cancer progression and reduce malignant transformation in a transgenic mouse model of HER2-positive mammary cancer [58]. Although chronic use of BAPN is contraindicated for long term clinical use, a LOX function blocking antibody has been developed. However, initial clinical trial results have been disappointing and attributed to inefficient enzymatic inhibition and poor tumor penetration of the inhibitory antibodies. Importantly, epithelial LOX has other functions including anti-Ras activity of the pro-peptide that is released following LOX activation that would be prevented when LOX activity is inhibited and this could impede its anti-tumor effect [153]. LOX-L2 inhibitory antibodies including simtuzumab, have also been developed, but these inhibitors have also proven to be unsuccessful in early clinical trials [154–156], possibly because this enzyme is primarily expressed by the tumor epithelium which preclinical studies in knock-in and knock-out transgenic mouse models of mammary tumors clearly demonstrate has little to no effect on the tumor ECM [157]. CAFs also express lysyl hydroxylases that modify fibrillar collagens prior to their secretion. CAF LH2 depletion prevented collagen gel stiffening and prevented collagen-mediated tumor cell invasion and LH2-mediated collagen cross-links correlated with human breast tumor aggression and poor patient survival [43,158]. Although no specific small molecule inhibitors against LH2 other than Minoxidil exist [159], the development of 3D structure models and a high throughput assay for LH2 activity should accelerate the identification of anti-LH2 compounds [160,161].
A stiff, dense tumor ECM compresses intratumoral blood and lymphatic vessels to increase interstitial tissue pressure, induce hypoxia and impede anti-cancer drug treatment delivery [55]. Much effort has been expended to reduce ECM density and stiffness to ameliorate the high interstitial tissue pressure and permit efficient drug delivery to the tumor. One example is the enzymatic degradation of HA that preclinical PDAC transgenic models demonstrated very efficiently attenuated interstitial pressure to facilitate Gemcitabine penetration, decrease metastatic burden and improve mouse survival [162,163]. Unfortunately, a phase Ib trial evaluating the PEGPH20 (PEGylated recombinant human hyaluronidase) plus gemcitabine combination demonstrated modest improvements in overall survival and progression free survival in patients selected for high HA content [164]. Nevertheless, PEGPH20 plus nab-paclitaxel/gemcitabine combination-therapy improved PFS in patients with untreated metastatic PDAC, especially in patients with high HA levels [165]. In contrast, a large multicenter phase III study on the combination of PEGPH20 plus nab-paclitaxel/gemcitabine failed to improve the median OS (NCT02715804, [166]). Moreover, a phase I/IIb evaluation of PEGPH2 with FOLFIRINOX in metastatic pancreatic cancer reported detrimental outcomes due to higher PFS and OS in the control arm, and increased toxicity rates in the combination arm [167]. Despite potential adverse effects the addition of PEGPH2 treatment may increase treatment efficacy in patients with high-HA tumors. Consistently, PEGPH20 in combination with anti-PD-L1 antibody immune therapy increased sensitivity towards PD-L1, reduced therapy resistance and increased survival in mouse models of breast cancer [163]. The findings suggest that HA targeting may comprise an effective strategy to improve drug delivery and enhance cancer therapy.
Recent studies suggest that the ECM may constitute a viable targeting strategy. For example, the collagen-binding properties of the protein lumican tethered to the cytokines IL-2 and IL-12 effectively increased cytokine retention and provided long-term therapeutic effects in combination with simultaneous dual checkpoint blockade using anti-PD-L1 treatment [168]. Clearly, targeting the ECM or its post translation modification or using it for a targeting vector are viable strategies to treat tumors.
6.2. Targeting mechanosensing and transduction
Mechano-sensing and -signaling through focal adhesions is increased in fibrotic tumors and fuels tumor cell proliferation, survival, migration and invasion. Nevertheless, and despite encouraging pre-clinical study results using α5β1and αvβ3 integrin function blocking antibodies and cyclic peptides or peptidomimetics, clinical trials failed to demonstrate significant therapeutic efficacy [169]. The search for effective anti-integrin function blocking antibodies continue with a recent focus on integrin α11, whose expression is high in stromal fibroblasts [170], and synergizes with PDGFβ to induce CAF-dependent breast cancer cell invasion [171].
Better success has been achieved by targeting integrin-dependent signaling. The dual focal adhesion kinase 1–2 (FAK1-FAK2) inhibitor VS-4718 not only repressed integrin-dependent mechanosignaling but also effectively decreased fibrosis in a transgenic mouse model of PDAC [23], and normalized tumor immunity in syngeneic PDACs and rendered them responsive to chemo- and immune-therapy [172]. Similarly, another FAK inhibitor Defactinib, effectively reduced the frequency of cancer stem cells in a pre-clinical breast cancer model [173]. Defactinib is current being investigated in a phase II clinical trial as combination therapy with anti-PD-1 immunotherapy and gemcitabine in patients with advanced pancreatic cancer, and the first reports are expected in 2020 [NCT02546531]. Notwithstanding these encouraging results, FAK inhibition can induce tumor-intrinsic STAT3 hyperactivation that promotes disease progression [174] but that could be ameliorated using combinatorial FAK and JAK/STAT3 inhibitors. Indeed, analogous to the complexity and resistance that routinely arises in tumor-targeted therapies, anti-stromal therapies will also likely induce resistance that can be addressed using combinatorial therapies.
