

A critical step in bacterial cytokinesis is the activation of septal peptidoglycan synthesis at the Z ring. Although FtsN is the trigger and acts through FtsQLB and FtsA to activate FtsWI the mechanism is unclear.
KEYWORDS: cell division, divisome, septal ring, septation
ABSTRACT
Spatiotemporal regulation of septal peptidoglycan (PG) synthesis is achieved by coupling assembly and activation of the synthetic enzymes (FtsWI) to the Z ring, a cytoskeletal element that is required for division in most bacteria. In Escherichia coli, the recruitment of the FtsWI complex is dependent upon the cytoplasmic domain of FtsL, a component of the conserved FtsQLB complex. Once assembled, FtsWI is activated by the arrival of FtsN, which acts through FtsQLB and FtsA, which are also essential for their recruitment. However, the mechanism of activation of FtsWI by FtsN is not clear. Here, we identify a region of FtsL that plays a key role in the activation of FtsWI which we designate AWI (activation of FtsWI) and present evidence that FtsL acts through FtsI. Our results suggest that FtsN switches FtsQLB from a recruitment complex to an activator with FtsL interacting with FtsI to activate FtsW. Since FtsQLB and FtsWI are widely conserved in bacteria, this mechanism is likely to be also widely conserved.
INTRODUCTION
Cell division in most bacteria is carried out by a large protein complex called the divisome or septal ring (1, 2). In Escherichia coli, it consists of 10 essential proteins, 2 quasi-essential proteins (FtsEX), and an ever-increasing number of nonessential proteins. The essential (and quasi-essential) proteins include FtsZ, which assembles into treadmilling filaments that are tethered to the membrane by FtsA and ZipA (Z ring), and 7 additional proteins which display the following dependency for recruitment: FtsE/X < FtsK < FtsQ < FtsL/B < FtsW < FtsI, and FtsN (Fig. 1) (1–4). Among these, FtsW is a newly described glycosyltransferase of the SEDS (septation, elongation, division, and sporulation) family that works in concert with a transpeptidase (FtsI [PBP3]) to synthesize septal peptidoglycan (PG) (5–8). A key step in cell division is the activation of these enzymes by FtsN, the last arriving essential protein (3, 9), which acts through FtsA and the FtsQLB complex (10–12).
FIG 1.

Model for recruitment and activation of FtsWI. FtsQLB localizes to the Z ring and recruits FtsW in a cytoFtsL-dependent manner with FtsW recruiting FtsI. FtsN arrives, and cytoFtsN interacts with FtsA in the cytoplasm, and the EFtsN domain acts in the periplasm, switching both FtsA and FtsQLB from the OFF to the ON conformation, resulting in activation of FtsWI. In the model, activation occurs when the AWI (activation of FtsWI) domain in FtsL (which overlaps the CCD [constriction control domain]) becomes available to contact FtsI. Activation mutations (*) in ftsA, ftsB, ftsL, and ftsW require less FtsN. FtsB* and FtsL* are thought to switch FtsQLB to the ON state. FtsA* may also do this, whereas FtsW* is likely to lead to an enzymatically active form of FtsW. For simplicity ZipA, FtsEX, and FtsK are not depicted. FtsZ, FtsA, FtsB, FtsQ, FtsL, FtsW, FtsI, and FtsN are indicated by single letters. The red rectangles highlight interactions between FtsL and FtsWI.
The FtsQLB complex is widely conserved among peptidoglycan-containing bacteria and links the Z ring to the septal PG synthesis machinery (FtsWI) (13). Each protein in the FtsQLB complex is a bitopic membrane protein with a short cytoplasmic region connected to a larger periplasmic domain by a single transmembrane domain. FtsQ targets the FtsQLB complex to the Z ring in an FtsK-dependent fashion, and the cytoplasmic domain of FtsL is required to recruit FtsW (3) (Fig. 1). FtsL and FtsB form a multimer with interactions occurring between their alpha-helical transmembrane domains as well as their putative periplasmic coiled-coil domains (14–17). They also interact with FtsQ through their C-terminal domains that lie beyond the coiled-coil domains forming a 1:1:1 complex which may dimerize (13, 15, 18). The structure of a peptide corresponding to the C-terminal region of FtsB bound to the periplasmic domain of FtsQ was recently determined (19, 20).
Activation of FtsWI by FtsN requires two domains of FtsN; the cytoFtsN domain acts on FtsA, and the EFtsN domain, a short putative helical segment in the periplasm, likely acts on FtsQLB (10, 21, 22, 36) (Fig. 1). In a proposed model, FtsN switches both FtsA and FtsQLB to an ON state which activates FtsWI (10, 11). This regulatory model is based in part upon the isolation of “activation (superfission)” mutations (requiring less FtsN) in ftsL and ftsB which identified a short periplasmic region in both proteins, designated CCD for constriction control domain (10). The CCD connects the coiled-coil domain of each protein to its distal C-terminal region, which binds to FtsQ (13, 18). It is not clear how these mutations work, but it is likely they mimic FtsN action, resulting in a change in conformation of the FtsQLB complex to the ON state that activates FtsWI (Fig. 1). Activation mutations have also been isolated in ftsA and ftsW (10, 12). Such mutations in ftsA could cause it to act on FtsQLB or FtsW, whereas such mutations in ftsW could lead to an enzymatically active conformation. To address the mechanism of FtsWI activation, we set out to isolate dominant negative mutations in ftsL and ftsB. Such mutations should yield an FtsQLB complex that no longer activates FtsWI and yield information about the activation mechanism. By exploring the effect of the dominant negative mutations, as well as the activation mutations, on the recruitment and activation of FtsWI, we find an essential role for FtsL in the activation of FtsWI.
RESULTS
Isolation of dominant negative mutations in ftsL but not ftsB.
To isolate dominant negative mutations in ftsL and ftsB, they were subjected to random mutagenesis, cloned into a plasmid downstream of an IPTG-inducible promoter, and introduced into a wild-type strain. Colonies were then picked, and dominant negative mutants were identified by screening for growth inhibition after streaking on plates containing increasing amounts of IPTG (isopropyl-β-d-thiogalactopyranoside). Three strong dominant negative mutations were obtained in ftsL (ftsLE87K, ftsLL86F, and ftsLA90E) as well as two weak mutations (ftsLR61C and ftsLL24K), but none were obtained in ftsB (Fig. 2A and Table 1). Changing ftsLR61C to ftsLR61E resulted in a stronger dominant negative mutant (Table 1), while ftsLL24K is discussed later. Induction of the ftsL alleles in liquid culture resulted in filamentation (Fig. 2B and Table 1). Complementation tests confirmed they were loss of function mutations, as they were unable to complement a ΔftsL strain (Fig. S1A, Table 1). Interestingly, three of these mutations overlapped the CCD domain, which was previously defined by activation mutations that decrease the dependency upon FtsN (10, 11) (Fig. 2C). Using site-directed mutagenesis, we altered additional residues around the CCD and isolated three additional dominant negative mutations (ftsLR82E, ftsLN83K, and ftsLL84K) (Fig. 2C and Table 1). However, extending the mutagenesis to flanking regions as well as the C-terminal region of ftsL did not yield any additional dominant negative mutations (Fig. 2C and Table 1). Although the residues we identified overlap the CCD, they are distinct from the residues involved in activation and lie mostly on the opposite side of a putative alpha helix. Since these mutations lead to a dominant negative effect, they behave as though they are nonresponsive to FtsN, just the opposite of activation mutations (Fig. 2D). We designate the region identified by the dominant negative residues as AWI (activation of FtsWI) based on the results described below.
FIG 2.

Isolation of dominant negative mutations in ftsL. (A) Spot test of dominant negative mutations in ftsL. ftsL was subjected to random PCR mutagenesis, cloned downstream of the tac promoter in an expression vector containing an IPTG-inducible promoter (pJF118EH), and transformed into JS238. Transformants were screened for sensitivity to IPTG. ftsLWT and ftsLE88K (an activation allele) were included as controls and are not toxic. Several strong dominant negative mutations (ftsLE87K, ftsLL86F, and ftsLA90E) and two weak mutations (ftsLR61C and ftsLE24K) were obtained in this way (Table 1). Additional mutations were obtained by site-directed mutagenesis. (B) Dominant negative mutants inhibit division. Phase contrast micrographs of JS238 expressing ftsL or ftsLE87K (derivatives of pKTP100 [Ptac::ftsL]) grown in liquid culture and induced with 50 μM IPTG for 2 h. Induction of the other alleles also inhibited division (Table 1). (C) FtsL, residues 54 to 99, was modeled (for illustration purposes) as an alpha helix since it is thought to form a continuous alpha helix with the TM, and this region is also thought to form a coiled coil with FtsB. Altering the residues in green leads to activation mutations, whereas altering those residues in red results in dominant negative mutations. Altering the residues in yellow had no effect. Note that the activation mutations affect residues that lie mostly on one side of the helix, whereas the dominant negative mutations affect residues that lie mostly on the other side. The red residues (including L86 and E87) identify a region designated AWI (activation of FtsWI). The positions of residues 24 and 28 in the cytoplasmic domain are indicated along with the transmembrane (TM) domain. The cytoplasmic domain of FtsL is required to recruit FtsW, which in turn recruits FtsI. (D) Cartoons depicting the effect of various mutations on the activation of FtsWI according to the model. Top, FtsN action makes AWI available; middle, FtsLE88K is less dependent upon FtsN as the E88K substitution makes AWI available; bottom, FtsLE87K is resistant to FtsN action, and AWI does not become available or is defective in interaction with FtsWI.
TABLE 1.
Summary of the point mutations in ftsL and ftsBa
| Mutations | Complementationb | DN | Relative strength of DNc | Suppression of DN by E88Kc | Suppression of DN by FtsN expressiond |
|---|---|---|---|---|---|
| FtsL mutations | |||||
| L24K | NT | Weak | + | NT | NT |
| L24K, I28K | No | Yes | ++ | NT | NT |
| R61C | NT | Weak | + | NT | NT |
| R61E | No | Yes | ++ | Yes | Yes |
| L77K | NT | No | |||
| D78K | NT | No | |||
| E80K | NT | No | |||
| W81A | NT | No | |||
| R82E | No | Yes | +++ | NT | NT |
| N83K | No | Yes | +++ | Yes | NT |
| L84K | No | Yes | +++ | NT | NT |
| L86F | No | Yes | ++++ | No | No |
| E87K | No | Yes | ++++ | No | No |
| A90E | No | Yes | +++ | Yes | Yes |
| L91K | Yes | No | |||
| R96E | Yes | No | |||
| A101K | Yes | No | |||
| L105D | Yes | No | |||
| M107K | Yes | No | |||
| E115K | Yes | No | |||
| P112-Stop | Yes | No | |||
| Q114-Stop | Yes | No | |||
| FtsB mutationse | |||||
| N43K | Yes | No | |||
| N50Kf | No | No | |||
| Q52K | Yes | No | |||
| F54K | Yes | No | |||
| I57K | Yes | No | |||
| L60K | Yes | No | |||
| A66K | Yes | No |
DN, dominant negative.
NT, not tested. Complementation and suppression tests were done in strain SD399.
Indicates IPTG concentration that inhibited colony formation: ++++, 25 μM; +++, 30 μM; ++, 50 to 100 μM; +, cells filamentous at 100 μM. Dominant negative tests were done in JS238 with derivatives of pKTP100 (Ptac::ftsL) and pKTP101 (Ptac::ftsB) carrying the indicated mutations.
Suppression by ftsN was done with strain SD399 (pSD256) containing plasmids pSD296 (ftsLm) and pSEB417 (ftsN).
For these strains, complementation was done using strain BL155/pBL194.
It is likely that this mutant is unstable.
