

Abstract
Many positive-sense RNA viruses, especially those infecting plants, are known to experience stringent, stochastic population bottlenecks inside the cells they invade, but exactly how and why these populations become bottlenecked are unclear. A model proposed ten years ago advocates that such bottlenecks are evolutionarily favored because they cause the isolation of individual viral variants in separate cells. Such isolation in turn allows the viral variants to manifest the phenotypic differences they encode. Recently published observations lend mechanistic support to this model and prompt us to refine the model with novel molecular details. The refined model, designated Bottleneck, Isolate, Amplify, Select (BIAS), postulates that these viruses impose population bottlenecks on themselves by encoding bottleneck-enforcing proteins (BNEPs) that function in a concentration-dependent manner. In cells simultaneously invaded by numerous virions of the same virus, BNEPs reach the bottleneck-ready concentration sufficiently early to arrest nearly all internalized viral genomes. As a result, very few (as few as one) viral genomes stochastically escape to initiate reproduction. Repetition of this process in successively infected cells isolates viral genomes with different mutations in separate cells. This isolation prevents mutant viruses encoding defective viral proteins from hitchhiking on sister genome-encoded products, leading to the swift purging of such mutants. Importantly, genome isolation also ensures viral genomes harboring beneficial mutations accrue the cognate benefit exclusively to themselves, leading to the fixation of such beneficial mutations. Further interrogation of the BIAS hypothesis promises to deepen our understanding of virus evolution and inspire new solutions to virus disease mitigation.
Keywords: viral population bottleneck, superinfection exclusion, positive-sense RNA virus, plant virus
Introduction
For free-living organisms, we define population bottlenecks as dramatic, but non-selective, decreases in the number of reproducing individuals in a population, often due to abrupt environmental changes, diseases, migration, geographic or sympatric isolation, resulting in stark loss of genetic diversity in subsequent generations. Population bottlenecks can lead to fixation of certain sub-optimal mutations that would have been eliminated through natural selection had the population size been larger. Severe and stochastic population bottlenecks have also been documented in infections of many viruses, occurring at various stages of viral reproduction, spread, and transmission. Examples of such viral population bottlenecks, along with insightful discussions of viral and host factors contributing to bottleneck sizes, and mathematical simulation models, have been extensively chronicled in a number of excellent reviews and research papers (Gutiérrez, Michalakis, and Blanc 2012; Zwart, Tromas, and Elena 2013; Zwart and Elena 2015), thus will not be repeated here. Instead, we wish to bring readers’ attention to more recent advances on viral population bottlenecks that occur inside virus-infected cells, especially those in cells infected by positive sense (+) RNA plant viruses.
In particular, we wish to highlight the potential adaptive value of these intracellular population bottlenecks in (+) RNA virus infections. At the first glance, this idea appears to be counterintuitive because one would ordinarily expect viral population bottlenecks to compromise virus viability and adaptability because they often lead to the fixation of mildly deleterious mutations in subsequent generations of viruses. With (+) RNA viruses this problem would be exacerbated by the high error rate of the replication processes, eventually leading to the corruption of virus populations (the error catastrophe or lethal mutagenesis) (Eigen 2002; Bull, Sanjuán, and Wilke 2007). However, several recent publications provide fresh experimental and mechanistic support for an unorthodox view that intracellular population bottlenecks, at least for (+) RNA viruses, manifest a virus-encoded trait that ensures constant selection of fitter genomes. We review these new findings and provide a mechanistic model that account for these observations.
It should be noted that the prototype of this model, which we now call Bottleneck, Isolate, Amplify, Select (BIAS), was proposed ten years ago (Miyashita and Kishino 2010). Similar concepts have also been proposed by Zwart and Elena in an earlier review (Zwart and Elena 2015). The current review aims to furnish the BIAS model with additional molecular details gleaned from the recent literature. We further argue that the same BIAS model provides a mechanistic explanation for superinfection exclusion between highly similar viruses. If validated, this model is expected to inspire novel virus control strategies.
Many (+) RNA viruses of plants bottleneck their populations inside virus-infected cells
The existence of intracellular population bottlenecks in (+) RNA virus infections was first hinted by Hall et al., when they observed that mixed infection with two different wheat streak mosaic virus (WSMV) variants led to their segregation in tiny adjacent cell clusters in systemically infected wheat leaves (Hall et al. 2001). These results indicated that even though both variants colonized the same wheat leaf, they rarely co-replicated in the same cells/cell clusters, suggesting the presence of stringent intracellular bottlenecks that restrict the number of reproductive genomes to no more than five per cell (French and Stenger 2003). Other authors have made similar observations by using viral variants tagged with different fluorescent proteins. Using two engineered plum pox virus (PPV) variants encoding a green fluorescent protein (GFP) and a red fluorescent protein is known as DsRed, Dietrich and Maiss (2003) observed that the two PPV variants formed separate, single-colored cell clusters in systemically infected tobacco leaves that are adjacent to each other. Co-infection by both occurred only in the one-cell-width borderlines between green and red cell clusters. The very fact that each of the cell clusters contained a single variant betrays highly stringent population bottlenecks that permitted just one variant to infect a group of connected cells. Strikingly analogous observations have since been made with other viruses, including WSMV, apple latent spherical virus (ALSV), tobacco etch virus (TEV), and a negative-strand RNA virus known as sonchus yellow net virus (SYNV) (Takahashi et al. 2007; Zwart, Daròs, and Elena 2011; Tatineni and French 2016; Zhou et al. 2019), underlining intracellular bottlenecks as a highly conserved characteristic shared by diverse virus populations.
The existence of intracellular population bottlenecks in systemically infected plant tissues was also corroborated by three elegant studies that examined individual cells derived from these tissues (González-Jara et al. 2009; Tromas et al. 2014; Donaire, Burgyán, and García-Arenal 2016). These authors infected plants with paired viral variants derived from three viruses: TEV, tobacco mosaic virus (TMV), and tomato bushy stunt virus (TBSV); and isolated single protoplast cells from infected leaves at multiple time points. They then subjected these cells to analyses that robustly differentiated cells that were infected by either, or both variants (González-Jara et al. 2009; Tromas et al. 2014; Donaire, Burgyán, and García-Arenal 2016). The authors established that, in the systemically infected plant leaves, successfully infected cells were mostly colonized by fewer than three founding genomes. In summary, stringent intracellular population bottlenecks constitute a conserved feature shared by nearly all (+) RNA viruses examined.