6.3. Targeting CAFs
CAFs comprise an attractive anti-tumor therapeutic target. CAF depletion using CD8+ T cell-mediated killing of FAP+ CAFs suppressed primary breast and colon tumor growth and metastasis in experimental models [175]. Similarly, adoptive transfer of FAP-specific chimeric antigen receptor (CAR) T cells, restrained the growth of desmoplastic human lung cancer xenografts and syngeneic murine pancreatic cancers [176]. However, and unfortunately, FAP is not exclusively found in CAFs, and its expression by multipotent bone marrow stem cells and skeletal muscle means that therapies targeting this protein can induce unwanted and potentially deleterious effects including cachexia and lethal bone toxicity [177]. One approach that could be used to avoid the toxicity associated with targeting conserved CAF proteins is the targeting of specific CAF subsets particularly if they are implicated in tumor phenotypes such as chemoresistance as was illustrated using a GPR77-neutralizing antibody to target CD + GPR77+ CAFs to reduce tumor stemness and enhance chemosensitivity in breast and lung cancer patient PDX xenografts [178].
The phenotype of tumor associated fibroblasts or myofibroblasts is quite plastic and they can spontaneously revert to an inactive state as has been observed following liver fibrosis regression [77]. This concept has prompted the development of therapeutic strategies to shift the CAF phenotype from tumor-promoting to tumor-suppressive as was demonstrated by replenishing the depleted retinoic acid stores in activated (PSCs) by administering all-trans retinoic acid (ATRA) to inactive them [179]. Similarly, treatment of PSCs with calcipotriol, the active component of vitamin D, suppressed pancreatitis, reduced PDAC growth and enhanced therapeutic efficacy by reprogramming the PSCs towards a quiescent state [180].
Strategies have also been developed to either deplete CAFs or prevent their induction. For instance, nanoparticles loaded with TRAIL can block TGFβ-mediated fibroblast differentiation into CAFs [181] and metronomic chemotherapy can limit CAF induction in tumors by decreasing chemokine expression [182].
7. Conclusions and future perspectives
There is increasing recognition that the density, organization and tensile properties of the ECM play an important role in tumor pathogenesis. ECM stiffness and tumor cell contractility can potentiate tumor cell growth and survival to drive malignant transformation, promote invasion and facilitate metastasis. Nevertheless, it is still unclear whether there exist cancer-specific ECM mechanophenotypes, and if so, what dictates these differences and their physiological relevance for tumor behavior. Are there qualitative differences in ECM composition, architecture and collagen cross-linking that distinguish chronic fibrosis from tumor fibrosis and if so, what regulates these differences, and do they contribute to cancer initiation? Do tissue specific differences in CAF subsets exist, and do they derive from distinct lineages and what role do they play? Are there distinct immune suppressive CAFs and is it possible to reprogram these CAFs to promote anti-tumor immunity and could this be used to prevent malignant transformation and/or metastasis? Clearly constructing a comprehensive atlas of ECM organization, mechanical phenotype and CAF subtype, function and origin will provide critical information with which to clarify the role of the ECM in malignancy help develop stromal-specific anti-cancer treatments.
Acknowledgements
The authors apologize to all colleagues whose work could not be cited due to space constraints. This work was supported by NIH/NCI grants U01CA202241, R01CA192914, R01CA222508, and R01CA174929 (V.M.W.).
Abbreviations:
- BM
basement membrane
- CAF
cancer-associated fibroblast
- DCIS
ductal carcinoma in situ
- ECM
extracellular matrix
- EMT
epithelial-to-mesenchymal transition
- FACIT
fibril associated collagens with interrupted triple helices
- FAP
fibroblast activation protein
- FAK
focal adhesion kinase
- GAG
glycosaminoglycan
- HA
hyaluronic acid
- HER2
human epidermal growth factor receptor 2
- HLCC
Hylald-derived cross-links
- ICAF
inflammatory CAF
- LCC
Lysald-derived collagen cross-links
- LH2
lysyl hydroxylase 2
- LOX
lysyl oxidase
- MEC
mammary epithelial cell
- MLC
myosin light chain
- MMP
matrix metalloproteinase
- MYCAF
Myofibroblastic CAF
- PDAC
pancreatic ductal carcinoma
- ROCK
rho-associated protein kinase
- SLRP
small leucine-rich repeat proteoglycan
- SMA
smooth muscle actin
- TACS
tumor-associated collagen signature
- TGF
transforming growth factor
- TME
tumor microenvironment
- TNBC
triple negative breast cancer
Footnotes
Declarations of Competing Interest
None.