Characterization of dominant negative mutations in ftsL. (A) Dominant negative mutations do not complement an ΔftsL strain. Two dominant negative alleles of ftsL were tested for their ability to complement an ftsL depletion strain by transforming SD439 (ftsL::kan)/pSD296 (Para::ftsL) with pKTP100 (Ptac::ftsL) derivatives carrying different ftsL alleles. The toxicity of these alleles was tested on plates containing both arabinose (induced WT ftsL) and IPTG (to induce the allele to be tested) (center panel). The ability to complement the ftsL depletion strain was assessed on plates without arabinose or IPTG (basal level of expression from pKTP100 (Ptac::ftsL) is sufficient for complementation) (right panel). (B) Lineup of a region (amino acids 57 to 110) of the periplasmic domain of FtsL. FtsLs from a variety of Gram-negative and Gram-positive bacteria were aligned using Cobalt. Residues in red are more conserved than residues in blue, which are more conserved than those in gray. E87 is indicated by the arrow. (C) Examination of additional substitutions at position 87 in FtsL on toxicity. Derivatives of pKT100 (Ptac::ftsL) carrying various alleles of ftsL were introduced into JS238 and tested for toxicity by performing spot tests on plates containing increasing amounts of IPTG. (D) Effect of the dominant negative ftsL mutations on the recruitment of GFP-FtsI. SD285 (leu::Tn10 P206::gfp-ftsI) was transformed with pSD296 (Para::ftsL) derivatives expressing ftsLE87K, ftsLA90E, or ftsL. Three hours after induction with 0.2% arabinose and 25 mM IPTG, samples were taken and examined by phase and fluorescent microscopy. Download FIG S1, TIF file, 9.03 MB (9MB, tif) .
Copyright © 2020 Park et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Of residues composing the CCD domain of FtsL, residue E88 is the most conserved, and mutational analysis indicated that loss of the negative charge results in the activation phenotype (10). The neighboring residue E87 is even more conserved (Fig. S1B) and was altered in one of our dominant negative mutants. Additional analysis indicates that changing this residue to amino acids other than aspartate produces a dominant negative phenotype (Fig. S1C). Thus, the loss of the negative charge in two neighboring glutamate residues yields contrasting phenotypes. Since loss of the negative charge in each case produced their respective phenotypes, it strongly suggests that these mutations disrupt rather than enhance interactions.
In our random mutagenesis screen, we did not isolate dominant negative mutations in ftsB; however, since six of the dominant negative mutations in ftsL overlapped the CCD, we used site-directed mutagenesis to alter the more conserved residues that overlap FtsB’s CCD domain. Seven residues flanking the CCD domain were altered, but none produced a dominant negative phenotype (Table 1). Six of these still complemented an ftsB deletion strain. This result suggests that the dominant negative mutations are unique to ftsL.
Dominant negative FtsL mutants are defective in activation of septal PG synthesis.
A dominant negative phenotype could result from incorporation of an FtsL mutant into the FtsQLB complex that fails to (i) recruit downstream proteins (FtsWI), (ii) respond to FtsN (FtsQLB locked in OFF state), or (iii) generate an output signal in response to FtsN (ON state but failure to interact with a downstream partner). To test the first possibility, we assessed the localization of green fluorescent protein (GFP)-FtsI, which depends upon FtsW (3, 23). It was present in crossbands within filamentous cells following expression of ftsLE87K or ftsLA90E, indicating recruitment to the Z ring (Fig. S1D). This result suggests that the ftsL mutations blocked either the response to FtsN or a downstream event such as interaction with FtsWI.
The dominant negative ftsL mutations were tested to see if they could be rescued by a strong activation mutation (ftsLE88K) in cis. While ftsLR61E and ftsLA90E were readily rescued by ftsLE88K, ftsLL86F and ftsLE87K were not (Fig. S2A). If we assume that ftsLE88K mimics FtsN action and switches FtsQLB to the ON state, it suggests that ftsLR61E and ftsLA90E are able to carry out steps downstream of FtsN action. Based on these results, we suspected overexpression of ftsN would also rescue ftsLA90E and ftsLR61E but not ftsLE87K or ftsLL86F. This, in fact, was the case (Fig. S2B and Table 1). Since ftsLR61E and ftsLA90E were rescued by enhancing the activation signal (by introducing an ftsL activation mutation or ftsN overexpression), it suggests they favor the OFF state (partially resistant to FtsN) but can carry out downstream events when activated. We therefore focused on ftsLL86F and ftsLE87K since it is unclear if they are locked in the OFF state or are unable to produce a signal in response to FtsN.
Test for rescue of dominant negative ftsL mutations by an ftsL activation mutation or by ftsN overexpression. (A) Rescue by an ftsL activation mutation in cis. The ftsLE88K mutation was added to various ftsL alleles in cis and then tested for complementation. To do this, SD439 (ftsL::kan/pSD296 [Para::ftsL]) was transformed with derivatives of pKTP100 (Ptac::ftsL) carrying the various mutations. The strains were spotted on plates at 30°C without arabinose to deplete WT ftsL, and IPTG was added to induce the various ftsL alleles. ftsLA90E and ftsLR61E were rescued, but ftsLE87K and ftsLL86F (Table 1) were not. (B) Rescue by overexpression of ftsN. To test if ftsN overexpression can rescue any of the dominant negative alleles of ftsL, SD399 (ftsL::kan/pSD256 [repATS Psyn135::ftsL]) containing plasmids expressing the ftsL alleles under an arabinose inducible promoter (derivatives of pSD296 [Para::ftsL]) was transformed with a plasmid with ftsN under IPTG control (pSEB417 [P204::ftsN]). The strains were spotted at 37°C to deplete WT ftsL, and arabinose was added to induce the ftsL allele, and IPTG added to induce ftsN. Download FIG S2, TIF file, 10.6 MB (10.8MB, tif) .
Copyright © 2020 Park et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Dominant negative FtsL mutants are rescued by FtsW activation mutants.
Based on our results, we hypothesized that activation of FtsWI requires a signal from the periplasmic domain of FtsL (AWI domain) which is made available by FtsN action or ftsL activation mutations. We also hypothesized that activated alleles of ftsW might rescue a strong dominant negative ftsL allele since they require less input from FtsN. Two such ftsW alleles exist: ftsWM269I, which weakly bypasses ftsN (12), and ftsWE289G, which was isolated as described in Materials and Methods and bypasses ftsN. The latter mutation was also isolated using another approach and shown to bypass ftsN (24).
To see if these ftsW alleles could rescue ftsLL86F or ftsLE87K, a plasmid with these alleles under an arabinose-inducible promoter (derivatives of pSD296 [Para::ftsL]), as well as a compatible plasmid with ftsW alleles under an IPTG-inducible promoter (derivatives of pSEB429 [P204::ftsW]), were introduced into SD399 (ftsL::kan/pSD256 [repAts::ftsL]). The resultant strains were tested on plates at 37°C to deplete wild-type (WT) ftsL, and arabinose and IPTG were added to induce the ftsL and ftsW alleles, respectively. Expression of ftsWM269I and ftsWE289G, but not ftsW, rescued the dominant negative ftsL alleles (Fig. 3). These ftsW activation alleles still required the presence of ftsL, as they could not bypass it (Fig. 3, right panel). Also, ftsWM269I was able to rescue an allele containing both mutations (ftsLL86F/E87K), whereas overexpression of ftsN could not (Fig. S3A). These results indicate that ftsLL86F/E87K cannot transmit the periplasmic signal in response to FtsN.
FIG 3.

Rescue of dominant negative mutations in ftsL by overexpression of active FtsW mutants. SD399 (ftsL::kan/pSD256 [repAts::ftsL]) containing derivatives of pSD296 (Para::ftsL) with different alleles of ftsL was transformed with derivatives of pSEB429 (P204::ftsW) carrying WT ftsW or either of two active alleles of ftsW. Transformants were spot tested at 37°C (to deplete WT FtsL) in the presence of arabinose (to induce the ftsL allele present on derivatives of pSD296) and increasing concentrations of IPTG to induce alleles of ftsW (ftsW, ftsWM269I or ftsWE289G). The cartoons below depict the interpretation of the results. On the left, FtsWI is not recruited in the absence of FtsL; center, FtsWI is recruited but not activated in the presence of a dominant negative FtsL mutant; right, active FtsW mutants suppress dominant negative FtsL mutants in one of two ways (see the text).
Effect of activation mutations in ftsW and ftsB as well as ftsN overexpression on the rescue of dominant negative ftsL mutations. (A) Overexpression of an activation allele of ftsW, but not ftsN, suppresses a dominant negative allele of ftsL (ftsLL86F/E87K). SD399 (ftsL::kan/pSD256 [repAts-ftsL]) was transformed with plasmids expressing either ftsL or a dominant negative allele of ftsL (ftsLE87K/L86F) under an arabinose-inducible promoter (derivatives of pSD296 [Para::ftsL]) and plasmids expressing ftsWM269I or ftsN under an IPTG-inducible promoter (pSEB429-I [P204::ftsWM269I] and pSEB417 [P204::ftsN], respectively). The strains were spotted on plates at 37°C (to deplete WT ftsL) containing 0.2% arabinose to induce the ftsL alleles, with increasing concentrations of IPTG to induce ftsWM269I or ftsN. The second panel lacks arabinose, demonstrating that the ftsW activation mutations cannot bypass ftsL. (B) An activated allele of ftsB cannot suppress a dominant negative ftsL mutation. Strain PK168-1 (ftsBE56A recA::aadA ftsL::kan/pSD296 [Para::ftsL]) containing pKTP100 (Ptac::ftsL) or a version expressing ftsLE87K was subjected to spot tests on plates without arabinose to deplete WT ftsL and with IPTG added to induce the ftsL allele cloned in derivatives of pKTP100. Download FIG S3, TIF file, 10.6 MB (10.8MB, tif) .
Copyright © 2020 Park et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Although the above-described results demonstrate that the two dominant negative mutations (ftsLL86F or ftsLE87K, alone or combined) block FtsN, they do not distinguish between whether they lock FtsQLB in the OFF state (nonresponsive to FtsN) or prevent a downstream step (responsive to FtsN but failing to interact with FtsWI). We suspect the latter for the following reasons. To rescue ftsLL86F or ftsLE87K, ftsWE289G has to be overexpressed, whereas the chromosomal level of ftsWE289G was sufficient to bypass ftsN (expression of ftsW or the activation alleles from the plasmids complement an ftsW depletion mutant in the absence of IPTG [Fig. S4A], whereas 15 to 30 μM is required to rescue ftsLL86F or ftsLE87K). Consistent with this, expression of ftsLE87K is toxic to a strain with ftsWM269I on the chromosome (Fig. S4B), highlighting that an active ftsW allele cannot bypass the dominant negative ftsL mutation at the chromosomal level. These results suggest that the dominant negative ftsL mutants are defective in interaction with FtsWI in the periplasm (lack of the periplasmic interaction necessitates overexpression of an active ftsW). Consistent with the ftsL mutations blocking a step downstream of FtsN action, an active ftsB mutation, ftsBE56A, which can also bypass ftsN (10), cannot suppress ftsLE87K (Fig. S3B). This result is also consistent with an activation mutation in ftsL or overexpression of ftsN being unable to rescue ftsLE87K (Fig. S3A). Furthermore, all substitutions in ftsLE87 that remove the negative charge are dominant negative (Fig. S1C), suggesting they disrupt, rather than enhance, an interaction. Therefore, we favor the idea that these mutations in the AWI domain abrogate FtsL’s interaction with FtsWI and that under physiological conditions, FtsWI is recruited by cytoFtsL and activated by FtsQLB when it is in the ON state (AWI available).
Effect of activation mutations in ftsW on complementation and rescue of a dominant negative ftsL allele. (A) Test of ftsW alleles for complementation of an ftsW depletion strain in the absence of IPTG. To test if the addition of IPTG was required for complementation of an ftsW depletion strain, EC912 (ftsW::kan/pDSW406 [Para::ftsW]) was transformed with derivatives of pSEB429 (P204::ftsW) carrying various alleles of ftsW. The strains were spot tested on plates without arabinose and with or without 200 μM IPTG. (B) Expression of ftsLE87K inhibits a strain with the ftsWM269I mutation on the chromosome. Strains KTP1 (ftsW+) and SD247-1 (ftsWM269I) were transformed with pKTP100 (Ptac::ftsL) or a derivative expressing ftsLE87K. The strains were spot tested on plates containing increasing concentrations of IPTG to test for toxicity. The strain containing the ftsWM269I is more resistant but still sensitive to ftsLE87K. Download FIG S4, TIF file, 10.6 MB (10.8MB, tif) .
Copyright © 2020 Park et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Loss of cytoFtsL function rescued by activation mutations in the CCD domain of FtsL.
