

Abstract
Objective
A spectrum of seizure disorders is linked to mutations in Kv7.2 and Kv7.3 channels. Linking functional effects of identified mutations to their clinical presentation requires ongoing characterization of newly identified variants. In this study, we identified and functionally characterized a previously unreported mutation in the selectivity filter of Kv7.3.
Methods
Next‐generation sequencing was used to identify the Kv7.3[T313I] mutation in a family affected by neonatal seizures. Electrophysiological approaches were used to characterize the functional effects of this mutation on ion channels expressed in Xenopus laevis oocytes.
Results
Substitution of residue 313 from threonine to isoleucine (Kv7.3[T313I]) likely disrupts a critical intersubunit hydrogen bond. Characterization of the mutation in homomeric Kv7.3 channels demonstrated a total loss of channel function. Assembly in heteromeric channels (with Kv7.2) leads to modest suppression of total current when expressed in Xenopus laevis oocytes. Using a Kv7 activator with distinct effects on homomeric Kv7.2 vs heteromeric Kv7.2/Kv7.3 channels, we demonstrated that assembly of Kv7.2 and Kv7.3[T313I] generates functional channels.
Significance
Biophysical and clinical effects of the T313I mutation are consistent with Kv7.3 mutations previously identified in cases of pharmacoresponsive self‐limiting neonatal epilepsy. These findings expand our description of functionally characterized Kv7 channel variants and report new methods to distinguish molecular mechanisms of channel mutations.
Keywords: epilepsy, KCNQ2, KCNQ3, Kv7, neonatal, potassium channel, seizure
Key Points.
The Kv7.3[T313I] mutation was identified in a family with heritable pharmacoresponsive self‐limiting neonatal seizures
Functional characterization revealed that Kv7.3[T313I] mutant subunits assemble with Kv.7.2 and suppress currents, but have little effect on gating parameters
These findings validate Kv7.3[T313I] as a pathogenic mutation in neonatal epilepsy
1. INTRODUCTION
Mutations of the KCNQ2 and KCNQ3 voltage‐gated potassium channel genes have been implicated in the pathophysiology of a spectrum of seizure disorders, ranging from pharmacoresponsive self‐limiting neonatal epilepsy to severe epileptic encephalopathy. 1 , 2 , 3 , 4 , 5 , 6 , 7 The milder end of this spectrum has been historically referred to as BFNE (Benign Familial Neonatal Epilepsy) and is characterized by seizures that begin in the first few postnatal days, which are typically pharmacoresponsive and self‐limiting within a few months and rarely to a few years, and do not interfere with structural or cognitive development. 8 , 9 The term BFNE has been discouraged in recent position statements because the term “Benign” may minimize the impact of the disease on patients and caregivers. 10 One issue often encountered clinically is how to best predict long‐term outcomes and approach management for a neonate with new‐onset seizures. Increased access to genetic sequencing has allowed clinicians to rapidly identify genetic forms of epilepsy, but there is often still insufficient information for an accurate prediction of phenotype from genotype, particularly for mutations in KCNQ2 and KCNQ3, which can present with a wide range of epileptic severity and may have overlapping features. 11 , 12
KCNQ2 and KCNQ3 gene products, Kv7.2 and Kv7.3, form heterotetrameric potassium channels that encode the neuronal M‐current, 13 which plays a modulatory role on threshold properties and repetitive burst firing in response to excitatory stimuli. 14 , 15 , 16 , 17 Heteromeric assembly of Kv7.2 and Kv7.3 subunits enhances expression of channels in heterologous systems by roughly 10‐fold relative to homomeric channels of either subtype alone, 18 and so dysfunction in either subunit can be linked to a pathological outcome. In general, Kv7.2 is more frequently implicated in both self‐limiting seizures and epileptic encephalopathy phenotypes. Pathogenic variants are less frequently identified in Kv7.3. 19
Kv7.3 mutations have been reported throughout the channel sequence, including C‐terminal domains associated with calmodulin or PIP2 association and the voltage‐sensing domain. This is highlighted in Figure 1A, illustrating the location of pathogenic point mutations of Kv7.3 that are reported in clinVar, on the structural template of a recently reported Kv7.1 cryo‐EM structure. 20 There is a cluster of validated disease‐linked mutations in the Kv7.3 selectivity filter region, which underlies ion selectivity and permeation. 5 , 21 , 22 , 23 , 24 , 25 The geometry and stability of the selectivity filter are determined by several intra‐ and intersubunit interactions that are widely conserved among K+ channels. 26 , 27 , 28 In some channel types prone to selectivity filter mediated inactivation, the disruption of these bonds leads to pronounced alterations of the kinetics of inactivation or a total loss of channel conductance. 29 , 30 , 31 , 32 In contrast, M‐channels exhibit no apparent inactivation, and selectivity filter mutations can lead to a range of effects on overall channel function. 13 , 16 , 33
Figure 1.
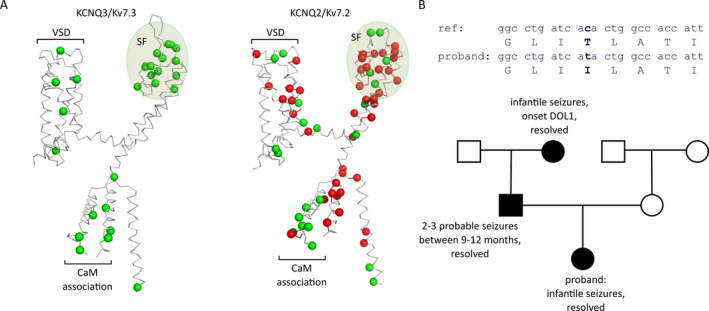
Inheritance of Kv7.2 and Kv7.3 mutations associated with epilepsy. A, Mutations in Kv7.2 or Kv7.3 associated with a documented case of epilepsy (compiled from ClinVar or RIKEE databases) are highlighted on molecular models of each channel. Mutations are color coded based on severity (green = BFNE, red = epileptic encephalopathy or other severe outcomes). Mutations that do not map to structural elements defined in the KCNQ1 cryo‐EM structure have been omitted (VSD, voltage‐sensing domain, SF, selectivity filter, CaM, Calmodulin). B, Pattern of inheritance of a neonatal seizure phenotype in a family carrying the Kv7.3[T313I] mutation. Upper, sequence alignment of the reference KCNQ3 gene and Kv7.3 protein in relation to the proband. The identified mutation [T313I] is highlighted in bold type. Lower, pedigree for the family characterized in our study with filled symbols indicating affected individuals
In this study, we report a family with a pattern of inheritance and symptoms indicating a diagnosis of pharmacoresponsive self‐limiting seizures (BFNE). The proband was identified to have a KCNQ3 variant, p.Thr313Ile, which is a previously unreported selectivity filter mutation in Kv7.3. A mutation has been reported in Kv7.2 channels at a homologous position (T274M), associated with a severe clinical outcome including profound global developmental delay, motor dysfunction, and remitting pharmacoresistant seizures. 7 , 33 , 34 , 35 We undertook the characterization of the electrophysiological effects of the T313I mutation in Kv7.3 homomeric and Kv7.2/Kv7.3 heteromeric channels, to describe in more detail the potential effects of selectivity filter mutations in this channel family. We also developed an assay using a subtype‐specific Kv7.2 activator (ICA‐069673) as a “fingerprinting” tool to distinguish functional Kv7.2/Kv7.3 heteromeric channels from Kv7.2 homomers. 36 , 37 , 38 Our findings highlight variable outcomes of mutations in this critical selectivity filter position on the overall function of Kv7.2/Kv7.3 heteromeric channels. We find that Kv7.2/Kv7.3 heteromers are relatively tolerant of the Kv7.3[T313I] mutation, and this may underlie the mild disease phenotype, in contrast to severe outcomes arising from Kv7.2[T274M]. 33
2. MATERIALS AND METHODS
2.1. Molecular biology and plasmid construction
The KCNQ3[T313I][A315T] and KCNQ3[T313I] mutation were generated by site‐directed mutagenesis in pSRC5 (gift from Dr. M. Taglialatela, University of Naples, Italy, and Dr. T. Jentsch, Max‐Delbrück‐Centrum für Molekulare Medizin, Germany) and pTLN plasmids encoding Kv7.3.