Direct evidence for intracellular population bottlenecking was provided by Miyashita et al. (2015). These authors constructed a population of tomato mosaic virus (ToMV) with more than a million variants by inserting randomized sequences composed of 10 randomized nucleotides into ToMV genomes. This ToMV population was then brought into individual protoplasts to launch viral replication. Fifteen infected protoplasts were then processed individually and subjected to deep sequencing to reveal the identity of ToMV variants in each of the cells. These experiments led to at least two important observations. (1) No more than seven variants replicated in each protoplast, even though the number of internalized ToMV genomes was estimated to be more than 5,000. (2) The identities of the ToMV genomes isolated from all these cells were different from each other. Together these data revealed highly stringent, yet stochastic population bottlenecks encountered by ToMV variants inside the cells they invaded.
To advocate for the relevance of population bottlenecks inside virus-infected cells, one must first establish that cells of naturally infected animals or plants are routinely invaded by large numbers of virions or viral genomes. Indeed this is what virologists are beginning to recognize in recent years. They found that many viruses invade cells in the form of so-called ‘collective infectious units’ (CIUs) (Sanjuán 2017; Leeks, Sanjuán, and West 2019). CIU can take many forms. HIV, for example, spreads between infected and uninfected T cells through intercellular connections known as virological synapses that transmit massive amounts of virions between two cells (Chen et al. 2007; Hübner et al. 2009; Del Portillo et al. 2011; Iwami et al. 2015; Law et al. 2016). By contrast, poliovirus and coxsackievirus particles are non-lytically released from infected cells in the form of lipid membrane vesicles containing dozens of virions (Robinson et al. 2014; Chen et al. 2015). Such vesicles then fuse with uninfected cells to deliver all the virions ‘en bloc’ (Chen et al. 2015). Even more dramatically, respiratory syncytial virus (RSV) induces the fusion of infected cell membrane with that of neighboring cells to form giant multinucleated cells (Cifuentes-Muñoz, Dutch, and Cattaneo 2018). Still, other viruses connect infected and uninfected cells through tubular or filamentous intercellular extensions (Martinez and Kielian 2016). Such multi-genome transmission is likely a more efficient mode of viral intercellular spread inside infected individuals (Feng et al. 2013; Santiana et al. 2018). Importantly, plant viruses spread cell-to-cell through plasmodesmata channels modified by virus-encoded movement proteins. Such modified intercellular channels are thought to shuffle large numbers of virions or viral genomes between adjacent cells.
Contrasting with the mass entry of viral genomes, only a small fraction of the entered genomes were found to embark on active replication. Such intracellular population bottlenecks have been observed not only in detached protoplast cells (Miyashita et al. 2015) but also in the cells of infected host tissues. Aside from the examples discussed earlier, one key study by Miyashita and Kishino (2010) used yellow and cyan fluorescent proteins (YFP and CFP) to differentially label soil-borne wheat mosaic virus (SBWMV) in order to follow the fate of the two viral variants, focusing only on the cells co-infected by both variants at the very beginning. They found that the virus-borne fluorescence markers began to segregate into cell clusters with a single FP after seven to nine cell-to-cell movement events. They further estimated that the progeny viruses in these cells must have descended from no more than six founding genomes.
Analogous investigations have also been carried out by Gutiérrez et al. (2015) using a different plant virus model (turnip mosaic virus, TuMV). Interestingly, these authors found that, in the systemically infected tobacco leaves, the first cells becoming infected mostly had one founding TuMV genome. By contrast, subsequent cells that became infected through cell-to-cell spread accommodated up to 40 different viral genomes. This study indicated that viral population bottleneck sizes could vary from virus to virus, and even between different types of cells of the same plant. However, one should also be aware that TuMV synthesizes the fluorescent marker proteins as part of the polyproteins directly translated from viral genomic RNAs. Therefore, expression of fluorescent markers may merely reflect the translation of the internalized genomes, rather than replicating genomes (see later). Nevertheless, variations in intracellular population sizes among different (+) RNA viruses should be reassessed through careful side-by-side comparisons.
Finally, it is probably worth noting that severe intracellular bottlenecks are not unique to plant viruses, or RNA viruses. Kobiler et al. (2010) engineered the PRV263 strain of pseudorabies virus, a herpesvirus with a large, double-stranded DNA genome, using a special tool called Brainbow cassette. The progeny genomes of the engineered PRV263 were each expected to encode one of the three fluorescent proteins: CFP, GFP, and RFP. Using the engineered PRV263 to infect culture cells, the authors observed the segregation of cell clusters containing single or double fluorescent proteins almost immediately after the viral spread into neighboring cells. These results led them to conclude that infections in new cells must have been founded with fewer than seven genomes.
How do viral populations become bottlenecked inside infected cells?
Answering this question requires an experimental approach capable of generating large intracellular virus populations. While a ToMV population comprising more than 1,000,000 (410) variants was constructed by Miyashita et al. (2015), that population was used to infect protoplast cells detached from plants. By contrast, it is not trivial to verify the internalization of multiple viral variants in cells of intact plants or animals. Fortunately, with (+) RNA viruses we can transfect host cells with transcribable viral cDNA. Viral cDNA of (+) RNA viruses, when equipped with promoters recognizable by RNA polymerases of the host cells (e.g. DNA-dependent RNA polymerase II, Pol II), directs the transcription of numerous viral genomic RNA copies that initiate potent viral infections. Indeed, plant virologists routinely use this approach to launch infections of (+) RNA viruses (Qu, Ren, and Morris 2003; Yi, Gopinath, and Kao 2007). It is commonly used in combination with the model plant Nicotiana benthamiana, thanks to the exceptional amenability of this plant to modified transfer DNA (T-DNA) exported from cells of Agrobacterium tumefaciens, a plant-infecting bacterium. This process is hence dubbed as ‘agro-infiltration’. An additional advantage of agro-infiltration is that multiple A.tumefaciens transformants, each carrying a different construct, can be mixed to deliver multiple viral cDNA into the same N.benthamiana cells with up to 100 per cent co-introduction efficiency.