One mutation from the random mutagenesis screen altered a residue in the cytoFtsL domain (ftsLL24K). Although weak, adding a second mutation that altered a conserved residue in this domain (ftsLI28K) yielded a stronger dominant negative phenotype (Fig. S5A). Since cytoFtsL is required for FtsW recruitment (13), it suggests that FtsLL24K, FtsLI28K, and the double mutant assemble into a complex with FtsQ and FtsB that poorly recruits FtsW. Consistent with this, deletion of the cytoplasmic domain of FtsL (FtsLΔ1-30) produced a strong dominant negative phenotype (Fig. S5B) resulting in filamentation and a failure to recruit FtsI (Fig. S5C).
Mutations in the cytoplasmic domain of ftsL result in a dominant negative phenotype and fail to recruit GFP-FtsI. (A) Mutations (L24K/I28K) in the cytoftsL domain lead to a dominant negative phenotype. JS238 containing pKTP100 (Ptac::ftsL) derivatives expressing different alleles of ftsL were tested for toxicity following induction with IPTG. (B) Deletion of the cytoplasmic domain of FtsL (ftsLΔ1-30) results in a dominant negative allele. JS238 containing pKTP104 (PT5::ftsL) or pKTP105 (PT5::ftsLΔ1-30) was tested for toxicity following induction with IPTG. (C) FtsLL24K/I28K fails to recruit GFP-FtsI. SD285 (leu::Tn10 P206::gfp-ftsI) was transformed with pSD296 (Para::ftsLL24/I28K). Three hours after induction with 0.2% arabinose and 25 mM IPTG, samples were taken and examined by phase and fluorescent microscopy. Download FIG S5, TIF file, 10.6 MB (10.8MB, tif) .
Copyright © 2020 Park et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Since FtsN is proposed to switch FtsQLB to the ON state to activate FtsWI (10, 11), we speculated above that this switch involves a conformational change that exposes AWI to activate FtsWI. If this is the case, the activation mutations may compensate for the loss of cytoFtsL by making the AWI domain available, which recruits FtsWI as well as activating it. As expected, ftsLΔ1-30 failed to complement ΔftsL; however, ftsLΔ1-30 carrying two activation mutations (ftsLG92D and ftsLE88K) restored colony formation, indicating that both recruitment and activation of FtsW were restored (Fig. 4A). Further tests showed that both activation mutations were required for rescue (Fig. S6A). The rescue was fairly effective, as the average cell length of the strain expressing ftsLΔ1-30/G92D/E88K was only twice that of a strain expressing ftsL (Fig. S6B), whereas the strain expressing ftsLΔ1-30 was extremely filamentous. These two activation mutations also eliminated the toxicity of the ftsLL24K/I28K allele (Fig. S6C) and rescued its ability to complement (Fig. S6D). These results are consistent with a model in which the ftsL activation mutations cause a conformational change in FtsQLB that makes AWI available to recruit and activate FtsWI. It follows that under physiological conditions, the arrival of FtsN results in the exposure of AWIFtsL, which cooperates with cytoFtsL to recruit and activate FtsWI.
FIG 4.

Effect of ftsL activation and dominant negative mutations on the rescue of FtsLΔ1-30. (A) ftsLΔ1-30 is rescued by ftsL activation mutations, which is negated by an ftsL dominant negative mutation. SD439 (ftsL::kan/pSD296 [Para::ftsL]) was transformed with derivatives of pKTP105 (PT5::ftsL) carrying various alleles of ftsL inducible with IPTG. The strains were spotted on plates without arabinose (to deplete WT ftsL) but with IPTG (to induce the various alleles of ftsL present in derivatives of pKTP105). The cartoons on the right depict the interpretation of the results. (B) Overexpression of ftsWM269I rescues ftsLΔ1-30. SD399 (ftsL::kan/pSD256 [repATS Psyn135::ftsL]) carrying pKTP107 (Para::ftsLΔ1-30) was transformed with compatible plasmids expressing different alleles of ftsW (derivatives of pSEB429 [P204::ftsW]) under the control of an IPTG-inducible promoter. Transformants were spotted on plates at 37°C (to deplete WT ftsL) in the presence of 0.2% arabinose (to induce ftsL alleles contained on the plasmids) and increasing concentrations of IPTG (to induce ftsW alleles). The cartoon indicates that FtsWM269I is recruited by FtsLΔ1-30. (C) Dominant negative ftsL mutations negate rescue of ftsLΔ1-30 by ftsWM269I. Strain SD399 (ftsL::kan/pSD256 [repATS Psyn135::ftsL]) was transformed with a plasmid (derivatives of pKTP107 [Para::ftsLΔ1-30]) with ftsLΔ1-30 under arabinose promoter control and a compatible plasmid (pSEB429 [P204::ftsW]) that carries ftsW or ftsWM269I under the control of an IPTG-inducible promoter. The presence of a dominant negative ftsL mutation negates rescue by the activated FtsW.
Rescue of ftsL cytoplasmic mutations by ftsL activation mutations and ftsN overexpression. (A) Rescue of ftsLΔ1-30 requires two ftsL activation mutations. SD439 (ftsL::kan/pSD296 [Para::ftsL]) carrying pKTP105 (PT5::ftsLΔ1-30) derivatives expressing various alleles of ftsL were tested for their ability to complement an ftsL depletion strain. Complementation was tested by spotting strains on plates without arabinose (to deplete WT ftsL) and with IPTG to induce the various ftsL alleles. Loss of a functional cytoFtsL domain (and therefore the ability to recruit FtsW) is compensated for by the two activation mutations ftsLE88K/G92D. The cartoon depicts the defect caused by ftsLΔ1-30 and its rescue by the activation mutations. (B) Morphology of ΔftsL cells rescued by ftsLΔ1-30 carrying activation mutations. SD399 (ftsL::kan/pSD256 [Psyn135::ftsL]) carrying derivatives of pKTP107 [Para::ftsL] expressing various alleles of ftsLΔ1-30 were grown to the exponential phase at 30°C with antibiotics, centrifuged, washed, and resuspended in LB with 0.2% arabinose and antibiotics at 37°C. Samples were taken 2.5 h later for photography, and the cell length distributions were determined. (C) ftsL activation mutations reduce the toxicity of ftsLL24K/I28K. JS238 carrying derivatives of pKTP100 (Ptac::ftsL) expressing different alleles of ftsL was tested for toxicity by spotting on plates containing 200 μM IPTG to induce the ftsL alleles. (D) ftsL activation mutations rescue ftsLL24K/I28K for complementation. SD439 (ftsL::kan/pSD296 [Para::ftsL]) containing derivatives of pKTP100 [Ptac::ftsL] with various ftsL alleles was spotted on plates without arabinose (to deplete WT ftsL) and with IPTG to induce the mutant ftsL alleles. (E) Overexpression of ftsN suppresses ftsLΔ1-30. SD439 (ftsL::kan/pSD296 [Para::ftsL]) containing derivatives of pKTP105 (PT5::ftsLΔ1-30) with various alleles of ftsL was transformed with pBL154 (repATS Psyn135::ftsN) which constitutively expresses ftsN. Rescue of FtsLΔ1-30 was tested by spotting transformants on plates at 30°C without arabinose and with IPTG. The cartoon depicts that FtsN overexpression rescues FtsLΔcyto. (F) ftsL mutations do not affect interaction with FtsQ. To ensure that the ftsL mutations did not affect the stability of FtsL, we tested their effect on the FtsL-FtsQ interaction in the BACTH system. Strain DHM1 was transformed with plasmids carrying various alleles of ftsL (pUT18C derivatives) and a plasmid expressing ftsQ (pKT25-ftsQ). Three transformants were picked for each pair of constructs and spot tested. Column 1, FtsL; column 2, vector; column 3, FtsLΔcyto; column 4, FtsLΔcyto/E88K/G92D; column 5, FtsLΔcyto/E88K/G92D/E87K. (G) Interaction between FtsL and FtsWI assessed with the BACTH system. This test is similar to the test in Fig. 7 except that FtsLΔcyto was replaced with FtsLL24K/I28K. The ftsL alleles were contained in pUT18C, and the ftsW and ftsI alleles were in pKT25. The plates were photographed after overnight incubation. Column 1, FtsL versus FtsI; column 2, vector versus FtsI; column 3, FtsLL24K/I28K versus FtsW; column 4, FtsLL24K/I28K versus FtsWM269I; column 5, FtsLL24K/I28K versus FtsI; column 6, FtsLL24K/I28K/E88K/G92D versus FtsW; column 7, FtsLL24K/I28K/E88K/G92D versus FtsWM269I; and column 8, FtsLL24K/I28K/E88K/G92D versus FtsI. Download FIG S6, TIF file, 10.6 MB (10.8MB, tif) .
Copyright © 2020 Park et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Since the ftsL activation mutations appear to mimic FtsN action, we expected that overexpression of ftsN would also rescue ftsLΔ1-30. To test this, an ftsL depletion strain was transformed with a plasmid expressing ftsLΔ1-30 and a plasmid that overexpresses ftsN to a level that is sufficient to bypass zipA or ftsEX (21). The increased FtsN rescued ftsLΔ1-30 (Fig. S6E), suggesting that the excess FtsN caused AWI to be available to recruit and activate FtsWI, indicating that overexpression of ftsN is comparable to combining the two activation mutations (ftsLG92D and ftsLE88K) in rescuing ftsLΔ1-30.
Dominant negative ftsL mutations negate rescue by activation mutations.
If ftsL activation mutations rescue ftsLΔ1-30 by making AWI available to recruit and activate FtsWI, the dominant negative mutations should impair rescue by blocking the interaction. As seen in Fig. 4A, addition of ftsLE87K negated the rescue of ftsLΔ1-30 by the activation mutations, consistent with ftsLE87K blocking interaction between the AWI domain and FtsWI.
The FtsQLB complex probably exists in equilibrium between ON and OFF states, with the activation mutations and overexpression of FtsN favoring the ON state (AWI available). Overexpression of FtsW or FtsWM269I may also tip the equilibrium to the ON state and rescue ftsLΔ1-30, as the increased level of FtsW may promote capture of the ON state. Indeed, expression of ftsWM269I, even at low levels of induction, rescued ftsLΔ1-30, and at higher levels of induction, WT ftsW also started to rescue (Fig. 4B).
Earlier, we showed that overexpression of ftsWM269I and ftsWE289G, but not ftsW, rescued ftsL carrying dominant negative mutations (Fig. 3). This result is consistent with these activated mutants being recruited by the FtsL mutants (through cytoFtsL) but not requiring an activation signal from the AWI domain (via FtsN) (12). In the absence of cytoFtsL, however, our results suggest rescue requires a functional AWI in FtsLperi. If so, the dominant negative mutations should be detrimental in this context. As expected, the addition of either of two dominant negative mutations (ftsLL86F or ftsLE87K) to ftsLΔ1-30 prevented rescue by FtsWM269I (Fig. 4C). These results are consistent with AWI being required to recruit FtsWI in the absence of cytoFtsL. It is worth noting that when either of two FtsL domains is nonfunctional (due to either inactivation of the cytoplasmic domain or the presence of the dominant negative mutations [such as L86F and E87K] in full-length FtsL), the active FtsW mutants must be overexpressed to rescue growth (see Discussion).
Rescue of FtsLΔ1-30 by overexpression of FtsI.
In the hierarchical assembly pathway, FtsW is recruited in a cytoFtsL-dependent manner followed by FtsI, which is recruited by interaction between FtsW and the transmembrane segment of FtsI (23). However, we considered the possibility that with FtsLΔ1-30, the recruitment is reversed or FtsWI is recruited as a complex through interaction of AWI with FtsI. This thinking was driven in part by geometric constraints. The periplasmic domain of FtsL is thought to be a continuous alpha helix with its transmembrane domain such that the AWI domain would extend about ∼45 Å away from the cytoplasmic membrane (15) (Fig. S7). In the RodA-PBP2 structure (homologous to FtsW-FtsI), the non-penicillin-binding (nPB) or pedestal domain of PBP2 sits on top of RodA and extends into the periplasm (25). Assuming FtsW-FtsI adopts a similar structure, FtsI could contact AWI in FtsL.