Primers used for mutagenesis for the KCNQ3[T313I] construct were as follows (together with their reverse complement primer):
KCNQ3[T313I]: 5′‐GCCTGATCATACTGGCCACCATTGGC‐3′
KCNQ3[T313I]: 5′‐GCCTGATCATGCTGGCCACCATTGGC‐3′
KCNQ3[A315T][T131I]: 5′‐GCCTGATCATACTGACCACCATTGGC‐3′
KCNQ2[T274I]: 5′‐GGGGCCTGATCATCCTGACCACCATTG‐3′
KCNQ2[T274M]: 5′‐GGGGCCTGATCATGCTGACCACCATTG‐3′
2.2. Two‐electrode voltage‐clamp expression and recording
Complementary RNA was transcribed from cDNA to express the monomeric KCNQ3 constructs in Xenopus laevis oocytes using the mMessage mMachine kit (Ambion). Constructs expressed in pSRC5 (KCNQ3[T313I][A315T] or (KCNQ3[A315T])) were linearized with ApaL1 and transcribed using a T7 primer. Constructs in pTLN (KCNQ2, KCNQ3, and KCNQ3[T313I]) were linearized with HpaI and transcribed using an SP6 primer. Oocyte preparation and RNA injection were performed as described previously, 39 under the approval of the University of Alberta Animal Care protocol AUP00001752. After injection, oocytes were incubated for 48 hours at 18°C before electrophysiological recording. Voltage‐clamp recordings were obtained in modified Ringer’s solution (in mmol/L): 116 NaCl, 2 KCl, 1 MgCl2, 0.5 CaCl2, and 5 HEPES, pH adjusted to 7.4 with NaOH, using an OC‐725C voltage clamp (Warner). Microelectrodes were backfilled with 3 mol/L KCl to obtain resistance between 0.1 and 1 MΩ. Data were processed using a Digidata 1440A acquisition system controlled by pClamp 10 software. For experiments using ICA‐069673, oocytes were incubated in 100 μmol/L ICA‐069673 in modified Ringer's solution for 3‐4 minutes before recording.
2.3. Data analysis and statistics
Statistical tests used throughout the manuscript are described in the corresponding figure legends. Gating parameters describing voltage dependence of channel activation were determined by fitting with a standard single‐component Boltzmann equation of the form:
where V 1/2 is the voltage where channels exhibit half‐maximal activation, and k is a slope factor reflecting the voltage range over which an e‐fold change in open probability (Po) is observed. The extent of tail current deactivation in the presence of ICA‐069673 was inferred by the ratio of the magnitude of instantaneous: peak current after the repolarization interval, rather than fitting tail current kinetics directly (these are often too slow to generate a meaningful fit).
2.4. DNA sequencing
Patient samples were sequenced clinically by an external provider (CeGAT GmbH, Tubingen, Germany). The proband was initially investigated using a metabolic/mitochondrial epilepsy panel in order to rule out potentially treatable metabolic causes. Given the family history, some individual genes associated with pharmacoresponsive self‐limiting seizures were also included in addition to the standard panel. The genes examined as part of the standard metabolic epilepsy clinical panel were as follows: AARS2, ABAT, ABCC8, ACY1, ADCK3, ADK, ADSL, ALDH5A1, ALDH7A1, AMT, ATIC, AUH, BCKDHA, BCKDHB, BCKDK, BCS1L, BTD, C10ORF2, CAD, CARS2, CNNM2, COQ4, COX8A, CPT1A, CPT2, D2HGDH, DARS2, DBT, DHFR, DLD, DNM1L, DPYD, EARS2, ETFA, ETFB, ETFDH, ETHE1, FARS2, FOLR1, FOXRED1, GAMT, GATM, GCDH, GCH1, GCK, GCSH, GFM1, GLDC, GLUD1, GLUL, GPHN, HADH, HLCS, HPD, 1DH2, INSR, ITPA, IVD, KCNJ11, L2HGDH, LIAS, MDH2, MLYCD, MMACHC, MOCS1, MOCS2, MT‐ATP6, MT‐TK, MT‐TL1, MTHFR, NARS2, NDUFA1, PC, PCBD1, PCCA, PCCB, PDHA1, PDHX, PDSS2, PET100, PHGDH, PNPO, POLG, PROSC, PSAT1, PSPH, PTS, QDPR, SDHA, SLC16A1, SLC19A3, SLC1A2, SLC25A1, SLC2A1, SLC46A1, SLC6A8, SLC6A9, SUOX, SURF1, VARS2. In addition, KCNQ2, KCNQ3, and PRRT2 sequences were added for analysis based on a suspicion of a BFNE diagnosis. The T313I mutation was identified in the proband, and in silico predictors suggested the variant was damaging. The T313I variant was classified as a VUS because this position had not been previously associated with disease and was not present in the EVS, gnomAD, or RIKEE databases. 40 Targeted testing was then used to determine the presence of the variant in other family members. Targeted testing confirmed the presence of the variant in the father and paternal grandmother who also have a history of early seizures and absence of the variant in the mother who has no history of seizure.
3. RESULTS
3.1. Proband features
The proband was a term infant female born after an unremarkable pregnancy and uncomplicated vaginal delivery. At 4 days of age, she began having spells described as “not breathing properly” while “holding her arms and legs out (flexed or extended) with a mild tremor and at times hyperextension of her back”. The events increased in frequency, beginning to cluster, and she was admitted to the hospital for workup. Seizures were suspected and she was loaded with 20 mg/kg of IV levetiracetam and continued with 10 mg/kg bid of maintenance. Video‐EEG monitoring was initiated. Six discrete, self‐limited electroclinical seizures were recorded in sleep and wakefulness in the first 24 hours of recording. The seizures lasted between 43 and 69 seconds (mean 54 seconds) and while the semiology varied slightly, the typical pattern entailed three clearly discernible stages: (a) wide eye opening, with occasional gaze deviation to the left, (b) tonic posturing of limbs and torso (typically left arm extension followed by arching of back and flection or extension of legs), and (c) clonic movements of bilateral arms and legs until offset. The ictal EEG correlate consisted of symmetric background attenuation with overriding low amplitude fast beta frequencies for ~15 seconds (tonic phase) followed by bilateral high amplitude spike‐sharp delta‐theta frequencies in a crescendo‐decrescendo manner (clonic phase) until offset. Interictally, the EEG showed bilateral symmetric and continuous background activity with mixture of delta‐theta frequencies and rare (<1/min) non‐specific negative sharp waves with independent maximum amplitude over T7, T8, C3, C4, or Cz. Normal wake‐sleep cycling was also seen.