References
- [1].Neglia JP, et al. , The risk of cancer among patients with cystic fibrosis. Cystic fibrosis and cancer study group, N. Engl. J. Med 332 (8) (1995) 494–499. [DOI] [PubMed] [Google Scholar]
- [2].Sørensen HT, et al. , The risk of a diagnosis of cancer after primary deep venous thrombosis or pulmonary embolism, N. Engl. J. Med 338 (17) (1998) 1169–1173. [DOI] [PubMed] [Google Scholar]
- [3].Raimondi S, et al. , Pancreatic cancer in chronic pancreatitis; aetiology, incidence, and early detection, Best Pract. Res. Clin. Gastroenterol 24 (3) (2010) 349–358. [DOI] [PubMed] [Google Scholar]
- [4].Bataller R, Brenner DA, Liver fibrosis, J. Clin. Invest 115 (2) (2005) 209–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Koay EJ, et al. , A visually apparent and quantifiable CT imaging feature identifies biophysical subtypes of pancreatic ductal adenocarcinoma, Clin. Cancer Res 24 (23) (2018) 5883–5894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Liu T, et al. , Noninvasive in-vivo quantification of mechanical heterogeneity of invasive breast carcinomas, PLoS One 10 (7) (2015) e0130258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Paszek MJ, Weaver VM, The tension mounts: mechanics meets morphogenesis and malignancy, J. Mammary Gland Biol. Neoplasia 9 (4) (2004) 325–342. [DOI] [PubMed] [Google Scholar]
- [8].Mouw JK, Ou G, Weaver VM, Extracellular matrix assembly: a multiscale de-construction, Nat. Rev. Mol. Cell. Biol 15 (12) (2014) 771–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Yamauchi M, et al. , The fibrotic tumor stroma, J. Clin. Invest 128 (1) (2018) 16–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Tharp KM, Weaver VM, Modeling tissue polarity in context, J. Mol. Biol 430 (19) (2018) 3613–3628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Martino MM, Hubbell JA, The 12th-14th type III repeats of fibronectin function as a highly promiscuous growth factor-binding domain, FASEB J. 24 (12) (2010) 4711–4721. [DOI] [PubMed] [Google Scholar]
- [12].Gjaltema RA, Bank RA, Molecular insights into prolyl and lysyl hydroxylation of fibrillar collagens in health and disease, Crit. Rev. Biochem. Mol. Biol 52 (1) (2017) 74–95. [DOI] [PubMed] [Google Scholar]
- [13].Sottile J, Hocking DC, Fibronectin polymerization regulates the composition and stability of extracellular matrix fibrils and cell-matrix adhesions, Mol. Biol. Cell 13 (10) (2002) 3546–3559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Kubow KE, et al. , Mechanical forces regulate the interactions of fibronectin and collagen I in extracellular matrix, Nat. Commun 6 (2015) 8026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Klotzsch E, et al. , Fibronectin forms the most extensible biological fibers displaying switchable force-exposed cryptic binding sites, Proc. Natl. Acad. Sci. U. S. A 106 (43) (2009) 18267–18272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Dvorak HF, Tumors: wounds that do not heal-redux, Cancer Immunol. Res 3 (1) (2015) 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Rybinski B, Franco-Barraza J, Cukierman E, The wound healing, chronic fibrosis, and cancer progression triad, Physiol. Genomics 46 (7) (2014) 223–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Guerra L, et al. , Stromal microenvironment in type VII collagen-deficient skin: the ground for squamous cell carcinoma development, Matrix Biol. 63 (2017) 1–10. [DOI] [PubMed] [Google Scholar]
- [19].Karampitsakos T, et al. , Lung cancer in patients with idiopathic pulmonary fibrosis, Pulm. Pharmacol. Ther 45 (2017) 1–10. [DOI] [PubMed] [Google Scholar]
- [20].Barker TH, Engler AJ, The provisional matrix: setting the stage for tissue repair outcomes, Matrix Biol. 60–61 (2017) 1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Chen Y, et al. , Lysyl hydroxylase 2 induces a collagen cross-link switch in tumor stroma, J. Clin. Invest 125 (3) (2015) 1147–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Acerbi I, et al. , Human breast cancer invasion and aggression correlates with ECM stiffening and immune cell infiltration, Integr. Biol. (Camb.) 7 (10) (2015) 1120–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Laklai H, et al. , Genotype tunes pancreatic ductal adenocarcinoma tissue tension to induce matricellular fibrosis and tumor progression, Nat. Med 22 (5) (2016) 497–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Naba A, et al. , The extracellular matrix: tools and insights for the “omics” era, Matrix Biol. 49 (2016) 10–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Naba A, et al. , Extracellular matrix signatures of human mammary carcinoma identify novel metastasis promoters, Elife 3 (2014) e01308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Moriggi M, et al. , ECM remodeling in breast cancer with different grade: contribution of 2D-DIGE proteomics, Proteomics 18 (24) (2018) e1800278. [DOI] [PubMed] [Google Scholar]
- [27].Provenzano PP, et al. , Collagen reorganization at the tumor-stromal interface facilitates local invasion, BMC Med. 4 (1) (2006) 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Conklin MW, et al. , Aligned collagen is a prognostic signature for survival in human breast carcinoma, Am. J. Pathol 178 (3) (2011) 1221–1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Beck AH, et al. , The fibromatosis signature defines a robust stromal response in breast carcinoma, Lab. Investig 88 (6) (2008) 591–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Brisson BK, et al. , Type III collagen directs stromal organization and limits metastasis in a murine model of breast cancer, Am. J. Pathol 185 (5) (2015) 1471–1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Drifka CR, et al. , Highly aligned stromal collagen is a negative prognostic factor following pancreatic ductal adenocarcinoma resection, Oncotarget 7 (46) (2016) 76197–76213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Storm C, et al. , Nonlinear elasticity in biological gels, Nature 435 (7039) (2005) 191–194. [DOI] [PubMed] [Google Scholar]