Diagram indicating the position AWI of FtsL relative to the membrane and the RodA-PBP2 complex. Part of the periplasmic domain of FtsL was modeled as an alpha helix (residues 57 to 99) and positioned next to the structure of the RodA-PBP2 complex (PDB ID: 6PL6). The RodA-PBP2 complex is homologous to the FtsW-PBP3 (FtsI) complex. The non-penicillin binding domain (nPB) and penicillin binding domain (PD) are indicated. Key FtsL residues (E87 [AWI], E88 [CCD]) are about 4.3 nm from the membrane. The position of R61 is also indicated. Download FIG S7, TIF file, 10.6 MB (10.8MB, tif) .
Copyright © 2020 Park et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
If FtsI interacts with the AWI domain, overexpression of ftsI may rescue FtsLΔ1-30 by enhancing the interaction with AWIFtsL and shifting the equilibrium of FtsQLB from OFF to ON through mass action. To test this, we compared the ability of the overexpression of ftsI and ftsW to rescue FtsLΔ1-30. As shown in Fig. 5A, expression of ftsI was much more efficient than that of ftsW in rescuing FtsLΔ1-30. The efficient rescue of FtsLΔ1-30 by FtsI suggests that it captures the transient ON state of FtsQLB (AWI exposed) and converts FtsQLΔ1-30B into an active form similar to ftsL activation mutations (Fig. 4A). The rescue of FtsLΔ1-30 by overexpression of FtsW may involve the formation of an FtsWI complex that interacts with AWI, and the more efficient rescue of FtsLΔ1-30 by activated FtsW (compared to WT FtsW seen in Fig. 4B) may be due to it being active and more readily forming a complex with FtsI.
FIG 5.

Rescue of FtsLΔ1-30 by ftsI expression. (A) ftsI expression rescues FtsLΔ1-30. To test if overexpression of ftsI could rescue FtsLΔ1-30, PK4-1 (ftsL::kan/pKTP108 [repATS::ftsL]) was transformed with a plasmid expressing ftsLΔ1-30 (pKTP107X/Para::ftsLΔ1-30-6Xhis) and a plasmid expressing ftsI (pSEB420/P204::ftsI) or ftsW (pSEB429/P204::ftsW) under an IPTG-inducible promoter. The strains were spot tested on plates at 37°C to deplete ftsL. In addition, arabinose was added to induce ftsLΔ1-30, and increasing concentrations of IPTG were added to induce ftsI or ftsW. (B) ftsI overexpression cannot rescue a dominant negative ftsL allele. SD399 (ftsL::kan/pSD256 [repAts::ftsL]) was transformed with pSD296-2 (Para::ftsLL86F/E87K) and pSEB420 (P204::ftsI) or pSEB429 (P204::ftsW). Transformants were spot tested at 37°C (to deplete ftsL) on plates containing 0.2% arabinose (to induce ftsLL86F/E87K) and increasing concentrations of IPTG (to induce ftsI or ftsW).
The above-described results indicate that the signal from FtsN via the AWI domain goes through FtsI. As shown earlier, expression of activated alleles of ftsW suppressed ftsLL86F or ftsLE87K, as they no longer require the signal from AWI. In contrast, WT ftsW cannot suppress these alleles, as it still requires the AWI activation signal. Likewise, overexpression of ftsI would not be expected to rescue FtsL carrying the dominant negative ftsL mutations since the AWI activation signal would not be present. As expected, overexpression of ftsI was unable to suppress ftsLL86F/E87K, indicating the AWI signal was still required (Fig. 5B).
The possibility that AWI recruits and activates FtsWI by acting through FtsI was further examined by testing FtsI mutants isolated by the Weiss lab (26). These mutants localize to the division site but fail to complement a depletion strain and recruit FtsN. We reasoned that if an active FtsL acts directly on FtsW (to generate an active FtsW), an activated FtsL should have no more ability to rescue such mutants than an active FtsW mutant. However, if an activated FtsL acts on FtsI, it might have more ability to rescue FtsI mutants than an active FtsW. Therefore, each FtsI mutant was tested to see if it could be rescued by an active form of FtsL or FtsW (FtsLG92D/E88K and FtsWM269I, respectively). Of the seven FtsI mutants tested, two mutants (FtsIS61F and FtsIR210C) were rescued by both FtsWM269I and FtsLG92D/E88K (Fig. 6 and Fig. S8). However, FtsLG92D/E88K rescued two additional mutants (FtsIG57D and FtsIV86E; Fig. 6B, rows 5 and 9) not rescued by FtsWM269I (Fig. 6A, rows 3 and 5). The rescue of these two mutants by an activated FtsL (but not an activated FtsW) suggests that AWI acts through FtsI to activate FtsW rather than acting directly on FtsW.
FIG 6.

Rescue of FtsI mutants by activated FtsL and FtsW mutants. (A) Rescue of FtsI mutants by FtsWM269I. To test if the FtsI mutants could be rescued by an activated allele of ftsW, MCI23 (ftsI23ts recA::spc) was transformed with compatible plasmids expressing an activated allele of ftsW (pSEB429 [P204::ftsWM269I]) and ftsI alleles under arabinose promoter control (derivatives of pKTP109 [Para::ftsI]). Transformants were spot tested on plates at 37°C (to inactivate ftsI23ts) with arabinose added to induce the ftsI alleles and increasing concentrations of IPTG to induce ftsWM269I. Note: additional alleles of ftsI were not rescued by ftsWM269I (Fig. S8). (B) Rescue of FtsI mutants by ftsLE88K/G92D. To test rescue of FtsI mutants by activated FtsL, MCI23 (ftsI23ts recA::spc) was transformed with compatible plasmids expressing an activated allele of ftsL (pKTP100* [Ptac::ftsLE88K/G92D]) and the various ftsI alleles under arabinose promoter control (derivatives of pKTP109 [Para::ftsI]). Transformants were spot tested on plates at 37°C (to inactivate ftsI23ts), and arabinose was added to induce the ftsI alleles, and increasing concentrations of IPTG were added to induce ftsLE88K/G92D.
Testing the rescue of additional alleles of ftsI by an activated FtsW mutant. Additional ftsI alleles were tested to see if they were rescued by ftsWM269I expression. Some alleles were tested in Fig. 6A, and the others are tested here. Note that among the alleles tested here, only ftsIS61F is rescued. The bottom row of panels is also presented in Fig. 6A and was included here for comparison. MCI23 (ftsI23ts recA::aadA) was transformed with compatible plasmids expressing an activated allele of ftsW (pSEB429 [P204::ftsWM269I]) and the various ftsI alleles under arabinose promoter control (derivatives of pBAD33-ftsI). Transformants were spot tested on plates at 42°C (to inactivate ftsI23Ts), and arabinose was added to induce the ftsI alleles and increasing concentrations of IPTG to induce ftsWM269I. Download FIG S8, TIF file, 10.6 MB (10.8MB, tif) .
Copyright © 2020 Park et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Interaction between FtsL and FtsWI.
Our results point to an interaction between the cytoplasmic domain of FtsL and FtsW required for recruitment of FtsWI and between the periplasmic domain of FtsL with FtsI, which is required for activation of FtsWI. To obtain additional support for interactions between the various proteins, we tested the effect of these mutations using the bacterial two-hybrid (BACTH) system. We observed strong interactions between FtsL and FtsW and between FtsL and FtsI, which were eliminated when the cytoplasmic domain of FtsL was deleted, consistent with cytoFtsL being required for recruiting FtsWI (FtsLΔ1-30; Fig. 7A). Elimination of these interactions allowed us to use FtsLΔ1-30 to assess the effects of the activation mutations in ftsL and ftsW on the interactions. Although the ftsW activation mutation had little effect, the addition of two ftsL activation mutations resulted in a strong interaction between FtsLΔ1-30 and FtsI and a weaker interaction between FtsLΔ1-30 and FtsW (Fig. 7B). The strong interaction with FtsI suggests it interacts with FtsL, where the weak interaction with FtsW suggests that FtsW is an intermediate. Importantly, the further addition of a dominant negative mutation (ftsLE87K) eliminated the interaction conferred by the activation mutations. This FtsL variant with three amino acid substitutions was stable, as it interacted with FtsQ as well as the WT FtsL (Fig. S6F). These effects with FtsLΔ1-30 were also observed with FtsLL24K/I28K (Fig. S6G). The effects of these ftsL mutations in the BACTH system correlate with the effects these mutations have on the rescue of FtsLΔ1-30 and FtsLL24K/I28K; the ftsL activation mutations promote rescue which is negated by an ftsL dominant negative mutation (Fig. 4A and Fig. S6D, respectively).
FIG 7.
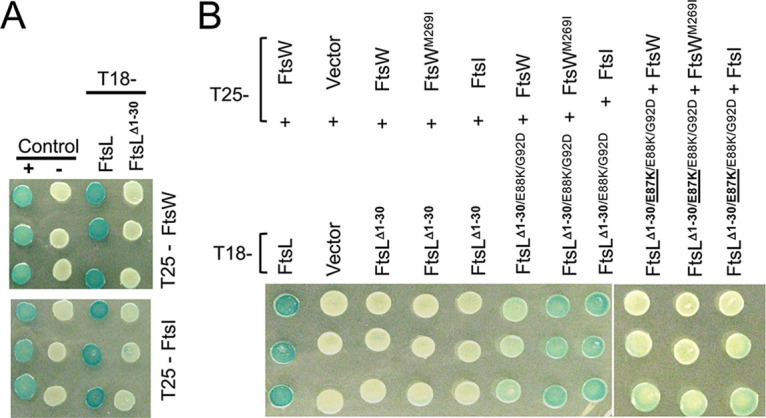
Interaction between FtsL and FtsWI assessed with the BACTH system. (A) Effect of cytoFtsL on the interaction between FtsL and FtsWI. Strain DHM1 was transformed with plasmids carrying various alleles of ftsL (pUT18C derivatives) and plasmids expressing ftsW or ftsI (pKT25 derivatives). Three transformants were picked and spot tested for each pair of constructs. The positive control contained plasmids pUT18C-zip and pKT25-zip, whereas the negative control contained the corresponding empty vectors. (B) The effect of activation and dominant negative mutations in ftsL on the interaction of FtsLΔ1-30 or FtsLL24K/I28K with FtsW and FtsI. Three transformants from each transformation of DHM1 with plasmids carrying the ftsL and ftsW or ftsI alleles were spotted on plates containing the color indicator. The ftsL alleles were contained in pUT18C, and the ftsW and ftsI alleles were in pKT25. The plates were photographed after overnight incubation.
Rescue of ΔftsL by MalF-FtsL and FtsW-FtsK fusions.
Next, we tested if the periplasmic portion of FtsL transported to the periplasm could activate FtsWI in the absence of full-length FtsL. To do this, a MalF-FtsL fusion was constructed under the control of an IPTG-inducible promoter in which the cytoplasmic and transmembrane (TM) domains of FtsL were replaced with the corresponding regions of MalF (cyto/TMMalF-periFtsL). In contrast to FtsLΔ1-30, this MalF-FtsL fusion was not dominant negative (Fig. S9A), indicating that the TM region of FtsL must be present for the fusion to displace FtsL from the FtsQLB complex and disrupt FtsW recruitment. This is consistent with the TM region of FtsL being unique (27) and the TMs of FtsL and FtsB being required for these proteins to interact (16, 18). Furthermore, the MalF-FtsL fusion was unable to complement an ftsL depletion strain even if the strain carried an ftsWM269I mutation and the ftsL construct carried the two activation mutations (Fig. S9B). This was expected since FtsW would not be recruited.
Characterization of FtsL fusions. (A) The MalF-FtsL fusion lacks toxicity and fails to complement an ftsL depletion strain. The left panel tests for complementation, and the right panel tests for toxicity. SD439 (ftsL::kan/pSD296 [Para::ftsL]) containing pKTP100 (Ptac::ftsL) derivatives expressing various alleles of ftsL were incubated without arabinose or IPTG (basal expression from pKTP100 [Ptac::ftsL] is sufficient for complementation) to test for complementation. In the right panel, both arabinose and IPTG were added. Arabinose induces WT ftsL from pSD296 (Para::ftsL), whereas IPTG induces the ftsL allele from pKTP100 (Ptac::ftsL). The ftsLL24K/I28K allele was added as a control to demonstrate toxicity. The lack of toxicity indicates that the MalF-FtsL fusion fails to form a complex with FtsQB. (B) FtsL fusions are unable to complement an ftsL-depleted strain. Fusion of the periplasmic domain of FtsL to the MalF cytoplasmic and transmembrane domains (with or without ftsL activation mutations) was tested for the ability to complement an ftsL depletion strain. To do this, PK247-4 (ftsWM269I ftsL::kan/pSD296 [Para::ftsL]) containing derivatives of pKTP100 (Ptac::ftsL) carrying various fusions in place of ftsL was spot tested on plates with IPTG. Download FIG S9, TIF file, 10.6 MB (10.8MB, tif) .