A neurological examination revealed a normal non‐dysmorphic infant without focal neurological deficits. Brain MRI showed normal anatomy. On review of the pregnancy history, the patient’s mother was healthy, with a history of 3 previous early pregnancy losses. There was hyperemesis in the first trimester but pregnancy was otherwise unremarkable with no drug or alcohol exposures, and the patient was delivered at 40 weeks gestation. On review of the family history, the infant’s father had a history of seizures in early infancy (9‐12 months) that were self‐limiting. He also has attention deficit hyperactivity disorder but normal cognition. The patient’s paternal grandmother had seizures in infancy, which were also self‐limiting and normal cognition. There is also a paternal cousin with epilepsy. There was no history of seizure in the patient’s mother or maternal relatives (Figure 1B).
With the continuation of her seizures, the maintenance dose of IV levetiracetam was increased to 15 mg/kg bid without effect. She was then loaded with 10 mg/kg of IV phenobarbital and the seizures stopped. Given the efficacy of phenobarbital, her levetiracetam was discontinued and she was kept on maintenance phenobarbital (2.5 mg/kg bid). Due to the initial uncertainty of the etiology of this patient’s seizures, effective control only after initiating phenobarbital, and that the patient’s father had sporadic seizures in infancy (9–12 months), a maintenance dose was kept for safety until a clear etiology was found. The patient remained event‐free and a repeat EEG at 6 months of age was normal. With the normal EEG, lack of clinical events, and normal development, phenobarbital was weaned at 8 months of age. Seizures had remained after levetiracetam initiation; therefore, seizure cessation was suspected to be due to phenobarbital effects. Nevertheless, one cannot rule out spontaneous seizure remission. Seizures did not relapse despite being off anti‐seizure therapy, and she remains seizure‐free for more than 3 years. Development also remained normal in all domains, suggesting a clinical presentation consistent with self‐limiting pharmacoresponsive seizures.
Metabolic investigations for this patient were unremarkable. Clinical genetic testing had been initiated for this patient at seizure onset. Array CGH (60K) was negative. A 114 gene epilepsy panel through the clinical commercial laboratory CeGAT (see Materials and Methods) identified a variant of uncertain significance in KCNQ3, c.938C>T, p.Thr313Ile (NM_004519) (Figure 1B). This variant was not identified in the patient’s mother, but was present in the patient’s father and paternal grandmother (Figure 1B). This variant is absent from gnomAD and was not previously reported in ClinVar, EVS, or RIKEE databases. 40 , 41
3.2. KCNQ3[T313I] mutation eliminates homomeric Kv7.3 function
We were intrigued by both the biophysical and physiological consequences of the T313I mutation. The T313 position is one of several positions identified in patients with self‐limiting neonatal seizures, clustered in the region of the selectivity filter (Figure 1A). Due to the critical functional importance of this region, variants in the vicinity of the selectivity filter are often linked to disease (D305, A306, W308, W309, G310, I317, Y319, G320 are listed in ClinVar and linked to BFNE). Despite the essential role of the selectivity filter, the pathology arising from these mutants is relatively mild and self‐resolving. A previous study of an analogous mutation in the widely studied Shaker potassium channel, a model system for understanding many basic principles of voltage‐dependent ion channel function, highlighted the importance of this particular site in maintaining the stability of the selectivity filter. 29 Kv7.3 position T313 is analogous to Shaker residue T439, which is predicted to form an intersubunit hydrogen bond with the selectivity filter residue Y445 (GYG), highlighted in Figure 2A (2 of 4 potential H‐bonds are shown). In Shaker channels, disruption of this H‐bond in even a single subunit caused a profound suppression of overall channel current and acceleration of inactivation by ~100‐fold. 29 These powerful effects were attributed to the intersubunit nature of the interaction, leading to a propagated effect of a mutation from a single subunit (Figure 2A). Similarly, the Kv7.2[T274M] mutation of an analogous position in Kv7.2 has been associated with severe epileptic encephalopathy and dominant negative effects when co‐expressed with Kv7.3. 33 However, the physiological outcomes in this family carrying the Kv7.3[T313I] mutation were much less severe. We initially characterized the effects of the T313I mutation in homomeric Kv7.3, using the A315T mutation to enable expression of functional homotetramers. 42 , 43 Consistent with prior studies in Shaker channels, we observed that the T313I mutation virtually abolished functional current relative to Kv7.3[A315T] channels (Figure 2B,C).
Figure 2.
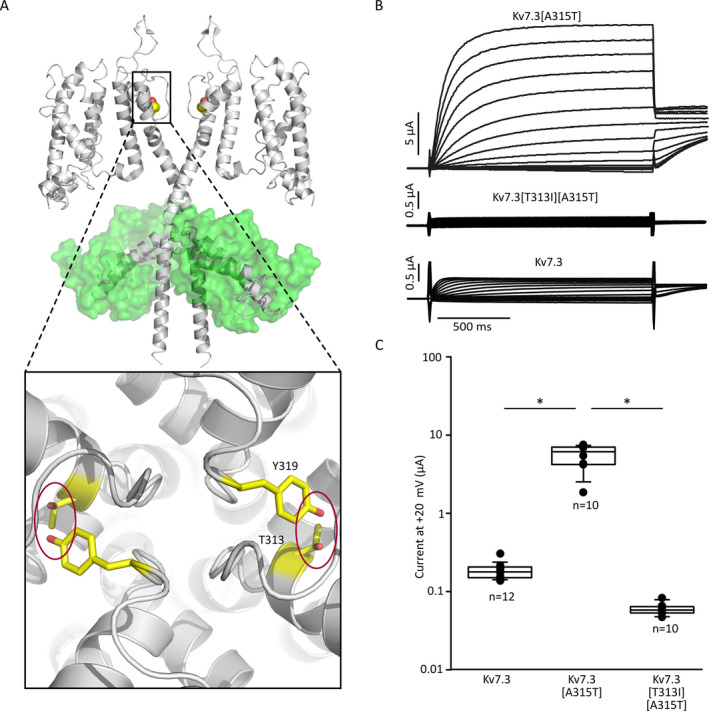
Kv7.3[T313I] abolishes Kv7.3 function. A, Molecular model of essential intersubunit hydrogen bond between conserved residues T313 and Y319 located in the selectivity signature sequence. B, Two‐electrode voltage‐clamp recordings from Xenopus laevis oocytes expressing Kv7.3[A315T] and Kv7.3[A315T][T313I]. Oocytes were held at −80 mV and depolarized for 1.5s to voltages between −140 mV and +40 mV (in 10‐mV steps) followed by repolarization to −20 mV test pulse. Current amplitudes at +20 mV of Kv7.3 [A315T] and Kv7.3[A315T] [T313I] were compared using a student’s t test (* indicates P<0.05 relative to A315T alone)
3.3. Kv7.3[T313I] attenuates currents in Kv7.2/Kv7.3 heteromers
Although this profound effect of Kv7.3[T313I] was consistent with previous reports in Shaker channels and Kv7.2, it does not address the effects of the mutation in heteromeric Kv7.2/Kv7.3 channels that predominate in vivo. 19 , 44 To investigate the effects of the T313I mutation in heteromeric channels, we co‐expressed different ratios of Kv7.2, Kv7.3, and Kv7.3[T313I] to simulate different heteromeric assemblies that may occur in vivo. Wild‐type Kv7.2/Kv7.3 heteromeric channels (1:1 mRNA injection ratio) generated larger currents relative to homomeric Kv7.2 channels. When Kv7.2 and Kv7.3[T313I] subunits were injected in a 1:1 ratio, the current was reduced significantly relative to the wild‐type heteromer (Figure 3A,B).