- [33].Wen Q, Janmey PA, Effects of non-linearity on cell-ECM interactions, Exp. Cell Res 319 (16) (2013) 2481–2489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Nam S, et al. , Strain-enhanced stress relaxation impacts nonlinear elasticity in collagen gels, Proc. Natl. Acad. Sci. U. S. A 113 (20) (2016) 5492–5497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Ma X, et al. , Fibers in the extracellular matrix enable long-range stress transmission between cells, Biophys. J 104 (7) (2013) 1410–1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Wang H, et al. , Long-range force transmission in fibrous matrices enabled by tension-driven alignment of fibers, Biophys. J 107 (11) (2014) 2592–2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Hall MS, et al. , Fibrous nonlinear elasticity enables positive mechanical feedback between cells and ECMs, Proc. Natl. Acad. Sci. U. S. A 113 (49) (2016) 14043–14048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Han YL, et al. , Cell contraction induces long-ranged stress stiffening in the extracellular matrix, Proc. Natl. Acad. Sci. U. S. A 115 (16) (2018) 4075–4080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Brauchle E, et al. , Biomechanical and biomolecular characterization of extracellular matrix structures in human colon carcinomas, Matrix Biol. 68–69 (2018) 180–193. [DOI] [PubMed] [Google Scholar]
- [40].Neesse A, et al. , Stromal biology and therapy in pancreatic cancer: a changing paradigm, Gut 64 (9) (2015) 1476–1484. [DOI] [PubMed] [Google Scholar]
- [41].Bailey AJ, Paul RG, Knott L, Mechanisms of maturation and ageing of collagen, Mech. Ageing Dev 106 (1–2) (1998) 1–56. [DOI] [PubMed] [Google Scholar]
- [42].Eyre DR, Weis MA, Wu JJ, Advances in collagen cross-link analysis, Methods 45 (1) (2008) 65–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Pankova D, et al. , Cancer-associated fibroblasts induce a collagen cross-link switch in tumor stroma, Mol. Cancer Res 14 (3) (2016) 287–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].van der Slot AJ, et al. , Identification of PLOD2 as telopeptide lysyl hydroxylase, an important enzyme in fibrosis, J. Biol. Chem 278 (42) (2003) 40967–40972. [DOI] [PubMed] [Google Scholar]
- [45].Malandrino A, et al. , Dynamic filopodial forces induce accumulation, damage, and plastic remodeling of 3D extracellular matrices, PLoS Comput. Biol 15 (4) (2019) e1006684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].van der Slot-Verhoeven AJ, et al. , The type of collagen cross-link determines the reversibility of experimental skin fibrosis, Biochim. Biophys. Acta 1740 (1) (2005) 60–67. [DOI] [PubMed] [Google Scholar]
- [47].Thomas W, Catch bonds in adhesion, Annu. Rev. Biomed. Eng 10 (2008) 39–57. [DOI] [PubMed] [Google Scholar]
- [48].Friedland JC, Lee MH, Boettiger D, Mechanically activated integrin switch controls alpha5beta1 function, Science 323 (5914) (2009) 642–644. [DOI] [PubMed] [Google Scholar]
- [49].Kong F, et al. , Demonstration of catch bonds between an integrin and its ligand, J. Cell Biol 185 (7) (2009) 1275–1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Vogel V, Sheetz MP, Cell fate regulation by coupling mechanical cycles to biochemical signaling pathways, Curr. Opin. Cell Biol 21 (1) (2009) 38–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].del Rio A, et al. , Stretching single Talin rod molecules activates vinculin binding, Science 323 (5914) (2009) 638–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Elosegui-Artola A, et al. , Mechanical regulation of a molecular clutch defines force transmission and transduction in response to matrix rigidity, Nat. Cell Biol 18 (5) (2016) 540–548. [DOI] [PubMed] [Google Scholar]
- [53].Tamada M, Sheetz MP, Sawada Y, Activation of a signaling cascade by cytoskeleton stretch, Dev. Cell 7 (5) (2004) 709–718. [DOI] [PubMed] [Google Scholar]
- [54].Sawada Y, et al. , Force sensing by mechanical extension of the Src family kinase substrate p130Cas, Cell 127 (5) (2006) 1015–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Schiller HB, Fässler R, Mechanosensitivity and compositional dynamics of cell-matrix adhesions, EMBO Rep. 14 (6) (2013) 509–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Schoen I, Beth P, Vogel V, The Yin-Yang of rigidity sensing: how forces and mechanical properties regulate the cellular response to materials, Annu. Rev. Mater. Res (2013) 589–618. [Google Scholar]
- [57].Paszek MJ, et al. , Tensional homeostasis and the malignant phenotype, Cancer Cell 8 (3) (2005) 241–254. [DOI] [PubMed] [Google Scholar]
- [58].Levental KR, et al. , Matrix crosslinking forces tumor progression by enhancing integrin signaling, Cell 139 (5) (2009) 891–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Buffone A, Weaver VM, Don’t sugarcoat it: how glycocalyx composition influences cancer progression, J. Cell Biol (2020) 219(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Paszek MJ, et al. , The cancer glycocalyx mechanically primes integrin-mediated growth and survival, Nature 511 (7509) (2014) 319–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Barnes JM, et al. , A tension-mediated glycocalyx-integrin feedback loop promotes mesenchymal-like glioblastoma, Nat. Cell Biol 20 (10) (2018) 1203–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Humphrey JD, Dufresne ER, Schwartz MA, Mechanotransduction and extracellular matrix homeostasis, Nat. Rev. Mol. Cell. Biol 15 (12) (2014) 802–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].DuFort CC, Paszek MJ, Weaver VM, Balancing forces: architectural control of mechanotransduction, Nat. Rev. Mol. Cell. Biol 12 (5) (2011) 308–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Yu H, Mouw JK, Weaver VM, Forcing form and function: biomechanical regulation of tumor evolution, Trends Cell Biol. 21 (1) (2011) 47–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Bonnans C, Chou J, Werb Z, Remodelling the extracellular matrix in development and disease, Nat. Rev. Mol. Cell. Biol 15 (12) (2014) 786–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Serini G, et al. , The fibronectin domain ED-A is crucial for myofibroblastic phenotype induction by transforming growth factor-beta1, J. Cell Biol 142 (3) (1998) 873–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Klingberg F, et al. , The fibronectin ED-A domain enhances recruitment of latent TGF-β-binding protein-1 to the fibroblast matrix, J. Cell Sci 131 (5) (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Kollmannsberger P, et al. , Tensile forces drive a reversible fibroblast-to-myofibroblast transition during tissue growth in engineered clefts, Sci. Adv 4 (1) (2018) (p. eaao4881). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Goffin JM, et al. , Focal adhesion size controls tension-dependent recruitment of alpha-smooth muscle actin to stress fibers, J. Cell Biol 172 (2) (2006) 259–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Castella LF, et al. , A new lock-step mechanism of matrix remodelling based on subcellular contractile events, J. Cell Sci 123 (Pt 10) (2010) 1751–1760. [DOI] [PubMed] [Google Scholar]