Copyright © 2020 Park et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Since the MalF-FtsL fusion cannot cooperate with FtsQB to recruit FtsW, we used an FtsW-cytoFtsK fusion which complements an ftsK deletion mutant, as well as a ftsW deletion mutant, indicating it is targeted directly to the Z ring and bypasses FtsQLB for recruitment (28 and data not shown). This MalF-FtsL fusion was unable to rescue the growth of a strain depleted for FtsL and containing FtsW-cytoFtsK, even if the fusion carried both ftsL mutations (Fig. 8, top panel). The inability to activate the FtsW-cytoFtsK fusion could be for a variety of reasons, including that FtsB is uncoupled from FtsL, and the FtsW-cytoFtsK likely competes with endogenous FtsW for FtsI. Nonetheless, the MalF-FtsL fusion with the two activation mutations was able to rescue an FtsL-depleted strain containing the FtsW-cytoFtsK fusion with the ftsWM269I mutation. (Fig. 8). Even the MalF-FtsL fusion without the ftsL activation mutations partially rescued growth at higher induction levels. These results suggest that MalF-FtsL acts on FtsI associated with the FtsWM269I-cytoFtsK fusion that is already at the Z ring to rescue growth. Since the activation mutations in ftsL potentiate MalF-FtsL activity, it suggests that in addition to making AWI available within the FtsQLB complex, they may also alter the structure of AWI to enhance its interaction with FtsWI.
FIG 8.

Activation mutations allow a malF-ftsL fusion to complement ΔftsL in the presence of a ftsW-cytoftsK fusion. Plasmid pKTP103 (Ptac::malF1-37-ftsL58-121-6xhis) was introduced into an ftsL depletion strain (SD439 ftsL::kan/pSD296 [Para::ftsL]) in the presence of a plasmid constitutively expressing a FtsW-FtsKcyto fusion without or with an activation mutation (pND16 [PftsK::ftsW-cytoftsK] or pND16* [PftsK::ftsWM269I-cytoftsK], respectively). The strains were spot tested on plates without arabinose (to deplete WT ftsL) and in the presence of IPTG (to induce malF-ftsL) (with or without the activation mutations [ftsLE88K and ftsLG92D]). The cartoon to the right depicts the activity of the FtsL constructs.
DISCUSSION
Here, we investigated how septal PG synthesis in the divisome is activated by FtsN and identified a critical and unique role for FtsL. Our results are consistent with the recruitment of FtsW requiring the cytoplasmic domain of FtsL and the activation of FtsWI being dependent upon AWI in the periplasmic domain of FtsL. Based upon the seminal work by the de Boer lab, which is supported by the work from the Bernhardt lab (10, 11) and our results (12) and those here, we propose that the arrival of FtsN leads to a conformational change in the FtsQLB complex that makes the AWI domain of FtsL, as defined by the dominant negative ftsL mutations, available to activate FtsWI by acting through FtsI. Furthermore, activation mutations in the CCD domain of FtsL as well as those in FtsB mimic FtsN action to cause a conformational change in FtsQLB to expose the AWI domain. This model is supported by the ability of activation mutations in ftsL to rescue FtsL mutations (FtsLΔ1-30 and FtsLL24K/I28K) deficient in FtsWI recruitment and by the dominant negative mutations in ftsL (ftsLL86F/E87K) negating the rescue. The effects of these ftsL mutations (both activation and dominant negative) on the rescue of the FtsL mutants correlates with their effects on the observed interaction between FtsL and FtsWI in the BACTH system. The model is also supported by the ability of the expression of ftsI to rescue FtsLΔ1-30 more efficiently than ftsW. Furthermore, FtsL acting on FtsI to activate FtsW is supported by the ability of an active FtsL mutant to rescue FtsI mutants not rescued by an activated FtsW. Thus, we propose that as a result of EFtsN action, the AWI domain of FtsL becomes available to interact with FtsI within the FtsWI complex to activate FtsW and synergizes with cytoFtsL in stabilizing the FtsWI complex in the divisome. Thus, FtsL within the FtsQLB complex functions as a clamp to maintain FtsWI in the divisome.
The AWI domain.
Altering seven residues in the periplasmic domain of FtsL produced a dominant negative phenotype. All, except for one, are clustered together around the CCD. We focused on L86 and E87 and believe these are central to the AWI domain. This suggestion is based upon the following: (i) L86 and E87 are relatively well conserved, and loss of the negative charge at E87 is sufficient to produce a dominant negative allele (suggesting disruption of an interaction); (ii) the ftsLL86F or ftsLE87K dominant negative mutations are not suppressed by activation mutations (ftsLE88K or ftsBE56A) or ftsN overexpression; (iii) cytoFtsL mutants that fail to recruit FtsWI are rescued by the addition of two ftsL activation mutations (ftsLE88K/G92D); (iv) the rescue of cytoFtsL mutants by ftsL activation mutations or overexpression of ftsWM269I is negated by adding dominant negative mutations (ftsLL86F or ftsLE87K); and (v) the effects of these mutations on the interaction of FtsL with FtsWI in the BACTH system correlate well with the effects of these mutations on the rescue of FtsLΔ1-30. It is likely that other regions of FtsL (and FtsB), such as the transmembrane domains (TM) and coiled coil domains, are also involved in interaction with FtsWI.
The dominant negative mutations in ftsL are less responsive to FtsN, and most overlap the CCD domain, which was defined by hyperactive mutations that are less dependent upon FtsN (10, 11). Despite the overlap, the residues comprising each domain mostly lie on opposite sides of a putative helix (Fig. 2C). The dominant negative mutations appear to be unique to ftsL, as we were unable to isolate any such mutations in ftsB. Although previous studies suggested that FtsN induces a change in FtsQLB from an OFF to ON conformation (10), it was not clear how this switch led to activation of FtsWI. Here, we identify the AWI domain of FtsL and suggest that the function of the conformational switch is to make AWI available to interact with FtsWI. Since FtsQLB may be a dimer, the conformational change could involve disruption of this dimer which makes AWI available; however, this will require further study (15, 16, 29).
Additional evidence for the unique importance of the periplasmic domain of FtsL comes from the ability of the MalF-periFtsL fusion to rescue an FtsW-cytoFtsK fusion when both are carrying activation mutations. The FtsW-cytoFtsK fusion is unable to support growth in the absence of FtsL even though it localizes. On the other hand, the MalF-periFtsL fusion does not form a complex with FtsQB, so it is not recruited to the divisome. Nonetheless, the ability of the MalF-periFtsL to collaborate with FtsW-cytoFtsK (when both are carrying activation mutations) to rescue growth suggests that the periplasmic domain of FtsL is able to act on FtsW-cytoFtsK complexed with FtsI.
While this paper was under review, Marmont and Bernhardt (30) reported that FtsLB was sufficient to activate PG synthesis by FtsWI in vitro, providing biochemical evidence for an activation model. They also isolated dominant negative mutations in ftsL which overlap those we isolated, even though their work was done in Pseudomonas aeruginosa and FtsL is not so highly conserved at the sequence level. Some, but not all, of the dominant negative mutants prevented activation in vitro. However, the in vitro system does not fully recapitulate the in vivo regulation, as FtsN was not required for activation.
Conditions that rescue FtsLΔ1-30 favor interaction between the AWI domain of FtsL and FtsI.
Surprisingly, loss of the cytoplasmic domain of FtsL, which prevents recruitment of FtsWI and blocks cell division, could be rescued by activation mutations in the periplasmic domain of FtsL as well as by overexpression of FtsN. We reasoned that these activation conditions expose an interaction that normally occurs when the divisome is activated and that this interaction is able to compensate for the loss of cytoFtsL to recruit FtsWI. In support of this model, ftsL activation mutations in ftsLΔ1-30 promoted interaction between FtsL and both FtsW and FtsI. Also, these interactions were negated by the addition of a dominant negative mutation. These results suggest that FtsL within the FtsQLB complex functions as a transmembrane clamp (Fig. 1) to stabilize the active FtsWI complex within the divisome. The cytoplasmic domain of FtsL is required to recruit FtsW, which in turn recruits FtsI. FtsN action then frees the AWI domain to interact with FtsI and, as we have shown here, this domain, when freed, is able to rescue ftsLΔ1-30, indicating FtsWI recruitment is restored.
Since it is likely FtsQLB exists in equilibrium between ON and OFF states, we reasoned that expression of the downstream partner might also rescue ftsLΔ1-30 by capturing the ON form and pulling the equilibrium in that direction. In fact, the active form of FtsW was effective in rescuing ftsLΔ1-30, much more so than FtsW. However, expression of FtsI was very effective in rescuing ftsLΔ1-30 and much more so than overexpression of FtsW, which barely rescued at high overexpression. This (i) suggested that FtsI is the direct downstream target of AWI, (ii) suggested that rescue by expression of FtsW likely involves formation of an FtsWI complex recruited by AWI, and (iii) raises the possibility that the activated form of FtsW interacts more strongly with FtsI. Consistent with the rescue of ftsLΔ1-30 by expression of FtsI or activated FtsW being dependent upon the interaction of AWI with FtsWI in the periplasm, it was prevented by the addition of the dominant negative ftsL mutations. This is in stark contrast to the suppression of the dominant negative mutations in full-length ftsL by activated FtsW. When full-length FtsL is present, an FtsW activated by mutation is recruited normally and no longer requires the activation signal so the dominant negative mutations do not prevent the rescue (although rescue is aided by overexpression of the activated FtsW). On the other hand, FtsW and FtsI are unable to rescue, as they still depend upon the AWI signal.
Our results suggest that FtsWI forms a dynamic complex, and it is this complex that is preferred by FtsL. If FtsWI formed a stable complex, then overexpression of FtsW would be toxic, as excess FtsW would titrate FtsI away from the division site inhibiting division. However, overexpression of ftsW is not toxic in WT cells and it only weakly rescued ftsLΔ1-30. Also, when FtsQLB is overexpressed and purified, FtsW and FtsI only copurify efficiently if they are both expressed, indicating that the FtsWI complex interacts more stably with FtsQLB than FtsW or FtsI alone (31). Thus, overexpression of FtsW may favor complex formation with FtsI and septal localization to rescue ftsLΔ1-30. More efficient rescue by an activated FtsW could be due to it favoring complex formation with FtsI. On the other hand, the rescue of ftsLΔ1-30 by FtsI expression is probably due to a direct interaction with AWI; otherwise, the rescue of ftsLΔ1-30 by FtsW and FtsI should be comparable, since overexpression of either should promote complex formation.
The product of FtsN action is an activated FtsWI complex in which both FtsW and FtsI are active. The ability of active FtsW mutants to suppress the dominant negative FtsL mutants (and bypass the periplasmic signal) indicates that an active FtsW leads to an active FtsWI complex. Among previously isolated FtsI mutants, we found some that were rescued by both an active FtsW mutant and an active FtsL mutant. However, an activated FtsL rescued two additional FtsI mutants that could not be rescued by an activated FtsW. This suggests that AWI acts on FtsI to activate FtsW and does not act directly on FtsW. In other words, the signal transmission from FtsN is from periFtsL → FtsI → FtsW and not periFtsL → FtsW → FtsI.
Although in vitro results suggest that FtsQLB acts as an inhibitor with FtsL inhibiting PBP1b and FtsQ inhibiting FtsI and therefore FtsW (31), our results are more compatible with a model in which AWI is sequestered within FtsQLB and becomes available upon FtsN action to activate FtsWI. The findings that ftsL activation mutations rescue FtsLΔ1-30 and promote interaction between FtsLΔ1-30 and FtsWI in the BACTH are consistent with the FtsL-FtsWI interaction activating FtsWI. This conclusion is also supported by the ftsL dominant negative mutations negating both of these activities.
Comparison of models for divisome and elongasome activation.