Figure 3.
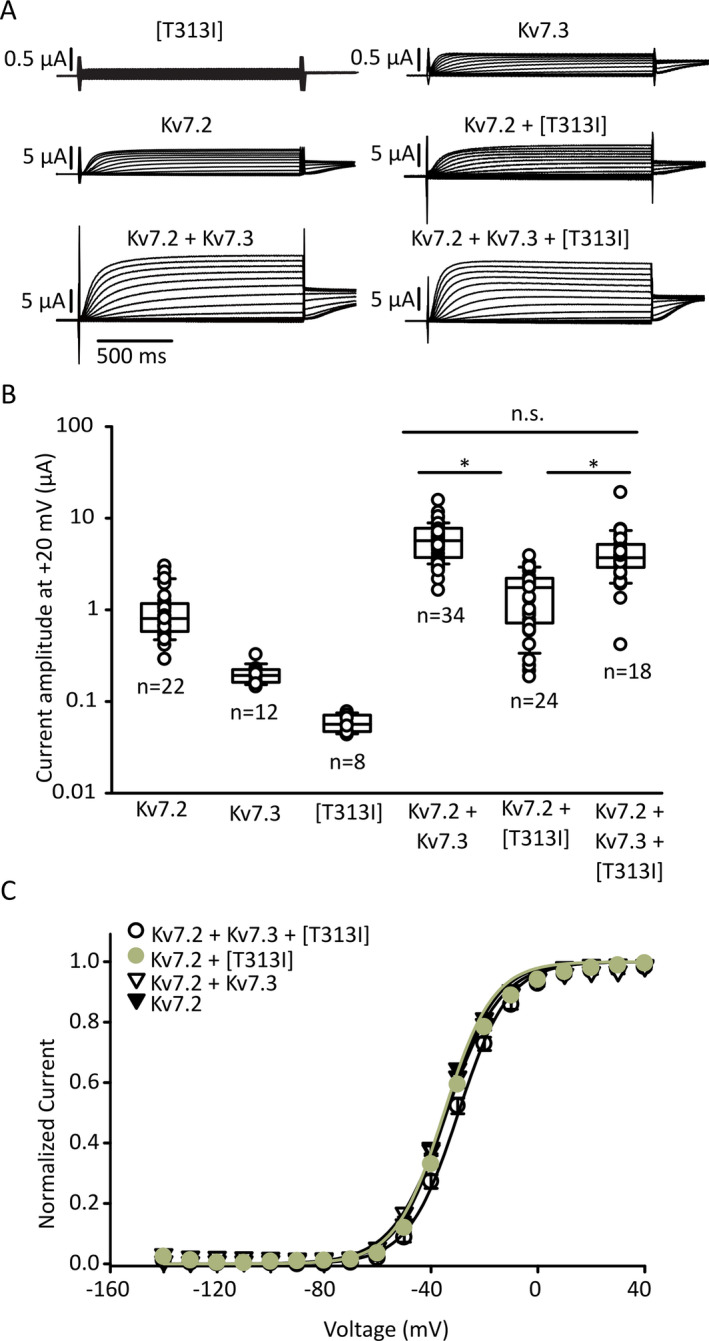
Co‐expression with Kv7.2 and Kv7.3[T313I] reduces heteromeric channel function with no effect on gating. A, Two‐electrode voltage‐clamp sample traces from oocytes expressing various combinations of Kv7.2 and Kv7.3 (injected with a total of 50 ng of mRNA per group). The voltage step protocol is the same as Figure 2B. B, Current amplitudes after a 1.5 s + 20 mV voltage step. Current magnitudes were compared using one‐way ANOVA, followed by Tukey’s post hoc test (* indicates P < 0.05). C, Conductance‐voltage relationships were collected using the protocol in panel (A), using tail current magnitudes (−20 mV) to assess the extent of channel opening during the conditioning step. Fitted gating parameters were (mean ± S.E.M.): for Kv7.2 + Kv7.3 +[T313I], k = 9.4 ± 0.3 mV, V1/2 = −30 ± 1 mV; for Kv7.2 + [T313I], k = 9.1 ± 0.3 mV, V1/2 = −35.1 ± 0.5 mV; for Kv7.2 + Kv7.3, k = 10.2 ± 0.3 mV, V1/2 = −34.2 ± 0.3 mV; for Kv7.2 homomers, k = 9.2 ± 0.2 mV, V1/2 = −34.2 ± 0.6 mV. No significant differences in gating parameters were detected
We also tested the mixed expression of WT Kv7.3 and Kv7.3[T313I] channels with Kv7.2, to mimic the heterozygous genotype of the proband (Figure 3A,B). A 1:0.5:0.5 (Kv7.2:Kv7.3:Kv7.3[T313I]) RNA injection ratio did not suppress current magnitude relative to WT Kv7.2/Kv7.3 (Figure 3A,B). However, current magnitudes in these experiments are variable and there may be assembly of various channel stoichiometries even with fixed RNA ratios. 45 No difference in voltage‐dependent activation was detected in oocytes expressing different ratios of Kv7.2, Kv7.3, and Kv7.3[T313I] (Figure 3C). Although these experiments illustrate modest current suppression by Kv7.3[T313I] in certain subunit combinations, they do not fully reveal the nature of Kv7.3[T313I] mutant subunits on function of heteromeric channels, particularly in light of the severe phenotype arising from the Kv7.2[T274M] mutation. 33 That is, it was not clear whether currents generated from co‐expression of Kv7.3[T313I] and Kv7.2 were generated by heteromeric channels, or from Kv7.2 homomers that did not assemble with Kv7.3[T313I].
3.4. Heteromeric composition determined by ICA‐069673 sensitivity
We recently recognized that the functional effects of the Kv7 activator ICA‐069673 depend strongly on subunit composition. 37 Kv7.2 homomeric channels exhibit profound deceleration of deactivation in the presence of ICA‐069673, whereas these effects are much weaker in homomeric Kv7.2/Kv7.3 channels (due to weak/absent ICA‐069673 sensitivity of Kv7.3). We used this pharmacological “fingerprint” to investigate whether currents observed in the Kv7.2/Kv7.3[T313I] condition were generated by Kv7.2/Kv7.3[T313I] heteromers or only arise from Kv7.2 homomers (due to strong current suppression by Kv7.3[T313I]).