Zhang et al. used agro-infiltration to generate large intracellular populations of turnip crinkle virus (TCV) (Zhang et al. 2017). To drive efficient transcription of TCV cDNA in agro-infiltrated cells, they adopted the duplicated 35S promoter (2X35S) of cauliflower mosaic virus (CaMV), which has the potential to drive the transcription of up to 10,000 RNA copies from each cDNA construct (Hull 2000). Furthermore, to facilitate the estimation of bottleneck sizes, they generated two different 2X35S-driven TCV cDNAs, tagged with GFP and mCherry, respectively, and delivered both into the same N.benthamiana cells, where thousands of copies of infectious RNA from both variants were expected to co-exist, making up the initial populations. Note that neither of the TCV variants could move cell to cell. Hence, their replication, and the replication-dependent fluorescent protein production, would occur only in the cells entered by the constructs.
This experimental setup predicted that, if just two of the thousands of TCV genomes replicated in each cell, up to 50 per cent of fluorescent cells would express both GFP and mCherry [(a + b)2 = a2+2ab+b2; with ‘2ab’ representing the fraction of cells replicating both]. Even assuming the transcription of the two variants was extremely biased at a nine-to-one ratio, one would still expect 18 per cent of fluorescent cells to express both proteins. Contrary to this prediction, fewer than 0.1 per cent of fluorescent cells expressed both proteins, even though cells expressing either GFP or mCherry were equally abundant, and frequently adjacent to each other. Note that the cDNA constructs for the two TCV variants should have been present in most of co-infiltrated cells because a control co-infiltration with a TCV construct and a carnation mottle virus (CarMV) construct resulted in more than 80 per cent of fluorescent cells replicating both TCV and CarMV (Zhang et al. 2017). It was hence concluded that the intracellular TCV populations faced extremely narrow bottlenecks that permitted the replication of just one copy of one variant in nearly all fluorescent cells (Zhang et al. 2017; Guo et al. 2020).
Strikingly, under this agro-infiltration-mediated cDNA delivery system, bottlenecking occurred even before the commencement of TCV replication. A time course experiment established that cells agro-infiltrated with the mCherry-encoding TCV replicon initiated viral replication in a much delayed, and gradual fashion, so that 3, 10, 23, and 33 per cent of the cells produced the replication-dependent red fluorescence at 48, 72, 96, and 120 h post infiltration, respectively. This contrasted with a co-delivered, non-viral, GFP-expressing construct that caused more than 90 per cent of the same cells to fluoresce green at 36 h post infiltration. Therefore, a bottlenecked state must have been in place almost immediately after the cellular entry of the TCV replicon construct, blocking the overwhelming majority of the transcribed TCV genomes from initiating replication. As a result, very few TCV genomes managed to overcome varying lengths of delay to escape from the bottlenecks in different cells. Such stochastic escapes accounted for asynchronous initiation of replication at the multicellular scale.
How were such pre-replication bottlenecks established? It turned out that the TCV-encoded p28 protein, when present at high intracellular concentrations, was alone sufficient to establish a polymeric state that repressed TCV replication (Zhang et al. 2017; Guo et al. 2020). Note that TCV p28 is encoded on the TCV genomic RNA, and directly translated from the internalized TCV genomes in the infected cells. Aside from its novel role in population bottlenecking, p28 was also known to have an enabling role in TCV replication, possibly at lower intracellular concentrations (White et al. 1995). Paradoxically, rapid accumulation of TCV genomic RNA in agro-infiltrated cells, propelled by the strong 2X35S promoter, could allow p28 protein to accumulate to high concentrations within a relatively short time, conditioning the swift establishment of the polymeric state that in turn blocked the replication of most of the TCV genomic RNA transcripts.
How did the p28 polymeric state block TCV replication? Zhang et al. (2017) demonstrated that when abundantly available, p28 protein molecules polymerized into self-perpetuating protein aggregates that actively captured p28 copies not already in the aggregates, through rapid intracellular movements. Since the replication of TCV likely requires p28 in a non-aggregating state, it cannot occur when all p28 molecules become trapped by the self-perpetuating aggregates. It was further speculated that the p28 aggregates could also trap TCV genomic RNAs through p28-TCV RNA interactions, thus solidifying the repressive state (Zhang et al. 2017, 2018).
Collectively these findings unveiled a novel molecular switch that routes TCV p28 to two opposite functions based on its concentration in the cell, providing a simple mechanism for the p28-mediated bottlenecking of TCV populations (Zhang et al. 2017, 2018). Consistent with this idea, Guo et al. recently reported that several single amino acid mutations in p28 caused the TCV population bottleneck to dramatically relax, permitting the GFP and mCherry-tagged mutant replicons to co-exist in 20 per cent of fluorescent cells (as opposed to <0.1% for wild-type replicons) (Guo et al. 2020). Such mutants invariably accumulated substantially less p28 proteins than wild-type p28 (Guo et al. 2020). These points will become important in the next section.
A mechanistic model for intracellular population bottlenecks of (+) RNA viruses
By now we hope that you are convinced that viruses frequently deliver large quantities of genomes into individual host cells, where only a small fraction of the entered genomes do replicate. In fact, new evidence suggests that some viruses with multiple genome segments may not even succeed in replicating all of their segments in every infected cell (McCrone et al. 2018; Sicard et al. 2019). It is also well-known that established cellular infections exclude secondary invasions by the same virus. Finally, such exclusion/repression has been shown to be actively enforced by virus-encoded proteins in several cases. Why would viruses actively block most copies of their own genomes from replicating in the same cell? The insights gleaned from the TCV-based system prompt us to update a model first put forward by Miyashita and Kishino (2010). That original model was based both on experimental findings derived from SBWMV infections, and subsequent simulation attempts. It predicted that small intracellular population sizes allow viruses to isolate both deleterious and adaptive mutations in trans-acting viral genes encoding viral proteins. For example, results of multiple simulation runs showed that an intracellular bottleneck restricting the number of replicating viral genomes to five would isolate and purge deleterious mutations within ten cell-to-cell movement events (Miyashita and Kishino 2010). Here we incorporate a mechanistic underpinning to this model, and formally name the model as BIAS. For now, we limit the applicability of the BIAS model to (+) RNA viruses, although it may prove to be more widely applicable as new data emerge.