- [71].Tomasek JJ, et al. , Contraction of myofibroblasts in granulation tissue is dependent on rho/rho kinase/myosin light chain phosphatase activity, Wound Repair Regen. 14 (3) (2006) 313–320. [DOI] [PubMed] [Google Scholar]
- [72].Liu F, et al. , Mechanosignaling through YAP and TAZ drives fibroblast activation and fibrosis, Am. J. Physiol. Lung Cell. Mol. Physiol 308 (4) (2015) L344–L357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Piersma B, et al. , YAP1 is a driver of Myofibroblast differentiation in Normal and diseased fibroblasts, Am. J. Pathol 185 (12) (2015) 3326–3337. [DOI] [PubMed] [Google Scholar]
- [74].Foster CT, Gualdrini F, Treisman R, Mutual dependence of the MRTF-SRF and YAP-TEAD pathways in cancer-associated fibroblasts is indirect and mediated by cytoskeletal dynamics, Genes Dev. 31 (23–24) (2017) 2361–2375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Darby I, Skalli O, Gabbiani G, Alpha-smooth muscle actin is transiently expressed by myofibroblasts during experimental wound healing, Lab. Investig 63 (1) (1990) 21–29. [PubMed] [Google Scholar]
- [76].Desmoulière A, et al. , Apoptosis mediates the decrease in cellularity during the transition between granulation tissue and scar, Am. J. Pathol 146 (1) (1995) 56–66. [PMC free article] [PubMed] [Google Scholar]
- [77].Kisseleva T, et al. , Myofibroblasts revert to an inactive phenotype during regression of liver fibrosis, Proc. Natl. Acad. Sci. U. S. A 109 (24) (2012) 9448–9453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Jamin Y, et al. , Exploring the biomechanical properties of brain malignancies and their pathologic determinants in vivo with magnetic resonance elastography, Cancer Res. 75 (7) (2015) 1216–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Costa A, et al. , Fibroblast heterogeneity and immunosuppressive environment in human breast cancer, Cancer Cell 33 (3) (2018) (463–479.e10). [DOI] [PubMed] [Google Scholar]
- [80].Hu G, et al. , Activated tumor-infiltrating fibroblasts predict worse prognosis in breast cancer patients, J. Cancer 9 (20) (2018) 3736–3742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Lazard D, et al. , Expression of smooth muscle-specific proteins in myoepithelium and stromal myofibroblasts of normal and malignant human breast tissue, Proc. Natl. Acad. Sci. U. S. A 90 (3) (1993) 999–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Sappino AP, et al. , Smooth-muscle differentiation in stromal cells of malignant and non-malignant breast tissues, Int. J. Cancer 41 (5) (1988) 707–712. [DOI] [PubMed] [Google Scholar]
- [83].Attieh Y, et al. , Cancer-associated fibroblasts lead tumor invasion through integrin-β3-dependent fibronectin assembly, J. Cell Biol 216 (11) (2017) 3509–3520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Erdogan B, et al. , Cancer-associated fibroblasts promote directional cancer cell migration by aligning fibronectin, J. Cell Biol 216 (11) (2017) 3799–3816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Calvo F, et al. , Mechanotransduction and YAP-dependent matrix remodelling is required for the generation and maintenance of cancer-associated fibroblasts, Nat. Cell Biol 15 (6) (2013) 637–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Lee HO, et al. , FAP-overexpressing fibroblasts produce an extracellular matrix that enhances invasive velocity and directionality of pancreatic cancer cells, BMC Cancer 11 (2011) 245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Gaggioli C, et al. , Fibroblast-led collective invasion of carcinoma cells with differing roles for RhoGTPases in leading and following cells, Nat. Cell Biol 9 (12) (2007) 1392–1400. [DOI] [PubMed] [Google Scholar]
- [88].Labernadie A, et al. , A mechanically active heterotypic E-cadherin/N-cadherin adhesion enables fibroblasts to drive cancer cell invasion, Nat. Cell Biol 19 (3) (2017) 224–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Gilkes DM, et al. , Hypoxia-inducible factor 1 (HIF-1) promotes extracellular matrix remodeling under hypoxic conditions by inducing P4HA1, P4HA2, and PLOD2 expression in fibroblasts, J. Biol. Chem 288 (15) (2013) 10819–10829. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [90].Chandler EM, et al. , Adipose progenitor cells increase fibronectin matrix strain and unfolding in breast tumors, Phys. Biol 8 (1) (2011) 015008. [DOI] [PubMed] [Google Scholar]
- [91].Smith NR, et al. , Tumor stromal architecture can define the intrinsic tumor response to VEGF-targeted therapy, Clin. Cancer Res 19 (24) (2013) 6943–6956. [DOI] [PubMed] [Google Scholar]
- [92].Biffi G, et al. , IL1-induced JAK/STAT signaling is antagonized by TGFβ to shape CAF heterogeneity in pancreatic ductal adenocarcinoma, Cancer Discov. 9 (2) (2019) 282–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Kalluri R, The biology and function of fibroblasts in cancer, Nat. Rev. Cancer 16 (9) (2016) 582–598. [DOI] [PubMed] [Google Scholar]
- [94].Bartoschek M, et al. , Spatially and functionally distinct subclasses of breast cancer-associated fibroblasts revealed by single cell RNA sequencing, Nat. Commun 9 (1) (2018) 5150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Öhlund D, et al. , Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer, J. Exp. Med 214 (3) (2017) 579–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Neuzillet C, et al. , Inter- and intra-tumoural heterogeneity in cancer-associated fibroblasts of human pancreatic ductal adenocarcinoma, J. Pathol 248 (1) (2019) 51–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Nia HT, et al. , Solid stress and elastic energy as measures of tumour mechanopathology, Nat. Biomed. Eng 1 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Servais C, Erez N, From sentinel cells to inflammatory culprits: cancer-associated fibroblasts in tumour-related inflammation, J. Pathol 229 (2) (2013) 198–207. [DOI] [PubMed] [Google Scholar]