Using oocytes continuously perfused with a saturating concentration of 100 μmol/L ICA‐069673, we delivered a voltage step protocol that depolarized channels to +20 mV for 1.5 seconds, followed by repolarization to a range of voltages for 12 seconds (Figure 4A). This was followed by a second step to +20 mV to assess the extent of channel closure during the repolarization interval, based on the instantaneous vs activating current fractions elicited by the voltage step (expanded time scales of the second depolarizing step are illustrated in the right hand panels of Figure 4A). Kv7.2 homomeric channels in 100 μmol/L ICA‐069673 are characterized by extremely slow channel closure, resulting in a large instantaneous current fraction, even after repolarization at very negative voltages. In contrast, heteromeric Kv7.2/Kv7.3 channels exhibit much more prominent deactivation during this interval, which results in smaller instantaneous currents (Figure 4A,B). In 100 μmol/L ICA‐069673, Kv7.2 homomeric channels exhibited ~70% instantaneous current after a 12 second repolarization at −120 mV, whereas heteromeric channels generated <20% instantaneous current (Figure 4B,C). Co‐expression of 1:1 Kv7.2:Kv7.3[T313I] channels exhibited an intermediate behavior, but was distinct from WT Kv7.2 homomers. This observation suggests that heteromeric Kv7.2:Kv7.3[T313I] channels retain function, but there is also likely a contribution of homomeric Kv7.2 channels (Figure 4C). Co‐expression to mimic the heterozygous genotype (1:0.5:0.5, Kv7.2:Kv7.3: Kv7.3[T313I]) displayed a similar ICA‐069673 response to the wild‐type heterozygous control (Figure 4C).
Figure 4.
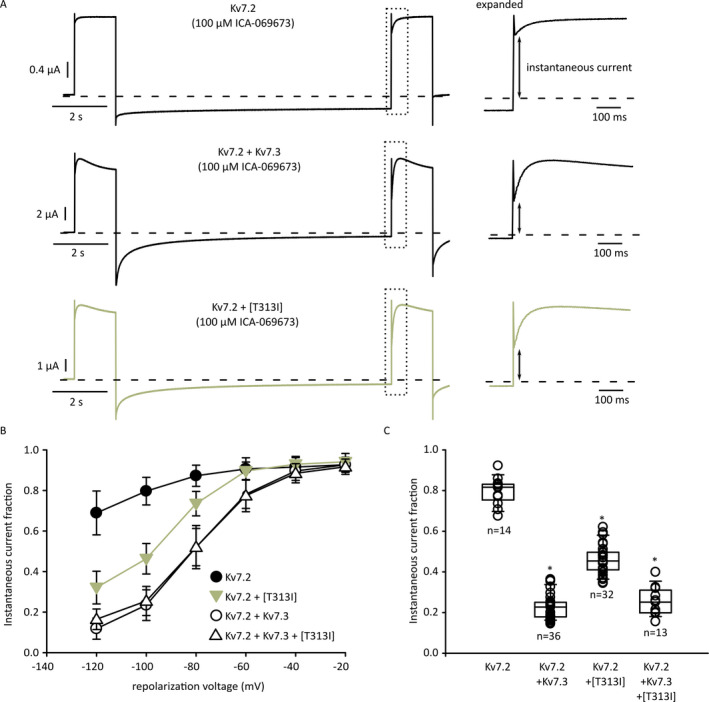
Reduced ICA‐069673 sensitivity of Kv7.2/Kv7.3[T313I] heteromeric channels. A, Example currents of two‐electrode voltage‐clamp recordings. Oocytes were depolarized to +20 mV and repolarized for 12 s in a step‐down manner (−20 mV per sweep), followed by another +20 mV depolarizing pulse to determine instantaneous current at −100 mV. Currents within the dashed box are illustrated on an expanded time scale in the right panels, showing the assessment of instantaneous current levels in different experimental conditions. B, Fractional instantaneous current after incubation with 100 μmol/L ICA‐069673 was measured as indicated by the arrows in panel (A), data are shown as mean ± SEM. C, Fractional instantaneous current (repolarization voltage of −100 mV) for various combinations of Kv7.2, Kv7.3, and Kv7.3[T313I], shown on a cell‐by‐cell basis (Kruskal‐Wallis one‐way ANOVA on ranks, * indicates P < 0.05 relative to Kv7.2 homomeric channels)
3.5. Distinct effects of Kv7.2 and Kv7.3 mutations
In order to investigate the functional basis for different clinical outcomes of Kv7.2 and Kv7.3 mutations reported at analogous positions, we also tested the effects of the Kv7.3[T313M] mutation (equivalent to Kv7.2[T274M]) and the Kv7.2[T274I/M] mutations (Figure 5). We measured current amplitude (Figure 5A) and also used the ICA‐069673 response described in Figure 4 to confirm assembly of Kv7.2/Kv7.3 heteromers (Figure 5B). Among these mutant channels, the Kv7.3[T313I] mutant stood out for its comparably mild effects on current magnitude. Co‐expression of Kv7.3[T313M] with Kv7.2 led to a significantly stronger suppression of current relative to Kv7.3[T313I]. Furthermore, co‐expression of Kv7.2[T274M] or Kv7.2[T274I] led to even further suppression of heteromeric Kv7.2/Kv7.3 channel currents. For most of these subunit combinations, we could detect currents resembling Kv7.2/Kv7.3 heteromeric channels based on their weak sensitivity to ICA‐069673 (Figure 5B), although the Kv7.2[T274I] + Kv7.3 combination did not generate sufficient currents in any oocytes to confidently assess any properties of the elicited currents. Overall, these findings confirm that the various Kv7.2 and Kv7.3 mutants can assemble with their WT partner in heteromeric channels, but with very markedly different outcomes on overall channel function.
Figure 5.
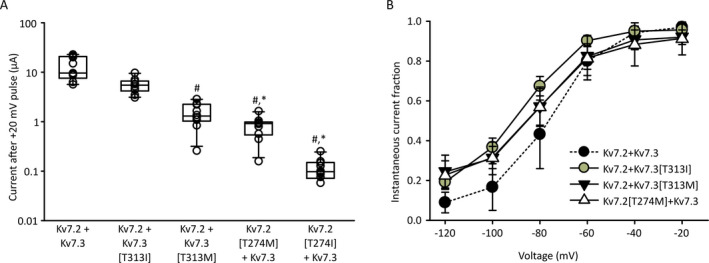
Variable outcomes of homologous selectivity filter mutations in Kv7.2 and Kv7.3. A, Current magnitude was measured at +20 mV in Xenopus oocytes injected with Kv7.2 + Kv7.3, or various combinations of channel mutants and their wild‐type counterpart, as indicated. Currents were recording 48‐56 h after injection (n = 10 per condition, one‐way ANOVA on ranks and Tukey post hoc test, # indicates P < 0.05 relative to Kv7.2/Kv7.3, * indicates P < 0.05 relative to Kv7.2/Kv7.3[T313I]). B, Instantaneous tail current magnitudes were recorded as described in Figure 4 in the presence of 100 μmol/L ICA‐069673, for all possible combinations of injected channels (note that Kv7.3 + Kv7.2[T274I] did not yield current sizes that could be confidently assessed or analyzed)
4. DISCUSSION
Determining genotype‐phenotype correlation of mutations in Kv7.2 and Kv7.3 is of significant interest in understanding the prognosis and pathogenesis of affected patients. In addition, characterization of variants is valuable for understanding assembly, function, and regulation of Kv7.2/Kv7.3 heteromeric channels, which play a significant modulatory role in the central nervous system. Delineation of how variants impact channels also informs development of targeted drug therapeutics. In this study, we report a novel mutation in the selectivity filter region of Kv7.3 channels, predicted to form an intersubunit interaction that has been previously shown to strongly disrupt the function of Shaker channels, and M‐channels when mutated in Kv7.2. 29 , 33 The T313I mutation in Kv7.3 was identified in a family presenting with neonatal self‐resolving pharmacoresponsive epilepsy (frequently referred to as BFNE) and was not previously reported in gnomAD, clinVar, or the KCNQ disease‐specific RIKEE databases. Although this variant was initially classified as a variant of uncertain significance, we feel our findings classify this mutation as pathogenic given its impact on channel function (Figure 3), and due to segregation of the variant with disease in the family.