First and foremost, the BIAS model addresses the need for separating viral genomes with varying potentials of replication and survival. The major replication enzyme encoded by (+) RNA viruses, known as RNA-dependent RNA polymerase (RdRp), introduces mutations into newly synthesized genomes at a rate of roughly 10−4 per nucleotide. This mutation rate translates into at least one error per genome for most (+) viruses, meaning nearly all viral genomes received by an uninfected cell contain errors. These errors are randomly distributed through the entire genome and their impacts on genome integrity range from lethal to highly beneficial. Mutations within cis-acting RNA elements yield phenotypes that are immediately acted upon by natural selection. For example, a mutation disrupting an RNA motif specifically recognized by the cognate RdRp leads to the immediate exclusion of the mutated genome from the replicable genome pool. By contrast, errors harming replication proteins, or other viral proteins for that matter, would have to be purged by a different mechanism. For simplicity, hereafter we limit the discussion to errors in the RdRp gene, although the general logic should apply for all viral protein-coding genes.
The central assumption of the BIAS hypothesis is that nearly all of the internalized viral genomes will template the translation of proteins directly translatable from the genomic RNA, even though a majority of these RNA molecules do not have the chance to replicate. This is not really controversial because all kinds of non-replicating RNAs are known to undergo protein translation once inside a living cell. Moreover, the genomes of (+) RNA viruses need the replication proteins to be translated first in order to initiate replication. The only new proposition here is that aside from replication proteins, another class of proteins, designated bottleneck-enforcing proteins (BNEPs), are also translated from these genomic RNA molecules. In some viruses, a single protein could act as both a replication protein and a BNEP (e.g. TCV p28). Another assumption, based on TCV p28 results, is that establishment of an operational bottleneck requires the viral BNEP to reach a certain concentration threshold (Zhang et al. 2017; Guo et al. 2020).
With these assumptions in mind, let us first consider a viral population that does not bottleneck. As depicted in the upper half of Fig. 1, this would mean virtually all genomes a cell receives could replicate themselves. Indeed even those containing fatal errors in RdRp could still replicate—by hitchhiking on functional RdRp produced by sister genomes in the same cell. These fatal errors are thus retained in the pool of progeny genomes (Fig. 1, upper half, cell 1, column 2). To aggravate this problem, genomes that contain no errors in RdRp replicate to incur new errors in the progeny. Bear in mind that the number of new errors is not insignificant. Considering that (1), almost every newly replicated viral genome will contain one new error (see earlier), and (2), a typical (+) RNA virus devotes at least one-third of its genome to encode RdRp; we can expect that more than 30 per cent of the newly synthesized genomes contain errors in RdRp. Since a substantial proportion of the errors will be deleterious (Elena and Sanjuán 2005; Duffy 2018), it would be rather conservative to assume that one-fifth of the errors in RdRp cause the protein to malfunction. At this rate, approximately 6 per cent of the newly synthesized genomes would be predicted to encode a defective RdRp. Therefore, even if there is just one cycle of replication in each cell, the repetition of replication in consecutively infected cells would still lead to rapid increase of defective viral genomes harboring deleterious RdRp errors (Fig. 1, upper half, cells 2 and 3), and simultaneous decrease of those still encoding intact RdRp. It is easy to see why such a population cannot survive for long, even if the effect of host defense is not considered (Fig. 1, upper half).
Figure 1.

Failure to establish population bottlenecks leads to rapid accumulation of deleterious mutations as viral infection progresses in consecutive cells. Meanings of the symbols used are given on the very top. Abbreviations: RdRp, RNA-dependent RNA polymerase; ORF, open reading frame. The three gray rectangular boxes immediately underneath the symbols depict three cells consecutively infected by a virus population, unconstrained by population bottlenecks. Absence of bottlenecks allows all internalized genomes, including the one with a deleterious mutation in RdRp (red star), to replicate, leading to the retention of existing mutation, as well as emergence of new mutations. As a result, the viral population entering the second round cells contains higher proportion of mutant genomes encoding defective RdRp than does the initial population. The correspondingly lower proportion of genomes encoding functional RdRp is predicted to lead to less robust replication. This trend continues in subsequent cells, eventually causing the viral population to collapse. By contrast, the three green rectangles at the bottom depict different primary cells invaded by viral populations with essentially the same composition, but this time the populations are bottlenecked. As a result, only one of the internalized genomes stochastically escapes the bottleneck to replicate. In both the left and right cells, the genomes succeeding in replication encode functional RdRp, producing progeny genomes that are mostly capable of launching renewed replication in the next set of cells. In the middle cell, the stochastically escaping genome encodes a defective RdRp but can nevertheless replicate using RdRps encoded by other genome copies. However, the defective progenies are unable to replicate in the next set of cells they invade. For simplicity, only five founding genomes were drawn to represent the dozens, if not hundreds, of viral genomes received by most of the susceptible cells in a typical host individual. Furthermore, our discussions also omitted detrimental, yet non-lethal mutations.
At the bottom half of Fig. 1, we consider the opposite scenario where the viral population is tightly bottlenecked in every cell it enters. To simplify the discussion we focus our attention on genomes that either maintain steady RdRp function or contain deleterious mutations that abolishes the RdRp function, while ignoring all intermediary mutations that do not seriously handicap RdRp functions. We further limit the discussion to very narrow bottlenecks that permit the escape of just one genome. As depicted in the three green rectangles at the bottom half of Fig. 1, in most cells (represented by the left and right cells) such bottlenecks serve to maintain a constant level of mutated genomes as the infection progresses cell to cell. In cells where the genome encoding a defective RdRp escapes the bottleneck, such genome should be able to replicate by utilizing the functional RdRps produced by sister genomes in the same cell (Fig. 1, bottom half, middle cell). However, its progenies are then prevented from repeating the replication process in the next set of cells, where they can no longer count on sister genomes to provide functional RdRps (because now all sister genomes translate defective RdRps).