It is interesting to compare our model for FtsWI activation with the model proposed for activation of the RodA-PBP2 pair that are part of the elongasome (homologous to FtsW-FtsI [PBP3]). That model is based upon (i) the structure of the MreC-PBP2 complex (32) and (ii) the finding that mutations that bypass mreC and activate RodA-PBP2 map to the nonpenicillin (nPD) or pedestal domain of PBP2 (33). It is thought that these mutations mimic the binding of MreC to PBP2, altering the conformation of PBP2, which results in the activation of RodA. In this way, the activity of RodA and PBP2 are coupled to ensure RodA only makes glycan strands when its cognate PBP is present. This is remarkably similar to our model for FtsW-FtsI (PBP3) activation with FtsL (with possibly a supporting role for FtsB) being analogous to MreC. The isolation of FtsW activation mutants that bypass FtsN suggests that an activated FtsW results in an active FtsI. Furthermore, an active FtsW mutant can rescue dominant negative FtsL mutants (i.e., bypass the signal from FtsN), indicating FtsI is also activated. Thus, we propose that FtsN action alters the conformation of FtsQLB so that AWI becomes available to interact with FtsI, leading to conformational change in FtsI that activates FtsWI’s enzymatic activities.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
Bacterial strains are listed in Table S1A. JS238 [MC1061, araD Δ(ara leu) galU galK hsdS rpsL Δ(lacIOPZYA)X74 malP::lacIQ srlC::Tn10 recA1] was primarily used for screening for ftsL and ftsB dominant negative mutations and as a host for most cloning experiments. W3110 was used to generate SD399, SD439, and SD285. To construct SD399 [W3110, ftsL::kan/pSD256], P1 phage grown on BL156 [ftsL::kan/pJH2] was used to transduce ftsL::kan into W3110/pSD256 by selecting for Kan resistance on LB agar plates containing 25 μg/ml kanamycin, 50 μg/ml spectinomycin, and 8 mM sodium citrate at 30°C. Several colonies were subcloned onto fresh plates of the same composition at 30°C and were further screened for temperature sensitivity at 42°C. SD439 was created by transforming SD399 with pSD296 (Para::ftsL) and selecting the transformants that grow at 42°C (to remove pSD256) in the presence of 10 μg/ml chloramphenicol and 0.2% arabinose. Colonies were streaked and further tested for spectinomycin sensitivity (indicating loss of pSD256). Construction of SD285 [leu::Tn10 bla lacIq P207‐gfp‐ftsI] involved transduction with P1 phage grown on EC436 [MC4100 Δ(λattL‐lom)::bla lacIq P207‐gfp‐ftsI] into S3 (W3110 leu::Tn10). Transductants were selected on LB agar plates containing 25 μg/ml ampicillin and 10 μg/ml tetracycline. Expression of GFP-FtsI was confirmed in the transductant clones by induction with 10 to 20 μM IPTG. SD247 (W3110 ftsWM269I) was previously described (12), and PK247-4 [SD247 ftsL::kan/pSD296] was generated by P1 transduction of ftsL::kan from the SD399 donor to the recipient strain SD247/pSD296 [Para::ftsL] and by selecting Kan resistance and screening for arabinose dependency. PK4-1 (ftsL::kan/pKTP108 [Para::ftsL]) was generated by using the same procedure described above. Unless stated otherwise, Luria-Bertani broth (LB) medium containing 0.5% NaCl was used at the indicated temperatures. For selection on LB agar and growth in LB broth, the following antibiotics and reagents were added at the indicated final concentrations as necessary: ampicillin, 100 μg/ml; spectinomycin, 50 μg/ml; kanamycin, 25 μg/ml; chloramphenicol, 10 μg/ml; tetracycline, 10 μg/ml; IPTG, 10 to 200 μM; glucose, 0.2%; and arabinose, 0.2%.
Strains and plasmids used in this study. Download Table S1, DOCX file, 0.04 MB (37.3KB, docx) .
Copyright © 2020 Park et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Plasmids.
The plasmids are listed in Table S1B. Genomic DNA extracted from the W3110 strain was used as a template to obtain PCR fragments to generate expression plasmids for ftsL. To construct the plasmids pKTP100 (Ptac::ftsL) and pKTP103 [Ptac::malF1-37 ftsL58-121-6xhis], the ftsL open reading frame (ORF) was PCR amplified incorporating a strong ribosome binding site in the forward primers targeting ftsL, which included sequences for ftsL and malF1-37, respectively. The PCR fragments were digested with EcoRI and HindIII and ligated into the same sites in pJF118EH. Construction of pKTP104 (PT5::ftsL) and pKTP105 (PT5::ftsL30-121) involved PCR amplification of the ftsL ORF, digestion with BamHI and HindIII, and ligation into the same sites in the pQE80L vector (Qiagen). The construction of pKTP108 [repAts Psyn135::ftsL] employed a similar approach to that used for pSD256 (12) except that a strong ribosome binding site was added and the XbaI site was used instead of EcoRI. To create pKTP109, the ftsI ORF was PCR amplified and digested with SacI and HindIII, followed by ligation into pBAD33 using sites with compatible overhangs. To generate plasmid pSD296 (Para::ftsL), the ftsL ORF and its flanking sequences (250 bp) were PCR-amplified, digested with XbaI and HindIII, and ligated into the same sites in the pBAD33 vector. Plasmids pKTP106 (Para::ftsL) and pKTP107 (Para::ftsL30-121) were created by PCR amplification of ftsL and ftsL30-121, respectively, using the primers that contain the same ribosome binding site as in pKTP100. The two PCR fragments were cut with SacI and HindIII and cloned into sites in pBAD33 with compatible overhangs. To create pKTP101 (Ptac::ftsB), the ORF was PCR amplified and digested with EcoRI and HindIII followed by ligation into pJF118EH cut with the same enzymes. The pND16 [PftsK::ftsW-ftsK179-1329] plasmid constitutively expresses the FtsW-FtsK C-terminal fusion protein, and pBL154 (repATS Psyn135::ftsN) was previously described (10, 34). The overexpression plasmids for FtsN, FtsI, and FtsW and pSEB417 (P204::ftsN), pSEB420 (P204::ftsI), and pSEB429 (P204::ftsW), respectively, were previously described (21, 34). Note that these genes are expressed from their endogenous ribosome binding sites.
The bacterial two-hybrid (BACTH) vectors, pUT18C (cyaAT18 fragment) and pKT25 (cyaAT25 fragment), were described previously (35). The pUT18C-ftsL (cyaT18-ftsL) and pUT18C-ftsL30-121 (cyaT18-ftsL30-121) plasmids were generated by ligating PCR-amplified ftsL and ftsL30-121 into pUT18C (cyaT18) digested with BamHI and EcoRI, respectively. Construction of pUT18C-ftsW (cyaT18-ftsW) and pKT25-ftsW (cyaT25-ftsW) involved PCR amplification of E. coli ftsW ORF and digestion of the fragments with BamHI and KpnI, followed by ligation into the BACTH vectors digested with the same enzymes. pKT25-ftsI (cyaT25-ftsI) was created by similar procedures, but BamHI and EcoRI were used for digestion of PCR fragment and vector. For construction of pKT25-ftsQ (cyaT25-ftsQ), the ftsQ ORF was PCR amplified, digested with XbaI and EcoRI, and ligated into pKT25 cut with the same enzymes. All primers are available on request.
Random and site-directed mutagenesis.
To obtain the ftsL and ftsB mutant libraries (with a single missense mutation per ORF) an optimal mutation rate (0.3 to 1 base/kb) for 1 μg of template was adopted as recommended in the GeneMorph II random mutagenesis kit (Agilent Technologies). The PCR products were then digested with EcoRI and HindIII and ligated into the pJF118EH vector using the same restriction enzymes. A ligation pool of pJF118EH-ftsL or pJF118EH-ftsB containing putative mutations was transformed into JS238 by electroporation, and transformants were selected on LB plates containing ampicillin (100 μg/ml) at 37°C. A dominant negative phenotype was screened for by screening sensitivity to IPTG. Specific point mutations in ftsL, ftsL30-121, and ftsW were introduced into some plasmids by using the QuikChange site-directed mutagenesis kit according to the manufacturer’s instructions (Agilent Technologies).
Isolation of an allele of ftsW that bypasses ftsN.
To generate a library of random ftsW mutations, ftsW was subjected to random PCR mutagenesis and cloned into plasmid pSEB429 (P204::ftsW) to replace the WT ftsW. The mutagenized library (pSEB429M) was transformed into strain SD399 [ftsL::kan/pSD256 (repAts::ftsL)] harboring plasmid pSD296-E87K (Para::ftsLE87K), and suppressors of FtsLE87K were selected on LB plates with 0.2% arabinose (to induce ftsLE87K) and 60 μM IPTG (to induce ftsW) at 37°C. Fourteen of the surviving clones were purified, retested, and sequenced. Eleven contained a single mutation (E289G), while 3 contained this mutation plus other mutations. The ftsWE289G mutation was introduced into S3 (W3110, leu::Tn10) by recombineering. P1 transduction of ftsN::kan from strain CH34/pMG20 (ftsN::kan/Para::SStorA-bfp ftsN71-105) into SD488 (leu::Tn10, ftsWE289G) was done using a standard procedure. The KanR transductants had a slightly longer phenotype than a WT strain.
Helix modeling of the FtsL periplasmic domain.
A secondary structure of FtsL was generated for illustrative purposes. To do this, a crude model of the putative coiled coil region of FtsL was modeled on the coiled coil structure (tropomyosin, 1IC2). Structures were visualized using PyMOL (Molecular Graphics System version 1.2r3pre; Schrödinger, LLC).
Bacterial two-hybrid analysis.
The cya null strain DHM1 [F-, cya-854, recA1, endA1, gyrA96 (Nalr), thi1, hsdR17, spoT1, rfbD1, glnV44(AS)] was simultaneously transformed with plasmids pKT25-ftsW or pKT25-ftsI and pUT18C-ftsL (or-ftsL30-121), carrying wild-type or mutant ftsW and ftsL alleles, and grown overnight at 30°C on LB plates containing 0.2% glucose, 25 μg/ml kanamycin, and 100 μg/ml ampicillin. Colonies from the LB plates were diluted in 300 μl volume of LB broth and spotted onto fresh LB plates supplemented with 25 μg/ml kanamycin, 100 μg/ml ampicillin, 40 μg/ml 5-bromo-4-chloro-3-indoyl-β-d-galactopyranoside (X-Gal), and 0.5 mM IPTG. The color changes were recorded after overnight incubation at room temperature at 30°C.
Microscopy.
The dominant negative effects of the FtsL mutants on cell division were assessed using phase-contrast microscopy by monitoring the degree of filamentation. JS238 containing pKTP100 or derivatives carrying ftsL mutations was grown overnight at 30°C in the presence of 100 μg/ml ampicillin and 0.2% glucose. The cultures were diluted 1/200 to 1/500 in fresh LB medium containing 100 μg/ml ampicillin at 30°C. At an optical density at 540 nm (OD540) of ∼0.02, 50 μM IPTG was added, and cell morphologies were analyzed 2 h later.
To visualize GFP-FtsI localization, SD285 (leu::Tn10 bla lacIq P207‐gfp‐ftsI) containing pKTP106 (Para::ftsL) or derivatives with the ftsLE87K or ftsLA90E mutations was grown overnight at 30°C in LB medium containing 50 μg/ml ampicillin and 10 μg/ml chloramphenicol. The overnight cultures were diluted 1/200 to ∼1/500 in fresh LB medium containing the same antibiotics, 0.2% arabinose, and 10 to 20 μM IPTG and were incubated at 37°C until the OD540 was ∼0.4. Cells were immobilized on an LB agarose pad, and the localization of GFP-FtsI was recorded using a cooled charge-coupled-device (CCD) camera and processed using Metamorph (Molecular Devices) and Adobe Photoshop.
ACKNOWLEDGMENTS
We thank Piet de Boer and David Weiss for strains and plasmids and Scott Lovell for generating the model of the FtsL alpha helix. This study was supported by NIH grant GM29746 to J.L.
K.-T.P. and J.L. designed the research; K.-T.P. and S.D. performed the research; K.-T.P., S.D., and J.L. analyzed data and wrote the manuscript.