Previous work in Shaker channels demonstrated the contribution of this position to channel function, based on formation of a H‐bond between T439 (equivalent to Kv7.3[T313I]) and Y445 (in the GYG selectivity filter sequence of an adjacent subunit). An important comparison was drawn to a commonly used non‐conducting mutant of Shaker (W434F), illustrating much greater tolerance of the W434F relative to either T439V or Y445F mutations, which both cause a dramatic loss of function even when expressed in a single channel subunit. 29 , 30 , 31 This difference was attributed to the intersubunit nature of the T439 interaction. This previous finding of an essential structural role for this amino acid position motivated us to investigate the generality of this finding and the differing clinical outcomes in patients with mutations at this position in Kv7.3 in our study, versus previously described mutations at this position in Kv7.2 with severe clinical outcomes. 33
The overall importance of position T313 (and its likely H‐bond interaction with Y319) is highlighted by the observation that Kv7.3[T313I] subunits cannot form functional homomeric channels, even using a high‐expressing Kv7.3[A315T] background channel (Figure 2). However, the effects of the T313I mutation in heteromeric channels (co‐expressed with Kv7.2) are much less pronounced (Figure 3). Modestly smaller current magnitudes are observed in the Xenopus oocyte system used for this study, and no significant differences in gating properties were observed between Kv7.2/Kv7.3[T313I] heteromeric channels and Kv7.2/Kv7.3 wild‐type channels (Figure 3). This observation is a departure from the powerful effects of disruption of intersubunit selectivity filter interactions in Shaker channels and likely highlights that other intersubunit interactions contribute significantly to stabilizing the selectivity filter of Kv7 channels.
Our findings also indicate variable outcomes of mutations at this position, depending on the amino acid substitution and mutated Kv7 subtype. For example, a Kv7.2 mutation at the analogous position (Kv7.2[T274M]) is linked to severe epileptic encephalopathy and global delay. 7 , 33 , 34 , 35 Functional characterization of this mutation in Xenopus oocytes results in a more pronounced effect on current suppression of heteromeric channels, relative to Kv7.3[T313I] (Figure 5). 33 This difference in functional effects in heteromeric channels correlates with the severity of clinical outcomes. We investigated this difference in more detail by comparing equivalent mutations in both Kv7.2 and Kv7.3 and demonstrated non‐equivalent effects of these mutations in these different subtypes. Overall, mutations in Kv7.3 (T313I or T313M) were less severe than their comparators in Kv7.2 (Figure 5). In a broad context, this finding is consistent with reports that neonatal epilepsy (including severe epileptic encephalopathy) is far more commonly reported to arise from Kv7.2 mutations as opposed to Kv7.3 (Figure 1A). 7 , 40 , 46 However, in terms of biophysical effects on channel function, there is not an obvious explanation for these diverging effects of Kv7.2 and Kv7.3. In addition to this apparent subtype‐dependent effect of certain mutations, other differences in physiological regulation of Kv7.2 and Kv7.3 likely contribute to the general differences in severity of diseases between these subtypes. For instance, Kv7.2 expression is more prominent than Kv7.3 in early development and infancy, 47 , 48 and these subtypes may contribute differently to channels with alternative stoichiometries (perhaps Kv7.2 homomeric channels, or other assemblies). 49 , 50 , 51 , 52 Another possibility is more penetrant effects of Kv7.2 versus Kv7.3 mutations on channel function may translate into stronger effects on structural and electrical remodeling of the axon initial segment, as has been observed in the presence of Kv7 inhibitors. 53
Lastly, we hope to highlight the use of subtype‐selective Kv7 activators such as ICA‐069673 for investigating the assembly of heteromeric channels and inferring the functional impact of disease‐linked mutations in heteromeric channels. Since ICA‐069673 is strongly selective for Kv7.2 over Kv7.3 subunits, Kv7.2/Kv7.3 heteromeric channels have a distinct response from Kv7.2 homotetramers (Figure 4). This property reveals the impact of mutated versions of Kv7.3 on heteromeric channel function and clearly distinguishes homomeric Kv7.2 channels versus heteromeric assembly of Kv7.2/Kv7.3. Another useful approach used previously has been to exploit the differential sensitivity of Kv7.2 and Kv7.3 to extracellular tetraethylammonium. 54 , 55 , 56 Our alternative approach using a more specific pharmacological agent is useful for resolution of distinct Kv7.2/Kv7.3 stoichiometries. Our findings also further validate prior reports of the stoichiometry‐dependent effects of ICA‐069673 and related compounds.
In summary, our study reports a novel variant in the Kv7.3 selectivity filter, linked to BFNE. The biophysical consequences of this mutation primarily involve suppression of function, without pronounced effects on voltage‐dependent gating. The assembly of functional channels comprising Kv7.2 and Kv7.3[T313I] subunits is unambiguously demonstrated based on sensitivity to a Kv7.2‐specific activator compound. Interestingly, the biophysical consequences of the Kv7.3[T313I] are less severe than reported in the prototypical Shaker potassium channel.
CONFLICT OF INTEREST
None of the authors have any conflicts of interest to disclose. We confirm that we have read the Journal’s position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
AUTHOR CONTRIBUTIONS
J. Maghera, J. Li, and S.M. Lamothe collected experimental data, performed molecular biology, and analyzed data. M. Braun, J.P. Appendino, and BPY Au collected clinical data and interacted with patients. All authors approved the final version of the manuscript.
ACKNOWLEDGMENTS
This work was funded by CIHR operating grant MOP‐97988 to H.T. Kurata. J. Li was supported by a CIHR CGS‐M graduate award. S.M. Lamothe was supported by the Dr. Rowland and Muriel Haryett Neuroscience Fellowship, University of Alberta Neuroscience and Mental Health Institute. H.T. Kurata was supported by a CIHR New Investigator Salary Award and the Alberta Diabetes Institute. We are also very grateful for the essential contributions of genetic counselors Setareh Ashtiani and Heather Barnes, for assistance with handling of genetic samples and liaising with the affected family.