Now imagine the intracellular bottlenecking is actively enforced by a BNEP (Fig. 2). As discussed in the previous paragraph, in many cells the genome(s) that escape from bottlenecks would probably contain no deleterious error in the RdRp, allowing replication to reiterate in successive cells (Fig. 2A and B). In these cells, the defective genomes would be purged simply due to their failure to escape from the bottleneck (those with a red star in Fig. 2A and B). On the other hand, if a genome encoding a defective RdRp happens to pass through the bottleneck, it could still be amplified to high numbers in this cell because it has access to functional RdRp produced by sister genomes present in the same cell (Fig. 2C1). However, the defective genome will become isolated in the next batch of cells because its progeny will now be the exclusive residents of these new cells. It is in this next batch of invaded cells that the defective genomes are eliminated by natural selection, as they can no longer hitchhike via functional RdRP produced by other genomes (Fig. 2C2).
Figure 2.

Anticipated fates for different mutations in the RdRp gene incurred during viral replication under the BIAS model. The meanings of all symbols are given in the top left box, as well as in cell A. Rectangles with rounded corners represent different cells, with A as the primarily infected cell, B, C1, D1 as representatives of second-round cells, and C2, D2, as cells infected by progenies of C1 and D1, respectively. Note that replication in all cells gives rise to new mutations that are depicted as either red (deleterious) or yellow (beneficial) mutations at different positions of the RdRp ORF.
Importantly, this BIAS model can be further extrapolated to predict the flourishing of beneficial mutations. Extending from the logic of the previous paragraph, viral genomes with beneficial mutations are blocked from replication most of the time as well (Fig. 2B and C). However, should one of such genomes escape from the bottleneck in one of the cells, its progeny will likewise become the exclusive residents of the next batch of cells becoming infected. Assuming this beneficial mutation enables the mutated RdRp to synthesize more genomic RNA copies than the original RdRp or enhances its ability to withstand cellular level innate immunity, in these cells only the genomic RNA harboring this mutation benefits from the mutation by propagating more progeny genomes carrying the same mutation (Fig. 2D1 and D2). In short, the BIAS model predicts that the intracellular bottlenecks of viral populations create the environment in which both purifying and positive selections can take place, ensuring the long-term survival of the viral genomes.
The prototype of this model was first described by Miyashita and Kishino (2010). These authors used data-based simulation to predict that restricting the number of replicating viral genomes in each cell to five would allow for relatively swift purging of deleterious mutations, and the fixation of adaptive mutations, in virus-encoded trans-acting factors, namely proteins. While in the current review we limited the discussion to viral RdRp, the same model remains valid for other viral proteins. In summary, the BIAS hypothesis stipulates that (+) RNA viruses encode BNEPs that act in a concentration-dependent manner to establish intracellular population bottlenecks in order to limit the number of reproducing viral genomes in each cell. Counterintuitively, this model predicts that the more copies of viral genomes (of the same virus) entering a host cell, the more stringent is the bottleneck in this cell. Reiteration of such population bottlenecks in successive virus-infected cells isolates individual viral variants with mutations that either thwart or bolster the relative competitiveness of the virus. The isolated variant genomes can then be directly selected based on their ability to decrease or increase their own copy numbers. Such a mechanism synchronizes the unit of viral selection in most of the infected cells, and purges cheaters in the population in almost real-time. In conclusion, the BIAS model postulates that viral intracellular population bottlenecks serve as a natural selection mechanism that operates in each of the infected cells to constantly surveil viral genome integrity and competitiveness.
Viral population dynamics in cells invaded by varying numbers of virions
Until now we simplified the discussion to focus on cells receiving relatively large numbers of genomes of the same (+) RNA virus. These viral genomes together translate BNEPs in amounts sufficient for forming protein aggregates within a relative short time, thereby preventing most of the internalized viral genomes from participating in replication. The exact number of the internalized genomes needed for establishing a tight bottleneck may vary from virus to virus but probably amounts to hundreds for many common (+) RNA viruses. According to the BIAS model, the narrow bottlenecks are only evaded by a tiny fraction of the internalized genomes, which parent the progeny viral genomes in these cells. It is further important to recognize that the bottlenecks lack the specificity needed to differentiate between parental and progeny genomes, hence are predicted to also exclude the progeny genomes from repeating the replication in the cells of their parent(s). Indeed, the bottlenecks are expected to be reinforced by new BNEP copies translated from progeny genomes.
However, what actually occurs in virus-infected organisms is probably much more complicated. While most cells in an infected plant/animal probably become infected by virions in the form of CIU that were amplified elsewhere in the same body, the very first cell(s) of a newly infected host possibly receive just a few virions (e.g. <10). One can also foresee cells that are invaded by an intermediate number of virions (e.g. between 20 and 50). What would happen in these cells if the BIAS model holds?
The BNEP concentration threshold probably could not be reached fast enough to form the bottlenecking protein aggregate if a cell contained just five copies of BNEP-translating viral genomes. The absence of bottlenecks in these cells would ironically permit the replication of possibly all five genomes (Fig. 3). Recombination between these replicating genomes would also be frequent in these cells. However, one must also recognize that viruses in these cells probably face a treacherous fate. This is because fewer viral genomes also equate slower accumulation of viral replication proteins, thus delayed commencement of replication, giving the host cells more time to activate innate antiviral defenses.
Figure 3.
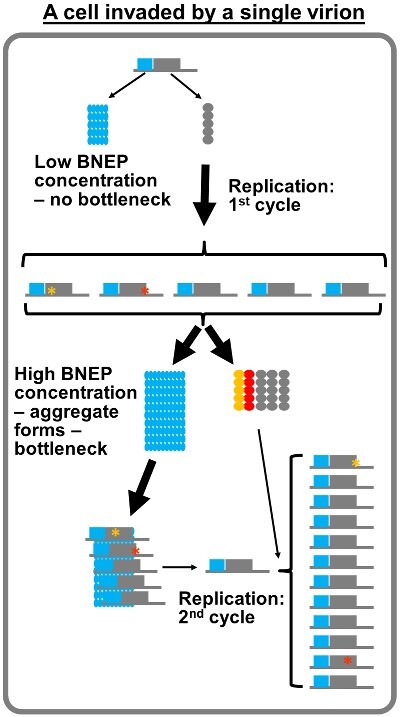
Limited second cycle replication in cells entered by a very small number (<10) of viral genomes.