Footnotes
This article is a direct contribution from Joseph Lutkenhaus, a Fellow of the American Academy of Microbiology, who arranged for and secured reviews by Piet de Boer, Case Western Reserve University School of Medicine, and William Margolin, McGovern Medical School.
Citation Park K-T, Du S, Lutkenhaus J. 2020. Essential role for FtsL in activation of septal peptidoglycan synthesis. mBio 11:e03012-20. https://doi.org/10.1128/mBio.03012-20.
REFERENCES
- 1.de Boer PA. 2010. Advances in understanding E. coli cell fission. Curr Opin Microbiol 13:730–737. doi: 10.1016/j.mib.2010.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Du S, Lutkenhaus J. 2017. Assembly and activation of the Escherichia coli divisome. Mol Microbiol 105:177–187. doi: 10.1111/mmi.13696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Goehring NW, Gonzalez MD, Beckwith J. 2006. Premature targeting of cell division proteins to midcell reveals hierarchies of protein interactions involved in divisome assembly. Mol Microbiol 61:33–45. doi: 10.1111/j.1365-2958.2006.05206.x. [DOI] [PubMed] [Google Scholar]
- 4.Yang X, Lyu Z, Miguel A, McQuillen R, Huang KC, Xiao J. 2017. GTPase activity-coupled treadmilling of the bacterial tubulin FtsZ organizes septal cell wall synthesis. Science 355:744–747. doi: 10.1126/science.aak9995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Spratt BG. 1975. Distinct penicillin binding proteins involved in the division, elongation, and shape of Escherichia coli K12. Proc Natl Acad Sci U S A 72:2999–3003. doi: 10.1073/pnas.72.8.2999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Meeske AJ, Riley EP, Robins WP, Uehara T, Mekalanos JJ, Kahne D, Walker S, Kruse AC, Bernhardt TG, Rudner DZ. 2016. SEDS proteins are a widespread family of bacterial cell wall polymerases. Nature 537:634–638. doi: 10.1038/nature19331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cho H, Wivagg CN, Kapoor M, Barry Z, Rohs PDA, Suh H, Marto JA, Garner EC, Bernhardt TG. 2016. Bacterial cell wall biogenesis is mediated by SEDS and PBP polymerase families functioning semi-autonomously. Nat Microbiol 1:16172. doi: 10.1038/nmicrobiol.2016.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Taguchi A, Welsh MA, Marmont LS, Lee W, Sjodt M, Kruse AC, Kahne D, Bernhardt TG, Walker S. 2019. FtsW is a peptidoglycan polymerase that is functional only in complex with its cognate penicillin-binding protein. Nat Microbiol 4:587–594. doi: 10.1038/s41564-018-0345-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Addinall SG, Cao C, Lutkenhaus J. 1997. FtsN, a late recruit to the septum in Escherichia coli. Mol Microbiol 25:303–309. doi: 10.1046/j.1365-2958.1997.4641833.x. [DOI] [PubMed] [Google Scholar]
- 10.Liu B, Persons L, Lee L, de Boer PA. 2015. Roles for both FtsA and the FtsBLQ subcomplex in FtsN-stimulated cell constriction in Escherichia coli. Mol Microbiol 95:945–970. doi: 10.1111/mmi.12906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tsang MJ, Bernhardt TG. 2015. A role for the FtsQLB complex in cytokinetic ring activation revealed by an ftsL allele that accelerates division. Mol Microbiol 95:925–944. doi: 10.1111/mmi.12905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Du S, Pichoff S, Lutkenhaus J. 2016. FtsEX acts on FtsA to regulate divisome assembly and activity. Proc Natl Acad Sci U S A 113:E5052–E5061. doi: 10.1073/pnas.1606656113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gonzalez MD, Akbay EA, Boyd D, Beckwith J. 2010. Multiple interaction domains in FtsL, a protein component of the widely conserved bacterial FtsLBQ cell division complex. J Bacteriol 192:2757–2768. doi: 10.1128/JB.01609-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Robichon C, Karimova G, Beckwith J, Ladant D. 2011. Role of leucine zipper motifs in association of the Escherichia coli cell division proteins FtsL and FtsB. J Bacteriol 193:4988–4992. doi: 10.1128/JB.00324-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Condon SGF, Mahbuba D-A, Armstrong CR, Diaz-Vazquez G, Craven SJ, LaPointe LM, Khadria AS, Chadda R, Crooks JA, Rangarajan N, Weibel DB, Hoskins AA, Robertson JL, Cui Q, Senes A. 2018. The FtsLB subcomplex of the bacterial divisome is a tetramer with an uninterrupted FtsL helix linking the transmembrane and periplasmic regions. J Biol Chem 293:1623–1641. doi: 10.1074/jbc.RA117.000426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Khadria AS, Senes A. 2013. The transmembrane domains of the bacterial cell division proteins FtsB and FtsL form a stable high-order oligomer. Biochemistry 52:7542–7550. doi: 10.1021/bi4009837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Buddelmeijer N, Beckwith J. 2004. A complex of the Escherichia coli cell division proteins FtsL, FtsB and FtsQ forms independently of its localization to the septal region. Mol Microbiol 52:1315–1327. doi: 10.1111/j.1365-2958.2004.04044.x. [DOI] [PubMed] [Google Scholar]
- 18.Gonzalez MD, Beckwith J. 2009. Divisome under construction: distinct domains of the small membrane protein FtsB are necessary for interaction with multiple cell division proteins. J Bacteriol 191:2815–2825. doi: 10.1128/JB.01597-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kureisaite-Ciziene D, Varadajan A, McLaughlin SH, Glas M, Montón Silva A, Luirink R, Mueller C, den Blaauwen T, Grossmann TN, Luirink J, Löwe J. 2018. Structural analysis of the interaction between the bacterial cell division proteins FtsQ and FtsB. mBio 9:e01346-18. doi: 10.1128/mBio.01346-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Choi Y, Kim J, Yoon H-J, Jin KS, Ryu S, Lee HH. 2018. Structural insights into the FtsQ/FtsB/FtsL complex, a key component of the divisome. Sci Rep 8:18061. doi: 10.1038/s41598-018-36001-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pichoff S, Du S, Lutkenhaus J. 2015. The bypass of ZipA by overexpression of FtsN requires a previously unknown conserved FtsN motif essential for FtsA-FtsN interaction supporting a model in which FtsA monomers recruit late cell division proteins to the Z ring. Mol Microbiol 95:971–987. doi: 10.1111/mmi.12907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Busiek KK, Eraso JM, Wang Y, Margolin W. 2012. The early divisome protein FtsA interacts directly through its 1c subdomain with the cytoplasmic domain of the late divisome protein FtsN. J Bacteriol 194:1989–2000. doi: 10.1128/JB.06683-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mercer KL, Weiss DS. 2002. The Escherichia coli cell division protein FtsW is required to recruit its cognate transpeptidase, FtsI (PBP3), to the division site. J Bacteriol 184:904–912. doi: 10.1128/jb.184.4.904-912.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang X, McQuillen R, Lyv Z, Phillips-Mason P, Cruz A, McCausland J, Liang H, DeMeester K, Grimes C, Boer P, Xiao J. 2019. FtsW exhibits distinct processive movements driven by either septal cell wall synthesis or FtsZ treadmilling in E. coli. bioRxiv doi: 10.1101/850073. [DOI]
- 25.Sjodt M, Rohs PDA, Gilman MSA, Erlandson SC, Zheng S, Green AG, Brock KP, Taguchi A, Kahne D, Walker S, Marks DS, Rudner DZ, Bernhardt TG, Kruse AC. 2020. Structural coordination of polymerization and crosslinking by a SEDS-bPBP peptidoglycan synthase complex. Nat Microbiol 5:813–820. doi: 10.1038/s41564-020-0687-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wissel MC, Weiss DS. 2004. Genetic analysis of the cell division protein FtsI (PBP3): amino acid substitutions that impair septal localization of FtsI and recruitment of FtsN. J Bacteriol 186:490–502. doi: 10.1128/jb.186.2.490-502.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guzman LM, Weiss DS, Beckwith J. 1997. Domain-swapping analysis of FtsI, FtsL, and FtsQ, bitopic membrane proteins essential for cell division in Escherichia coli. J Bacteriol 179:5094–5103. doi: 10.1128/jb.179.16.5094-5103.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dubarry N, Barre FX. 2010. Fully efficient chromosome dimer resolution in Escherichia coli cells lacking the integral membrane domain of FtsK. EMBO J 29:597–605. doi: 10.1038/emboj.2009.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.LaPointe LM, Taylor KC, Subramaniam S, Khadria A, Rayment I, Senes A. 2013. Structural organization of FtsB, a transmembrane protein of the bacterial divisome. Biochemistry 52:2574–2585. doi: 10.1021/bi400222r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Marmont LS, Bernhardt TG. 2020. A conserved subcomplex within the bacterial cytokinetic ring activates cell wall synthesis by the FtsW-FtsI synthase. Proc Natl Acad Sci U S A 117:23879–23885. doi: 10.1073/pnas.2004598117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Boes A, Olatunji S, Breukink E, Terrak M. 2019. Regulation of the peptidoglycan polymerase activity of PBP1b by antagonist actions of the core divisome proteins FtsBLQ and FtsN. mBio 10:e01912-18. doi: 10.1128/mBio.01912-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Contreras-Martel C, Martins A, Ecobichon C, Trindade DM, Matteï P-J, Hicham S, Hardouin P, Ghachi ME, Boneca IG, Dessen A. 2017. Molecular architecture of the PBP2-MreC core bacterial cell wall synthesis complex. Nat Commun 8:776. doi: 10.1038/s41467-017-00783-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rohs PDA, Buss J, Sim SI, Squyres GR, Srisuknimit V, Smith M, Cho H, Sjodt M, Kruse AC, Garner EC, Walker S, Kahne DE, Bernhardt TG. 2018. A central role for PBP2 in the activation of peptidoglycan polymerization by the bacterial cell elongation machinery. PLoS Genet 14:e1007726. doi: 10.1371/journal.pgen.1007726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dubarry N, Possoz C, Barre FX. 2010. Multiple regions along the Escherichia coli FtsK protein are implicated in cell division. Mol Microbiol 78:1088–1100. doi: 10.1111/j.1365-2958.2010.07412.x. [DOI] [PubMed] [Google Scholar]
- 35.Karimova G, Pidoux J, Ullmann A, Ladant D. 1998. A bacterial two-hybrid system based on a reconstituted signal transduction pathway. Proc Natl Acad Sci U S A 95:5752–5756. doi: 10.1073/pnas.95.10.5752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gerding MA, Liu B, Bendezú FO, Hale CA, Bernhardt TG, de Boer PAJ. 2009. Self-enhanced accumulation of FtsN at division sites and roles for other proteins with a SPOR domain (DamX, DedD, and RlpA) in Escherichia coli cell constriction. J Bacteriol 191:7383–7401. doi: 10.1128/JB.00811-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Characterization of dominant negative mutations in ftsL. (A) Dominant negative mutations do not complement an ΔftsL strain. Two dominant negative alleles of ftsL were tested for their ability to complement an ftsL depletion strain by transforming SD439 (ftsL::kan)/pSD296 (Para::ftsL) with pKTP100 (Ptac::ftsL) derivatives carrying different ftsL alleles. The toxicity of these alleles was tested on plates containing both arabinose (induced WT ftsL) and IPTG (to induce the allele to be tested) (center panel). The ability to complement the ftsL depletion strain was assessed on plates without arabinose or IPTG (basal level of expression from pKTP100 (Ptac::ftsL) is sufficient for complementation) (right panel). (B) Lineup of a region (amino acids 57 to 110) of the periplasmic domain of FtsL. FtsLs from a variety of Gram-negative and Gram-positive bacteria were aligned using Cobalt. Residues in red are more conserved than residues in blue, which are more conserved than those in gray. E87 is indicated by the arrow. (C) Examination of additional substitutions at position 87 in FtsL on toxicity. Derivatives of pKT100 (Ptac::ftsL) carrying various alleles of ftsL were introduced into JS238 and tested for toxicity by performing spot tests on plates containing increasing amounts of IPTG. (D) Effect of the dominant negative ftsL mutations on the recruitment of GFP-FtsI. SD285 (leu::Tn10 P206::gfp-ftsI) was transformed with pSD296 (Para::ftsL) derivatives expressing ftsLE87K, ftsLA90E, or ftsL. Three hours after induction with 0.2% arabinose and 25 mM IPTG, samples were taken and examined by phase and fluorescent microscopy. Download FIG S1, TIF file, 9.03 MB (9MB, tif) .