Maghera J, Li J, Lamothe SM, et al. Familial neonatal seizures caused by the Kv7.3 selectivity filter mutation T313I. Epilepsia Open. 2020;5:562–573. 10.1002/epi4.12438
REFERENCES
- 1. Rogawski MA. KCNQ2/KCNQ3 K+ channels and the molecular pathogenesis of epilepsy: implications for therapy. Trends Neurosci. 2000;23:393–8. [DOI] [PubMed] [Google Scholar]
- 2. Castaldo P, del Giudice EM, Coppola G, Pascotto A, Annunziato L, Taglialatela M. Benign familial neonatal convulsions caused by altered gating of KCNQ2/KCNQ3 potassium channels. J Neurosci. 2002;22:RC199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bassi MT, Balottin U, Panzeri C, Piccinelli P, Castaldo P, Barrese V, et al Functional analysis of novel KCNQ2 and KCNQ3 gene variants found in a large pedigree with benign familial neonatal convulsions (BFNC). Neurogenetics. 2005;6:185–93. [DOI] [PubMed] [Google Scholar]
- 4. Singh NA, Charlier C, Stauffer D, DuPont BR, Leach RJ, Melis R, et al A novel potassium channel gene, KCNQ2, is mutated in an inherited epilepsy of newborns. Nat Genet. 1998;18:25–9. [DOI] [PubMed] [Google Scholar]
- 5. Charlier C, Singh NA, Ryan SG, Lewis TB, Reus BE, Leach RJ, et al A pore mutation in a novel KQT‐like potassium channel gene in an idiopathic epilepsy family. Nat Genet. 1998;18:53–5. [DOI] [PubMed] [Google Scholar]
- 6. Biervert C, Schroeder BC, Kubisch C, Berkovic SF, Propping P, Jentsch TJ, et al A potassium channel mutation in neonatal human epilepsy. Science. 1998;279:403–6. [DOI] [PubMed] [Google Scholar]
- 7. Weckhuysen S, Mandelstam S, Suls A, Audenaert D, Deconinck T, Claes LRF, et al KCNQ2 encephalopathy: emerging phenotype of a neonatal epileptic encephalopathy. AnnNeurol. 2012;71:15–25. [DOI] [PubMed] [Google Scholar]
- 8. Pressler R, Cilio M, Mizrahi E, Moshé S, Nunes M, Plouin P, et al.The ILAE Classification of Seizures & the Epilepsies: Modification for Seizures in the Neonate. Proposal from the ILAE Task Force on Neonatal Seizures. Epilepsia. https://www.ilae.org/guidelines/definition‐and‐classification/neonatal‐seizure‐classification [DOI] [PubMed]
- 9. Fisher RS, Acevedo C, Arzimanoglou A, Bogacz A, Cross JH, Elger CE, et al ILAE official report: a practical clinical definition of epilepsy. Epilepsia. 2014;55:475–82. [DOI] [PubMed] [Google Scholar]
- 10. Scheffer IE, Berkovic S, Capovilla G, Connolly MB, French J, Guilhoto L, et al ILAE classification of the epilepsies: position paper of the ILAE Commission for Classification and Terminology. Epilepsia. 2017;58:512–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Miceli F, Soldovieri MV, Joshi N, Weckhuysen S, Cooper E, Taglialatela M. KCNQ2‐related disorders In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJ, Stephens K, et al., editors. GeneReviews® [Internet]. Seattle, WA: University of Washington, Seattle; 1993. [PubMed] [Google Scholar]
- 12. Miceli F, Soldovieri MV, Joshi N, Weckhuysen S, Cooper EC, Taglialatela M. KCNQ3‐related disorders In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJ, Stephens K, et al., editors. GeneReviews® [Internet]. Seattle, WA: University of Washington, Seattle; 1993. [PubMed] [Google Scholar]
- 13. Wang HS, Pan Z, Shi W, Brown BS, Wymore RS, Cohen IS, et al KCNQ2 and KCNQ3 potassium channel subunits: molecular correlates of the M‐channel. Science. 1998;282:1890–3. [DOI] [PubMed] [Google Scholar]
- 14. Shah M, Mistry M, Marsh SJ, Brown DA, Delmas P. Molecular correlates of the M‐current in cultured rat hippocampal neurons. J Physiol. 2002;544:29–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Adams PR, Brown DA. Synaptic inhibition of the M‐current: slow excitatory post‐synaptic potential mechanism in bullfrog sympathetic neurones. J Physiol. 1982;332:263–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Brown DA, Adams PR. Muscarinic suppression of a novel voltage‐sensitive K+ current in a vertebrate neurone. Nature. 1980;283:673–6. [DOI] [PubMed] [Google Scholar]
- 17. Selyanko AA, Hadley JK, Wood IC, Abogadie FC, Jentsch TJ, Brown DA. Inhibition of KCNQ1‐4 potassium channels expressed in mammalian cells via M1 muscarinic acetylcholine receptors. J Physiol. 2000;522(Pt 3):349–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yang W‐P, Levesque PC, Little WA, Conder ML, Ramakrishnan P, Neubauer MG, et al Functional expression of two KvLQT1‐related potassium channels responsible for an inherited idiopathic epilepsy. J Biol Chem. 1998;273:19419–23. [DOI] [PubMed] [Google Scholar]
- 19. Jentsch TJ. Neuronal KCNQ potassium channels:physislogy and role in disease. Nat Rev Neurosci. 2000;1:21–30. [DOI] [PubMed] [Google Scholar]
- 20. Sun J, MacKinnon R. Cryo‐EM structure of a KCNQ1/CaM complex reveals insights into congenital long QT syndrome. Cell. 2017;169(6):1042–1050.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hirose S, Zenri F, Akiyoshi H, Fukuma G, Iwata H, Inoue T, et al A novel mutation of KCNQ3 (c.925T–>C) in a Japanese family with benign familial neonatal convulsions. Ann Neurol. 2000;47:822–6. [PubMed] [Google Scholar]
- 22. Singh NA, Westenskow P, Charlier C, Pappas C, Leslie J, Dillon J, et al KCNQ2 and KCNQ3 potassium channel genes in benign familial neonatal convulsions: expansion of the functional and mutation spectrum. Brain J Neurol. 2003;126:2726–37. [DOI] [PubMed] [Google Scholar]
- 23. Schroeder BC, Kubisch C, Stein V, Jentsch TJ. Moderate loss of function of cyclic‐AMP‐modulated KCNQ2/KCNQ3 K+ channels causes epilepsy. Nature. 1998;396:687–90. [DOI] [PubMed] [Google Scholar]
- 24. Soldovieri MV, Boutry‐Kryza N, Milh M, Doummar D, Heron B, Bourel E, et al Novel KCNQ2 and KCNQ3 mutations in a large cohort of families with benign neonatal epilepsy: first evidence for an altered channel regulation by syntaxin‐1A. Hum Mutat. 2014;35:356–67. [DOI] [PubMed] [Google Scholar]
- 25. Sands TT, Balestri M, Bellini G, Mulkey SB, Danhaive O, Bakken EH, et al Rapid and safe response to low‐dose carbamazepine in neonatal epilepsy. Epilepsia. 2016;57:2019–30. [DOI] [PubMed] [Google Scholar]
- 26. MacKinnon R, Yellen G. Mutations affecting TEA blockade and ion permeation in voltage‐activated K+ channels. Science. 1990;250:276–9. [DOI] [PubMed] [Google Scholar]
- 27. Doyle DA, Morais Cabral J, Pfuetzner RA, Kuo A, Gulbis JM, Cohen SL, et al The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science. 1998;280:69–77. [DOI] [PubMed] [Google Scholar]
- 28. Zhou Y, Morais‐Cabral JH, Kaufman A, MacKinnon R. Chemistry of ion coordination and hydration revealed by a K+ channel‐Fab complex at 2.0 A resolution. Nature. 2001;414:43–8. [DOI] [PubMed] [Google Scholar]