Following the logic of the BIAS model a step further, in cells invaded by very few virions, some of the newly synthesized progeny genomes are predicted to re-enter the replication cycle before the bottleneck is firmly established. Eventually, the bottlenecks will be established with the help of BNEPs translated from progeny genomes, which then block a majority of progeny genomes from repeating the replication cycle more than once (Fig. 3). We must note that this prediction is contradicted by findings of one published study (Schulte et al. 2015), which found that poliovirus could undergo five cycles of replication in each of the infected cells. The authors went on to speculate that multiple replication cycles in each cell allow viral genomes to quickly accumulate mutations, creating the sequence space upon which selection could act. Poliovirus is a human-infecting virus for which the existence of intracellular bottlenecks remains to be determined. In addition, these results were obtained from cultured cells adapted for optimal poliovirus propagation, rather than host individuals infected by the virus. Nevertheless, we recognize that these differences do not satisfactorily reconcile the contradiction. Additional investigations will be needed to rigorously test the BIAS model against more viruses.
What happens in the cells invaded by an intermediate number of virions? The BIAS model predicts that in these cells the bottlenecks would initially be relatively loose, due to inefficient polymerization of BNEPs at concentrations near the necessary threshold. This in turn is predicted to permit the escape of more viral genomes than firmly established bottlenecks. These cells are also expected to accommodate robust recombination between viral genomes. Together these dynamic scenarios highlight the capacity of the BIAS model to account for viral adaptions to changing priorities in different cellular environments. Specifically, in cells invaded by many virions, the priority for the virus would be to isolate and eliminate deleterious mutations through bottlenecks; whereas in cells invaded by very few virions, the BNEP inaction early on allows the virus to prioritize genome multiplication over integrity surveillance.
Additional questions, counter arguments, and outlook
In this review, we focused our attention on the intracellular population bottlenecks observed in (+) RNA virus infections. Contrary to the prevailing view of population bottlenecks as being detrimental to viral long-term survival, we argue that the intracellular bottlenecks of (+) RNA virus populations are likely selected for by viruses. We further propose a specific mechanism, in the form of the BIAS model, to account for the establishment of these intracellular bottlenecks. Central to the BIAS hypothesis is the debut of BNEP, a virus-encoded protein which, upon reaching a certain concentration threshold, enforces intracellular population bottlenecks by aggregating into large protein complexes that trap viral replication proteins and/or viral genomes, excluding them from active replication. Consequently, only a few random viral genomes that escape the bottlenecks manage to launch productive replication, possibly before the bottlenecks are firmly established. Importantly, we advocate that the viral genomic RNA that failed to replicate nevertheless contribute the establishment of the bottleneck by serving as the mRNA for BENP translation. Finally, we reason that the BNEP coding capacity in viral genomes is likely preserved by natural selection because the resulting bottlenecks, by limiting the number of replicating genomes in each cell, act to isolate mutant genomes with either deleterious or advantageous mutations in separate cells, forcing the mutants to manifest their phenotypes independent of their alleles, ensuring prompt phenotype-based selection.
We hasten to note that while logically satisfactory, the mechanistic support for the BIAS model is provided primarily by investigations of one virus—TCV. Consequently, we urge fellow virologists to subject their favorite viruses to similar investigations, helping us evaluate the applicability of the BIAS model in other (+) RNA viruses, or even viruses with other types of genomes. Along this line, we would particularly welcome scrutiny from colleagues studying animal-infecting (+) RNA viruses. Unlike their plant cousins, it is yet unknown whether animal-infecting (+) RNA viruses are also constrained by intracellular population bottlenecks. Below we discuss a few pressing questions concerning the BIAS model, and potential directions of future research.
The first question concerns the possible mechanistic link between intracellular viral population bottlenecks and superinfection exclusion (SIE). The BIAS model predicts SIE as one of the manifestations of the intracellular viral population bottlenecks. This is because the narrow bottlenecks in most of the infected cells allow only very few viral genomes to replicate, even if they enter the cells at the same time. The bottlenecks are further tightened by active genome replication that produces more progeny genomes that in turn template the translation of more BNEPs. It is then no surprise that the superinfecting genomes will be excluded from replication in these cells. We hasten to note that other mechanisms for SIE have been proposed (Ziebell and Carr 2010; Bergua et al. 2014). It would be interesting to examine how these mechanisms interact with each other to shape virus infections in plants and animals.
Next, we address why a large amount of the viral genomes need to enter a cell, if most of them do not embark on replication anyway. The idea of enlisting all genomic RNAs in a cell to enforce a bottleneck that blocks most of them from reproduction is not untenable. Indeed the entry of multiple genomes not only accelerates the establishment of bottlenecks—it also enables the rapid construction of intracellular structures needed for replication, known as replication organelles (Xu and Nagy 2016; Ertel et al. 2017). In short, collaboration among multiple internalized genomes powers the swift mobilization of both the replication machine and the bottleneck, ensuring efficient and faithful reproduction of the virus. It follows that genes encoding such collaborative traits, including BNEP genes, but also those facilitating multi-virion entry, must be selected for by virus evolution. By contrast, since nearly all internalized genomes (except for those containing eventful mutations) encode genes that are phenotypically identical, exactly which one of them gets the chance to replicate is inconsequential to the evolutionary persistence of the virus. Indeed similar bottlenecks are also evident in single-celled bacteria like Agrobacterium tumefaciens—an overwhelming majority of bacterial cells that participate in the induction of crown galls become trapped inside the galls, with no chance to ever pass their genes to future generations (Dawkins 1989).
Why should we care about virus population bottlenecks? If high intracellular concentration of BNEPs is indeed the culprit that causes viral populations to bottleneck, and such bottlenecks are essential for keeping viral mutation rate low, targeting BNEPs for destruction or inactivation constitutes a previously unrecognized antiviral treatment strategy. Alternatively, the functionality of BNEPs could be defeated by purposely introducing mutations into BNEP coding sequence. The resulting mutant viruses would be predicted to undergo transient infections in host cells before being overburdened by replication errors. Such viral mutants might work as live attenuated vaccines. Therefore, it is vitally important for us to have a thorough understanding of viral population bottlenecks in order to gain an upper hand in the constant battle against virus infections in plants, animals, and humans.
Acknowledgements
We are thankful to current and former members of Qu lab for stimulating discussions. We also thank Drs Marilyn Roossinck, Andrew K. White, and Aiming Wang for critically reading the manuscript, and providing insightful suggestions.