Copyright © 2020 Park et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Test for rescue of dominant negative ftsL mutations by an ftsL activation mutation or by ftsN overexpression. (A) Rescue by an ftsL activation mutation in cis. The ftsLE88K mutation was added to various ftsL alleles in cis and then tested for complementation. To do this, SD439 (ftsL::kan/pSD296 [Para::ftsL]) was transformed with derivatives of pKTP100 (Ptac::ftsL) carrying the various mutations. The strains were spotted on plates at 30°C without arabinose to deplete WT ftsL, and IPTG was added to induce the various ftsL alleles. ftsLA90E and ftsLR61E were rescued, but ftsLE87K and ftsLL86F (Table 1) were not. (B) Rescue by overexpression of ftsN. To test if ftsN overexpression can rescue any of the dominant negative alleles of ftsL, SD399 (ftsL::kan/pSD256 [repATS Psyn135::ftsL]) containing plasmids expressing the ftsL alleles under an arabinose inducible promoter (derivatives of pSD296 [Para::ftsL]) was transformed with a plasmid with ftsN under IPTG control (pSEB417 [P204::ftsN]). The strains were spotted at 37°C to deplete WT ftsL, and arabinose was added to induce the ftsL allele, and IPTG added to induce ftsN. Download FIG S2, TIF file, 10.6 MB (10.8MB, tif) .
Copyright © 2020 Park et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Effect of activation mutations in ftsW and ftsB as well as ftsN overexpression on the rescue of dominant negative ftsL mutations. (A) Overexpression of an activation allele of ftsW, but not ftsN, suppresses a dominant negative allele of ftsL (ftsLL86F/E87K). SD399 (ftsL::kan/pSD256 [repAts-ftsL]) was transformed with plasmids expressing either ftsL or a dominant negative allele of ftsL (ftsLE87K/L86F) under an arabinose-inducible promoter (derivatives of pSD296 [Para::ftsL]) and plasmids expressing ftsWM269I or ftsN under an IPTG-inducible promoter (pSEB429-I [P204::ftsWM269I] and pSEB417 [P204::ftsN], respectively). The strains were spotted on plates at 37°C (to deplete WT ftsL) containing 0.2% arabinose to induce the ftsL alleles, with increasing concentrations of IPTG to induce ftsWM269I or ftsN. The second panel lacks arabinose, demonstrating that the ftsW activation mutations cannot bypass ftsL. (B) An activated allele of ftsB cannot suppress a dominant negative ftsL mutation. Strain PK168-1 (ftsBE56A recA::aadA ftsL::kan/pSD296 [Para::ftsL]) containing pKTP100 (Ptac::ftsL) or a version expressing ftsLE87K was subjected to spot tests on plates without arabinose to deplete WT ftsL and with IPTG added to induce the ftsL allele cloned in derivatives of pKTP100. Download FIG S3, TIF file, 10.6 MB (10.8MB, tif) .
Copyright © 2020 Park et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Effect of activation mutations in ftsW on complementation and rescue of a dominant negative ftsL allele. (A) Test of ftsW alleles for complementation of an ftsW depletion strain in the absence of IPTG. To test if the addition of IPTG was required for complementation of an ftsW depletion strain, EC912 (ftsW::kan/pDSW406 [Para::ftsW]) was transformed with derivatives of pSEB429 (P204::ftsW) carrying various alleles of ftsW. The strains were spot tested on plates without arabinose and with or without 200 μM IPTG. (B) Expression of ftsLE87K inhibits a strain with the ftsWM269I mutation on the chromosome. Strains KTP1 (ftsW+) and SD247-1 (ftsWM269I) were transformed with pKTP100 (Ptac::ftsL) or a derivative expressing ftsLE87K. The strains were spot tested on plates containing increasing concentrations of IPTG to test for toxicity. The strain containing the ftsWM269I is more resistant but still sensitive to ftsLE87K. Download FIG S4, TIF file, 10.6 MB (10.8MB, tif) .
Copyright © 2020 Park et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Mutations in the cytoplasmic domain of ftsL result in a dominant negative phenotype and fail to recruit GFP-FtsI. (A) Mutations (L24K/I28K) in the cytoftsL domain lead to a dominant negative phenotype. JS238 containing pKTP100 (Ptac::ftsL) derivatives expressing different alleles of ftsL were tested for toxicity following induction with IPTG. (B) Deletion of the cytoplasmic domain of FtsL (ftsLΔ1-30) results in a dominant negative allele. JS238 containing pKTP104 (PT5::ftsL) or pKTP105 (PT5::ftsLΔ1-30) was tested for toxicity following induction with IPTG. (C) FtsLL24K/I28K fails to recruit GFP-FtsI. SD285 (leu::Tn10 P206::gfp-ftsI) was transformed with pSD296 (Para::ftsLL24/I28K). Three hours after induction with 0.2% arabinose and 25 mM IPTG, samples were taken and examined by phase and fluorescent microscopy. Download FIG S5, TIF file, 10.6 MB (10.8MB, tif) .
Copyright © 2020 Park et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Rescue of ftsL cytoplasmic mutations by ftsL activation mutations and ftsN overexpression. (A) Rescue of ftsLΔ1-30 requires two ftsL activation mutations. SD439 (ftsL::kan/pSD296 [Para::ftsL]) carrying pKTP105 (PT5::ftsLΔ1-30) derivatives expressing various alleles of ftsL were tested for their ability to complement an ftsL depletion strain. Complementation was tested by spotting strains on plates without arabinose (to deplete WT ftsL) and with IPTG to induce the various ftsL alleles. Loss of a functional cytoFtsL domain (and therefore the ability to recruit FtsW) is compensated for by the two activation mutations ftsLE88K/G92D. The cartoon depicts the defect caused by ftsLΔ1-30 and its rescue by the activation mutations. (B) Morphology of ΔftsL cells rescued by ftsLΔ1-30 carrying activation mutations. SD399 (ftsL::kan/pSD256 [Psyn135::ftsL]) carrying derivatives of pKTP107 [Para::ftsL] expressing various alleles of ftsLΔ1-30 were grown to the exponential phase at 30°C with antibiotics, centrifuged, washed, and resuspended in LB with 0.2% arabinose and antibiotics at 37°C. Samples were taken 2.5 h later for photography, and the cell length distributions were determined. (C) ftsL activation mutations reduce the toxicity of ftsLL24K/I28K. JS238 carrying derivatives of pKTP100 (Ptac::ftsL) expressing different alleles of ftsL was tested for toxicity by spotting on plates containing 200 μM IPTG to induce the ftsL alleles. (D) ftsL activation mutations rescue ftsLL24K/I28K for complementation. SD439 (ftsL::kan/pSD296 [Para::ftsL]) containing derivatives of pKTP100 [Ptac::ftsL] with various ftsL alleles was spotted on plates without arabinose (to deplete WT ftsL) and with IPTG to induce the mutant ftsL alleles. (E) Overexpression of ftsN suppresses ftsLΔ1-30. SD439 (ftsL::kan/pSD296 [Para::ftsL]) containing derivatives of pKTP105 (PT5::ftsLΔ1-30) with various alleles of ftsL was transformed with pBL154 (repATS Psyn135::ftsN) which constitutively expresses ftsN. Rescue of FtsLΔ1-30 was tested by spotting transformants on plates at 30°C without arabinose and with IPTG. The cartoon depicts that FtsN overexpression rescues FtsLΔcyto. (F) ftsL mutations do not affect interaction with FtsQ. To ensure that the ftsL mutations did not affect the stability of FtsL, we tested their effect on the FtsL-FtsQ interaction in the BACTH system. Strain DHM1 was transformed with plasmids carrying various alleles of ftsL (pUT18C derivatives) and a plasmid expressing ftsQ (pKT25-ftsQ). Three transformants were picked for each pair of constructs and spot tested. Column 1, FtsL; column 2, vector; column 3, FtsLΔcyto; column 4, FtsLΔcyto/E88K/G92D; column 5, FtsLΔcyto/E88K/G92D/E87K. (G) Interaction between FtsL and FtsWI assessed with the BACTH system. This test is similar to the test in Fig. 7 except that FtsLΔcyto was replaced with FtsLL24K/I28K. The ftsL alleles were contained in pUT18C, and the ftsW and ftsI alleles were in pKT25. The plates were photographed after overnight incubation. Column 1, FtsL versus FtsI; column 2, vector versus FtsI; column 3, FtsLL24K/I28K versus FtsW; column 4, FtsLL24K/I28K versus FtsWM269I; column 5, FtsLL24K/I28K versus FtsI; column 6, FtsLL24K/I28K/E88K/G92D versus FtsW; column 7, FtsLL24K/I28K/E88K/G92D versus FtsWM269I; and column 8, FtsLL24K/I28K/E88K/G92D versus FtsI. Download FIG S6, TIF file, 10.6 MB (10.8MB, tif) .
Copyright © 2020 Park et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Diagram indicating the position AWI of FtsL relative to the membrane and the RodA-PBP2 complex. Part of the periplasmic domain of FtsL was modeled as an alpha helix (residues 57 to 99) and positioned next to the structure of the RodA-PBP2 complex (PDB ID: 6PL6). The RodA-PBP2 complex is homologous to the FtsW-PBP3 (FtsI) complex. The non-penicillin binding domain (nPB) and penicillin binding domain (PD) are indicated. Key FtsL residues (E87 [AWI], E88 [CCD]) are about 4.3 nm from the membrane. The position of R61 is also indicated. Download FIG S7, TIF file, 10.6 MB (10.8MB, tif) .
Copyright © 2020 Park et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Testing the rescue of additional alleles of ftsI by an activated FtsW mutant. Additional ftsI alleles were tested to see if they were rescued by ftsWM269I expression. Some alleles were tested in Fig. 6A, and the others are tested here. Note that among the alleles tested here, only ftsIS61F is rescued. The bottom row of panels is also presented in Fig. 6A and was included here for comparison. MCI23 (ftsI23ts recA::aadA) was transformed with compatible plasmids expressing an activated allele of ftsW (pSEB429 [P204::ftsWM269I]) and the various ftsI alleles under arabinose promoter control (derivatives of pBAD33-ftsI). Transformants were spot tested on plates at 42°C (to inactivate ftsI23Ts), and arabinose was added to induce the ftsI alleles and increasing concentrations of IPTG to induce ftsWM269I. Download FIG S8, TIF file, 10.6 MB (10.8MB, tif) .
Copyright © 2020 Park et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Characterization of FtsL fusions. (A) The MalF-FtsL fusion lacks toxicity and fails to complement an ftsL depletion strain. The left panel tests for complementation, and the right panel tests for toxicity. SD439 (ftsL::kan/pSD296 [Para::ftsL]) containing pKTP100 (Ptac::ftsL) derivatives expressing various alleles of ftsL were incubated without arabinose or IPTG (basal expression from pKTP100 [Ptac::ftsL] is sufficient for complementation) to test for complementation. In the right panel, both arabinose and IPTG were added. Arabinose induces WT ftsL from pSD296 (Para::ftsL), whereas IPTG induces the ftsL allele from pKTP100 (Ptac::ftsL). The ftsLL24K/I28K allele was added as a control to demonstrate toxicity. The lack of toxicity indicates that the MalF-FtsL fusion fails to form a complex with FtsQB. (B) FtsL fusions are unable to complement an ftsL-depleted strain. Fusion of the periplasmic domain of FtsL to the MalF cytoplasmic and transmembrane domains (with or without ftsL activation mutations) was tested for the ability to complement an ftsL depletion strain. To do this, PK247-4 (ftsWM269I ftsL::kan/pSD296 [Para::ftsL]) containing derivatives of pKTP100 (Ptac::ftsL) carrying various fusions in place of ftsL was spot tested on plates with IPTG. Download FIG S9, TIF file, 10.6 MB (10.8MB, tif) .
Copyright © 2020 Park et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Strains and plasmids used in this study. Download Table S1, DOCX file, 0.04 MB (37.3KB, docx) .
Copyright © 2020 Park et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.