- 29. Pless SA, Galpin JD, Niciforovic AP, Kurata HT, Ahern CA. Hydrogen bonds as molecular timers for slow inactivation in voltage‐gated potassium channels. eLife. 2013;2:e01289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yang Y, Yan Y, Sigworth FJ. How does the W434F mutation block current in Shaker potassium channels? J Gen Physiol. 1997;109:779–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Perozo E, MacKinnon R, Bezanilla F, Stefani E. Gating currents from a nonconducting mutant reveal open‐closed conformations in Shaker K+ channels. Neuron. 1993;11:353–8. [DOI] [PubMed] [Google Scholar]
- 32. Kurata HT, Fedida D. A structural interpretation of voltage‐gated potassium channel inactivation. Prog Biophys Mol Biol. 2006;92:185–208. [DOI] [PubMed] [Google Scholar]
- 33. Orhan G, Bock M, Schepers D, Ilina EI, Reichel SN, Löffler H, et al Dominant‐negative effects of KCNQ2 mutations are associated with epileptic encephalopathy. Ann Neurol. 2014;75:382–94. [DOI] [PubMed] [Google Scholar]
- 34. Milh M, Boutry‐Kryza N, Sutera‐Sardo J, Mignot C, Auvin S, Lacoste C, et al Similar early characteristics but variable neurological outcome of patients with a de novo mutation of KCNQ2. Orphanet J Rare Dis. 2013;8:80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Milh M, Lacoste C, Cacciagli P, Abidi A, Sutera‐Sardo J, Tzelepis I, et al Variable clinical expression in patients with mosaicism for KCNQ2 mutations. Am J Med Genet A. 2015;167A:2314–8. [DOI] [PubMed] [Google Scholar]
- 36. Wang AW, Yau MC, Wang CK, Sharmin N, Yang RY, Pless SA, et al Four drug‐sensitive subunits are required for maximal effect of a voltage sensor–targeted KCNQ opener. J Gen Physiol. 2018;150:1432–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wang CK, Lamothe SM, Wang AW, Yang RY, Kurata HT. Pore‐ and voltage sensor‐targeted KCNQ openers have distinct state‐dependent actions. J Gen Physiol. 2018;150:1722–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Li J, Maghera J, Lamothe SM, Marco EJ, Kurata HT. Heteromeric assembly of truncated neuronal Kv7 channels: implications for neurological disease and pharmacotherapy. Mol Pharmacol. 2020;98(3):192‐202. [DOI] [PubMed] [Google Scholar]
- 39. Shin TM, Smith RD, Toro L, Goldin AL. [29] High‐level expression and detection of ion channels in Xenopus oocytes In: Methods in enzsymology [Internet]. Academic Press; 1998:529–56. (Ion Channels Part B; vol. 293). [DOI] [PubMed] [Google Scholar]
- 40. Millichap JJ, Park KL, Tsuchida T, Ben‐Zeev B, Carmant L, Flamini R, et al KCNQ2 encephalopathy: Features, mutational hot spots, and ezogabine treatment of 11 patients. Neurol Genet. 2016;2:e96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, et al The mutational constraint spectrum quantified from variation in 141,456 humans. bioRxiv. 2020;531210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Etxeberria A, Santana‐Castro I, Regalado MP, Aivar P, Villarroel A. Three mechanisms underlie KCNQ2/3 heteromeric potassium M‐channel potentiation. J Neurosci Off J Soc Neurosci. 2004;24:9146–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Gomez‐Posada JC, Etxeberria A, Roura‐Ferrer M, Areso P, Masin M, Murrell‐Lagnado RD, et al A pore residue of the KCNQ3 potassium M‐channel subunit controls surface expression. J Neurosci. 2010;30:9316–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Delmas P, Brown DA. Pathways modulating neural KCNQ/M (Kv7) potassium channels. Nat Rev Neurosci. 2005;6:850–62. [DOI] [PubMed] [Google Scholar]
- 45. Stewart AP, Gómez‐Posada JC, McGeorge J, Rouhani MJ, Villarroel A, Murrell‐Lagnado RD, et al The Kv7.2/Kv7.3 heterotetramer assembles with a random subunit arrangement. J Biol Chem. 2012;287:11870–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Goto A, Ishii A, Shibata M, Ihara Y, Cooper EC, Hirose S. Characteristics of KCNQ2 variants causing either benign neonatal epilepsy or developmental and epileptic encephalopathy. Epilepsia. 2019;60:1870–80. [DOI] [PubMed] [Google Scholar]
- 47. Kanaumi T, Takashima S, Iwasaki H, Itoh M, Mitsudome A, Hirose S. Developmental changes in KCNQ2 and KCNQ3 expression in human brain: possible contribution to the age‐dependent etiology of benign familial neonatal convulsions. Brain Dev. 2008;30:362–9. [DOI] [PubMed] [Google Scholar]
- 48. Tinel N, Lauritzen I, Chouabe C, Lazdunski M, Borsotto M. The KCNQ2 potassium channel: splice variants, functional and developmental expression. Brain localization and comparison with KCNQ3. FEBS Lett. 1998;438:171–6. [DOI] [PubMed] [Google Scholar]
- 49. Tzingounis AV, Heidenreich M, Kharkovets T, Spitzmaul G, Jensen HS, Nicoll RA, et al The KCNQ5 potassium channel mediates a component of the afterhyperpolarization current in mouse hippocampus. Proc Natl Acad Sci U S A. 2010;107:10232–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Soh H, Park S, Ryan K, Springer K, Maheshwari A, Tzingounis AV. Deletion of KCNQ2/3 potassium channels from PV+ interneurons leads to homeostatic potentiation of excitatory transmission. eLife. 2018;7:e38617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Bal M, Zhang J, Zaika O, Hernandez CC, Shapiro MS. Homomeric and heteromeric assembly of KCNQ (Kv7) K+ channels assayed by total internal reflection fluorescence/fluorescence resonance energy transfer and patch clamp analysis. J Biol Chem. 2008;283:30668–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Robbins J, Passmore GM, Abogadie FC, Reilly JM, Brown DA. Effects of KCNQ2 gene truncation on M‐type Kv7 potassium currents. PLoS One. 2013;8:e71809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lezmy J, Lipinsky M, Khrapunsky Y, Patrich E, Shalom L, Peretz A, et al M‐current inhibition rapidly induces a unique CK2‐dependent plasticity of the axon initial segment. Proc Natl Acad Sci U A. 2017;114:E10234–E10243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Hadley JK, Noda M, Selyanko AA, Wood IC, Abogadie FC, Brown DA. Differential tetraethylammonium sensitivity of KCNQ1‐4 potassium channels. Br J Pharmacol. 2000;129:413–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Lauritano A, Moutton S, Longobardi E, Tran Mau‐Them F, Laudati G, Nappi P, et al A novel homozygous KCNQ3 loss‐of‐function variant causes non‐syndromic intellectual disability and neonatal‐onset pharmacodependent epilepsy. Epilepsia Open. 2019;4:464–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Soldovieri MV, Ambrosino P, Mosca I, De Maria M, Moretto E, Miceli F, et al. Early‐onset epileptic encephalopathy caused by a reduced sensitivity of Kv7.2 potassium channels to phosphatidylinositol 4,5‐bisphosphate. Sci Rep. 2016;6:38167. [DOI] [PMC free article] [PubMed] [Google Scholar]