- [99].Cohen N, et al. , Fibroblasts drive an immunosuppressive and growth-promoting microenvironment in breast cancer via secretion of Chitinase 3-like 1, Oncogene 36 (31) (2017) 4457–4468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Erez N, et al. , Cancer-associated fibroblasts are activated in incipient neoplasia to orchestrate tumor-promoting inflammation in an NF-kappaB-dependent manner, Cancer Cell 17 (2) (2010) 135–147. [DOI] [PubMed] [Google Scholar]
- [101].Chomarat P, et al. , IL-6 switches the differentiation of monocytes from dendritic cells to macrophages, Nat. Immunol 1 (6) (2000) 510–514. [DOI] [PubMed] [Google Scholar]
- [102].Fujii N, et al. , Cancer-associated fibroblasts and CD163-positive macrophages in oral squamous cell carcinoma: their clinicopathological and prognostic significance, J. Oral Pathol. Med 41 (6) (2012) 444–451. [DOI] [PubMed] [Google Scholar]
- [103].Herrera M, et al. , Cancer-associated fibroblast and M2 macrophage markers together predict outcome in colorectal cancer patients, Cancer Sci. 104 (4) (2013) 437–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Chen W, et al. , Conversion of peripheral CD4+CD25− naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3, J. Exp. Med 198 (12) (2003) 1875–1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Nazareth MR, et al. , Characterization of human lung tumor-associated fibroblasts and their ability to modulate the activation of tumor-associated T cells, J. Immunol 178 (9) (2007) 5552–5562. [DOI] [PubMed] [Google Scholar]
- [106].Li T, et al. , Hepatocellular carcinoma-associated fibroblasts trigger NK cell dys-function via PGE2 and IDO, Cancer Lett. 318 (2) (2012) 154–161. [DOI] [PubMed] [Google Scholar]
- [107].Salmon H, et al. , Matrix architecture defines the preferential localization and migration of T cells into the stroma of human lung tumors, J. Clin. Invest 122 (3) (2012) 899–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Chakravarthy A, et al. , TGF-β-associated extracellular matrix genes link cancer-associated fibroblasts to immune evasion and immunotherapy failure, Nat. Commun 9 (1) (2018) 4692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Mariathasan S, et al. , TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells, Nature 554 (7693) (2018) 544–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Büll C, et al. , Sialic acids sweeten a tumor’s life, Cancer Res. 74 (12) (2014) 3199–3204. [DOI] [PubMed] [Google Scholar]
- [111].McAndrews KM, et al. , Enhanced adhesion of stromal cells to invasive cancer cells regulated by cadherin 11, ACS Chem. Biol 10 (8) (2015) 1932–1938. [DOI] [PubMed] [Google Scholar]
- [112].Lodyga M, et al. , Cadherin-11-mediated adhesion of macrophages to myofibroblasts establishes a profibrotic niche of active TGF-β, Sci. Signal 12 (564) (2019). [DOI] [PubMed] [Google Scholar]
- [113].Hinz B, Myofibroblasts, Exp. Eye Res 142 (2016) 56–70. [DOI] [PubMed] [Google Scholar]
- [114].Parker MW, et al. , Fibrotic extracellular matrix activates a profibrotic positive feedback loop, J. Clin. Invest 124 (4) (2014) 1622–1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Samuel MS, et al. , Actomyosin-mediated cellular tension drives increased tissue stiffness and β-catenin activation to induce epidermal hyperplasia and tumor growth, Cancer Cell 19 (6) (2011) 776–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Rubashkin MG, et al. , Force engages vinculin and promotes tumor progression by enhancing PI3K activation of phosphatidylinositol (3,4,5)-triphosphate, Cancer Res. 74 (17) (2014) 4597–4611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Wei SC, et al. , Matrix stiffness drives epithelial-mesenchymal transition and tumour metastasis through a TWIST1-G3BP2 mechanotransduction pathway, Nat. Cell Biol 17 (5) (2015) 678–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Omelchenko T, et al. , Contact interactions between epitheliocytes and fibroblasts: formation of heterotypic cadherin-containing adhesion sites is accompanied by local cytoskeletal reorganization, Proc. Natl. Acad. Sci. U. S. A 98 (15) (2001) 8632–8637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Stylianopoulos T, et al. , Causes, consequences, and remedies for growth-induced solid stress in murine and human tumors, Proc. Natl. Acad. Sci. U. S. A 109 (38) (2012) 15101–15108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Fernández-Sánchez ME, et al. , Mechanical induction of the tumorigenic β-catenin pathway by tumour growth pressure, Nature 523 (7558) (2015) 92–95. [DOI] [PubMed] [Google Scholar]
- [121].Lo CM, et al. , Cell movement is guided by the rigidity of the substrate, Biophys. J 79 (1) (2000) 144–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].DuChez BJ, et al. , Durotaxis by human cancer cells, Biophys. J 116 (4) (2019) 670–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Sunyer R, et al. , Collective cell durotaxis emerges from long-range intercellular force transmission, Science 353 (6304) (2016) 1157–1161. [DOI] [PubMed] [Google Scholar]
- [124].Gopal S, et al. , Fibronectin-guided migration of carcinoma collectives, Nat. Commun 8 (2017) 14105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Miyazaki K, et al. , Cancer cell migration on elongate protrusions of fibroblasts in collagen matrix, Sci. Rep 9 (1) (2019) 292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Glentis A, et al. , Cancer-associated fibroblasts induce metalloprotease-independent cancer cell invasion of the basement membrane, Nat. Commun 8 (1) (2017) 924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Keating M, et al. , Spatial distributions of pericellular stiffness in natural extracellular matrices are dependent on cell-mediated proteolysis and contractility, Acta Biomater. 57 (2017) 304–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Naba A, et al. , The matrisome: in silico definition and in vivo characterization by proteomics of normal and tumor extracellular matrices, Mol. Cell. Proteomics 11 (4) (2012) (p. M111.014647). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Alibert C, Goud B, Manneville JB, Are cancer cells really softer than normal cells? Biol. Cell 109 (5) (2017) 167–189. [DOI] [PubMed] [Google Scholar]