Funding
Research in Qu and Slot labs are supported by grants from the National Science Foundation, the U.S. Department of Agriculture Hatch funds, as well as grants from the United Soybean Board, the North Central Soybean Research Program, the Ohio Soybean Council, and the SEEDS awards of the Ohio Agricultural Research and Development Center. Research by Miyashita was supported by JSPS KAKENHI grant numbers JP16H06185, JP17K19257, and 19H02953; MEXT ‘Scientific Research on Innovative Areas’ grant numbers 16H06429, 16K21723, and 16H06435; JSPS Core-to-Core Program (Advanced Research Networks) entitled ‘Establishment of international agricultural immunology research-core for a quantum improvement in food safety’.
Conflict of interest: None declared.
Contributor Information
Feng Qu, Department of Plant Pathology and; Ohio Agricultural Research and Development Center, The Ohio State University, Wooster, OH 44691, USA.
Limin Zheng, Department of Plant Pathology and; Ohio Agricultural Research and Development Center, The Ohio State University, Wooster, OH 44691, USA.
Shaoyan Zhang, Department of Plant Pathology and; Ohio Agricultural Research and Development Center, The Ohio State University, Wooster, OH 44691, USA.
Rong Sun, Department of Plant Pathology and; Ohio Agricultural Research and Development Center, The Ohio State University, Wooster, OH 44691, USA.
Jason Slot, Department of Plant Pathology and.
Shuhei Miyashita, Graduate School of Agricultural Science, Tohoku University, 468-1 Aramaki-aoba, Aoba-ku, Sendai 980-0845, Japan.
References
- Bergua M. et al. (2014) ‘ A Viral Protein Mediates Superinfection Exclusion at the Whole-Organism Level but Is Not Required for Exclusion at the Cellular Level’, Journal of Virology, 88: 11327–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bull J. J., Sanjuán R., Wilke C. O. (2007) ‘ Theory of Lethal Mutagenesis for Viruses’, Journal of Virology, 81: 2930–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen P. et al. (2007) ‘ Predominant Mode of Human Immunodeficiency Virus Transfer between T Cells Is Mediated by Sustained Env-Dependent Neutralization-Resistant Virological Synapses’, Journal of Virology, 81: 12582–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y.-H. et al. (2015) ‘ Phosphatidylserine Vesicles Enable Efficient En Bloc Transmission of Enteroviruses’, Cell, 160: 619–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cifuentes-Muñoz N., Dutch R. E., Cattaneo R. (2018) ‘ Direct Cell-to-Cell Transmission of Respiratory Viruses: The Fast Lanes’, PLoS Pathogens, 14: e1007015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawkins R. (1941/1989) The Selfish Gene, New edition. Oxford; New York: Oxford University Press; <https://search.library.wisc.edu/catalog/999612476002121> accessed 29 Oct 2020. [Google Scholar]
- Del Portillo A. et al. (2011) ‘ Multiploid Inheritance of HIV-1 during Cell-to-Cell Infection’, Journal of Virology, 85: 7169–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietrich C., Maiss E. (2003) ‘ Fluorescent Labelling Reveals Spatial Separation of Potyvirus Populations in Mixed Infected Nicotiana benthamiana Plants’, Journal of General Virology, 84: 2871–6. [DOI] [PubMed] [Google Scholar]
- Donaire L., Burgyán J., García-Arenal F. (2016) ‘ RNA Silencing May Play a Role in but Is Not the Only Determinant of the Multiplicity of Infection’, Journal of Virology, 90: 553–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffy S. (2018) ‘ Why Are RNA Virus Mutation Rates So Damn High?’, PLoS Biology, 16: e3000003–e3000003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eigen M. (2002) ‘ Error Catastrophe and Antiviral Strategy’, Proceedings of the National Academy of Sciences of the United States of America, 99: 13374–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elena S. F., Sanjuán R. (2005) ‘ Adaptive Value of High Mutation Rates of RNA Viruses: Separating Causes from Consequences’, Journal of Virology, 79: 11555–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ertel K. J. et al. (2017) ‘ Cryo-Electron Tomography Reveals Novel Features of a Viral RNA Replication Compartment’, eLife, 6 <https://elifesciences.org/download/aHR0cHM6Ly9jZG4uZWxpZmVzY2llbmNlcy5vcmcvYXJ0aWNsZXMvMjU5NDAvZWxpZmUtMjU5NDAtdjIucGRm/elife-25940-v2.pdf?_hash=8VcHktmBASivleN0OBYcdFBZ3SQRKAOdziKyK6%2F9ntI%3D> accessed 18 Sep 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Z. et al. (2013) ‘ A Pathogenic Picornavirus Acquires an Envelope by Hijacking Cellular Membranes’, Nature, 496: 367–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- French R., Stenger D. C. (2003) ‘ Evolution of Wheat Streak Mosaic Virus: Dynamics of Population Growth within Plants May Explain Limited Variation’, Annual Review of Phytopathology, 41: 199–214. [DOI] [PubMed] [Google Scholar]
- González-Jara P. et al. (2009) ‘ The Multiplicity of Infection of a Plant Virus Varies during Colonization of Its Eukaryotic Host’, Journal of Virology, 83: 7487–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Q. et al. (2020) ‘ Superinfection Exclusion by p28 of Turnip Crinkle Virus Is Separable from Its Replication Function’, Molecular Plant–Microbe Interactions, 33: 364–75. [DOI] [PubMed] [Google Scholar]
- Gutiérrez S., Michalakis Y., Blanc S. (2012) ‘ Virus Population Bottlenecks during within-Host Progression and Host-to-Host Transmission’, Current Opinion in Virology, 2: 546–55. [DOI] [PubMed] [Google Scholar]
- Gutiérrez S. et al. (2015) ‘ The Multiplicity of Cellular Infection Changes Depending on the Route of Cell Infection in a Plant Virus’, Journal of Virology, 89: 9665–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall J. S. et al. (2001) ‘ Three Distinct Mechanisms Facilitate Genetic Isolation of Sympatric Wheat Streak Mosaic Virus Lineages’, Virology, 282: 230–6. [DOI] [PubMed] [Google Scholar]
- Hübner W. et al. (2009) ‘ Quantitative 3D Video Microscopy of HIV Transfer across T Cell Virological Synapses’, Science, 323: 1743–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hull S. P. D. (2000) ‘ Genetically Modified Plants and the 35S Promoter: Assessing the Risks and Enhancing the Debate’, Microb Ecol Health Dis, 12: 1–5. [Google Scholar]