- [130].Swaminathan V, et al. , Mechanical stiffness grades metastatic potential in patient tumor cells and in cancer cell lines, Cancer Res. 71 (15) (2011) 5075–5080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Plodinec M, et al. , The nanomechanical signature of breast cancer, Nat. Nanotechnol 7 (11) (2012) 757–765. [DOI] [PubMed] [Google Scholar]
- [132].Denais CM, et al. , Nuclear envelope rupture and repair during cancer cell migration, Science 352 (6283) (2016) 353–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Tice JA, et al. , Benign breast disease, mammographic breast density, and the risk of breast cancer, J. Natl. Cancer Inst 105 (14) (2013) 1043–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Martin LJ, Boyd NF, Mammographic density. Potential mechanisms of breast cancer risk associated with mammographic density: hypotheses based on epidemiological evidence, Breast Cancer Res. 10 (1) (2008) 201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Boyd NF, et al. , Evidence that breast tissue stiffness is associated with risk of breast cancer, PLoS One 9 (7) (2014) e100937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Hasebe T, et al. , Fibrotic focus in invasive ductal carcinoma of the breast: a histopathological prognostic parameter for tumor recurrence and tumor death within three years after the initial operation, Jpn. J. Cancer Res 88 (6) (1997) 590–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Kreike B, et al. , Gene expression profiling and histopathological characterization of triple-negative/basal-like breast carcinomas, Breast Cancer Res. 9 (5) (2007) R65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Provenzano PP, et al. , Collagen density promotes mammary tumor initiation and progression, BMC Med. 6 (2008) 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Allinen M, et al. , Molecular characterization of the tumor microenvironment in breast cancer, Cancer Cell 6 (1) (2004) 17–32. [DOI] [PubMed] [Google Scholar]
- [140].Ma XJ, et al. , Gene expression profiling of the tumor microenvironment during breast cancer progression, Breast Cancer Res. 11 (1) (2009) R7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Finak G, et al. , Stromal gene expression predicts clinical outcome in breast cancer, Nat. Med 14 (5) (2008) 518–527. [DOI] [PubMed] [Google Scholar]
- [142].Chang HY, et al. , Robustness, scalability, and integration of a wound-response gene expression signature in predicting breast cancer survival, Proc. Natl. Acad. Sci. U. S. A 102 (10) (2005) 3738–3743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Farmer P, et al. , A stroma-related gene signature predicts resistance to neoadjuvant chemotherapy in breast cancer, Nat. Med 15 (1) (2009) 68–74. [DOI] [PubMed] [Google Scholar]
- [144].Ren B, Liu X, Suriawinata AA, Pancreatic ductal adenocarcinoma and its precursor lesions: histopathology, cytopathology, and molecular pathology, Am. J. Pathol 189 (1) (2019) 9–21. [DOI] [PubMed] [Google Scholar]
- [145].Lafaro KJ, Melstrom LG, The paradoxical web of pancreatic cancer tumor microenvironment, Am. J. Pathol 189 (1) (2019) 44–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Neesse A, et al. , Emerging concepts in pancreatic cancer medicine: targeting the tumor stroma, Onco Targets Ther. 7 (2013) 33–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Liang C, et al. , Complex roles of the stroma in the intrinsic resistance to gemcitabine in pancreatic cancer: where we are and where we are going, Exp. Mol. Med 49 (12) (2017) e406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Coppola S, et al. , A mechanopharmacology approach to overcome chemoresistance in pancreatic cancer, Drug Resist. Updat 31 (2017) 43–51. [DOI] [PubMed] [Google Scholar]
- [149].Bachem MG, et al. , Pancreatic carcinoma cells induce fibrosis by stimulating proliferation and matrix synthesis of stellate cells, Gastroenterology 128 (4) (2005) 907–921. [DOI] [PubMed] [Google Scholar]
- [150].Mollenhauer J, Roether I, Kern HF, Distribution of extracellular matrix proteins in pancreatic ductal adenocarcinoma and its influence on tumor cell proliferation in vitro, Pancreas 2 (1) (1987) 14–24. [DOI] [PubMed] [Google Scholar]
- [151].Durymanov M, et al. , Subcutaneous inoculation of 3D pancreatic cancer spheroids results in development of reproducible stroma-rich tumors, Transl. Oncol 12 (1) (2019) 180–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Öhlund D, et al. , Type IV collagen stimulates pancreatic cancer cell proliferation, migration, and inhibits apoptosis through an autocrine loop, BMC Cancer 13 (2013) 154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Palamakumbura AH, et al. , Lysyl oxidase propeptide inhibits prostate cancer cell growth by mechanisms that target FGF-2-cell binding and signaling, Oncogene 28 (38) (2009) 3390–3400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Grossman M, et al. , Tumor cell invasion can be blocked by modulators of collagen fibril alignment that control assembly of the extracellular matrix, Cancer Res. 76 (14) (2016) 4249–4258. [DOI] [PubMed] [Google Scholar]
- [155].Benson AB, et al. , A phase II randomized, double-blind, placebo-controlled study of Simtuzumab or placebo in combination with gemcitabine for the first-line treatment of pancreatic adenocarcinoma, Oncologist 22 (3) (2017) 241–e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Hecht JR, et al. , A phase II, randomized, double-blind, placebo-controlled study of Simtuzumab in combination with FOLFIRI for the second-line treatment of metastatic, Oncologist 22 (3) (2017) 243–e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [157].Salvador F, et al. , Lysyl oxidase-like protein LOXL2 promotes lung metastasis of breast cancer, Cancer Res. 77 (21) (2017) 5846–5859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Maller, et al. , Inflammation Promotes Tumor Aggression by Stimulating Stromal Cell-Dependent Collagen Crosslinking and Stromal Stiffening, (2020).
- [159].Piersma B, Bank RA, Collagen cross-linking mediated by lysyl hydroxylase 2: an enzymatic battlefield to combat fibrosis, Essays Biochem. 63 (3) (2019) 377–387. [DOI] [PubMed] [Google Scholar]
- [160].Scietti L, Campioni M, Forneris F, SiMPLOD, a structure-integrated database of collagen Lysyl hydroxylase (LH/PLOD) enzyme variants, J. Bone Miner. Res 34 (7) (2019) 1376–1382. [DOI] [PubMed] [Google Scholar]
- [161].Devkota AK, et al. , Development of a high-throughput Lysyl hydroxylase (LH) assay and identification of small-molecule inhibitors against LH2, SLAS Discov. 24 (4) (2019) 484–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].Provenzano PP, et al. , Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma, Cancer Cell 21 (3) (2012) 418–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [163].Maloney E, et al. , Non-invasive monitoring of stromal biophysics with targeted depletion of hyaluronan in pancreatic ductal adenocarcinoma, Cancers (Basel) (2019) 11(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [164].Hingorani SR, et al. , Phase Ib study of PEGylated recombinant human hyaluronidase and gemcitabine in patients with advanced pancreatic cancer, Clin. Cancer Res 22 (12) (2016) 2848–2854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].Hingorani SR, et al. , HALO 202: randomized phase II study of PEGPH20 plus nab-paclitaxel/gemcitabine versus nab-paclitaxel/gemcitabine in patients with untreated, metastatic pancreatic ductal adenocarcinoma, J. Clin. Oncol 36 (4) (2018) 359–366. [DOI] [PubMed] [Google Scholar]
- [166].Doherty GJ, Tempero M, Corrie PG, HALO-109–301: a phase III trial of PEGPH20 (with gemcitabine and nab-paclitaxel) in hyaluronic acid-high stage IV pancreatic cancer, Future Oncol. 14 (1) (2018) 13–22. [DOI] [PubMed] [Google Scholar]
- [167].Ramanathan RK, et al. , Phase IB/II randomized study of FOLFIRINOX plus pegylated recombinant human hyaluronidase versus FOLFIRINOX alone in patients with metastatic pancreatic adenocarcinoma: SWOG S1313, J. Clin. Oncol 37 (13) (2019) 1062–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [168].Momin N, et al. , Anchoring of intratumorally administered cytokines to collagen safely potentiates systemic cancer immunotherapy, Sci. Transl. Med (2019) 11(498). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [169].Alday-Parejo B, Stupp R, Rüegg C, Are integrins still practicable targets for anti-cancer therapy? Cancers (Basel) 11 (7) (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [170].Schnittert J, et al. , Integrin α11 in pancreatic stellate cells regulates tumor stroma interaction in pancreatic cancer, FASEB J. 33 (5) (2019) 6609–6621. [DOI] [PubMed] [Google Scholar]
- [171].Primac I, et al. , Stromal integrin α11 regulates PDGFR-β signaling and promotes breast cancer progression, J. Clin. Invest 130 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [172].Jiang H, et al. , Targeting focal adhesion kinase renders pancreatic cancers responsive to checkpoint immunotherapy, Nat. Med 22 (8) (2016) 851–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [173].Kolev VN, et al. , Inhibition of FAK kinase activity preferentially targets cancer stem cells, Oncotarget 8 (31) (2017) 51733–51747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [174].Jiang H, et al. , Development of resistance to FAK inhibition in pancreatic cancer is linked to stromal depletion, Gut 69 (1) (2019) 122–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [175].Loeffler M, et al. , Targeting tumor-associated fibroblasts improves cancer chemotherapy by increasing intratumoral drug uptake, J. Clin. Invest 116 (7) (2006) 1955–1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [176].Lo A, et al. , Tumor-promoting Desmoplasia is disrupted by depleting FAP-expressing stromal cells, Cancer Res. 75 (14) (2015) 2800–2810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [177].Tran E, et al. , Immune targeting of fibroblast activation protein triggers recognition of multipotent bone marrow stromal cells and cachexia, J. Exp. Med 210 (6) (2013) 1125–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [178].Su S, et al. , CD10, Cell 172 (4) (2018) (841–856.e16). [DOI] [PubMed] [Google Scholar]
- [179].Froeling FE, et al. , Retinoic acid-induced pancreatic stellate cell quiescence reduces paracrine Wnt-β-catenin signaling to slow tumor progression, Gastroenterology 141 (4) (2011) (p. 1486–97, 1497.e1–14). [DOI] [PubMed] [Google Scholar]
- [180].Sherman MH, et al. , Vitamin D receptor-mediated stromal reprogramming suppresses pancreatitis and enhances pancreatic cancer therapy, Cell 159 (1) (2014) 80–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [181].Miao L, et al. , Targeting tumor-associated fibroblasts for therapeutic delivery in desmoplastic tumors, Cancer Res. 77 (3) (2017) 719–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [182].Chan TS, et al. , Metronomic chemotherapy prevents therapy-induced stromal activation and induction of tumor-initiating cells, J. Exp. Med 213 (13) (2016) 2967–2988. [DOI] [PMC free article] [PubMed] [Google Scholar]