- Iwami S. et al. (2015) ‘ Cell-to-Cell Infection by HIV Contributes over Half of Virus Infection’, eLife, 4: e08150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobiler O. et al. (2010) ‘ Herpesviruses Carrying a Brainbow Cassette Reveal Replication and Expression of Limited Numbers of Incoming Genomes’, Nature Communications, 1: 146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law K. M. et al. (2016) ‘ In Vivo HIV-1 Cell-to-Cell Transmission Promotes Multicopy Micro-Compartmentalized Infection’, Cell Reports, 15: 2771–83. [DOI] [PubMed] [Google Scholar]
- Leeks A., Sanjuán R., West S. A. (2019) ‘ The Evolution of Collective Infectious Units in Viruses’, Virus Research, 265: 94–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez M. G., Kielian M. (2016) ‘ Intercellular Extensions Are Induced by the Alphavirus Structural Proteins and Mediate Virus Transmission’, PLoS Pathogens, 12: e1006061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCrone J. T. et al. (2018) ‘ Stochastic Processes Constrain the within and between Host Evolution of Influenza Virus’, eLife, 7: e35962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyashita S. et al. (2015) ‘ Viruses Roll the Dice: The Stochastic Behavior of Viral Genome Molecules Accelerates Viral Adaptation at the Cell and Tissue Levels’, PLoS Biology, 13: e1002094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyashita S., Kishino H. (2010) ‘ Estimation of the Size of Genetic Bottlenecks in Cell-to-Cell Movement of Soil-Borne Wheat Mosaic Virus and the Possible Role of the Bottlenecks in Speeding up Selection of Variations in Trans-Acting Genes or Elements’, Journal of Virology, 84: 1828–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu F., Ren T., Morris T. J. (2003) ‘ The Coat Protein of Turnip Crinkle Virus Suppresses Posttranscriptional Gene Silencing at an Early Initiation Step’, Journal of Virology, 77: 511–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson S. M. et al. (2014) ‘ Coxsackievirus B Exits the Host Cell in Shed Microvesicles Displaying Autophagosomal Markers’, PLoS Pathogens, 10: e1004045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjuán R. (2017) ‘ Collective Infectious Units in Viruses’, Trends in Microbiology, 25: 402–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santiana M. et al. (2018) ‘ Vesicle-Cloaked Virus Clusters Are Optimal Units for Inter-Organismal Viral Transmission’, Cell Host & Microbe, 24: 208–20.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulte M. B. et al. (2015) ‘ Experimentally Guided Models Reveal Replication Principles That Shape the Mutation Distribution of RNA Viruses’, eLife, 4: e03753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sicard A. et al. (2019) ‘ A Multicellular Way of Life for a Multipartite Virus’, eLife, 8: e43599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi T. et al. (2007) ‘ Analysis of the Spatial Distribution of Identical and Two Distinct Virus Populations Differently Labeled with Cyan and Yellow Fluorescent Proteins in Coinfected Plants’, Phytopathology, 97: 1200–6. [DOI] [PubMed] [Google Scholar]
- Tatineni S., French R. (2016) ‘ The Coat Protein and NIa Protease of Two Potyviridae Family Members Independently Confer Superinfection Exclusion’, Journal of Virology, 90: 10886–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tromas N. et al. (2014) ‘ Within-Host Spatiotemporal Dynamics of Plant Virus Infection at the Cellular Level’, PLoS Genetics, 10: e1004186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White K. A. et al. (1995) ‘ Immunodetection, Expression Strategy and Complementation of Turnip Crinkle Virus p28 and p88 Replication Components’, Virology, 211: 525–34. [DOI] [PubMed] [Google Scholar]
- Xu K., Nagy P. D. (2016) ‘ Enrichment of Phosphatidylethanolamine in Viral Replication Compartments via Co-Opting the Endosomal Rab5 Small GTPase by a Positive-Strand RNA Virus’, PLoS Biology, 14: e2000128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi G., Gopinath K., Kao C. C. (2007) ‘ Selective Repression of Translation by the Brome Mosaic Virus 1a RNA Replication Protein’, Journal of Virology, 81: 1601–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X.-F. et al. (2017) ‘ A Self-Perpetuating Repressive State of a Viral Replication Protein Blocks Superinfection by the Same Virus’, PLOS Pathogens, 13: e1006253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X.-F. et al. (2018) ‘ A New Mechanistic Model for Viral Cross Protection and Superinfection Exclusion’, Front Plant Sci, 9. doi:10.3389/fpls.2018.00040 <http://journal.frontiersin.org/article/10.3389/fpls.2018.00040/full> accessed 11 Mar 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X. et al. (2019, Jul 24) ‘ The Matrix Protein of a Plant Rhabdovirus Mediates Superinfection Exclusion by Inhibiting Viral Transcription’, Journal of Virology, 93: JVI.00680–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziebell H., Carr J. P. (2010) ‘Cross-Protection’, in Advances in Virus Research, vol. 76, pp. 211–64. Elsevier; <http://linkinghub.elsevier.com/retrieve/pii/S0065352710760061> accessed 8 Jun 2017, Amsterdam: Elsevier Inc. [DOI] [PubMed] [Google Scholar]
- Zwart M. P., Daròs J.-A., Elena S. F. (2011) ‘ One Is Enough: In Vivo Effective Population Size Is Dose-Dependent for a Plant RNA Virus’, PLoS Pathogens, 7: e1002122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwart M. P., Elena S. F. (2015) ‘ Matters of Size: Genetic Bottlenecks in Virus Infection and Their Potential Impact on Evolution’, Annual Review of Virology, 2: 161–79. [DOI] [PubMed] [Google Scholar]
- Zwart M. P., Tromas N., Elena S. F. (2013) ‘ Model-Selection-Based Approach for Calculating Cellular Multiplicity of Infection during Virus Colonization of Multi-Cellular Hosts’, PLoS One, 8: e64657. [DOI] [PMC free article] [PubMed] [Google Scholar]